

Abstract
Background:
Two major forms of inherited J wave syndrome (JWS) are recognized: early repolarization syndrome (ERS) and Brugada syndrome (BrS).
Objectives:
We sought to assess the distinct features between ERS and BrS patients carrying pathogenic variants in SCN5A.
Methods:
Clinical evaluation and next-generation sequencing were performed in 262 BrS and 104 ERS probands. Nav1.5 and Kv4.3 channels were studied using patch-clamp techniques. A computational model was employed to investigate the protein structure.
Results:
The SCN5A+ yield in ERS was significantly lower than in BrS (9.62% vs. 22.90%, p=0.004). Patients diagnosed with ERS displayed shorter QRS and QTc than BrS patients. More than 2 pathogenic SCN5A variants were found in five probands. These patients displayed longer PR intervals, QRS duration and experienced more major arrhythmia events (MAE) compared to those carrying only a single pathogenic variant. SCN5A-L1412F, detected in a fever-induced ERS patient, led to total loss-of-function, destabilized the Nav1.5 structure, and showed a dominant-negative effect, which was accentuated during a febrile state. ERS-related SCN5A-G452C did not alter INa when SCN5A was expressed alone, but reduced peak INa by 44.52% and increased Ito by 106.81% when co-expressed with KCND3.
Conclusion:
Our findings point to SCN5A as a major susceptibility gene in ERS as it is in BrS.whereas the lower SCN5A+ ratio in ERS indicates the difference in underlying electrophysiology. We also identify the first case of fever-induced ERS, demonstrate a critical role of Ito in JWS and a higher risk for MAE in JWS probands carrying multiple pathogenic variants in SCN5A.
Keywords: J wave syndrome, Genetics, Sodium channel, Sudden Cardiac Death, Risk stratification
Condensed Abstract:
We compared clinical and electrophysiologic characteristics of 104 ERS and 262 BrS probands carrying pathogenic variants in SCN5A. Ten new variants were uncovered in ERS and two in BrS. The yield of SCN5A variants in ERS is significantly lower than in BrS. ERS patients displayed shorter QRS and QTc. Five probands with multiple SCN5A pathogenic variants displayed longer PR interval, QRS duration, and experienced more major arrhythmia events. Our findings point to SCN5A+ as a major susceptibility gene in ERS as in BrS, identify the first fever-induced ERS case, and demonstrate a critical role for Ito in JWS.
Introduction
J wave syndromes (JWSs), consisting of the Brugada syndrome (BrS) and early repolarization syndrome (ERS), are associated with vulnerability to the development of polymorphic ventricular tachycardia and ventricular fibrillation (VT/VF) leading to sudden cardiac death (SCD)(1,2). BrS is characterized by the appearance of a coved-type ST-segment elevation usually limited to the right precordial ECG leads, whereas ERS is characterized by an early repolarization pattern (ERP) in the ECG, consisting of a distinct J-wave or J-point elevation, or a notch or slur of the terminal part of the QRS of in 2 or more contiguous inferolateral ECG leads.
The appearance of J waves on the ECG is due to the presence of a prominent Itomediated action potential notch in the ventricular epicardium, but not endocardium causing a transmural voltage gradient during early ventricular repolarization. Augmentation of net repolarization current, due either to an increase of outward currents (Ito, IK-ATP) or decrease of inward current (ICa, INa), accentuates the electrocardiographic manifestation of the J wave(3,4). Meanwhile, it is notable that delayed depolarization also plays a key role in the pathogenesis of JWS(5,6). The SCN5A gene encodes the α subunit of the voltage-gated Nav1.5 cardiac sodium channel. Loss-of-function in SCN5A has been associated with the development of JWS. Since first identified in BrS in 1998, SCN5A accounts for 20–30% of BrS probands, and more than 350 pathogenic variants have been reported(7). While the relationship between SCN5A and ERS has been known for nearly a decade(8,9), far fewer disease-causing pathogenic variants are recognized, and the percentage of ERS patients with SCN5A genetic background is not clear. In the present study, we seek to compare the prevalence of pathogenic variants in SCN5A in ERS and BrS patients and characterize their respective phenotypes.
Methods
See additional details in Supplemental Materials.
Study population and enrollment criteria
A total of 366 JWS probands, including 262 BrS and 104 ERS cases, were enrolled in the study. BrS was diagnosed if the value in the Shanghai BrS scoring system was ≥3.5. The diagnosis of ERS was made base on a value of ≥3.0 in the Shanghai ERS scoring system(1,10). ERS was divided into three types: type 1, which exhibited early repolarization pattern (ERP) in only lateral precordial leads; type 2, which displayed ERP in the inferior or inferolateral leads; type 3, which showed ERP globally in the inferior, lateral, and right precordial leads(1,10). We collected the clinical profile of all probands, documenting symptoms including syncope, VT/VF, SCD, and aborted SCD (ASCD), etc. Major arrhythmic events (MAE) were defined as sustained VT, VF, SCD, ASCD, and appropriate shock in patients with ICD. All subjects or their guardians signed informed consent for genetic and clinical evaluation studies. This study was reviewed and approved by the Medical Ethical Committee of Renmin Hospital of Wuhan University and the review board of each participated institution. It was in accordance with the declaration of Helsinki.
Electrocardiographic characteristics
The ECG parameters were measured in lead II, which includes P-wave and QRS duration and PR interval. QT and Tpeak-Tend (Tp-e) were measured in leads II and V5. The corrected QT was calculated by the Bazett’s formula. Tp-e was defined as the interval from the peak of a positive T-wave to the end of T-wave. In the case of a negative or biphasic T wave, Tp-e was measured from the nadir of the T-wave(11). A global J wave was defined as type 1 Brugada ECG (coved type ST-segment elevation) in the right precordial leads plus J waves in two or more inferolateral leads or J waves in the anterior and inferolateral leads.
Genetic analysis
DNA analysis was conducted on genomic DNA extracted from peripheral leukocytes using standard protocols. All exon and intron borders of susceptibility genes for BrS and ERS were analyzed (Supplemental Table 1.). Variants in SCN5A were determined using the splice variant with 2016 amino acids (Pubmed Accession No. NM198056). The protein topology was annotated according to the Swissprot database (http://ca.expasy.org/uniprot/). In order to be classified as pathogenic, a variant was required to meet the criteria specified in Supplemental Methods in accordance with ACMG guidelines using the VarSome classifier.
Site-Directed Mutagenesis and Transfection of the Cell
Site-directed mutagenesis was performed on full-length human wild-type (WT) and mutant SCN5A cDNA cloned in pCDNA3.1 vector, as well as WT-SCN1B. For the patch-clamp study, they were transfected into HEK293 cells using Fugene6. In addition, CD8 cDNA was co-transfected as a reporter gene to visually identify transfected cells using Dynabeads. In some experiments, pCDNA3.1-GFP-SCN5A and pCDNA3.1-KCND3 were co-transfected. Cells were placed in a 5% CO2 incubator at 37°C for 24 to 48 hours prior to the study.
Patch Clamp Method
Membrane currents were measured using whole-cell patch-clamp techniques. All recordings were obtained at room temperature using an Axopatch 200B amplifier equipped with a CV-201A head stage. The holding potential for INa was maintained at −120 mV. Patch pipettes were fabricated from borosilicate glass capillaries (1.5 mm O.D., Fisher Scientific, Pittsburgh, PA). They were pulled using a gravity puller (Model PP-89, Narishige Corp, Japan) to obtain a resistance between 0.8 – 2.8 MΩ when filled with the solution. Currents were filtered with a four-pole Bessel filter at 5 kHz and digitized at 50 kHz. Series resistance was electronically compensated at 70–85%. In some experiments, temperature was increased to 38°C from room temperature (22°C). All data acquisition and analysis were performed using pCLAMP V10, EXCEL, and ORIGIN 7.5.
Computational modeling
Many disease-associated variants cause amino acid substitutions causing a significant change in the thermodynamic stability (ΔΔG) of the protein structure. To investigate the effect of SCN5A-L1412F on protein structure, we evaluated the ΔΔG of SCN5A-L1412F relative to the reference (WT) structure with a computational ΔΔG protocol(12), following a procedure previously described(13). Variant-induced ΔΔG was computed as the energy difference between the refined variant structure and the refined reference structure: ΔΔreference → variant = ΔGvariant − ΔGreference,, using the Rosetta macromolecular modeling software suite.
Statistical analysis
Continuous variables were expressed as mean ± standard deviation. Continuous variables with normal distribution were compared using the Student t-test. Categorical variables were presented as absolute values and percentages. The Chi-square test or Fisher’s exact test were used to compare categorical variables. A two-tailed p-value of <0.05 was considered statistically significant. Statistical analyses were conducted using the SPSS version 26.
Results
Clinical Characteristics
We identified SCN5A pathogenic variants in 70 probands (60 BrS and 10 ERS cases) from China, USA, Canada and Japan, including 51 Caucasians, 14 Asians, 3 African Americans, 2 Hispanics, providing an overall yield of 19.13%. The demographics for SCN5A+ probands are summarized in Table 1. Most patients were male (n=51, 72.86%) with a mean age at diagnosis of 36.50±18.74 years. Thirty-eight probands (54.29%) had a history of syncope, and MAE occurred in 22 patients (31.43%). Of all the SCN5A+ probands, 24 (34.29%) presented with a family history of unexplained SCD.
Table 1.
Clinical characteristics and demographics of probands with JWS
| BrS | ERS | p-Value | |
|---|---|---|---|
|
| |||
| Total Probands | |||
|
| |||
| Diagnosis NO. of Proband, n | 262 | 104 | |
| Age at diagnosis (years) | 38.26±17.65 | 30.86±14.45 | <0.001 |
| Sex, n (male %) | 197 (75.19%) | 89 (85.58%) | 0.03 |
|
| |||
| Probands with SCN5A pathogenic variants | |||
|
| |||
| No. of patients with SCN5A | 60 | 10 | |
| Age at diagnosis (years) | 36.82±19.17 | 34.00±16.73 | 0.579 |
| Range (years) | 0–72 | 14–64 | |
| Sex, n (male %) | 45 (75.00%) | 6 (60.00%) | 0.443 |
| Symptomatic patients, n (%) | 44 (73.33%) | 7 (70.00%) | 1.000 |
| Patients with syncope, n (%) | 31 (51.67%) | 7 (70.00%) | 0.326 |
| MAE, n (%) | 16 (27.67%) | 6 (60.00%) | 0.061 |
| Atypical symptoms, n (%) | 13 (21.67%) | 0 (0.00%) | 0.190 |
| Family history, n (%) | 22 (36.67%) | 2 (20.00%) | 0.476 |
| fQRS, n (%) | 14 (23.33%) | 3 (30.00%) | 0.718 |
| CCD, n (%) | 22 (36.67%) | 6 (60.00%) | 0.183 |
| Bradycardia, n (%) | 11 (18.33%) | 6 (60.00%) | 0.042 |
| Pore region variants in SCN5A, n (%) | 23 (38.33%) | 3 (30.00%) | 0.734 |
| HR (bpm) | 76.49±18.56 | 65.15±16.20 | 0.106 |
| QRS duration (ms) | 105.49±19.08 | 90.40±19.97 | 0.018 |
| PR interval (ms) | 180.10±31.48 | 197.40±34.69 | 0.121 |
| P wave duration (ms) | 106.18±21.94 | 110.03±13.57 | 0.597 |
| QTc interval II (ms) | 425.13±42.62 | 393.63±40.04 | 0.025 |
| QTc interval V5 (ms) | 427.35±47.24 | 390.47±29.56 | 0.012 |
| Tp-e II (ms) | 92.80±19.10 | 87.40±20.62 | 0.398 |
| Tp-e /QT II | 0.239±0.038 | 0.228±0.044 | 0.412 |
| Tp-e V5 (ms) | 94.95±24.01 | 90.60±16.93 | 0.589 |
| Tp-e /QT V5 | 0.242±0.045 | 0.238±0.039 | 0.816 |
Among SCN5A+ BrS probands, type 1 Brugada ECG was observed spontaneously in 22 patients (36.67%, Figure 1A), occurred after drug challenge in 28 patients (46.66%, Figure 1B) or during fever in 10 patients (16.67%, Figure 1C). Among the drug-induced BrS patients, the MAE ratio was similar among patients with or without global J wave manifestation (28.57% vs.23.81%, p>0.05). However, the occurrence of syncope was significantly higher in patients displaying global J waves (100.00% vs. 52.38%, p<0.05). An inferolateral J wave was identified in 9 BrS patients (15.00%, Figure 1D). Of all SCN5A+ ERS probands, ERS3 ECG was observed in 8 ERS patients (80.00%, Figure 1E), the remaining (20.00%) ERS probands presented with ERS2 ECG (Figure 1F); none of ERS probands presented with an ERS1 ECG. Figure1G–I shows representative pedigrees carrying pathogenic variants in SCN5A. Pedigree G displays a father and son carrying SCN5A-Q646RfsX6. The father presented with ERS, whereas the son presented with a BrS phenotype. In Pedigree H, the twins carrying SCN5A-K249X both exhibited global J waves (ERS3). Unfortunately, their genotype-positive brother was clinically unknown. In Pedigree I, family members with SCN5A-G1408R showed distinct mixed phenotypes among the 10 carriers, including BrS, ERS, cardiac conduction disease (CCD), and bradycardia.
Figure 1. Representative cases of different JWS phenotypes with SCN5A pathogenic variants.
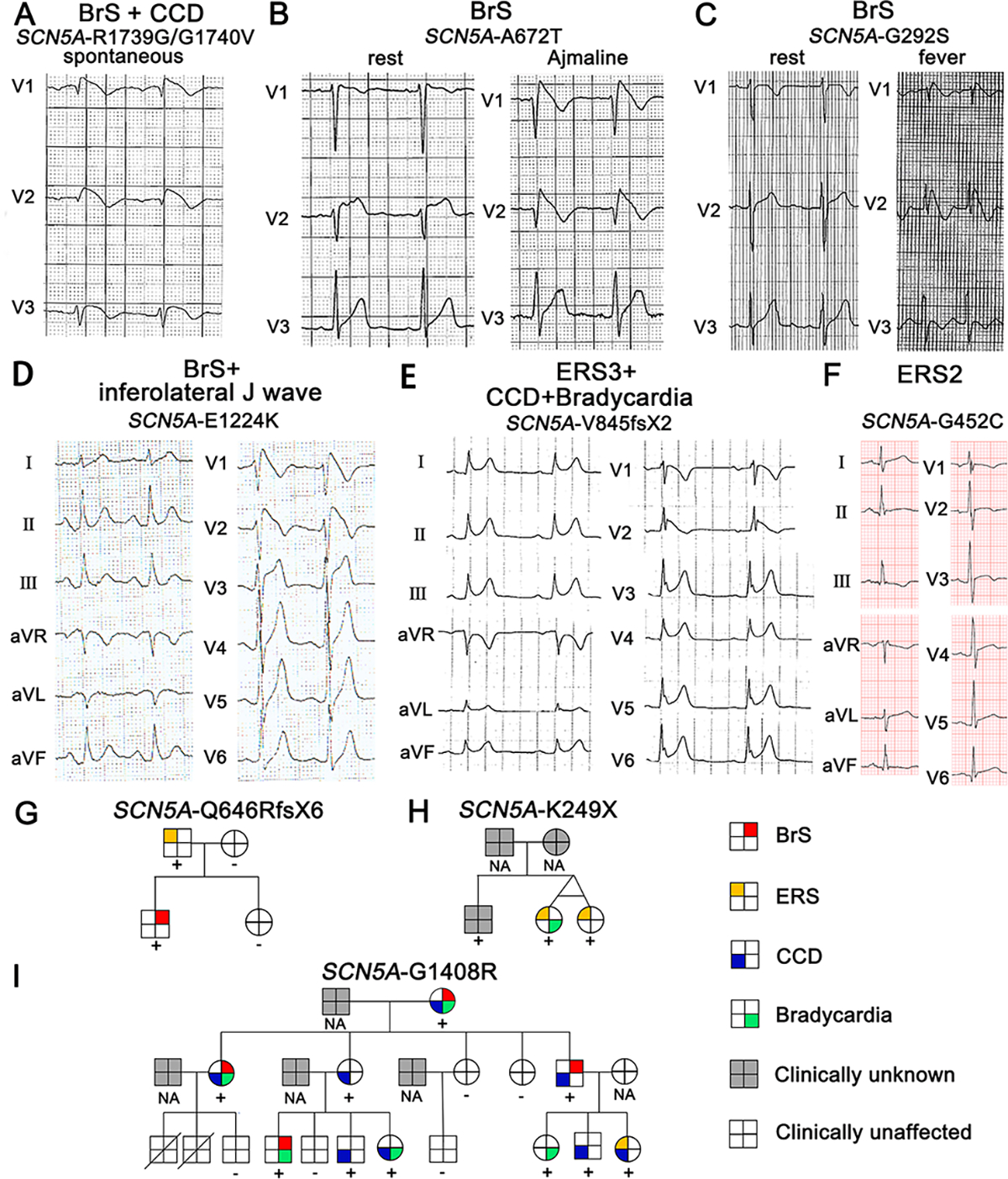
A-F: Each panel shows an ECG of different JWS phenotypes. G-I: Pedigrees of representative families with SCN5A pathogenic variants. For the pedigrees in panels G-I, male and female subjects are denoted by squares and circles; phenotypes presented by the family members are depicted by a red top right square for BrS; yellow top left square for ERS; blue bottom left square for cardiac conduction defects; green bottom right square for bradycardia; gray background for clinically unknown; blank background for clinically unaffected.
Pathogenic variant yield and analysis
In 70 JWS probands, 5 BrS cases carried two SCN5A pathogenic variants. This was not observed in any of the ERS cases. Eight SCN5A variants were found in 2 unrelated probands each (D356N, Q646RfsX6, L1308F, G1408R, L1412F, S1787N, E1784K, P2006A). In total, 67 SCN5A pathogenic variants were identified, including 51 missense ones (76.12%). The remaining were radical pathogenic variants (6 nonsense, 6 frameshift, 2 splice variants, 2 inframe deletion/insertion, Figure 2B). Most pathogenic variants were localized in transmembrane regions (26.86% in S1-S4, 37.31% in S5-S6, Figure 2C).
Figure 2. Clinical and genetic prevalence of SCN5A pathogenic variants in JWS probands.
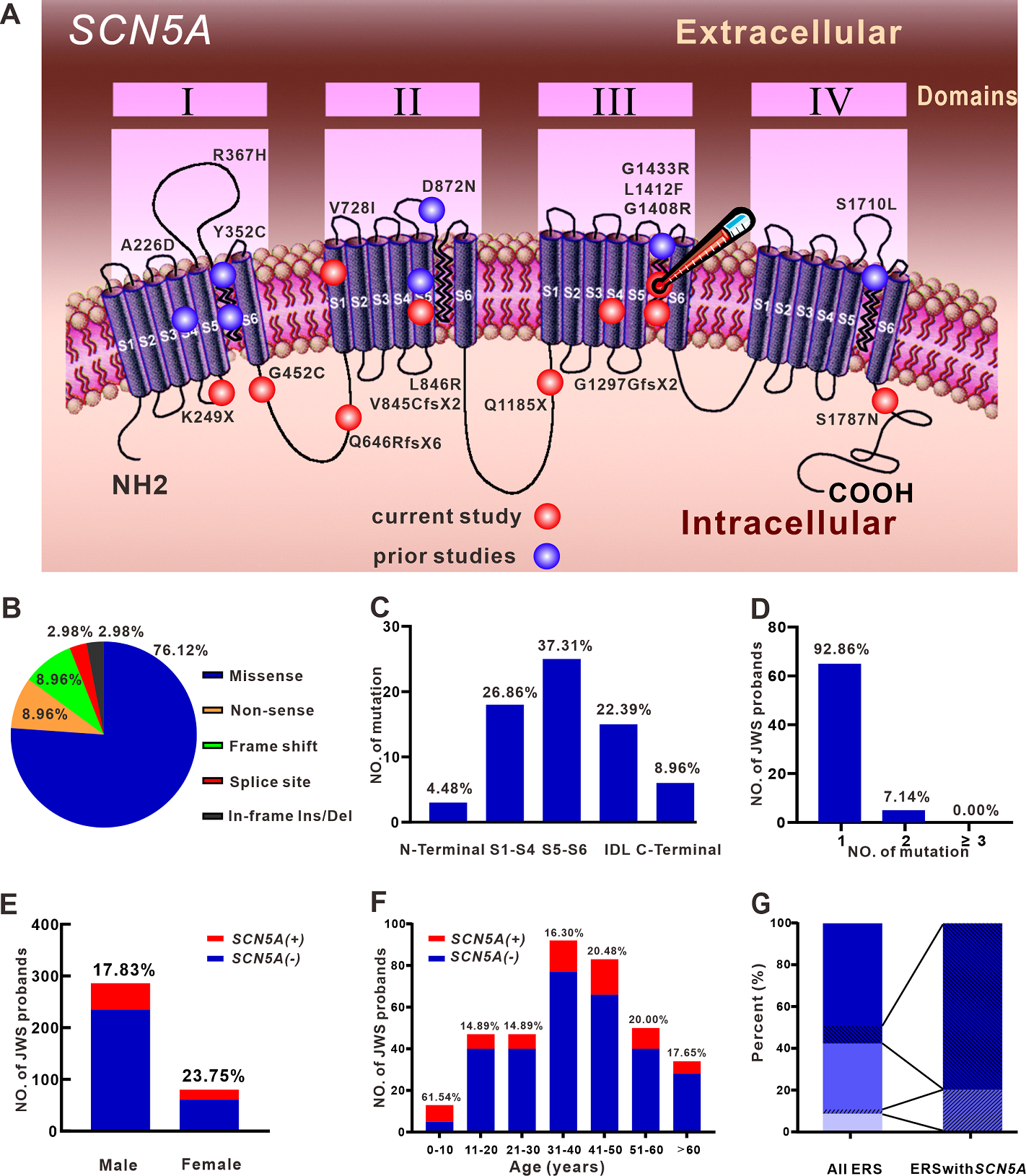
A: Topology of SCN5A and location of pathogenic variants revealed in ERS probands. The red ball delineates pathogenic variants revealed in the current study; those previously reported are delineated by a blue ball. The red ball with thermometer indicates the L1412F variant of the ERS patient unmasked by fever. B & C: Proportion of pathogenic variants in JWS probands by pathogenic variant types and location. D: Percentage of JWS probands by SCN5A+ pathogenic variant number. E & F: Fraction of SCN5A distribution in JWS probands according to sex and age. G: Proportion of ERS types in all ERS probands and ERS probands with SCN5A pathogenic variants. Light blue, medium blue, and dark blue successively represent ERS1, ERS2, and ERS3. SCN5A+ ERS2 cases are presented as lower left diagonal lines in the medium blue bar and SCN5A+ ERS3 cases as lower right diagonal lines in the dark blue bar.
Sixty-five (92.86%) probands carried only a single pathogenic SCN5A variant, whereas the rest carried multiple SCN5A variants (Figure 2D, Table 2). The yield of SCN5A+ was slightly greater in females than in males (Figure 2E). The number and percentage of SCN5A+ JWS probands at different age groups are presented in Figure 2F. The SCN5A+ yield was highest in the group aged ≤10 years; the yield among the other age groups was not significantly different.
Table 2.
JWS probands with multiple pathogenic variants in SCN5A
| NO. | Diagnosis | Sex | Age at diagnosis (years) | First SCN5A variant |
Second SCN5A variant |
||||
|---|---|---|---|---|---|---|---|---|---|
| Exon | Amino acid change | Nucleotide change | Exon | Amino acid change | Nucleotide change | ||||
|
| |||||||||
| 1 | BrS | Male | 11 | 6 | A226V | c.677C>T | 28 | R1629X | c.4885C>T |
| 2 | BrS | Male | 40 | 9 | P336L | c.1007C>T | 28 | I1660V | c.4978A>G |
| 3 | BrS | Male | 45 | 22 | L1308F | c.3922C>T | 6 | V232I | c.694G>A |
| 4 | BrS | Female | 0.42 | 26 | R1512W | c.4534C>T | 28 | S1710L | c.5129C>T |
| 5 | BrS | Male | 56 | 28 | R1739G | c.5215C>G | 28 | G1740V | c.5219G>T |
We identified two novel pathogenic variants in BrS probands, p.Q1695H (c.5085G>A) and p.R1826P (c.5477G>C). Both were predicted to be deleterious using the bioinformatics tools: SIFT (0.021 and 0.001) and PolyPhen-2 (0.985 and 0.974).
Ten SCN5A pathogenic variants were found in ERS probands, all with minor allele frequency (MAF)of 0 to 0.000007 in the GnomAD database (Table 3 and Figure 2A). In all ERS probands, SCN5A+ yield in ERS2 patients was 5.71%, whereas the percentage of SCN5A+ in ERS3 cases was 13.33% (p > 0.05, Figure 2G). Of these 10 pathogenic variants, 5 (50.00%) were radical variants; the rest were missense variants. All of the missense variants were damaging based on in silico predictions. Except for SCN5A-G452C, the other 9 variants were all pathogenic or likely pathogenic according to ACMG in VarSome. It is noteworthy that the SCN5A-G452C could be upgraded to “likely pathogenic” in ACMG with the addition of the functional expression assessment in the current study.
TABLE 3.
SCN5A Pathogenic Variants in ERS
| Amino Acid Change | rs Number | Nucleotide Change | Variant Type | Exon | Location | Ethnicity | MAFa | SIFT |
PolyPhen-2 |
MetaLR |
ACMG in VarSome | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | Prediction | Score | Prediction | Score | Prediction | |||||||||
|
| ||||||||||||||
| p.K249X | rs1553705562 | c.745A>T | Nonsense | 7 | DI-S4/5 | White | 0 | NA | NA | NA | NA | NA | NA | Pathogenic |
| p.G452C | NA | c.1354G>T | Missense | 11 | DI/DII | Asian | 0 | 0 | Damaging | 0.960 | Probably damaging | 0.921 | Damaging | VUSb |
| p.Q646RfsX6 | rs727505158 | c.1936delC | Frameshift | 13 | DI/DII | Asian | 0 | NA | NA | NA | NA | NA | NA | Pathogenic |
| p.V728I | rs958480279 | c.2182G>A | Missense | 14 | DII-S1 | White | 0.000007 | 0.013 | Damaging | 0.305 | benign | 0.857 | Damaging | Likely pathogenic |
| p.V845CfsX2 | rs794728912 | c.2533delG | Frameshift | 16 | DII-S5 | Black | 0 | NA | NA | NA | NA | NA | NA | Pathogenic |
| p.Q1185X | rs794728873 | c.3553C>T | Nonsense | 20 | DII/DIII | White | 0 | NA | NA | NA | NA | NA | NA | Pathogenic |
| p.G1297GfsX22 | NA | c.3891insA | Frameshift | 22 | DIII-S4 | White | 0 | NA | NA | NA | NA | NA | NA | Pathogenic |
| p.G1408R | rs137854612 | c.4222G>A | Missense | 23 | DIII-S5/6 | White | 0 | 0 | Damaging | 1 | Probably damaging | 0.975 | Damaging | Pathogenic |
| p.L1412F | rs199473241 | c.4234C>T | Missense | 23 | DIII-S5/6 | White | 0 | 0 | Damaging | 1 | Probably damaging | 0.981 | Damaging | Likely pathogenic |
| p.S1787N | rs199473316 | c.5360G>A | Missense | 28 | C-terminal | Asian | 0 | 0 | Damaging | 0.99 | Probably damaging | 0.939 | Damaging | Likely pathogenic |
Minor allele frequency (GnomAD) according to ethnicity of the specific case.
Variant of uncertain significance. With the functional expression results of the present study, G452C could be upgraded to “likely pathogenic” according to the American College of Medical Genetics and Genomics.
ERS = early repolarization syndrome.
Comparison between BrS and ERS
The BrS group had strikingly longer QRS duration (105.49±19.08 ms vs. 90.40±19.97 ms, p=0.018, Table 1, Figure 3A), longer QTc interval in lead II (425.13±42.62 ms vs. 393.64±40.04 ms, p=0.025, Table 1) and V5 (427.35±47.24 ms vs. 390.47±29.56 ms, p=0.012, Table 1, Figure 3B) when compared to the ERS group. Furthermore, the SCN5A+ yieldin BrS probands was significantly higher than that in ERS probands (22.90% vs. 9.62%, p=0.004, Figure 3C). ERS probands were more likely to present with bradycardia than BrS probands (60.00% vs. 18.33%, p=0.042, Table 1). Other variables, including heart rate (HR), PR interval, P-wave duration, syncope, family history of unexplained SCD, and MAE, did not differ significantly between BrS and ERS cohorts.
Figure 3. Differences between BrS and ERS probands.
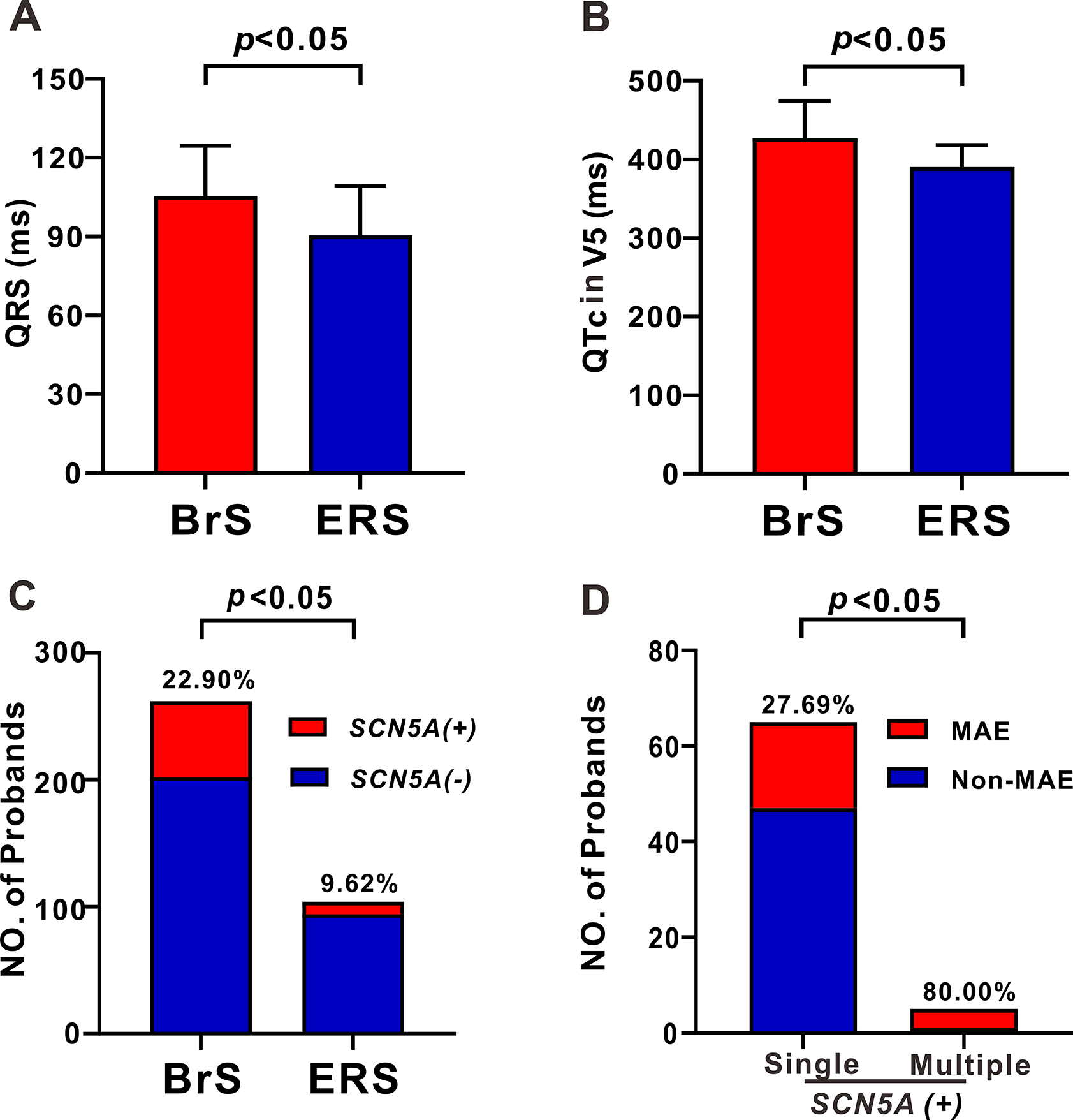
A-B: Bar graph of QRS duration (A), QTc V5 (B) between BrS and ERS probands. C: The yield of SCN5A+ pathogenic variants in BrS and ERS probands. D: The number of probands exhibiting MAE among JWS probands carrying single and multiple pathogenic variants limited to the SCN5A gene.
Comarison between probands with a single (SPV) vs. multiple (MPV) pathogenic variants in SCN5A
In all SCN5A+ probands, 5 carried more than 1 pathogenic variant in SCN5A. Patients with multiple SCN5A variants displayed longer PR intervals (214.50±16.24 ms vs. 181.24±32.31 ms, p<0.001) and QRS duration (123.56±11.03 ms vs. 102.23±19.68 ms, p=0.041, Table 4) than those with SPV. Patients with MPV in SCN5A also experienced more MAE (80.00% vs. 27.69%, p=0.031, Figure 3D).
Table 4.
Clinical characteristic of probands with single or multiple pathogenic variants in SCN5A
| SPV (N=65) | MPV (N=5) | p-value | |
|---|---|---|---|
|
| |||
| Age (years) | 37.31±18.64 | 26.08±18.82 | 0.199 |
| Children (age<16 years) | 10 (15.38%) | 2 (40.00%) | 0.201 |
| Family history, n (%) | 22 (33.85%) | 2 (40.00%) | 1.000 |
| CCD, n (%) | 25 (38.46%) | 3 (60.00%) | 0.383 |
| Bradycardia, n (%) | 15 (23.08%) | 2 (40.00%) | 0.589 |
| Syncope, n (%) | 35 (53.85%) | 3 (60.00%) | 1.000 |
| fQRS, n (%) | 15 (23.08%) | 2 (40.00%) | 0.589 |
| Global J wave, n (%) | 16 (24.62%) | 1 (20.00%) | 1.000 |
| MAE, n (%) | 18 (27.69%) | 4 (80.00%) | 0.031 |
| HR (bpm) | 74.63±18.62 | 75.99±21.38 | 0.936 |
| QRS duration (ms) | 102.23±19.68 | 123.56±11.03 | 0.041 |
| PR interval (ms) | 181.24±32.31 | 214.50±16.24 | <0.001 |
| P wave duration (ms) | 106.79±20.86 | 108.92±18.97 | 0.912 |
| QTc interval (II, ms) | 419.95±44.26 | 427.13±25.09 | 0.574 |
| QTc interval (V5, ms) | 421.43±47.81 | 428.77±11.92 | 0.611 |
| Tp-e (II, ms) | 91.63±19.62 | 97.42±5.77 | 0.320 |
| Tp-e /QT (II, ms) | 0.237±0.040 | 0.235±0.020 | 0.982 |
| Tp-e (V5, ms) | 93.78±23.17 | 105.92±3.653 | 0.224 |
| Tp-e /QT (V5, ms) | 0.241±0.044 | 0.253±0.004 | 0.179 |
Note: SPV and MPV represent single and multiple pathogenic variant(s), respectively.
Fever-induced ERS
Fever is known to unmask the BrS phenotype, but this is generally not so with ERS. Among the cases of ERS, we identified the first one associated with fever. This was a 54-year-old female presenting with pneumonia, cough, and shortness of breath. Her baseline ECG revealed slurred J waves in leads I, II, III, aVF, V5, V6 with a horizontal ST segment, bradycardia (HR=53bpm), and first-degree atrioventricular block (PR interval = 280 ms) (Figure 4A). When her body temperature increased to 39.4°C, HR increased to 94 bpm, and the amplitude of the J waves in leads II, III and aVF increased from 0.16 mV to 0.35mV (Figure 4A). She had a history of syncope. Genetic analysis revealed a heterozygous missense pathogenic variant of c.4234C>T p.L1412F (Figure 4B). Leucine 1412 in SCN5A was highly conserved among species (Figure 4C). Interestingly, p.L1412F variant was also uncovered in a fever-induced Chinese BrS proband (Supplemental Figure 1).
Figure 4. Functional expression studies of SCN5A-L1412F and SCN5A-G452C.
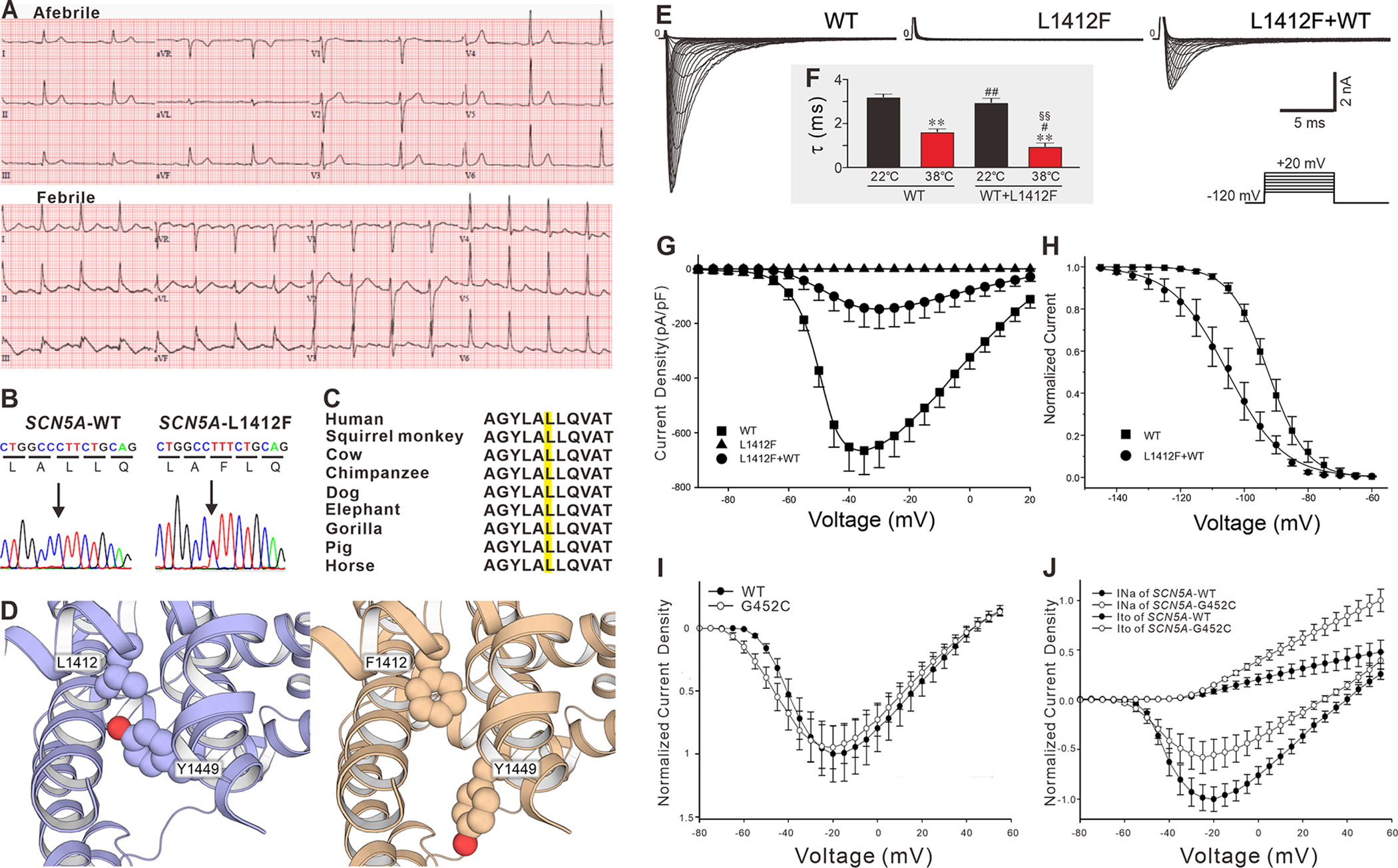
A: ECG at baseline (top) and during a febrile episode (bottom). B: Electropherogram of SCN5A-WT and SCN5A-L1412F. C: Amino acid sequence alignment of SCN5A-L1412 D: Computational model of L1412F. E: Representative sodium current traces recorded from HEK293 cells transfected with SCN5A-WT, SCN5A-L1412F, SCN5A-L1412F+WT. The protocol is presented at the inset panel. F: The decay (τ) of WT and WT+L1412F at room temperature (22.0°C) and 38.0°C. * and **: p<0.05 and 0.01 vs. WT at 22.0°C; # and ##: p<0.05 and 0.01 vs. WT at 38.0°C; § and §§: p<0.05 and 0.01 vs. WT+L1412F at 22.0°C. G: Current-voltage relationship for WT (squares, n=23), L1412F (triangles, n=12) and L1412F+WT (circles, n=22). H: Steady-state inactivation in WT (squares, n=22) and L1412F+WT Channels (circles, n=6). I: Normalized current-voltage relationship recorded from HEK293 cell for WT and G452C. J: Normalized current-voltage relationship in HEK293 cells stably expressing SCN1B+KCNIP2 transfected with SCN5A-G452C/KCND3 vs. SCN5A-WT/KCND3.
Functional expression and computer model studies
In heterologous expression studies, the homozygously expressed SCN5A-L1412F channel failed to generate any sodium channel current (n = 12). When co-expressed with WT, it showed a dominant-negative effect (peak INa, 665.1±87.6 pA/pF in WT vs. 147.8±71.0 pA/pF in L1412F+WT, n=23 and 22 respectively, P<0.01, Figure4E & 4G). The voltage dependence of steady-state inactivation of L1412F+WT was significantly shifted by −13 mV (V1/2: WT, −92.48±0.82 mV; L1412F+WT, −105.25±2.65 mV; P<0.05), and the slope factor was significantly larger in L1412F (k: WT, 5.98±0.30; L1412F+WT, 8.80±0.97 mV; P<0.05, Figure 4H). At 22.0°C, there was no difference in the current decay (τ) of INa between SCN5A-WT+L1412F and WT at a voltage of −20mV. Increasing temperature to 38.0°C significantly accelerated current decay in the WT and more so in the case of SCN5A-WT+L1412F (Figure 4F). Our computational ΔΔG calculation suggests that L1412F destabilizes Nav1.5 structure by 1.04±0.04 kcal/mol. Inspection of the modeled protein structure of the channel indicated that in the L1412F variant structure, residue Y1449 moved away from its conformation in the wild-type structure to make room for the bulkier phenylalanine residue. This conformational change of Y1449 broke the hydrogen bond between Y1449 and F1348 (Figure 4D). While the destabilization effect of L1412F is likely due to this conformational rearrangement, further in-depth investigation is needed to ascertain how the structural effects are related to the electrophysiology phenotype of L1412F.
The case illustrated in Figure 1F is a 27-year-old Chinese male who presented with ASCD. His ECG displayed slurred J waves in leads II (0.25mV), III (0.5mV), aVF (0.4mV, Figure 1F). Genetic testing identified a heterozygous pathogenic variant in SCN5A-G452C (c.1354G>T), which was not reported in any population databases. The glycine residue was highly conserved. Furthermore, SCN5A-G452C was predicted to be damaging by in silico prediction tools. When SCN5A-G452C was transfected in HEK293 cells alone, it did not significantly alter INa density compared to that of SCN5A-WT (Figure 4I). However, when co-expressed with KCND3-WT, SCN5A-G452C not only significantly decreased peak INa by 44.52% at −20 mV but also caused a 106.81% increase in Ito density at +40 mV (Figure 4J).
Discussion
The principal aim of the current study was to assess the extent to which SCN5A pathogenic variants contribute to the two principal forms of JWS and to characterize their phenotypic expression. Our results indicate that ERS probands carry SCN5A+ variants less often than BrS probands, but SCN5A is nevertheless a major susceptibility gene in ERS as it is in BrS (Central Illustration). Our study uncovered 10 novel SCN5A pathogenic variants in ERS and 2 in BrS. Moreover, we identified the first case of fever-induced ERS and the associated SCN5A variant. Our functional expression studies also provide further evidence in support of a prominent role for Ito in the pathogenesis of JWS. Last, but not least, our findings show that patients with MPV display longer PR intervals, QRS durations and experience more MAE than those carrying only a SPV in SCN5A (Central Illustration).
Central Illustration: Distinct Features of J Wave Syndrome (JWS) Carrying SCN5A Pathogenic Variants.

A: Topology of SCN5A and location of pathogenic variants revealed in Early Repolarization Syndrome (ERS) probands of the present study. The ball with a thermometer indicates mutant carrier is unmasked by fever. B: The percentage yield of SCN5A+ pathogenic variants in BrS and ERS probands. SCN5A is a major susceptibility gene in ERS as in BrS, whereas the yield in ERS is significantly lower than in BrS. C: The percentage yield of probands exhibiting major arrhythmic events (MAE) among JWS probands carrying single pathogenic variant and multiple pathogenic variants limited to SCN5A gene.
SCN5A is responsible for initiating the cardiac action potential. Loss-of-function of SCN5A reduces the excitability of myocardial cells and slows impulse conduction throughout the myocardium(14). The SCN5A+ yield of 22.90% in our BrS probands is consistent with previously reported studies (18%~28%)(1). The SCN5A+ yield in ERS has not been systematically studied(6,9,10). In addition to the 7 SCN5A pathogenic variants previously reported for ERS, we identified 10 novel pathogenic variants, extending the limited genetic knowledge of ERS. To the best of our knowledge, we enroll the largest ERS cohort undergoing clinical and genetic analysis, and are the first to compare the SCN5A+ yield in ERS probands with that of BrS. Dr. Nademanee and Haissaguerre find the distinct dissimilarity of late potential prevalence between BrS and ERS in epicardial mapping, which is comparable to our data(6,15,16) The difference in SCN5A+ yield likely underlies the difference in electrophysiological mechanism between BrS and ERS. We found SCN5A pathogenic variants in 13.33%, 5.71%, and 0.00% of ERS3, ERS2, and ERS1 probands, respectively. These results are consistent with reports that loss-of-function pathogenic variants in SCN5A are prevalent in cases of ERS3(9). The results suggest that SCN5A loss of function contributes more prominently to ERS3 than to ERS1 or ERS2.
At odds with a previous report(17), we found a strikingly shorter QTc intervals in ERS probands (shortest in ERS3) than in probands diagnosed with BrS. Activation recovery intervals (ARI) is surrogate for local action potential duration. Our results are consistent with those of Rudy and colleagues who identified regions with prominent J-wave have abnormally short ARI in the epicardium of ERS patients using noninvasive electrocardiographic imagining(18). Nonetheless, prolonged ARI has been described in the right ventricular outflow tract (RVOT) of BrS patients(3). Therefore, our ERS patients, especially in type 3, may harbor extensive regions with short ARI, which results in the shorter QT interval.
The development of the electrocardiographic and arrhythmic manifestations of ERS are known to be much more sensitive to hypothermia than those of BrS. (1) BrS, however, is more sensitive to hyperthermia or fever. Fever is a common predisposing factor unmasking BrS and promoting MAE. A recent consensus statement points out that fever rarely augments the amplitude of J wave in ERS(1), but there is no study thus far delineated fever-induced ERS.
We provide the first evidence of fever-induced ERS associated with a SCN5A pathogenic variant. When this patient’s body temperature increased to 39.4°C, the amplitude of J waves in the inferior leads were as greatly amplified. The associated SCN5A-L1412F variant was judged to be the causative pathogenic variant based on the following. SCN5A-L1412F is absent in our >200 healthy ethnicity-matched controls. It is located in the pore region of domain III, a highly conserved region in mammals; is predicted to be damaging by in silico tools; and is “likely pathogenic” by ACMG guidelines in VarSome. Moreover, the variant destabilizes the Nav1.5 structure by 1.04 ± 0.04 kcal/mol by altering the tertiary structure in computational modeling. Lastly, the functional expression study demonstrates that it is associated with a significantly reduced INa density and a remarkable negative shift of steady-state inactivation.
The SCN5A+ yield ranges from 26.4% to 32.7% in febrile BrS patients, and 50% to 75% in those with fever-induced MAE, increasing to 100% in cases of pediatric BrS with fever-induced MAE (19, 20). The electrophysiological characteristics of the cardiac sodium channel are known to be temperature sensitive. Dumaine et al. reported that higher temperature exacerbates the unique gating properties of defective channels, leading to accelerated decay, slowed recovery from inactivation, and a positive shift of steady-state activation(4). SCN5A-L1412F significantly accelerated the decay at 38°C compared to SCN5A-WT. This is also consistent with the results of Abriel and colleagues, who reported that the loss of function of SCN5A-L325R and SCN5A-R535X was accentuated at high temperature (21). These findings support loss-of-function in sodium channel may be particularly relevant to fever-induced BrS and ERS, and the latter should be recognized as a new clinical entity. SCN5A-L1412F was also found in a fever-induced BrS case in the present study, indicating fever is an intimate trigger to unmask JWS in this genetic locus.
As illustrated in Figure 1G–I, pathogenic variants in SCN5A cause different phenotypes among different carriers of the same family and overlapping phenotypes in the same patient. These results indicate SCN5A can have a pleiotropic effect leading to a distinct phenotype spectrum, consistent with previous studies (22). At the same time, incomplete penetrance in families with causative pathogenic variants, sporadic cases, and failure to identify novel pathogenic genes in familial linkage analysis have all been shown in JWS in current research and previous studies (23,24). Wijeyeratne et al. recently reported that in 312 individuals from families carrying SCN5A pathogenic variants, BrS-GRS (BrS-genetic risk score) ≥4 risk alleles correlates with BrS regardless of the presence of SCN5A pathogenic variant(25). These facts illustrate that other factors, including the possibility of genetic and environmental modifiers, may underline the pathophysiology regionalization of J wave abnormalities.
Patients with multiple pathogenic SCN5A variants experienced more MAE than those with single pathogenic variants, suggesting that the number of rare variants influences disease manifestation and severity. Compound variants in SCN5A causing more severe phenotypes in BrS patients have been previously reported. Tan et al. showed that a boy carrying paternal (A226V) and maternal (R1629X) experienced syncope and PVT while his parents were both asymptomatic(26). Sacilotto et al. identified a child carrying compound SCN5A mutations (G400R and T1461S) that showed a more severe phenotype, including VT/VF(27). Inheritance of >1 pathogenic variant in ≥1 genes has been reported to cause more severe phenotype in LQTS and HCM(23). SCN5A-T1620M and R1232W are two of the first pathogenic variants associated with BrS. When separately expressed, they produced subtle biophysical abnormalities. However, when co-expressed, they rendered the channels non-functional due to trafficking defects(28). Our previous research also demonstrates an additive effect of 2 missense mutations (SCN5A-V232I and L1308F) to sensitize the sodium channel to lidocaine leading to potent use-dependent inhibition of INa(29). Other examples are reports in which SCN5A-H558R, a common polymorphism, is shown to aggravate the dysfunction of specific SCN5A pathogenic variants (30, 31, 32). More recently, 3 BrS probands with dual SCN5A and CACNA1C mutations, SCN5A and TRPM4 mutations, SCN5A and ABCC9 mutations, all experiencing ASCD or syncope at young ages, have also been reported (33, 34, 35).SCN5A BrS patients who are young at diagnosis have been reported to be at higher risk(32). Consistent with this observation, our MRV group was diagnosed with JWS at a significantly younger age suggesting that JWS probands carrying MRV are at increased risk for MAE.
When SCN5A-G452C was expressed alone, there was no influence on INa compared to SCN5A-WT. However, when co-expressed with KCND3, SCN5A-G452C exhibited reduced INa compared to SCN5A-WT in addition to a robust Ito, which highlights inter-regulation of the two channels and a case in which expression of Ito can exacerbate the severity of ion channel defect leading to a more severe phenotype. These observations also provide further evidence supporting the critical role of Ito in the pathogenesis of JWS.
Limitations
Because this is a retrospective analysis from multicenter registries, and the ECGs in JWS probands are dynamic, selection bias is inevitable. A second limitation is the lack of long-term follow-up. Consequently, the prognostic value of the risk factors identified needs to be assessed in prospective studies. Third, we only present functional expression data for 2 of 10 pathogenic variants identified in the ERS probands. However, among the other 8, 5 are radical variants causing a total loss-of-function, and 2 have been previously validated via functional expression studies. Only one novel variant awaits the completion of the functional genomic study.
Conclusion
We compare clinical and electrophysiologic characteristics of 104 patients diagnosed with ERS and 262 diagnosed with BrS carrying pathogenic variants in SCN5A. ERS patients displayed shorter QRS and QTc interval. Our study extends knowledge of the genetics of JWS, uncovering 10 novel SCN5A variants in ERS and 2 in BrS. Probands with MPV in SCN5A displayed longer PR intervals, QRS durations, and experienced MAE more often. Our findings point to SCN5A as a major susceptibility gene in ERS as in BrS, however the yield in ERS is significantly lower than in BrS. This study also identifies the first case of fever-induced ERS and demonstrates a critical role for Ito in JWS induced by an SCN5A loss of function variant.
Supplementary Material
Clinical Perspectives:
Competency in Medical Knowledge:
Patients with early repolarization syndrome (ERS) are more often male and present at younger ages but less often carry pathogenic SCN5A genetic variants than those with Brugada syndrome (BrS).
Translational Outlook:
Further research is needed to define the specific arrhythmic risks associated with the various pathogenic SCN5A variants alone and in combination, and their interactions with physiological perturbations such as fever, autonomic nervous system activity and metabolic derangements.
Acknowledgments
The authors are grateful to Bo Cheng from Renmin Hospital of Wuhan University, China; Yi Guo from Center for Evidence-based and Translational Medicine, Zhongnan Hospital of Wuhan University; and Ryan Pfeiffer from Masonic Medical Research Institute, USA, for technical assistance.
Source of Funding
The current work was supported by the National Natural Science Foundation Project of China (Grant No. 81670304); the National Institutes of Health of USA (NIH R56 - HL47678], NIH R01 [HL138103]), NIH R01 [HL152201], the W.W. Smith Charitable Trust and the Wistar and Martha Morris Fund, and the American Heart Association (Postdoctoral Fellowship grant 20POST35220002).
ABBREVIATIONS AND ACRONYMS
- ARI
activation recovery interval
- ASCD
aborted sudden cardiac death
- BrS
Brugada syndrome
- CCD
cardiac conduction disease
- ECG
electrocardiogram
- ERP
Early repolarization pattern
- ERS
early repolarization syndrome
- HR
heart rate
- JWS
J wave syndrome
- MAE
major arrhythmic events
- MAF
minor allele frequency
- MPV
multiple pathogenic variant
- RVOT
right ventricular outflow tract
- SCD
sudden cardiac death
- SPV
single pathogenic variant
- Tp-e
Tpeak-Tend
- VT/VF
ventricular tachycardia/fibrillation
Footnotes
Disclosures
Dr. Antzelevitch served as a consultant and received grant funds from Novartis and Trevena Inc. All other authors report no relationships to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Antzelevitch C, Yan G-X, Ackerman MJ et al. J-wave syndromes expert consensus conference report:Emerging concepts and gaps in knowledge. Heart Rhythm 2016;13:e295–e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haïssaguerre M, Derval N, Sacher F et al. Sudden Cardiac Arrest Associated with Early Repolarization. N Engl J Med 2008;358:2016–23. [DOI] [PubMed] [Google Scholar]
- 3.Di Diego JM, Antzelevitch C. J wave syndromes as a cause of malignant cardiac arrhythmias. Pacing Clin Electrophysiol 2018;41:684–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dumaine R, Towbin JA, Brugada P et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999;85:803–9. [DOI] [PubMed] [Google Scholar]
- 5.Behr ER, Ben-Haim Y, Ackerman MJ, Krahn AD, Wilde AAM. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: a final common pathway? Eur Heart J 2021;42:1073–1081. [DOI] [PubMed] [Google Scholar]
- 6.Nademanee K, Haissaguerre M, Hocini M et al. Mapping and ablation of ventricular fibrillation associated with early repolarization syndrome. Circulation 2019;140:1477–1490. [DOI] [PubMed] [Google Scholar]
- 7.Glazer AM, Wada Y, Li B et al. High-Throughput Reclassification of SCN5A Variants. Am J Hum Genet 2020;107:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu D, Barajas-Martinez H, Pfeiffer R, Burashnikov E, Caceres G, Antzelevitch C. Abstract 18124: The Role of SCN5A Mutations in J Wave Syndromes. Circulation 2010;122:A18124–A18124. [Google Scholar]
- 9.Watanabe H, Nogami A, Ohkubo K et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol 2011;4:874–81. [DOI] [PubMed] [Google Scholar]
- 10.Voskoboinik A, Hsia H, Moss J et al. The many faces of early repolarization syndrome: A single-center case series. Heart Rhythm 2020;17:273–281. [DOI] [PubMed] [Google Scholar]
- 11.Castro Hevia J, Antzelevitch C, Tornes Barzaga F et al. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J Am Coll Cardiol 2006;47:1828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park H, Bradley P, Greisen P Jr. et al. Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J Chem Theory Comput 2016;12:6201–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leaver-Fay A, Tyka M, Lewis SM et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 2011;487:545–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park DS, Cerrone M, Morley G et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest 2015;125:403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nademanee K, Veerakul G, Chandanamattha P et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011;123:1270–9. [DOI] [PubMed] [Google Scholar]
- 16.Haissaguerre M, Nademanee K, Hocini M et al. Depolarization versus repolarization abnormality underlying inferolateral j-wave syndromes: New concepts in sudden cardiac death with apparently normal hearts. Heart Rhythm 2019;16:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawata H, Noda T, Yamada Y et al. Effect of sodium-channel blockade on early repolarization in inferior/lateral leads in patients with idiopathic ventricular fibrillation and Brugada syndrome. Heart Rhythm 2012;9:77–83. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Hocini M, Strom M et al. The Electrophysiological Substrate of Early Repolarization Syndrome: Noninvasive Mapping in Patients. JACC Clin Electrophysiol 2017;3:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizusawa Y, Morita H, Adler A et al. Prognostic significance of fever-induced Brugada syndrome. Heart Rhythm 2016;13:1515–20. [DOI] [PubMed] [Google Scholar]
- 20.Wu CI, Postema PG, Arbelo E et al. SARS-CoV-2, COVID-19 and inherited arrhythmia syndromes. Heart Rhythm 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller DI, Rougier JS, Kucera JP et al. Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5 A mutations. Cardiovasc Res 2005;67:510–9. [DOI] [PubMed] [Google Scholar]
- 22.Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res 2011;108:884–97. [DOI] [PubMed] [Google Scholar]
- 23.Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res 2015;116:1919–36. [DOI] [PubMed] [Google Scholar]
- 24.Burashnikov E, Pfeiffer R, Barajas-Martinez H et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 2010;7:1872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wijeyeratne YD, Tanck MW, Mizusawa Y et al. SCN5A Mutation Type and a Genetic Risk Score Associate Variably With Brugada Syndrome Phenotype in SCN5A Families. Circ Genom Precis Med 2020;13:e002911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan BY, Yong RY, Barajas-Martinez H et al. A Brugada syndrome proband with compound heterozygote SCN5A mutations identified from a Chinese family in Singapore. Europace 2016;18:897–904. [DOI] [PubMed] [Google Scholar]
- 27.Sacilotto L, Epifanio HB, Darrieux FC et al. Compound Heterozygous SCN5A Mutations in a Toddler - Are they Associated with a More Severe Phenotype? Arq Bras Cardiol 2017;108:70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baroudi G, Acharfi S, Larouche C, Chahine M. Expression and intracellular localization of an SCN5A double mutant R1232W/T1620M implicated in Brugada syndrome. Circ Res 2002;90:E11–6. [PubMed] [Google Scholar]
- 29.Barajas-Martínez HM, Hu D, Cordeiro JM et al. Lidocaine-Induced Brugada Syndrome Phenotype Linked to a Novel Double Mutation in the Cardiac Sodium Channel. Circulation Research 2008;103:396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veltmann C, Barajas-Martinez H, Wolpert C et al. Further Insights in the Most Common SCN5A Mutation Causing Overlapping Phenotype of Long QT Syndrome, Brugada Syndrome, and Conduction Defect. J Am Heart Assoc 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu D, Viskin S, Oliva A et al. Novel mutation in the SCN5A gene associated with arrhythmic storm development during acute myocardial infarction. Heart Rhythm 2007;4:1072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarawate P, Chaosuwannakit N, Vannaprasaht S et al. SCN5A Genetic Polymorphisms Associated With Increased Defibrillator Shocks in Brugada Syndrome. J Am Heart Assoc 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gualandi F, Zaraket F, Malagù M et al. Mutation Load of Multiple Ion Channel Gene Mutations in Brugada Syndrome. Cardiology 2017;137:256–260. [DOI] [PubMed] [Google Scholar]
- 34.Béziau DM, Barc J, O'Hara T et al. Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic Res Cardiol 2014;109:446. [DOI] [PubMed] [Google Scholar]
- 35.Hu D, Barajas-Martinez H, Terzic A et al. ABCC9 is a novel Brugadaand early repolarization syndrome susceptibility gene. Int J Cardiol 2014;171:431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.