

Abstract
Motivation
Testing for pathway enrichment is an important aspect in the analysis of untargeted metabolomics data. Due to the unique characteristics of untargeted metabolomics data, some key issues have not been fully addressed in existing pathway testing algorithms: (i) matching uncertainty between data features and metabolites; (ii) lacking of method to analyze positive mode and negative mode liquid chromatography–mass spectrometry (LC/MS) data simultaneously on the same set of subjects; (iii) the incompleteness of pathways in individual software packages.
Results
We developed an innovative R/Bioconductor package: metabolic pathway testing with positive and negative mode data (metapone), which can perform two novel statistical tests that take matching uncertainty into consideration—(i) a weighted gene set enrichment analysis-type test and (ii) a permutation-based weighted hypergeometric test. The package is capable of combining positive- and negative-ion mode results in a single testing scheme. For comprehensiveness, the built-in pathways were manually curated from three sources: Kyoto Encyclopedia of Genes and Genomes, Mummichog and The Small Molecule Pathway Database.
Availability and implementation
The package is available at https://bioconductor.org/packages/devel/bioc/html/metapone.html.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Untargeted metabolomics, which measures of abundance of small molecules in an unbiased manner, is gaining wider applications in biomedical research (Jacob et al., 2019). Pathway testing is a critical step in data interpretation for untargeted metabolomics data. Many works have been done on metabolic pathway testing, but there is no consensus on the best approach (Karnovsky and Li, 2020). The current leading methods, e.g. Mummichog and MetaboAnalyst (Chong et al., 2018; Li et al., 2013), use incomplete pathway information. More importantly, pathway testing is hampered by matching uncertainty between extracted features from LC/MS data and known metabolites, as possible correspondences are largely identified by comparison of feature m/z values and the theoretical m/z values of common adduct ions of known metabolites, and many metabolites share the same molecular composition. Furthermore, when multiple adduct ions are considered, the issue becomes more complicated. Although efforts were made to annotate LC/MS data features to known metabolites in a more reliable manner, currently the majority of features are matched with uncertainty, i.e. a feature could potentially be derived from multiple metabolites (Chaleckis et al., 2019; Kuhl et al., 2012; Uppal et al., 2017). This uncertainty heavily impacts pathway testing, as in many cases, we cannot be sure a differentially abundant feature is truly derived from a certain pathway.
Some methods use extra information in pathway testing, such as methods for longitudinal metabolomics data (Ebrahimpoor et al., 2021), matching features to metabolites by utilizing the similarity of MS2 in reaction-paired neighborhood (Shen et al., 2019), and combining feature matching with predictive modeling (Cai et al., 2017). However, for untargeted data collected using the most common case–control study design, such methods are not suitable. And since not all matched metabolites can be found in the known metabolic network, some information may be lost when using network-based methods. Therefore, a method that can be widely used with relatively complete pathway information is urgently needed.
In this work, we developed an R/Bioconductor package named ‘Metapone’. It facilitates pathway testing by making improvements in three areas:
We developed two novel tests to address the matching uncertainty issue. The first follows the concept of gene set enrichment analysis (GSEA) (Subramanian et al., 2005), but uses weighted features or metabolites based on multiple-matching status. The second is a permutation-based weighted hypergeometric test which also accounts for matching uncertainty.
We compiled pathways from three established database sources by computationally removing (partial) overlaps—Kyoto Encyclopedia of Genes and Genomes (KEGG) (Ogata et al., 1998), Mummichog (Li et al., 2013) and The Small Molecule Pathway Database (SMPDB) (Frolkis et al., 2010).
Metapone can jointly analyze positive- and negative-ion mode data produced in the same study, which can help avoid double counting and to generate a more integrated and comprehensive view of metabolic perturbations.
2 Materials and methods
2.1 Combining pathways from different databases
All metabolites were mapped using the human metabolome database (HMDB) IDs. We first collected all pathways from the three established sources. Then between any pair of pathways, if the number of overlapping metabolites is of the size of both pathways, we removed the pathway with smaller number of metabolites. A total of 372 pathways were included in the Metapone package.
2.2 Testing procedures
Metapone can use data from a single-ion mode, and it can also handle the case of jointly testing both positive- and negative-ion mode data. In the joint analysis scenario, data from the two ion modes are first individually mapped to HMDB metabolites based on the listed of allowed adduct ions, which the user can select. We then combine the feature-metabolite matching from both modes in the pathway testing. Metapone allows two approaches for pathway testing.
(1) Weighted GSEA test.
This is a modified GSEA-type test based on Fast Gene Set Enrichment Analysis (fgsea) approach (Korotkevich et al., 2021), using weighted features/metabolites. The features are denoted as , the metabolites as , the potential matching between features and metabolites as , where if there is potential matching between and , and 0 otherwise. For feature , we define the feature weight as:
where is a cap of the number of matchings to a feature to limit the penalty, and is the power term which tunes the penalty on multiple matching. The recommended range for parameter is between 0 and 1, with a default value of 0.5. The higher the value of d, the higher the penalty on multiply matched features. Users can adjust the parameters according to the scope of matchings and specific task requirements.
We then take steps to limit the total contribution of a single feature/metabolite. If a feature is connected to too many metabolites such that , we limit its total contribution by replacing with . And if the total weight of a metabolite’s matched features is larger than 1, we divide each of the weights by the square-root of the total weight. The procedure avoids the weights of single metabolites being too large and dominating the pathway test.
For GSEA calculation, the feature importance is defined as the weighted negative log P-value:
For metabolites , we define its importance as the sum of the importance of its associated features:
In combination with pathway assignments, the importance scores of metabolites are used in fgsea. Alternatively, the testing can be conducted using fgsea with respect to features, with the relevant pathways for each feature being determined by the pathways of its matched metabolites.
(2) Permutation-based weighted hypergeometric test.
The procedure is similar to the regular hypergeometric test of gene sets in GOstats (Falcon and Gentleman, 2007). The difference is we factor the matching uncertainty into the test statistics by using as the fractional counts of feature i. For each pathway, we first find the total fractional counts of significant features assigned to pathway k:
where denotes the collection of features associated with pathway k through their matched metabolites, denotes the P-value threshold, and is the identity function. To assign pathway significance, we use a permutation test. In the nth permutation, we permute the original feature P-values, and calculate the total fractional counts of significant features . After N permutations, we assign the pathway P-value by .
Following either testing procedure, the pathway-level P-values are then transformed to local false discovery rate and cumulative false discovery rate (Strimmer, 2008). The output contains a table of pathway testing results and a list of the mapped significant features in each pathway.
3 Example analysis using metapone
As an illustration, we performed metapone analysis on data from the Metabolome Atlas of the Aging Mouse Brain (ST001888) dataset downloaded from the Metabolomics Workbench (https://www.metabolomicsworkbench.org/data/DRCCMetadata.php?Mode=Study&StudyID=ST001888). The F-test results of metabolites between prime-age and aging mice were used. Using weighted hypergeometric test with 2000 permutations and parameters , set to 10 and 0.5, we obtained 9 pathways with P-value and the weighted number of significant metabolites (Fig. 1a). The majority of the selected pathways were associated important lipid species in the brain. The remaining pathways include retinol metabolism, which is known to be critical to the nervous system development, and pentose glucuronate interconversions, which was linked to Alzheimer’s disease in gene expression analysis (Chen et al., 2016). We also conducted pathway testing using only positive- (Fig. 1b) or negative- (Fig. 1c) ion mode data, using the same criteria with P-value and the weighted number of significant metabolites . Less significant pathways were found by comparing both ion modes. Similar results using weighted GSEA test are shown in Supplementary Figure S1. Therefore, the results indicate analyzing both ion modes data together yields higher power than analyzing individual ion mode data separately.
Fig 1.
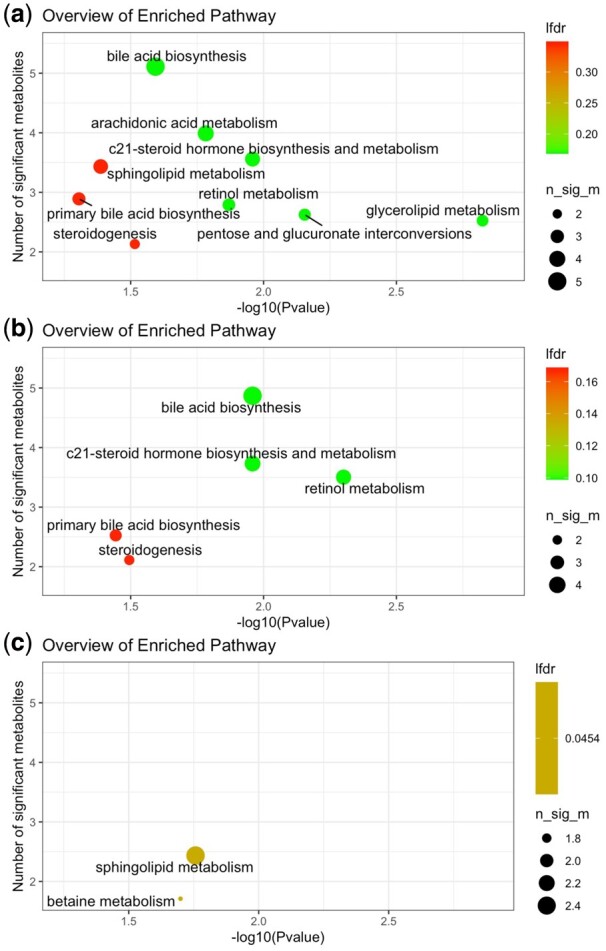
Example weighted hypergeometric testing result on the ST001888 dataset. (a) 2D plot of significant pathways using both positive- and negative-ion mode data. (b) and (c) are results using positive- or negative-ion mode data, respectively
For comparison, we conducted testing using Mummichog and MetaboAnalyst with both ion modes data, through the webserver of MetaboAnalyst 5.0. Only four and three pathways were found with P-value respectively (Supplementary Figs S2 and S3). Unlike Metapone, Mummichog mostly found core amino acid metabolism pathways (Supplementary Fig. S4). The results indicated the benefit of using more complete pathway information.
Metapone requires the setting of a few parameters. To assess the robustness of the model against parameter settings, we conducted a series of weighted GSEA tests with 2000 permutations using different parameters, i.e. 5, 10, 20 for , and 0.25, 0.50, 1.00 for . The Pearson correlation coefficients of the estimated pathway P-values are in the range of 0.94 to 1 (Fig. 2).
Fig 2.
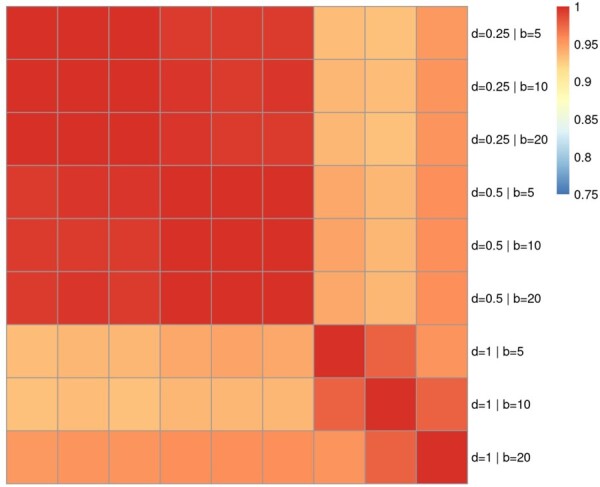
Heatmap of the correlation of pathway P-values with different model parameters on ST001888 dataset
Overall, metapone can analyze positive- and negative-ion mode data simultaneously and test against a comprehensive list of pathways, yielding rich functional results for untargeted metabolomics studies.
Supplementary Material
Acknowledgement
The authors thank Drs Shuzhao Li and Dean Jones for helpful discussions.
Funding
This work was partially supported by National Institutes of Health [R21ES032117, P30ES019776 and 1R01GM124061]; Shenzhen Research Institute of Big Data and the University Development Fund of CUHK-Shenzhen.
Conflict of Interest: none declared.
Contributor Information
Leqi Tian, Shenzhen Research Institute of Big Data, Shenzhen 518712, China; School of Data Science, The Chinese University of Hong Kong – Shenzhen, Shenzhen 518712, China.
Zhenjiang Li, Gangarosa Department of Environmental Health, Emory University, Atlanta, GA 30322, USA.
Guoxuan Ma, School of Data Science, The Chinese University of Hong Kong – Shenzhen, Shenzhen 518712, China; Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, USA.
Xiaoyue Zhang, Gangarosa Department of Environmental Health, Emory University, Atlanta, GA 30322, USA.
Ziyin Tang, Gangarosa Department of Environmental Health, Emory University, Atlanta, GA 30322, USA.
Siheng Wang, School of Data Science, The Chinese University of Hong Kong – Shenzhen, Shenzhen 518712, China.
Jian Kang, Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, USA.
Donghai Liang, Gangarosa Department of Environmental Health, Emory University, Atlanta, GA 30322, USA.
Tianwei Yu, Shenzhen Research Institute of Big Data, Shenzhen 518712, China; School of Data Science, The Chinese University of Hong Kong – Shenzhen, Shenzhen 518712, China; Warshel Institute, Shenzhen, Guangdong 518712, China.
References
- Cai Q. et al. (2017) Network marker selection for untargeted LC–MS metabolomics data. J. Proteome Res., 16, 1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaleckis R. et al. (2019) Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol., 55, 44–50. [DOI] [PubMed] [Google Scholar]
- Chen J. et al. (2016) Gene expression analysis reveals the dysregulation of immune and metabolic pathways in Alzheimer's disease. Oncotarget, 7, 72469–72474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J. et al. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res., 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimpoor M. et al. (2021) Pathway testing for longitudinal metabolomics. Stat. Med., 40, 3053–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S., Gentleman R. (2007) Using GOstats to test gene lists for GO term association. Bioinformatics, 23, 257–258. [DOI] [PubMed] [Google Scholar]
- Frolkis A. et al. (2010) SMPDB: the small molecule pathway database. Nucleic Acids Res., 38, D480–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob M. et al. (2019) Metabolomics toward personalized medicine. Mass Spectrom. Rev., 38, 221–238. [DOI] [PubMed] [Google Scholar]
- Karnovsky A., Li S. (2020) Pathway analysis for targeted and untargeted metabolomics. Comput. Methods Data Anal. Metab., 387–400. [DOI] [PubMed] [Google Scholar]
- Korotkevich G. et al. (2021) Fast gene set enrichment analysis. BioRxiv 060012.
- Kuhl C. et al. (2012) CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem., 84, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. (2013) Predicting network activity from high throughput metabolomics. PLoS Comput. Biol., 9, e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H. et al. (1998) Computation with the KEGG pathway database. Biosystems, 47, 119–128. [DOI] [PubMed] [Google Scholar]
- Shen X. et al. (2019) Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat. Commun., 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K. (2008) fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics, 24, 1461–1462. [DOI] [PubMed] [Google Scholar]
- Subramanian A. et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppal K. et al. (2017) xMSannotator: an R package for network-based annotation of high-resolution metabolomics data. Anal. Chem., 89, 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.