

Abstract
Motivation
Accurately predicting drug–target interaction (DTI) is a crucial step to drug discovery. Recently, deep learning techniques have been widely used for DTI prediction and achieved significant performance improvement. One challenge in building deep learning models for DTI prediction is how to appropriately represent drugs and targets. Target distance map and molecular graph are low dimensional and informative representations, which however have not been jointly used in DTI prediction. Another challenge is how to effectively model the mutual impact between drugs and targets. Though attention mechanism has been used to capture the one-way impact of targets on drugs or vice versa, the mutual impact between drugs and targets has not yet been explored, which is very important in predicting their interactions.
Results
Therefore, in this article we propose MINN-DTI, a new model for DTI prediction. MINN-DTI combines an interacting-transformer module (called Interformer) with an improved Communicative Message Passing Neural Network (CMPNN) (called Inter-CMPNN) to better capture the two-way impact between drugs and targets, which are represented by molecular graph and distance map, respectively. The proposed method obtains better performance than the state-of-the-art methods on three benchmark datasets: DUD-E, human and BindingDB. MINN-DTI also provides good interpretability by assigning larger weights to the amino acids and atoms that contribute more to the interactions between drugs and targets.
Availability and implementation
The data and code of this study are available at https://github.com/admislf/MINN-DTI.
1 Introduction
In drug discovery and design, verifying whether a drug interacts with a certain target is a key step to prove the effectiveness of the drug. Since large-scale in vitro and in vivo experiments are high-cost and time consuming, computational methods for drug–target interaction (DTI in short) prediction have received increasing attention. However, traditional computational methods have obvious limitations. For example, the widely used molecular docking is inefficient and sometimes ineffective because of its huge amount of computation and inaccurate scoring function (Su et al., 2019). On the other hand, traditional machine learning models such as Random Forest (RF) and Support Vector Machine (SVM) have also been used for DTI prediction (Ballester and Mitchell, 2010; Bleakley and Yamanishi, 2009; Liu et al., 2015). These methods are generally simple and efficient, but the performance is far from satisfaction. Recently, the introduction of deep learning models to DTI prediction has greatly advanced this area (Bagherian et al., 2021; Tian et al., 2016). In deep learning-based works, DTI prediction can be characterized as a binary classification, a ranking task or a regression task for binding affinity. Usually, deep learning models for DTI prediction are composed of a target feature extraction module, a drug feature extraction module and a prediction module. These modules are carefully designed according to various practical factors including the input representations.
The most commonly used representations of drugs and targets are one-dimensional (1D) sequences such as Simplified Molecular Input Line Entry Specification (SMILES) strings for drugs and amino acid sequences for targets (Karimi et al., 2019; Liu et al., 2020; Peng et al., 2020; Tsubaki et al., 2019; Zheng et al., 2020). The models with 1D sequences as input generally use Convolutional Neural network (CNN), Recurrent Neural Network (RNN), Long Short-Term Memory (LSTM) blocks or Gated Recurrent Unit (GRU) blocks to extract drug and target features. For instance, Ozturk et al. used CNN modules to extract hidden representations of amino acid sequences and SMILES strings, which were later combined and input to a multi-layer perceptron to perform the prediction (Ozturk et al., 2018). Besides sequences, feature vectors representing physicochemical properties of targets/drugs and drug molecular fingerprints are also common forms of 1D input to DTI prediction models (Lee et al., 2019; Lenselink et al., 2017; Rifaioglu et al., 2021). Slightly different from the work of Ozturk et al., instead of SMILES strings, Lee et al. used molecular fingerprints with fully connected networks to extract drug features to predict DTI prediction (Lee et al., 2019). According to the research of Lenselink et al. (2017), with feature vectors composed of physicochemical properties of targets/drugs and molecular fingerprints as input, Deep Neural Network (DNN) models outperform Naive Bayes, RF, SVM, logistic regression models in DTI prediction. Two-dimensional (2D) paired feature map has also been used for DTI prediction, which uses a matrix of a specific property calculated for amino acid pairs in the corresponding protein sequence to represent a target (Rifaioglu et al., 2021; Zheng et al., 2020). For example, Rifaioglu et al. used multi-channel protein feature maps involving sequence, structural, evolutionary and physicochemical properties to represent proteins for proteochemometric protein-drug binding affinity prediction (Rifaioglu et al., 2021). 2D structural images are intuitive representations of drugs and have been used in DTI prediction (Rifaioglu et al., 2020; Wang et al., 2021). Wang et al. proposed an efficient DTI prediction system using only 2D images as input, which includes CNN-based models for 704 targets (Rifaioglu et al., 2020). Two-dimensional (2D) molecular graph is another effective representation of drugs, which has been widely used in Graph Neural Network (GNN)-based models for predicting molecular properties (Gilmer et al., 2017; Song et al., 2020; Xiong et al., 2020; Yang et al., 2019; Zhang et al., 2021). Recently, molecular graph has also been increasingly used in DTI prediction (Nguyen et al., 2021; Torng and Altman, 2019; Tsubaki et al., 2019). Tsubaki et al. used GNN to extract the information of a small molecule as a feature vector, which is then concatenated with the target feature vector extracted by CNN from amino acid sequence to make prediction (Tsubaki et al., 2019). Compared with SMILES-based models, graph-based models can readily exploit topological information of molecules, which show obvious advantages in the task of DTI prediction.
Although 1D and 2D-based DTI prediction models have made significant progress recently, as DTI is in essence three-dimensional (3D) physical interaction, so it is natural and reasonable to predict DTI using three-dimensional structural information. Many studies directly use 3D Cartesian coordinates to represent the 3D structures of targets, but the limited samples cannot cover such a huge space, which leads to poor performance (Ragoza et al., 2017; Wallach et al., 2015; Zheng et al., 2019). Some models apply 3D voxel grids to represent targets and use 3D-CNN to extract target features, their accuracy is limited because the grid coordinates are not accurate enough to represent the spatial positions of targets’ atoms. On the other hand, 2D paired distance map represents 3D structure of each target by a matrix of internal pairwise distances between the amino acids of the target, which has been mainly used in protein structure prediction (Skolnick et al., 1997). Recently, Zheng et al. proposed an advanced model called drugVQA to predict DTI, where LSTM and dynamic 2D-CNN were used to extract features from SMILES strings and distance maps, respectively (Zheng et al., 2020), and achieved satisfactory performance. Though 2D paired distance map and molecular graph are promising representations of targets and drugs, respectively. However, they have not yet been jointly used for DTI prediction.
After extracting feature vectors from the drug and the target, respectively, the features are usually concatenated and input to a MLP to predict DTI. In most existing models, targets and drugs are represented and processed separately, they can hardly capture the interacting context between targets and drug molecules. The attention mechanism is often used to acquire the contributions of different components of a drug or target to the interaction (Karimi et al., 2019; Tsubaki et al., 2019; Zheng et al., 2020), which has been recently used to characterize the interactions between targets and drugs (Chen et al., 2020, 2021). The attention mechanism was found to be able to better capture the impact of targets on drugs and vice versa, so as to obtain better representations. However, these models pay attention only to the impact of one participant of a DTI on the counterpart, such as target on drug, but ignore the reverse impact. According to the induced-fit theory (Johnson, 2008), interacting drug molecules and targets are mutually impacted. Therefore, it is natural and reasonable to consider their mutual impacts when learning to represent the drugs and targets for DTI prediction.
In this article, to overcome the above-mentioned drawbacks of existing DTI prediction models, we propose a new model for DTI prediction. In this model, 2D paired distance maps of proteins and molecular graphs are served as inputs for targets and drugs, respectively. To capture the interactive impacts between targets and drugs, we design a mutual interaction neural network (MINN) by innovatively combining two interacting-transformers (Interformer in short) with an improved Communicative Message Passing Neural Network (CMPNN) (called Inter-CMPNN). In the experiments, our model achieves better performance than state-of-the-art methods on DUD-E, human and BindingDB benchmark datasets. Case studies show that individual contributions of residues in the target and atoms in the drug to the formation of DTI can be inferred from learned attention weights, which indicates that our proposed model is interpretable and can help to explain the drug action mechanism, and suggests the direction of drug optimization in the future.
2 Materials and methods
2.1 Overview
The architecture of our proposed model MINN-DTI is shown in Figure 1. It consists of three modules: a target preprocessing network (TPN), a MINN and an interaction prediction network (IPN). MINN is the core component of our model, which consists of an Interformer module and an Inter-CMPNN module. The Interformer module is constructed by two interacting transformer decoders, and the Inter-CMPNN module is a variant of CMPNN. With these two modules, we can extract the latent vectors of targets and drugs while considering their interacting contexts.
Fig. 1.

Architecture of MINN-DTI
The 2D distance map and 2D molecular graph are served as the input representation of a given target and a molecule, respectively. The molecular graph is directly used, while the distance map is firstly preprocessed by the target preprocessing network (TPN). The latent feature vectors of both the target and the molecule are extracted by MINN. These latent feature vectors are then concatenated and fed to the interaction prediction network (IPN) to predict DTI. The details of these modules are presented in the following sections.
2.2 Target preprocessing network
Given a target, we calculate a 2D paired distance map (Skolnick et al., 1997), which is subsequently preprocessed by the target preprocessing network (TPN) into a fixed-size matrix following drugVQA (Zheng et al., 2020). TPN is implemented by a dynamic attentive CNN. As shown in Figure 2, the dynamic attentive CNN is composed of a Dynamic CNN (DyCNN) block and a Sequential Self-Attention (SSA) block. The DyCNN block contains a number of residual blocks and an average pooling layer as ResNet (He et al., 2016). To handle targets with different lengths, the pooling layers between the residual blocks are eliminated. Through the DyCNN, a 2D paired distance map is transformed into a feature map , where f is the number of filters of the residual block. With the SSA block, a weight matrix is derived by a two-layer perceptron without bias from :
where and are learnable parameters. This multilayer perceptron (MLP) block can be regarded as a multi-head attention where the number of neurons r in the last layer is interpreted as the number of attentional heads. The attentional feature map is derived by multiplying and , which indicates the relative importance of amino acid sites.
Fig. 2.
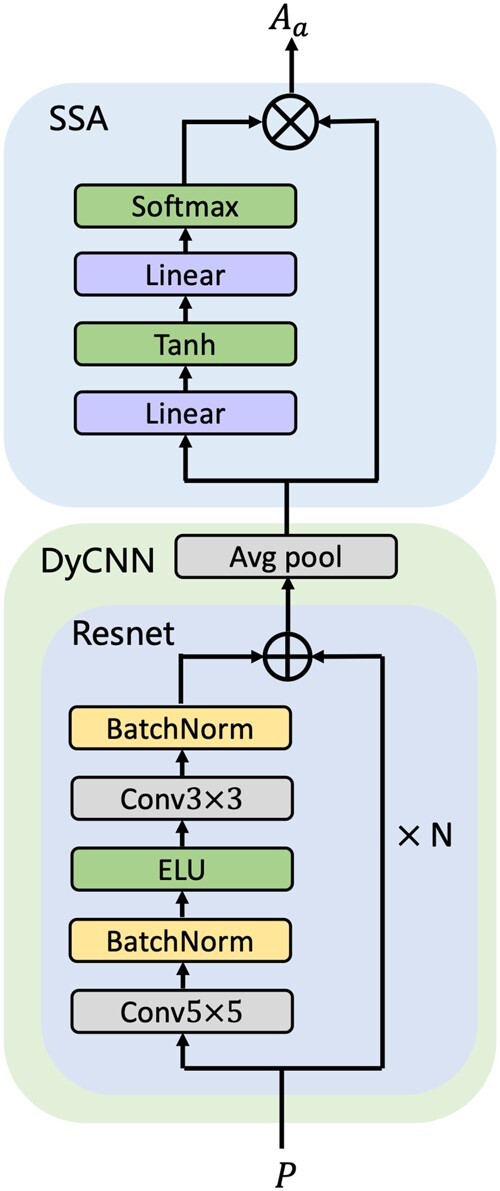
The structure of target preprocessing network
2.3 Mutual interaction neural network (MINN)
The goal of MINN is to learn the representations of targets and drugs while considering their interacting contexts. For this purpose, we design an Interformer module to interact the information extracted from targets and small molecules and an Inter-CMPNN module to support information interacting from the drug side. Thus, we can get more comprehensive representations of the targets and drugs, and consequently boost DTI prediction.
2.3.1. Interformer
Here, we use two interacting transformer decoders to extract feature vectors of targets and drugs, which is called Interformer in short. The structure of Interformer is shown in Figure 3. Each decoder of Interformer consists of one or more identical layers, similar to transformer (Vaswani et al., 2017). Each layer of Interformer consists of three sublayers: a multi-head self-attention layer, an interaction attention layer and a fully connected feed-forward network. The multi-head self-attention sublayer and the feed-forward sublayer are essentially consistent with transformer, except that the mask operation is eliminated to leverage complete drug and target information following the work of Chen et al. (2020).
Fig. 3.
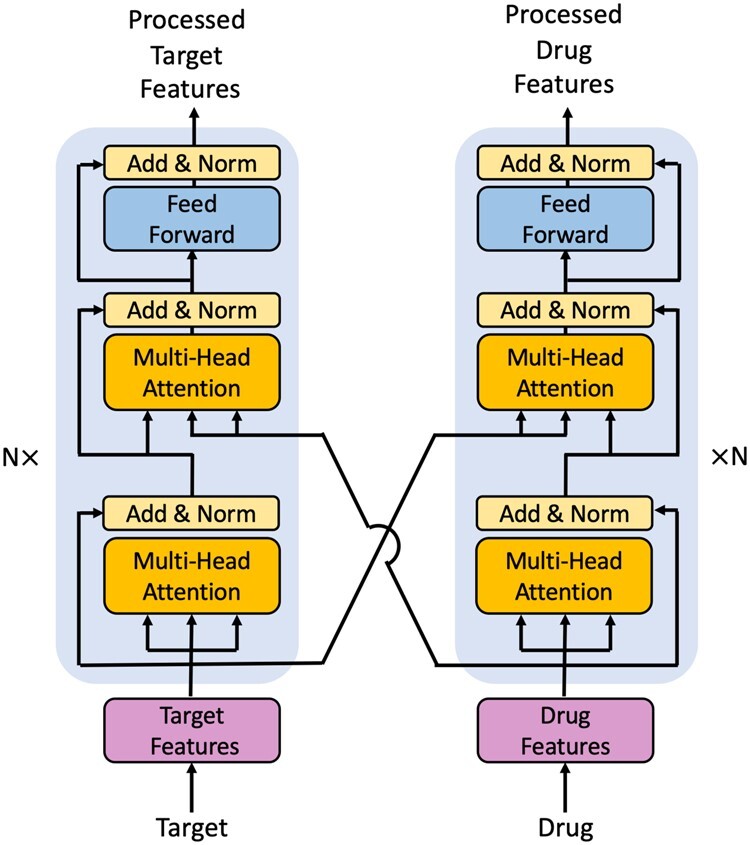
The structure of Interformer
The interaction attention layer in each decoder of Interformer adopts a multi-head scaled dot attention block to receive the external information from another decoder. The source of external information is the biggest difference between interaction attention layer of Interformer and encode-decoder layer of transformer, where the source of external information is the encoder. A scaled-dot attention block can be expressed as:
where Q is the linearly transformed output of the multi head self-attention layer of the decoder, K and V are linearly transformed outputs of the last layer of another transformer decoder, and is the dimension of K and V.
where , , and are parameter matrices. With the Interformer, the representation of each small molecule/target is involved with the information of the corresponding interacted target/drug, which conforms to the real situation of DTI.
2.3.2. Inter-CMPNN
The Inter-CMPNN module is implemented by an improved CMPNN, which is a variant of message passing neural network based on directed graph (Song et al., 2020). CMPNN strengthens the message interaction between nodes and edges through three well designed modules (AGGREGATE, COMMUNICATE, UPDATE) for L iterations:
where is the message obtained by node v in iteration k, is the hidden representation of node v in iteration , is the hidden representation of edge e in iteration . After L iterations, one more iteration is executed to exchange information more thoroughly:
where is the raw features of atoms. The module incorporates a message booster, which processes the information of edges with a maximum pooling layer and calculates the element-wise product of the maximum pooling layer’s result and the sum of the hidden representations of edges. The module is a single-layer neural network with a skip connection, and the module takes the form of a multilayer perception.
Here, an Interformer is adopted after the function to completely exploit the mutual impacts between targets and drug molecules:
where is the target feature map in iteration . Similarly, one more iteration is executed:
where and is the final drug graph feature and target feature map. The schematic diagram of message interaction between Interformer and Inter-CMPNN is shown in Figure 4. The last hidden atom representations of each molecular graph and the feature map vectors of each target are averaged to obtain fix-sized vectors of the target and the small molecule, which are then fed to the interaction prediction network.
Fig. 4.
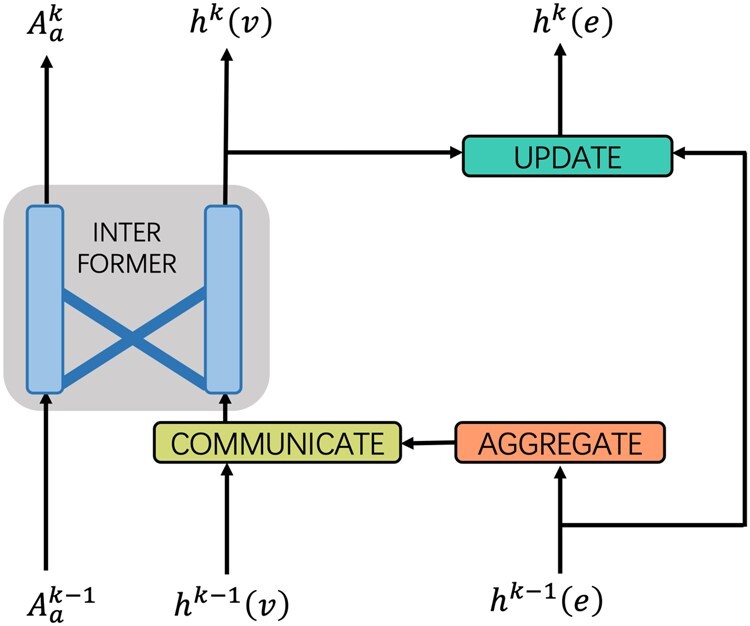
Message interaction between Interformer and Inter-CMPNN
2.4 Interaction prediction network (IPN)
The obtained target feature vector T and small molecule feature vector D are concatenated and fed to a two-layer perceptron without bias to obtain the prediction result:
where and are learnable weight parameters. Since the prediction of DTI is regarded as a binary classification problem, cross entropy is used as the loss function to train the model.
3 Experiments and results
3.1 Datasets
We evaluated our model MINN-DTI and compared it with state-of-the-art DTI prediction methods on three widely used public datasets, human dataset, DUD-E dataset and BindingDB dataset.
3.1.1 The DUD-E dataset
The DUD-E dataset consists of 22 886 active compounds against 102 targets. For each active compound, 50 decoys are generated, which have similar physico-chemical properties but dissimilar 2-D topologies to the active compound (Mysinger et al., 2012). We processed the DUD-E dataset following the works of Zheng et al. (2020) and Ragoza et al. (2017), we obtained 22 645 positive examples and 1 407 145 negative examples, which were split according to a threefold cross-validation strategy. Ligands for the targets belonging to the same target family are put into the same fold. We randomly selected the same number of negative samples as the active samples in training to obtain a balanced model, but we used unbalanced data in model evaluation.
3.1.2. The human dataset
The human dataset contains highly credible positive and negative CPI samples extracted by a systematic screening framework according to a similarity rule (Liu et al., 2015). Following the works of Zheng et al. (2020) and Tsubaki et al. (2019), we used a dataset with equal number of positive and negative samples, forming 3369 positive interactions between 1052 unique compounds and 852 unique targets. Then, the dataset was randomly divided into a training set, a validation set and a test set according to the ratio of 8:1:1. To evaluate the generalization power of our model on different data, we redivided the human dataset according to molecular scaffold similarities between drugs. The scaffold-based split method implemented in the open source DeepChem package of MoleculeNet (Wu et al., 2018) was used to divide structurally different molecules into training/validation/test sets according to the ratio of 8:1:1.
3.1.3. The BindingDB dataset
The BindingDB dataset (Gao et al., 2018) was a customized subset of the Binding database (Gilson et al., 2016), which is a publicly accessible database that mainly contains the interaction affinities between targets and drug-like small molecules. The BindingDB dataset contains 39 747 positive samples and 31 218 negative samples, which was divided into a large training set (50 155 samples), a validation set (5607 samples) and a test set (5508 samples). To evaluate the generalization power of our model on novel targets, the data of the test set were divided into two parts to check the model performance according to whether the targets appear in the training set.
3.2 Implementation details and experimental settings
We implemented MINN-DTI with Pytorch 1.7.1 (Paszke et al., 2019). The Adam optimizer was used in training and the learning rate was set to 0.0001 (Kingma and Ba, 2015). The number of residual blocks, the number of filters and the dimension of molecular graph features were set to 32. We explored hyperparameters on the human dataset, and the best parameters are listed in Table 1. Following the work of Zheng et al. (2020), hyperparameter optimization was not performed for the DUD-E and BindingDB datasets. All experiments were conducted on NVIDIA RTX3090 GPUs.
Table 1.
Hyperparameter setting in MINN-DTI
| Hyperparameter | Value |
|---|---|
| Learning rate | 0.0001 |
| Number of residual blocks | 32 |
| Number of filters | 32 |
| Graph feature size | 32 |
| Attention heads | 8 |
| Hidden size of Decoder | 32 |
| Iterations of message passing | 4 |
| Dropout | 0.2 |
3.3 Performance metrics
Here, different metrics were used on different datasets following previous works (Chen et al., 2020; Zheng et al., 2020). The area under the receiver operating characteristic curve (AUC) was used as the main metric to evaluate our model. Besides, the ROC enrichment metric (RE) that describes the ratio of the true positive rate (TPR) to the false positive rate (FPR) at a given FPR threshold was used for performance assessment on the DUD-E dataset, where FPR was set to 0.5%, 1%, 2% and 5%, respectively as the threshold. Moreover, recall and precision were used for performance evaluation on the human dataset, while the area under precision recall curve (PRC) was applied to performance evaluation on the BindingDB dataset. We repeated each experiment three times with different seeds to calculate the mean and the standard deviation as in the work of Zheng et al. (2020).
3.4 Results
3.4.1. Performance on the DUD-E dataset
Here, we compared our model with different types of existing methods on DUD-E. These compared methods include two traditional machine learning-based methods NNscore (Durrant and McCammon, 2011) and RF-score (Ballester and Mitchell, 2010), a docking-based method Vina (Trott and Olson, 2009) and three recent deep learning-based methods 3D-CNN (Ragoza et al., 2017), PocketGCN (Torng and Altman, 2019) and DrugVQA (Zheng et al., 2020). As shown in Table 2, MINN-DTI has significant advantage in terms of AUC and RE. The AUC of MINN-DTI is about 2.5% higher than that of the state-of-the-art method DrugVQA, and over 10% higher than that of the other methods. In terms of RE, MINN-DTI is at least twice as high as all the other methods. These results show that our method is more effective in drug screening, since the number of positive samples in DUD-E is much less than that of negative samples, which is close to the actual situation of virtual screening. In addition, deep learning-based methods are obviously superior to the descriptor-based traditional machine learning methods and the docking-based method in terms of AUC and RE, which suggests that deep learning-based methods are more effective in learning the representations of drugs and targets.
Table 2.
Performance comparison between our model and existing methods on the DUD-E dataset
| Model | AUC | 0.5% RE | 1.0% RE | 2.0% RE | 5.0% RE |
|---|---|---|---|---|---|
| NNscorea | 0.584 | 4.166 | 2.980 | 2.460 | 1.891 |
| RF-scorea | 0.622 | 5.628 | 4.274 | 3.499 | 2.678 |
| Vinaa | 0.716 | 9.139 | 7.321 | 5.811 | 4.444 |
| 3D-CNNa | 0.868 | 42.559 | 26.655 | 19.363 | 10.710 |
| PocketGCNa | 0.886 | 44.406 | 29.748 | 19.408 | 10.735 |
| DrugVQAa | 0.972 ± 0.003 | 88.17 ± 4.88 | 58.71 ± 2.74 | 35.06 ± 1.91 | 17.39 ± 0.94 |
| MINN-DTI | 0.992 ± 0.007 | 175.89 ± 12.02 | 90.77 ± 5.81 | 46.49 ± 2.63 | 19.10 ± 0.71 |
Note: The first row, the percentage before RE is the given threshold of FPR. Best results of the corresponding experiments were represented in bold.
Means results obtained from the article (Zheng et al., 2020).
3.4.2. Performance on the human dataset
Here, we compared our method with nine existing methods on the human dataset to further evaluate our model, including k-nearest neighbor (k-NN), random forest (RF), L2-logistic (L2), support vector machine (SVM), graph neural network (GNN) (Tsubaki et al., 2019), graph convolution network (GCN), GraphDTA (Nguyen et al., 2021), TransformerCPI (Chen et al., 2020) and DrugVQA (Zheng et al., 2020). The results are presented in Table 3, from which we can see that our model is 0.2% better than the state-of-the-art method DrugVQA in AUC. However, in terms of recall and precision, SVM is the best. Actually, it is difficult to objectively and completely evaluate the performance of a model by using only recall and precision, as the performance of SVM is generally inferior to deep learning models according to many existing works. Given the reliability of AUC, we can still claim that our method is the best one.
Table 3.
Performance comparison between our model and existing methods on the random splitting human dataset
| Model | AUC | Recall | Precision |
|---|---|---|---|
| k-NNa | 0.860 | 0.927 | 0.798 |
| RFa | 0.940 | 0.897 | 0.861 |
| L2a | 0.911 | 0.913 | 0.861 |
| SVMb | 0.910 | 0.966 | 0.969 |
| GNNa | 0.970 | 0.918 | 0.923 |
| GCNb | 0.956 ± 0.004 | 0.862 ± 0.006 | 0.928 ± 0.010 |
| GraphDTAb | 0.960 ± 0.005 | 0.882 ± 0.040 | 0.912 ± 0.040 |
| TransformerCPIb | 0.973 ± 0.002 | 0.916 ± 0.006 | 0.925 ± 0.006 |
| DrugVQAa | 0.979 ± 0.003 | 0.961 ± 0.002 | 0.954 ± 0.03 |
| MINN-DTI | 0.981 ± 0.003 | 0.945 ± 0.030 | 0.902 ± 0.045 |
Means results obtained from the article (Zheng et al., 2020).
Means results obtained from the article (Chen et al., 2020).
Best results of the corresponding experiments were represented in bold.
A newly constructed dataset with a scaffold-based split was also used to train and test the above-mentioned methods. As shown in Table 4, our model consistently exceeds all the competitors in AUC. Compared to the results on the random splitting human dataset, the performance of all models on the scaffold splitting human dataset degrades. However, our model has the slightest drop in AUC, i.e. 1.4%, about half of that of the second-place DrugVQA. These results suggest that more comprehensive extraction of DTI information could help enhance our model's ability to identify novel interactions.
Table 4.
Performance comparison between our model and existing methods on the scaffold-based splitting human dataset
| Model | AUC | Recall | Precision |
|---|---|---|---|
| k-NN | 0.841 | 0.803 | 0.892 |
| RF | 0.885 | 0.890 | 0.832 |
| L2 | 0.881 | 0.832 | 0.827 |
| SVM | 0.892 | 0.857 | 0.883 |
| GNN | 0.921 ± 0.002 | 0.843 ± 0.004 | 0.855 ± 0.003 |
| GCN | 0.905 ± 0.010 | 0.823 ± 0.012 | 0.902 ± 0.008 |
| GraphDTA | 0.926 ± 0.008 | 0.855 ± 0.004 | 0.898 ± 0.006 |
| TransformerCPI | 0.948 ± 0.005 | 0.930 ± 0.003 | 0.933 ± 0.005 |
| DrugVQA | 0.952 ± 0.002 | 0.925 ± 0.004 | 0.928 ± 0.006 |
| MINN-DTI | 0.967 ± 0.003 | 0.922 ± 0.013 | 0.931 ± 0.021 |
Best results of the corresponding experiments were represented in bold.
3.4.3. Performance on the BindingDB dataset
We also compared our model with GCN, GNN (Tsubaki et al., 2019), GraphDTA (Nguyen et al., 2021), DrugVQA (Zheng et al., 2020) and TransfomerCPI (Chen et al., 2020) on the BindingDB dataset. As shown in Table 5, our model achieves the highest AUC and PRC, which is in line with our expectation. Although this advantage is not as obvious as that on the DUD-E dataset, considering that AUC has less room to improve at the level of more than 95% on these datasets, such progress is still considerable.
Table 5.
Performance comparison between our model and existing methods on the BindingDB dataset
| Task | AUC | PRC |
|---|---|---|
| GNN | 0.909 ± 0.002 | 0.901 ± 0.005 |
| GCN | 0.912 ± 0.003 | 0.907 ± 0.002 |
| GraphDTA | 0.923 ± 0.003 | 0.916 ± 0.004 |
| DrugVQA | 0.936 ± 0.005 | 0.928 ± 0.007 |
| TransformerCPI | 0.950 ± 0.002 | 0.949 ± 0.005 |
| MINN-DTI | 0.961 ± 0.009 | 0.956 ± 0.002 |
Best results of the corresponding experiments were represented in bold.
We further investigated the generalization capability of our models on the test subsets with/without training targets, the results are shown in Figure 5. We can see that our model performs better than the other models regardless of whether or not the tested targets are in the training set. Our model achieves an AUC of 0.972 ± 0.008 and a PRC of 0.957 ± 0.002 when test targets are in the training set. While for new targets, our model still maintains an AUC of 0.957 ± 0.009 and a PRC of 0.951 ± 0.002. However, the other methods all show a larger drop of performance than MINN-DTI on the subset without the training targets, which means that the performance gap between MINN-DTI and these methods is widened. These results suggest that the generalization ability of our model on new targets is higher than that of these existing methods.
Fig. 5.
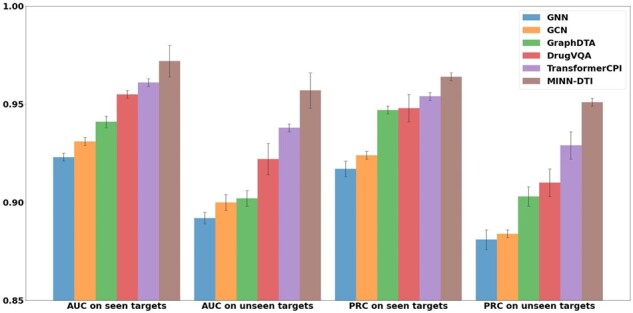
Performance comparisons on seen and unseen protein targets from the BindingDB dataset. Error bars indicate the standard deviations
3.5 Ablation study
We conducted ablation studies on the DUD-E and human datasets to check the effectiveness of different configurations of MINN-DTI. All experiments were repeated three times on the divided datasets used above with different seeds to calculate the mean and the standard deviation. Models with different configurations and their performance are presented in Table 6. The first model concatenates the output vectors of DynCNN and CMPNN and uses MLP to predict DTI. The second model replaces Interformer in MINN-DTI with transformer. There are two possible settings: the encoder of transformer receives the information of protein, the decoder of transformer receives the information of drug, or vice versa. For mimicking and competing with Inter-CMPNN, the first setting was used to extract drug information during each round of message passing with the decoder in second model. For the third model, only an Interformer is deployed behind the whole CMPNN module instead of deploying an Interformer during each round of message passing using multiple Interformer as MINN-DTI. As shown in Table 6, MINN-DTI maintains the best performance on both datasets and the model without Interformer module (the first model) performs worst. The performance of the third model is close to the second model, with a difference of only 0.002/0.003 in AUC. However, the results are different on the two datasets. The second model is better than the third model on the human dataset, while the opposite is true on the DUD-E dataset. We speculate that the main reason for the different results may be due to the different data distribution of the two datasets: the test set of DUD-E has unbalanced positive and negative samples, while human dataset is balanced. The second and third models achieve significantly improved performance compared with the first model, which indicates that adopting transformer or Interformer to extract the interaction information between drug and target is helpful for DTI prediction. The second model is still much worse than MINN-DTI, which shows that the Interformer model is more effective than transformer in DTI prediction. As MINN-DTI shows considerable performance advantage over the third model, we believe that the iterative use of Interformer in Inter-CMPNN is beneficial to the extraction of drug and protein interaction information. The ablation results show that MINN-DTI combining Interformer with Inter-CMPNN can indeed improve the prediction performance to a considerable extent.
Table 6.
Ablation results on the DUD-E and human datasets
| Model | DUD-E | Human |
|---|---|---|
| Without Interformer | 0.953 ± 0.003 | 0.967 ± 0.004 |
| Transformer & CMPNN | 0.975 ± 0.004 | 0.971 ± 0.007 |
| Single Interformer | 0.978 ± 0.006 | 0.969 ± 0.005 |
| MINN-DTI | 0.992 ± 0.007 | 0.981 ± 0.003 |
Best results of the corresponding experiments were represented in bold.
3.6 Interpretability and visualization
Two representative DTIs were selected to illustrate the interpretability of our model. By inputting the target distance map and the compound graph representations, the model generates multi-head attentions of the target and the compound, and the weights of each amino acid residue and each atom in the target and drug molecule are calculated to identify their importance in the prediction. Protein distance maps and attention bars representing attention weights of residues and ligand atoms are shown in Figure 6. Here, we visualize the top-5 weighted residues of the example target as pink skeletons, and highlight the top-10 weighted atoms by red dots. We found that the significantly weighted residues and atoms are highly consistent with the actual interacting residues and atoms. In CXCR4 protein co-crystal complex 3ODU, GLU288, ASP97 and CYS186 form polar interactions with the drug molecule, and HIS113 and TYR116 form hydrophobic interactions with the drug molecule. These residues and atoms that form the interactions have larger attention weights, which can also be seen in the target ALK5 with co-crystal 3HMM. Meanwhile, there are also residues or atoms with low weight, which are considered to have low contribution to the formation of interactions. For example, in the drug attention bar of complex 3ODU, atoms 21–26 of ligand have small weights, and this group is actually exposed to the solvent without participating in the formation of interaction. The above exploration shows that our model can learn and highlight the important target amino acid residues and drug atoms. This helps us to understand DTIs more comprehensively, which is beneficial to the study on the structure-activity relationship and action mechanism of drugs.
Fig. 6.

Attention visualization of DTIs. Left: Protein distance maps are displayed in the form of heat maps. The corresponding targets’ attention bars are shown. Middle: Ligands and predicted important residues are represented as green and pink skeletons, respectively. Predicted important atoms of ligands are highlighted in red. Known hydrogen bonds are marked with yellow dashed lines. Local target structures are painted grey as the background. Right: Ligands are represented by 2D Kekule formula. The corresponding predicted important atoms are highlighted by light red dots. Ligands’ attention bars are shown (A color version of this figure appears in the online version of this article.)
4 Conclusion
In this article, we proposed a novel model MINN-DTI to boost DTI prediction by comprehensively mining the mutual impacts between the targets and drugs, which are represented by protein distance maps and molecular graphs, respectively. To this end, a MINN was designed by combining a newly designed Interformer with an improved CMPNN (called Inter-CMPNN) to capture the interacting context of drugs and targets. Compared with the existing models, MINN-DTI achieves the best performance on three public datasets, due to the fact that MINN-DTI can more effectively exploit the interacting information between targets and drugs. We believe that the Interformer and Inter-CMPNN-based MINN should be also effective in other related tasks, including target–target, target–peptide and drug–drug interaction prediction.
Funding
F.L., Z.Z. and S.Z. were partially supported by 2021 Tencent AI Lab Rhino-Bird Focused Research Program [JR202104] and National Natural Science Foundation of China (NSFC) [61972100]; J.G. was supported by NSFC [61772367].
Conflict of Interest: none declared.
Contributor Information
Fei Li, School of Computer Science, Fudan University, Shanghai 200438, China.
Ziqiao Zhang, School of Computer Science, Fudan University, Shanghai 200438, China.
Jihong Guan, Department of Computer Science and Technology, Tongji University, Shanghai 201804, China.
Shuigeng Zhou, School of Computer Science, Fudan University, Shanghai 200438, China; Shanghai Key Lab of Intelligent Information Processing, Shanghai 200438, China.
Reference
- Bagherian M. et al. (2021) Machine learning approaches and databases for prediction of drug–target interaction: a survey paper. Brief Bioinform., 22, 247–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester P.J., Mitchell J.B. (2010) A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics, 26, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleakley K., Yamanishi Y. (2009) Supervised prediction of drug–target interactions using bipartite local models. Bioinformatics, 25, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. et al. (2020) TransformerCPI: improving compound-protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics, 36, 4406–4414. [DOI] [PubMed] [Google Scholar]
- Chen W. et al. (2021) Predicting drug–target interactions with deep-embedding learning of graphs and sequences. J. Phys. Chem. A, 125, 5633–5642. [DOI] [PubMed] [Google Scholar]
- Durrant J.D., McCammon J.A. (2011) NNScore 2.0: a neural-network receptor–ligand scoring function. J. Chem. Inf. Model., 51, 2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K.Y. et al. (2018) Interpretable drug target prediction using deep neural representation. In: Proceedings of the Twenty-Seventh International Joint Conference on Artificial Intelligence (IJCAI-18), Stockholm, Sweden, pp. 3371–3377. [Google Scholar]
- Gilmer J. et al. (2017) Neural message passing for quantum chemistry. Proc. Mach. Learn. Res., 70, 1263–1272. [Google Scholar]
- Gilson M.K. et al. (2016) BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res., 44, D1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K. et al. (2016) Deep residual learning for image recognition. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, Nevada. pp. 770–778. [Google Scholar]
- Johnson K.A. (2008) Role of induced fit in enzyme specificity: a molecular forward/reverse switch. J. Biol. Chem., 283, 26297–26301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M. et al. (2019) DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics, 35, 3329–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P. and Ba J. (2015) Adam: a method for stochastic optimization. In: Proceedings of the 3rd International Conference on Learning Representations (ICLR 2015), San Diego, CA. [Google Scholar]
- Lee I. et al. (2019) DeepConv-DTI: prediction of drug–target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol., 15, e1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenselink E.B. et al. (2017) Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform., 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2015) Improving compound-protein interaction prediction by building up highly credible negative samples. Bioinformatics, 31, i221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2020) HNet-DNN: inferring new drug–disease associations with deep neural network based on heterogeneous network features. J. Chem. Inf. Model., 60, 2367–2376. [DOI] [PubMed] [Google Scholar]
- Mysinger M.M. et al. (2012) Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem., 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. et al. (2021) GraphDTA: predicting drug–target binding affinity with graph neural networks. Bioinformatics, 37, 1140–1147. [DOI] [PubMed] [Google Scholar]
- Ozturk H. et al. (2018) DeepDTA: deep drug–target binding affinity prediction. Bioinformatics, 34, i821–i829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszke A. et al. (2019) PyTorch: an imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. (Nips 2019), 32, 8026–8037. [Google Scholar]
- Peng Y. et al. (2020) TOP: a deep mixture representation learning method for boosting molecular toxicity prediction. Methods, 179, 55–64. [DOI] [PubMed] [Google Scholar]
- Ragoza M. et al. (2017) Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model., 57, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifaioglu A.S. et al. (2020) DEEPScreen: high performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem Sci., 11, 2531–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifaioglu A.S. et al. (2021) MDeePred: novel multi-channel protein featurization for deep learning-based binding affinity prediction in drug discovery. Bioinformatics, 37, 693–704. [DOI] [PubMed] [Google Scholar]
- Skolnick J. et al. (1997) MONSSTER: a method for folding globular proteins with a small number of distance restraints. J. Mol. Biol., 265, 217–241. [DOI] [PubMed] [Google Scholar]
- Song Y. et al. (2020) Communicative representation learning on attributed molecular graphs. In: Proceedings of the 29th International Joint Conference on Artificial Intelligence (IJCAI 2020), Yokohama, Japan, pp. 2831–2838. [Google Scholar]
- Su M. et al. (2019) Comparative assessment of scoring functions: the CASF-2016 update. J. Chem. Inf. Model., 59, 895–913. [DOI] [PubMed] [Google Scholar]
- Tian K. et al. (2016) Boosting compound–protein interaction prediction by deep learning. Methods, 110, 64–72. [DOI] [PubMed] [Google Scholar]
- Torng W., Altman R.B. (2019) Graph convolutional neural networks for predicting drug–target interactions. J. Chem. Inf. Model., 59, 4131–4149. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubaki M. et al. (2019) Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics, 35, 309–318. [DOI] [PubMed] [Google Scholar]
- Vaswani A. et al. (2017) Attention is all you need. In: Proceedings of 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA. [Google Scholar]
- Wallach I. et al. (2015) AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv preprint arXiv:1510.02855.
- Wang X. et al. (2021) CSConv2d: a 2-D structural convolution neural network with a channel and spatial attention mechanism for protein–ligand binding affinity prediction. Biomolecules, 11, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. et al. (2018) MoleculeNet: a benchmark for molecular machine learning. Chem. Sci., 9, 513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z. et al. (2020) Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem., 63, 8749–8760. [DOI] [PubMed] [Google Scholar]
- Yang K. et al. (2019) Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model., 59, 3370–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. et al. (2021) FraGAT: a fragment-oriented multi-scale graph attention model for molecular property prediction. Bioinformatics, 37, 2981–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S. et al. (2019) Identifying structure-property relationships through SMILES syntax analysis with self-attention mechanism. J. Chem. Inf. Model., 59, 914–923. [DOI] [PubMed] [Google Scholar]
- Zheng S. et al. (2020) Predicting drug–protein interaction using quasi-visual question answering system. Nat. Mach. Intell., 2, 134–140. [Google Scholar]