

Abstract
In this Letter, we adapt a recently reported Pd-catalyzed transannular C(sp3)–H arylation of alicyclic amines for applications in fragment-based drug discovery (FBDD). We apply this method to the synthesis of a series of 6-arylated 3-azabicyclo[3.1.0]hexanes that are rule-of-three compliant fragments. Several modifications were made to the Pd-catalyzed C–H arylation method to enhance its utility in fragment synthesis. These include the use of microwave heating to shorten reaction times to under 1 h and the development of new approaches for directing group cleavage. Finally, we demonstrate that this fragment library falls within desirable physicochemical space for FBDD applications.
Keywords: directed C–H functionalization, fragment based drug discovery, amines, palladium, arylation, microwave, directing group
Graphical Abstract
We recently reported a Pd-catalyzed method for the remote C(sp3)–H arylation of alicyclic amines.1,2 This method was applied to the selective functionalization of a variety of bioactive molecules, and could thus find application in medicinal chemistry for the late-stage modification of drug candidates. Another potential application in the earlier stages of drug discovery would involve the rapid assembly of small molecules for fragment-based drug discovery (FBDD). Over the past decade, FBDD has become an increasingly important approach for the identification of pharmaceutical lead compounds.3 However, a key limitation of current fragment libraries is that they are dominated by sp2-rich, planar compounds. As such, there is significant demand for fragments containing three-dimensional character, and alicyclic amine-based fragments would be particularly desirable in this context.4,5
We sought to apply our Pd-catalyzed C(sp3)–H arylation of alicyclic amines to the assembly of 3D fragments for FBDD.1 However, in order to rapidly access the desired fragments, we needed to address several challenges associated with the existing method. First, the current method requires relatively long reaction times (18 h), which impedes the rapid synthesis of analogues. Second, reaction conditions must be identified that are compatible with diverse coupling partners, particularly those containing heteroaromatics and halogenated functional groups. Finally, conditions must be established to rapidly remove the directing group tethered to nitrogen in order to release the desired amines.
Herein, we focus on addressing these challenges in the context of the transannular C(sp3)–H functionalization of 3-azabicyclo[3.1.0]hexane to generate derivatives bearing aryl groups at the C-6 position. Notably, 3-azabicyclo[3.1.0]hexane serves as the core of a variety of bioactive molecules.6 C-6-arylated derivatives are attractive fragments for FBDD, as they are rule-of-three compliant (MW < 300; ClogP < 3; hydrogen bond donors/acceptors < 3; rotatable bonds < 3, and TPSA < 60 A2)7 (Scheme 1), and they have a high degree of saturation and 3-D topology. Traditionally, the assembly of 3-azabicyclo[3.1.0]hexane derivatives bearing functional groups at C-6 required long synthetic sequences.6b,d In contrast, the Pd-catalyzed transannular C–H arylation method enables selective functionalization at C-6 in just three steps: (1) directing group installation, (2) transannular C(sp3)–H arylation, and (3) directing group removal.
Scheme 1.

Proposed synthesis of fragments through C–H functionalization
This work aims to streamline the assembly of 3-azabicyclo[3.1.0]hexane fragments by: (1) decreasing reaction times in order to increase reaction throughput and (2) increasing the efficiency and practicality of directing group removal. These advances have enabled the preparation of a variety of derivatives, whose physicochemical properties have been calculated.
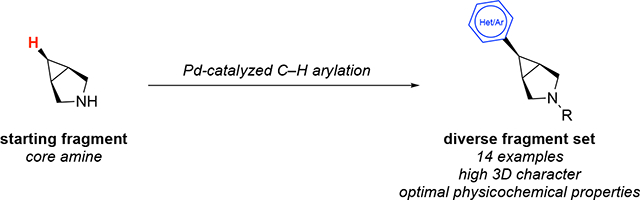
The original conditions for the C–H arylation of the 3-azabicyclo[3.1.0]hexane substrate S1 required 18 h at 130 °C and also involved reaction set-up in a N2-atmosphere glovebox. Thus, we first sought to develop conditions to accelerate these transformations and to eliminate the need for a glovebox.1a,8 As shown in Figure 1, we found that the Pd-catalyzed C–H arylation of S1 proceeds in less than 1 h at 180 °C with microwave heating to afford 1 in 68% yield. Importantly, this reaction could be set up on the bench-top and then flushed with N2 for 1 min prior to heating. The yield under these conditions compares favorably to that obtained in the original report (74%).1a Furthermore, an improved yield of 83% was obtained when the scale was doubled from 0.52 to 1.04 mmol of S1.9
Figure 1.
Substrate scope with modified reaction conditions. aReaction conducted with S-1 (0.52 mmol). b Reaction conducted with S-1 (1.04 mmol). c Yields from the original conditions reported in reference 1a. d Reaction conducted for 100 min.
The scope of aryl and heteroaryl iodide coupling partners was next explored under these modified reaction conditions. A variety of aryl iodides proved compatible, providing 6-aryl-3-azabicyclo[3.1.0]hexanes 2-9 in moderate to good yields (Figure 1). Aryl iodides containing halogen, ether, and unprotected phenol groups could be employed under the microwave conditions. A variety of substituted pyridyl and quinolinyl iodides also reacted to afford modest to good yields of 10-16 (Figure 1).
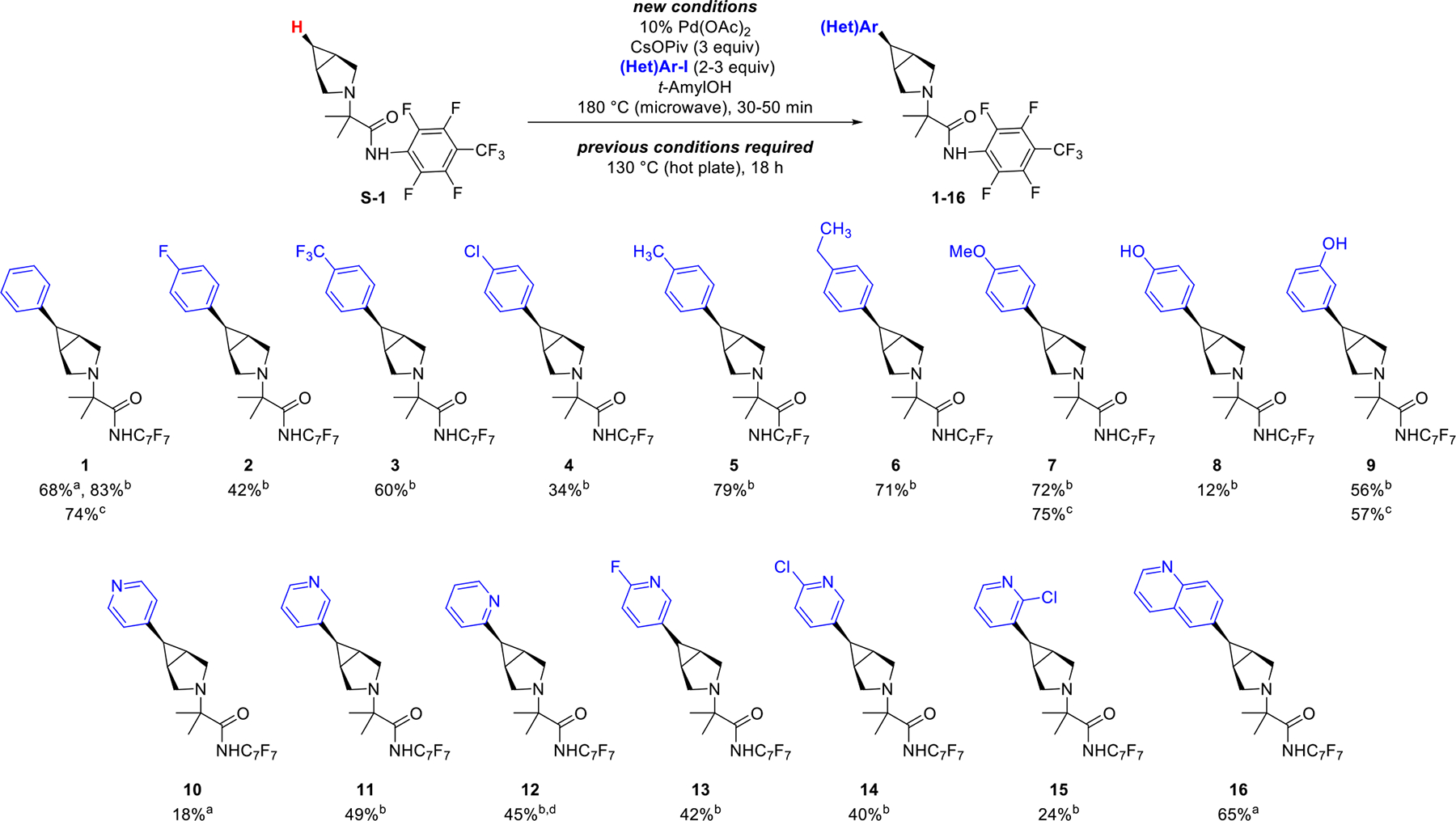
Products 1–16 would be highly attractive scaffolds for FBDD following cleavage of the fluoroamide directing group. As such, we next focused on developing practical methods for directing group removal. The original procedure involved reductive cleavage with SmI2, subsequent in situ protection of the 2° amine with pivaloyl chloride, and then isolation of the resultant amide.1a A first issue with these conditions is the requirement for superstoichiometric quantities of toxic HMPA as an activator for SmI2.10 An second limitation is that this process yields amide products rather than the target amines. As such, we first targeted a modified procedure that replaces HMPA with a less toxic additive and also enables the direct isolation of amine products.
We explored alternative activators for SmI2 and identified tripyrrolidinophosphoric acid triamide (TPPA) as a viable replacement for HMPA.11 With this new activator, the directing group cleavage proceeds within 3 h at room temperature, compared to 24 h under the previous conditions. Further experimentation revealed that the 2° amine product can be isolated by changing the work-up procedure. Instead of adding pivaloyl chloride at the end of the SmI2 reaction, the crude mixture was subjected to an aqueous work-up, and the product was then purified via reverse-phase HPLC. This procedure enabled isolation of a variety of 2° amine products in yields ranging from 34–51% (Table 1).12,13 Notably, a slightly modified work-up involving a Boc-protection step was required for the highly polar product 9A.
Table 1.
Removal of directing group via reductive cleavage with SmI2
| ||
|---|---|---|
| Entry | Product | Yield |
| 1 | 4-Ph (1A) | 34% |
| 2 | 4-F-Ph (2A) | 48% |
| 3 | 4-CF3-Ph (3A) | 44% |
| 4 | 4-Me-Ph (5A) | 49% |
| 5 | 4-Et-Ph (6A) | 39% |
| 6 | 4-MeO-Ph (7A) | 51% |
| 7 | 3-OH-Ph (9A) | 38%a |
| 8 | 3-Pyr (11A) | 0% |
Deprotection followed by protection Boc2O and deprotection.
A limitation of the SmI2-mediated directing group cleavage process is that it is incompatible with substrates containing aryl halide functional groups.14 Additionally, pyridine derivatives such as 11 undergo heteroarene reduction in the presence of SmI2.15 As such, we developed a complementary procedure to remove the directing group from these substrates. We hypothesized that acylation of the basic nitrogen could facilitate subsequent dealkylation of the fluoroamide directing group.16 A major challenge for this process is that the nitrogen center is very sterically hindered; as such, acylation requires relatively forcing reaction conditions. However, after some optimization, we found that the treatment of 1 with neat acetyl chloride under microwave heating at 150 °C for 3 h affords the amide product 1B in 35% yield as determined by 1H NMR spectroscopic analysis of the crude reaction mixture. 1B was isolated in 25% yield from this reaction.17
These conditions proved effective for cleaving the directing group from 1 and 4 as well as many of the pyridine-containing derivatives in modest yields (12–25%, Table 2). In the case of 10, complete consumption of starting material was observed, but the desired product 10B was not detected. This appears to be due to decomposition of 10 under these conditions, as the solution turned dark upon the addition of the acylating reagent. Notably, the aryl-Cl bonds of 4B, 14B, and 15B were compatible with these conditions (Table 2). However, product 13B was not isolable due to competing nucleophilic aromatic substitution to generate the 2-chloro product 14B.
Table 2.
Removal of directing group through acylative dealkylation
| ||
|---|---|---|
| Entry | Product | Yield |
| 1 | 4-Ph (1B) | 25% |
| 2 | 4-Cl-Ph (4B) | 25% |
| 3 | 4-Pyr (10B) | 0% |
| 4 | 3-Pyr (11B) | 19% |
| 5 | 2-Pyr (12B) | 19% |
| 6 | 2-F-5-Pyr (13B) | 0% |
| 7 | 2-Cl-5-Pyr (14B) | 19% |
| 8 | 2-Cl-3-Pyr (15B) | 12% |
| 9 | 6-quinoline (16B) | 23% |
We next assessed the physicochemical properties of the molecules in Tables 1 and 2 to assess their suitability for applications in FBDD.18 The calculations confirm that these 14 molecules fall within the ideal range for fragments, as defined by the restrictions set by the Rule of 3 and guidelines by Astex Pharmaceuticals (Table 3).7 The fragments have a low mean molecular weight (204), while maintaining high levels of Fsp3 (Fraction aromatic = 0.48). In Figure 2 the molecular weights of the fragments are plotted against their ClogP values, demonstrating that most fall within the desirable range of 0 to 2.
Table 3.
Physicochemical Properties of Fragment Set
| Property | Ideal6 | Fragment Set |
|---|---|---|
| ClogP a | 0–2 | 0.92 |
| Molecular Weight | 140–230 | 204 |
| Polar Surface Area b | ≤60 | 22.8 |
| Hydrogen Bond Acceptor | ≤3 | 1.5 |
| Hydrogen Bond Donor | ≤3 | 0.57 |
| Rotatable Bond Count | -- | 1.2 |
| Fraction Aromatic | -- | 0.48 |
cLogP =calculated octanol/ water partition coefficient
(A2)
Figure 2.
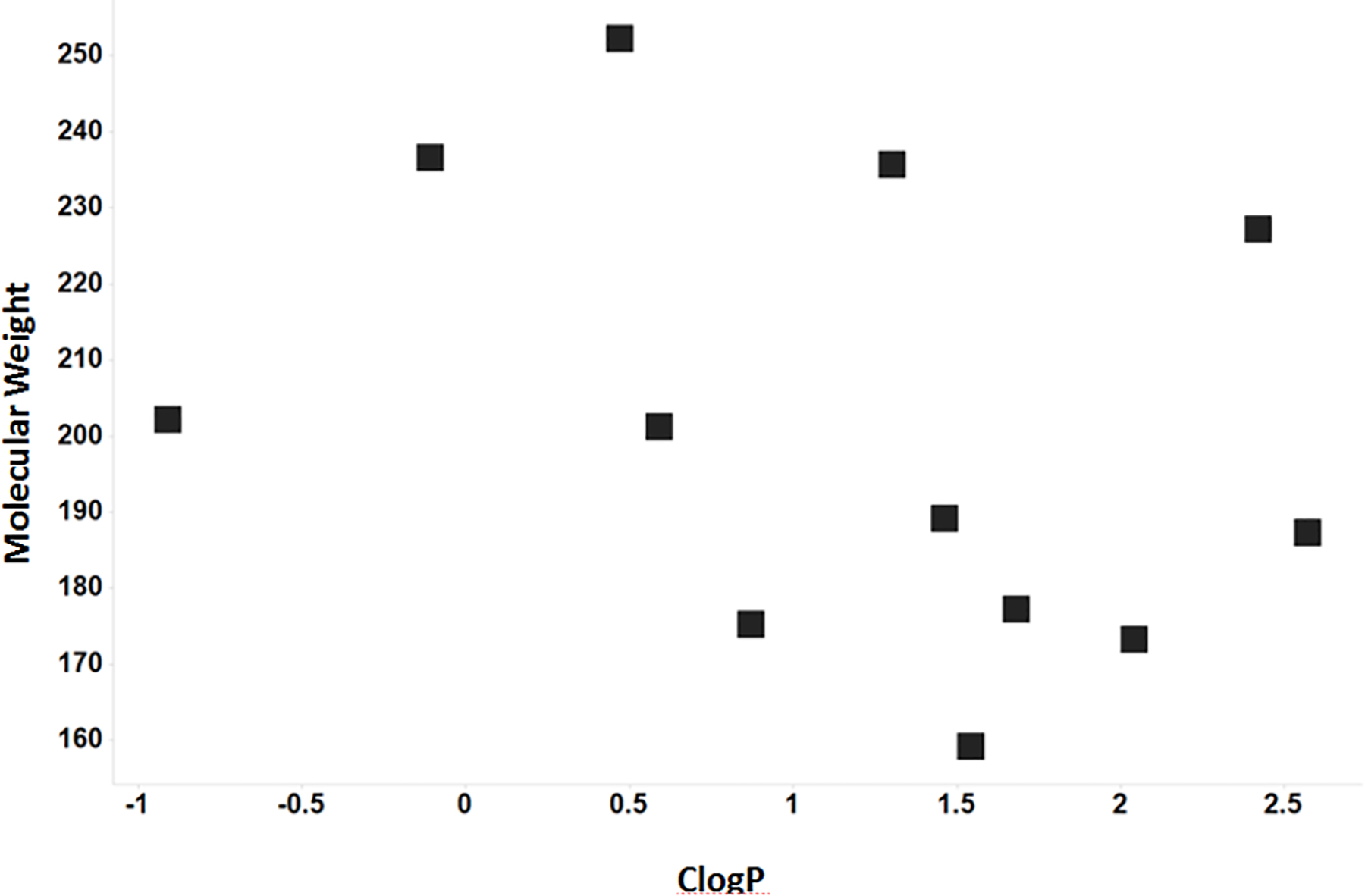
Plot of molecular weight versus ClogP
We also assessed two orthogonal topological descriptors for this set of fragments: the Principal Moments of Inertia (PMI)19 and the Plane of Best Fit score (PBF)20. These descriptors were calculated using AbbVie’s internal design platform from Pipeline Pilot protocols employing published calculation methods.18–20 Based on the plot of the normalized principle moments of inertia (NPR1 and NPR2), the shapes of the molecules can be characterized as being rods, disks, or spheres (Figure 3). The combined PMI plot demonstrates that three of the compounds possess disc-like characteristics while the other 11 possess a rod- or sphere-like shape. The calculated mean NPR1+NPR2 (normalized principle moments of inertia) for this set of compounds is 1.24.
Figure 3.
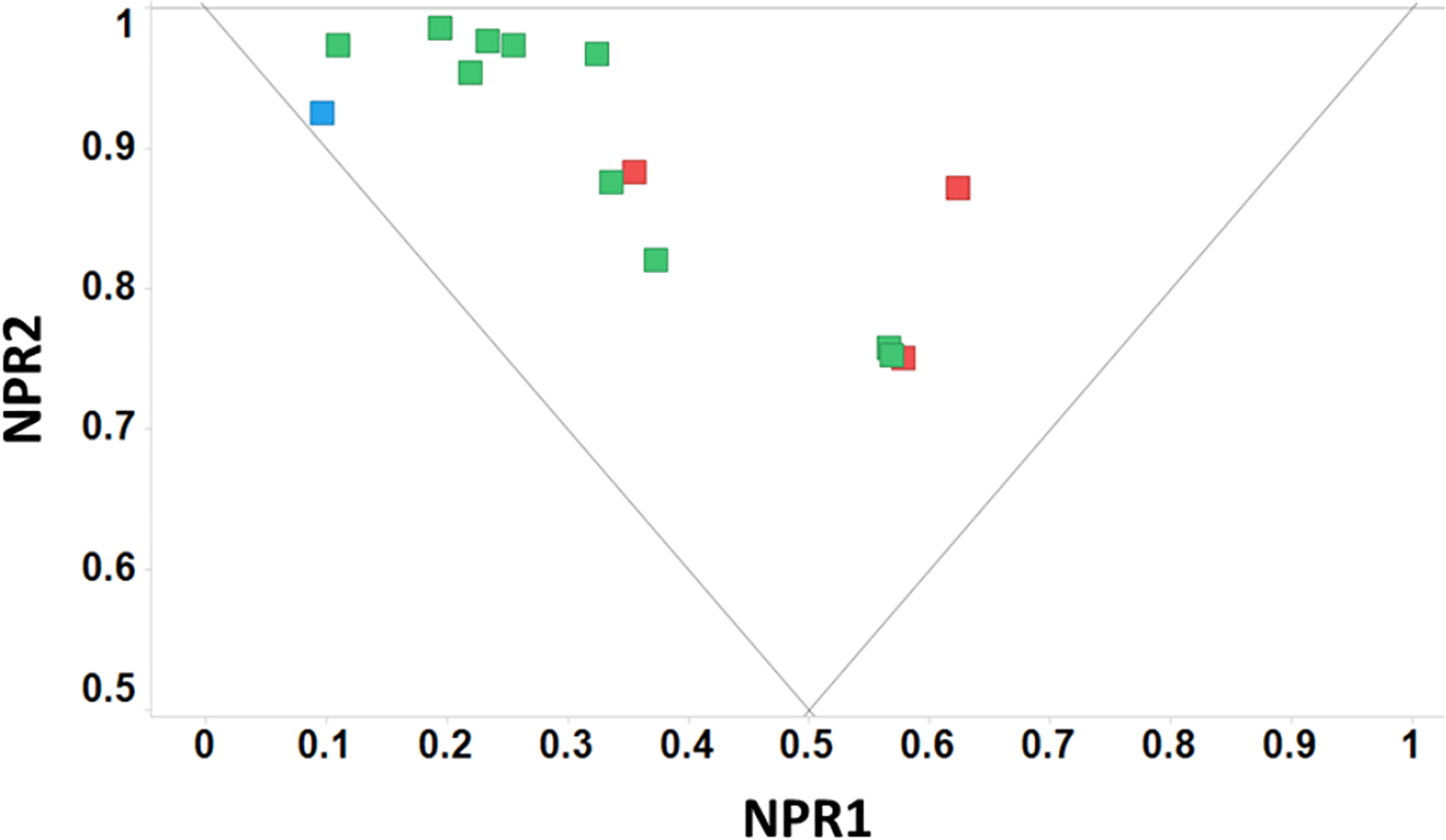
PMI plot for the C-6 arylated amines and amides synthesized. The average distance from the plane of best fit (PBF score) is denoted by the color of the markers (blue ≤ 0.50, 0.50 < green ≤ 0.75, 0.75 < red ≤ 1.00)
An arguably more granular topological descriptor for determining shape and, in particular, the “three dimensionality” of molecules is the Plane of Best Fit (PBF) score introduced by Firth et al.20 The PBF score calculates the distance from the theoretical plane of best fit to the furthest atom of a molecule. The larger the distance (score), the more shape or three dimensionality a molecule possesses. The average PBF score for the synthesized fragments is 0.69, indicating a high degree of three dimensionality. The PBF score of 15B is the highest at 0.87, due to the substitution pattern on the pendant pyridyl ring. Molecules with combined PMI (NPR1 +NPR2) > 1.07 and a PBF > 0.6 are considered to possess a high degree of 3D character.20 By this definition, all 14 fragments have a high degree of 3D-character. Given that the majority of commercially available fragments are outside this area of 3D topological space (around 10% of AbbVie’s fragments are in this 3D compound space), these fragments represent a unique set of compounds with optimal physicochemical properties and atypical topology.
In conclusion, we demonstrate that the Pd-catalyzed C–H arylation of the azabicyclo[3.1.0]hexane core provides rapid access to three-dimensional fragments with attractive physicochemical properties for FBDD. Several key modifications were made to the catalysis conditions, including microwave heating to reduce reaction times and nitrogen purging to eliminate the need for a glove box. These changes increase the practicality of this method for library synthesis. In addition, new procedures were developed that increase the safety and practicality of removing the directing group while maintaining the integrity of the newly installed aryl/heteroaryl substituents. Computational analysis of the 14 amines/amides synthesized demonstrate that these compounds possess optimal physicochemical properties, including a high degree of saturation and 3D character. As such, they should serve as valuable additions to FBDD libraries and as potential scaffolds for early stage drug discovery efforts.
Supplementary Material
Acknowledgment
We thank Dr. Pablo Cabrera for helpful discussions on the C−H arylation and protecting group removal procedures. We also thank the Abbvie high throughput purification team (Harry Spiwek, Tomas Galicia, and Anita McGreal) and Lei Shi for their assistance with purification as well as Noel Wilson for his assistance with the microwave instrument.
Funding Information
This work was supported by Abbvie as well as by the NIH NIGMS (GM073836). ML thanks the National Science Foundation and Rackham Graduate School for graduate fellowships. The authors declare the following competing financial interest(s): Ashley Adams and Phil Cox are employees of AbbVie. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
Footnotes
Supporting Information
YES (this text will be updated with links prior to publication)
Primary Data
YES (this text will be updated with links prior to publication)
References and Notes
- (1).(a) Topczewski JJ; Cabrera PJ; Saper NI; Sanford MS Nature 2016, 531, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cabrera PJ; Lee M; Sanford MS J. Am. Chem. Soc 2018, 140, 5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Daugulis O; Roane J; Tran LD Acc. Chem. Res 2015, 48, 1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) For papers and reviews on FBDD see: Hann MM; Leach AR; Harper G J. Chem. Inf. Comput. Sci 2001, 41, 856. [DOI] [PubMed] [Google Scholar]; (b) Leach AR; Hann MM Curr. Opin. Chem. Bio 2011, 15, 489. [DOI] [PubMed] [Google Scholar]; (c) Scott DE; Coyne AG; Hudson SA; Abell C Biochemistry 2012, 51, 4990. [DOI] [PubMed] [Google Scholar]; (d) Erlanson DA; Fesik SW; Hubbard RE; Jahnke W; Jhoti H Nature Reviews Drug Discovery 2016, 15, 605. [DOI] [PubMed] [Google Scholar]; (d) Johnson CN; Erlanson DA; Jahnke W; Mortenson PN; Rees DC J. Med. Chem 2018, 61, 1774. [DOI] [PubMed] [Google Scholar]
- (4).(a) Lawrence SA Amines: Synthesis, Properties and Applications; Cambridge University Press: Cambridge, U.K., 2004. [Google Scholar]; (b) Dorwald FZ Lead Optimization for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds, first edition; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]; (c) Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (5).(a) Murray CW; Rees DC Angew. Chem. Int. Ed 2016, 55, 488. [DOI] [PubMed] [Google Scholar]; (b) Foley DJ; Nelson A; Marsden SP Angew. Chem. Int. Ed 2016, 55, 13650. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morely AD; Pugliese A; Birchall K; Bower J; Brennan P; Brown N; Chapman T; Drysdale M; Gilbert IH; Hoelder S; Jordan A; Ley SV; Merritt A; Miller D; Swarbrick ME; Wyatt PG Drug Discovery Today 2013, 18, 1221. [DOI] [PubMed] [Google Scholar]
- (6).(a) Epstein JW; Brabander HJ; Fanshawe WJ; Hofmann CM; McKenzie TC; Safir SR; Osterberg AC; Cosulich DB; Lovell FM J. Med. Chem 1981, 24, 481. [DOI] [PubMed] [Google Scholar]; (b) Lunn G; Roberts EL; Content S; Critcher SD; Fenwick AE; Gethin DM; Goodwin G; Greenway D; Greenwood S; Hall K; Thomas M; Thompson S; Williams D; Wood G; Wylie A Bioorg. Med. Chem. Lett 2012, 22, 2200. [DOI] [PubMed] [Google Scholar]; (c) Bakonyi B; Furegati M; Kramer C; Vecchia LL; Ossola F J. Org. Chem 2013, 78, 9328. [DOI] [PubMed] [Google Scholar]; (d) Runyon SP; Kormos CM; Gichinga MG; Mascarella SW; Navarro HA; Deschamps JR; Imler GH; Carroll FI J. Org. Chem 2016, 81, 10383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jones S; Ahmet J; Ayton K; Ball M; Cockerill M; Fairweather E; Hamilton N; Harper P; Hitchin J; Jordon A; Levy C; Lopez R; McKenzie E; Packer M; Plant D; Simpson I; Simpson P; Sinclair I; Somervaille TCP; Small H; Spencer GJ; Thomson G; Tonge M; Waddell I; Walsh J; Waszkowycz B; Wigglesworth M; Wiseman DH; Ogilvie D J. Med. Chem 2016, 59, 11120. [DOI] [PubMed] [Google Scholar]; (f) Harris MR; Li Q; Lian Y; Xiao J; Londregan AT Org. Lett 2017, 19, 2450. [DOI] [PubMed] [Google Scholar]
- (7).(a) Congreve M; Carr R; Murray C; Jhoti H Drugs Discovery Today. 2003, 8, 876–877. [DOI] [PubMed] [Google Scholar]; (b) Palmer N; Peakman TM; Norton D; Rees DC Org. Biomol. Chem 2016, 14, 1599. [DOI] [PubMed] [Google Scholar]
- (8).Lew A; Krutzik PO; Hart ME; Chamberlin RA J. Comb. Chem 2002, 4, 95. [DOI] [PubMed] [Google Scholar]
- (9).General procedure for arylation of of S-1: To a large microwave tube (Biotage®, 10–20 mL) equipped with a stir bar was added Pd(OAc)2 (23.4 mg, 0.10 mmol, 10 mol %), S-1 (400 mg, 1.04 mmol, 1 equiv), cesium pivalate (731 mg, 3.12 mmol, 3 equiv), aryl iodide (2–3 equiv), and anhydrous tert-amyl alcohol (9.6 mL). The cap was crimped, and the vessel was flushed with nitrogen. The microwave tube was heated with the following parameters: 1 min pre-stirring, followed by a ramp (normal) to 180 °C and held at temperature for 40 to 50 min. Hydrazine (500 μL of 35% aqueous) was added to the reaction and allowed to stir at 60 °C for 1 h or at room temperature overnight. The tert-amyl alcohol was removed in vacuo, and the resulting residue was dissolved in EtOAc, filtered through a plug of celite, and concentrated under vacuum. The crude reaction was purified via flash column chromatography, eluting with EtOAc/heptanes. Isolated yield of compound 2: 42% (210.2 mg). LC-MS: APCI+ (m/z): [M+H]+ calcd for C22H19F8N2O: 479.129; found: 479.119. 1H NMR (CDCl3, 500 MHz): δ 7.27 (m, 2H), 6.78 (t, J = 8.5 Hz, 2H), 6.48 (br s, 1H), 2.94 (d, J = 9.0 Hz, 2H), 2.89 (ddd, J = 9.0, 2.0 Hz, 1.0 Hz, 2H), 2.04 (t, J = 8.0 Hz, 1H), 1.87 (m, 2H), 1.14 (s, 6 H). 13C NMR (CDCl3, 126 MHz): δ 175.7, 161.1 (d, JC–F = 246 Hz), 133.7, 129.6 (d, JC–F = 7.6 Hz) Hz), 61.1, 45.1, 27.1, 22.2, 20.9. 20.1. The carbon resonances of the directing group (perfluoroarene, C7F7) appear as complex multiplets and are not listed. 19F NMR (CDCl3, 376 MHz): δ −56.1 (t, J = 21.8 Hz, 3F), −116.4 (m, 1F), −141.4 (m, 2F), −143.1 (m, 2F).
- (10).Kimbrough R; Gaines TR Nature 1966, 211, 146. [DOI] [PubMed] [Google Scholar]
- (11).McDonald CE; Ramsey JD; Sampsell DG; Butler JA; Cecchini MR Org. Lett 2010, 12, 5178. [DOI] [PubMed] [Google Scholar]
- (12).General procedure for directing group removal with SmI2: To a round bottom flask under a flow of nitrogen was added the starting material (1 equiv), 0.1 M SmI2 (12 equiv), anhydrous triethylamine (80 equiv), anhydrous methanol (40 equiv), and tris(N,N-tetramethylene)phosphoric acid triamide (5.5 equiv). The reaction was allowed to stir at room temperature for 3 h. The reaction vessel was then exposed to air, and a white precipitate was observed within 30 min. The reaction was quenched with 1 N HCl. To this mixture was added ethyl acetate, and the product was extracted into the aqueous acidic layer. The organic layer was set aside, and the aqueous layer was basified with solid NaOH until pH 11–12. The aqueous layer was extracted with ethyl acetate (3 × 100 mL) and dried over sodium sulfate. After the volatiles were removed, a viscous yellow oil remained and was purified by reverse-phase HPLC [Waters XBridge™ C-18 column, 5 μm, 30×100 mm, flow rate 40 mL/minute, 5–100% gradient of acetonitrile in buffer (0.025 M aqueous ammonium bicarbonate, adjusted to pH 10 with ammonium hydroxide or 0.1% TFA)]. Isolated yield of compound 2A: 49% (57.9 mg). LC-MS: APCI+ (m/z): [M+H]+ calcd for C11H13FN: 178.103; found: 178.069. 1H NMR (CDCl3, 400 MHz): δ 10.85 (br s, 1H), 7.19 (t, J = 8.4 Hz, 2H), 7.08 (t, J = 8.4 Hz, 2H), 5.94 (br s, 1H), 3.58 (d, J = 11.2 Hz, 2H), 3.30 (d, J = 11.2 Hz, 2H), 2.39 (t, J = 8.0 Hz, 1H), 2.22 (m, 2H). 13C NMR (CDCl3, 101 MHz): δ 162.6 (d, JC–F = 249 Hz), 131.2 (d, JC–F = 8.1 Hz), 127.2 (d, JC–F = 3.0 Hz), 116.9 (d, JC–F = 22.2 Hz), 45.0, 23.3. 22.0. 19F NMR (CDCl3, 376 MHz): −75.8 (s, 3F), −113.2 (s, 1F).
- (13).Low yields for the directing group removal procedure with SmI2 are attributed to the double extraction process carried out before the reverse-phase HPLC purification.
- (14).(a) Procter DJ; Flowers RA; Skrydstrup F Organic Synthesis using Samarium Diiodide: A Practical Guide; Royal Society of Chemistry: Cambridge, U.K., 2010. [Google Scholar]
- (15).(a) Kamochi Y; Kudo T Heterocycles 1993, 36, 2383. [Google Scholar]; (b) Kuishima M; Hoiki K; Kono K; Takayuki S; Tani S Chem. Pharm. Bull 1994, 42, 2190. [Google Scholar]
- (16).Dave PR; Kumar KA; Duddu RJ Org. Chem 2000, 65, 1207. [DOI] [PubMed] [Google Scholar]
- (17).General procedure for directing group removal with acetyl chloride: To a medium microwave tube (Biotage®, 2–5 mL vial) equipped with a stir bar was added the starting material (0.217 mmol, 1 equiv) and acetyl chloride (neat, 3.0 mL). The reaction was heated to 150 °C for 3 h. The acetyl chloride was removed under reduced pressure, and the product was redissolved in DCM (10 mL). NaOH (1 M, 10 mL) and was added. The product was extracted with DCM (2 × 10 mL). The volatiles were removed in vacuo, and the product was purified by reverse phase HPLC. Isolated yield of compound 1B: 25% yield (11 mg). LC-MS: APCI+ (m/z): [M+H]+ calcd for C13H16NO: 202.123; found: 202.153. 1H NMR (CDCl3, 400 MHz): δ 7.30 (t, J = 7.6 Hz, 2H), 7.21 (multiple peaks, 3H), 4.92 (d, J = 12.4 Hz, 1H), 3.59 (dd, J = 11.0, 4.0 Hz, 1H), 3.32 (d, J = 11.0 Hz, 1H), 3.39 (dd, J = 12.4, 4.0 Hz, 1H), 2.23 (t, J = 8.0 Hz, 1H), 1.97 (m, 2H), 1.47 (s, 3H). Hindered rotation of acetyl group breaks symmetry of molecule 13C NMR (CDCl3, 101 MHz): δ 168.9, 133.8, 128.8, 128.5, 126.9, 46.8, 44.6, 22.5, 21.6, 20.6, 20.1.
- (18).Physicochemical properties were calculated using ChemAxon (ChemAxon Component collection for Pipeline Pilot version 1.9_j55) for cLogD and BioVia’s Pipeline Pilot version 9.1 calculators for NRB, NAR, HBA, HBD, MW, TPSA, N+O, Fsp3, NPR, and PBF, with minor customization as needed. Additionally we used BioByte’s cLogP calculator for calculating the octanol–water partition coefficient. Additional calculations and visualizations were conducted using PerkinElmer’s Tibco Spotfire.
- (19).Sauer WHB; Schwarz MK J. Chem. Inf. Comput. Sci 2003, 43, 987. [DOI] [PubMed] [Google Scholar]
- (20).Firth NC; Brown N; Blagg JJ Chem. Inf. Model 2012, 52, 2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.