

Summary
Ferroptosis, a form of cell death driven by iron-dependent lipid peroxidation, was identified as a distinct phenomenon and named a decade ago. Ferroptosis has been implicated in a broad set of biological contexts, from development to aging, immunity, and cancer. This review describes key regulators of this form of cell death within a framework of metabolism, ROS biology, and iron biology. Key concepts and major unanswered questions in the ferroptosis field are highlighted. The next decade promises to yield further breakthroughs in the mechanisms governing ferroptosis, and additional ways of harnessing ferroptosis for therapeutic benefit.
Introduction to Ferroptosis
The concept of a distinct form of regulated, iron-dependent cell death driven by lipid peroxidation, along with the term ferroptosis, was introduced by my lab a decade ago (Dixon et al., 2012) (Figure 1). Ferroptosis is distinct from apoptosis and other forms of cell death (Dixon et al., 2012). The discovery of ferroptosis emerged from our identification in 2003 of small molecules that induced a non-apoptotic form of cell death (Dolma et al., 2003), which we found to be regulated in an iron-dependent manner (Yang and Stockwell, 2008), and to be highly modulatable with molecular perturbations (Wolpaw et al., 2011), as well as the finding by Marcus Conrad and colleagues in 2008 that genetic modulation of key genes controlling redox state drove non-apoptotic cell death (Banjac et al., 2008); Seiler et al., 2008).
Figure 1. Key milestones and growth in the literature of ferroptosis over time.
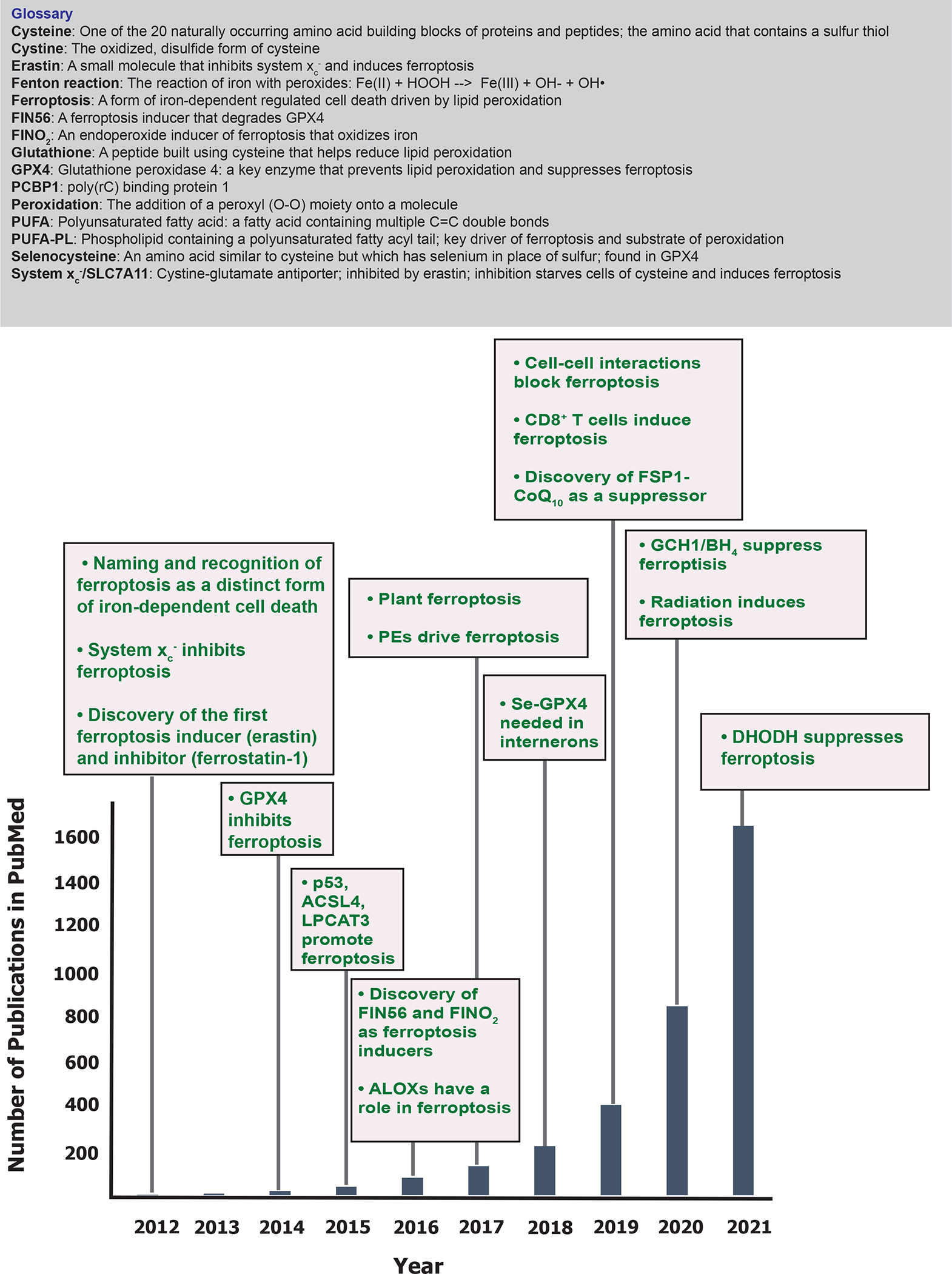
The number of publications listed in PubMed in each year is indicated, as well as key discoveries related to ferroptosis in each year. A glossary of terms related to ferroptosis is also shown.
Prior to the introduction of the concept of ferroptosis, there were three major areas of research that converged to provide the foundational understanding for what we now refer to as the field of ferroptosis—(i) the mechanisms of metabolism, (ii) the control of reactive oxygen species (ROS), and (iii) the regulation of iron.
First, the mechanisms of amino acid and lipid metabolism provided a foundation for ferroptosis. In 1955, Harry Eagle reported that the amino acid cysteine was essential for the survival and growth of the mouse fibroblast strain L and for the HeLa cell line (Eagle, 1955), and in 1973, Jerry Mitchell found that acetaminophen caused hepatic necrosis in rats that depended on glutathione and cysteine (Mitchell et al., 1973). Shiro Bannai reported in 1977 that withdrawal of cystine (see Figure 1 for glossary of key terms in ferroptosis), the oxidized form of cysteine, caused cell death that correlated with glutathione depletion and could be prevented by treatment with vitamin E (Bannai et al., 1977).
Numerous researchers elucidated key mechanisms of lipid metabolism. Michel Eugène Chevreul in 1823 reported the isolation of fatty acids (Chevreul, 1823), and in 1929, husband and wife George and Mildred Burr reported that polyunsaturated fatty acids (PUFAs, see Figure 2 for a glossary), such as linoleic acid (18:2, where 18 represents number of carbons in the fatty acid and 2 represents the number of double bonds) and linolenic acid (18:3), are essential components of the diet of rats (Burr and Burr, 1929). The studies of Nicolas-Louis Vauquelin in 1813, Théodore-Nicolas Gobley in 1846, and Johann Wilhelm Thudichum in 1874 led to the discovery of phospholipids (Sourkes, 2004), key building blocks of membranes. PUFA moieties serve as the essential peroxidation substrates for ferroptosis, when present in membrane lipids, such as phospholipids.
Figure 2. Mechanisms of ferroptosis.
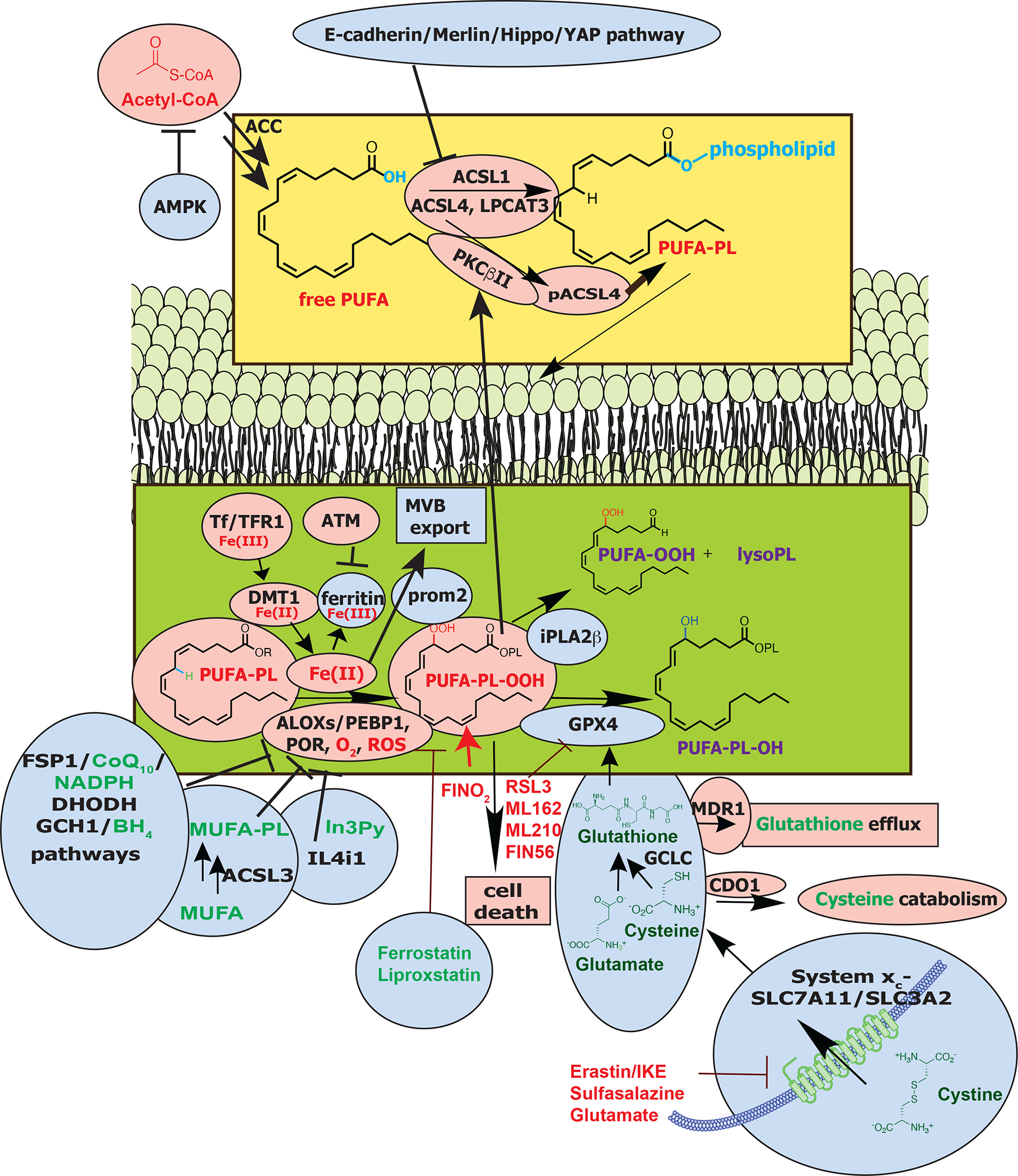
Protein names are in black, those that facilitate ferroptosis are in red circles, and those that suppress ferroptosis in blue circles. Mechanisms are in rectangles. Small molecule inhibitors, lipids and metabolite structures are shown and labeled in green for those that suppress ferroptosis and red for those that induce ferroptosis, and purple for those that neither promote not suppress ferroptosis. The bottom right membrane is the plasma membrane, where system xc− imports cystine, which is reduced in the cell to the amino acid cysteine. Cysteine and glutamate are used in the biosynthesis of reduced glutathione, which is in turn used by GPX4 to reduce reactive PUFA phospholipid hydroperoxides (PUFA-PL-OOH) to non-reactive and non-lethal PUFA phospholipid alcohols (PUFA-PL-OH). Alternatively, the oxidized PUFA-OOH tail can be cleaved from a phospholipid by the action of iPLA2β, suppressing death. The membrane in the middle represents cellular membranes that experience lipid peroxidation, such as the ER. PUFA-PLs are oxidized by labile Fe(II) and Fe(II)-dependent enzymes, such as ALOXs in conjunction with PEBP1, and POR. Fe(III) is imported into cells by Tf through TfR1, after which it is reduced to Fe(II) and imported via DMT1. Iron is stored as Fe(III) in ferritin, where it is not available to promote ferroptosis. Export of iron in MVBs is promoted by prom2, which suppresses ferroptosis. In the top panel, acetyl CoA is used to make free PUFAs, which are activated by ACSL4, LPCAT3 and ACSL1 to generate PUFA-PLs; ACSL4 can be phosphorylated by PKCβII to further activate it. The yellow box shows the process of generating PUFA-PLs and the green box shows how PUFA-PLs are oxidized. Abbreviations: ALOXs, lipoxygenases; AMPK, adenosine-monophosphate-activated protein kinase; ACC, acetyl coenzyme A carboxylase; ACSL, acyl-CoA synthetase long chain family member; ATM, ATM serine/threonine kinase; BH4, tetrahydrobiopterin; CDO1, Cysteine dioxygenase type 1; CoA, coenzyme A; CoQ10, coenzyme Q10; cys, cysteine; DHODH, dihydroorotate dehydrogenase (quinone); DMT1, ferrous ion membrane transport protein DMT1; FSP, ferroptosis suppressor protein 1/ AIFM2; GCH1, GTP cyclohydrolase 1; GCLC, glutamate-cysteine ligase catalytic subunit; Glu, glutamate; GPX4, glutathione peroxidase 4; GSH, glutathione; IL4i1, interleukin-4-induced 1; In3Py, indole-3-pyruvate; iPLA2β, phospholipase A2 group VI; LPCAT3, lysophosphatidylcholine acyltransferase 3; lysoPL, lysophospholipid; MDR1, ATP binding cassette subfamily B member 1/ABCB1; MUFA, monounsaturated fatty acid; MVB, multivesicular body; NADPH, reduced nicotinamide adenine dinucleotide phosphate; PEBP1, phosphatidylethanolamine binding protein 1; PKCβII, protein kinase C beta type isoform 2; PL, phospholipid; POR, Cytochrome p450 oxidoreductase; prom2, prominin-2; PUFA, polyunsaturated fatty acid; PUFA-PL-OOH, phospholipid with peroxidized polyunsaturated fatty acyl tail; ROS, reactive oxygen species; system xc−, sodium-independent, anionic amino acid transport system; Tf, transferrin; TfR1, transferrin receptor protein 1; YAP, Yes1 associated transcriptional regulator
Second, researchers illuminated the biological significance of oxidative damage to biomolecules. The oxidant hydrogen peroxide was synthesized by Louis Jacques Thénard in 1818, and biological mechanisms for decomposing H2O2 were elucidated by Christian Friedrich Schonbein in 1863, Oscar Loew in 1900, Otto Warburg in 1923, Kurt Stern in the 1930s, and Gordon Mills in 1965 (Flohe, 2020). In 1980, Shiro Bannai and Emi Kitamura discovered system xc− as an antiporter that imports cystine (Bannai and Kitamura, 1980), a key amino acid building block for glutathione (Figure 2), which blocks the ability of oxidants such as hydrogen peroxide to cause “oxidative stress”, a term coined in 1985 by Helmut Sies (Sies, 1985). Moreover, a key axis of protection from oxidative stress involves selenium; selenium was discovered in 1818 by Jöns Jacob Berzelius (Flohe, 2020). In 1982, Fulvio Ursini and colleagues discovered GPX4, a selenoprotein that functions as a glutathione-dependent peroxidase to counter the oxidation of lipids in membranes (Ursini et al., 1982) (Figure 2); replacement of the selenocysteine active site residue in GPX4 with cysteine in C57BL/6J x 129S6SvEv mice affects, however, only specific interneurons in the brain, which Marcus Conrad and colleagues reported in 2018 (Ingold et al., 2018). Of note, replacement of the GPX4 active site selenocysteine with cysteine causes embryonic lethality in C57BL6 mice, suggesting a context-dependence to the requirement for selenocysteine in GPX4.
A third community of researchers discovered the biological importance and metabolism of iron. Iron was found in the human body by Lemery and Geoffroy in 1713 (Vannotti and Delachaux, 1949), and in 1876 Henry John Horstman Fenton reported that iron salts react with peroxides to yield hydroxyl radicals, proposing the reaction that now bears his name (Fe2+ + HOOH → Fe3+ + OH− + OH•)(Fenton, 1876). In 1937, Vilém Laufberger discovered and crystallized an essential iron storage protein, ferritin (Laufberger, 1937), which sequesters iron as Fe(III), preventing the Fenton reaction. In 1945, Carl Holmberg and Carl-Bertil Laurell found a major mechanism of iron transport in the form of transferrin (Holmberg and Laurell, 1945). Discovery of the DMT1 iron transporter emerged from the finding that resistance of mice to intracellular pathogens was controlled by a gene termed NRAMP1 for “natural resistance-associated macrophage protein” (Vidal et al., 1993); a related gene was termed NRAMP2 (Gruenheid et al., 1995). Nancy Andrews found that NRAMP2, later renamed DMT1, was the genetic cause of microcytic anemia in mice and a long-sought iron transporter (Fleming et al., 1997); Gunshin et al., 1997). Together with the discovery of the iron-export protein ferroportin, these studies laid the foundation for our understanding of iron homeostasis.
In parallel, mechanisms underlying the form of cell death known as apoptosis were elucidated. Regulated, non-apoptotic cell death was then identified in the form of inflammatory cell death, termed pyroptosis (Cookson and Brennan, 2001), and necroptosis, a type of regulated necrosis (Degterev et al., 2005). At the same time, a high-throughput screen for HRASV12-selective lethal small molecules in my lab in 2001–2003 revealed a new compound, which we termed erastin (for eradicator of RAS-transformed cells), that induced non-apoptotic cell death (Dolma et al., 2003) that depended on accumulation of oxidative stress and cellular iron levels (Yagoda et al., 2007). We performed a larger screen that yielded additional compounds, including RAS-selective-lethal-3 (RSL3) that similarly induced non-apoptotic, iron-dependent oxidative cell death (Yang and Stockwell, 2008). In 2008, Marcus Conrad and his colleagues reported that genetic inactivation of GPX4 induced a non-apoptotic cell death driven by lipid peroxidation that could be suppressed by alpha-tocopherol (Seiler et al., 2008), and that overexpression of xCT protected cells from lipid-peroxidation-induced non-apoptotic cell death (Banjac et al., 2008). In 2012, my labmembers and I, including Scott Dixon, proposed the concept of an iron-dependent form of regulated cell death, which we termed ferroptosis, that was distinct from apoptosis, unregulated necrosis, and necroptosis (Dixon et al., 2012).
We found that erastin induces ferroptosis by blocking uptake of cystine through system xc−, resulting in depletion of cysteine and glutathione (Dixon et al., 2014) and that the molecular target of RSL3 was GPX4 (Yang et al., 2014), consistent with the earlier report of GPX4 controlling a non-apoptotic form of oxidative cell death (Seiler et al., 2008) and placing GPX4 at the center of ferroptosis regulation (Figure 2).
Ferroptosis is defined as a form of cell death that depends on iron-dependent lipid peroxidation. This can be assessed in cell culture by testing whether iron chelators and lipophilic antioxidants suppress cell death. In human and animal tissue samples, the presence of markers of iron-dependent lipid peroxidation can be used to identify the presence of ferroptosis, along with the absence of other forms of cell death (see below).
Since 2012, ferroptosis has been detected in a variety of biological systems—as a driver of organ injury (Friedmann Angeli et al., 2014), as an ancient form of cell death kept in check by endogenous repair and protection systems (Jiang et al., 2021), and as a mode of cell death activated upon culturing mouse embryo fibroblasts (MEFs) in the absence of cysteine that depends on the presence of transferrin and the amino acid glutamine (Gao et al., 2015); Gao et al., 2019), to name a few. The recurrent observation of ferroptosis in diverse systems shows the myriad contexts in which this type of cell death occurs.
In this review, I provide a framework for understanding the vast emerging literature, which has been growing exponentially for the last decade (Figure 1), on this form of iron-dependent oxidative cell death: ferroptosis is at the intersection of metabolism, ROS biology, and iron regulation, and each of these fields contributes to our understanding of ferroptosis mechanisms, biological significance, and therapeutic relevance. I describe within this framework the latest advances in the mechanisms governing ferroptosis, including the subcellular organelles that control ferroptosis, and the normal physiological functions of ferroptosis. In addition, I summarize key pathological contexts involving ferroptosis, and emerging therapeutic applications of modulating ferroptosis. Finally, I explain useful concepts for understanding ferroptosis, as well as key unanswered questions that will likely drive the next decade of ferroptosis studies.
Mechanisms of ferroptosis
There have been major advances in the last decade, and especially in the last few years, in our understanding of the mechanisms that govern ferroptosis, which in turn has influenced how we conceptualize the role of ferroptosis in biology and medicine. As described below, numerous researchers illuminated the specific lipids that undergo oxidation during ferroptosis and drive the cell death process, endogenous mechanisms of suppressing ferroptosis, key facets of iron regulation that control ferroptosis sensitivity, and organelles that contribute to ferroptosis. I summarize the paradigm of ferroptosis within the framework of its centrality at the intersection of metabolism, ROS biology, and iron regulation.
Metabolism generates the substrates for, and natural inhibitors of, ferroptosis
For ferroptosis to occur, specific lipids must undergo oxidation, and natural mechanisms for blocking accumulation of oxidized lipids must become compromised (Figure 2). Normal metabolism generates the lipid substrates and oxidants that drive ferroptosis, and the endogenous inhibitors that prevent lipid peroxidation.
Lipid metabolism: ACSL4 and LPCAT3 generate the lipid drivers of ferroptosis
Ferroptosis is ultimately driven by the peroxidation of specific membrane lipids (Figure 2). Only specific carbon atoms within a lipid are susceptible to peroxidation, as this reaction involves replacing a hydrogen atom attached to a carbon atom with a peroxyl group (O-O): the propensity of a lipid to undergo peroxidation therefore depends on the strength of its carbon-hydrogen bonds (Figure 2). PUFAs have been known since their discovery to be highly susceptible to peroxidation due to the presence of exceptionally weak C-H bonds in between adjacent C=C double bonds, and initially it seemed plausible that free PUFAs could be drivers of ferroptosis. However, in 2015–2017, several papers demonstrated that free fatty acids are not drivers of ferroptosis, but rather that polyunsaturated fatty acids (PUFAs) need to be activated and incorporated into membrane lipids, such as phospholipids, in in order to exert their lethal effects upon peroxidation. Identification of the specific lipids that drive the execution of cell death, as well as the enzymes that promote their generation and incorporation into cell membranes has been one of the important discoveries in the last decade of ferroptosis research.
An insertional mutagenesis screen in the predominantly haploid cell line KBM7 revealed that inactivation of ACSL4 and LPCAT3 rendered these cells resistant to two different GPX4 inhibitors—the first GPX4 inhibitor RSL3, as well as an additional compound named ML162 (Dixon et al., 2015). Moreover, analysis of ferroptosis-resistant cell lines and a CRISPR suppression screen independently yielded ACSL4 inactivation as a key mechanism for suppressing ferroptosis in diverse contexts (Doll et al., 2017); overexpression of ACSL4 also sensitized to ferroptosis in these contexts (Doll et al., 2017). Given that ACSL4 and LPCAT3 are involved in activating and incorporating PUFAs, such as arachidonic acid, into membrane-localized lipids (Doll et al., 2017), these results suggested that PUFAs need to be present in their membrane-bound context in order to exhibit lethality after peroxidation (Figure 2). Thus, while system xc− and GPX4 normally function as strong suppressors of ferroptosis, ACSL4 and LPCAT3 were the first identified pro-ferroptotic gene products, by virtue of their role in facilitating incorporation of PUFAs into membrane lipids (Figure 2). A recent report demonstrated that ACSL4 also participates actively in a feed-forward loop to execute ferroptosis: PKCβII senses initial lipid peroxidation events, and phosphorylates ACSL4 on Thr328 to drive phospho-ACSL4 (pACSL4) activation and promote PUFA incorporation into phospholipids and drive subsequent cell death (Zhang et al., 2022) (Figure 2). ACLS4 is also a point of regulating ferroptosis through signaling pathways (Wu et al., 2019a): E-cadherin acts through the Merlin-Hippo-Yap pathway to control sensitivity to ferroptosis by regulating expression of ACSL4 in response to cell-cell contacts. Thus, ACSL4 may be more akin to caspase-3, the executioner of apoptosis, than to a housekeeping protein.
Other ACSL enzymes can also regulate ferroptosis: conjugated linolenic acids, such as α-eleostearic acid, which is produced by some plants, require ACSL1 in order to exert their pro-ferroptotic activity (Beatty et al., 2021)(Figure 2). Moreover, monounsaturated fatty acids, such as oleic acid, require ACSL3 in order to exert anti-ferroptotic effects (Magtanong et al., 2019). Presumably, these ACSL-dependent ferroptosis-modulating effects require fatty acids to be incorporated into membrane-bound lipids, such as phospholipids. These studies have identified the ACSL enzyme family as being crucial for ferroptosis, showing that activation of fatty acids into coenzyme A esters is a key regulated step for ferroptosis.
Several studies have sought to identify the specific membrane lipids that drive ferroptosis. The discovery of ACSL4 and LPCAT3 as genetic suppressors of ferroptosis, as noted above, indicated that free PUFAs are not themselves drivers of ferroptosis. Another recent finding consistent with this conclusion was that the phospholipase iPLA2β suppresses p53-driven ferroptosis by removing oxidized PUFA tails from phospholipids (Chen et al., 2021b). Thus, oxidized PUFA tails must remain in the context of membrane-bound phospholipids to contribute to the execution of ferroptosis; intriguingly, once cleaved from a phospholipid, the oxidized PUFA tail no longer drives cell death (Figure 2). This highlights that oxidized PUFAs are not intrinsically toxic to cells. Excessive accumulation of oxidized PUFA-containing lipids within specific cell membranes is the event that drives ferroptosis. Inactivation or deficiency of iPLA2β (also known as PLA2G6) is associated with Parkinson’s disease and motor deficits (Sun et al., 2021), as well as with excessive ferroptosis in placental trophoblasts, causing a risk of poor fetal development (Beharier et al., 2020). Thus, accumulation of oxidized PUFA-containing membrane lipids drives pathological ferroptosis in at least two disease contexts. Therefore, ferroptosis should not be thought of as a general type of oxidative stress, but rather the accumulation of lethal membrane-localized lipid peroxides.
Not all PUFA-containing membrane-localized lipids contribute to ferroptosis. In 2017, it was reported that specific phospholipids, namey phosphatidylethanolamines (PEs) with one arachidonyl (20:4) or one adrenyl (22:4) PUFA tail, are more tightly associated with ferroptosis than other phospholipids (Kagan et al., 2017). While 350 different phospholipids could be detected in this study by LC-MS/MS, including 130 oxygenated lipids, those with 20:4 and 22:4 acyl tails on PE head groups were most correlated with susceptibility to ferroptosis.
Phospholipids have two fatty acyl tails, one typically derived from a saturated fatty acid, such as palmitic acid (C16:0), and one derived from any type of fatty acid, such as a monounsaturated fatty acid, a polyunsaturated fatty acid, or a saturated fatty acid. Unusual phospholipids can be detected, however, with two PUFA tails—these are particularly relevant to ferroptosis (Kraft et al., 2020). PUFA-containing ether lipids, such as plasmalogens, are also associated with ferroptosis sensitivity (Zou et al., 2020a), as is the lipid 2-arachidonoyl glycerol results (Kathman et al., 2020). Thus, several different lipids with PUFA tails contribute to driving ferroptosis. Moreover, PUFA biosynthesis is itself a means of regulating sensitivity to ferroptosis: energy stress drives resistance to ferroptosis through activation of AMPK and its effects on limiting PUFA biosynthesis through control of acetyl-coenzyme A carboxylase (ACC) (Lee et al., 2020a) (Figure 2). In summary, PUFA-containing membrane-localized lipids are the drivers of ferroptosis, and these include phospholipids, ether lipids, and other glycerol-derived lipids.
ROS biology
GPX4 and glutathione suppress lipid ROS accumulation
Erastin and RSL3 represent the first two classes of ferroptosis-inducing compounds, acting by inhibiting cystine uptake through system xc− and by inhibiting GPX4, respectively (Figure 2). Depleting glutathione through other mechanisms can also sensitize to ferroptosis: the multidrug resistance gene MDR1 drives increased sensitivity to ferroptosis by causing efflux of glutathione (Cao et al., 2019), and the cysteine catabolic enzyme cysteine dioxygenase 1 (CDO1) drives sensitivity to ferroptosis by depleting cysteine, and in turn glutathione (Hao et al., 2017). A recent report demonstrated that glutamate-cysteine ligase (the catalytic subunit of which is encoded by GCLC) contributes to ferroptosis resistance not only by synthesizing glutathione, but also by restricting levels of glutamate through its conversion to γ-glutamyl peptides, suggesting that glutamate can promote ferroptosis (Kang et al., 2021) (Figure 2).
GPX4 degradation, rather than GPX4 enzymatic inhibition, can also promote ferroptosis. In 2016, my lab identified a new ferroptosis-inducing compound that acts through GPX4 degradation: FIN56 emerged from a screen for caspase-independent lethal compounds (Shimada et al., 2016b). The most notable hit compound from the screen was termed Caspase-Independent Lethal Compound 56 (CIL56). Optimization of the structure around this scaffold yielded a compound that selectively induced ferroptosis, without activating necrosis. This compound, which was termed Ferroptosis-Inducer-56 (FIN56), induces the degradation of GPX4 and also acts through the mevalonate pathway to drive increased sensitivity to ferroptosis by depleting CoQ10. Other mechanisms for controlling degradation of GPX4 also modulate sensitivity to ferroptosis: chaperone-mediated autophagy, for example, induces degradation of GPX4 and contributes to the execution of ferroptosis (Wu et al., 2019b).
GPX4-independent control of lipid ROS accumulation
GPX4 is a central inhibitor of ferroptosis. However, three GPX4-independent systems for suppressing ferroptosis have been identified in the last few years. FSP1/coenzyme Q10, dihydroorotate dehydrogenase (DHODH), and GCH1/BH4 act independent of GPX4 to suppress ferroptosis. In 1956, coenzyme Q10 (CoQ10), originally known as vitamin Q10 or ubiquinone due its vitamin-like structure and ubiquity, was first purified from cow heart (Crane, 2007); it was shown in the subsequent decades to have a crucial electron-carrying role in energy production in mitochondria. While GPX4 serves as the major defender against lipid peroxidation, CoQ10 serves as a second endogenous mechanism for protecting against the lipid peroxidation that drives ferroptosis. Indeed, CoQ10 is found in diverse membranes throughout cells, not just in mitochondria. Two groups reported that the gene product formerly known AIFM2, and renamed Ferroptosis Suppressor Protein 1 (FSP1), functions to regenerate the reduced form of CoQ10 through the use of NADPH, the cell’s major source of reducing power (Bersuker et al., 2019); Doll et al., 2019). The scavenging of lipid peroxidation intermediates by reduced CoQ10 causes CoQ10 to be oxidized. NADPH is then consumed as it regenerates the reduced form of CoQ10. Indeed, NADPH was found to be a biomarker predicting resistance to ferroptosis across the NCI60 set of cancer cell lines (Shimada et al., 2016a), and the cytosolic phosphatase MESH1 was reported to control sensitivity to ferroptosis through its effect on NADPH abundance (Ding et al., 2020).
Two additional GPX4-independent systems for suppressing the lipid peroxidation that drives ferroptosis were reported in 2020 and 2021. First, GTP cyclohydrolase 1 (GCH1), which generates the endogenous metabolite tetrahydrobiopterin (BH4), was discovered in a CRISPR activation screen as a suppressor of ferroptosis (Kraft et al., 2020), as well as an enhancer of ferroptosis in a CRISPR loss of function screen (Soula et al., 2020). GCH1 expression suppresses ferroptosis by a two-pronged mechanism. First, GCH1 generates the lipophilic antioxidant BH4, which functions analogously to CoQ10 to prevent lipid peroxidation. Second, GCH1 causes remodeling of the lipid membrane environment to increase the abundance of reduced CoQ10, and to deplete the PUFA-PLs that drive sensitivity to ferroptosis. Second, dihydroorotate dehydrogenase (DHODH) was identified as a mitochondrial suppressor of ferroptosis that functions through reducing mitochondrial CoQ10, analogous to the function of FSP1 in extramitochondrial membranes (Mao et al., 2021). Cells with high expression of GCH1 or DHODH are more resistant to ferroptosis, and those with low expression more sensitive to ferroptosis.
Finally, a recent report indicated that there are yet other mechanisms for suppressing ferroptosis, independent of glutathione/GPX4, FSP1/DHODH/CoQ10 and GCH1/BH4. The amino acid oxidase interleukin-4-induced-1 (IL4i1), originally discovered as a gene induced in B cells in response to IL-4, generates the metabolite indole-3-pyruvate, which suppresses ferroptosis both through a radical scavenging mechanism and by orchestrating a gene expression profile that attenuates ferroptosis (Zeitler et al., 2021) (Figure 2). Other endogenous metabolites may act similarly to suppress ferroptosis, either by radical intermediates required for lipid peroxidation or controlling expression of genes that regulate lipid peroxidation.
Iron regulation: Iron-driven lipid peroxidation
The peroxidation of membrane-bound, PUFA-containing lipids is driven by both the labile iron pool facilitating the Fenton reaction, which propagates lipid peroxidation (Shah et al., 2018), and by iron-dependent enzymes, such as lipoxygenases, that initiate the formation of lipid hydroperoxides that are substrates for the Fenton reaction (Yang et al., 2016) (Figure 3). 15-lipoxygenase can complex with phosphatidylethanolamine binding protein 1 (PEBP1), switching the substrate specificity of the enzyme from free PUFAs to PUFA tails of phospholipids (Wenzel et al., 2017). Additionally, 12-lipoxygenase is required for p53-dependent ferroptosis (Chu et al., 2019). Cytochrome P450 oxidoreductase (POR) also contributes to lipid peroxidation during ferroptosis (Zou et al., 2020b), demonstrating that several iron-containing enzymes have this capability to promote the lipid peroxidation that drives ferroptosis (Figure 2).
Figure 3. Role of organelles and organs in ferroptosis.
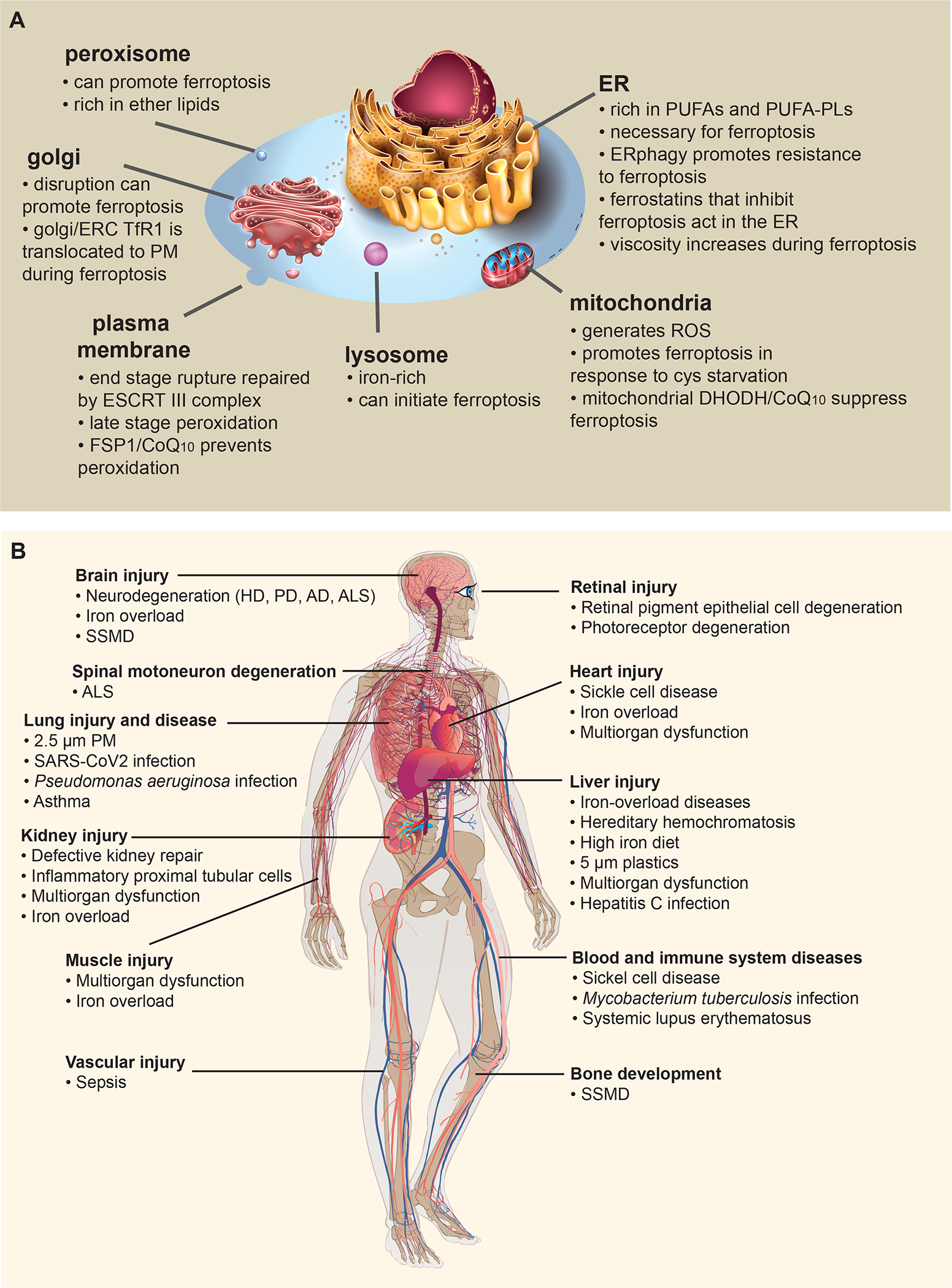
(A) The contributions of major organelles in ferroptosis are shown. (B) Ferroptosis has been implicated in a variety of diseases, organs and tissues, as shown. Abbreviations: AD, Alzheimer’s Disease; ALS, amyotrophic lateral sclerosis; HD, Huntington Disease; PD, Parkinson’s Disease; PM, particulate matter; SARS-CoV2, severe acute respiratory syndrome, coronavirus 2; SSMD, Sedaghatian-type Spondylometaphyseal Dysplasia
The regulation of iron-abundance through controlling the level of the iron-storage protein ferritin via ferritinophagy dictates sensitivity to ferroptosis (Gao et al., 2016); Hou et al., 2016), as the availability of ferritin determines the size of the labile iron pool. The kinase ATM also controls sensitivity to ferroptosis through regulation of ferritin abundance (Chen et al., 2020b). Other mechanisms for controlling cellular abundance of iron influence the sensitivity of cells to ferroptosis: iron export through ferroportin, or through prominin2-mediated ferritin-containing multivesicular bodies (MVBs) and exosomes drives resistance to ferroptosis (Brown et al., 2019), as this depletes the cellular pool of iron, and its ability to promote lipid peroxidation.
Reaction of endoperoxide compounds with iron can also trigger ferroptosis. FINO2, representing the fourth class of ferroptosis-inducing compounds, was discovered by Keith Woerpel and coworkers in 2016, by synthesis and analysis of a series of 1,2-dioxolane-containing compounds (Abrams et al., 2016). FINO2 does not inhibit system xc−, deplete glutathione, directly inhibit GPX4, or induce GPX4 degradation. Thus, FINO2 represents an additional means of inducing ferroptosis. FINO2 oxidizes Fe(II) to Fe(III): its lethal activity is more potently suppressed by iron chelators than is the case for other ferroptosis-inducing compounds. The profile of oxidized lipids detected after FINO2 treatment of cells is more widespread than after treatment with erastin, suggesting that FINO2 undergoes a Fenton reaction with Fe(II), generating alkoxyl radical that directly initiate lipid peroxidation. Another possibility is that FINO2 binds to and activates lipoxygenases or other iron-dependent enzymes by oxidizing the non-heme iron cofactor; the active form of lipoxygenases is the ferric Fe(III) form (Schilstra et al., 1994). Other endoperoxides have also been reported to induce a degree of ferroptosis, such as artemisinin-related compounds (Chen et al., 2020a). Thus, endoperoxides represent a source of ferroptosis-inducing compounds that act in a manner distinct from those such as erastin, RSL3, and FIN56, which disable the defense mechanisms that protect against lipid peroxides. It is thus possible that endogenous endoperoxides such as prostaglandins could act as natural triggers for ferroptosis in some contexts.
Fe(II) is maintained in cells in the form of the labile iron pool, bound to low molecular weight compounds, including glutathione (Patel et al., 2019). Depletion of glutathione can not only inactivate GPX4, but can also mobilize Fe(II) for Fenton chemistry, promoting propagation of lipid peroxides and ultimately ferroptosis. In addition, iron storage in ferritin requires formation of the glutathione-iron complex, which is delivered to ferritin via the chaperone poly(rC) binding protein 1 (PCBP1) (Patel et al., 2021). Thus, depletion of glutathione promotes the availability of labile iron. However, glutathione depletion is not necessary for ferroptosis, as direct inhibition or deletion of Gpx4 without glutathione depletion is sufficient to trigger ferroptosis.
Mechanisms of resistance to ferroptosis integrate metabolism, ROS biology, and iron regulation
At least three major mechanisms of ferroptosis resistance have been identified—the antioxidant regulator NRF2, the transsulfuration pathway, and mTOR. NRF2, a master regulator of the antioxidant response, drives transcriptional responses that suppress ferroptosis (Sun et al., 2016); the mechanism of NRF2-mediated protection varies by cell and tissue context: in pancreatic cancer cells, for example, NRF2 suppresses ferroptosis by activating microsomal glutathione S-transferase 1 (MGST1) (Kuang et al., 2021), which inhibits lipoxygenase 5 (ALOX5). In hepatocarcinoma cells, NRF2 drives resistance to ferroptosis through the control of ferritin (Sun et al., 2016).
The transsulfuration pathway drives resistance to ferroptosis by producing cysteine from methionine, overcoming the cysteine starvation produced by inhibition of system xc−. An siRNA screen for suppressors of ferroptosis in 2016 revealed that the top hit was knockdown of CARS, the cysteinyl-tRNA synthetase, which suppressed erastin-induced ferroptosis through activating the transsulfuration pathway (Hayano et al., 2016). The transsulfuration pathway metabolite homocysteine was reported to enhance ferroptosis rather than drive resistance to ferroptosis when supplemented in the culture medium (Wallis et al., 2021). This suggests that homocysteine can act independently of serving as a source of cysteine.
The mechanistic target of rapamycin (mTOR) pathway promotes resistance to ferroptosis through increased GPX4 protein synthesis (Zhang et al., 2021) and increased SREBP-mediated lipogenesis (Yi et al., 2020). Thus, ferroptosis resistance can arise through control of iron homeostasis, ROS abundance, and metabolism.
The subcellular organelles that drive ferroptosis
Ferroptosis is executed by accumulation of peroxidized lipids. The subcellular membranes containing these oxidized lipids have been explored in recent years, illuminating the contributions of different organelles to ferroptosis (Figure 3).
Plasma membrane rupture is a late event in ferroptosis
The end stage of ferroptotic cell death involves permeabilization of the plasma membrane, as is common in diverse types of cell death. The ESCRT III complex repairs damage to the plasma membrane and slows death by ferroptosis (Pedrera et al., 2021). In addition, ferroptosis has been shown to propagate between cells, but this propagation does not depend on rupture of the plasma membrane (Riegman et al., 2020). The propagation of ferroptosis to neighboring cells instead may depend on release of oxidized lipids with intact plasma membranes, perhaps through exocytic vesicles; released oxidized lipids may then initiate ferroptosis in neighboring cells. FSP1, described above, exerts its protective function against ferroptosis via reduction of plasma membrane CoQ10 (Bersuker et al., 2019); Doll et al., 2019), arguing that plasma membrane lipid peroxidation is an essential late step in ferroptosis.
Endoplasmic reticulum is an essential site of lipid peroxidation during ferroptosis
Imaging the localization of inhibitors of ferroptosis, namely alkyne-tagged ferrostatin analogs using stimulated Raman scattering (SRS) microscopy, revealed that these compounds accumulate in lysosomes, mitochondria and the ER (Gaschler et al., 2018). However, accumulation of such compounds in lysosomes and mitochondria does not influence their ability to induce ferroptosis, suggesting that suppressing lipid peroxidation in the ER is sufficient to block ferroptosis. Compounds that modulate ferroptosis by affecting lipid peroxidation primarily localize to the ER, suggesting that the ER is the most critical site of lipid peroxidation during ferroptosis (Figure 3). While PUFAs can also localize to lipid droplets, the presence of PUFAs in lipid droplets doesn’t contribute to ferroptosis.
The volume of ER present in cells may be a key determinant of sensitivity to ferroptosis. Consistent with this, the viscosity of the ER was shown to increase during ferroptosis (Hao et al., 2021), likely due to clustering of oxidized PUFA-PLs (Agmon et al., 2018), which causes stiffening of the ER membrane (Chng et al., 2021).
Mitochondria can act as initiators and amplifiers of ferroptosis induced by cysteine starvation
Several studies have shown the mitochondria can act as initiators or amplifiers of ferroptosis initiated by cysteine starvation and glutathione depletion. The ability of mitochondrial DHODH and mitochondrial CoQ10 to suppress ferroptosis triggered in this manner (Mao et al., 2021), for example, highlights this function of mitochondria. Inhibiting the mitochondrial electron transport chain also attenuates ferroptosis induced by cysteine starvation, as does depletion of mitochondria (Gao et al., 2019) (Figure 3). The role of the electron transport chain may be due to the leakage of electrons that produce superoxide and H2O2, which can then react with Fe(II) to drive Fenton chemistry and lipid peroxidation.
Peroxisomes can drive ferroptosis through peroxidation of ether lipids
Peroxisomal ether lipids, such as plasmalogens, have been implicated as drivers of ferroptosis, through the discovery in a CRISPR screen of genes involved in peroxisomal functions (Zou et al., 2020a). While peroxisomal ROS have not been directly examined during ferroptosis, it is possible that H2O2 produced in peroxisomes initiates lipid peroxidation, possibly in conjunction with peroxisomal Fe(II).
Lysosomes are reservoirs of iron, and can initiate ferroptosis
Ferrostatins accumulate in lysosomes, but their lysosomal localization does not contribute to their ability to suppress ferroptosis induced by erastin or RSL3. Thus, lysosomes do not normally contribute to ferroptosis in this context. However, lysosomes are reservoirs of iron and in principle could initiate ferroptosis. Indeed, re-localizing FINO2-type endoperoxides to lysosomes can initiate ferroptosis, although propagation through the ER is still required (Figure 3). In addition, knockout of the lysosomal protein prosaponsin caused lipofuscin accumulation, which drives lysosomal iron and ROS accumulation, and subsequent ferroptosis in neurons (Tian et al., 2021).
Golgi stress can promote ferroptosis
Triggering Golgi stress with agents such as brefeldin A triggers ferroptosis in some contexts (Alborzinia et al., 2018). In addition, the transferrin receptor TfR1 is translocated to the plasma membrane from its intracellular localization in the Golgi and in the closely associated endosomal recycling compartment; this re-localization can be used as a ROS-independent marker of ferroptosis and may also further enhance ferroptosis as a kill switch by promoting additional iron-loaded transferrin uptake (Feng et al., 2020) (Figure 3).
In summary, the ER is a central hub that drives the lipid peroxidation that promotes cell death through ferroptosis. This is logical, given that much lipid and PUFA metabolism occurs in the ER. However other organelles, such as mitochondria, lysosomes, peroxisomes, and the Golgi can initiate or enhance the susceptibility of cells to ferroptosis by initiating lipid peroxidation or enhancing ROS production. Ultimately, lipid peroxides spread to the plasma membrane, where they trigger rupture of the plasma membrane (Figure 3). How ROS and lipid peroxides are transported between organelles to promote ferroptosis remains largely unexplored. In addition, how lipid peroxides ultimately damage membranes and result in cell death is still unknown. Nonetheless, we know that the process of lipid-peroxidation-driven ferroptosis has been harnessed by nature in a number of normal physiological contexts, as described in the next section.
Physiological Functions of Ferroptosis
When we first proposed the concept of ferroptosis in 2012, we found that it was implicated in glutamate toxicity (Dixon et al., 2012), but whether ferroptosis might be exploited naturally for normal physiology was unclear—recent data have revealed that indeed ferroptosis is involved a number of physiological processes (Figure 4).
Figure 4. Physiological functions of ferroptosis.
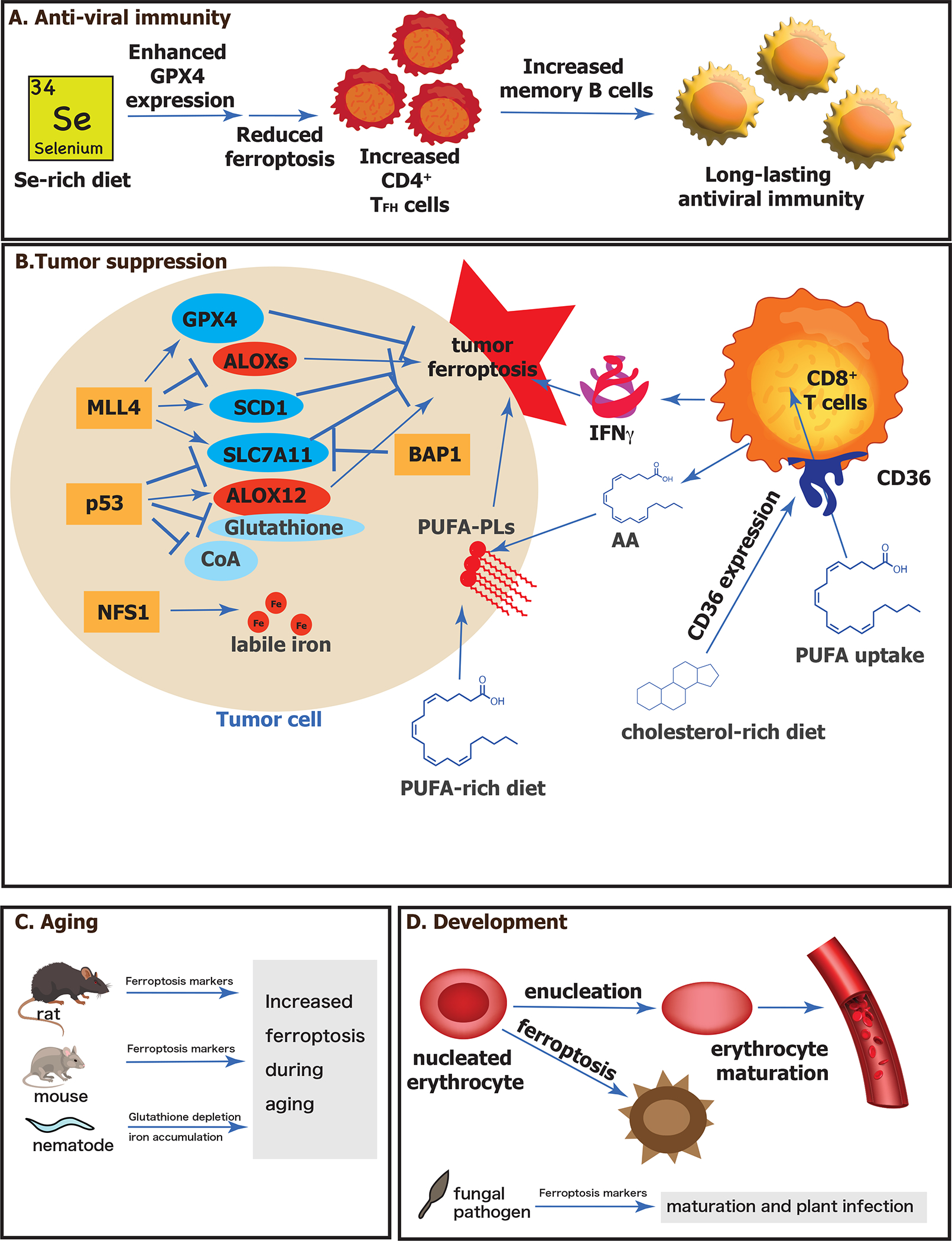
(A) Anti-viral immunity is promoted by selenium supplementation. A selenium-rich diet enhances expression of the selenoprotein GPX4, which suppresses ferroptosis in CD4+ TFH cells, which in turn promotes increased memory B cells and long-lasting viral immunity. (B) Role of ferroptosis in tumor suppression. Tumor suppressors are indicated in orange boxes, proteins that suppress ferroptosis are in blue circles and those that promote ferroptosis are in red circles. Small molecules that suppress ferroptosis are in light blue circles. A PUFA-rich diet promotes production of PUFA-PLs, which promote tumor ferroptosis. CD8+ T cells further promote tumor cell ferroptosis by releasing IFNγ and AA. A cholesterol-rich diet promotes increased expression of CF36 on CD8+ T cells, which caused them to take up PUFAs themselves, leading to their death by ferroptosis and promoting tumor formation. (C) Ferroptosis increases with aging. Rats and mice experience increased ferroptosis markers as they age. Nematodes (C. elegans) experience increased iron content and depletion of glutathione as they age, leading to increased ferroptosis. (D) Ferroptosis is involved in development. Nucleated erythrocytes undergo ferroptosis prior to enucleation and erythrocyte maturation. A fungal pathogen has evidence of ferroptosis during its maturation required for plant infection. Abbreviations: AA, arachidonic acid; ALOXs, lipoxygenases; BAP1, ubiquitin carboxyl-terminal hydrolase; CoA, coenzyme A; GPX4, glutathione peroxidase 4; IFNγ, interferon gamma; MLL4, histone-lysine N-methyltransferase 2B; NFS1, cysteine desulfurase, mitochondrial; p53, p53 tumor suppressor protein; PUFA, polyunsaturated fatty acid; PUFA-PL, phospholipid with polyunsaturated fatty acid tail; SCD1, stearoyl-coenzyme A desaturase 1; SLC7A11, solute carrier family 7 member 11; TFH, Follicular helper T cells
A key advance in the search for biological processes in which ferroptosis is involved was the discovery of markers for detecting ferroptosis (Figure 5). Given that ferroptosis is driven by iron-dependent lipid peroxidation, detecting such lipid peroxidation events during ferroptosis is essential (Figure 5). In addition, mitochondria typically exhibit shrunken, dense morphologies during ferroptosis, specific gene expression changes can be detected, and TfR1 upregulation and movement to the plasma membrane marks ferroptotic cells (Figure 5). Using such markers, numerous natural physiological functions of this ancient form of cell death have been uncovered.
Figure 5. Markers of ferroptosis.
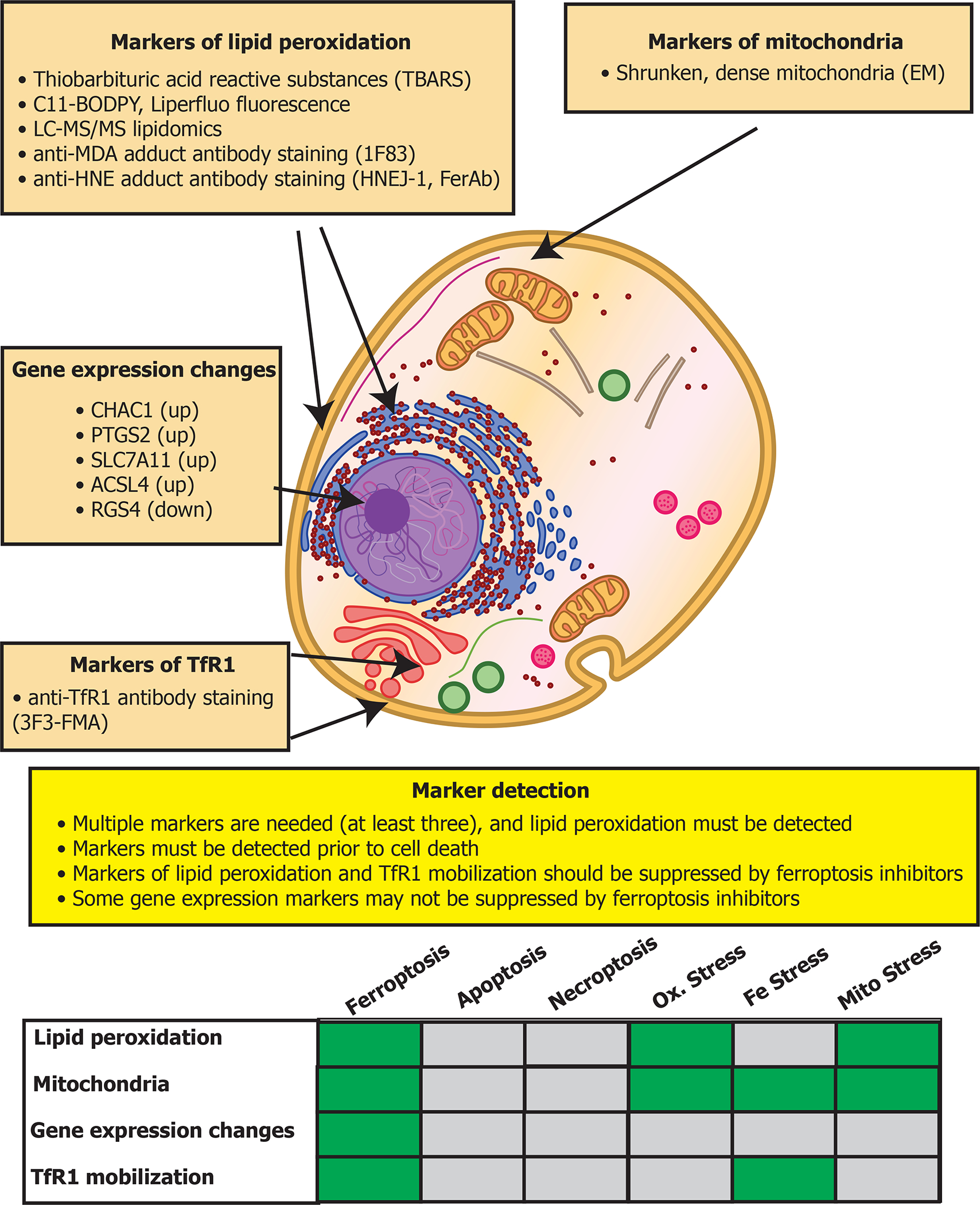
Four classes of markers for detecting ferroptotic cells are shown, along with recommendations for their use. The table on the bottom compares how these markers may appear in various contexts: during ferroptosis, apoptosis, necroptosis, oxidative stress, iron stress, or mitochondrial stress. While some markers may be activated by these other types of stress, if at least three markers are used, or a suitable combination of two markers (such as lipid peroxidation and TfR1 mobilization) ferroptosis can be distinguished from these other stress conditions.
Tumor suppression and immune functions
A number of tumor suppressors exert part of their tumor suppression function through induction of ferroptosis (Figure 4). The most commonly mutated tumor suppressor protein, p53, suppresses SLC7A11 expression (Jiang et al., 2015), which is one of the two genes that encode the components of system xc−. p53 also activates ALOX12, which is on a region of chromosome 17p that is often lost in tumors (Chu et al., 2019). Induction of p53 in the presence of t-butylhydroperoxide, an initiator of lipid peroxidation, causes ALOX12-dependent cell death that is inhibitable by ferrostatin-1 and involves expression of the ferroptosis marker gene Ptgs2, suggesting that this paradigm is a variant of ferroptosis, although the iron dependence remains to be tested. Moreover, a variant of p53 at codon 47 (S47) found in African-descent populations alters the ability of p53 to induce cell death and to suppress tumor formation (Jennis et al., 2016). This variant causes glutathione and coenzyme A accumulation (Leu et al., 2019), as well as iron accumulation and increased risk of infection, but resistance to the malaria toxin hemozoin, perhaps explaining its selection in regions with a high risk for malarial infection (Singh et al., 2020).
Knockout in mice of the epigenetic regulator MLL4, which is commonly altered in cutaneous squamous cell carcinomas in the skin, caused the development of pre-cancerous skin lesions (Egolf et al., 2021). The loss of MLL4 in the skin of these mice drives transcriptional changes that suppress ferroptosis, including increased expression of SLC7A11, GPX4, and SCD1, all of which drive resistance to ferroptosis, and loss of expression of the lipoxygenases ALOX12, ALOX12B and ALOXE3; as noted above, these lipoxygenases promote ferroptosis in some contexts (Figure 4). Similarly, BAP1 (Zhang et al., 2018) and NFS1 (Alvarez et al., 2017) exert tumor suppressive effects that depend on regulating components of the ferroptosis network (Figure 4).
Uptake of PUFAs by tumor cells from the extracellular milieu suppresses tumor growth, and a diet rich in PUFAs slows growth of HCT116 xenograft tumors in mice compared to a diet rich in monounsaturated fatty acids (Dierge et al., 2021). Thus, dietary PUFAs may naturally promote tumor suppression through this mechanism (Figure 4). On the other hand, cholesterol in the tumor microenvironment leads to increased CD36 expression on CD8+ T cells and causes these cells to take up PUFAs and undergo ferroptosis (Ma et al., 2021). Thus, excess cholesterol accumulation may promote tumor formation by ablating CD8+ tumor-infiltrating lymphocytes (Figure 4).
CD8+ T cells have also been reported to exert a tumor suppressive role through induction of ferroptosis in tumor cells; this activity is mediated by IFNγ secretion from activated CD8+ cells and results in tumor cell ferroptosis via downregulation of SLC7A11 (Wang et al., 2019). IFNγ also upregulates ACSL4, which aids in the incorporation of PUFAs into phospholipids (Liao et al., 2022). Addition of exogenous arachidonic acid (AA), a PUFA, together with IFNγ, was sufficient to induce tumor ferroptosis. Moreover, excess AA was detected in the tumor microenvironment, suggesting that AA + IFNγ derived from CD8+ T cells may be the first natural ferroptosis trigger identified (Figure 4). The lack of other known physiological inducers of ferroptosis may reflect simply a gap in our knowledge, or it may be that the consequences of triggering ferroptosis in tissues are damaging, making ferroptosis an inefficient means of cell death beyond tumor suppression.
Long-lasting immune responses after exposure to an infection or vaccine requires germinal-center-derived mature, high-affinity plasma cells and memory B cells. Follicular CD4+ helper T cells (TFH) promote the production of such long-lasting immunological B cell responses. The activity of these TFH cells must be tightly regulated under normal conditions to prevent autoimmunity or re-infection by pathogens. The number of TFH cells is regulated by GPX4-controlled cell death, and enhancing GPX4 abundance through selenium supplementation increases antibody responses after vaccination against influenza (Yao et al., 2021)(Figure 4). However, some other studies have suggested limited benefits of selenium supplementation; further validation of the role of selenium and GPX4 in TFH cell number is needed.
Development and aging
A recent study reported the generation of an antibody (HNEJ-1) that recognizes proteins modified with the end product of lipid peroxidation, 4-hydroxy-2-nonenal (HNE) and demonstrated that it recognizes ferroptotic cells (Zheng et al., 2021). Rats of varying ages from E9.5 to 2.5 years of age, exhibited this marker in numerous tissues and cells, including nucleated erythrocytes at E13.5; inhibiting ferroptosis also caused a delay in enucleation of erythrocytes. Markers of ferroptosis were also increased in the Senescence Accelerated Mouse-Prone 8(SAMP8) mouse strain, which naturally exhibits accelerated aging. Thus, ferroptosis may be involved in the normal process of erythropoiesis, as well as in aging in numerous organs (Figure 4).
Ferroptosis has a role in aging and development across species and kingdoms. For example, aging in the nematode C. elegans was reported to be associated with elevated levels of iron, depletion of glutathione, and increased sensitivity to ferroptosis (Jenkins et al., 2020) (Figure 4). Ferroptosis is also implicated in developmental cell death in the rice blast fungal pathogen Magnaporthe Oryzae and found to be required for its infection (Shen et al., 2020).
Pathological contexts involving ferroptosis
In addition to the normal, physiological functions of ferroptosis described above, ferroptosis has been implicated in many pathologies (Figure 3). It should be noted that the specific cellular and molecular pathophysiological mechanisms triggering ferroptosis, and the mechanisms by which ferroptosis triggers cause pathological sequelae are in many cases not known.
Iron-overload diseases
Given the critical role of iron in driving lipid peroxidation, it is not surprising that numerous diseases involving iron overload are associated with ferroptosis. One such iron-overload disease is hereditary hemochromatosis, which is caused by inherited mutations in iron regulatory genes. A recent report found that mice fed a high iron diet, and mice with mutations associated with hereditary hemochromatosis, develop liver damage with markers of ferroptosis, which was reversed by ferroptosis inhibition with ferrostatin-1 (Wang et al., 2017); moreover, a low iron diet reversed markers of ferroptosis, even in the hereditary hemochromatosis model mice. This highlights that dietary changes can influence sensitivity to ferroptosis.
Brain trauma has been associated with iron overload, and can be modeled by injection of FeCl3 into the somatosensory cortex of rats, causing seizures and reduced cognitive functioning. Ferrostatin-1 rescues these effects of iron overload, suggesting the involvement of excessive lipid peroxidation, and possibly ferroptosis (Chen et al., 2021c) (Figure 3).
Sedaghatian-type spondylometaphyseal dysplasia (SSMD)
SSMD is a rare disorder in newborn children that manifests as severe respiratory distress, metaphyseal chondrodysplasia, and other developmental issues. Four SSMD-linked truncating mutations in GPX4 have been described, associated with complete loss of GPX4 protein, and death shortly after birth. Recently a partial loss-of-function point mutation in GPX4 (R152H) was identified, which results in impaired enzymatic activity, but not a complete loss of function (Liu et al., 2022). This variant, unlike previous GPX4 variants associated with SSMD, is compatible with survival, albeit with a developmental impact (Figure 3). The variant also stabilizes the GPX4 protein against degradation by preventing ubiquitination of two lysine residues on GPX4.
Organ injury
A variety of models have been reported over the past decade in which ferroptosis is activated during organ injury (Stockwell et al., 2017) (Figure 3). Recently, 5 μm microplastics were found to induce markers of ferroptosis in mouse liver (Mu et al., 2021), 2.5 μm particulate matter (PM2.5) was reported to induce ferroptosis in mouse lung (Guohua et al., 2021), and ferroptosis was found to cause defective kidney repair and the formation of inflammatory proximal tubular cells (Ide et al., 2021). In addition, the vascular leakage that occurs during sepsis, which can result in injury of multiple organs, is associated with gene expression, metabolomic changes, and markers characteristic of ferroptosis (She et al., 2021). Sickle cell disease is associated with accumulation of heme in plasma due to lysis of erythrocytes that can damage organs. Sickle cell disease model mice were reported to have elevated serum heme and cardiac markers of ferroptosis, and cardiomyopathy that was attenuated by ferroptosis inhibition (Menon et al., 2021). Other diseases involving leakage of heme into organs are likely to involve ferroptosis, as this may be a common mechanism by which ferroptosis is pathologically triggered.
A recent study reported that multiorgan dysfunction syndrome (MODS), which is common in critically ill patients in intensive care units (ICUs), involves ferroptosis (Van Coillie et al., 2022). The level of catalytic iron and malondialdehyde, a marker of lipid peroxidation, in plasma among 176 critically ill adult patients was positive correlated with patient sequential organ failure assessment (SOFA) score. In addition, administration of iron sulfate to mice caused multiorgan injury, including markers of damage in kidney, liver, muscle, heart, and plasma that was attenuated by ferroptosis inhibitors, such as vitamin E, GPX4 and radical trapping antioxidants in the ferrostatin class of compounds. Thus, ferroptosis inhibition may be a viable strategy to prevent multi-organ injury in the critical care setting.
Retinal degeneration
Retinal pigment epithelial cells undergo degeneration and death during retinal diseases, such as age-related macular degeneration. Death of these cells can be suppressed by Prussian Blue nanoparticles, which reduce available ferrous iron (Tang et al., 2021), suggesting that ferroptosis may be involved. When clearance of all-trans retinal is impaired, photoreceptor cells undergo ferroptosis, and ferroptosis inhibition prevents their death (Chen et al., 2021a). Thus, retinal diseases may involve activation of ferroptosis (Figure 3).
Neurodegeneration
Numerous reports implicate ferroptosis in a variety of neurodegenerative diseases, including Huntington Disease, Alzheimer’s Disease, Parkinson’s Disease, and ALS (Stockwell et al., 2017) (Figure 3). Recent data extend these findings: GPX4 is depleted in post-mortem spinal cords of ALS patients and mouse models, GPX4 overexpression extends survival and delays disease onset in the G93A SOD1 model (Chen et al., 2021d); Wang et al., 2021b), and a pre-clinically and clinically effective treatment for ALS, CuII(atsm), inhibits ferroptosis (Southon et al., 2020).
Glutamate toxicity may involve ferroptosis. In 2001, the term oxytosis was introduced to describe oxidative cell death in neurons triggered in response to glutamate, and that involves protein synthesis, eIF2α, ROS production, cGMP-gated channels, and calcium influx, as well as lipoxygenases and glutathione depletion (Tan et al., 2001). The extent to which mechanisms governing the concept of oxytosis overlap with those governing ferroptosis in various cell types and tissues remains unclear. Although both can involve system xc− inhibition and glutathione depletion, oxytosis involves excess calcium influx, cGMP-gated channels and eIF2α, and mitochondrial swelling and DNA fragmentation (Albrecht et al., 2010) whereas these features are not observed in ferroptosis (Jiang et al., 2021). To define the relationship between oxytosis and ferroptosis, a clear set of markers, assays, and reagents for inducing and inhibiting oxytosis and ferroptosis are needed, to enable a systematic side-by-side comparison of similarities and differences between these modes of cell death; such a side-by-side comparison would also be valuable for other modes of cell death (Hadian and Stockwell, 2021). Thus, the extent to which glutamate toxicity involves oxytosis, ferroptosis, and/or other mechanisms remains unclear.
Infectious diseases
Ferroptosis has been implicated in the response to infectious agents. Numerous viruses influence host iron metabolism, iron transport, ROS production, and anti-oxidant defenses. Hepatitis C is a leading cause of liver disease, and hepatitis C viral replication is restricted by activation of ferroptosis in the host cell, and specifically under the control of Fatty Acid Desaturase 2 (FADS2) (Yamane et al., 2021). FADS2 participates in the biosynthesis of highly unsaturated fatty acids such as arachidonic acid (20:4) and docosahexaenoic acid (22:6) from linoleic acid (18:2) and α-linolenic acid (18:3). The more highly desaturated PUFAs promote ferroptosis through their susceptibility to peroxidation. Knockdown of FADS2 using RNA interference enhances replication of HCV, consistent with the conclusion that suppressing ferroptosis favors HCV replication. Thus, in the case of HCV, ferroptosis suppresses viral replication (Figure 3).
SARS-CoV-2 may activate ferroptosis during infection. Patients with COVID-19 have high serum ferritin levels, suggesting high iron exposure in tissues. SARS-Cov-2 infection of Syrian Golden Hamsters causes features of COVID-19 in humans, such as acute lung injury, inflammation and hypoxemia. In this model, lipid changes typical of ferroptosis, as well as induction of the ferroptosis marker TfR1 were reported (Bednash et al., 2021). In this hamster model, it isn’t clear if ferroptosis contributes to restricting infection, as in the HCV model, or contributes to inflammation, or is a byproduct of infection without significant consequence (Figure 3).
Mycobacterium marinum infection in zebrafish is suppressed by heme oxygenase 1 (HMOX1), which regulates iron availability from heme, and by treatment with ferrostatin-1 (Luo et al., 2021), suggesting that ferroptosis promotes infection. Consistent with this, Mycobacterium tuberculosis (Mtb) infection of macrophages induces ferroptosis, and in mice, infection by Mtb is restricted when ferroptosis is inhibited (Amaral et al., 2019). Thus, ferroptosis promotes Mtb infection (Figure 3).
Pseudomonas aeruginosa is a common bacterial infection in hospitalized patients and patients with cystic fibrosis. Pseudomonas aeruginosa secrete a lipoxygenase (pLoxA) to induce ferroptosis in host lungs, which is associated with worse clinical outcomes (Dar et al., 2018). Ferroptosis enhances Pseudomonas aeruginosa infection, as is the case with Mtb (Figure 3). Some infections might also be amenable to treatment with ferroptosis inducers. For example, trypanosomes are sensitive to ferroptosis induced by loss of the trypanosome homolog of GPX4, tryparedoxin (Bogacz and Krauth-Siegel, 2018); tryparedoxin inhibitors might be effective in treating trypanosome infections, if a sufficient therapeutic window can be found versus host GPX4 (Figure 3).
Autoimmune diseases
Activation of ferroptosis has been detected in models of autoimmune disorders. Systemic lupus erythematosus (SLE) is such an autoimmune disease that was linked in 2021 to ferroptosis activation in neutrophils (Li et al., 2021). Patients with SLE were found to have low neutrophil counts, and serum from SLE patients promoted neutrophil death via ferroptosis driven by downregulation of GPX4 that was mediated by CaMKIV-CREMα. In addition, mice with neutrophil-specific knockout of GPX4 had features of SLE. Thus, activation of ferroptosis in neutrophils promotes autoimmune disease, perhaps by releasing autoantigens. Similarly, ferroptosis in human airway epithelial cells driven by 15-lipoxygenase was reported to be associated with the release of mitochondrial DNA and consequent worsening of asthma (Nagasaki et al., 2022). Asthma, which involves overactivation of immune responses, thus may be triggered or worsened by ferroptosis in airway epithelial cells that releases immunogenic mitochondria DNA. Ferroptosis, when it occurs in neutrophils or airway epithelial cells can thus cause excessive immune activation, leading to autoimmune disease (Figure 3).
Tumorigenesis
While ferroptosis is reported to function as a tumor-suppression mechanisms, loss of ferroptosis can drive tumorigenesis. Recent studies found that selenoproteins are predictors of cancer risk and that selenium is elevated in tumor tissue (Wu et al., 2021), implicating elevated GPX4 abundance and activity in tumorigenesis. Thus, genetic and environmental perturbations that inactivate ferroptosis may drive tumorigenesis in some contexts.
Finally, the lymph environment protects invasive melanoma cells from ferroptosis, allowing increased metastatic spread. Metastasis of tumor cells through blood leads to tumor cell ferroptosis, but metastasis through the lymphatic system protects metastatic tumor cells from ferroptosis, perhaps due to high levels of oleic acid in lymph (Ubellacker et al., 2020).
Response to environmental heat stress
With ongoing climate change driving elevated global temperatures, heat stress is likely to become an increasingly common agricultural problem. Heat stress has been shown to induce ferroptosis in numerous organisms, including the plant Arabidopsis thaliana (Distefano et al., 2017) and in the photosynthetic cyanobacteria Synechocystis sp. PCC 6803 (Aguilera et al., 2022).
Therapeutic applications of ferroptosis
Given the wealth of recent studies implicating ferroptosis in the diseases described above, there may be opportunities to treat these diseases through modulating ferroptosis. In addition to developing potent and selective compounds and biologics that can regulate ferroptosis through specific mechanisms, it will be important to expand the set of tools for reliably detecting ferroptosis and other cell death modalities (Hadian and Stockwell, 2021). Currently, monitoring lipid peroxidation is one means of identifying the presence of ferroptosis (Figure 5); methods of detecting lipid peroxidation include use of the thiobarbituric acid reactive substances (TBARS) assay, detection of isoprostanes or lipid peroxidation products directly by LC-MS/MS, oxidation of the C11-BODIPY fluorescent probe, and reactivity with antibodies that detect adducts formed by products of lipid peroxidation, such as with the anti-HNE FerAb antibody (Kobayashi et al., 2021), the HNEJ-1 antibody (Zheng et al., 2021), and the anti-MDA adduct 1F83 antibody (Yamada et al., 2001) (Figure 5).
My lab recently reported that increased abundance and plasma localization of the transferrin receptor TfR1 is a marker for ferroptosis, which can be detected with the newly identified 3F3-FMA antibody, as well as with additional anti-TfR1 antibodies (Feng et al., 2020). We then found that TfR1 staining could be coupled with machine learning to distinguish cells undergoing ferroptosis from those undergoing apoptosis (Jin et al., 2022). Several genes, such as CHAC1, PTGS2, SLC7A11, ACSL4, are induced during ferroptosis, and RGS4 is downregulated during ferroptosis; altered expression of these genes can be detected by qPCR as indicators of ferroptosis (Figure 5).
While these markers distinguish ferroptosis from apoptosis and necroptosis, certain stress conditions can activate some of these markers without activating ferroptosis (Figure 5). Thus, detecting multiple markers is important to definitively identify ferroptosis—markers of lipid peroxidation and TfR1 mobilization is potential means of detecting ferroptosis, while excluding other stress conditions. Nonetheless, the definitive criteria for detecting ferroptosis continue to evolve as the community’s understanding of ferroptosis and its relationship to related biological processes deepens.
Inducers of ferroptosis
As a cell death modality, ferroptosis can potentially be used to eliminate problematic cell types, such as cancer cells, inflammatory cells, or activated fibroblasts. It is critical to determine which cell types are amenable to induction to ferroptosis, and through which specific mechanisms, and whether induction mechanisms would also activate ferroptosis in other cell types.
There are four broad mechanisms that have been identified for inducing ferroptosis—(1) inhibition of system xc−, (2) inhibition/degradation/inactivation of GPX4, (3) depletion of reduced CoQ10, and (4) induction of lipid peroxidation through peroxides, iron, or PUFA overload.
Several lines of evidence validate that inhibition of system xc− is a potent mechanism for inducing ferroptosis—multiple structurally distinct small molecule inhibitors of this antiporter, such as erastin, sulfasalazine, and glutamate, inhibit system xc− and consequently induce ferroptosis (Figure 2); moreover, cystine depletion removes the extracellular substrate for system xc− and also induces ferroptosis. Similarly, genetic inactivation or small-molecule-mediated inhibition or degradation of GPX4 induces ferroptosis in many cell types. Inhibition of CoQ10 biosynthesis through the mevalonate pathway or inactivation of CoQ10 reductases, such as AIFM2/FSP1 or DHODH, induces ferroptosis in settings where GPX4 is not present. Finally, ferroptosis is induced by treatment with excess iron, PUFAs, or with peroxides, such as tBOOH or FINO2. All four of these mechanisms are quite specific for inducing ferroptosis versus other cell death mechanisms, as their lethal effects are mostly or completely suppressed by ferroptosis-specific inhibitors, and markers of other types of cell death are not activated.
There are numerous contexts in which ferroptosis inducers may be beneficial, such as infectious diseases and fibrosis. The most well-studied application is elimination of various types of cancer cells. Inducers of ferroptosis, such as GPX4 inhibitors and system xc− inhibitors, have shown promise, for example, in treating specific tumor types: a diffuse large B cell lymphoma xenograft was sensitive to treatment with the system xc− inhibitor imidazole ketone erastin (IKE), which activated markers of ferroptosis in the xenografted tumors (Zhang et al., 2019). Knockout of SLC7A11 provided significant benefit in a mouse genetic model of pancreatic cancer, without inducing markers of other types of cell death beyond ferroptosis (Badgley et al., 2020).
There are additional targets beyond GPX4 and system xc− for inducing ferroptosis in tumor cells. Pancreatic cancers cells are dependent on the aspartate aminotransaminase GOT1, and knockdown of GOT1 sensitizes to ferroptosis (Kremer et al., 2021). In addition, lung tumor spheroids are dependent on NRF2 for suppressing ferroptosis in order to survive (Takahashi et al., 2020).
Beyond their use in specific tumor contexts, such as lymphoma and pancreatic cancer, ferroptosis inducers may be beneficial in the context of radiotherapy and immunotherapy. Radiation can induce ferroptosis, and ferroptosis inducers can act as radiosensitizers (Lei et al., 2020); Ye et al., 2020) alone or in combination with immunotherapy (Lang et al., 2019). Aggressive cancers that have undergone epithelial-to-mesenchymal transition are more susceptible to ferroptosis (Viswanathan et al., 2017), and resistance to targeted agents that induce apoptosis is associated with increased sensitivity to ferroptosis (Hangauer et al., 2017); BRAF amplification is one mechanism that shifts sensitivity from BRAF inhibitors to ferroptosis inducers (Song et al., 2021).
Identifying the specific contexts in which ferroptosis-inducing drugs would be effective is aided by the use of predictive biomarkers. Numerous biomarkers have been reported for selecting patients most likely to respond to ferroptosis-inducing anti-cancer therapy, such as low levels of transsulfuration metabolites (Hayano et al., 2015), low levels of NADPH (Shimada et al., 2016a), susceptibility to photochemical lipid peroxidation (Wang et al., 2021a), absence of PI3K/mTOR pathway activity (Yi et al., 2020), mutation of the Hippo/Yap pathway (Wu et al., 2019a), expression of the PUFA biosynthesis genes ELOVL5 and FADS1 (Lee et al., 2020b), amplification of MDM2/MDMX (Venkatesh et al., 2020), and clear cell cancer cell morphology (Zou et al., 2019). In summary ferroptosis inducers are being explored for their ability to eliminate specific cell populations, such as specific tumor types, although further pre-clinical research is needed to validate this concept.
Inhibitors of ferroptosis
There are several key control points that may allow for potent and selective therapeutic suppression of ferroptosis. The process of ferroptosis is driven by peroxidation of specific lipids, and directly blocking this peroxidation process is a key control point. Upregulation of GPX4 through selenium supplementation is an indirect means of intervening in ferroptosis. Generation of the substrate lipids for peroxidation through the action of lipid biosynthesis and ACSL4 is an additional control point, and controlling iron availability is a third control point.
Early after the discovery of ferroptosis, several potential uses of the first inhibitors of this process were reported: ferrostatin-1 and liproxstatin were found to act as radical trapping agents to inhibit propagation of lipid peroxidation (Zilka et al., 2017), and to be effective in models of glutamate toxicity (Dixon et al., 2012), Huntington Disease (Skouta et al., 2014), premature ventricular leukomalacia (Skouta et al., 2014), kidney injury (Friedmann Angeli et al., 2014); Skouta et al., 2014) and liver injury (Carlson et al., 2016).
In addition to ferrostatins and liproxstatins, additional inhibitors of ferroptosis have been identified. The hypocholesterolemic drug probucol and its analogs have been reported to suppress ferroptosis and to be effective in models of glutamate toxicity (Bueno et al., 2020). Of note, necrostatin-1 (nec-1), which is a RIPK1 inhibitor that suppresses necroptosis, has an off-target effect in suppressing ferroptosis at high concentrations and thus must be used with care. Selenium delivery suppresses ferroptosis during stroke (Alim et al., 2019). The mitochondrially-targeted nitroxide XJB-5–131 suppresses both apoptosis and ferroptosis and is an effective treatment in models of traumatic brain injury and Huntington Disease (Krainz et al., 2016).
Control of ferroptosis through diet
A number of studies suggested that dietary factors can promote or inhibit ferroptosis. For example, dietary PUFAs promote ferroptosis in C. elegans (Perez et al., 2020). Dietary vitamin E suppresses the impact of loss of GPX4, and as noted above, dietary selenium, and iron influence susceptibility to ferroptosis. These findings imply that the diet of model organisms may be an important variable influencing susceptibility to ferroptosis and should be carefully controlled. In addition, it may be possible to create optimized diets for controlling ferroptosis in animals and in humans.
Conclusion and Future Directions
To summarize the implications of the above studies, there are six key aspects of ferroptosis that are important to fully appreciate the mechanisms governing this form of cell death (Figure 6). First, ferroptosis has not been synonymous with generalized production of reactive oxygen species (ROS); indeed, it is possible to have ROS production without ferroptosis, and ferroptosis generally involves the oxidation of specific lipids rather than generalized accumulation of ROS. Second and similarly, the search for the specific lipids that promote ferroptosis in a variety of contexts is ongoing, but it is clear that oxidized lipids have different capacities to promote ferroptosis, whereas some lipids, such as MUFAs, suppress ferroptosis.
Figure 6. Key concepts in ferroptosis.
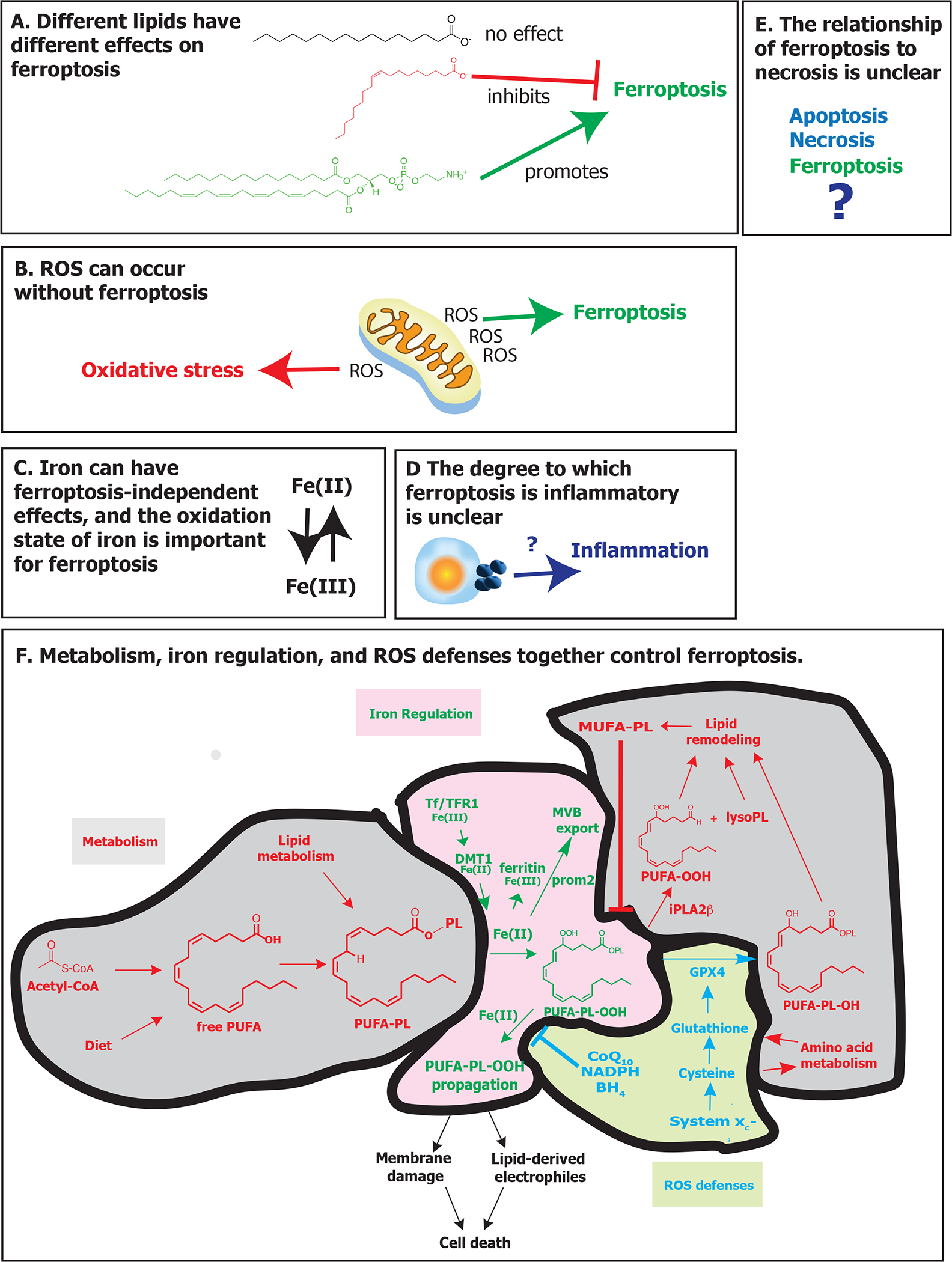
Six key concepts that aid in understanding ferroptosis are described. A. Different lipids have different effects on ferroptosis. Lipids with saturated fatty acids generally don’t affect ferroptosis, whereas those with monounsaturated fatty acids suppress ferroptosis and those with polyunsaturated fatty acids promote ferroptosis. (B) Reactive oxygen species (ROS) can occur without inducing ferroptosis. In some cases, soluble ROS accumulate and cause oxidative stress without inducing lipid peroxidation or ferroptosis. (C) Iron can induce effects unrelated to ferroptosis, and the oxidation state of iron is critical for ferroptosis. Some iron toxicity does not depend on lipid peroxidation. The ability of iron to promote ferroptosis mostly depends on it being in the Fe(II) state, although lipoxygenase generally require iron to be in Fe(III) to be activated. (D) The degree to which ferroptosis is inflammatory is unclear. Ferroptosis involves the release of some factors, which can be inflammatory, but perhaps not to the same extent as during classic necrosis or pyroptosis. (E) The relationship of ferroptosis to necrosis is unclear. Whether necrosis is a broader umbrella or a distinct process remains uncertain. (F) Metabolism, iron regulation, and ROS defenses together control ferroptosis. Free polyunsaturated fatty acids can be obtained through biosynthesis or diet, and are converted into membrane-localized phospholipids. Iron promotes oxidation of PUFA-PLs to PUFA-PL hydroperoxides, which can be eliminated through reduction by glutathione peroxidase 4, using cysteine-derived glutathione, yielding non-toxic lipid alcohols, or by iPLA2β, yielding free oxidized PUFA and lysolipids, which undergo further lipid remodeling. Oxidized PUFA-PLs can propagate in the presence of Fe(II), eventually yielding membrane damage and/or generation of reactive lipid-derived electrophiles. CoQ10 and BH4 and the pathways that generate them can block propagation of oxidized PUFA-PLs. Metabolism-related pathways are shown in red, iron regulation pathways are shown in green, and ROS defense pathways are shown in blue. Abbreviations: BH4, tetrahydrobiopterin; CoA, coenzyme A; CoQ10, coenzyme Q10; cys, cysteine; DMT1, ferrous ion membrane transport protein DMT1; GPX4, glutathione peroxidase 4; iPLA2β, phospholipase A2 group VI; lysoPL, lysophospholipid; MUFA, monounsaturated fatty acid; MVB, multivesicular body; NADPH, reduced nicotinamide adenine dinucleotide phosphate; PL, phospholipid; prom2, prominin-2; PUFA, polyunsaturated fatty acid; PUFA-PL, phospholipid containing a polyunsaturated fatty acyl lipid tail; PUFA-PL-OOH, phospholipid with peroxidized polyunsaturated fatty acyl tail; ROS, reactive oxygen species; system xc−, sodium-independent, anionic amino acid transport system; Tf, transferrin; TfR1, transferrin receptor protein 1.
Third, iron can have numerous effects beyond inducing ferroptosis, such that the accumulation of iron is not synonymous with ferroptosis. Moreover, the oxidation state of iron is important for its ability to contribute to ferroptosis—Fe(II) promotes ferroptosis, whereas Fe(III) is generally inert and stored in ferritin, except in the active site of lipoxygenases, where Fe(III) is the active form of the enzyme. Thus, determining the redox state of iron and whether it contributes specifically to ferroptosis in a given context is important.
Fourth, the degree to which ferroptosis is a necrotic death remains uncertain; ferroptosis is clearly distinguishable from apoptosis, pyroptosis, necroptosis, and unregulated necrosis. However, whether ferroptosis can be classified as a type of necrotic death will require a better definition and clear functions associated with the term necrotic death.
Fifth, the degree to which ferroptosis is inflammatory and/or immunogenic in different contexts remains uncertain. A number of studies have suggested that ferroptosis contributes to inflammation or immunogenicity, but the degree to which this is generally the case and how ferroptosis compares in this regard to pyroptosis, necroptosis, or apoptosis remains uncertain. Sixth and finally, metabolism, iron regulation and ROS defenses together control sensitivity to ferroptosis. Therefore, perturbing metabolism, iron homeostasis and ROS levels are key means of modulating the susceptibility of cells to ferroptosis. The field of ferroptosis has been rapidly accelerating, in part due to the increasing synergies and insights obtained by cross-fertilization of ideas, findings, and methods within the fields of metabolism, iron regulation, and ROS biology. These three fields each yield insight into distinct aspects of ferroptosis: metabolism explains how the key substrates of ferroptosis are generated and remodeled, iron regulation reveals how availability of Fe(II) is controlled, and ROS biology shows how endogenous defenses against lipid peroxidation function (Figure 6).
Nonetheless, our understanding of ferroptosis remains incomplete—there are at least three key unanswered questions that are likely to drive advances in ferroptosis over the next decade. First, what is the execution mechanism of ferroptosis? As described above, ferroptosis is driven by peroxidation of specific PUFA-containing lipids in particular organelles, such as the ER. How, where, and when this leads to cell death per se is unknown.
Second, identifying the diverse triggers for ferroptosis will shed light on the mechanisms and contexts in which this form of cell death is activated physiologically and pathologically. It is clear that glutamate, iron overload, SLC7A11 suppression, GPX4 depletion and PUFA uptake can act as triggers for ferroptosis. Defining the full array of initiating mechanisms for ferroptosis and the contexts in which they are used will greatly enrich our understanding of how ferroptosis fits into the broader world of biology and medicine.
Third, mechanisms and methods for the selective control of ferroptosis remain elusive. Such selective activation or inhibition in specific tissues, cells, and/or disease contexts may be critical for translating knowledge of ferroptosis into therapies. For example, targeting GPX4 systematically is likely to cause toxicities, such as kidney damage and neurodegeneration, among other organ damage. However, selective targeting of persister tumor cells could be impactful and may offer a therapeutic safety window. There are a variety of means by which selective control of ferroptosis might be realized using drug delivery, optimized biodistribution and pharmacokinetics, and the selection of targets and mechanisms that are used in specific contexts for the control of ferroptosis. Elucidating these mechanisms and strategies may be critical for leveraging knowledge of ferroptosis for therapeutic benefit. Addressing these unanswered questions promises to yield important new insights not only into ferroptosis, but into the increasingly diverse areas of biology to which it is connected.
Acknowledgements
I am grateful to the many members of my laboratory and our network of generous and supportive collaborators over the last decade who have helped to launch and sustain the new field of ferroptosis. Research in the Stockwell lab on ferroptosis is generously supported by grants from the National Cancer Institute (NCI) (P01CA87497 and R35CA209896), the National Institute of Neurological Disorders and Stroke (NINDS) (R61NS109407 and R33NS109407), and NIH grant UG3CA256962.
Footnotes
Declaration of Interests
B.R.S. is an inventor on patents and patent applications involving small molecule drug discovery and ferroptosis, and co-founded and serves as a consultant to Inzen Therapeutics, Nevrox Limited, Exarta Therapeutics, and ProJenX, Inc., serves as a consultant to Weatherwax Biotechnologies Corporation, and Akin Gump Strauss Hauer & Feld LLP, and receives sponsored research support from Sumitomo Dainippon Pharma Oncology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams RP, Carroll WL, and Woerpel KA (2016). Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem Biol 11, 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agmon E, Solon J, Bassereau P, and Stockwell BR (2018). Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep 8, 5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, Berdun F, Bartoli C, Steelheart C, Alegre M, Bayir H, Tyurina YY, Kagan VE, Salerno G, Pagnussat G, et al. (2022). C-ferroptosis is an iron-dependent form of regulated cell death in cyanobacteria. J Cell Biol 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alborzinia H, Ignashkova TI, Dejure FR, Gendarme M, Theobald J, Wolfl S, Lindemann RK, and Reiling JH (2018). Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun Biol 1, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, and Methner A (2010). Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc-as a neuroprotective drug target. CNS & neurological disorders drug targets 9, 373–382. [DOI] [PubMed] [Google Scholar]
- Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ, et al. (2019). Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 177, 1262–1279 e1225. [DOI] [PubMed] [Google Scholar]
- Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K, and Possemato R (2017). NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB, and Sher A (2019). A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med 216, 556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, Kolle P, Tschoep K, Issels RD, Daniel PT, et al. (2008). The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene 27, 1618–1628. [DOI] [PubMed] [Google Scholar]
- Bannai S, and Kitamura E (1980). Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem 255, 2372–2376. [PubMed] [Google Scholar]
- Bannai S, Tsukeda H, and Okumura H (1977). Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochemical and Biophysical Research Communications 74, 1582–1588. [DOI] [PubMed] [Google Scholar]
- Beatty A, Singh T, Tyurina YY, Tyurin VA, Samovich S, Nicolas E, Maslar K, Zhou Y, Cai KQ, Tan Y, et al. (2021). Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat Commun 12, 2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednash JS, Kagan VE, Englert JA, Farkas D, Tyurina YY, Tyurin VA, Samovich SN, Farkas L, Elhance A, Johns F, et al. (2021). Syrian hamsters as a model of lung injury with SARS-CoV-2 infection: Pathologic, physiologic, and detailed molecular profiling. Transl Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharier O, Tyurin VA, Goff JP, Guerrero-Santoro J, Kajiwara K, Chu T, Tyurina YY, St Croix CM, Wallace CT, Parry S, et al. (2020). PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc Natl Acad Sci U S A 117, 27319–27328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks J, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogacz M, and Krauth-Siegel RL (2018). Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, Baer CE, Dixon SJ, and Mercurio AM (2019). Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell 51, 575–586 e574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno DC, Canto RFS, de Souza V, Andreguetti RR, Barbosa FAR, Naime AA, Dey PN, Wullner V, Lopes MW, Braga AL, et al. (2020). New Probucol Analogues Inhibit Ferroptosis, Improve Mitochondrial Parameters, and Induce Glutathione Peroxidase in HT22 Cells. Mol Neurobiol 57, 3273–3290. [DOI] [PubMed] [Google Scholar]
- Burr GO, and Burr MM (1929). A new defi ciency disease produced by the rigid exclusion of fat from the diet. J Biol Chem 82, 345–367. [Google Scholar]
- Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, Dovey CM, Wang J, Locasale JW, Stone E, et al. (2019). A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell reports 26, 1544–1556 e1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL, and Conrad M (2016). Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol 9, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Chen J, Wang Y, Liu Z, and Wu Y (2021a). Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance. J Biol Chem 296, 100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, Rabadan R, Jiang X, Stockwell BR, and Gu W (2021b). iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun 12, 3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, and Jiang X (2020a). Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ 27, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KN, Guan QW, Yin XX, Wang ZJ, Zhou HH, and Mao XY (2021c). Ferrostatin-1 obviates seizures and associated cognitive deficits in ferric chloride-induced posttraumatic epilepsy via suppressing ferroptosis. Free Radic Biol Med 179, 109–118. [DOI] [PubMed] [Google Scholar]
- Chen L, Na R, Danae McLane K, Thompson CS, Gao J, Wang X, and Ran Q (2021d). Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci Rep 11, 12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, Xu Y, Yang WH, Mathey-Prevot B, and Chi JT (2020b). Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ 27, 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreul ME (1823). Sur les causes des differences que l’on observe dans les savons. Annales de Chimie et de Physique 23, 16–32. [Google Scholar]
- Chng CP, Sadovsky Y, Hsia KJ, and Huang C (2021). Site-Specific Peroxidation Modulates Lipid Bilayer Mechanics. Extreme Mech Lett 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, and Gu W (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson BT, and Brennan MA (2001). Pro-inflammatory programmed cell death. Trends Microbiol 9, 113–114. [DOI] [PubMed] [Google Scholar]
- Crane FL (2007). Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion 7 Suppl, S2–7. [DOI] [PubMed] [Google Scholar]
- Dar HH, Tyurina YY, Mikulska-Ruminska K, Shrivastava I, Ting HC, Tyurin VA, Krieger J, St Croix CM, Watkins S, Bayir E, et al. (2018). Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest 128, 4639–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, and Yuan J (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1, 112–119. [DOI] [PubMed] [Google Scholar]
- Dierge E, Debock E, Guilbaud C, Corbet C, Mignolet E, Mignard L, Bastien E, Dessy C, Larondelle Y, and Feron O (2021). Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell metabolism 33, 1701–1715 e1705. [DOI] [PubMed] [Google Scholar]
- Ding CC, Rose J, Sun T, Wu J, Chen PH, Lin CC, Yang WH, Chen KY, Lee H, Xu E, et al. (2020). MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat Metab 2, 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distefano AM, Martin MV, Cordoba JP, Bellido AM, D’Ippolito S, Colman SL, Soto D, Roldan JA, Bartoli CG, Zabaleta EJ, et al. (2017). Heat stress induces ferroptosis-like cell death in plants. J Cell Biol 216, 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch ME, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti N, Slusher BS, et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife, doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel C, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S, Lessnick SL, Hahn WC, and Stockwell BR (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. [DOI] [PubMed] [Google Scholar]
- Eagle H (1955). Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514. [DOI] [PubMed] [Google Scholar]
- Egolf S, Zou J, Anderson A, Simpson CL, Aubert Y, Prouty S, Ge K, Seykora JT, and Capell BC (2021). MLL4 mediates differentiation and tumor suppression through ferroptosis. Sci Adv 7, eabj9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka JM, et al. (2020). Transferrin Receptor Is a Specific Ferroptosis Marker. Cell reports 30, 3411–3423 e3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton HJH (1876). On a new reaction or tartaric acid. Chemical News 33, 190. [Google Scholar]
- Fleming MD, Trenor CC 3rd, Su MA, Foernzler D, Beier DR, Dietrich WF, and Andrews NC (1997). Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 16, 383–386. [DOI] [PubMed] [Google Scholar]
- Flohe L (2020). Looking Back at the Early Stages of Redox Biology. Antioxidants (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, and Jiang X (2016). Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, and Jiang X (2019). Role of Mitochondria in Ferroptosis. Mol Cell 73, 354–363 e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Hu F, Feng H, Linkermann A, Min W, and Stockwell BR (2018). Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, Cellier M, Vidal S, and Gros P (1995). Identification and characterization of a second mouse Nramp gene. Genomics 25, 514–525. [DOI] [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, and Hediger MA (1997). Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388, 482–488. [DOI] [PubMed] [Google Scholar]
- Guohua F, Tieyuan Z, Xinping M, and Juan X (2021). Melatonin protects against PM2.5-induced lung injury by inhibiting ferroptosis of lung epithelial cells in a Nrf2-dependent manner. Ecotoxicol Environ Saf 223, 112588. [DOI] [PubMed] [Google Scholar]
- Hadian K, and Stockwell BR (2021). A roadmap to creating ferroptosis-based medicines. Nat Chem Biol 17, 1113–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L, Zhong YM, Tan CP, and Mao ZW (2021). Quantitative tracking of endoplasmic reticulum viscosity during ferroptosis by an iridium complex via TPPLM. Chem Commun (Camb) 57, 5040–5042. [DOI] [PubMed] [Google Scholar]
- Hao S, Yu J, He W, Huang Q, Zhao Y, Liang B, Zhang S, Wen Z, Dong S, Rao J, et al. (2017). Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia 19, 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano M, Yang WS, Corn CK, Pagano NC, and Stockwell BR (2015). Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ, Jul 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano M, Yang WS, Corn CK, Pagano NC, and Stockwell BR (2016). Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ 23, 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg CG, and Laurell C-B (1945). Studies on the capacity of serum to bind iron — a contribution to our knowledge of the regulation mechanism of serum iron. Acta Physiol Scand 10, 307. [Google Scholar]
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, and Tang D (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide S, Kobayashi Y, Ide K, Strausser SA, Abe K, Herbek S, O’Brien LL, Crowley SD, Barisoni L, Tata A, et al. (2021). Ferroptotic stress promotes the accumulation of pro-inflammatory proximal tubular cells in maladaptive renal repair. eLife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421. [DOI] [PubMed] [Google Scholar]
- Jenkins NL, James SA, Salim A, Sumardy F, Speed TP, Conrad M, Richardson DR, Bush AI, and McColl G (2020). Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. (2016). An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30, 918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, and Gu W (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Stockwell BR, and Conrad M (2021). Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Schorpp K, Samaga D, Unger K, Hadian K, and Stockwell BR (2022). Machine Learning Classifies Ferroptosis and Apoptosis Cell Death Modalities with TfR1 Immunostaining. ACS Chem Biol 17, 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, Harris IS, and DeNicola GM (2021). Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell metabolism 33, 174–189 e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathman SG, Boshart J, Jing H, and Cravatt BF (2020). Blockade of the Lysophosphatidylserine Lipase ABHD12 Potentiates Ferroptosis in Cancer Cells. ACS Chem Biol 15, 871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Harada Y, Homma T, Yokoyama C, and Fujii J (2021). Characterization of a rat monoclonal antibody raised against ferroptotic cells. J Immunol Methods 489, 112912. [DOI] [PubMed] [Google Scholar]
- Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kossl J, et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainz T, Gaschler MM, Lim C, Sacher JR, Stockwell BR, and Wipf P (2016). A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent Sci 2, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, Sajjakulnukit P, Myers A, Thurston G, Hou SW, et al. (2021). GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun 12, 4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang F, Liu J, Xie Y, Tang D, and Kang R (2021). MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol 28, 765–775 e765. [DOI] [PubMed] [Google Scholar]
- Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A, et al. (2019). Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufberger V (1937). Sur la cristallisation de la ferritine. Bull Soc Chim Biol 19., 1575–1582. [Google Scholar]
- Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al. (2020a). Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 22, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nam M, Son HY, Hyun K, Jang SY, Kim JW, Kim MW, Jung Y, Jang E, Yoon SJ, et al. (2020b). Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc Natl Acad Sci U S A 117, 32433–32442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H, et al. (2020). The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res 30, 146–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Murphy ME, and George DL (2019). Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A 116, 8390–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, Xu Y, Krishfield S, Lipsky PE, Tsokos GC, et al. (2021). Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol 22, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, Sell A, Wei S, Grove S, Johnson JK, et al. (2022). CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Forouhar F, Seibt T, Saneto R, Wigby K, Friedman J, Xia X, Shchepinov MS, Ramesh SK, Conrad M, et al. (2022). Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol 18, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Stocker R, Britton WJ, Kikuchi K, and Oehlers SH (2021). Haem oxygenase limits Mycobacterium marinum infection-induced detrimental ferrostatin-sensitive cell death in zebrafish. Febs J. [DOI] [PubMed] [Google Scholar]
- Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, Wang Q, Yang M, Qian J, and Yi Q (2021). CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell metabolism 33, 1001–1012 e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432 e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon AV, Liu J, Tsai HP, Zeng L, Yang S, Asnani A, and Kim J (2021). Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, and Brodie BB (1973). Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 187, 211–217. [PubMed] [Google Scholar]
- Mu Y, Sun J, Li Z, Zhang W, Liu Z, Li C, Peng C, Cui G, Shao H, and Du Z (2021). Activation of pyroptosis and ferroptosis is involved in the hepatotoxicity induced by polystyrene microplastics in mice. Chemosphere, 132944. [DOI] [PubMed] [Google Scholar]
- Nagasaki T, Schuyler AJ, Zhao J, Samovich SN, Yamada K, Deng Y, Ginebaugh SP, Christenson SA, Woodruff PG, Fahy JV, et al. (2022). 15LO1 dictates glutathione redox changes in asthmatic airway epithelium to worsen type 2 inflammation. J Clin Invest 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SJ, Frey AG, Palenchar DJ, Achar S, Bullough KZ, Vashisht A, Wohlschlegel JA, and Philpott CC (2019). A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat Chem Biol 15, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SJ, Protchenko O, Shakoury-Elizeh M, Baratz E, Jadhav S, and Philpott CC (2021). The iron chaperone and nucleic acid-binding activities of poly(rC)-binding protein 1 are separable and independently essential. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J, Hailfinger S, von Karstedt S, and Garcia-Saez AJ (2021). Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ 28, 1644–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez MA, Magtanong L, Dixon SJ, and Watts JL (2020). Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells. Dev Cell 54, 447–454 e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, Wiesner U, Bradbury MS, Niethammer P, Zaritsky A, et al. (2020). Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol 22, 1042–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilstra MJ, Veldink GA, and Vliegenthart JF (1994). The dioxygenation rate in lipoxygenase catalysis is determined by the amount of iron (III) lipoxygenase in solution. Biochemistry 33, 3974–3979. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell metabolism 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Shah R, Shchepinov MS, and Pratt DA (2018). Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She H, Hu Y, Zhou Y, Tan L, Zhu Y, Ma C, Wu Y, Chen W, Wang L, Zhang Z, et al. (2021). Protective Effects of Dexmedetomidine on Sepsis-Induced Vascular Leakage by Alleviating Ferroptosis via Regulating Metabolic Reprogramming. J Inflamm Res 14, 6765–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Liang M, Yang F, Deng YZ, and Naqvi NI (2020). Ferroptosis contributes to developmental cell death in rice blast. New Phytol 227, 1831–1846. [DOI] [PubMed] [Google Scholar]
- Shimada K, Hayano M, Pagano NC, and Stockwell BR (2016a). Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol 23, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016b). Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat Chem Biol 12, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H (1985). Oxidative stress (London: Orlando: : Academic Press; ). [Google Scholar]
- Singh KS, Leu JI, Barnoud T, Vonteddu P, Gnanapradeepan K, Lin C, Liu Q, Barton JC, Kossenkov AV, George DL, et al. (2020). African-centric TP53 variant increases iron accumulation and bacterial pathogenesis but improves response to malaria toxin. Nat Commun 11, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg P, Lo D, Weinberg J, Linkermann A, et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136, 4551–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Minami JK, Huang A, Dehkordi SR, Lomeli SH, Luebeck J, Goodman MH, Moriceau G, Krijgsman O, Dharanipragada P, et al. (2021). Plasticity of extrachromosomal and intrachromosomal BRAF amplifications in overcoming targeted therapy dosage challenges. Cancer discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA, and Birsoy K (2020). Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 16, 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourkes TL (2004). The discovery of lecithin, the first phospholipid. Bull Hist Chem 29, 9–15. [Google Scholar]
- Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, Barnham KJ, Crouch PJ, Ayton S, Donnelly PS, et al. (2020). Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharmacol 177, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WY, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai YJ, Pan MH, Gong HB, Lu DH, Sun J, et al. (2021). Phospholipase iPLA2beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol 17, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Cho P, Selfors LM, Kuiken HJ, Kaul R, Fujiwara T, Harris IS, Zhang T, Gygi SP, and Brugge JS (2020). 3D Culture Models with CRISPR Screens Reveal Hyperactive NRF2 as a Prerequisite for Spheroid Formation via Regulation of Proliferation and Ferroptosis. Mol Cell 80, 828–844 e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Schubert D, and Maher P (2001). Oxytosis: A novel form of programmed cell death. Current topics in medicinal chemistry 1, 497–506. [DOI] [PubMed] [Google Scholar]
- Tang Z, Huo M, Ju Y, Dai X, Ni N, Liu Y, Gao H, Zhang D, Sun H, Fan X, et al. (2021). Nanoprotection Against Retinal Pigment Epithelium Degeneration via Ferroptosis Inhibition. Small Methods 5, e2100848. [DOI] [PubMed] [Google Scholar]
- Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Drager N, Leng K, Nalls MA, Singleton AB, Xu K, et al. (2021). Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat Neurosci 24, 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, Gu Z, McCormick ML, Durham AB, Spitz DR, et al. (2020). Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini F, Maiorino M, Valente M, Ferri L, and Gregolin C (1982). Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta 710, 197–211. [DOI] [PubMed] [Google Scholar]
- Van Coillie S, Van San E, Goetschalckx I, Wiernicki B, Mukhopadhyay B, Tonnus W, Choi SM, Roelandt R, Dumitrascu C, Lamberts L, et al. (2022). Targeting ferroptosis protects against experimental (multi)organ dysfunction and death. Nat Commun 13, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannotti A, and Delachaux A (1949). Iron Metabolism and its Clinical Significance (New York: Grune & Stratton; ). [Google Scholar]
- Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, Kengmana ES, Aron AT, Klein AM, Csuka JM, et al. (2020). MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev 34, 526–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal SM, Malo D, Vogan K, Skamene E, and Gros P (1993). Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell 73, 469–485. [DOI] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis KF, Morehead LC, Bird JT, Byrum SD, and Miousse IR (2021). Differences in cell death in methionine versus cysteine depletion. Environ Mol Mutagen 62, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Graham ET, Naowarojna N, Shi Z, Wang Y, Xie G, Zhou L, Salmon W, Jia JM, Wang X, et al. (2021a). PALP: A rapid imaging technique for stratifying ferroptosis sensitivity in normal and tumor tissues in situ. Cell Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, et al. (2017). Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66, 449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Tomas D, Perera ND, Cuic B, Luikinga S, Viden A, Barton SK, McLean CA, Samson AL, Southon A, et al. (2021b). Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, et al. (2017). PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 171, 628–641 e626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Pe’er D, Shaik F, Bulinski JC, and Stockwell BR (2011). Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc Natl Acad Sci U S A 108, E771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN, and Jiang X (2019a). Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Li D, Feng X, Zhao F, Li C, Zheng S, and Lyu J (2021). A pan-cancer study of selenoprotein genes as promising targets for cancer therapy. BMC Med Genomics 14, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, Shan B, Pan H, and Yuan J (2019b). Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A 116, 2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Kumazawa S, Ishii T, Nakayama T, Itakura K, Shibata N, Kobayashi M, Sakai K, Osawa T, and Uchida K (2001). Immunochemical detection of a lipofuscin-like fluorophore derived from malondialdehyde and lysine. J Lipid Res 42, 1187–1196. [PubMed] [Google Scholar]
- Yamane D, Hayashi Y, Matsumoto M, Nakanishi H, Imagawa H, Kohara M, Lemon SM, and Ichi I (2021). FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by Gpx4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, Wei Y, Wan Y, Wang N, Zhou M, et al. (2021). Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol 22, 1127–1139. [DOI] [PubMed] [Google Scholar]
- Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang Y, et al. (2020). Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Zhu J, Wu J, Thompson CB, and Jiang X (2020). Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A 117, 31189–31197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitler L, Fiore A, Meyer C, Russier M, Zanella G, Suppmann S, Gargaro M, Sidhu SS, Seshagiri S, Ohnmacht C, et al. (2021). Anti-ferroptotic mechanism of IL4i1-mediated amino acid metabolism. eLife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, Peng XD, Li X, Huang Y, Zhu XY, et al. (2022). PKCbetaII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol 24, 88–98. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al. (2018). BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20, 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, Lei G, Mao C, Koppula P, Cheng W, et al. (2021). mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun 12, 1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, and Stockwell BR (2019). Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633 e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang L, Tsuduki T, Conrad M, and Toyokuni S (2021). Embryonal erythropoiesis and aging exploit ferroptosis. Redox Biol 48, 102175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, and Pratt DA (2017). On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci 3, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, Paradkar S, Boehnke N, Deik AA, Reinhardt F, et al. (2020a). Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, and Schreiber SL (2020b). Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dancik V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 10, 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]