

Abstract
Aims:
Electronic cigarette (ECIG) has been used as an alternative to tobacco smoking as it lacks the majority of toxicants found in tobacco smoke. However, the effect of ECIG aerosol inhalation on cardiac health are not well studied. The present study aimed to compare the effects of ECIGs with that of combustible tobacco cigarette (T-Cigs) and waterpipe (WP) smoke on cardiac biomarkers of oxidative stress, inflammation, and fibrosis.
Main Methods:
Rats were randomized into control (fresh air, n=12), ECIG aerosol (n=12), T-Cig smoke (n=15), or WP (n=13) smoke conditions in which they were exposed 1 h/daily, 6 day/week for 4 weeks. Cardiac biomarkers of oxidative stress, inflammation, and remodeling were assessed.
Key Findings:
Relative to control, significant increase in heart to body weight ratio was observed in all exposed groups. Cardiac endothelin-1 and myeloperoxidase were increased for ECIG and T-Cig. Cardiac nitrite and TBARS were increased in all exposed groups, but activity of superoxide dismutase was increased for ECIG and T-Cig only while glutathione levels increased for ECIG only. No changes were observed for cardiac C-reactive protein and catalase activity. Cardiac fibrosis was observed in all exposed groups coupled with an increase in the transforming growth factor beta protein that was significant for ECIG only.
Significance:
ECIG aerosol may promote cardiac alterations in similar manner to tobacco smoke by promoting myocardial oxidative stress and inflammation leading to fibrosis. With regard to cardiac health, exposure to ECIG aerosol and combustible T-Cig smoke may lead to similar adverse outcomes.
Keywords: Electronic cigarette, cigarette tobacco smoking, waterpipe tobacco smoking, inflammation, oxidative stress, fibrosis
1. Introduction
Cardiovascular disease (CVD) is the leading cause of death globally [1]. Tobacco smoking is a common and significant risk factor for the initiation and progression of CVD, particularly atherosclerosis. Tobacco smoking is a serious public health concern, contributing to increased risk of thrombosis, atherosclerosis, and death [2]. Tobacco cigarettes (T-Cigs), cigars, and waterpipes (WP) (aka “shisha”, “hookah”, “narghile”) are examples of diverse methods for smoking tobacco. Waterpipe use, once limited to certain world regions (e.g., Eastern Mediterranean, Indian subcontinent) has gained popularity globally [3, 4].
Electronic cigarettes (ECIGs) have been proposed as a less lethal alternative to combustible tobacco smoking and, though evidence of efficacy is limited, as a potential method for smoking cessation [3, 5, 6]. ECIGs use an electronically powered heating element to aerosolize a liquid that usually contains propylene glycol, vegetable glycerin, and the psychoactive stimulant drug nicotine; the user inhales the resulting aerosol [3]. Claims that ECIG use is safer than combustible tobacco smoking are based on the fact that ECIG aerosols lack the majority of toxicants found in tobacco smoke [4]. However, more recent results are not so supportive of claims of decreased health risks and there is much debate on this issue [1, 5]. While the health effects of T-Cig smoking, and to a lesser extent WP use, have received considerable empirical study, the health effects of the much newer ECIGs require further investigation.
Several factors contribute to the development of CVD. These factors include oxidative stress, inflammation, and remodeling [7]. Previous studies have shown that tobacco consumption contributes to CVD risk via activation of neutrophils, monocytes and platelets in addition to cytokines production, generation of reactive oxygen and nitrogen species, and smooth muscle and fibroblast proliferation [7]. Tobacco smoking has been documented to promote the production of high amount of free radicals among smokers. These free radicals oxidize lipids, proteins and DNA, resulting in tissue damage [8]. We have found previously that T-Cig smoke exposure augments myocardial inflammatory levels of endothelin 1 (ET-1), interlukins 6 (IL-6) and myeloperoxidase (MPO), and promotes generation of nitrites, lipid peroxides, and fibrosis in a rat model of diabetes [9]. Interestingly, it has been found that e-cigarettes fluids and vapor promoted generation of oxidant radicals and inflammatory cytokines of IL-6 and 8 in lung cells and tissues [10]. Recently, Reinikovaite et al [11] documented that exposure to e-cigarettes vapor for 5 weeks can cause destruction of lung structure and vasculature just as toxic as tobacco cigarettes. In this study, we will examine and compare for the first time the impact of ECIG aerosol exposure with that of T-Cig and WP smoke exposure on cardiac health. We hypothesized that ECIG aerosol exposure will cause elevations in cardiac biomarkers of inflammation, oxidative stress and remodeling similar to T-Cig and WP smoke.
2. Methods
2.1. Animals and Treatment
Male Wistar rats (weight=200–250g, 8–9 weeks old) were housed in the animal house at room temperature with a 12 hour dark/light photoperiod, and receive adequate amounts of food and water. Each cage included 4–5 rats. All procedures were performed in accordance with the guidelines of Animal Care and Use Committee (The Institutional Animal Care and Use Committee Guidebook, 2ndedition of 2002 [12]) at Jordan University of Science and Technology (JUST).
Rats were randomly assigned into 4 groups (12–15 in each group):
Fresh air exposure only (Control, n=12).
Electronic cigarette aerosol exposed (ECIG, n=12).
Tobacco cigarette smoke exposed (T-Cig, n=15).
Waterpipe smoke exposed (WP, n=13).
For ECIG, T-Cig, and WP, a whole body exposure system was used for one hour/day, 6 days/week for 4 weeks as described previously [13]. Control animals were sham exposed to fresh air in the exposure system. Each exposure chamber included 4–5 rats during the light cycle. We have four exposure chambers; one for each group. Both ECIG and T-Cig rats were exposed simultaneously in different rooms, followed by WP and control exposed rats in the same order daily.
2.2. Electronic cigarette aerosol exposure system
The ECIG group was exposed to ECIG aerosol from an 18 mg/ml nicotine solution that was 70% (by volume) propylene glycol (PG) and 30% vegetable glycerin (VG) using a reverse puffing machine manufactured by the Aerosol Research Lab of the American University of Beirut. The solution was prepared using free base nicotine and analytical grade PG and VG obtained from Sigma Aldrich, USA. ECIG aerosol was emitted by a 1.8 Ω single coil ECIG (Mini Protank2, KangerTech, Shenzhen, China) powered at 5.76 W. The computer-controlled reverse-puffing machine automatically activated a pump to force lab air through the air inlet ports of the ECIG. The resulting aerosol exited through the ECIG mouthpiece directly into the exposure chamber (38×25×25 cm, L×W×H). The machine was programmed to produce a 4-second puff of volume 116.7 mL with a 10 second interpuff interval, for the duration of the 1 hour exposure session. About 2 ml of ECIG solution is consumed each hour (~36 mg nicotine/hr). The exposure chamber was coupled with a fresh air ventilation system to maintain a net air change rate of 1.75 L/min. On average, the aerosol concentration in the chamber was approximately 1.33 g/m3. The exposure chamber was made from crystalline polycarbonate, and had a removable ceiling with two inlets (one for aerosol and the other for fresh air) and one outlet for the excess flow.
2.3. Waterpipe and cigarette smoking exposure systems
The WP smoke exposure system described in Khabour et al.[14] was used for waterpipe exposure. In brief, this system utilizes a transfer pump to simultaneously draw smoke from the WP and inject it into the exposure chamber described above. The volume of the WP puff was monitored and recorded using a puff topography instrument during each exposure session and the diaphragm pump flow rate was adjusted to maintain the 530 mL puff volume in accordance of the Beirut Method [13]. This regimen is chosen because it is similar on average to human puff topography during WP smoking [13]. An electrochemical sensor (Bacharach Monoxor II) was used to monitor carbon monoxide (CO). The average concentration of CO (mean±sem) was 514±14.72 ppm for all WP exposed rats throughout the study. For each session, 10 g of ma’assel (0.5% nicotine, Nakhla Brand, Cairo, Egypt) was loaded into the WP with total nicotine content of 50 mg/hr. The Ma’assel was heated using lighted charcoal briquettes (Shaban Company, Cairo, Egypt). Water in the WP bowl was changed every time the water pipe machine was used.
For T-Cig, rats were exposed to side-stream smoke by placing a smoldering cigarette in the upper middle half of the chamber as previously described [9, 15]. Marlboro™ Red (Philip Morris, USA) cigarettes were used for the purpose of this study and approximately 5–6 cigarettes were consumed over a 1-hr session. Each cigarette is ~ 10.9 mg nicotine with total content of 55–66 mg/hr. The average concentration of CO (mean±sem) was 237.1±2.40 ppm for all T-Cig exposed rats throughout the study. Control rats were placed in the same exposure chamber exposed to fresh air only.
2.4. Molecular analysis
Rats were sacrificed by decapitation. Hearts were detached immediately, washed with normal saline, weighed and dissected into two parts; one part (small part of the right side of the heart) was fixed in formalin for histopathology (n=5–6 each group) and the second part (left side of the heart and remaining right side, n=12–15 each group) was stored at −80°C until time of homogenization. Cardiac tissues were homogenized in a cold phosphate buffer saline containing protease inhibitors (Sigma, St. Louis, MO, USA,). Centrifugation of the homogenate was carried out at 15000×g for 15 min at 4 °C and the supernatant was collected for measurements of cardiac markers. Sample aliquots were stored at −80°C until time of molecular analysis.
Cardiac contents of nitrite were measured by the total nitric oxide and nitrate/nitrite parameter assay kit with an assay sensitivity range of 0.09–0.78 μmol/L (R&D Systems, MA, USA). Measurements of thiobarbituric acid reactive substances (TBARS) were performed using the TBARS assay kit with a sensitivity range of 0.007–0.005 μM (R&D Systems, MA, USA). Total glutathione levels and the activities of superoxide dismutase (SOD) were assessed by standard colorimetric assays (Sigma-Aldrich Corp, St. Louis, MO, USA). The activity of catalase was measured by catalase assay kit with a sensitivity range of 2–35 nmol/min/ml (Cayman Chemical, MI, USA). Levels of myeloperoxidase (MPO) and C reactive protein (CRP) were determined by ELISA kits (Rat CRP and MPO ELISA kits, MyBioSource, Inc. CA, USA) with a sensitivity of 2.0 U/L and up to 0.5 ng/ml; respectively. Measurements of endothelin-1 (ET-1), the transforming growth factor-beta (TGF-β1) and matrix metalloproteinase −2 (MMP-2) were carried out using specific ELISA assays (Quantikine ELISA, R&D Systems, MA, USA). The sensitivity range was 0.031–0.207 pg/ml, 1.7–15.4 pg/ml, and 0.014–0.082 ng/ml for ET-1, TGF-β1, and MMP-2; respectively. Plates were read at the specified wavelength determined in each kit using Epoch Biotek microplate reader (BioTek, Winooski, VT, USA). All analyses were normalized to total cardiac protein contents.
2.5. Myocardial fibrosis
Right sided cardiac tissues were fixed in 10% formalin and cut into 4μm thick sections. Paraffin embedded sections were deparaffinized by xylene followed by graded alcohol. Sections were washed with deionized water and incubated with Bouin’s solution overnight and stained with Masson’s Trichrome according to kit instructions (Sigma, St. Louis, MO, USA). Sections were viewed using high resolution light microscope (Nikon, Shinagawa, Japan) at 10x and 40x magnification. The percentage area of fibrosis was quantified by NIH Image J software (version 1.50i, Maryland, USA) at 10x magnification (100x original magnification). For each slide, three representative images were taken, analyzed, and averaged.
2.6. Statistical analysis
Data are presented as means ±SEM for continuous data and as percentages for categorical data. Because molecular data were not normally distributed, the Kruskal-Wallis test was used to assess presence of statistical differences in measurements among study groups, and the Dunn’s post hoc test was used for evaluation of statistical differences for each pair of groups. Two-way ANOVA was used to assess changes in body weight over time across study groups. A probability value (p)<0.05 was considered statistically significant. Statistical analysis was carried out using GraphPad Prism 7.
3. Results
3.1. Heart and Body Weight
A significant increase in body weight was observed for control throughout the study with increases of less magnitude observed for ECIG, T-Cig, and WP (Figure 1A). By the end of the study, ECIG, T-Cig, and WP had lower body weight relative to control. Heart weight was not changed at the end of the study (p=0.1742, Figure 1B), but the ratio of heart to body weight was higher for ECIG, T-Cig and WP compared to control (Figure 1C).
Figure 1: Heart and body weight.
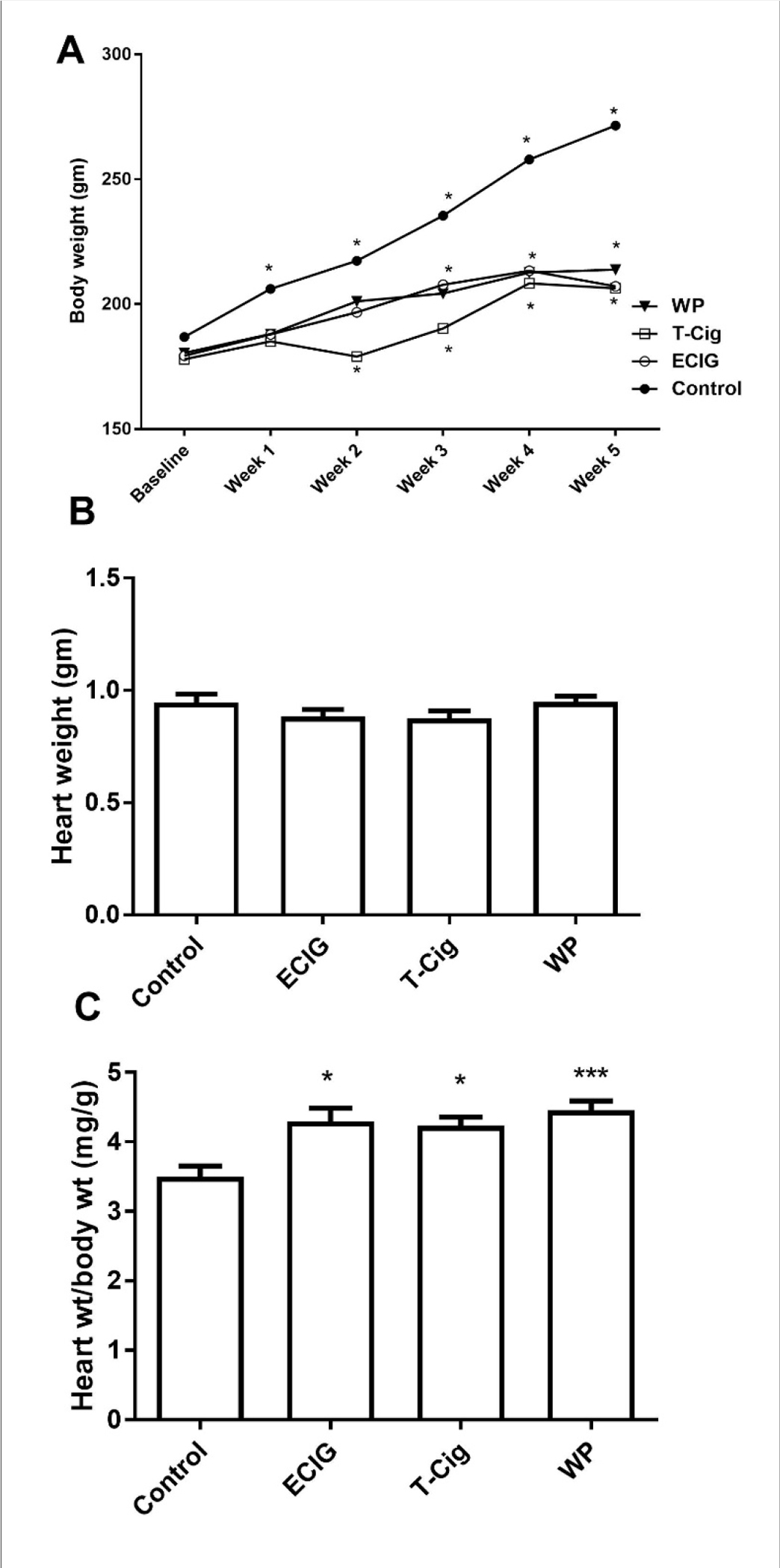
Changes in body weight (A), heart weight (B) and heart to body weight ratio (C) for control, ECIG, T-Cig, and WP. P*<0.05, ***p<0.001 vs. Control; Dunn’s post hoc test, n=12–15 each group.
3.2. Changes in Cardiac Inflammatory Factors
Relative to control, cardiac content of ET-1 was increased in ECIG and T-Cig, with a trend toward an increase in WP (Figure 2A). A similar trend of increase relative to control was also found for cardiac MPO, however, a statistical significance was only found for ECIG (p<0.01, Figure 2 B). Compared to control, no significant increases in cardiac CRP were observed among ECIG, T-Cig, or WP (Figure 2 C). However, levels of cardiac CRP and ET-1 were significantly higher for ECIG than for WP (Figure 2).
Figure 2: Cardiac biomarkers of inflammation.
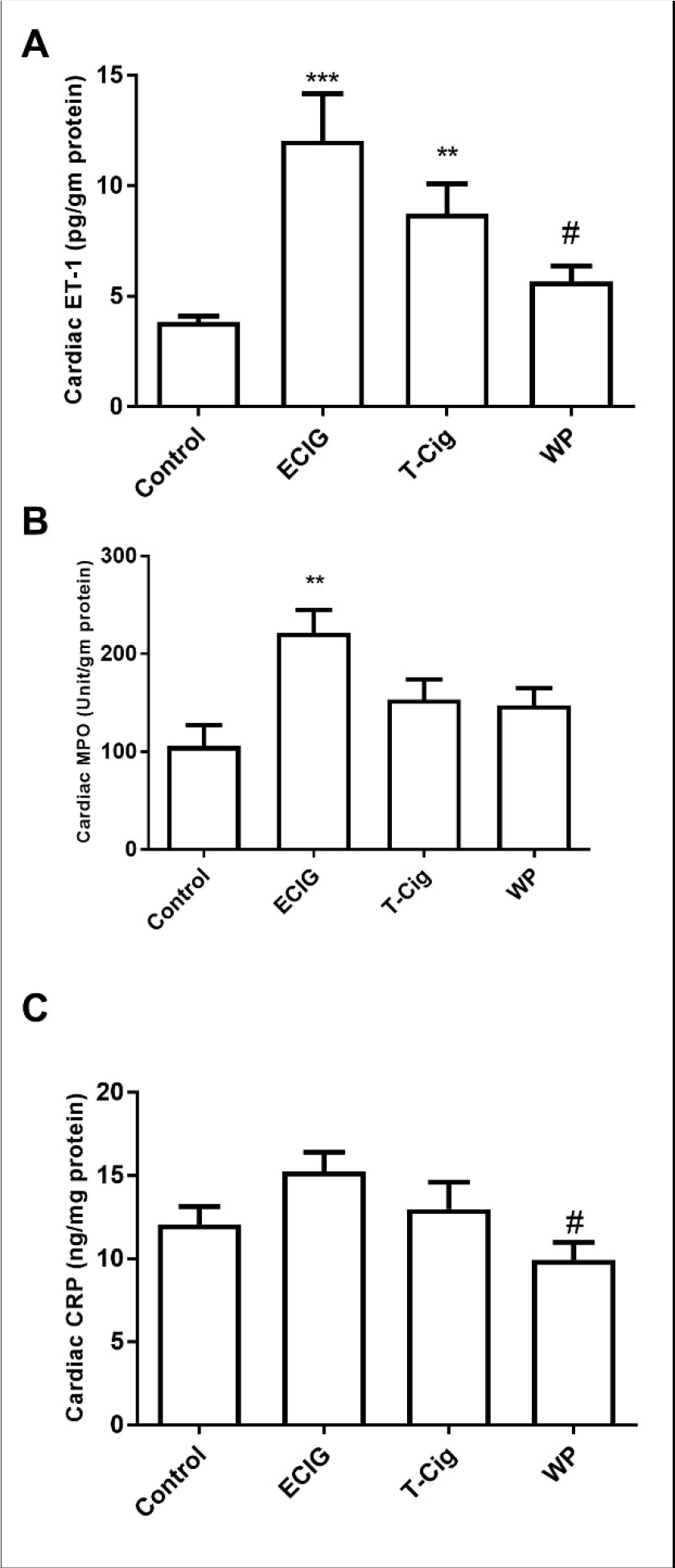
Levels of cardiac of endothelin-1 (ET-1, A), Myeloperoxidase (MPO, B) and C-reactive protein (CRP, C) in the control, ECIG, T-Cig, and WP. **p<0.01, ***p<0.001 vs. Control; #p<0.05, vs. ECIGs; Dunn’s post hoc test, n=11–15 each group.
3.3. Levels of Cardiac Oxidants and Antioxidants
Cardiac oxidant levels are presented in Figure 3. Relative to control, cardiac levels of total nitrites were increased for ECIG and T-Cig (Figure 3A). On the other hand, relative to control, cardiac TBARS substances were increased significantly for ECIG, T-Cig, and WP (Figure 3B). Cardiac SOD activity was significantly increased for ECIG and T-Cig relative to control, and levels were statistically lower in WP relative to ECIG and T-Cig (Figure 3C). No differences in cardiac catalase activities were observed among groups (p=0.6753, Figure 3D). A trend of increase in cardiac GSH was noted for T-Cig and WP, relative to control, with significant results obtained for ECIG only (Figure 3E).
Figure 3: Cardiac biomarkers of oxidative status.
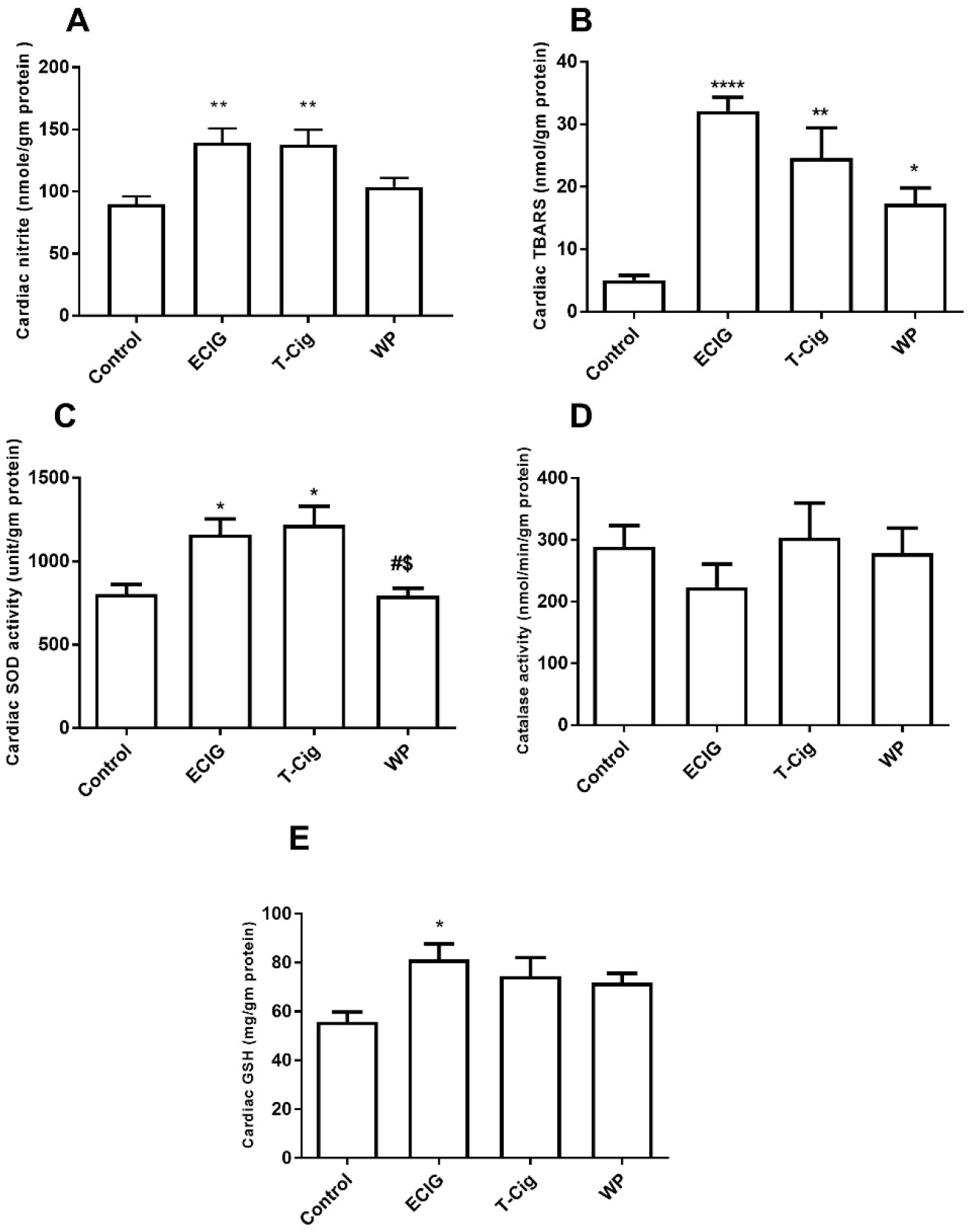
Levels of cardiac nitrites (A), thiobarbituric acid reactive substances (TBARS, B), Super oxide dismutase (SOD) activities (C), catalase activities (D) and glutathione levels (GSH, E) in the control, ECIG, T-Cig, and WP. *p<0.05, **p<0.01, ****p<0.0001 vs. Control; #p<0.05 vs. ECIGs; $p<0.05 vs. T-Cigs; Dunn’s post hoc test, n=11–15 each group.
3.4. Levels of Cardiac Pro-Fibrotic Factors
Relative to control, a trend of an increase in cardiac TGF beta content was observed for T-Cig and WP, with statistically significant results obtained for ECIG only (Figure 4A). Cardiac MMP-2 content was significantly increased in ECIG and T-Cig, relative to control (Figure 4 B).
Figure 4: Cardiac biomarkers of fibrosis.
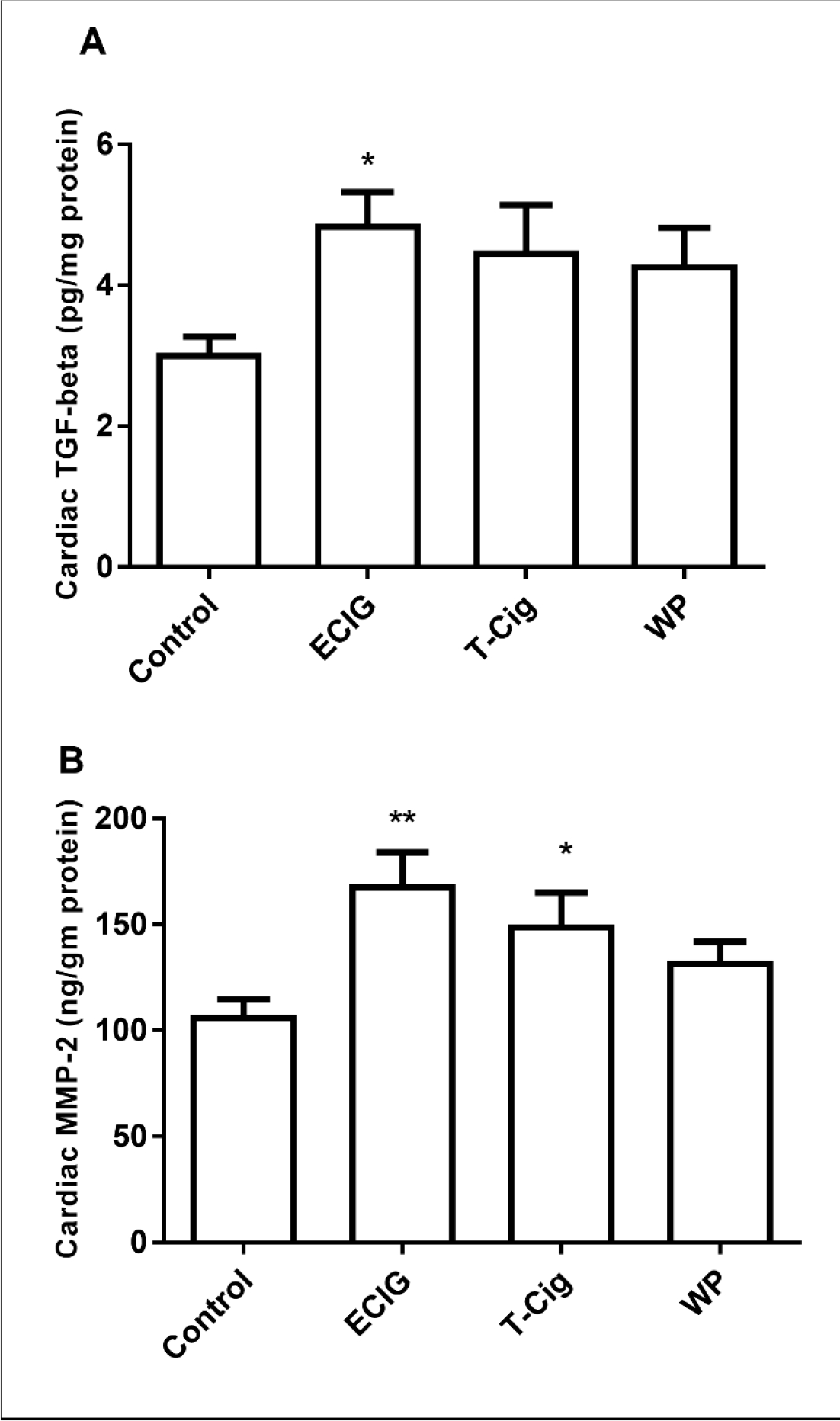
Cardiac contents of transforming growth factor-beta (TGF-beta, A) and matrix metalloproteinase −2 (MMP-2, B) in the control, ECIG, T-Cig, and WP. *p<0.05, **p<0. 01 vs. Control; Dunn’s post hoc test, n=11–13 each group.
3.5. Extent of Cardiac Fibrosis
Masson’s Trichrome staining revealed that myocardial fibrosis (Blue) was increased in ECIG, T-Cig, and WP relative to control (Figure 5 A–D). Quantification of extent of cardiac fibrosis at 10x magnification documented statistical increase in the percentage area of fibrosis in ECIG, T-Cig, and WP relative to control (Figure 5E).
Figure 5: Extent of cardiac interstitial fibrosis.
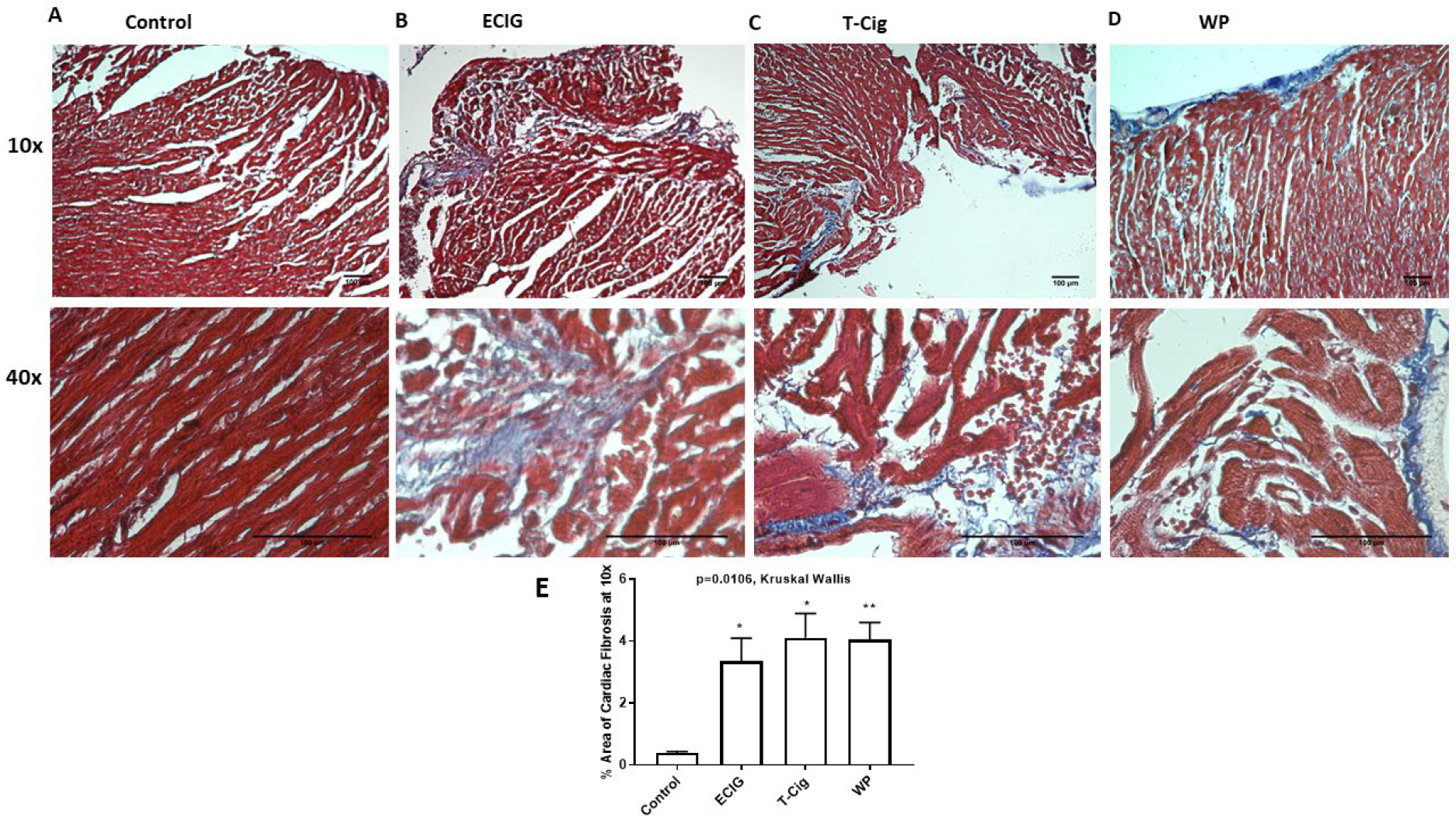
Masson’s Trichrome staining of right sided cardiac sections shows extent of fibrosis (blue), cardiac myocyte (red) in control (A), ECIG (B), T-Cig (C), WP (D). Sections were examined at 10x and 40x magnification. Figure 5E is quantitative analysis of % area of fibrosis at 10x. *p<0.05, **p<0. 01 vs. Control.
4. Discussion
Smoking is a major risk factor for development and progression of coronary artery diseases (CAD) and other CVDs. The impact of tobacco smoke on CVD risk has been widely studied. Both T-Cigs and WP are popular worldwide, especially in the Middle East, while ECIG use is becoming more prevalent everywhere. Current studies suggest that ECIGs are not a harm-free alternative method to tobacco smoke and could be of potential concern on cardiovascular health. However, these findings are mainly derived from acute studies and data on chronic ECIG exposure are not available. The mechanisms underlying their increased risk of cardiac disease are not fully known. In this study, we compare for the first time the effect of ECIGs aerosol exposure with T-Cig or WP smoke exposure on outcomes related to myocardial oxidative stress, inflammation and fibrosis.
Oxidative stress is associated with the onset of CVD and heart failure [17]. Oxidative stress is defined as a process of injury that occurs when there is an imbalance between reactive oxygen species (ROS)/reactive nitrogen species (RNS) production and antioxidant body defenses [18]. Tobacco smoking produces metabolites [19] that increase the production of oxidant free radicals leading to oxidative damage and remodeling. Tobacco smoking has been shown to promote myocardial oxidative stress, characterized by an increase in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity leading to an enhanced superoxide production, increased levels of lipid hydroperoxide and decreased antioxidant enzymes such as catalase and superoxide dismutase (SOD) [19]. Interestingly, exposure to ECIG aerosol in mice has been shown to increase vascular, cerebral, and pulmonary oxidative stress via NADPH oxidase indicating its potential to promote CVDs and pulmonary cerebrovascular disease [20]. Myocardial oxidative stress might be also driven by the mitochondria which play a critical role in cell regulation, ATP generation and apoptosis [21]. Myocardial mitochondrial damage and dysfunction is associated with cardiac disease [21]. The activity of the myocardial cytochrome oxidase is declined following single and prolonged exposure to secondhand smoke in rats [22]. In the present study, we observed increased cardiac levels of TBARS in all aerosol/smoke exposed groups (i.e., T-Cig, WP, and ECIG), and the results were greater for ECIG and T-Cig suggesting that these two products do not differ in risk of myocardial oxidative damage. Total nitrite levels were increased in the ECIG and T-Cig but not in the WP group. These changes were paralleled with an increase in cardiac activities of SOD probably to neutralize excess of ROS and RNS. Cardiac glutathione level was significantly increased in ECIG rats with marginal increases in other groups, clearly suggesting that ECIG aerosol promotes marked changes in myocardial oxidant status that at least as similar as T-Cig effects and more profound than WP effects. Burning charcoal briquettes is the specific source of toxic materials that are unique to WP smoking, some materials differ in their amounts from cigarettes with unknown cardiovascular effects [23].
ROS production leads to activation of nuclear factor (NF)-kappa B signaling pathways. The NF-kappa B signaling promotes regulation various inflammatory factors, including the tumor necrosis factor-alpha (TNF-α) and C reactive protein (CRP), in addition to the decrease in nitric oxide (NO) production [24]. Thus, oxidative stress causes sustained inflammation, in addition to fibrosis contributing to CVD and other disease processes [25]. High inflammatory states also are associated with smoking [26]. Tobacco smoking is associated with increased levels of aortic expression of interleukins and TNFα [26] and myeloperoxidase (MPO), a neutrophil-derived enzyme [27] contributing to the initiation and progression of vascular inflammatory diseases [28, 29]. Endothelial dysfunction, defined as the reduced release of vasodilators like NO or increased vasoconstrictors like ET-1, is an early sign of CAD [30]. The increased ET-1 expression is associated with tobacco smoking [31]. Endothelin-1 is a potent vasoconstrictor, inflammatory and mitogenic factor [31, 32]. We previously documented increased cardiac ET-1, MPO and interleukin-6 in diabetic hearts exposed to tobacco smoke [9]. Here, we also found that cardiac ET-1 but not CRP levels were increased in ECIG and T-Cig groups and not in WP group. Interestingly, cardiac MPO content was markedly elevated in the ECIG-exposed group only. Recently, it has been found that vaping with nicotine in ECIG smokers increased plasma MPO [33], an effect that was not observed in vaping vehicle in absence of nicotine. Overheated propylene glycol/vegetable glycerin produces free radicals and volatile carbonyls (i.e. acrolein) by thermal degradation, which are potent vasoconstrictors and oxidative stressors. MPO is primarily stored in the neutrophils and monocytes granules. Neutrophils are activated in the sputum of e-cigarettes smokers, but whether this can be attributed to the presence of nicotine is unclear [34]. However, a comparison between nicotine doses per hour exposure between study groups suggest that ECIG contains the least amount of nicotine with comparable content of nicotine in T-Cig and WP groups, indicating that effects on cardiac biomarkers may not be promoted by nicotine. Lack of significant changes in the WP group may be due to the differential composition of other toxic materials in WP relative to other groups or that ECIG aerosol induces cardiac alteration by different mechanisms than tobacco smoke. In addition, we do not know if a single constituent of the liquid mixture or the constituents as a whole could be responsible for the observed changes.
Cardiac fibrosis adversely affects the myocardial structure and function [35]. Tobacco smoking accelerates atrial collagen accumulation, leading to symptomatic atrial fibrosis [36]. Interestingly, tobacco smoking has been shown to promote expression of cardiac remodeling and hypertrophic factors such as fibronectin [26]. It has been found that exposure to ECIG aerosol in mice induced systemic inflammation and multi-organ fibrosis, including the kidney, liver and the heart [37]. In our study, cardiac fibrosis was observed in all exposed groups, including in animals exposed to ECIG aerosol. This was paralleled by a similar trend of increase in TGF beta growth factor in all exposed groups that was significant in the ECIG group, indicating remodeling. Interestingly, contents of cardiac MMP-2 were significantly increased in ECIG and T-Cig groups in a manner that is similar to changes in oxidant and inflammatory biomarkers, indicating that these responses might promote expression of MMP-2 resulting in fibrosis. However, no changes were observed in the WP group. As myocardial fibrosis was also found in WP group, our data might suggest presence of other fibrotic molecules that are differentially expressed in WP group.
Together, our findings suggest that ECIG aerosol promotes marked cardiac alterations that at least as similar as T-Cig and more profound than WP effects, suggesting that ECIG vapor or bi products of heated solvent produce chemicals and free radicals that promote inflammation and oxidative stress similar to T-Cig. The differential composition of WP versus T-Cig may explain the less significant changes seen in WP as compared to T-Cig and ECIG. Differences in the delivery system and rate of nicotine administration, as well as the evidence that the effects of nicotine are highly variable in function of its pharmacokinetics and pharmacodynamics, could perhaps explain these differences [7, 11, 33].
5. Limitations of the study
This study used a relatively short exposure period and only one ECIG liquid was used to produce aerosol from only one device. Future work may involve longer exposure periods and a variety of liquid/device combinations and nicotine contents, as ECIGs are a heterogeneous product class with great variety in device construction, electrical power output, and liquid constituents [3]. Future studies should evaluate levels of nicotine and its metabolites in serum and cardiac tissues to test if differential effects of ECIG versus tobacco smoke are nicotine dependent.
6. Conclusion
Exposure to ECIG aerosol can lead to cardiac inflammation and oxidative stress, in a manner similar to tobacco cigarette and more profound than waterpipe smoking. Exposure to ECIG aerosol, T-CIG and WP smoke led to cardiac fibrosis. Based on work published previously and the current results [1, 5], ECIGs aerosol and combustible T-Cig smoke may lead to similar adverse outcomes and cannot at this time be considered a less harmful method of nicotine administration, relative to tobacco cigarettes.
Funding Acknowledgment
The present study was funded by the deanship of research at Jordan University of Science and Technology (grant number 350/2018).
Funding:
Drs. Eissenberg and Shihadeh are supported by the National Institute on Drug Abuse of the U.S. National Institutes of Health (NIH) and the Center for Tobacco Products of the U.S. Food and Drug Administration (FDA) under Award Number U54DA036105. This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or FDA.
Footnotes
Conflict of interest: Dr. Eissenberg is a paid consultant in litigation against the tobacco industry and also the electronic cigarette industry and he and Dr. Shihadeh are named on a patent for a device that measures the puffing behavior of electronic cigarette users. The other authors declare that there is no conflict of interest
References
- [1].Buchanan ND, Grimmer JA, Tanwar V, Schwieterman N, Mohler PJ, Wold LE. Cardiovascular risk of electronic cigarettes: a review of preclinical and clinical studies. Cardiovasc Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Eriksen M, Mackay J, Schluger N. The Tobacco Atlas, 5th edn, Atlanta, GA, American Cancer Socieity. Inc; 2015. [Google Scholar]
- [3].Breland A, Soule E, Lopez A, Ramoa C, El-Hellani A, Eissenberg T. Electronic cigarettes: what are they and what do they do? Ann N Y Acad Sci. 2017;1394:5–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Burstyn I Peering through the mist: systematic review of what the chemistry of contaminants in electronic cigarettes tells us about health risks. BMC public health. 2014;14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gotts JE, Jordt SE, McConnell R, Tarran R. What are the respiratory effects of e-cigarettes? BMJ. 2019;366:l5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Walker N, Parag V, Verbiest M, Laking G, Laugesen M, Bullen C. Nicotine patches used in combination with e-cigarettes (with and without nicotine) for smoking cessation: a pragmatic, randomised trial. Lancet Respir Med. 2019. [DOI] [PubMed] [Google Scholar]
- [7].Control CfD Prevention. How tobacco smoke causes disease: the biology and behavioral basis for smoking-attributable disease: a report of the surgeon general. 2010. [PubMed]
- [8].Khabour OF, Alzoubi KH, Bani-Ahmad M, Dodin A, Eissenberg T, Shihadeh A. Acute exposure to waterpipe tobacco smoke induces changes in the oxidative and inflammatory markers in mouse lung. Inhalation toxicology. 2012;24:667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mayyas F, Alzoubi KH. Cardiac effects of cigarette tobacco smoking in rat model of diabetes. Life Sci. 2018;211:279–85. [DOI] [PubMed] [Google Scholar]
- [10].Lerner CA, Sundar IK, Yao H, Gerloff J, Ossip DJ, McIntosh S, et al. Vapors produced by electronic cigarettes and e-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10:e0116732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Reinikovaite V, Rodriguez IE, Karoor V, Rau A, Trinh BB, Deleyiannis FW, et al. The effects of electronic cigarette vapour on the lung: direct comparison to tobacco smoke. Eur Respir J. 2018;51. [DOI] [PubMed] [Google Scholar]
- [12].The Institutional Animal Care and Use Committee Guidebook, 2nd edition. ARENA and OLAW. 2002. [Google Scholar]
- [13].Alzoubi KH, Khabour OF, Alharahshah EA, Alhashimi FH, Shihadeh A, Eissenberg T. The effect of waterpipe tobacco smoke exposure on learning and memory functions in the rat model. Journal of Molecular Neuroscience. 2015;57:249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Khabour OF, Alzoubi KH, Al-Sawalha N, Ahmad MB, Shihadeh A, Eissenberg T. The effect of chronic exposure to waterpipe tobacco smoke on airway inflammation in mice. Life Sci. 2018;200:110–4. [DOI] [PubMed] [Google Scholar]
- [15].Ho YS, Yang X, Yeung SC, Chiu K, Lau CF, Tsang AW, et al. Cigarette smoking accelerated brain aging and induced pre-Alzheimer-like neuropathology in rats. PLoS One. 2012;7:e36752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yasuda M, Ota T, Morikawa A, Mawatari K, Fukuuchi T, Yamaoka N, et al. Simultaneous determination of nicotine and cotinine in serum using high-performance liquid chromatography with fluorometric detection and postcolumn UV-photoirradiation system. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;934:41–5. [DOI] [PubMed] [Google Scholar]
- [17].Taub PR, Gabbai-Saldate P, Maisel A. Biomarkers of heart failure. Congestive heart failure. 2010;16 Suppl 1:S19–24. [DOI] [PubMed] [Google Scholar]
- [18].Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M, et al. Diabetic Cardiovascular Disease Induced by Oxidative Stress. International journal of molecular sciences. 2015;16:25234–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rafacho BP, Azevedo PS, Polegato BF, Fernandes AA, Bertoline MA, Fernandes DC, et al. Tobacco smoke induces ventricular remodeling associated with an increase in NADPH oxidase activity. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2011;27:305–12. [DOI] [PubMed] [Google Scholar]
- [20].Kuntic M, Oelze M, Steven S, Kroller-Schon S, Stamm P, Kalinovic S, et al. Short-term e-cigarette vapour exposure causes vascular oxidative stress and dysfunction: evidence for a close connection to brain damage and a key role of the phagocytic NADPH oxidase (NOX-2). Eur Heart J. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang Z, Harrison CM, Chuang GC, Ballinger SW. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutat Res. 2007;621:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gvozdjak J, Gvozdjakova A, Kucharska J, Bada V. The effect of smoking on myocardial metabolism. Czech Med. 1987;10:47–53. [PubMed] [Google Scholar]
- [23].Rezk-Hanna M, Benowitz NL. Cardiovascular Effects of Hookah Smoking: Potential Implications for Cardiovascular Risk. Nicotine Tob Res. 2019;21:1151–61. [DOI] [PubMed] [Google Scholar]
- [24].Cooper SA, Whaley-Connell A, Habibi J, Wei Y, Lastra G, Manrique C, et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. American journal of physiology Heart and circulatory physiology. 2007;293:H2009–23. [DOI] [PubMed] [Google Scholar]
- [25].Taye A, Abouzied MM, Mohafez OM. Tempol ameliorates cardiac fibrosis in streptozotocin-induced diabetic rats: role of oxidative stress in diabetic cardiomyopathy. Naunyn-Schmiedeberg’s archives of pharmacology. 2013;386:1071–80. [DOI] [PubMed] [Google Scholar]
- [26].Al Hariri M, Zibara K, Farhat W, Hashem Y, Soudani N, Al Ibrahim F, et al. Cigarette Smoking-Induced Cardiac Hypertrophy, Vascular Inflammation and Injury Are Attenuated by Antioxidant Supplementation in an Animal Model. Frontiers in pharmacology. 2016;7:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lavi S, Prasad A, Yang EH, Mathew V, Simari RD, Rihal CS, et al. Smoking is associated with epicardial coronary endothelial dysfunction and elevated white blood cell count in patients with chest pain and early coronary artery disease. Circulation. 2007;115:2621–7. [DOI] [PubMed] [Google Scholar]
- [28].Zhang R, Brennan M-L, Fu X, Aviles RJ, Pearce GL, Penn MS, et al. Association between myeloperoxidase levels and risk of coronary artery disease. Jama. 2001;286:2136–42. [DOI] [PubMed] [Google Scholar]
- [29].Rudolph TK, Rudolph V, Baldus S. Contribution of myeloperoxidase to smoking-dependent vascular inflammation. Proceedings of the American Thoracic Society. 2008;5:820–3. [DOI] [PubMed] [Google Scholar]
- [30].Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407–18. [DOI] [PubMed] [Google Scholar]
- [31].Mayyas F, Niebauer M, Zurick A, Barnard J, Gillinov AM, Chung MK, et al. Association of left atrial endothelin-1 with atrial rhythm, size, and fibrosis in patients with structural heart disease. Circulation Arrhythmia and electrophysiology. 2010;3:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–5. [DOI] [PubMed] [Google Scholar]
- [33].Chaumont M, de Becker B, Zaher W, Culie A, Deprez G, Melot C, et al. Differential Effects of E-Cigarette on Microvascular Endothelial Function, Arterial Stiffness and Oxidative Stress: A Randomized Crossover Trial. Sci Rep. 2018;8:10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Reidel B, Radicioni G, Clapp PW, Ford AA, Abdelwahab S, Rebuli ME, et al. E-Cigarette Use Causes a Unique Innate Immune Response in the Lung, Involving Increased Neutrophilic Activation and Altered Mucin Secretion. Am J Respir Crit Care Med. 2018;197:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis & tissue repair. 2012;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Goette A, Lendeckel U, Kuchenbecker A, Bukowska A, Peters B, Klein HU, et al. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart. 2007;93:1056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Crotty Alexander LE, Drummond CA, Hepokoski M, Mathew D, Moshensky A, Willeford A, et al. Chronic inhalation of e-cigarette vapor containing nicotine disrupts airway barrier function and induces systemic inflammation and multiorgan fibrosis in mice. Am J Physiol Regul Integr Comp Physiol. 2018;314:R834–R47. [DOI] [PMC free article] [PubMed] [Google Scholar]