

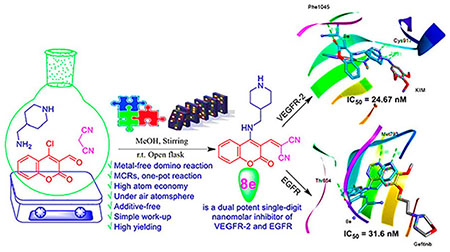
Abstract
A metal-free, atom-economy and simple work-up domino amination-Knoevenagel condensation approach to construct new coumarin analogous (4a-f and 8a-e) was described. Further, new formyl (5a,d-f) and nitro (9a,d-f) coumarin derivatives were synthesized via C-N coupling reaction of various cyclic secondary amines and 4-chloro-3-(formyl-/nitro)coumarins (1a,c), respectively. The confirmed compounds were screened for their in vitro anti-proliferative activity against KB-3-1, A549 and PC3 human cancer cell lines using resazurin cellular-based assay. Among them, coumarin derivatives 4e and 8e displayed the best anti-cervical cancer potency (KB-3-1) with IC50 values of 15.5 ± 3.54 and 21 ± 4.24 μM, respectively. Also, 4e showed the most promising cytotoxicity toward A549 with IC50 value of 12.94 ± 1.51 μM. As well, 9d presented a more significant impact of potency against PC3 with IC50 7.31 ± 0.48 μM. Moreover, 8d manifested selectivity against PC3 (IC50 = 20.16 ± 0.07 μM), while 8e was selective toward KB-3-1 cell line (IC50 = 21 ± 4.24 μM). Matching with docking profile, the enzymatic assay divulged that 8e is a dual potent single-digit nanomolar inhibitor of VEGFR-2 and EGFR with IC50 values of 24.67 nM and 31.6 nM that were almost equipotent to sorafenib (31.08 nM) and erlotinib (26.79 nM), respectively.
Keywords: Coumarin, metal-free domino reaction, C–N coupling, Knoevenagel condensation, VEGFR-2, EGFR
Graphical Abstract
1. Introduction
Cancer remains one of the most daunting diseases to treat, thus, the development of new antitumor agents is still a very critical research domain. Amongst the attractive therapeutic targets for cancer, protein tyrosine kinases (PTKs) that regulate the biological potency of proteins by phosphorylation process that play a crucial role in signal transduction mechanisms by which inter-cellular signals regulate significance intra-cellular functions such as ion transport, cellular proliferation, differentiation, angiogenesis, and hormone responses. Among them, the epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR) are receptor protein tyrosine kinases (RPTKs) that overexpressed or mutated in several tumors due to a mutation of a normal gene to an oncogene [1]. EGFR (a dimer of HER-1 and HER-2) and VEGFR-2 (KDR, a type of VEGFRs) are pro-angiogenic growth factor receptors that induce angiogenesis process in order to establish new blood vessels that have an important role in tumor growth and metastasis; therefore, they are attractive therapy targets and dominant strategy for the treatment of cancer [1–6].
VEGFR-2 type II inhibitors shared essential pharmacophoric features encompassing: (i) terminal heteroaromatic ring that occupies the ATP binding pocket (hinge region) via H-bond with cysteine acid (like Cys917); (ii) oxygen (N or S) linker that occupies the gatekeeper region between the hinge and DFG domains; (iii) urea or amide moiety spacer as H-bond acceptor–donor pair (HBA–HBD) that binds to DFG motif via H-bonds with glutamic (Glu833) and aspartic (Asp1044) acids; (iv) terminal lipophilic group that occupies the allosteric hydrophobic pocket (DFG-out) via hydrophobic interactions [7–11]. The well-defined EGFR inhibitors consists of: (i) central hetero aromatic unit that fits the adenine binding site through H-bond with methionine amino acid residue (Met793); (ii) hydrophobic head is a phenyl binding group with various hydrophobic substituents to interact with the hydrophobic region I; (iii) nitrogen spacer to link the hinge-binding central moiety with the terminal fragment that occupies the hydrophobic region I; (iv) hydrophobic tail is directly linked to the central heteroaromatic core and induced fit to the hydrophobic region II [12].
Coumarins are versatile oxygen-containing heterocyclic molecules in the medicinal chemistry domain due to their aspirational biological applications [13,14]. Particularly, they have anti-coagulant, anti-oxidant [15], anti-microbial, anti-viral [16–18], anti-cancer [19–24], anti-diabetic, analgesic, anti-neurodegenerative, and anti-inflammatory properties [17,25]. Interestingly, several coumarin derivatives obtained via Knoevenagel condensation reactions have been proved to be potent enzymes inhibitors [26–28]. Also, Knoevenagel reaction products are significant key intermediates for the synthesis of many marketed drugs such as atorvastatin (Lipitor®) and pioglitazone (Actos®) [29].
The recently developed transition-metal-catalyzed reactions have been confirmed as useful protocols for the synthesis of heterocyclic compounds as well. However, most of these approaches suffer from some drawbacks encompassing complex procedures (e.g. high expense, moisture sensitivity and toxicity nature of many of these metal catalysts and ligands). Consequently, studies of eco-friendly organic transformations using commercially accessible and cost-efficient reagents have also been recently extended [30–33]. Multi-component transformation domino reactions are a formidable and significant method in the organic synthesis domain due to their benefits to our environment and natural resources [34].
Synthesis and investigation of heterocyclic/aryl amines still cornerstone research area in medicinal chemistry and its neighboring disciplines. In particular, due to their high prospective to demonstrate potent biological activity and their application as lead compounds in drug design, as well as they are vital building blocks of various organic compounds [30]. The most effective methodology for the construction of heterocyclic/aryl amines is the C–N cross-coupling reaction between heterocyclic/aryl halides and primary or secondary amines via the Buchwald–Hartwig reaction using high-expensive phosphine-ligated palladium precatalysts [35]. As a result, a daunting application in the pharmaceutical industry. Hence, the development of metal-free C–N coupling reactions is a surrogate trend and hold a prominent position [30,31].
Spur by the aforementioned facts and our persistent research efforts to explore highly effective and convenient synthetic routes for the synthesis of bioactive molecules [22,24], we present in this research paper an efficient cascade amination-Knoevenagel strategy for the construction of new coumarin derivatives under metal-free conditions. Their anti-proliferative activity was evaluated against KB-3-1, A549 and PC3 cell lines using resazurin cellular assay. Moreover, we highlighted the in silico computational predictions including physicochemical features and biological targets of the active compounds. Furthermore, molecular docking of the most robust compounds 4e, 8e and 9d as potential inhibitors of vascular endothelial growth factor receptor (VEGFR-2) and epidermal growth factor receptor (EGFR) biomolecular targets was performed. Experimentally, an enzymatic assay for the promising dual inhibitors of VEGFR-2 and EGFR was investigated as well, delivering very potent inhibitory activities.
2. Results and discussion
2.1. Chemistry
In the preliminary investigation, we tried a model reaction of 4-chloro-3-formylcoumarin (1a) as an example of α,β-unsaturated-β-haloaldehydes [36,37], acidic CH2 called methyl cyanoacetate (2a), and a cyclic secondary amine named pyrrolidine (3a) in ethanol (EtOH) at room temperature. As a result, compound 4a was generated with 65% yield in 40 min (Table 1, entry 1). As previously reported [38,39], in nucleophilic aromatic substitution (SNAr)-Knoevenagel condensation domino reaction, the amine plays twofold role as a nucleophilic and base, hence, the produced HCl was neutralized with an excess of amine or additional base to perform the reaction in smoothly way. Consequently, the reactants ratio of 1a (1 equiv), 2a (1 equiv), and 3a (2.5 equiv) was used as an optimized reaction condition (Table 1). Also, the yield was further investigated by utilizing different solvents such as methanol (MeOH), dichloromethane (DCM), and water. We observed that MeOH was premium for the reaction and afforded 4a in the best yield (Table 1, entry 3). All the optimization trials were carried out under oxygen and air atmospheres (open flask).
Table 1.
Optimization of domino amination-Knoevenagel condensation reaction to form 4aa.
| Entry | Solvent | Temp (°C) | Time (min) | Yield (%)b |
|---|---|---|---|---|
| 1 | EtOH | r.t. | 40 | 65 |
| 2 | DCM | r.t. | 60 | 50 |
| 3 | MeOH | r.t. | 30 | 93 |
| 4 | H2O | 90 | 60 | 28 |
Reaction conditions: 1a (1.0 mmol, 1.0 equiv), 2a (1.0 mmol, 1.0 equiv), and 3a (2.5 mmol, 2.5 equiv) in solvent (2 mL) using open flask.
Isolated yield.
Once the optimal reaction conditions had been established (Table 1, entry 3), we explored the scope and generality of the tandem approach (Scheme 1). A variety of cyclic secondary amines [piperidine (3b), 4-hydroxypiperidine (3c), and morpholine (3d)] were used and to our delight, all the corresponding desired products 4b-d were obtained in excellent yields (91–96%) and purified by simple recrystallization/washing with hot MeOH where we did not need to perform the column chromatography work-up as in the previously reported methods [38,39].
Scheme 1.
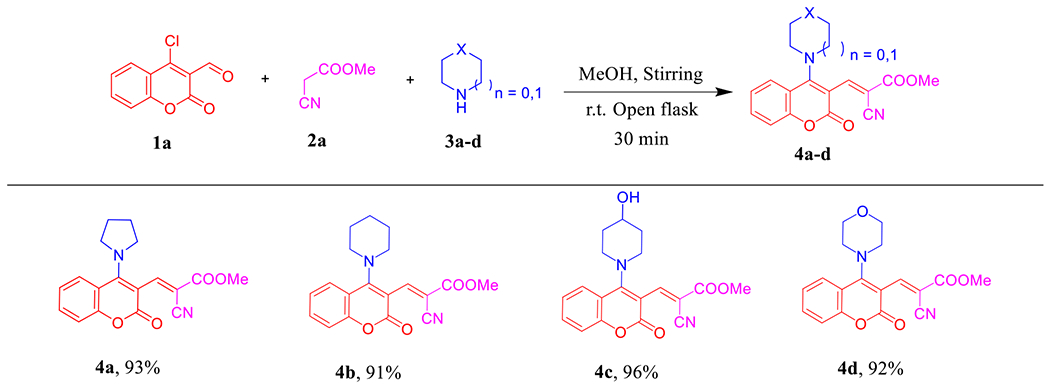
Scope of various cyclic secondary amines for the synthesis of 4a–d. Reagents and conditions: 1a (1.0 mmol, 1.0 equiv), 2a (1.0 mmol, 1.0 equiv), and 3a–d (2.5 mmol, 2.5 equiv) in MeOH (2 mL) using open flask at room temperature under stirring conditions.
In the case of the bifunctional amine that called; 4-(aminomethyl)piperidine (4-AMP, 3e), we got 4e as an exclusive and regiospecific product [40–43] in high yield instead of 4e′ (Scheme 2). Our interpretation based on the concept; nucleophilicity is much more sensitive to steric effects than basicity, thus in this case primary amine (−CH2NH2) is more nucleophilic than the secondary one (−CH2NHCH2−) and consider as the preference orientation. The obtained secondary amine 4e can be easily identified from the 1H NMR spectrum by the characteristic 1H chemical shift of aromatic triplet NH at δ 8.76 ppm (see Figure S13, Supporting Information). Also, it seems worthwhile to point out that 4e among this series showed [M + H]+ adduct ion in (+)-ESI-MS, while all the others exhibited [M + Na]+ molecular ion peaks.
Scheme 2.
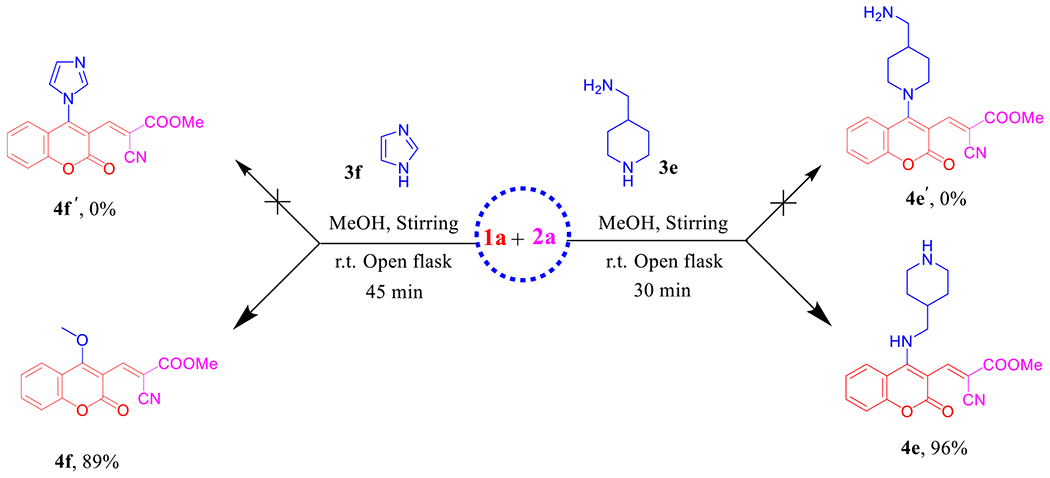
Synthesis of 4e and 4f.
Under the aforementioned conditions, our reconnaissance was expanded to utilize imidazole (3f) instead of the above-mentioned amines in an attempt to get the desirable product 4f′, astonishingly, 4-methoxy coumarin derivative 4f was isolated in 89% yield (Scheme 2). Unambiguously, imidazole dealing with the reaction as a nucleophilic organocatalyst (covalent catalyst) by forming a covalent bond with 1a [44] to produce a reactive intermediate because the positively charged nitrogen makes imidazole a very good leaving group and subsequently promoted the MeOH to attack the intermediate. Hence, imidazole hydrochloride may be catalyzed Knoevenagel condensation reaction in the next step to furnish 4f (Scheme 3). NMR data of 4f manifested the new methoxy 1H resonance at δ 4.13 and the corresponding 13C chemical shift at δ 63.8 ppm. The (+)-ESI-MS analysis of compound 4f proved its molecular weight as 285 Da, where it showed the molecular ion peak at m/z 308 [M + Na]+ (see Supporting Information, Figs. S16–S18).
Scheme 3.
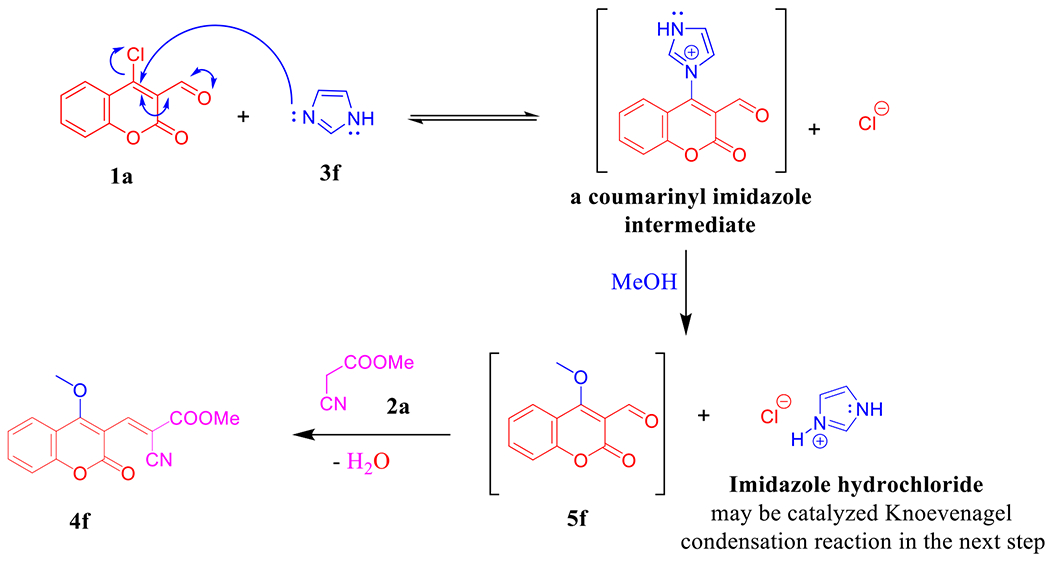
Plausible reaction mechanism of the imidazole-catalyzed C–O bond formation in 4f.
Having in mind the previous studies [38,45], we propose that the cascade one-pot reaction mechanism could be implemented via two routes as depicted in Scheme 4. First, 1a underwent SNAr reaction with pyrrolidine (3a) through 1,4-addition/elimination pathway to afford 5a that condensed with methyl cyanoacetate (2a) to generate 4a. As an alternative strategy, Knoevenagel product 6a was formed by pyrrolidine-catalyzed condensation reaction between 1a and 2a. Hence, subsequent SNAr reaction between 6a and 3a furnished 4a.
Scheme 4.
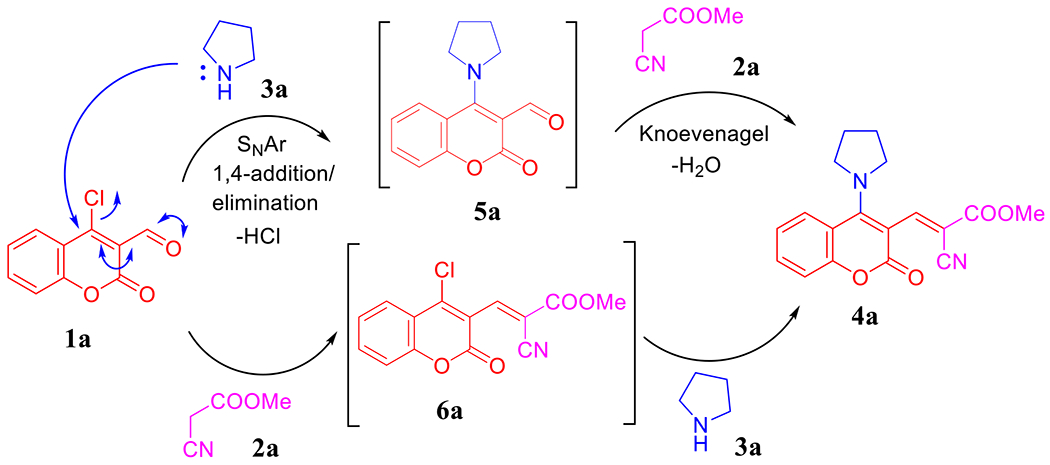
Plausible reaction mechanism.
Besides methyl cyanoacetate (2a), ethyl nitroacetate or cyanoacetic acid were used as a source of active methylene in this reaction with pyrrolidine (3a) and our substrate 1a. Under the optimized conditions, we obtained a mixture of products that may be involved amine salt and SNAr product 5a. Instead, we worked on expanding the application of this approach to involve malononitrile (2b) as a suitable starting material for the Knoevenagel condensation. Hereupon, we used the above-mentioned reaction conditions albeit with one-pot, two-step domino strategy. First, base (1.0 equiv, 3a/3b)-catalyzed dimerization of malononitrile (2.0 equiv, 2b) into non-isolated 2-amino-1,1,3-tricyanopropene (7) [ 46] that has drawn tremendous attention of interest due to its comprehensive implementations in the synthesis of heterocyclic compounds that manifest diverse biological and pharmaceutical properties [47]. At the same time, in another conical flask, suspension solution of 4-(pyrrolidin-1-yl)-3-formylcoumarin (5a)/4-(piperidin-1-yl)-3-formylcoumarin (5b) in MeOH (2 mL) were prepared from direct amination of 1a (1.0 equiv) and 3a/3b (1.5 equiv). Next, sequential addition of 7 to 5a/5b afforded the corresponding products 8a/8b in excellent isolated yields (Scheme 5).
Scheme 5.
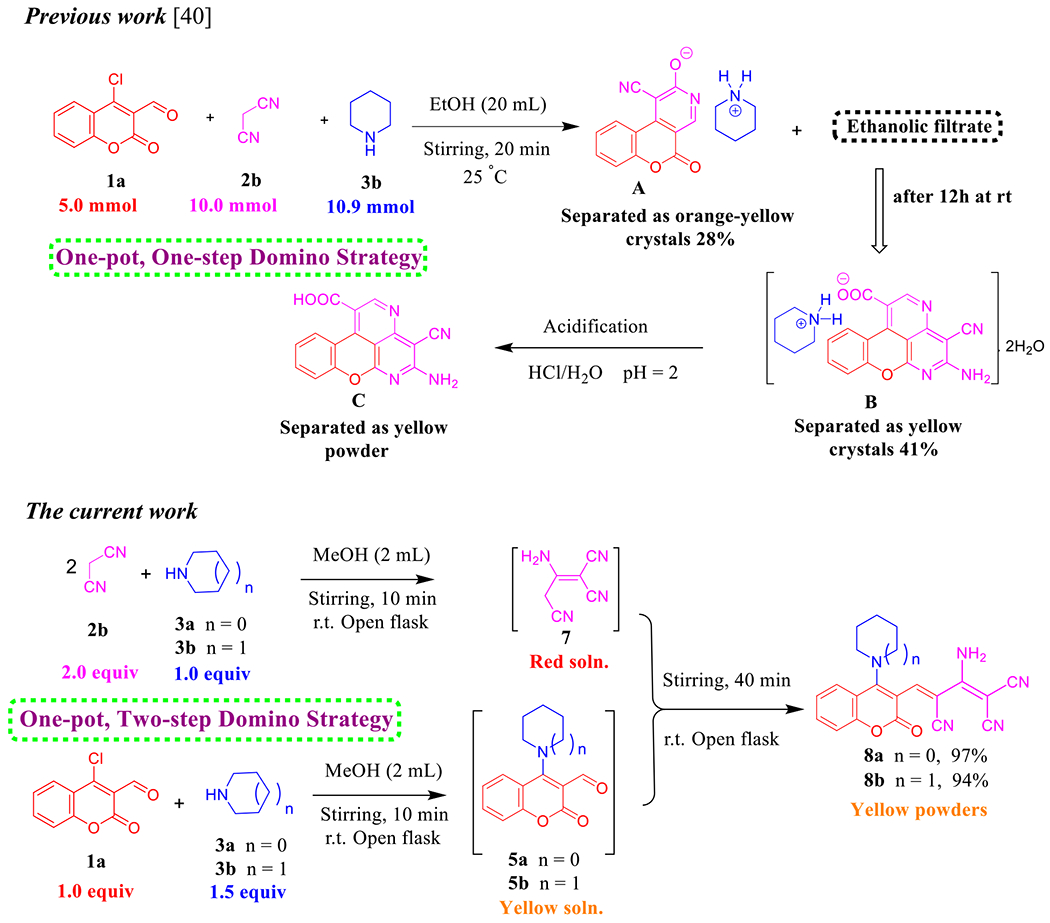
Previous work by Angelova group and synthesis of 8a,b.
As delineated in Scheme 5, in contrast to the published research paper by Angelova et al. [46] who described the synthesis of aminium salt A and piperidinium 5-amino-4-cyanochromeno[4,3,2-de]-1,6-naphthyridine-1-carboxylate dihydrate (B) that converted to the acidic form C via one-pot, one-step cascade reaction of starting materials 1a, 2b, and 3b as developed route to generate polyfunctionality substituted heterocyclic compounds with anticipated biological performance. Herein, our chemistry proposal was aimed to synthesize buta-1,3-diene-1,1,3-tricarbonitrile derivatives 8a,b with potential anti-proliferative activity and act as a key intermediate to construct privileged molecules. Therefore, we developed a striped pathway to achieve our target but, unfortunately, we failed to expand the scope of this methodology to include other bases such as 4-hydroxypiperidine (3c), morpholine (3d), and 4-AMP (3e), where the corresponding methylene malononitrile derivatives 8c–e (8d [39]) were produced in high yields 88–98% (Figure 1). We postulate this contradiction is attributed to the difference in basicity power between 3a,b and 3c–e.
Figure 1.
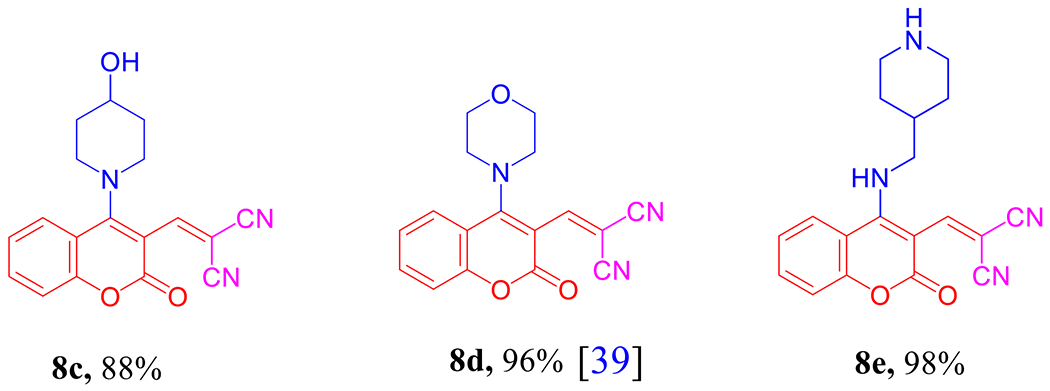
Chemical structures of the prepared methylene malononitrile derivatives 8c–e.
Next, we turned our concern to confirm the amination occurs via 1,4-addition/elimination SNAr reaction as a part of the mechanistic study (Scheme 4) and control experiments. Earlier study by Yang et al. [48] has been showed that the reaction between 3-chloro-3-phenylacrylaldehyde 1b and 1,2,3,4-tetrahydroisoquinoline (THIQ, 3g) exhibited predominant product D through Et3N-catalyzed 1,4-addition/elimination followed by intramolecular cyclization process (Scheme 6). In 2019, Vyasamudri and Yang [45] have reported that condensation of 4-chloro-3-formylcoumarin (1a) and THIQ (3g) using 4-dimethylaminopyridine (DMAP) as an organic base afforded E in good yield 78% and the reaction goes forward with 1,2-addition/elimination then cyclization process through removing of HCl. On the other hand, when Cs2CO3 was utilized as the base, the reaction proceeds via 1,4-addition/elimination then annulation fashion to furnish F with moderate yield 53% (Scheme 6). In our manner, the amination process of 1a and 3a,d,e passed through Et3N-promoted1,4-addition/elimination at room temperature in the open flask using MeOH to supply 5a,d,e with high yields and the subsequent intramolecular cyclization did not take place. We likewise sought to expand this fashion to include 4-chloro-3-nitrocoumarin (1c) with 3a,d,e. The desired corresponding amination products 9a,d,e were isolated in excellent yields and shorter time. As produced in the above series, we got the corresponding methoxy coumarin analogs 5f and 9f when imidazole (3f) was utilized as a reactant base (Scheme 6).
Scheme 6.
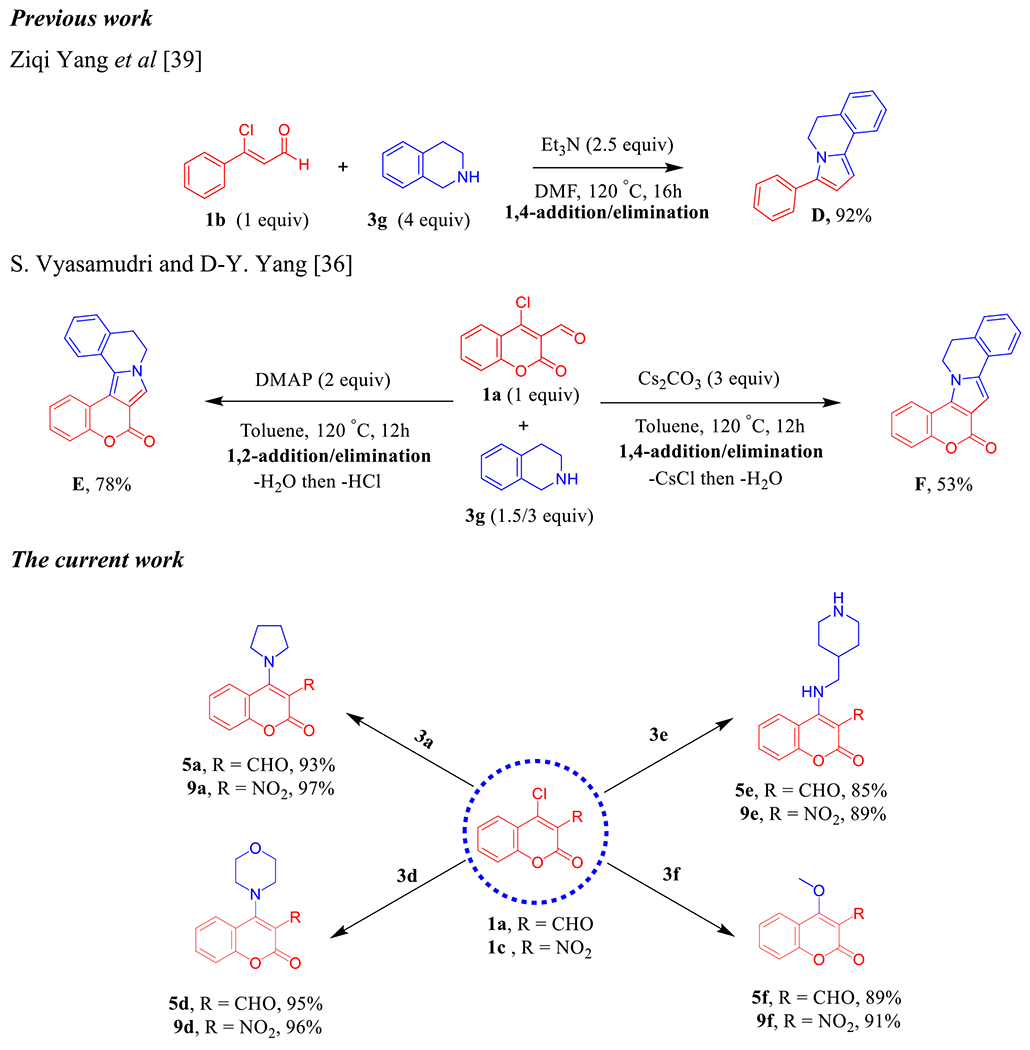
Previous work by Yang and Vyasamudri groups as well as scope of various cyclic secondary amines for the synthesis of 5a, d–f and 9a,d–f. Reagents and conditions: 1a,c (1.0 mmol, 1.0 equiv), 3a,d–f (1.0 mmol, 1.0 equiv), and Et3N (1 mmol, 1 equiv) in MeOH (2 mL) using open flask at room temperature under stirring conditions, R = CHO, 15 min, R= NO2, 5 min.
2.2. Biological evaluation
2.2.1. In vitro anti-proliferative activity
Anti-cervical cancer activity of all the synthesized compounds was evaluated for their in vitro cytotoxicity toward KB-3-1 cell line by resazurin-based assay [49] using (+)-griseofulvin as a positive control. The results were expressed as IC50 values that outlined in Table 2, Figure 2, and the values are an average ± SD of at least two separate experiments. Table 2 and Figure 2 show that the domino amination-Knoevenagel product 4e that containing 4-AMP core displayed superior potency (IC50 = 15.5 ± 3.54 μM) than (+)-griseofulvin (IC50 = 19 ± 2.83 μM) while 8e is the closer one (IC50 = 21 ± 4.24 μM). The amination products 5e and 9d showed moderately convergent activity with IC50 > 70 μM. On the other hand, compounds 4c,d, 5d, 8d, and 9a indicated low activity with IC50 values over than 100 μM, whilst molecules 4a,b, 4f, 5a,f, 8a–c, and 9e,f showed no activity against KB-3-1. The results manifested that the presence of 4-AMP fragment was benefit for the anti-cervical cancer activity.
Table 2.
IC50 (μM) of all the synthesized compounds against KB-3-1(cervix), A549 (non-small cell lung), PC3 (prostate) human cancer cell lines.
| Compounds | IC50 (μM) |
||
|---|---|---|---|
| KB-3-1a | A549b | PC3b | |
| 4a | — | >80 | >80 |
| 4b | — | >80 | >50 |
| 4c | >100 | >80 | >50 |
| 4d | >100 | >80 | >50 |
| 4e | 15.5 ± 3.54 | 12.94 ± 1.51 | 11.28 ± 0.06 |
| 4f | — | >80 | 38.90* |
| 5a | — | >80 | >80 |
| 5d | >100 | >50 | 31.12* |
| 5e | >70 | >80 | >80 |
| 5f | — | >80 | >80 |
| 8a | — | >80 | >80 |
| 8b | — | >80 | >80 |
| 8c | — | >80 | >80 |
| 8d | >100 | >80 | 20.16 ± 0.07 |
| 8e | 21 ± 4.24 | >50 | >50 |
| 9a | >100 | >80 | >80 |
| 9d | >70 | 19.78 ± 0.56 | 7.31 ± 0.48 |
| 9e | — | >80 | >80 |
| 9f | — | NDc | ND |
| (+)-Griseofulvin | 19 ± 2.83 | ND | ND |
| DMSOd | — | — | — |
Anti-cervical cancer activity was done at Organic and Bioorganic Chemistry, Faculty of Chemistry, Bielefeld University, Germany. IC50 values are the mean ± SD of two independent determinations, (+)-griseofulvin was used as positive control with IC50 = 19 ± 2.83 μM.
Cytotoxicity IC50 values (mean ± SD of triplicate wells) against A549 (non-small lung) and PC3 (prostate) human cancer cell lines were tested at the Center for Pharmaceutical Research and Innovation, University of Kentucky, Lexington, USA, and obtained after 72 h incubation. Actinomycin D and H2O2 were used as positive control at 20 μM and 2 mM concentration, respectively (0% viable cells) without dose–response curves (IC50 not determined) and just utilized to make sure the cytotoxic assay is working [50–52].
ND, not determined.
Negative control; 0.1% dimethyl sulphoxide was used as negative control (100% live cells).
‘—’ Means no obvious inhibitory effect.
Could not get +/− values due to the low toxicity.
Figure 2.
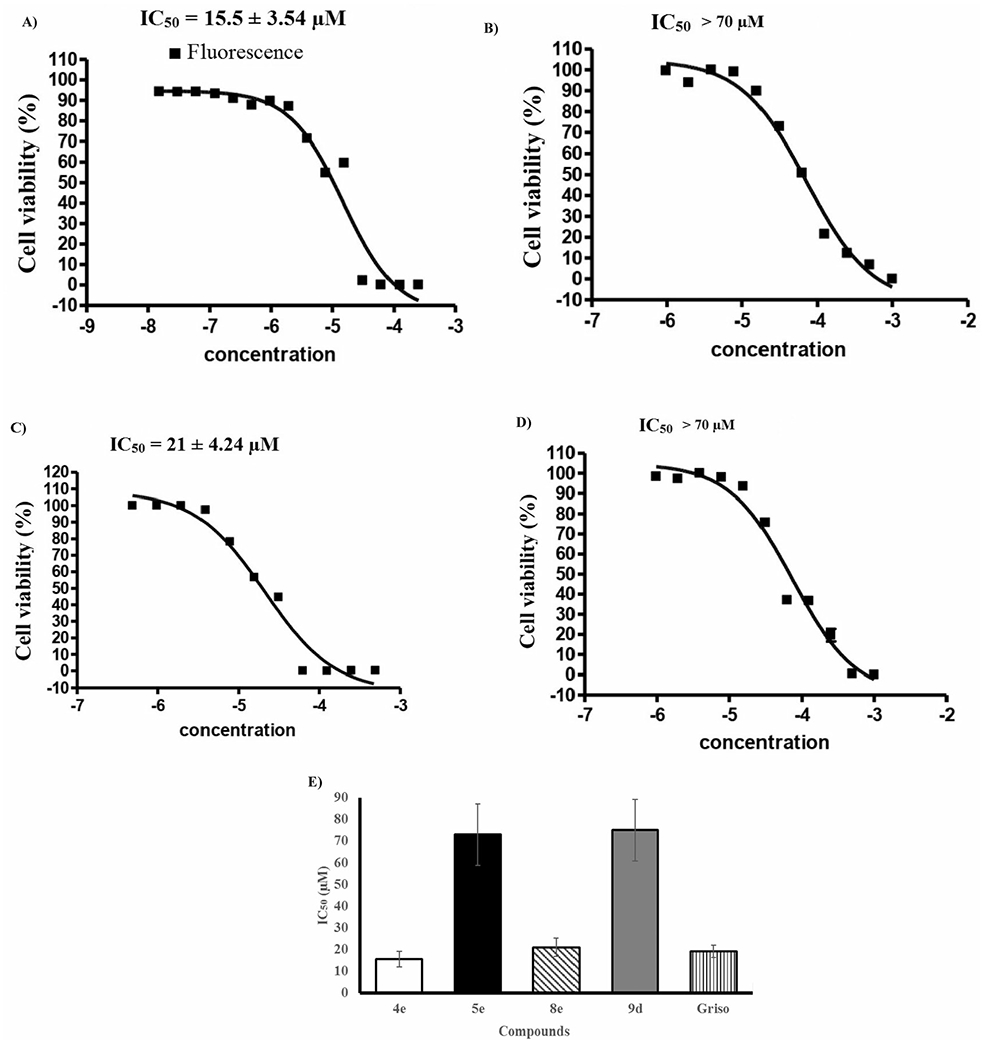
Sigmoidal dose–response against KB-3-1 cell line; (A) 4e, (B) 5e, (C) 8e, (D) 9d, and (E) graph show the IC50 values of the tested compounds 4e, 5e, 8e, 9d, and (+)-griseofulvin as a positive control.
Next, anti-proliferative potency of our synthesized compounds was tested against A549 (non-small lung) and PC3 (prostate) human cancer cell lines using also resazurin-based cytotoxicity assay as reported by Thorson research group [50–52] (Table 2, Figure 3). Similar trend was detected with 4e (the most active) against A549 (IC50 = 12.94 ± 1.51 μM), whilst 9d was found to be the best toward PC3 (IC50 = 7.31 ± 0.48 μM). From overall cytotoxicity results, compound 8d that bearing malononitrile fragment and morpholino unit showed selectivity against PC3 (IC50 = 20.16 ± 0.07 μM), while 8e that embraced 4-AMP core instead of morpholine ring was selective toward KB-3-1 cell line. The remaining molecules pretended with low cytotoxic effect. Grounded on these outcomes, we concluded that the 4-AMP and morpholine moieties would be the optimal cyclic secondary amines in this study.
Figure 3.
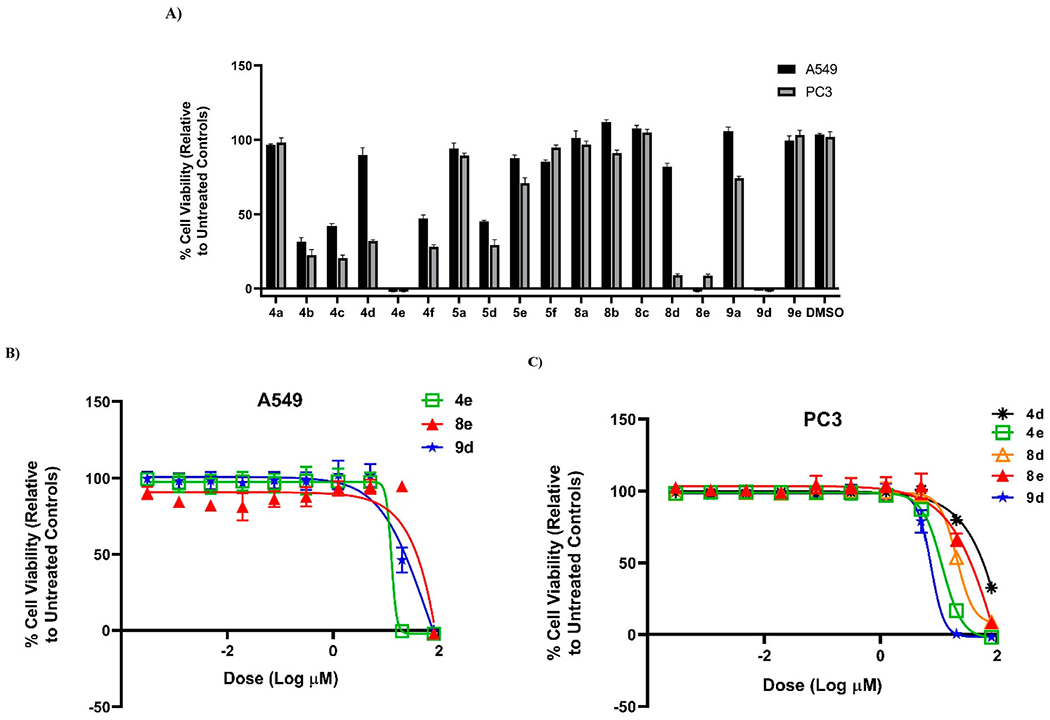
(A) % Viability of A549 (non-small lung) and PC3 (prostate) human cancer cell lines (after 72 h) at 80 μM concentration of compounds 4a–f, 5a, 5d–f, 8a–e, 9a, and 9d–e. (B) Dose–response of compounds 4e, 8e, and 9d against A549 (non-small cell lung) human cancer cell line (72 h). (C) Dose–response of compounds 4d, 4e, 8d, 8e, and 9d against PC3 (prostate) human cancer cell line (72 h).
2.2.2. VEGFR-2 and EGFR inhibitory activity
Overexpression and/or hyperactivation of VEGFR-2 and EGFR kinases are strongly associated with several malignant cancers such as non-small cell lung carcinoma (NSCLC), breast, ovarian, colon, and prostate cancers [53]. Targeting one or both of them is a promising pathway to explore more anti-angiogenic inhibitors [54,55]. In the present study (Table 3), our examined compounds; 4e, 5e, 8e, and 9d were more convenient to VEGFR-2 than EGFR. They showed VEGFR-2 inhibitory activities ranging from 24 to 88 nM while IC50 values with EGFR were 32–165 nM. Figure 4 and Table 3 display remarkable inhibition potency toward VEGFR-2 by 8e (IC50 = 24.67 ± 1.1 nM) and 9d (IC50 = 24.26 ± 1.1 nM). They are able to inactivate VEGFR-2 by 20% enhancement (0.8-fold) over the standard sorafenib (IC50 = 31.08 ± 1.8 nM). Concerning the EGFR inhibitory activity, the power of 8e was also comparable to the standard erlotinib (1.2-fold). Among the tested compounds, 9d recorded the lower inhibitory activity against EGFR (IC50 = 165.00 ± 8.0 nM), confirming the selectivity of 9d toward VEGFR-2.
Table 3.
VEGFR-2 and EGFR inhibitory activities by compounds 4e, 5e, 8e, and 9d with their fold inactivation relative to the standard.
| Compound | VEGFR-2 |
EGFR |
||
|---|---|---|---|---|
| IC50 (nM) | Fold to sorafenib | IC50 (nM) | Fold to erlotinib | |
| 4e | 36.76 ± 1.8 | 1.2 | 67.09 ± 3.5 | 2.5 |
| 5e | 88.08 ± 4.5 | 2.8 | 96.25 ± 4.7 | 3.6 |
| 8e | 24.67 ± 1.1 | 0.8 | 31.60 ± 1.5 | 1.2 |
| 9d | 24.26 ± 1.1 | 0.8 | 165.00 ± 8.0 | 6.2 |
| Standard* | 31.08 ± 1.8 | 1.0 | 26.79 ± 1.2 | 1.0 |
VEGFR-2, vascular endothelial growth factor receptor 2; EGFR, epidermal growth factor receptor.
The standard is the sorafenib as inhibitor to VEGFR-2 enzyme while it is the erlotinib in the case of the EGFR.
Figure 4.
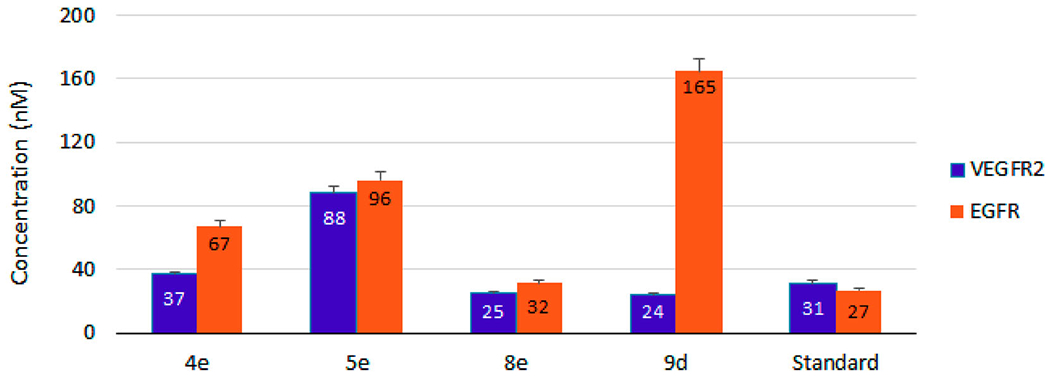
IC50 of compounds 4e, 5e, 8e, and 9d as inhibitors to the VEGFR-2 and EGFR human kinases. Sorafenib is the standard inhibitor to VEGFR-2 enzyme, while erlotinib is the equivalent in the case of the EGFR.
The most potent one against KB-3-1 and A549 cell lines (Table 2), 4e showed 1.2 and 2.5-fold inactivation toward VEGFR-2 and EGFR, respectively. Moderate activities were also obtained by 5e against the examined kinases. Regarding COVID-19, in silico studies have predicted higher affinity between the viral spike glycoprotein (S) and the inhibitors of the hepatocyte growth factor receptor (HGFR/c-MET), VEGFR and EGFR [56]. Consequently, exploring more agents against the above receptors is more than welcome.
2.3. In silico studies
2.3.1. Physicochemical descriptors, drug-likeness, and medicinal chemistry friendliness
In silico computational prediction represents a robust, fast, and cost-effective approach to find new ligands or drug leads as a part in drug discovery and development strategy. Motivated by the aforementioned biological results and to explore the drug-likeness, lead-likeness and biological target of the active molecules (4e, 5e, 8d,e, and 9d), in silico studies using SwissADME platform tools were effectuated [57]. To our delight, the estimated compounds show the optimal range for each physicochemical property where, the molecular weight as an indication for the size parameter is between 150 and 500 g/mol (MW, 276.24–367.40), flexibility: no more than 9 rotatable bonds (nROTB, 2-6), saturation: fraction of carbons in the sp3 hybridization not less than 0.25 (Fraction Csp3, 0.31–0.38), except 8d is very close (Fraction Csp3 = 0.24), polarity: topological polar surface area is between 20 and 130 Å2 (TPSA, 71.34–104.36) (Table 4).
Table 4.
Physicochemical properties, drug-likeness, and medicinal chemistry parameters of compounds 4e, 5e, 8d,e, and 9d.
| Compounds |
|||||
|---|---|---|---|---|---|
| Predictive models and their parameters | 4e | 5e | 8d | 8e | 9d |
| MW (g/mol) | 367.40 | 286.33 | 307.30 | 334.37 | 276.24 |
| nROTBa (≠) | 6 | 4 | 2 | 4 | 2 |
| Fraction Csp3 | 0.35 | 0.38 | 0.24 | 0.32 | 0.31 |
| HBAb (≠) | 6 | 4 | 6 | 5 | 5 |
| HBDc (≠) | 2 | 2 | 0 | 2 | 0 |
| MRd | 105.97 | 85.81 | 87.41 | 99.63 | 77.03 |
| TPSAe (Å2) | 104.36 | 71.34 | 90.26 | 101.85 | 88.50 |
| Log Po/wf XLOGP3 | 2.14 | 2.15 | 1.46 | 2.01 | 1.42 |
| WLOGP | 1.60 | 1.45 | 1.57 | 1.95 | 1.16 |
| MLOGP | 1.18 | 1.20 | 0.47 | 0.94 | 0.37 |
| Log Sg (ESOL) | −3.34 | −3.06 | −2.85 | −3.21 | −2.69 |
| Quantitative solubility (mg/mL) | 1.66e–01 | 2.50e–01 | 4.29e–01 | 2.06e–01 | 5.70e–01 |
| Qualitative solubility | Soluble | Soluble | Soluble | Soluble | Soluble |
| Lipinski (Pfizer filter, RO5)h | Yes | Yes | Yes | Yes | Yes |
| Ghosei (Amgen filter) | Yes | Yes | Yes | Yes | Yes |
| Veberj (GSK filter) | Yes | Yes | Yes | Yes | Yes |
| Egank (Pharmacia filter) | Yes | Yes | Yes | Yes | Yes |
| Mueggel (Bayer filter) | Yes | Yes | Yes | Yes | Yes |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| PAINS | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert |
| Lead-likeness (RO3)m | No; 1 violation: MW>350 | Yes | Yes | Yes | Yes |
| Target prediction | Kinase 73.3% | Kinase 53.3% | Kinase 26.7% | Nuclear receptor 20.0% | Protease 33.3% |
nROTB, no. of rotatable bonds.
HBA, hydrogen bond acceptor.
HBD, hydrogen bond donor.
MR, molar refractivity.
TPSA, topological polar surface area.
Log Po/w, the partition coefficient between n-octanol and water.
Log S, the decimal logarithm of the molar solubility in water.
Lipinski (RO5) criteria range are: lipophilicity (Log Po/w) ≤ 5, MW ≤ 500, H-bond donors ≤ 5, and H-bond acceptors ≤ 10.
Ghose filter criteria range are: Log Po/w in −0.4 to +5.6 range, MR from 40 to 130, MW from 180 to 480, no. of atoms from 20 to 70.
Veber rule criteria range are: RB ≤ 10 and TPSA ≤ 140 Å2.
Egan rule criteria range are: WLOGP ≤ 5.88, TPSA ≤ 131.6 Å2.
Muegge rule criteria range are: 200 ≤ MW ≤ 600, −2 ≤ XLOGP3 ≤ 5, TPSA = 150 Å2, no. of rings ≤ 7, no. of heteroatoms > 1, nROTB ≤ 15, HBA ≤ 10, HBD ≤ 5.
RO3 criteria range are: XLOGP3 ≤ 3.5, MW ≤ 350, H-bond donors ≤ 3, H-bond acceptors ≤ 3, and RB ≤ 3 [57].
Lipophilicity is a significant physicochemical parameter quantified by the partition coefficient Log Po/w between water and n-octanol that shows a good indicator of permeability across the cell wall [58]. Our estimated compounds manifested in XLOGP3 model Log Po/w values between −0.7 and +5.0, ranging from 1.42 to 2.15, suggesting good permeability and absorption through the cell membrane of infected cells. Moreover, solubility is one pivotal property affecting the absorption and impacts many processes in drug development activities such as handling and formulation [59]. Concerning qualitative water solubility, all the predicted molecules based on ESOL topological model are soluble (Table 4).
In light of the above informative predictions, the estimated compounds (4e, 5e, 8d,e, and 9d) passed all the drug-likeness metrics (Lipinski, Ghose, Veber, Egan, and Muegge) and have the same bioavailability score (0.55). As a consequence, they could be possible drug lead candidates. Regarding with medicinal chemistry parameters, recognition of potentially problematic fragments based on Pan Assay Interference Structures (PAINS) shows zero alerts for all the predicted compounds, confirming the safety and metabolic stability of them. According to the rule of three (RO3), all compounds are lead-likeness excepting compound 4e have one violation for this rule (MW>350), therefore, 4e as a lead hopping and potent compound will be subjected to lead optimization and extra studies in the future. In the quest for the biological target, we used SwissTarget-Prediction web tool to predict the most probable targets of our bioactive small molecules (4e, 5e, 8d,e, and 9d) [60]. The website performs target fishing using ligand-based target prediction method that based on the molecular similarity principle and the results are shown in Figure 5.
Figure 5.
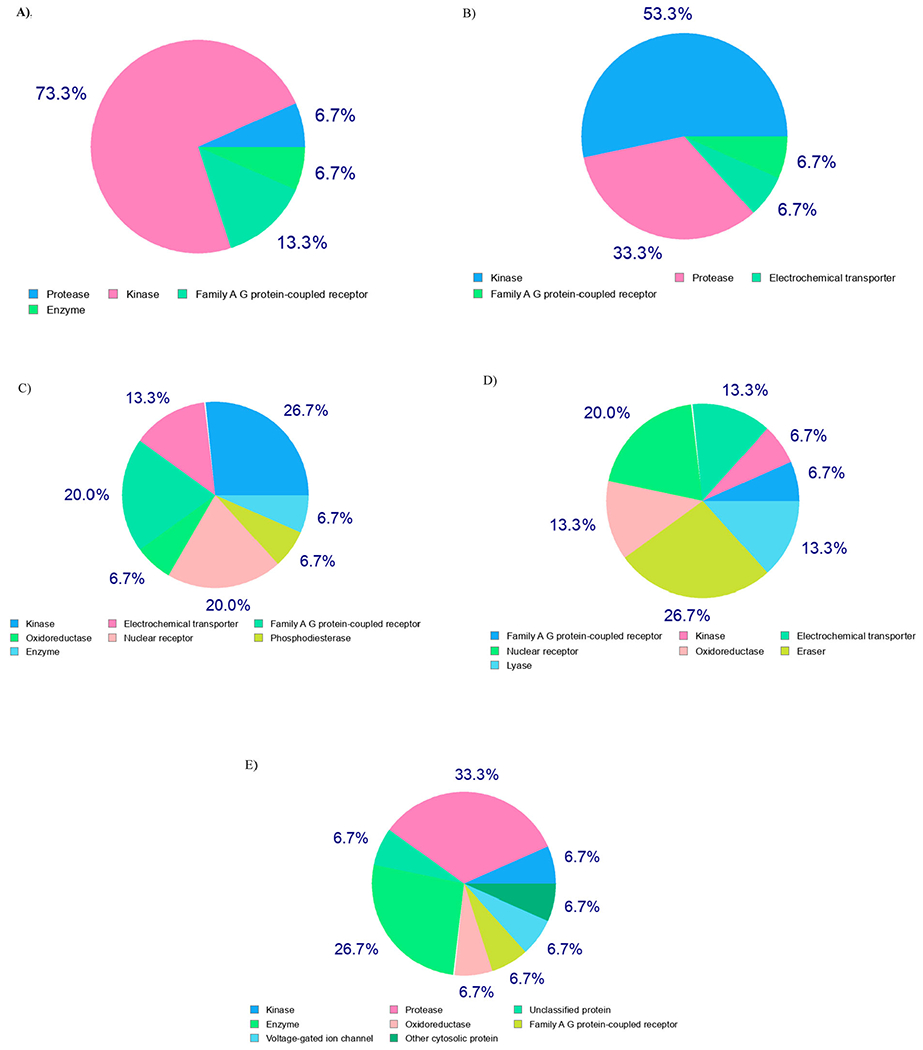
Ligand-based target prediction by SwissTarget-Prediction web tool; (A) 4e, (B) 5e, (C) 8d, (D) 8e, and (E) 9d.
2.3.2. Docking study
The results that emanated from the biological evaluation exhibit that 4e is the best in the cellular assay toward cancerous cell lines and 8e is the most potent against VEGFR-2 and EGFR in enzymatic assay as well as taking in our account the biological target prediction (Figure 5) and the previous literatures [4,5,61,62], docking study of 4e, 8e, and 9d against VEGFR-2 (KDR) and EGFR receptor tyrosine kinases as targeted therapy was implemented using iGEMDOCK program version 2.1 [63]. As a potent VEGFR-2 inhibitor, the crystal structure of VEGFR-2 kinase domains in complex with N4-methyl-N4-(3-methyl-1H-indazol-6-yl)-N2-(3,4,5-trimethoxy-phenyl)pyrimidine-2,4-diamine (three-letter code: KIM) (PDB ID: 3CJG), manifests that the indazole moiety fit well into the inside pocket of VEGFR-2. In the hinge region, the pyrimidine N-1 and the C-2 anilino N–H were predicted to make two significant hydrogen acceptor (2.86 Å) and donor (2.71 Å) bonds with the peptide backbone of Cys917, respectively (Figure 6A). Gefitinib (Iressa®) is a selective inhibitor of EGFR tyrosine kinase and an oral bioavailable drug utilized for certain breast, lung and other cancers [6,64,65]. The docked pose of gefitinib bound to EGFR (PDB ID: 4WKQ) shows that the quinazoline ring occupies the same region as the ATP purine ring (ATP-competitive inhibitor), and its ring N-1 makes an important hydrogen bond with the backbone nitrogen of Met793 (2.98 Å) (Figure 6B).
Figure 6.
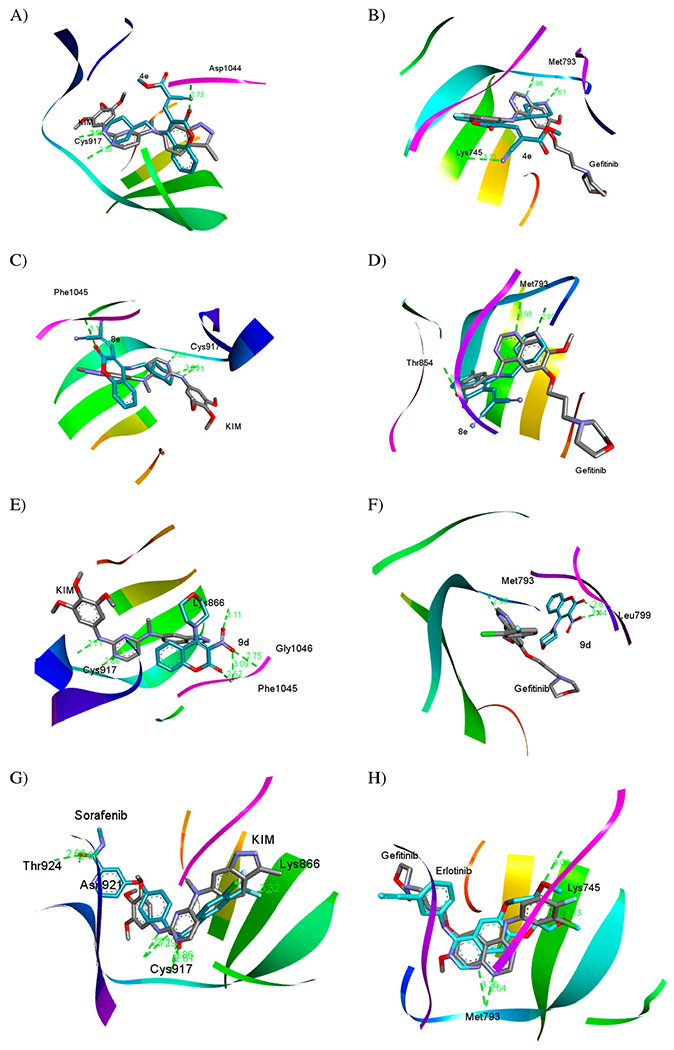
(A, C, E and G) Three-dimensional docking poses of 4e, 8e, 9d, and sorafenib (cyan), respectively, with KIM (grey) within the binding site of VEGFR-2 (PDB ID: 3CJG). (B, D, F and H) Three-dimensional binding modes of 4e, 8e, 9d, and erlotinib (cyan), respectively, with gefitinib (grey) within the active site of EGFR (PDB ID: 4WKQ). H-bonds are denoted by dashed lines in green. All pictures were prepared with Discovery Studio Visualizer Client 2020, and are simple for clarity of presentation.
Thereafter, we docked 4e, 8e, and 9d with VEGFR-2 and EGFR binding sites to compare the docking pose with co-crystallized ligands KIM and gefitinib, respectively. Table 5 and Figure 6A demonstrate that compound 4e is mostly overlapped with co-crystallized KIM into the binding site of VEGFR-2 by predicted fitness (total energy) value of – 104.204 kcal/mol comparable to KIM with the fitness value of −102.401 kcal/mol. Also, 4e docking mode with binding pocket of VEGFR-2 produced two conventional H-bonds, one among them in the hinge region between donor NH-1 of 4-AMP unit and carbonyl oxygen of the key amino acid residue Cys917 (2.60 Å), while the second one is observed between C-2 oxygen of coumarin moiety and peptide backbone of Asp1044 (2.73 Å) as a part of the DFG cavity. Consequently, these interactions proving the importance of 4-AMP and coumarin fragments in 4e.
Table 5.
Binding fitness and no. of H-bonds of 4e, 8e, and 9d to VEGFR-2 and EGFR in comparison with KIM, sorafenib, gefitinib, and erlotinib as the reference drugs.
| Compounds | Fitness (kcal/mol) |
H-bonds (≠) |
||
|---|---|---|---|---|
| VEGFR-2 | EGFR | VEGFR-2 | EGFR | |
| 4e | −104.204 | −112.020 | 2 | 2 |
| 8e | −100.502 | −113.558 | 2 | 2 |
| 9d | −89.051 | −94.544 | 4 | 2 |
| KIM | −102.401 | — | 2 | — |
| Sorafenib | −107.572 | — | 5 | — |
| Gefitinib | — | −104.299 | — | 1 |
| Erlotinib | − | −102.323 | — | 3 |
As amply illustrated in Figure 6B, 4e is overlapped with gefitinib in the active binding site of EGFR (PDB ID: 4WKQ) by the fitness value of −112.020 kcal/mol comparable to gefitinib with the fitness value of −104.299 kcal/mol (Table 5). Analysis of 4e docking mode in the binding pocket of EGFR shows two conventional H-bonds. In the same manner, NH-1 of 4-AMP core is stabilized by hydrogen bond with the carbonyl oxygen of the key amino acid residue Met793 (2.61 Å) in the hinge region. The second one is generated between cyano nitrogen (CN) in the side chain as a proton acceptor and the distal NH2 of Lys745 (3.22 Å). Moreover, the coumarin moiety inserts into a hydrophobic pocket.
Like 4e, NH-1 of 4-AMP fragment in compound 8e interacts with Cys917 (3.35 Å) in the binding cavity of VEGFR-2 (−100.502 kcal/mol) and Met793 (2.69 Å) in EGFR (−113.558 kcal/mol) via hydrogen bonding. At the same time, coumarin moiety plays an important role for interaction stabilizing between 8e and kinases, where its carbonyl oxygen forms H-bond with Phe1045 (3.18 Å) in VEGFR-2 and O-1 act as a proton acceptor from Thr854 (3.08 Å) in EGFR (Figure 6C and D).
The binding mode of compound 9d against VEGFR-2 (Figure 6E) and EGFR (Figure 6F) explains why 9d was a selective and highly potent nanomolar inhibitor of VEGFR-2 with the IC50 value of 24.26 ± 1.1 nM (Table 3). Figure 6E shows that 9d was overlapped with co-crystallized ligand KIM via coumarin unit only and the nitro group fit into the outside pocket of VEGFR-2. Thus, carbonyl coumarin and nitro group as hydrogen acceptors were predicted to form new four H-bonds with amino acid residues Lys866, Phe1045, and Gly1046. Noteworthy, 9d did not form the foremost H-bond with Cys917, confirming the importance of 4-AMP unit and establishing a new pathway to suppress VEGFR-2. On the contrary, Figure 6F displays that 9d did not overlap with gefitinib within the binding site of EGFR, thus, this interaction pattern dramatically decreases the activity of 9d toward EGFR kinase (IC50= 165.00 ± 8.0 nM).
Finally, the docking modes of sorafenib and erlotinib as the reference drugs in VEGFR-2 and EGFR inhibitory activity assay, respectively, were predicted (Table 5, Figure 6G,H). Sorafenib and erlotinib demonstrated high binding affinity toward VEGFR-2 and EGFR, respectively, as well displayed the major H-bonds in the hinge region with the key amino acid residues Cys917 and Met793, respectively, as generated with our compounds binding modes.
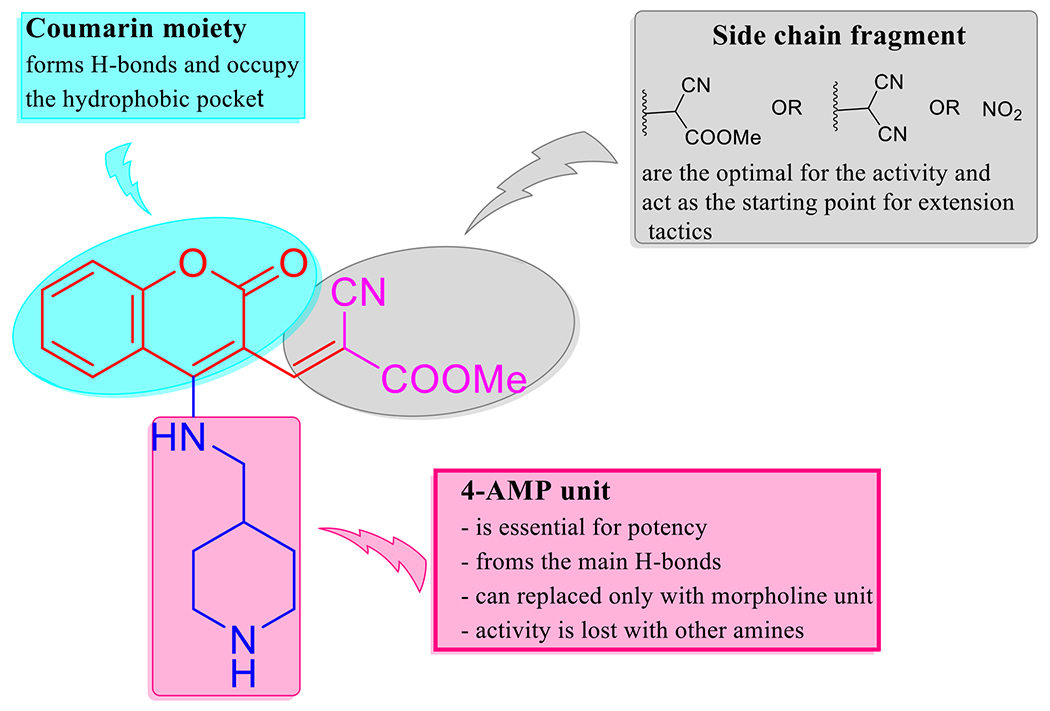
2.4. Structure–activity relationship
As stated above in the biological evaluation and predicted docking modes of compounds 4e, 8e, and 9d, structure–activity relationship (SAR) study on the cyclic secondary amine fragment shows that 4-AMP and morpholine units are the optimal for cellular and enzymatic potencies. Compound 4e with 4-AMP and methyl cyanoacrylate cores has good anti-proliferative activity toward all the three tumor cell lines and more active against VEGFR-2 than EGFR. When methyl cyanoacrylate fragment is replaced by malononitrile and 4-AMP is retained, the generated compound 8e is selective toward KB-3-1 and a promising dual inhibitor VEGFR-2 and EGFR. Compound 9d with morpholine and nitro groups is effective against A549 and PC3 cell lines, but selective inhibitor of VEGFR-2 (IC50 = 24.26 ± 1.1 nM) in comparison with EGFR (IC50= 165.00 ± 8.0 nM). When 3-NO2 group in 9d is changed by methylene malononitrile unit, the corresponding compound 8d is selective toward PC3 cell line (Table 2). It should be noted that, when methylene malononitrile unit in 8e is substituted by formyl group, the obtained small molecule 5e showed lower inhibitory activity against VEGFR-2 and EGFR kinases (Table 3).
In cellular anti-proliferative assay, a big drop-in activity is detected when 4-AMP and morpholine units are replaced with other amines. As illustrated by the docking study, coumarin framework is an important for potent activity, where it forms H-bonds and suitable to occupy the hydrophobic pocket, thereby improving the binding affinity of the hit molecules to the therapeutic targets. Through the inclusive valuation, cyclic secondary amines including 4-AMP and morpholine are crucial for maintaining the potency of the target small molecules in our study. As well side chain fragments encompassing methyl cyanoacetate, malononitrile and nitro group have an obviously impact on cellular and enzymatic activities. Coumarin system can be considered good bioisostere of quinazoline scaffold to discovery and development of EGFR potent inhibitors. These finding tell us the useful pathway to create a rational design using extension tactics in order to achieve the optimal binding interactions, hence get more potent VEGFR-2 and EGFR inhibitors (Figure 7).
Figure 7.
The SAR study of the synthesized compounds.
3. Materials and methods
3.1. Materials and instruments
Melting points were determined on a BÜCHI Melting Point B-540 apparatus in open capillaries and are uncorrected (BÜCHI Germany). NMR spectra (1H NMR and 13C NMR) were recorded on Bruker Avance DRX 500HD MHz (1H: 500 MHz, 13C: 125 MHz) spectrometer (Bruker, USA) at 298 K. Tetramethylsilane (TMS) is used for internal calibration (1H NMR and 13C NMR: 0.00 ppm). Chemical shifts were reported in parts per million (ppm) on the δ scale and relative to residual solvent peaks (DMSO-d6: 1H: 2.50 ppm, 13C: 39.5 ppm). Coupling constants (J) are reported in Hz with the following abbreviations used to indicate splitting: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad signal. ESI mass spectra were recorded using an ion trap mass spectrometer equipped with a standard ESI/APCI source. Samples were introduced by direct infusion with a syringe pump. Nitrogen served both as the nebulizer gas and the dry gas. Nitrogen was generated by a nitrogen generator. Helium served as cooling gas for the ion trap and collision gas for MSn experiments (Bruker Daltonik GmbH, Bremen, Germany). Starting materials and reagents were obtained from commercial sources and used without further purification, unless otherwise indicated. Solvents were dried and purified following standard procedures in organic chemistry. The purity of the synthesized compounds was investigated by TLC, performed on Merck precoated silica gel 60 F254 aluminum sheets with a solvent mixture of DCM-MeOH (982) as eluent. Spots were visualized under UV illumination at 254 and 366 nm.
3.2. Synthesis and characterization
3.2.1. Synthesis of 4-chloro-3-formylcoumarin (1a)
Our substrate la was synthesized from 4-hydroxycoumarin according to the Vilsmeier–Haack protocol [36].
3.2.2. General procedure for the synthesis of methyl (E)-2-cyano-3-(4-substitutedcoumarinyl)acrylates 4a–f
Methyl cyanoacetate (2a, 1 mmol) was treated with 4-chloro-3-formylcoumarin (la, 1.0 mmol) and cyclic secondary amines (3a–f, 2.5 mmol) in MeOH (2 mL). Using the open flask, the mixture was stirred at room temperature for 30-45 min. After the reaction was completed (TLC), the solvent was removed under reduced pressure and the crude product was purified by simple recrystallization (MeOH) or washing with hot methanol to afford the desired compounds.
Methyl (E)-2-cyano-3-(2-oxo-4-(pyrrolidin-1-yl)-2H-chromen-3-yl)acrylate (4a)
Yield 93%; yellow solid; mp 230–232 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.44 (s, 1H), 8.04 (dd, J = 8.1, 1.5 Hz, 1H), 7.67 (ddd, J = 8.5, 7.3, 1.4 Hz, 1H), 7.43–7.36 (m, 2H), 3.77 (s, 3H), 3.34–3.33 (m, 4H, overlapped with water of DMSO), 2.05–2.01 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ 164.7 (C4—N), 161.6 (C = O ester), 158.7 (C = O lactone), 151.5 (C—O lactone), 151.1 (CH olefinic), 133.2, 126.7, 124.1, 118.6, 117.7, 116.5, 93.9, 90.1, 56.1 (OCH3), 52.8 (2CH2N), 25.0 (2CH2); (+)-ESI-MS, m/z (%): 347 [M + Na]+ (100), 671 [2M + Na]+ (31). See Supporting Information, Figs. S1–S3.
Methyl (E)-2-cyano-3-(2-oxo-4-(piperidin-1-yl)-2H-chromen-3-yl)acrylate (4b)
Yield 91%; faint orange solid; mp 192–194 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.17 (s, 1H), 7.93 (dd, J = 8.2, 1.5 Hz, 1H), 7.70 (ddd, J = 8.5, 7.2, 1.4 Hz, 1H), 7.44–7.38 (m, 2H), 3.85 (s, 3H), 3.68–3.64 (m, 4H), 1.77–1.73 (m, 6H); 13C NMR (125 MHz, DMSO-d6) δ 163.5 (C4—N), 163.1 (C = O ester), 158.9 (C = O lactone), 153.4 (C—O lactone), 152.5 (CH olefinic), 134.0, 127.3, 124.6, 118.0, 117.5, 115.8, 102.8, 100.9, 54.8 (2CH2N), 53.5 (OCH3), 27.0 (2CH2), 23.6 (CH2); (+)-ESI-MS, m/z (%): 361 [M + Na]+ (100), 699 [2M + Na]+ (6). See Supporting Information, Figs. S4–S6.
Methyl (E)-2-cyano-3-(4-(4-hydroxypiperidin-1-yl)-2-oxo-2H-chromen-3-yl)acrylate (4c)
Yield 96%; orange solid; mp 194–196 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.16 (s, 1H), 7.92 (dd, J = 8.2, 1.6 Hz, 1H), 7.70 (ddd, J = 8.4, 7.2, 1.4 Hz, 1H), 7.44–7.38 (m, 2H), 4.97 (d, J = 3.7 Hz, 1H, OH), 3.89 (dq, J = 7.8, 3.8 Hz, 1H), 3.85 (s, 3H, OCH3), 3.81 (ddd, J = 12.7, 6.3, 3.0 Hz, 2H), 3.58 (ddd, J = 12.7, 8.6, 3.3 Hz, 2H), 2.00–1.93 (m, 2H), 1.69–1.60 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 163.5 (C4—N), 162.9 (C = O ester), 158.9 (C = O lactone), 153.3 (C—O lactone), 152.5 (CH olefinic), 134.0, 127.2, 124.6, 118.0, 117.5, 115.8, 102.8, 100.9, 64.7 (CHOH), 53.5 (OCH3), 51.2 (2CH2N), 35.4 (2CH2); (+)-ESI-MS, m/z (%): 377 [M + Na]+ (100), 731 [2M + Na]+ (12). See Supporting Information, Figs. S7–S9.
Methyl (E)-2-cyano-3-(4-morpholino-2-oxo-2H-chromen-3-yl)acrylate (4d)
Yield 92%; yellow solid; mp 218–220 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.19 (s, 1H), 7.95 (dd, J = 8.3, 1.5 Hz, 1H), 7.71 (ddd, J = 8.5, 7.2, 1.4 Hz, 1H), 7.45 (dd, J = 8.3, 1.2 Hz, 1H), 7.40 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 3.86 (s, 3H), (t, J = 4.6 Hz, 4H), 3.72 (t, J = 5.5, 3.7 Hz, 4H); 13C NMR (125 MHz, DMSO-d6) δ 163.4 (C4—N), 162.4 (C = O ester), 158.8 (C = O lactone), 153.4 (C—O lactone), 151.9 (CH olefinic), 134.1, 127.3, 124.7, 118.0 117.1, 115.7, 103.8, 101.3, 67.22 (2CH2O), 53.7 (2CH2N), 53.5 (OCH3); (+)-ESI-MS, m/z (%): 363 [M + Na]+ (100), 703 [2M + Na]+ (25). See Supporting Information, Figs. S10–S12.
Methyl (E)-2-cyano-3-(2-oxo-4-((piperidin-4-ylmethyl)amino)-2H-chromen-3-yl)acrylate (4e)
Yield 96%; yellow solid; mp 172–174 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.76 (t, J = 5.8 Hz, 1H), 8.64 (s, 1H), 8.34 (dd, J = 7.9, 1.7 Hz, 1H), 7.68 (ddd, J = 8.6, 7.3, 1.7 Hz, 1H), 7.44 (td, J = 7.6, 1.1 Hz, 1H), 7.39 (dd, J = 8.2, 1.0 Hz, 1H), 3.90 (s, 3H), 3.59 (t, J = 6.2 Hz, 2H), 2.95 (dt, J = 12.1, 3.4 Hz, 2H), 2.44 (td, J = 12.0, 2.5 Hz, 2H), 1.82–1.72 (m, 2H), 1.66 (dd, J = 12.6, 3.5 Hz, 2H), 1.23–1.17 (m, 1H), 1.17–1.12 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 166.5 (C4—NH), 160.1 (C=O ester), 159.71 (C=O lactone), 155.0 (C—O lactone), 153.9 (CH olefinic), 142.1, 133.8, 125.25, 125.17, 118.8, 117.5, 107.8, 105.8, 53.0 (OCH3), 47.1 (CH2NH), 46.3 (2CH2NH), 36.7 (CH aliphatic), 31.3 (2CH2); (+)-ESI-MS, m/z (%): 368 [M + H]+ (100), 735 [2M + H]+ (6). See Supporting Information, Figs. S13–S15.
Methyl (E)-2-cyano-3-(4-methoxy-2-oxo-2H-chromen-3-yl)acrylate (4f)
Yield 89%; yellow solid; mp 148–150 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.34 (s, 1H), 7.97 (dd, J = 8.0, 1.6 Hz, 1H), 7.78 (ddd, J = 8.7, 7.2, 1.6 Hz, 1H), 7.49 (dd, J = 8.4, 1.1 Hz, 1H), 7.46 (ddd, J = 8.3, 7.3, 1.1 Hz, 1H), 4.13 (s, 3H), 3.89 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 167.2, 162.0, 159.9, 150.5, 149.3, 133.2, 126.2, 125.1, 117.9, 117.2, 116.7, 103.9, 99.2, 63.8, 52.3; (+)-ESI-MS, m/z (%): 308 [M + Na]+ (100), 593 [2M + Na]+ (22). See Supporting Information, Figs. S16–S18.
3.2.3. General procedure for the synthesis of buta-1,3-diene-1,1,3-tricarbonitrile derivatives 8a,b and methylene malononitrile analogs 8c–e
Using open flask, a solution of malononitrile (2b, 2 mmol) and cyclic secondary amines (3a–e, 1 mmol) in MeOH (2 mL) was stirred at room temperature for 10 min. At the same time, anther mixture of 4-chloro-3-formylcoumarin (1a, 1.0 mmol) and the same cyclic secondary amines (3a–e, 1.5 mmol) in MeOH (2 mL) was also produced by stirring at ambient temperature for 10 min. Next, the two mixtures were combined together and sequential stirring was continued at room temperature until a new product was formed (TLC). Hence, the solvent was removed under reduced pressure using the rotary evaporator and the crude product was purified by simple recrystallization (MeOH) or only washing with hot methanol to get the desired compound.
(Z)-2-amino-4-(2-oxo-4-(pyrrolidin-1-yl)-2H-chromen-3-yl)buta-1,3-diene-1,1,3-tricarbonitrile (8a)
Yield 97%; yellow solid; mp > 320 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.72 (s, 1H), 7.85 (dd, J = 8.2, 1.5 Hz, 1H), 7.73 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 7.63 (s, 2H), 7.56 (dd, J = 8.4, 1.3 Hz, 1H), 7.45 (ddd, J = 8.3, 7.2, 1.3 Hz, 1H), 3.72–3.58 (m, 2H), 3.29–2.95 (m, 2H), 1.97–1.86 (m, 2H), 1.86–1.62 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 166.51(C—NH2), 161.8 (C4—N), 159.5, 155.9, 155.3, 152.4, 134.0, 133.5, 126.1, 125.9, 120.6, 118.9, 116.7, 116.6, 103.7, 76.4, 48.1, 46.1, 25.9, 24.6; (+)-ESI-MS, m/z (%): 737 [2M + Na]+ (90), 1094 [3M + Na]+ (2). See Supporting Information, Figs. S19–S21.
(Z)-2-amino-4-(2-oxo-4-(piperidin-1-yl)-2H-chromen-3-yl)buta-1,3-diene-1,1,3-tricarbonitrile (8b)
Yield 94%; yellow solid; mp 308–310 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.67 (s, 1H), 7.92 (dd, J = 8.3, 1.5 Hz, 1H), 7.73 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 7.65–7.60 (m, 2H), 7.56 (dd, J = 8.4, 1.2 Hz, 1H), 7.46 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 3.84–3.73 (m, 2H), 3.30–3.21 (m, 1H), 3.21–3.13 (m, 1H), 1.69–1.58 (m, 4H), 1.46–1.36 (m, 1H), 1.26–1.16 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 166.8, 161.7, 159.5, 155.9, 155.0, 152.4, 134.0, 133.9, 126.5, 125.8, 119.6, 118.9, 116.7, 116.5, 103.9, 76.3, 47.8, 26.3, 24.4; (+)-ESI-MS m/z (%): 372 [M + H]+. See Supporting Information, Figs. S22–S24.
2-((4-(4-Hydroxypiperidin-1-yl)-2-oxo-2H-chromen-3-yl)methylene)malononitrile (8c)
Yield 88%; yellow solid; mp 294–296 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.97 (dd, J = 8.3, 1.5 Hz, 1H), 8.76 (s, 1H), 7.81 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 7.53 (ddd, J = 8.4, 7.3, 1.3 Hz, 1H), 7.47 (dd, J = 8.4, 1.3 Hz, 1H), 5.77–5.76 (m, 2H), 3.18–3.17 (m, 4H), 2.10–2.09 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ 161.9, 158.4, 153.3, 148.7, 147.9, 135.8, 126.0, 125.6, 118.9, 117.3, 115.0, 103.0, 93.5, 55.4, 49.1, 31.2; (+)-ESI-MS, m/z (%): 344 [M + Na]+ (100), 665 [2M + Na]+ (12). See Supporting Information, Figs. S25–S27.
2-((4-Morpholino-2-oxo-2H-chromen-3-yl)methylene)malononitrile (8d) [39]
Yield 96%; yellow solid; mp 196–198 °C; (from literature [39], yield 52%; orange solid, mp 193–195 °C). (+)-ESI-MS, m/z (%):637 [2M + Na]+ (100). See Supporting Information, Figure S28.
2-((2-Oxo-4-((piperidin-4-ylmethyl)amino)-2H-chromen-3-yl)methylene)malononitrile (8e)
Yield 98%; yellow solid; mp 154–156 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.32 (dd, J = 7.9, 1.7 Hz, 1H), 7.69 (ddd, J = 8.7, 7.4, 1.8 Hz, 2H), 7.48–7.43 (m, 1H), 7.40 (d, J = 8.3 Hz, 1H), 3.51 (d, J = 6.7 Hz, 2H), 2.98 (dt, J = 12.4, 3.3 Hz, 2H), 2.50–2.43 (m, 2H), 1.89–1.81 (m, 1H), 1.71–1.63 (m, 3H), 1.23–1.11 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 159.6, 159.4, 154.5, 153.8, 145.4, 134.0, 125.3, 118.7, 117.5, 116.6, 115.9, 106.2, 93.4, 47.5, 46.0, 36.2, 30.8; (+)-ESI-MS, m/z (%): 335 [M + H]+ (100), 669 [2M + H]+ (1). See Supporting Information, Figs. S29–S31.
3.2.4. General procedure for the synthesis of 4-substituted-3-formyl/nitro-2H-chromen-2-ones 5a, d–f and 9a,d–f
Cyclic secondary amines (3a,d-f, 1 mmol) was added to a stirred solution of 4-chloro-3-formyl/nitrocoumarin (1a,c, 1.0 mmol) and triethylamine (Et3N, 1 mmol) in MeOH (2 mL). The reaction mixture was stirred at room temperature for 5–15 min. After the reaction was completed (TLC), the solvent was removed under reduced pressure and the crude product was purified by simple recrystallization (MeOH) or washing with hot methanol to afford the desired compound.
4-Pyrrolidino-3-formyl-2H-chromen-2-one (5a)
Yield 93%; white solid; mp 190–192 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.67 (s, 1H), 8.06 (dd, J = 8.1, 1.5 Hz, 1H), 7.63 (ddd, J = 8.5, 7.3, 1.4 Hz, 1H), 7.36 (dd, J = 8.3, 1.2 Hz, 1H), 7.33 (ddd, J = 8.4, 7.3, 1.3 Hz, 1H), 3.85 (s, 4H), 1.99–1.93 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ 183.4 (CHO), 163.7 (C4—N), 158.0 (C=O lactone), 152.2 (C—O lactone), 133.0, 127.9, 123.5, 118.1, 117.5, 97.4, 56.7 (2CH2N), 24.9 (2CH2); (+)-ESI-MS, m/z (%): 266 [M+ Na+ (100), 509 [2M + Na]+ (35). See Supporting Information, Figs. S32–S34.
4-Morpholino-3-formyl-2H-chromen-2-one (5d)
Yield 95%; yellow solid; mp 150–152 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.85 (s, 1H), 7.99 (dd, J = 8.2, 1.5 Hz, 1H), 7.69 (ddd, J = 8.5, 7.2, 1.4 Hz, 1H), 7.41 (dd, J = 8.4, 1.3 Hz, 1H), 7.37 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 3.90–3.86 (m, 4H), 3.65 (t, J = 4.7 Hz, 4H); 13C NMR (125 MHz, DMSO-d6) δ 186.5 (CHO), 176.5 (C4—N), 162.5 (C=O lactone), 154.4 (C—O lactone), 134.3, 126.3, 124.1, 121.3, 117.0, 95.3, 66.8 (2CH2O), 54.5 (2CH2N); (+)-ESI-MS, m/z(%): 282 [M + Na]+ (35), 541 [2M + Na]+ (100). See Supporting Information, Figs. S35–S37.
4-((Piperidin-4-ylmethyl)amino)-3-formyl-2H-chromen-2-one (5e)
Yield 85%; yellow solid; mp 208–210 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.94 (t, J = 5.1 Hz, 1H, NHAr), 9.94 (s, 1H), 8.28 (d, J = 8.2 Hz, 1H), 7.78 (t, J = 7.8 Hz, 1H), 7.46–7.39 (m, 2H), 3.91 (t, J = 5.9 Hz, 2H), 3.33–3.23 (m, 3H), 2.90 (td, J = 12.8, 2.9 Hz, 2H), 2.04 (ttt, J = 10.4, 6.6, 3.4 Hz, 1H), 1.95 (d, J = 13.7 Hz, 2H), 1.58–1.44 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 191.0 (CHO), 162.2 (C4—NH), 159.9 (C=O lactone), 155.2 (C—O lactone), 135.6, 129.3, 124.6, 118.5, 114.0, 96.3, 52.2 (CH2NH), 43.1 (2CH2NH), 34.6 (CH), 26.7 (2CH2); (+)-ESI-MS, m/z (%): 287 [M+ H]+. See Supporting Information, Figs. S38–S40.
4-Methoxy-3-formyl-2H-chromen-2-one (5f)
Yield 89%; pale yellow solid; mp 118–120 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.12 (s, 1H), 7.99 (dd, J = 8.0, 1.6 Hz, 1H), 7.79 (ddd, J = 8.7, 7.2, 1.6 Hz, 1H), 7.48–7.41 (m, 2H), 4.19 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 189.2 (CHO), 170.0 (C4—O), 162.3 (C=O lactone), 153.6 (C—O lactone), 135.5, 125.8, 125.4, 117.3, 117.1, 106.7, 65.6 (OCH3); (+)-ESI-MS, m/z(%): 227 [M + Na]+ (100), 431 [2M + Na]+ (7)+. See Supporting Information, Figs. S41–S43.
4-Pyrrolidino-3-nitro-2H-chromen-2-one (9a)
Yield 97%; yellow solid; mp 204–206 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.18 (dd, J = 8.1, 1.4 Hz, 1H), 7.69 (ddd, J = 8.5, 7.2, 1.4 Hz, 1H), 7.41–7.36 (m, 2H), 3.83–3.77 (m, 4H), 2.01–1.95 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ 155.2 (C4—N), 154.4 (C=O lactone), 151.2 (C—O lactone), 133.6, 128.1, 123.8, 117.6, 117.0, 114.6, 55.6 (2 CH2N), 24.7 (2CH2); (+)-ESI-MS, m/z(%): 283 [M+ Na]+ (100), 543 [2M + Na]+ (17). See Supporting Information, Figs. S44–S46.
4-Morpholino-3-nitro-2H-chromen-2-one (9d)
Yield 96%; yellow solid; mp 210–212 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.90 (dd, J = 8.1, 1.5 Hz, 1H), 7.76 (ddd, J = 8.6, 7.2, 1.5 Hz, 1H), 7.50–7.45 (m, 2H), 3.88–3.83 (m, 4H), 3.39–3.33 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ 155.5 (C4—N), 153.1 (C=O lactone), 152.5 (C—O lactone), 134.6, 127.7, 125.8, 125.5, 118.1, 116.1, 66.2 (2 CH2O), 51.5 (2 CH2N); (+)-ESI-MS, m/z (%): 299 [M+ Na]+ (100), 575 [2M+ Na]+ (25). See Supporting Information, Figs. S47–S49.
4-((Piperidin-4-ylmethyl)amino)-3-nitro-2H-chromen-2-one (9e)
Yield 89%; yellow solid; mp 208–210 °C; 1H NMR (500 MHz, DMSO-d6) δ 15.82 (s, 1H, NHAr), 7.22–7.18 (m, 2H), 6.78–6.75 (m, 1H), 6.69 (td, J = 7.5, 1.2 Hz, 1H), 3.32 (d, J = 6.5 Hz, 2H), 3.28 (dt, J = 12.4, 3.4 Hz, 3H), 2.88 (td, J = 12.8, 3.0 Hz, 2H), 1.98–1.88 (m, 1H), 1.84 (d, J = 14.6 Hz, 2H), 1.36 (qd, J = 12.5, 4.0 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 170.0, 163.4, 162.0, 131.7, 130.0, 120.4, 117.6, 117.3, 103.3, 55.7, 49.9, 43.7, 34.5, 27.5; (+)-ESI-MS, m/z (%): 304 [M+ H]+. See Supporting Information, Figs. S50–S52.
4-Methoxy-3-nitro-2H-chromen-2-one (9f)
Yield 91%; white solid; mp 110–112 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.98 (dd, J = 8.1, 1.6 Hz, 1H), 7.82 (ddd, J = 8.7, 7.3, 1.6 Hz, 1H), 7.55–7.48 (m, 2H), 4.18 (s, 3H), 13C NMR (125 MHz, DMSO-d6) δ 167.8 (C4—O), 158.7 (C=O lactone), 151.7 (C—O lactone), 135.4, 125.5, 123.4, 117.3, 116.5, 115.5, 60.7 (OCH3); (+)-ESI-MS, m/z (%): 244 [M+ Na]+ (100), 465 [2M+ Na]+ (6). See Supporting Information, Figs. S53–S55.
3.3. Resazurin cellular-based bioassay
Anti-cervical cancer activity of all the synthesized compounds were evaluated for the in vitro cytotoxicity toward KB-3-1 cell line (was obtained from the American Type Culture Collection, ATCC, Rockville, MD, USA) by resazurin-based assay [49] using (+)-griseofulvin as a positive control. Next, anti-proliferative potency of our synthesized compounds was tested against A549 (non-small lung) and PC3 (prostate) human cancer cell lines using the same assay as reported by Thorson research group [50–52].
3.4. Enzyme inhibitory bioassay
In the case of the VEGFR-2 enzymatic assay, we followed the instruction manual of VEGFR-2 (KDR) Kinase Assay Kit Catalog # 40325. On the other hand, EGFR Kinase Assay Ki Catalog # 40321 was followed to examine the capability of the selected compounds to inactivate the enzyme EGFR kinase. A series of 10-fold dilutions was prepared from each compound (4e, 5e, 8e, and 9d) while sorafenib (Cat no.284461-73-0, Santa Cruz) and erlotinib (Cat no.183321-74-6, Cayman) were matched as the positive controls in synchronize to VEGFR-2 and EGFR assays, respectively. At the end of the experiment, the luminescence was measured by Tecan spark microplate reader. The compounds concentration that inactivate 50% of the measured kinases was separately, calculated using a curve fitting software; Prism, version 6.
3.5. In silico computational predictions
3.5.1. Physicochemical descriptors, drug-likeness, and medicinal chemistry parameters
The physicochemical properties of compounds 4e, 5e, 8d,e, and 9d were predicted using SwissADME [57] as formerly described (Table 4).
3.5.2. Biological target prediction
The biomolecular target of compounds 4e, 5e, 8d,e, and 9d were estimated through the SwissTarget-Prediction web tool [60] that using ligand-based target prediction approach which based on the molecular similarity principle (Table 4, Figure 5).
3.5.3. Molecular docking simulation
In silico flexible ligand docking study has been performed by iGEMDOCK program version 2.1 (Department of Biological Science and Technology & Institute of Bioinformatics, National Chiao Tung University, Taiwan) [63] that uses a generic evolutionary approach (GA) and an empirical scoring function to explore the interaction modes between the hit molecules (4e, 8e, and 9d) and VEGFR-2/KDR (PDB ID: 3CJG) and EGFR (PDB ID: 4WKQ) as a biological targets. Firstly, the three-dimensional (3D) structures of VEGFR-2 with KIM complex and EGFR with gefitinib were obtained from RCSB Protein Data Bank (https://www.rcsb.org) and the co-crystallized ligand, was extracted and docked back to the corresponding binding site, to define the ability of docking protocol to reproduce the docking mode of the inhibitor observed in the crystal structure (iGEMDOCK validation) (Figure 8).
Figure 8.
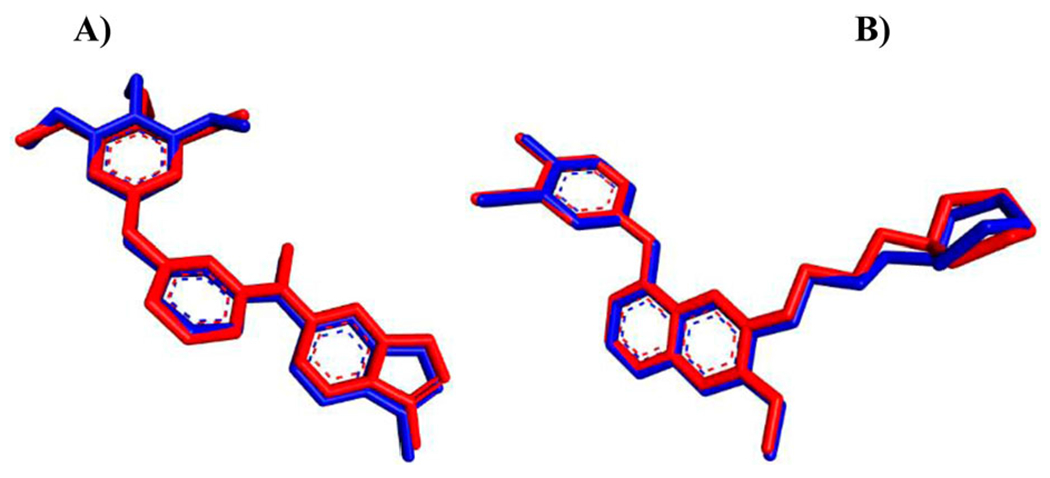
iGEMDOCK validation. (A) Crystal KIM (red) and the docked one (blue) display similar binding orientation in the binding pocket of VEGFR-2 with RMSD 0.4355 Å. (B) Crystal gefitinib (red) with the docked one (blue) are superimposed in EGFR binding cavity with RMSD 0.3462 Å.
Secondly, the two-dimensional (2D) structure of hit compounds were drawn by ChemBioDraw Ultra 14.0 (PerkinElmer Informatics, Waltham, MA, USA) and converted to 3D structure by ChemBio3D Ultra 14.0 then saved as mol format after energy minimized and Molecular Dynamic (MD) performed using MMFF94 (Merck molecular force field) method [66]. Finally, the docking process has been done by uploading the protein pdb and ligand mol files to the iGEMDOCK program and the result has been analyzed with the Discovery Studio Visualizer Client 2020 (BIOVIA, San Diego, CA, USA). The docking study of compounds 4e, 8e, and 9d toward VEGFR-2 and EGFR have been achieved in comparison with known inhibitors including KIM, gefitinib, sorafenib, and erlotinib (Figure 6). The fitness value (Table 5) is the total energy of a predicted pose in the binding site. The empirical scoring function of iGEMDOCK is estimated as [63]: Fitness = vdW + H-bond + Elec; where, the vdW term is pointed out to van der Waal energy. H-bond and Elec terms are denoted hydrogen bonding energy and electro statistic energy, respectively. As illustrated in Figure 8, the docked ligands (KIM and gefitinib) pretend the same binding mode as the crystal one and the Root Mean Square Distance (RMSD) was within the reliable range (≤2 Å), confirming the robustness of this approach.
Conclusions
A simple and an effective methodology without addition of metal catalyst has been developed for mild construction of new coumarin analogous (4a–f, 5a,d–f, 8a–e, and 9a,d–f) with high yield via domino amination-Knoevenagel condensation approach. All the synthesized compounds were characterized by various spectral methods. The anti-proliferative activity of hit compounds was performed using resazurin assay. Among them, compounds 4e and 8e that bearing 4-AMP unit displayed the best anti-cervical cancer activity with IC50 values of 15.5 ± 3.54 and 21 ± 4.24 μM, respectively. Similar way was observed with 4e that showed the most optimistic cytotoxicity result toward A549 with the IC50 value of 12.94 ± 1.51 μM. Also, 9d displayed a more significant impact of activity against PC3 with the IC50 value of 7.31 ± 0.48 μM. Moreover, the pooled results from the cellular anti-proliferative potency revealed that 8d demonstrated selectivity against PC3 (IC50 = 20.16 ± 0.07 μM), while 8e was selective toward KB-3-1 cell line with the IC50 value of 21 ± 4.24 μM. Enzymatic inhibitory activity disclosed that 8e is a dual inhibitor of VEGFR-2 and EGFR with IC50 values of 24.67 and 31.6 nM, which were almost equipotent to sorafenib (31.08 nM) and erlotinib (26.79 nM), respectively. In silico studies showed that the estimated compounds (4e, 5e, 8d,e, and 9d) passed all the drug-likeness metrics and they could be valuable lead compounds for further investigation in the future.
Supplementary Material
Acknowledgements
The authors are grateful to the NMR and MS Departments at Bielefeld University for spectral measurements. We would like to thank Carmela Michalek for biological activity testing and Marco Wißbrock with Anke Nieß for technical assistance. The authors also would like to thank the National Research Centre, Egypt for the laboratory facilities during VEGFR-2 and EGFR inhibitory studies. This work was also supported by National Institutes of Health grant R01 GM115261 (JST), the Center of Biomedical Research Excellence (COBRE) in Pharmaceutical Research and Innovation (CPRI, NIH P20 GM130456), the University of Kentucky College of Pharmacy, the University of Kentucky Markey Cancer Center, and the National Center for Advancing Translational Sciences (UL1TR000117 and UL1TR001998).
Funding
This work was supported by German Academic Exchange Service (DAAD): [Grant Number ID57166072].
Footnotes
Supplemental data for this article can be accessed at https://doi.org/10.1080/17518253.2021.1981462.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Avendano C; Carlos Menendez J Medicinal Chemistry of Anticancer Drugs, 1st ed.; Elsevier: 2008; pp 1–7. [Google Scholar]
- [2].Boraei ATA; Ashour HK; El-Tamany E-SH; Abdelmoaty N; El-Falouji AI; Gomaa MS Design and Synthesis of New Phthalazine-Based Derivatives as Potential EGFR Inhibitors for the Treatment of Hepatocellular Carcinoma. Bioorg. Chem 2019, 85, 293–307. doi: 10.1016/j.bioorg.2018.12.039. [DOI] [PubMed] [Google Scholar]
- [3].Amin KM; Barsoum FF; Awadallah FM; Mohamed NE Identification of New Potent Phthalazine Derivatives with VEGFR-2 and EGFR Kinase Inhibitory Activity. Eur. J. Med. Chem 2016, 123, 191–201. doi: 10.1016/j.ejmech.2016.07.049. [DOI] [PubMed] [Google Scholar]
- [4].Zhang H-Q;Gong F-H; Fei H-J; Li C-G;Zhang C; Wang Y-J; Xu Y-G; Hu R; Suna L-P Design and Discovery of 4-Anilinoquinazoline-Acylamino Derivatives as EGFR and VEGFR-2 Dual TK Inhibitors. Eur. J. Med. Chem 2016, 109, 371–379. doi: 10.1016/j.ejmech.2015.12.032. [DOI] [PubMed] [Google Scholar]
- [5].Modi SJ; Kulkarni VM Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Medicine in Drug Discovery 2019, 2, 100009. doi: 10.1016/j.medidd.2019.100009. [DOI] [Google Scholar]
- [6].Sordella R; Bell DW; Haber DA; Settleman J Gefitinib-sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- [7].El-Metwally SA; Abou-El-Regal MM; Eissa IH; Mehany ABM; Mahdy HA; Elkady H; Elwan A; Elkaeed EB Discovery of Thieno[2,3-d]Pyrimidine-Based Derivatives as Potent VEGFR-2 Kinase Inhibitors and Anti-Cancer Agents. Bioorg. Chem 2021, 112, 104947. doi: 10.1016/j.bioorg.2021.104947. [DOI] [PubMed] [Google Scholar]
- [8].Alanazi MM; Mahdy HA; Alsaif NA; Obaidullah AJ; Alkahtani HM; Al-Mehizia AA; Alsubaie SM; Dahab MA; Eissa IH New bis([1,2,4]Triazolo)[4,3-a:3′,4′-c]Quinoxaline Derivatives as VEGFR-2 Inhibitors and Apoptosis Inducers: Design, Synthesis, in Silico Studies, and Anticancer Evaluation. Bioorg. Chem 2021, 112, 104949. doi: 10.1016/j.bioorg.2021.104949. [DOI] [PubMed] [Google Scholar]
- [9].Eissa IH; Ibrahim MK; Metwaly AM; Belal A; Mehany ABM; Abdelhady AA; Elhendawy MA; Radwan MM; ElSohly MA; Mahdy HA Design, Molecular Docking, in Vitro, and in Vivo Studies of new Quinazolin-4(3H)-Ones as VEGFR-2 Inhibitors with Potential Activity Against Hepatocellular Carcinoma. Bioorg. Chem 2021, 707, 104532. doi: 10.1016/j.bioorg.2020.104532. [DOI] [PubMed] [Google Scholar]
- [10].Aziz MA; Serya RAT; Lasheen DS; Abdel-Aziz AK; Esmat A; Mansour AM; Singab AB; Abouzid KAM Discovery of Potent VEGFR-2 Inhibitors Based on Furopyrimidine and Thienopyrimidne Scaffolds as Cancer Targeting Agents. Sci. Rep 2016, 6, 24460. doi: 10.1038/srep24460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Saleh NM; Abdel-Rahman AA-H; Omar AM; Khalifa MM; El-Adl K Pyridine-Derived VEGFR-2 Inhibitors: Rational Design, Synthesis, Anticancer Evaluations, in Silico ADMET Profile, and Molecular Docking. Arch. Pharm 2021,354, 2100085. doi: 10.1002/ardp.202100085. [DOI] [PubMed] [Google Scholar]
- [12].Gabera AA; Bayoumia AH; El-morsya AM; Sherbinya FF; Mehanyb ABM; Eissa IH Design, Synthesis and Anticancer Evaluation of 1H-Pyrazolo[3,4-d] Pyrimidine Derivatives as Potent EGFRWT and EGFRT790M Inhibitors and Apoptosis Inducers. Bioorg. Chem 2018, 80, 375–395. doi: 10.1016/j.bioorg.2018.06.017. [DOI] [PubMed] [Google Scholar]
- [13].Kurt BZ; Sonmez F; Ozturk D; Akdemir A; Angeli A; Supuran CT Synthesis of Coumarin-Sulfonamide Derivatives and Determination of Their Cytotoxicity, Carbonic Anhydrase Inhibitory and Molecular Docking Studies. Eur. J. Med. Chem 2019, 783, 111702. doi: 10.1016/j.ejmech.2019.111702. [DOI] [PubMed] [Google Scholar]
- [14].Wang Z-C; Qin Y-J; Wang P-F; Yang Y-A; Wen Q; Zhang X; Qiu H-Y; Duan Y-T; Wang Y-T; Sang Y-L; Zhu H-L Sulfonamides Containing Coumarin Moieties Selectively and Potently Inhibit Carbonic Anhydrases II and IX: Design, Synthesis, Inhibitory Activity and 3D-QSAR Analysis. Eur. J. Med. Chem 2013, 66, 1–11. doi: 10.1016/j.ejmech.2013.04.035. [DOI] [PubMed] [Google Scholar]
- [15].Bubols GB; Vianna DR; Medina-Remon A; Poser GV; Lamuela-Raventos RM, Eifler-Lima VL, et al. The Antioxidant Activity of Coumarins and Flavonoids. Mini Rev. Med. Chem 2013, 73, 318–334. [DOI] [PubMed] [Google Scholar]
- [16].Matos MJ; Vazquez-Rodriguez S; Santana L; Uriarte E; Fuentes-Edfuf C, Santos Y, et al. Synthesis and Structure-Activity Relationships of Novel Amino/Nitro Substituted 3-Arylcoumarins as Antibacterial Agents. Molecules 2013, 18, 1394–1404. doi: 10.3390/molecules18021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Matos MJ; Vina D; Vazquez-Rodriguez S; Uriarte E; Santana L Focusing on New Monoamine Oxidase Inhibitors: Differently Substituted Coumarins as an Interesting Scaffold. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2012, 72, 2210–2239. doi: 10.2174/1568026611212200008. [DOI] [PubMed] [Google Scholar]
- [18].Hwu JR; Huang W-C; Lin S-Y; Tan K-T; Hu Y-C; Shieh F-K; Bachurin SO; Ustyugov A; Tsay S-C Chikungunya Virus Inhibition by Synthetic Coumarin-Guanosine Conjugates. Eur. J. Med. Chem 2019, 166, 136–143. doi: 10.1016/j.ejmech.2019.01.037. [DOI] [PubMed] [Google Scholar]
- [19].Kapoor S The Anti-Neoplastic Effects of Coumarin: An Emerging Concept. Cytotechnology 2013, 65, 787–788. doi: 10.1007/s10616-013-9538-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Diana CGAP; Artur MSS Anticancer Natural Coumarins as Lead Compounds for the Discovery of New Drugs. Curr. Top. Med. Chem 2017, 17, 3190–3198. [DOI] [PubMed] [Google Scholar]
- [21].Emami S; Dadashpour S Current Developments of Coumarin-Based Anti-Cancer Agents in Medicinal Chemistry. Eur. J. Med. Chem 2015, 102, 611–630. doi: 10.1016/j.ejmech.2015.08.033. [DOI] [PubMed] [Google Scholar]
- [22].Halawa AH; Abd El-Gilil SM; Bedair AH; Eliwa EM; Frese M; Sewald N; Shaaban M; El-Agrody AM Synthesis of Diverse Amide Linked Bis-indoles and Indole Derivatives Bearing Coumarin-Based Moiety: Cytotoxicity and Molecular Docking Investigations. Med. Chem. Res 2018, 27, 796–806. doi: 10.1007/s00044-017-2103-7. [DOI] [Google Scholar]
- [23].Zhang L; Xu Z Coumarin-containing Hybrids and Their Anticancer Activities. Eur. J. Med. Chem 2019, 187, 111587. doi: 10.1016/j.ejmech.2019.111587. [DOI] [PubMed] [Google Scholar]
- [24].Halawa AH; Eliwa EM; Hassan AA; Nassar HS; El-Eisawy RA; Ismail M; Frese M; Shaaban M; El-Agrody AM; Bedair AH; Sewald N Synthesis, in Vitro Cytotoxicity Activity Against the Human Cervix Carcinoma Cell Line and in Silico Computational Predictions of New 4-Arylamino-3-Nitrocoumarin Analogues. J. Mol. Struct 2020, 1200, 127047. doi: 10.1016/j.molstruc.2019.127047. [DOI] [Google Scholar]
- [25].Bansal Y; Sethi P; Bansal G Coumarin: A Potential Nucleus for Anti-Inflammatory Molecules. Med. Chem. Res 2013, 22, 3049–3060. doi: 10.1007/s00044-012-0321-6. [DOI] [Google Scholar]
- [26].Raman N; Pravin N DNA Fastening and Ripping Actions of Novel Knoevenagel Condensed Dicarboxylic Acid Complexes in Antitumor Journey. Eur. J. Med. Chem 2014, 80, 57–70. doi: 10.1016/j.ejmech.2014.04.032. [DOI] [PubMed] [Google Scholar]
- [27].Biradar JS; Sasidhar BS Solvent-free, Microwave Assisted Knoevenagel Condensation of Novel 2,5-Disubstituted Indole Analogues and Their Biological Evaluation. Eur. J. Med. Chem 2011, 46, 6112–6118. doi: 10.1016/j.ejmech.2011.10.004. [DOI] [PubMed] [Google Scholar]
- [28].Ryabukhin SV; Plaskon AS; Volochnyuk DM; Pipko SE; Shivanyuk AN; Tolmachev AA Combinatorial Knoevenagel Reactions. J. Comb. Chem 2007, 9, 1073–1078. doi: 10.1021/cc070073f. [DOI] [PubMed] [Google Scholar]
- [29].Menegatti R Green Chemistry - Aspects for the Knoevenagel Reaction, In Green Chemistry Environmentally Benign Approaches, Kidwai M, Ed.; InTech, 2012, ISBN: 978-953-51-0334-9. [Google Scholar]
- [30].Senoo T; Inoue A; Mori K Facile Synthesis of π-Conjugated Heteroaromatic Compounds via Weak-Base-Promoted Transition-Metal-Free C-N Coupling. Synthesis. (Mass) 2020, 52. [Google Scholar]
- [31].Chikayuki Y; Miyashige T; Yonekawa S; Kirita A; Matsuo N; Teramoto H; Sasaki S; Higashiyama K; Yamauchi T Transition-Metal-Free Synthesis of Pyridine Derivatives by Thermal Cyclization of N-Propargyl Enamines. Synthesis. (Mass) 2020, 52, A–I. [Google Scholar]
- [32].Sahu PK; Sahu PK; Kaurav MS; Messali M; Almutairi SM; Sahu PL; Agarwal DD Metal-Free Construction of Fused Pyrimidines via Consecutive C-C and C-N Bond Formation in Water. ACS Omega 2018, 3, 15035–15042. doi: 10.1021/acsomega.8b01993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gupta PK; Hussain MK; Asad M; Kant R; Mahar R; Shukla SK; Hajela K A Metal-Free Tandem Approach to Prepare Structurally Diverse N-Heterocycles: Synthesis of 1,2,4-Oxadiazoles and Pyrimidinones. New J. Chem 2014, 38, 3062–3070. doi: 10.1039/c4nj00361f. [DOI] [Google Scholar]
- [34].Tietze LF Domino Reactions: Concepts for Efficient Organic Synthesis; Wiley-VCH, March 2014. ISBN: 978-3-527-33432-2. [Google Scholar]
- [35].Bruneau A; Roche M; Alami M; Messaoudi S 2-Aminobiphenyl Palladacycles: The “Most Powerful” Precatalysts in C-C and C-Heteroatom Cross-Couplings. ACS Catal. 2015, 5, 1386–1396. doi: 10.1021/cs502011x. [DOI] [Google Scholar]
- [36].Ivanov IC; Angelova VT; Vassilev N; Tiritiris I; Iliev B Synthesis of 4-Aminocoumarin Derivatives with N-Substitutents Containing Hydroxy or Amino Groups. Z. Naturforsch 2013, 68, 1031–1040. doi: 10.5560/ZNB.2013-3102. [DOI] [Google Scholar]
- [37].Sulaiman Al-Ayed A Synthesis of New Substituted Chromen[4,3-c]Pyrazol-4-Ones and Their Antioxidant Activities. Molecules 2011, 16, 10292–10302. doi: 10.3390/molecules161210292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xu Z; Ge J; Wang T; Luo T; Liu H; Yu X Highly Stereoselective Synthesis of 2-Aminobenzylidene Derivatives by a Convergent 3-Component Approach. Synlett. 2014, 25, 2913–2917. doi: 10.1055/s-0034-1378906. [DOI] [Google Scholar]
- [39].Vishnu S; Srikanth P; Tiwari AK; Kumar DA; Raju BC Carbohydrate Hydrolyzing Enzymes Inhibitory and Free Radical Scavenging Activities of Substituted 2H-Chromene-3-Carboxylates, 2H-Chromenyl Knoevenagel Derivatives and 6H-Chromenoquinolinones. Indian J. Chem 2015, 54B, 1332–1341. http://hdl.handle.net/123456789/33034. [Google Scholar]
- [40].Vasbinder MM; Alimzhanov M; Augustin M; Bebernitz G; Bell K; Chuaqui C; Deegan T; Ferguson AD; Goodwin K; Huszar D; Kawatkar A; Kawatkar S; Read J; Shi J; Steinbacher S; Steuber H; Su Q; Toader D; Wang H; Woessner R; Wu A; Ye M; Zinda M Identification of Azabenzimidazoles as Potent JAK1 Selective Inhibitors. Bioorg. Med. Chem. Lett 2016, 26, 60–67. doi: 10.1016/j.bmcl.2015.11.031. [DOI] [PubMed] [Google Scholar]
- [41].Song P; Peng P; Han M; Cao X; Ma X; Liu T;Zhou Y; Hu Y Design, Synthesis and Biological Evaluation of Thienopyridinones as Chk1 Inhibitors. Bioorg. Med. Chem 2014,22, 4882–4892. doi: 10.1016/j.bmc.2014.06.044. [DOI] [PubMed] [Google Scholar]
- [42].Pastor J; Oyarzabal J; Saluste G; Alvarez RM; Rivero V; Ramos F; Cendón E; Blanco-Aparicio C; Ajenjo N; Cebriá A; Albarrán MI; Cebrián D; Corrionero A; Fominaya J; Montoya G; Mazzorana M Hit to Lead Evaluation of 1,2,3-Triazolo[4,5-b]Pyridines as PIM Kinase Inhibitors. Bioorganic Med. Chem. Lett 2012, 22, 1591–1597. doi: 10.1016/j.bmcl.2011.12.130. [DOI] [PubMed] [Google Scholar]
- [43].Renhowe PA; Pecchi S; Shafer CM; Machajewski TD; Jazan EM; Taylor C; Antonios-McCrea W; McBride CM; Frazier K; Wiesmann M; Lapointe GR; Feucht PH; Warne RL; Heise CC; Menezes D; Aardalen K; Ye H; He M; Le V; Vora J; Jansen JM; Wernette-Hammond ME; Harris AL Design, Structure-Activity Relationships and in Vivo Characterization of 4-Amino-3-Benzimidazol-2-Ylhydroquinolin-2-Ones: A Novel Class of Receptor Tyrosine Kinase Inhibitors. J. Med. Chem 2009, 52, 278–292. doi: 10.1021/jm800790t. [DOI] [PubMed] [Google Scholar]
- [44].Silvaa VB; Orth ES Are Imidazoles Versatile or Promiscuous in Reactions with Organophosphates? Insights from the Case of Parathion. J. Braz. Chem. Soc 2019, 30,2114–2124. [Google Scholar]
- [45].Vyasamudri S; Yang D-Y Base-Dependent Divergent Annulation of 4-Chloro-3-Formyl-Coumarin and Tetrahydroisoquinoline: Application to the Synthesis of Isolamellarins and Hydroxypyrrolones. J. Org. Chem 2019, 84, 3662–3670. doi: 10.1021/acs.joc.8b03259. [DOI] [PubMed] [Google Scholar]
- [46].Angelova VT; Frey W; Ivanov IC; Vassilev N; Glasnov TN One-pot Synthesis of a Chromeno-[4,3,2-de]-1,6-Naphthyridine Derivative from 4-Chlorocoumarin-3-Carbaldehyde. Bulg. Chem. Commun 2014, 46, 53–58. [Google Scholar]
- [47].Shaabani A; Hooshmand SE Malononitrile Dimer as a Privileged Reactant in Design and Skeletal Diverse Synthesis of Heterocyclic Motifs. Mol. Diversity 2018, 22, 207–224. doi: 10.1007/s11030-017-9807-y. [DOI] [PubMed] [Google Scholar]
- [48].Yang Z; Lu N; Wei Z; Cao J; Liang D; Duan H; Lin Y Base-Promoted Intermolecular Cyclization of Substituted 3-Aryl(Heteroaryl)-3-Chloroacrylaldehydes and Tetrahydroisoquinolines: An Approach to Access Pyrrolo [2,1-a]Isoquinolines. J. Org. Chem 2016, 87, 11950–11955. doi: 10.1021/acs.joc.6b01781. [DOI] [PubMed] [Google Scholar]
- [49].Sammet B; Bogner T; Nahrwold M; Weiss C; Sewald N Approaches for the Synthesis of Functionalized Cryptophycins. J. Org. Chem 2010, 75, 6953–6960. doi: 10.1021/jo101563s. [DOI] [PubMed] [Google Scholar]
- [50].Shaaban KA; Elshahawi SI; Wang X; Horn J; Kharel MK; Leggas M; Thorson JS Cytotoxic Indolocarbazoles from Actinomadura Melliaura ATCC 39691. J. Nat. Prod 2015, 78, 1723–1729. doi: 10.1021/acs.jnatprod.5b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shaaban KA; Wang X; Elshahawi SI; Ponomareva LV; Sunkara M; Copley GC; Hower JC; Morris AJ; Kharel MK; Thorson JS; Herbimycins D-F Ansamycin Analogues from Streptomyces sp. RM-7–15. J. Nat. Prod 2013, 76, 1619–1626. doi: 10.1021/np400308w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang X; Shaaban KA; Elshahawi SI; Ponomareva LV; Sunkara M; Zhang Y; Copley GC; Hower JC; Morris AJ; Kharel MK; Thorson JS Frenolicins C-G, Pyranonaphthoquinones from Streptomyces sp. RM-4-15. J. Nat. Prod 2013, 76, 1441–1447. doi: 10.1021/np400231r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mokhtari RB; Homayouni TS; Baluch N; Morgatskaya E; Kumar S; Das B; Yeger H Combination Therapy in Combating Cancer. Oncotarget. 2017, 8, 38022–38043. doi: 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bai ZG; Zhang ZT A Systematic Review and Meta-Analysis on the Effect of Angiogenesis Blockade for the Treatment of Gastric Cancer. OncoTargets Ther 2018, 11, 7077–7087. doi: 10.2147/OTT.S169484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shaaban M; Yassin FY; Soltan MM Calamusins J-K: new Anti-Angiogenic Sesquiterpenes from Sarcophyton Glaucum. Nat. Prod. Res 2020. in press. [DOI] [PubMed] [Google Scholar]
- [56].Khan I; Hatiboglu MA Can COVID-19 Induce Glioma Tumorogenesis Through Binding Cell Receptors? Med. Hypotheses 2020, 144, 110009. doi: 10.1016/j.mehy.2020.110009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Daina A; Michielin O; Zoete V SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep 2017, 7, 42717–42730. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Arnott JA; Planey SL The Influence of Lipophilicity in Drug Discovery and Design. Expert Opin. Drug Discov 2012, 7,863–875. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- [59].Savjani KT; Gajjar AK; Savjani JK Drug Solubility: Importance and Enhancement Techniques. ISRN. Pharm 2012, 2012, 195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Daina A; Michielin O; Zoete V SwissTargetPrediction: Updated Data and new Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Vora C; Gupta S Targeted Therapy in Cervical Cancer. ESMO Open 2019, 3, e000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Batran RZ; Dawood DH; El-Seginy SA; Ali MM; Maher TJ; Gugnani KS; Rondon-Ortiz AN New Coumarin Derivatives as Anti-Breast and Anti-Cervical Cancer Agents Targeting VEGFR-2 and p38a MAPK. Arch. Pharm. Chem. Life Sci 2017,350, e1700064. doi: 10.1002/ardp.201700064. [DOI] [PubMed] [Google Scholar]
- [63].Hsu K; Chen Y; Lin S; Yang J iGEMDOCK: a Graphical Environment of Enhancing GEMDOCK Using Pharmacological Interactions and Post-Screening Analysis. BMC Bioinformatics 2011, 12, S33–S44. doi: 10.1126/science.1101637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pao W; Miller V; Zakowski M; Doherty J; Politi K; Sarkaria I; Singh B; Heelan R; Rusch V; Fulton L; Mardis E; Kupfer D; Wilson R; Kris M; Varmus H EGF Receptor Gene Mutations Are Common in Lung Cancers from “Never Smokers” and are Associated with Sensitivity of Tumors to Gefitinib and Erlotinib. Proc. Natl Acad. Sci. USA 2004, 101, 13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lynch TJ; Bell DW; Sordella R; Gurubhagavatula S; Okimoto RA; Brannigan BW; Harris PL; Haserlat SM; Supko JG; Haluska FG; Louis DN; Christiani DC; Settleman J; Haber DA Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med 2004, 350, 2129–2139. doi: 10.1056/nejmoa040938. [DOI] [PubMed] [Google Scholar]
- [66].Halgren TA Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem 1996, 17, 490–519. doi: 10.1002/(SICI)1096-987X(199604). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.