

Abstract
Asthma is a heterogeneous inflammatory airway disease that develops in response to a combination of genetic predisposition and environmental exposures. Patients with asthma are grouped into phenotypes with shared clinical features and biomarker profiles to help tailor specific therapies. However, factors driving development of specific phenotypes are poorly understood. Prenatal exposure to maternal asthma is a unique risk factor for childhood asthma. Here we tested whether maternal asthma skews asthma phenotypes in offspring. We compared airway hyperreactivity and inflammatory and neurotrophin lung signatures before and after allergen challenge in offspring born to mice exposed to house dust mite (HDM) or vehicle during pregnancy. Maternal HDM exposure potentiated offspring responses to HDM allergen, significantly increasing both airway hyperreactivity and airway eosinophilia compared with control mice. Maternal HDM exposure broadly skewed the offspring cytokine response from a classic allergen-induced T-helper cell type 2 (Th2)-predominant signature in HDM-treated offspring of vehicle-exposed mothers, toward a mixed Th17/Th1 phenotype in HDM-treated offspring of HDM-exposed mothers. Morphologic analysis determined that maternal HDM exposure also increased airway epithelial sensory nerve density and induced distinct neurotrophin signatures to support airway hyperinnervation. Our results demonstrate that maternal allergen exposure alters fetal lung development and promotes a unique inflammatory phenotype at baseline and in response to allergen that persists into adulthood.
Keywords: asthma, maternal asthma, airway hyperreactivity, eosinophil, sensory nerve
Asthma often begins in childhood and is characterized by reversible airflow obstruction, airway hyperreactivity, inflammation, and structural remodeling. Children born to parents with asthma have a greater risk of developing asthma themselves (1–3); however, shared genetics account for only part of this risk. Maternal asthma confers more risk than paternal asthma (4), and better asthma control during pregnancy reduces a child’s asthma risk (5), indicating that exposure to maternal factors in utero uniquely influences childhood asthma risk and is potentially modifiable.
One mechanism by which maternal asthma increases childhood asthma risk is by potentiating airway responses to inhaled allergens. Mice born to dams exposed to allergens during pregnancy have heightened immune responses and airway hyperreactivity after allergen sensitization later in life (6). Dams with elevated IL-5 during pregnancy similarly give birth to offspring with potentiated airway inflammation and airway hyperreactivity after allergen exposure (7). Notably, the mechanism of allergen-induced airway hyperreactivity in mice exposed to IL-5 in utero is distinct from control mice. Offspring born to dams with elevated IL-5 during pregnancy have sensory airway hyperinnervation with marked increases in substance P, a tachykinin that causes bronchoconstriction and increases immune cell recruitment and activation via its receptor NK1 (neurokinin-1) (8, 9). Mice born with airway hyperinnervation responded to nerve-targeted therapies to reduce allergen-induced airway hyperreactivity, whereas mice born to control dams did not, suggesting these biologic features may represent unique phenotypes.
Patients with asthma are broadly categorized into disease phenotypes based on shared clinical features and biomarkers (10). Type 2 high asthma is the most common phenotype and is marked by airway and blood eosinophilia; elevated T-helper cell type 2 (Th2)-related cytokines, including IL-5, IL-4, and IL-13; and, often, the presence of atopy. Several targeted therapies have been introduced against IL-5, IL-4, and IL-13 that have significantly improved exacerbation rates for patients with type 2 high asthma, reinforcing the key role of cytokines in this phenotype. In contrast, type 2 low (alternatively termed non–type 2 or non-Th2) asthma is characterized by low or absent airway eosinophilia, elevated IL-17, and frequently by steroid insensitivity (11). Less is known about mechanisms driving type 2 low asthma, which contributes to the lack of effective targeted therapies for this phenotype.
Inciting factors for development of unique asthma phenotypes are unclear. A few specific exposures are linked to development of an asthma phenotype, such as the association of allergen sensitization with development of early-onset asthma (12, 13). However, the role of most risk factors and early-life exposures in development of asthma phenotypes has not been defined. Identifying mechanisms driving phenotypic differentiation will improve treatment options, predict disease severity, and identify new modifiable factors that can alter disease trajectory.
In this study, we tested whether mice born to allergen-exposed dams exhibit unique inflammatory and physiologic phenotypes before and after allergen exposure. Some data from these studies have been previously reported as an abstract (14).
Methods
Mice
Male and female C57Bl/6J mice (Jackson Laboratories) 8 weeks of age and older were used for experiments. Mice were housed with ad libitum access to food and water on a 12-hour light and dark cycle. Oregon Health and Science University’s Institutional Animal Care and Use Committee approved all experimental protocols.
Maternal Allergen Sensitization and Challenge
Nulliparous female mice were sedated with 5% isoflurane and intranasally exposed to 25 μg house dust mite (HDM, dissolved in 25 μl PBS, LPS content 83,250 EU/vial or 3,857 EU/mg protein, Greer Laboratories, n = 5 mothers) or vehicle (VEH) (PBS, n = 5 mothers) for 5 consecutive days per week followed by 2 days of rest for 4 weeks total (Figure 1). At the end of the fourth week of allergen exposure, female mice were mated with nonsensitized wild-type male mice who had received no treatment. Female HDM exposure then resumed during gestation with intranasal delivery of 25 μg HDM or VEH (5 days per week, from protocol Week 5 until delivery). Intranasal delivery was performed using one-handed manual restraint without isoflurane sedation during pregnancy, because pilot studies demonstrated that daily isoflurane exposure caused fetal death. Once offspring were born, maternal exposures were stopped.
Figure 1.

Breeding scheme and house dust mite (HDM) exposure protocol. Female mice were exposed to HDM or vehicle (VEH) for 5 d/wk × 4 weeks before breeding, paired with male mice, and then exposed 5 d/wk until delivery. After reaching 8 weeks of age, offspring were sensitized to HDM or VEH on protocol Days 0 and 1 and then challenged with HDM on protocol Days 14–17. All experiments were conducted on protocol Day 18.
Offspring Allergen Sensitization and Challenge
Adult offspring mice were anesthetized with 5% isoflurane and sensitized intranasally with 50 μg HDM on Days 0 and 1, followed by challenge with 25 μg HDM daily on Days 14–17. Control mice received intranasal PBS (VEH). Measurement of airway physiology and inflammation were performed 24 hours after the final HDM challenge (Day 18).
Airway Physiology
Mice were sedated with xylazine (10 mg/kg i.p.) and ketamine (100 mg/kg i.p.), tracheotomized, and mechanically ventilated via a 21-gauge catheter. The ventilator system and settings have been previously described (7, 15). Airway resistance was measured at baseline and after increasing doses of inhaled serotonin as detailed previously (7) and in the data supplement.
Tissue Collection
Tissue collection is detailed in the data supplement.
Cytokine and Chemokine Analysis
RNA was extracted from whole lung using RNeasy Mini Kit (Qiagen). cDNA synthesis and real-time PCR were performed using the RT2 Profiler PCR Array system (Qiagen) according to manufacturer’s instructions and ABI-7500 (Applied Biosystems) real-time machine. Targets are detailed in the data supplement.
Epithelial Nerve Modeling
Epithelial nerve density was quantified using tissue optical clearing, confocal microscopy, and three-dimensional nerve modeling, as detailed previously (7, 15).
Statistical Analysis and Reproducibility
Data are graphed as mean ± SEM and analyzed using GraphPad Prism 7. Dose–response curves were analyzed with a repeated-measures two-way ANOVA. Lavage and nerve data were analyzed with two-way ANOVA with Bonferroni post hoc test. RNA data were analyzed with one-way ANOVA with Bonferroni correction for multiple comparisons and further detailed in the data supplement. Each experimental group contained animals derived from at least four separate litters and was limited to three littermates per treatment group to prevent overrepresentation of single pregnancies.
Results
Maternal HDM Exposure Increases Offspring Airway Hyperreactivity and Inflammation
Baseline airway resistance, measured before administering serotonin, was similar between offspring from HDM- and VEH-exposed mothers irrespective of offspring treatment group. In VEH-treated offspring from HDM-exposed (HDM/VEH) and VEH-exposed (VEH/VEH) mothers, bronchoconstriction to inhaled serotonin was also similar (Figure 2A). In contrast, acute HDM sensitization and challenge significantly increased airway reactivity in all offspring compared with VEH control mice (VEH/HDM vs. VEH/VEH; P = 0.0269; HDM/VEH vs. HDM/HDM; P = 0.0044). Acute HDM-induced airway hyperreactivity was further potentiated in offspring born to HDM-exposed mothers compared with offspring born to VEH-exposed mothers (HDM/HDM vs. VEH/HDM, P = 0.0384) (Figure 2A).
Figure 2.
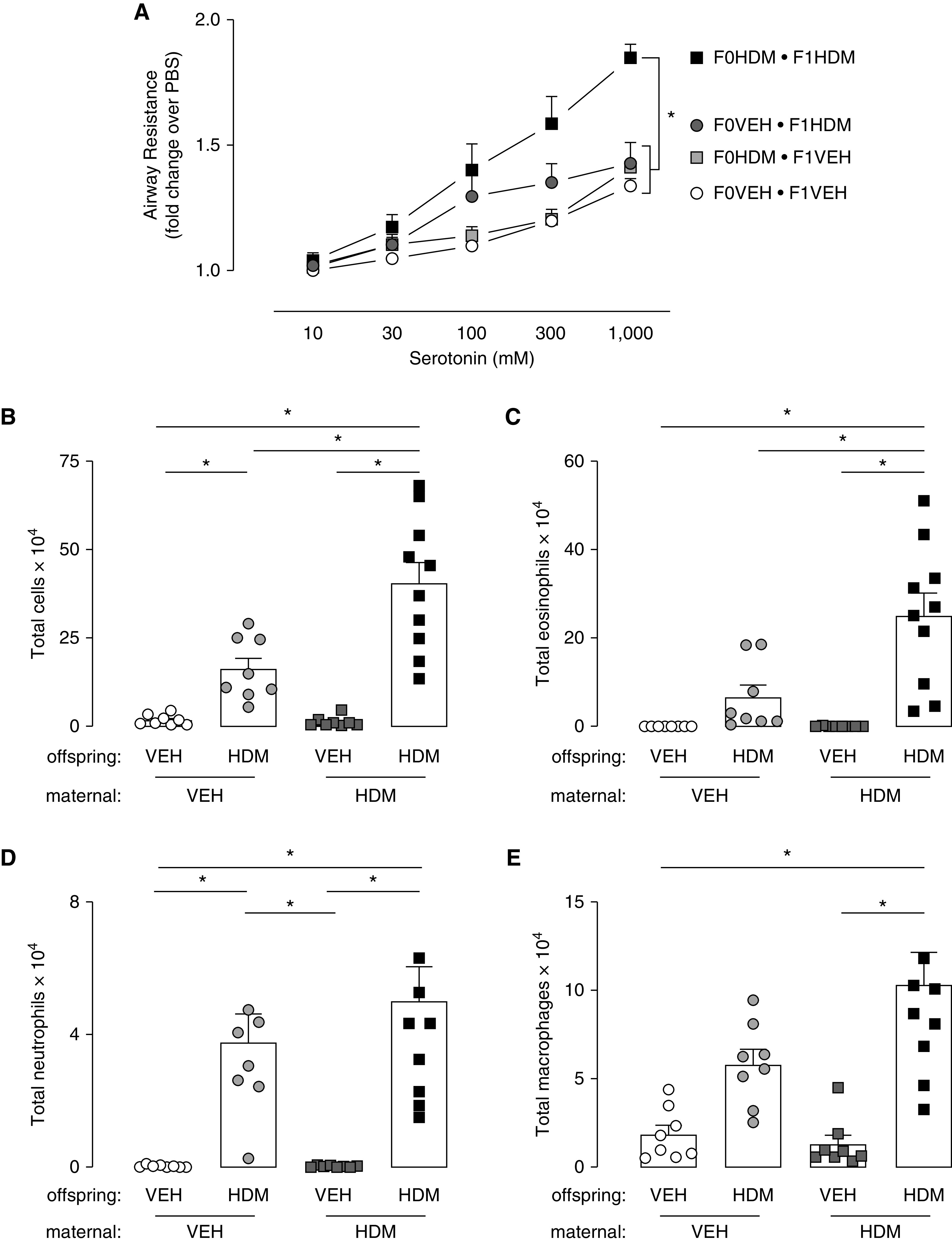
Maternal HDM exposure increased offspring airway hyperreactivity and inflammation. (A) Maternal HDM exposure did not change bronchoconstriction to inhaled serotonin in VEH-exposed offspring (VEH/VEH, open circles, n = 8; VEH/HDM, light gray squares, n = 8). Acute HDM sensitization and challenge caused airway hyperreactivity to inhaled serotonin in offspring compared with their VEH control mice independent of maternal treatment (VEH/VEH vs. VEH/HDM [dark gray circles, n = 8] and HDM/VEH vs. HDM/HDM [black squares, n = 10]). Allergen-induced airway hyperreactivity to serotonin was potentiated in offspring from HDM mothers (black squares) compared with VEH mothers (dark gray circles). (B) Airway inflammation increased in all offspring exposed to HDM compared with VEH control mice. (B and C) Maternal HDM exposure potentiated allergen-induced total (B) and eosinophilic (C) airway inflammatory cells in offspring. (D) Airway neutrophils and (E) macrophages increased in mice exposed to HDM compared to VEH control mice. *P < 0.05.
Acute HDM sensitization and challenge increased inflammatory cells in BAL fluid of offspring from both VEH-exposed (VEH/HDM) and HDM-exposed (HDM/HDM) mothers. However, HDM-treated offspring born to HDM-exposed mothers (HDM/HDM) had significantly potentiated inflammatory responses compared with HDM-treated offspring of VEH-exposed mothers (VEH/HDM), with more than three times as many eosinophils present in their airways (Figures 2B and 2C). To a lesser extent, airway macrophages and neutrophils were also increased (Figures 2D and 2E).
Maternal HDM Exposure Skews Lung Inflammatory Signature toward a Mixed Th17/Th1 Phenotype
Eighty chemokine and cytokine transcripts were measured in lungs of offspring from all treatment groups (Figure 3A). To determine effects of acute HDM exposure on lung inflammation, transcripts in HDM-treated offspring from VEH-exposed mothers (VEH/HDM) were compared with control mice (VEH/VEH) (Figure 3B). Twenty-one genes were upregulated by acute HDM treatment, corresponding to Th2 signaling (il4, il13, il21), T cell activation (cd70), Th1:Th2 polarization (il6), regulation of allergen sensitization (il1a), and chemoattractants that promote migration of eosinophils (ccl7, ccl11, ccl12, ccl24), neutrophils (cxcl1, cxcl3, cxcl5), B cells (cxcl13), macrophages (ccl2), and T lymphocytes (cxcl9, cxcl10, ccl1, ccl17) into lungs. Acute HDM also increased expression of il10, which promotes resolution of allergic airway inflammation, whereas genes that promote smooth muscle hypertrophy and airway remodeling (ctf1, tgfb2) and T-cell survival (cx3cl1) were suppressed. Two BMP (bone morphogenetic protein) transcripts were also decreased after acute HDM (bmp2, bmp6).
Figure 3.
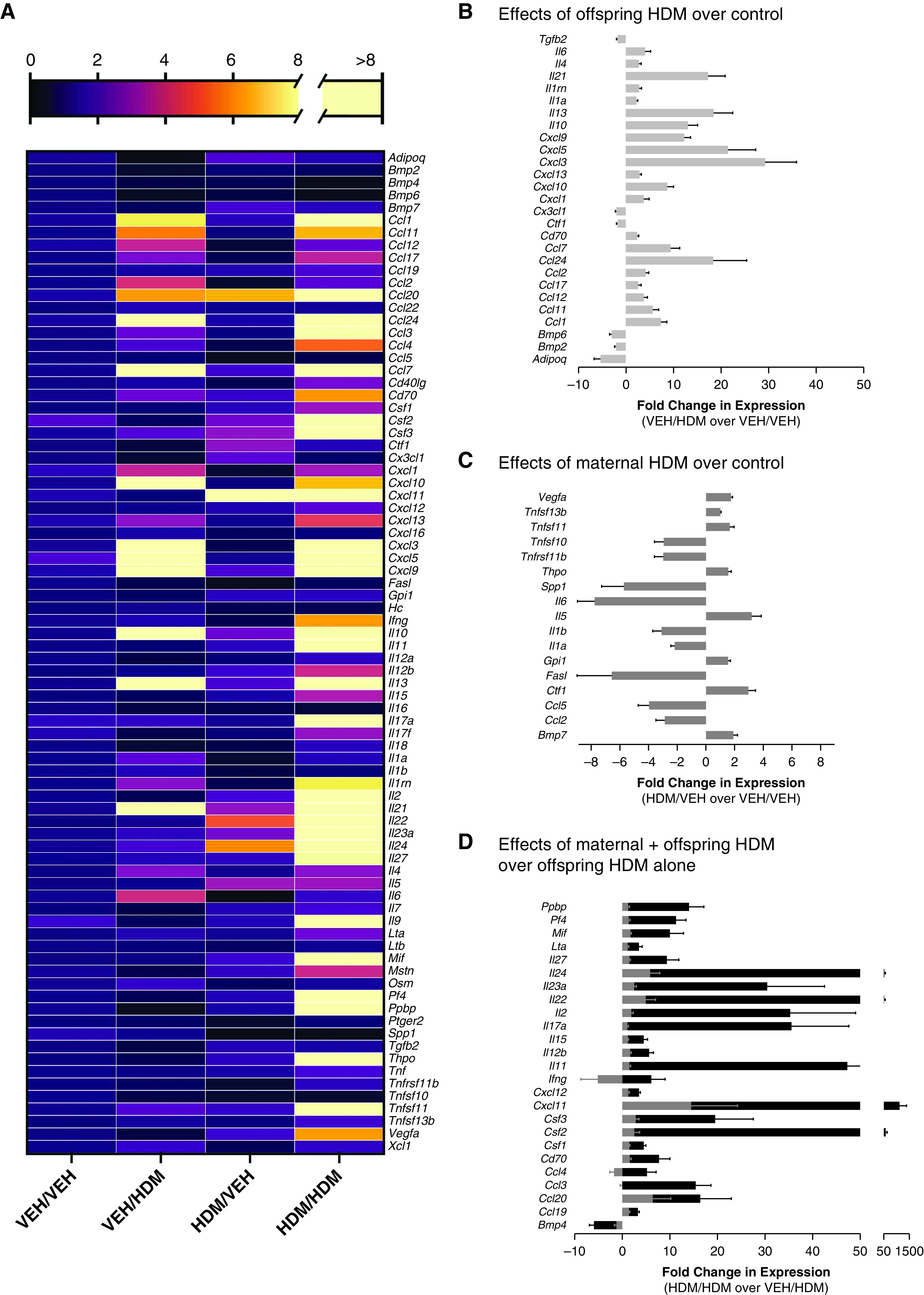
Allergen-induced inflammation signatures are unique in offspring from HDM-exposed mothers. (A) Fold change in expression of cytokines and chemokines using VEH-exposed offspring from VEH mothers (VEH/VEH) as baseline comparison group. (B) Acute HDM treatment increased expression of 21 genes and decreased expression of 6 genes in offspring from VEH-exposed mothers (VEH/HDM) compared with VEH-treated offspring from VEH-exposed mothers (VEH/VEH). (C) Maternal HDM exposure increased expression of eight genes and decreased expression of nine genes in VEH-treated offspring (HDM/VEH) compared with control mice (VEH/VEH). (D) Maternal HDM exposure potentiated expression of 25 genes in acute HDM-treated offspring (black bars, HDM/HDM) compared with VEH-treated offspring from HDM-exposed mothers (gray bars, HDM/VEH) and HDM-treated offspring from VEH-exposed mothers.
To test effects of maternal HDM exposure on lung inflammation at baseline, lung cytokine and chemokine transcripts in VEH-treated offspring from HDM-exposed mothers (HDM/VEH) were compared with control mice (VEH/VEH) (Figure 3C). Overall, 17 transcripts were differentially expressed after maternal HDM exposure, including increased expression of eight cytokines and chemokines involved in B-cell activation and proliferation (tnfsf13b), dendritic cell survival (tnfsf11 [tumor necrosis factor ligand superfamily member 11]), airway remodeling (vegfa, ctf1, bmp7), and thrombopoiesis (thpo). Nine transcripts were downregulated, including mediators involved in inflammatory cell apoptosis (tnfs10, fasl), Th1:Th2 polarization (il6) and allergen sensitization (il1a, il1b), dendritic cell survival (tnfrsf11b, the decoy receptor for tnfsf11), and lung development (spp1). Maternal HDM exposure had mixed effects on transcripts involved in inflammatory cell recruitment, both increasing (il5, vegfa) and decreasing (ccl5, ccl2) expression of cytokines/chemokines that promote eosinophil and macrophage infiltration into lungs.
To determine the effects of maternal HDM exposure on an offspring’s inflammatory response to acute HDM sensitization and challenge, lung cytokine and chemokine transcripts in HDM-treated offspring from HDM-exposed mothers (HDM/HDM) were compared with both VEH-treated offspring from HDM-exposed mothers (HDM/VEH) and HDM-treated offspring from VEH-exposed mothers (VEH/HDM) (Figure 3D). Overall, 25 genes were differentially regulated in HDM-treated offspring of HDM-exposed mothers (i.e., significantly different from both VEH/HDM and HDM/VEH), including a robust increase in gene transcripts promoting T-cell activation (cd70), survival (cd70, il15, il2), recruitment (cxcl12, cxcl11), and differentiation (il2); and a mixed Th1 (il12b, il27, ifng, ccl19), Th17 (il17a, il22, il23), and Th2 (il24, il11, pf4) polarization response. Gene transcripts promoting allergen sensitization (csf1); resistance to glucocorticoids (mif); IgE synthesis (lta); differentiation, survival, and recruitment of macrophages (csf1, ccl3, ccl4), dendritic cells (csf2, ccl19, ccl20, pf4), neutrophils (csf3, ccl3, ccl4, pf4, ppbp), and eosinophils (ccl3, ccl4) were also increased in lungs of HDM-treated offspring from HDM-exposed mothers. Only one gene, bmp4, was suppressed in HDM-treated offspring from HDM-exposed mothers.
Maternal HDM Exposure Increases Airway Innervation and Expression of Neurotrophic Receptors That Promote Sensory Nerve Growth and Survival
Three-dimensional models of substance P–expressing airway epithelial sensory nerves were generated from confocal image z-stacks to test whether maternal HDM exposure affected airway nerve morphology in offspring (Figure 4A). Epithelial sensory nerve density was significantly increased in offspring from HDM-exposed mothers compared with offspring from VEH-exposed mothers (Figures 4A–4C). Acute HDM sensitization and challenge in offspring had no effect on epithelial nerve density.
Figure 4.
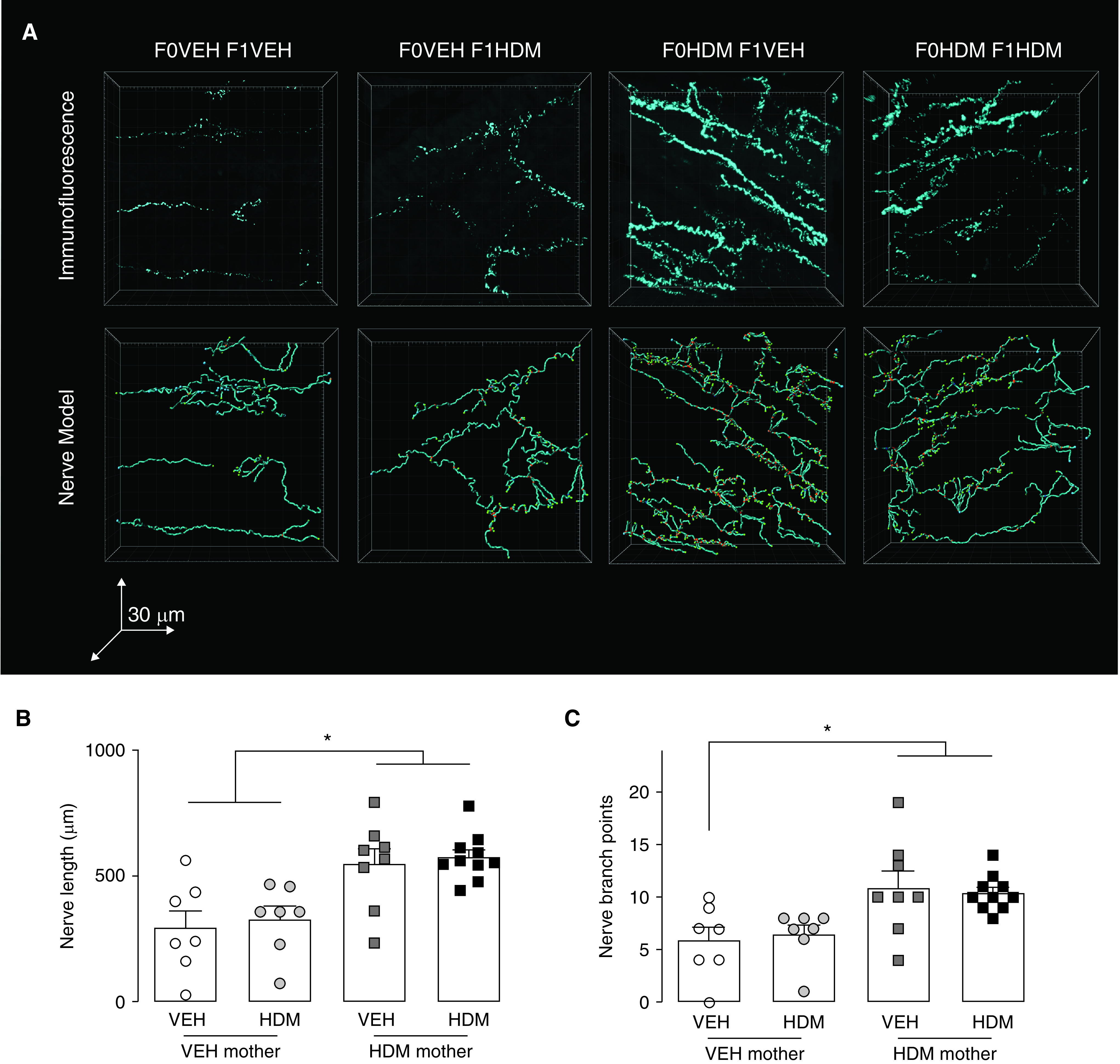
Maternal HDM exposure increased offspring airway sensory innervation. (A) Substance P–positive epithelial nerves in optically cleared airways (top) and three-dimensional computer models for quantification of nerve morphology (bottom). (B and C) Maternal HDM exposure increased offspring airway substance P–positive nerve length (B) and branching (C). Nerve length and branch points did not increase further with offspring HDM exposure. *P < 0.05.
Gene expression of neuropeptides, neurotrophins, and their respective receptors and downstream signaling pathways were measured in whole lungs of all offspring to identify potential mediators involved in development and/or maintenance of airway sensory hyperinnervation (Figure 5A). In acute HDM-treated offspring from VEH-exposed mothers (VEH/HDM), only expression of NK1 (tacr1), a substance P receptor, was increased in whole lung compared with control mice (VEH/VEH), whereas transcripts that promote neuronal proliferation (fgf2), sensory axon growth (nrg4), sensory nerve survival and regeneration after injury (lif, stat3, cntfr, mc2r), parasympathetic nerve development (gfra1), and Schwann cell differentiation and peripheral nerve myelination (fgf9) were all decreased (Figure 5B).
Figure 5.
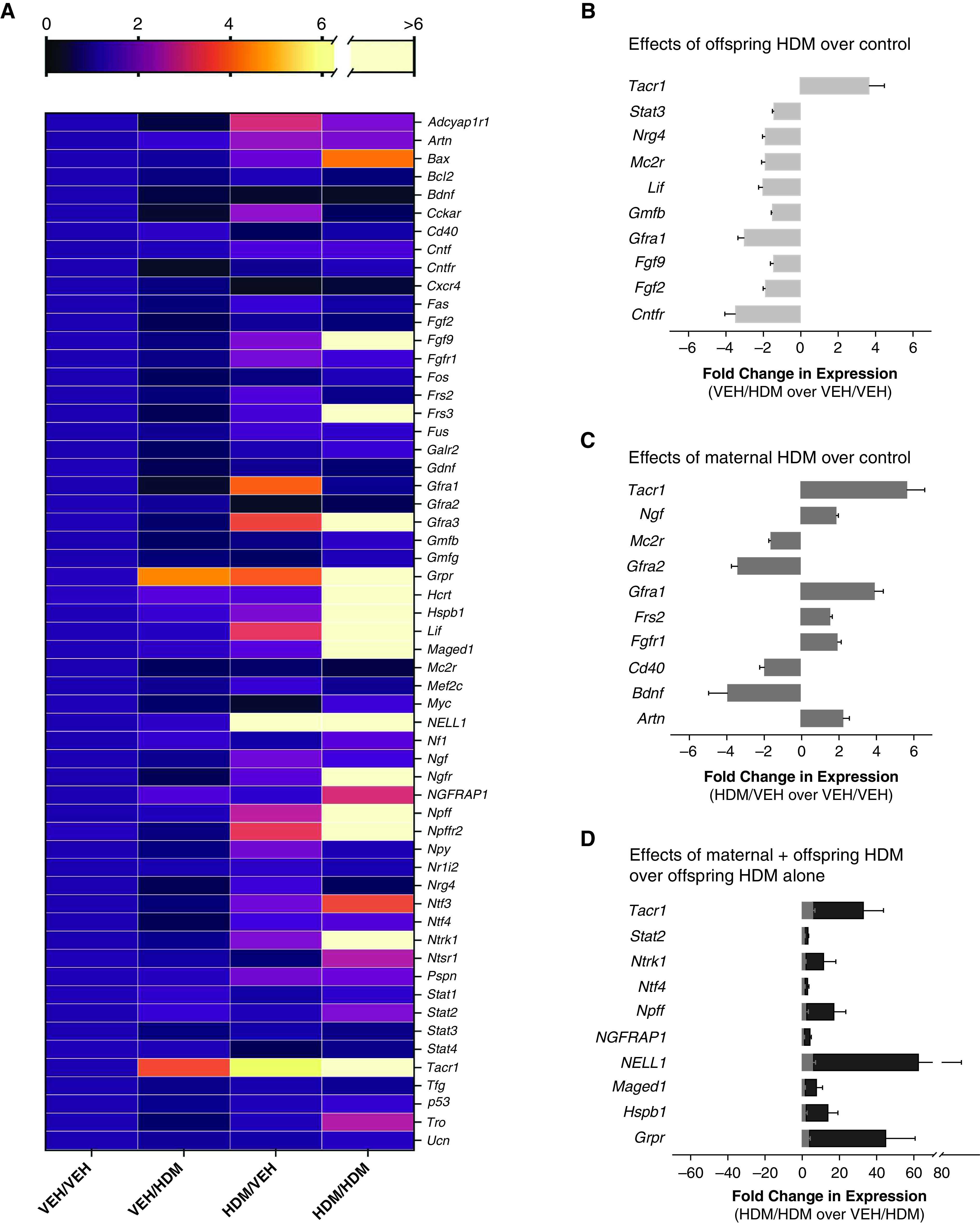
Allergen-induced neurotrophin signatures are unique in offspring from HDM-exposed mothers. (A) Fold change in expression of neuropeptides, neurotrophins, neurotrophin receptors, and modulating proteins using VEH-treated offspring from VEH-exposed mothers (VEH/VEH) as the baseline comparison group. (B) Acute HDM increased tacr1 expression and suppressed expression of nine genes in offspring from VEH-exposed mothers (VEH/HDM) compared with VEH-treated offspring from VEH-exposed mothers (VEH/VEH). (C) Maternal HDM exposure increased expression of six genes and decreased expression of four genes in VEH-treated offspring (HDM/VEH) compared with VEH-treated offspring from VEH-exposed mothers (VEH/VEH). (D) Maternal HDM exposure potentiated expression of 10 genes in HDM-treated offspring (black bars, HDM/HDM) compared with VEH-treated offspring from HDM-exposed mothers (gray bars, HDM/VEH) and HDM-treated offspring from VEH-exposed mothers.
Next, VEH-treated offspring from HDM-exposed mothers (HDM/VEH) were compared with control mice (VEH/VEH) to determine the effect of maternal HDM exposure on baseline neuropeptide and neurotrophin expression (Figure 5C). Similar to the effects of acute HDM, maternal HDM exposure increased RNA for NK1 receptors (tacr1), whereas expression of neurotrophins that promote sensory nerve growth and differentiation (ngf [nerve growth factor], artn, frs2, fgfr1) and parasympathetic nerve development (gfra1) were also increased. Maternal HDM exposure decreased expression of transcripts involved in sensory neuron repair (mc2r) and parasympathetic nerve survival (gfra2).
Last, acute HDM-treated offspring from HDM-exposed mothers (HDM/HDM) were compared with both VEH-treated offspring from HDM-exposed mothers (HDM/VEH) and to HDM-treated offspring from VEH-exposed mothers (VEH/HDM) to determine the effects of maternal HDM exposure on neuropeptide and neurotrophin expression after acute HDM sensitization and challenge in offspring (Figure 5D). Overall, 10 genes were differentially regulated in HDM-treated offspring of HDM-exposed mothers (i.e., significantly different from both VEH/HDM and HDM/VEH). Maternal HDM exposure significantly potentiated allergen-induced expression of NK1 receptors (tacr1) in HDM-treated offspring compared with VEH-treated offspring of HDM-exposed mothers (HDM/VEH) and HDM-treated offspring of VEH-exposed mothers (VEH/HDM). The combination of maternal HDM exposure and offspring HDM treatment also increased expression of transcripts that promote sensory nerve growth and survival (ntrk1, ntf4, ngfrap1), sensory neuron excitability (npff, grpr), and apoptosis (maged1).
Discussion
We have shown that airway inflammation and hyperreactivity induced by allergen sensitization and challenge is greatly increased in mice born to dams who were exposed to HDM exposure before and during pregnancy. We propose that potentiated airway inflammation and airway hyperreactivity in mice born to allergen-exposed dams represents a unique phenotype with distinct inflammatory and neurotrophic signatures in lungs.
Acute HDM exposure in offspring from VEH-exposed mothers led to a classic Th2-polarized inflammatory signature in the lung characterized by increased expression of chemokines involved in inflammatory cell recruitment in allergic asthma. In contrast, acute HDM exposure in offspring from HDM-exposed mothers caused a marked increase in cytokines and chemokines associated with mixed Th1/Th17 inflammation. In humans, mixed inflammatory asthma phenotypes are often associated with more severe disease, worse asthma control, and lower lung function (16). Th17 cells play a causal role in steroid-resistant inflammation and airway hyperreactivity in animal models of asthma (17), whereas elevated IL-17 directly correlates with more severe, steroid-resistant asthma in children (18). Skewing toward Th17 may be caused by increased expression of MIF (macrophage migration inhibitory factor) (mif), which increased after HDM exposure only in offspring from HDM-exposed mothers. MIF is a potent inducer of IL-17 secretion (19), as well as a strong countersignal that overcomes inhibitory effects of glucocorticoids (20). Our data suggest uncontrolled maternal asthma during pregnancy may increase the risk of severe asthma in children by skewing lung inflammation toward a steroid-resistant, Th1/Th17 phenotype.
Th2 and Th17 signaling reciprocally regulate each other (21, 22), with IL-13 suppressing IL-17 secretion and IL-17–related gene expression. Although Th2 and Th17 inflammatory signatures may be mutually exclusive (21), both have marked eosinophilia.
We found that despite a predominance of Th1/Th17 and few Th2 transcripts after HDM exposure in offspring from HDM mothers, these mice had significant eosinophilic airway inflammation. Eosinophils have multiple roles in asthma, including that they increase neurotransmitter release from parasympathetic nerves by blocking neuronal, inhibitory M2 muscarinic receptors (23–25) and are associated with hyperinnervation in human asthma (15). Multiple cytokines and chemokines induce eosinophil chemotaxis, including IL-5, CCL3, CCL4, CCL5, CCL7, CCL11 (eotaxin-1), CCL12, CCL13, CCL24 (eotaxin-2), and CCL26 (eotaxin-3). Acute HDM exposure in offspring from VEH-exposed mothers increased expression of ccl7, ccl11, ccl12, and ccl24, whereas acute HDM exposure in offspring from HDM-exposed mothers increased expression of ccl3 and ccl4, suggesting recruitment signals for eosinophils in Th2 versus Th1/Th17 inflammatory phenotypes may be distinct and that currently available anti-IL5–targeted treatments alone may be insufficient for reducing eosinophilic inflammation in this setting.
Expression of key regulators of dendritic cell survival and T cell activation may contribute to the potentiated immune response and Th1/Th17 phenotype observed in HDM-treated offspring from HDM-exposed mothers. Dendritic cells harvested from offspring of allergen-exposed mothers have increased antigen presentation capacity and increase offspring airway hyperreactivity and inflammation in adoptive transfer experiments (26), demonstrating that dendritic cells are important mediators in transmission of maternal-to-childhood asthma risk. In our study, maternal HDM exposure increased expression of tnfsf11 (also known as RANKL and TRANCE) in lungs of VEH-treated offspring. T cells express TNFSF11, a membrane-bound molecule that binds its cognate receptor on dendritic cells, ultimately promoting dendritic cell survival and serving as a feed-forward signal to amplify T-cell activation (27). Dendritic cells exposed to TNFSF11 secrete IL-12, which then directs Th1 polarization (28), and reverse signaling through the TNFSF11-receptor complex augments IFN-γ secretion by T cells (29). A soluble decoy receptor (tnfrsf11b, also known as osteoprotegerin) blocks the interaction of TNFSF11 with its receptor, and knockout of TNFSF11b promotes dendritic cell secretion of IL-12 and IL-23 (30). Expression of TNFRSF11b was suppressed by maternal HDM, presumably potentiating the effects of TNFSF11. The importance of this pathway is evident, as exogenous TNFRSF11b can inhibit allergen-induced airway inflammation in mouse models (31). Similarly, fasl expression was suppressed by maternal HDM exposure, which opposes TNFSF11-induced dendritic cell survival (32), demonstrating a likely net effect of maternal HDM exposure on promoting dendritic cell survival, antigen presentation, and T-cell activation that results in potentiated and skewed inflammatory responses after HDM exposure in offspring.
We also demonstrate that maternal HDM exposure increases airway epithelial substance P–positive sensory innervation in offspring that persists into adulthood. Increased epithelial innervation alone did not increase reflex bronchoconstriction at baseline, as VEH-treated offspring from HDM-exposed mothers had similar serotonin-induced bronchoconstriction as VEH-treated offspring from VEH-exposed mothers. It is likely hyperinnervation at baseline increases offspring susceptibility to potentiated airway hyperreactivity after allergen exposure by increased neuronal sensory field. Airway nerves are required for allergen-induced hyperreactivity (33), yet sensory nerve density did not further change after acute HDM exposure in adult offspring, suggesting that potentiated allergen-induced airway hyperreactivity in HDM-treated offspring from HDM-exposed mothers depended on preexisting neurons with either increased responsiveness to depolarizing stimuli or increased neurotransmitter expression. Similarly enhanced eosinophil recruitment to parasympathetic nerves and subsequently increased neurotransmitter release by blocking neuronal M2 muscarinic receptors (23, 34) may also contribute to the additive effects of acute antigen challenge and preexisting hyperinnervation on airway hyperreactivity.
Maternal HDM exposure potentiated allergen-induced expression of NK1 receptors (tacr1) in offspring. In mice with airway hyperinnervation due to maternal IL-5 exposure in utero (7), allergen-induced airway hyperreactivity was blocked by an NK1 antagonist, whereas in mice born to control dams, NK1 antagonism worsened airway hyperreactivity. Potentiated expression of NK1 receptors in our current study may explain why NK1 antagonists effectively reduce airway hyperreactivity in offspring with hyperinnervation due to in utero IL-5 exposure. HDM exposure in offspring from HDM mothers also increased expression of high-affinity NGF TrkA (tropomyosin receptor kinase A) receptor (ntrk1) as well as Bex3 (ngfrap1), a protein that regulates TrkA expression and is a signaling adaptor molecule for NGF p75NTR receptors, and for MAGED1, which controls NGF-p75NTR apoptosis signaling. Overexpression of NGF in airway epithelium results in sensory hyperinnervation and increases airway reactivity independently of allergen exposure in mice (35). Although increased expression of NGF receptors after acute HDM exposure in offspring is not associated with changed nerve density, it is possible the short time frame for acute HDM treatment is too brief to capture morphologic changes in nerve structure. Alternatively, neurons exposed to NGF have increased sensitivity to noxious stimuli, leading to airway hyperreactivity via a TrkA-dependent mechanism (36, 37).
Multiple neurotrophic factors increase airway innervation both during development and after early-life lung insults, including NGF, GDNF, and NT4 (neurotrophin 4) (35, 38, 39). Allergen exposure increases expression of most neurotrophic factors, and these are elevated in lavage samples from patients with asthma (40, 41). We previously demonstrated that exposure to maternal IL-5 during development increases lavage NGF, but not GDNF (glial cell–derived neurotrophic factor), BDNF (brain-derived neurotrophic factor), or NT4 (7), and here tested whether maternal HDM exposure increases neurotrophin expression. Similar to offspring exposed to maternal IL-5, offspring from HDM mothers had increased expression of ngf. Maternal HDM exposure also increased expression of intracellular adaptor protein frs2, which recruits protein complexes required for NGF signaling and, when combined with changes in TrkA (ntrk1), Bex3 (ngfrap1), and MAGED1, suggests the NGF signaling axis was uniquely and globally susceptible to maternal HDM exposure. Although we did not identify a source of increased NGF expression in offspring from HDM mothers, airway epithelium has previously been identified as a predominant source of NGF in inflamed lungs; thus, epithelial NGF likely contributes to airway hyperinnervation seen in offspring from HDM mothers. NGF, however, is not the only possible driver of airway hyperinnervation seen in these mice, as artn (artemin), a neurotrophic molecule that supports sensory neuron growth and increases afferent sensory nerve excitability, was also increased in VEH-treated offspring from HDM-exposed mothers.
We analyzed whole-lung transcripts. Although this permitted a more comprehensive assessment of lung chemokine and cytokine phenotypes, we are unable to identify specific cell types or lung niches contributing to expression patterns. Similarly, although we discussed chemokine and neurotrophic expression separately, neuro- and immune signaling axes in lungs are intimately associated and regulate each other. We also extrapolated potential effects from observed changes in transcript expression; however, many transcripts are subject to post-translational processing, and thus increased gene expression may not correlate with increased protein function.
Here we show that offspring born to allergen-exposed mothers develop unique inflammatory and neurotrophic lung transcript signatures with heightened airway hyperreactivity and inflammation after allergen challenge later in life. Inflammatory responses in offspring from HDM-exposed mothers skewed toward a mixed Th17/Th1 phenotype with prominent eosinophilia, as opposed to classic Th2-polarized inflammation found in control offspring. Our data suggest maternal asthma may change underlying mechanisms of airway inflammation in childhood asthma and that children born to mothers with asthma may not respond to currently available Th2-targeted therapies. Maternal HDM exposure also caused airway hyperinnervation in offspring at baseline, with increased expression of NGF, TrkA, and NK1 receptors and airway hyperreactivity after allergen challenge. Children with airway hyperinnervation born to mothers with asthma may therefore benefit from nerve-targeted therapies, such as NK1 antagonists or anticholinergic drugs, whereas children with asthma without airway hyperinnervation may not. In total, our data suggest exacerbations of maternal asthma during pregnancy is a modifiable early-life exposure that both worsens features of asthma (e.g.. airway reactivity and inflammation) and promotes phenotypic differentiation in children with asthma.
Acknowledgments
Acknowledgment
The authors thank Lauren Hales-Beck for technical help.
Footnotes
Supported by the National Institutes of Health National Heart, Lung, and Blood Institute grants HL132414 (K.M.L.), HL124165 (D.B.J.), HL121254 (M.G.D.), HL144008 (D.B.J.), and HL155623 (M.G.D.).
Author Contributions: K.M.L. and D.B.J. conceived of the study. K.M.L., M.G.D., A.B. Pincus, A.D.F., and D.B.J. designed experiments. K.M.L., M.G.D., A.B. Pincus, and A.B. Pierce performed experiments and analyzed data. All authors contributed to writing and editing the manuscript.
All data are available from the corresponding author upon reasonable request.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0535OC on April 1, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Kelly YJ, Brabin BJ, Milligan P, Heaf DP, Reid J, Pearson MG. Maternal asthma, premature birth, and the risk of respiratory morbidity in schoolchildren in Merseyside. Thorax . 1995;50:525–530. doi: 10.1136/thx.50.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paaso EM, Jaakkola MS, Rantala AK, Hugg TT, Jaakkola JJ. Allergic diseases and asthma in the family predict the persistence and onset-age of asthma: a prospective cohort study. Respir Res . 2014;15:152. doi: 10.1186/s12931-014-0152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Young S, Le Souëf PN, Geelhoed GC, Stick SM, Turner KJ, Landau LI. The influence of a family history of asthma and parental smoking on airway responsiveness in early infancy. N Engl J Med . 1991;324:1168–1173. doi: 10.1056/NEJM199104253241704. [DOI] [PubMed] [Google Scholar]
- 4. Lim RH, Kobzik L, Dahl M. Risk for asthma in offspring of asthmatic mothers versus fathers: a meta-analysis. PLoS One . 2010;5:e10134. doi: 10.1371/journal.pone.0010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morten M, Collison A, Murphy VE, Barker D, Oldmeadow C, Attia J, et al. Managing Asthma in Pregnancy (MAP) trial: FENO levels and childhood asthma. J Allergy Clin Immunol . 2018;142:1765–1772.e4. doi: 10.1016/j.jaci.2018.02.039. [DOI] [PubMed] [Google Scholar]
- 6. Richgels PK, Yamani A, Chougnet CA, Lewkowich IP. Maternal house dust mite exposure during pregnancy enhances severity of house dust mite-induced asthma in murine offspring. J Allergy Clin Immunol . 2017;140:1404–1415.e9. doi: 10.1016/j.jaci.2016.12.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lebold KM, Drake MG, Hales-Beck LB, Fryer AD, Jacoby DB. IL-5 exposure in utero increases lung nerve density and airway reactivity in adult offspring. Am J Respir Cell Mol Biol . 2020;62:493–502. doi: 10.1165/rcmb.2019-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Evans CM, Belmonte KE, Costello RW, Jacoby DB, Gleich GJ, Fryer AD. Substance P-induced airway hyperreactivity is mediated by neuronal M(2) receptor dysfunction. Am J Physiol Lung Cell Mol Physiol . 2000;279:L477–L486. doi: 10.1152/ajplung.2000.279.3.L477. [DOI] [PubMed] [Google Scholar]
- 9. Mauser PJ, Skeans S, Ritacco G, Fernandez X, House A, Chapman RW. Effect of tachykinins on airway function in cynomolgus monkeys. Pulm Pharmacol Ther . 2001;14:121–127. doi: 10.1006/pupt.2001.0278. [DOI] [PubMed] [Google Scholar]
- 10. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med . 2012;18:716–725. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 11. Sze E, Bhalla A, Nair P. Mechanisms and therapeutic strategies for non-T2 asthma. Allergy . 2020;75:311–325. doi: 10.1111/all.13985. [DOI] [PubMed] [Google Scholar]
- 12. Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, et al. National Heart, Lung, and Blood Institute’s Severe Asthma Research Program Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med . 2010;181:315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med . 2008;178:218–224. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pincus A, Fryer A, Jacoby D, Lebold K. Maternal allergen exposure increases offspring sensory airway innervation and airway hyperreactivity in mice (abstract) Eur Respir J . 2018;52:PA1380. [Google Scholar]
- 15. Drake MG, Scott GD, Blum ED, Lebold KM, Nie Z, Lee JJ, et al. Eosinophils increase airway sensory nerve density in mice and in human asthma. Sci Transl Med . 2018;10:eaar8477. doi: 10.1126/scitranslmed.aar8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, et al. National Heart, Lung, and Blood Institute Severe Asthma Research Program Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol . 2010;125:1028–1036.e13. doi: 10.1016/j.jaci.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol . 2008;181:4089–4097. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chien JW, Lin CY, Yang KD, Lin CH, Kao JK, Tsai YG. Increased IL-17A secreting CD4+ T cells, serum IL-17 levels and exhaled nitric oxide are correlated with childhood asthma severity. Clin Exp Allergy . 2013;43:1018–1026. doi: 10.1111/cea.12119. [DOI] [PubMed] [Google Scholar]
- 19. Stojanović I, Cvjetićanin T, Lazaroski S, Stosić-Grujicić S, Miljković D. Macrophage migration inhibitory factor stimulates interleukin-17 expression and production in lymph node cells. Immunology . 2009;126:74–83. doi: 10.1111/j.1365-2567.2008.02879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Calandra T, Bucala R. Macrophage migration inhibitory factor (MIF): a glucocorticoid counter-regulator within the immune system. Crit Rev Immunol . 2017;37:359–370. doi: 10.1615/CritRevImmunol.v37.i2-6.90. [DOI] [PubMed] [Google Scholar]
- 21. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med . 2015;7:301ra129. doi: 10.1126/scitranslmed.aab3142. [DOI] [PubMed] [Google Scholar]
- 22. Newcomb DC, Boswell MG, Zhou W, Huckabee MM, Goleniewska K, Sevin CM, et al. Human TH17 cells express a functional IL-13 receptor and IL-13 attenuates IL-17A production. J Allergy Clin Immunol . 2011;127:1006–1013. doi: 10.1016/j.jaci.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans CM, Fryer AD, Jacoby DB, Gleich GJ, Costello RW. Pretreatment with antibody to eosinophil major basic protein prevents hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. J Clin Invest . 1997;100:2254–2262. doi: 10.1172/JCI119763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fryer AD, Wills-Karp M. Dysfunction of M2-muscarinic receptors in pulmonary parasympathetic nerves after antigen challenge. J Appl Physiol (1985) . 1991;71:2255–2261. doi: 10.1152/jappl.1991.71.6.2255. [DOI] [PubMed] [Google Scholar]
- 25. Jacoby DB, Gleich GJ, Fryer AD. Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor. J Clin Invest . 1993;91:1314–1318. doi: 10.1172/JCI116331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fedulov AV, Kobzik L. Allergy risk is mediated by dendritic cells with congenital epigenetic changes. Am J Respir Cell Mol Biol . 2011;44:285–292. doi: 10.1165/rcmb.2009-0400OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Josien R, Wong BR, Li HL, Steinman RM, Choi Y. TRANCE, a TNF family member, is differentially expressed on T cell subsets and induces cytokine production in dendritic cells. J Immunol . 1999;162:2562–2568. [PubMed] [Google Scholar]
- 28. Dong C, Flavell RA. Cell fate decision: T-helper 1 and 2 subsets in immune responses. Arthritis Res . 2000;2:179–188. doi: 10.1186/ar85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen NJ, Huang MW, Hsieh SL. Enhanced secretion of IFN-gamma by activated Th1 cells occurs via reverse signaling through TNF-related activation-induced cytokine. J Immunol . 2001;166:270–276. doi: 10.4049/jimmunol.166.1.270. [DOI] [PubMed] [Google Scholar]
- 30. Chino T, Draves KE, Clark EA. Regulation of dendritic cell survival and cytokine production by osteoprotegerin. J Leukoc Biol . 2009;86:933–940. doi: 10.1189/jlb.0708419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregorczyk I, Maślanka T. Blockade of RANKL/RANK and NF-ĸB signalling pathways as novel therapeutic strategies for allergic asthma: a comparative study in a mouse model of allergic airway inflammation. Eur J Pharmacol . 2020;879:173129. doi: 10.1016/j.ejphar.2020.173129. [DOI] [PubMed] [Google Scholar]
- 32. Chen A, Xu H, Choi Y, Wang B, Zheng G. TRANCE counteracts FasL-mediated apoptosis of murine bone marrow-derived dendritic cells. Cell Immunol . 2004;231:40–48. doi: 10.1016/j.cellimm.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 33. Tränkner D, Hahne N, Sugino K, Hoon MA, Zuker C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc Natl Acad Sci USA . 2014;111:11515–11520. doi: 10.1073/pnas.1411032111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Costello RW, Schofield BH, Kephart GM, Gleich GJ, Jacoby DB, Fryer AD. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am J Physiol . 1997;273:L93–L103. doi: 10.1152/ajplung.1997.273.1.L93. [DOI] [PubMed] [Google Scholar]
- 35. Hoyle GW, Graham RM, Finkelstein JB, Nguyen KP, Gozal D, Friedman M. Hyperinnervation of the airways in transgenic mice overexpressing nerve growth factor. Am J Respir Cell Mol Biol . 1998;18:149–157. doi: 10.1165/ajrcmb.18.2.2803m. [DOI] [PubMed] [Google Scholar]
- 36. de Vries A, Engels F, Henricks PA, Leusink-Muis T, McGregor GP, Braun A, et al. Airway hyper-responsiveness in allergic asthma in guinea-pigs is mediated by nerve growth factor via the induction of substance P: a potential role for trkA. Clin Exp Allergy . 2006;36:1192–1200. doi: 10.1111/j.1365-2222.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 37. El-Hashim AZ, Jaffal SM. Nerve growth factor enhances cough and airway obstruction via TrkA receptor- and TRPV1-dependent mechanisms. Thorax . 2009;64:791–797. doi: 10.1136/thx.2009.113183. [DOI] [PubMed] [Google Scholar]
- 38. Patel KR, Aven L, Shao F, Krishnamoorthy N, Duvall MG, Levy BD, et al. Mast cell-derived neurotrophin 4 mediates allergen-induced airway hyperinnervation in early life. Mucosal Immunol . 2016;9:1466–1476. doi: 10.1038/mi.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tollet J, Everett AW, Sparrow MP. Development of neural tissue and airway smooth muscle in fetal mouse lung explants: a role for glial-derived neurotrophic factor in lung innervation. Am J Respir Cell Mol Biol . 2002;26:420–429. doi: 10.1165/ajrcmb.26.4.4713. [DOI] [PubMed] [Google Scholar]
- 40. Virchow JC, Julius P, Lommatzsch M, Luttmann W, Renz H, Braun A. Neurotrophins are increased in bronchoalveolar lavage fluid after segmental allergen provocation. Am J Respir Crit Care Med . 1998;158:2002–2005. doi: 10.1164/ajrccm.158.6.9803023. [DOI] [PubMed] [Google Scholar]
- 41. Noga O, Hanf G, Schäper C, O’Connor A, Kunkel G. The influence of inhalative corticosteroids on circulating nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3 in allergic asthmatics. Clin Exp Allergy . 2001;31:1906–1912. doi: 10.1046/j.1365-2222.2001.01249.x. [DOI] [PubMed] [Google Scholar]