

Abstract
Idiopathic pulmonary fibrosis (IPF) is the most common chronic interstitial lung disease and is characterized by progressive scarring of the lung. Transforming growth factor-β (TGF-β) signaling plays an essential role in IPF and drives fibroblast to myofibroblast transition (FMT). Dedicator of cytokinesis 2 (DOCK2) is known to regulate diverse immune functions by activating Rac and has been recently implicated in pleural fibrosis. We now report a novel role of DOCK2 in pulmonary fibrosis development by mediating FMT. In primary normal and IPF human lung fibroblasts (HLFs), TGF-β induced DOCK2 expression concurrent with FMT markers, smooth muscle α-actin (α-SMA), collagen-1, and fibronectin. Knockdown of DOCK2 significantly attenuated TGF-β-induced expression of these FMT markers. In addition, we found that the upregulation of DOCK2 by TGF-β is dependent on both Smad3 and ERK pathways as their respective inhibitors blocked TGF-β-mediated induction. TGF-β also stabilized DOCK2 protein, which contributes to increased DOCK2 expression. In addition, DOCK2 was also dramatically induced in the lungs of patients with IPF and in bleomycin, and TGF-β induced pulmonary fibrosis in C57BL/6 mice. Furthermore, increased lung DOCK2 expression colocalized with the FMT marker α-SMA in the bleomycin-induced pulmonary fibrosis model, implicating DOCK2 in the regulation of lung fibroblast phenotypic changes. Importantly, DOCK2 deficiency also attenuated bleomycin-induced pulmonary fibrosis and α-SMA expression. Taken together, our study demonstrates a novel role of DOCK2 in pulmonary fibrosis by modulating FMT and suggests that targeting DOCK2 may present a potential therapeutic strategy for the prevention or treatment of IPF.
Keywords: DOCK2, fibroblast to myofibroblast transition, lung, pulmonary fibrosis, TGF-β
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is the most common form of interstitial lung disease (ILD) and is characterized by progressive scarring of the lungs (1). This devastating lung disease affects ∼3 million people worldwide (2). Current therapies for this lethal lung disease have not been durable and successful, resulting in a median survival time of only 3 to 5 years following diagnosis (3). A striking, key feature of IPF is the appearance of smooth muscle α-actin (α-SMA)-positive myofibroblasts. These cells are considered the central effectors responsible for deposition of extracellular matrix (ECM) including fibronectin, laminin, and collagen in the pathogenesis of pulmonary fibrosis (4, 5). Extensive studies have demonstrated that lung fibroblast (LF) activation, particularly fibroblast to myofibroblast transition (FMT), damages the lung parenchyma, and contributes to IPF (6). Understanding of the underlying mechanisms remains incomplete.
FMT is a process by which fibroblasts acquire new phenotypic features characteristic of myofibroblasts. These include enhanced expression of α-SMA and production of ECM proteins including fibronectin and collagen. It is well recognized that transforming growth factor-β (TGF-β) and associated signaling plays a central role in pulmonary fibrosis. TGF-β is also known to drive the FMT process (7, 8). In the presence of TGF-β, quiescent fibroblasts differentiate into myofibroblasts with increased production of α-SMA and ECM proteins. Overexpression of TGF-β in the lung leads to pulmonary fibrosis in rodents (9–11), supporting the profibrotic effect of locally expressed TGF-β. Conversely, Smad3 deficiency abolishes bleomycin-induced pulmonary fibrosis in mice (12). TGF-β is known to transmit signals via Smad-dependent canonical and independent noncanonical pathways but, the downstream mediators of these signaling pathways remain unclear.
Dedicator of cytokinesis 2 (DOCK2) was primarily found to be expressed in hematopoietic cells under physiological conditions. DOCK2 plays a critical role in chemotaxis of neutrophils, lymphocytes, and plasmacytoid dendritic cells mainly by its Rac-specific guanine nucleotide exchange factor (GEF) activity (13–15). It is also critical for lymphocyte migration by mediating cytoskeletal reorganization through Rac activation (15), as well as differentiation of CD4+ T cells to T helper type 2 cells (16) and B cells to plasma cells (17, 18). Studies from our group and others found that DOCK2 knockdown/knockout (DOCK2−/−) inhibited vascular remodeling after injury by regulating smooth muscle cell phenotype (19) and proliferation (20). DOCK2−/− also blocks high-fat diet-induced obesity with reduced proinflammatory cytokines/chemokines in adipose tissue and the peripheral circulation (21). Furthermore, our recent work shows that DOCK2−/−attenuates lung inflammation and early organization that occurs in mice fed a chronic (5 mo) high-fat high-fructose diet (22). We also reported a critical role of DOCK2 in the development of pleural fibrosis by modulating mesothelial to mesenchymal transition (23). Based on these several lines of evidence, we speculated that DOCK2 plays a broad role in the development of pulmonary fibrosis, which is explored in the present study.
In this study, we found that DOCK2 expression is notably increased in the lungs of patients with IPF, and in the bleomycin, and TGF-β induced pulmonary fibrosis in mice model. Furthermore, DOCK2 is induced by TGF-β in primary human LFs (HLFs) and is required in TGF-β-induced FMT, a critical event in the development of IPF. Our study demonstrates that DOCK2 is a novel regulator of the FMT process and is implicated in the development of IPF. Targeting DOCK2, therefore, may represent a promising therapeutic option for patients with IPF.
MATERIALS AND METHODS
Reagents and Antibodies
Recombinant human transforming growth factor-β1 (TGF-β) was purchased from R&D Systems (Minneapolis, MN). Actinomycin D (Act D) was purchased from LC laboratories (Woburn, MA). Bleomycin (15 unit/vial, Teva) was purchased from the Pharmacy at the University of Texas Health Science Center at Tyler. Small molecular inhibitors including SIS3, Ly294002, U0126, SP600125, SB203580, and cycloheximide (CHX) were purchased from MedChemExpress (Monmouth Junction, NJ). The primary antibodies against DOCK2 (09–454) and α-SMA (A2547) were purchased from Sigma (St. Louis, MO). Antifibronectin antibody (Ab, ab2413) was purchased from Abcam (Cambridge, MA). Collagen type 1 Ab (1310-08) was purchased from SouthernBiotech (Birmingham, AL). GAPDH Ab (60004-1-Ig) was purchased from Proteintech (Rosemont, IL). α-Tubulin Ab (T5168) and β-actin (A1978) Ab were purchased from Sigma. Secondary antibody against collagen type 1, i.e., peroxidase-conjugated streptavidin (016–030-084) was purchased from Jackson ImmunoResearch (West Grove, PA). Secondary horseradish peroxidase (HRP)-conjugated antibodies including goat anti-rabbit IgG peroxidase-conjugated (PI31460) and goat anti-mouse IgG peroxidase-conjugated (PI31430) were purchased from Thermo Fisher Scientific (Waltham, MA). Secondary fluorescence labeled antibodies including goat anti-rabbit IgG-Texas Red (T6391), goat anti-mouse IgG-Texas Red (T862), and goat anti-rabbit IgG-Alexa 488 (A11034) were purchased from Invitrogen (Carlsbad, CA). These primary and secondary Abs have been validated in previous publications (19, 24–27).
Cell Culture and Treatment
Primary HLFs were isolated from histologically normal nonfibrotic lungs (Donors: a male, 60 yr old, non-smoker; and a male, 21 yr old, nonsmoker) and lung tissues from patients with IPF (a female, 62 yr old, non-smoker; and a male, 55 yr old, smoker) provided by the University of Michigan (Ann Arbor, MI) under a material transfer agreement (MTA), as previously described (10, 28). Briefly, fibroblasts were isolated from the periphery of the resected lung in patients undergoing surgical resection of lung nodules. Lung tissues were confirmed by clinical pathologists to be either histologically normal without evidence of fibrosis, cancer, or usual interstitial pneumonia (UIP), the most common histopathological pattern of IPF. Lung tissue specimens were isolated under sterile conditions. Fragments (1 mm3) from the same donor were pooled and maintained in DMEM (supplemented with 10% FBS, 100 U/mL penicillin/streptomycin, and 250 μg of fungizone) to allow fibroblasts to proliferate to 80% confluence before passage. These cells were passaged every 3–5 days and used for experiments within 10 passages. Cell starvation was performed using DMEM without FBS. Informed consent was obtained from all patients and protocols were approved by the University of Michigan Internal Review Board.
Western Blotting Analysis
The methods for protein extraction and Western blotting procedure were described previously (23, 29). Briefly, whole cell lysates were collected from cells or tissues in RIPA (radioimmunoprecipitation assay) lysis buffer containing proteinase inhibitor cocktail and phosphatase inhibitor, all purchased from Thermo Fisher Scientific (Waltham, MA). The BCA protein assay kit (Thermo Fisher Scientific) was used to determine total protein concentrations according to the manufacturer’s guide using the Infinite M Plex plate reader from Tecan (Männedorf, Switzerland). The denatured proteins (10 µg) were used for SDS-PAGE gel electrophoresis. After transferring to PVDF membrane, the membrane was blocked with 5% nonfat milk in phosphate-buffered saline with 0.1% Tween 20 (PBST), followed by incubation with diluted primary antibodies at 4°C, overnight. Appropriately diluted horseradish peroxidase (HRP)-labeled secondary antibodies were used to detect target proteins using an enhanced chemiluminescence kit (Thermo Fisher Scientific). The dilution of primary antibodies against the target proteins is as follows: DOCK2 (1:1,000), α-SMA (1:2,000), collagen type I (1:3,000), fibronectin (1:1,000), and GAPDH (1:8,000). The dilution of secondary antibodies was as follows: goat anti-rabbit IgG-HRP (1:3,000 for DOCK2, 1:5,000 for fibronectin), and goat anti-mouse IgG-HRP (1:3,000 for α-SMA and 1:5,000 for GAPDH). Secondary antibody against collagen type 1 (1:8,000) was used.
Quantitative PCR Analysis
Total RNA was extracted from cells after treatment using TRIzol reagent according to the manufacturer’s guide. RNA concentration was determined using the Infinite M Plex plate reader from Tecan, and total RNA (1 µg) was used for reverse transcription to obtain cDNA using a reverse transcription kit from Bio-Rad (Hercules, CA). Then, qPCR analysis was performed to detect target gene expression using the SYBR green reagent from Bio-Rad. GAPDH was used as the internal reference, and each analysis was run in triplicate. The primers of genes tested were described as reported previously (23, 30) and were shown in Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.19169975.
Gene Knockdown with Small Interfering RNA
DOCK2 expression was knocked down in primary HLFs via siRNA. The scramble control (SIC001) and siRNA targeting human DOCK2 (siDOCK2, SASI_Hs01_00204031) were purchased from Sigma (St. Louis, MO). Cells at a confluence of approximately 60% were transfected with scramble or siDOCK2 using the jetPRIME transfection reagent from Polyplus transfection (New York, NY) according to the manufacturer’s guide (22).
Cycloheximide Chase Assay
The CHX chase assay was conducted similarly as previously described (31, 32). After treatment with vehicle or TGF-β (5 ng/mL), HLFs grown in 6-cm dishes were exposed to 10 μg/mL CHX, followed by whole cell lysates harvest at different times for Western blotting analysis of DOCK2. Band intensities were quantified by the National Institutes of Health (NIH) ImageJ software. For each treatment group, the levels of DOCK2 protein after adjusting by internal reference GAPDH were presented as a percentage of levels at 0-time post CHX treatment. To compare protein expression in Western blotting, each protein level (band density) was normalized with its corresponding loading control. Then the protein fold changes were calculated by dividing the normalized protein levels (to loading control) of different treatment groups by that of the control group. The normalized protein level in the control group was accordingly set as 1. The protein fold changes were used for statistical comparison.
Animals
Male C57BL/6 mice (12–16 wk old) were purchased from Jackson Laboratory (Bar Harbor, ME). Male DOCK2 knockout (DOCK2−/−) mice in the C57BL/6 background were provided under an MTA with Dr. Fukui at Kyushu University and were reported previously (15, 19, 24, 25). All animals were housed under conventional conditions in the animal care facility and housed in compliance with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals. All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at Tyler and conducted according to relevant guidelines.
Human Lung Tissues from Normal Subjects and Patients with IPF
The lung tissue sections from normal subjects and patients with IPF were provided by the University of Michigan (Ann Arbor, Michigan) under an MTA. The normal subjects were defined as those who have histologically nonfibrotic lungs. Three normal and three IPF lung tissue sections were used for the comparison of DOCK2 expression. The information of patients with IPF: patient No. 1 (male: 55 yr old, smoker), patient No. 2 (male: 65 yr old, smoker), and patient No. 3 (male: 63 yr old, nonsmoker). The control donors’ information: donor No. 1 (male: 60 yr old, nonsmoker), donor No. 2 (male: 21 yr old, nonsmoker), and donor No. 3 (male: 59 yr old, smoker).
Bleomycin and TGF-β Induced Pulmonary Fibrosis in Mice
Male 12–16-wk-old WT C57BL/6 mice and DOCK2−/− mice were administered bleomycin (1 U/kg body wt, Teva) or 0.9% saline once through intratracheal instillation as previously reported (10). Bleomycin or saline was given at a volume of 40 µL/mouse and the mice were maintained for up to 4 wk. Animals were euthanized, and lung tissues were collected and fixed in 4% paraformaldehyde for histological analyses. The lung tissues from TGF-β adenovirus-induced pulmonary fibrosis were adopted from a previous study (10).
Lung Histology, Masson’s Trichrome Staining, and Immunofluorescence Staining
Histological and immunological analyses were performed using formalin-fixed and paraffin-embedded (FFPE) lung sections from human donors (normal controls and patients with IPF), and murine models of pulmonary fibrosis. Collagen deposition was determined using Masson’s Trichrome staining method according to the manufacturers’ guide. The immunohistochemical (IHC) staining of DOCK2 and the immunofluorescence (IF) staining of DOCK2 and α-SMA were performed similarly as previously reported (19, 23). Briefly, the hydrated slides were quenched in 3% H2O2 for 15 min, followed by boiling in EDTA buffer for 10 min for antigen retrieval. The sections were then blocked with 10% goat serum before incubation with primary antibodies against DOCK2 (1:100) or α-SMA (1:100) overnight at 4°C. Secondary antibodies used (1:100) were goat anti-rabbit (A11034) or anti-mouse (A11029) Alexa Fluor Plus 488, and goat anti-rabbit (T6391) or anti-mouse (T862) Texas Red purchased from Invitrogen (Waltham, MA). Control normal rabbit IgG (3703, ProSci) was used (1:100) as the negative control. Images were taken using a Nikon NiU microscope.
Statistical Analysis
The data in the study were shown as means ± SD and analyzed using the GraphPad Prism 5 software. Data were evaluated with a two-tailed, unpaired Student’s t test or one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference test. A P value less than 0.05 was considered statistically significant.
RESULTS
TGF-β Induces DOCK2 Expression in Primary Normal and IPF HLFs
Recently we reported the increased expression of DOCK2 in primary HLFs upon TNF-α treatment (22). In this study, we sought to determine whether TGF-β induces DOCK2 expression in both normal and IPF HLFs. We found that TGF-β significantly upregulated DOCK2 expression at 5 and 10 ng/mL in both normal (Fig. 1, A and B) and IPF (Fig. 1, C and D) HLFs. There was minimal effect in DOCK2 induction by further increasing the concentration of TGF-β (Fig. 1). FMT markers, including α-SMA, collagen type 1 (Col-1), and fibronectin (FN) were likewise induced by TGF-β in primary HLFs from normal subjects and patients with IPF. Because the minimum dose of 5 ng/mL of TGF-β treatment significantly induced DOCK2 and FMT markers’ expression, it was selected for the following experiments. To validate these findings, we included additional lines of normal and IPF HLFs for the assay (Supplemental Fig. S1, A and B). Similar results were found showing that TGF-β induces DOCK2 expression along with FMT markers.
Figure 1.

DOCK2 expression was induced by TGF-β in primary normal and IPF human lung fibroblasts (HLFs). A: serum-starved primary normal HLFs were treated with different doses of TGF-β for 24 h, followed by Western blotting (WB) analysis of DOCK2 and FMT markers including α-SMA, fibronectin (FN), and collagen type I (Col-1). GAPDH is a loading control. B: quantification of A based on three independent experiments. C: serum-starved primary IPF HLFs were treated with different doses of TGF-β, followed by WB analysis of DOCK2 and FMT markers. D is the quantification of C based on three independent experiments. ImageJ software was used to quantify the band intensity. *P < 0.05 compared with vehicle control, n = 3 independent experiments using the same donor fibroblasts. One-way ANOVA followed by Fisher’s least significant difference test was used for the multiple comparisons. α-SMA, smooth muscle α-actin; DOCK2, dedicator of cytokinesis 2; FMT, fibroblast to myofibroblast transition; IPF, idiopathic pulmonary fibrosis; TGF-β, transforming growth factor-β.
Next, we determined the time-dependent induction of DOCK2 by TGF-β in HLFs. The results show that DOCK2 expression was significantly increased by TGF-β as early as 2 h in normal (Fig. 2, A and B) and 4 h in IPF (Fig. 2, C and D) HLFs. Such inductions continue to peak at 24 h after TGF-β treatment (Fig. 2, A and B). Meanwhile, the time-dependent induction of FMT markers, α-SMA, Col-1, and FN, were also observed in both normal and IPF HLFs, which seems to occur after induction of DOCK2. A similar trend was observed in normal and IPF HLFs. In additional lines of primary normal and IPF HLFs, the time-dependent induction of DOCK2 was also observed (Supplemental Fig. S1, C and D). Together, these data indicate that DOCK2 expression is markedly induced by TGF-β preceding that of FMT markers in HLFs, suggesting a role for DOCK2 in mediating FMT induced by TGF-β.
Figure 2.

DOCK2 expression was induced by TGF-β in a time-dependent manner in primary normal and IPF HLFs. A: serum-starved primary normal HLFs were treated with TGF-β (5 ng/mL) for various times, followed by WB analysis of DOCK2 and FMT markers including α-SMA, FN, and Col-1. GAPDH is a loading control. B: quantification of A based on three independent experiments. C: serum-starved primary IPF HLFs were treated with TGF-β (5 ng/mL) for various times, followed by WB analysis of DOCK2 and FMT markers. D is the quantification of C, based on three independent experiments. ImageJ software was used to quantify the band intensity. *P < 0.05 compared with vehicle control, n = 3 independent experiments using the same donor fibroblasts. One-way ANOVA followed by Fisher’s least significant difference test was used for the multiple comparisons. α-SMA, smooth muscle α-actin; Col-1, collagen type I; DOCK2, dedicator of cytokinesis 2; HLFs, human lung fibroblasts; FMT, fibroblast to myofibroblast transition; FN, fibronectin; IPF, idiopathic pulmonary fibrosis; TGF-β, transforming growth factor-β; WB, Western blotting.
DOCK2 Is Required in TGF-β-Induced FMT
To determine whether DOCK2 is required in TGF-β-induced FMT, DOCK2 was knocked down using siRNA in primary normal and IPF HLFs. Serum-starved cells were then treated with TGF-β (5 ng/mL) for an additional 24 h. In primary normal HLFs, TGF-β significantly induced the expression of FMT markers including α-SMA, Col-1, and FN in the scramble group (Fig. 3, A and B). Conversely, DOCK2 knockdown significantly blocked the induction of these molecules by TGF-β in primary normal HLFs (Fig. 3, A and B, Supplemental Fig. S2A). Similar results were found in primary IPF HLFs (Fig. 3, C and D, Supplemental Fig. S2B). We also determined whether DOCK2 knockdown attenuates α-SMA expression using IF staining. Consistently, IF staining showed that knockdown of DOCK2 suppressed TGF-β-induced α-SMA expression (Supplemental Fig. S3). These data strongly suggest that DOCK2 is required in TGF-β-induced FMT.
Figure 3.
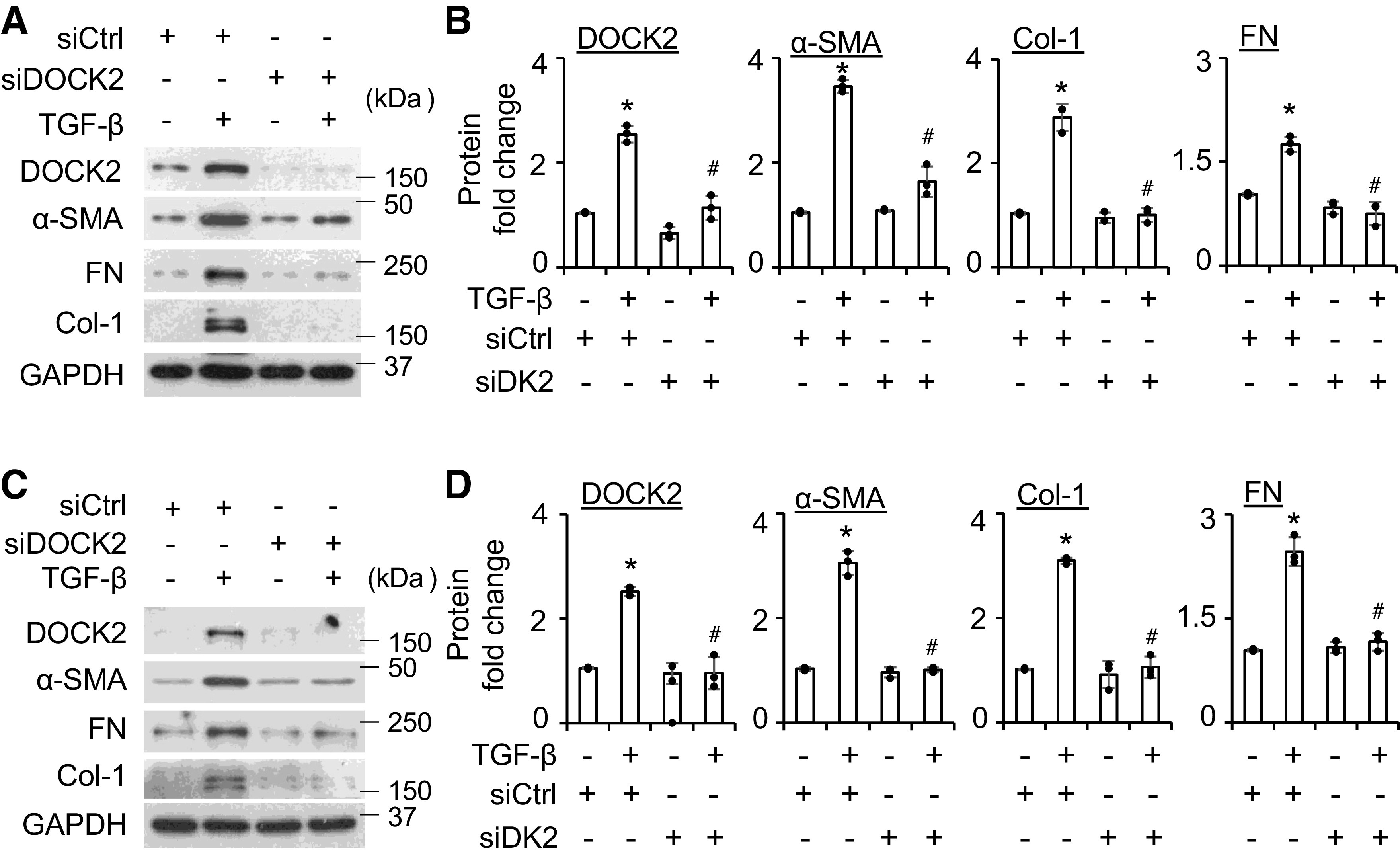
DOCK2 knockdown significantly attenuates TGF-β-induced FMT markers in primary normal and IPF HLFs. A and C: primary normal (A) and IPF (C) HLFs were transfected with scramble (siCtrl) or DOCK2 siRNA (siDOCK2) (40 nM), followed by treatment with TGF-β (5 ng/mL) for additional 24 h to detect DOCK2 and α-SMA, Col-1, and FN levels by WB. B and D are the quantifications of A and C, respectively, based on three independent experiments. ImageJ software was used to quantify the band intensity. *P < 0.05 vs. control group; #P < 0.05 vs. TGF-β-treated scramble control, n = 3 independent experiments using the same donor fibroblasts. One-way ANOVA followed by Fisher’s least significant difference test was used for the multiple comparisons. α-SMA, smooth muscle α-actin; Col-1, collagen type I; DOCK2, dedicator of cytokinesis 2; HLFs, human lung fibroblasts; FMT, fibroblast to myofibroblast transition; FN, fibronectin; IPF, idiopathic pulmonary fibrosis; TGF-β, transforming growth factor-β; WB, Western blotting.
DOCK2 Is Increased by TGF-β at Both the Transcriptional and the Posttranslational Levels
In these experiments, we first determined whether DOCK2 induction by TGF-β is transcriptionally regulated. The qPCR data show that TGF-β at 5 ng/mL and above significantly increased DOCK2 mRNA levels (Fig. 4A). Time-dependent induction was also observed in normal HLFs (Fig. 4B). Similar induction was found in IPF HLFs with some variation (Supplemental Fig. S4). Pretreatment of HLFs with the transcription inhibitor Act D (1 μg/mL) blocked the induction of DOCK2 by TGF-β in both normal (Fig. 4, C and E) and IPF (Fig. 4, D and F) HLFs, which was observed at the protein (Fig. 4, C and D) and mRNA (Fig. 4, E and F) levels. These results support the transcriptional regulation of DOCK2 by TGF-β.
Figure 4.

DOCK2 is regulated by TGF-β at both the transcriptional level and the posttranslational level. A and B: qPCR analysis showed DOCK2 mRNA levels were significantly increased by TGF-β in dose-dependent (A, 12 h) and time-dependent (B, 5 ng/mL TGF-β) manners in primary normal HLFs. GAPDH is an internal control, n = 3 replicates using the same donor HLFs. C and D: pretreatment with 1 μg/mL Actinomycin D (Act D) for 30 min significantly blocked TGF-β (5 ng/mL, 16 h)-induced DOCK2 protein levels in normal (C) and IPF HLFs. E and F: Act D (1 μg/mL) pretreatment for 30 min significantly blocked TGF-β (5 ng/mL, 4 h) induced DOCK2 mRNA levels, GAPDH is an internal control, n = 3 replicates using the same donor HLFs. G: primary normal HLFs were treated with TGF-β (5 ng/mL) overnight, followed by 10 μg/mL cycloheximide (CHX) treatment for the indicated times. Whole cell lysates were collected for WB analysis of DOCK2 degradation. H: DOCK2 levels were plotted relative to those at time 0 of CHX treatment (G) after being quantified by ImageJ software and normalized to GAPDH. *P < 0.05 vs. control group. #P < 0.05 vs. TGF-β-treated alone control. One-way ANOVA followed by Fisher’s least significant difference test was used for the multiple comparisons. DOCK2, dedicator of cytokinesis 2; HLFs, human lung fibroblasts; IPF, idiopathic pulmonary fibrosis; TGF-β, transforming growth factor-β; WB, Western blotting.
Posttranslational regulation represents an important mechanism that affects TGF-β signaling and pulmonary fibrosis (33). As such, we next sought to determine whether TGF-β also affects DOCK2 protein stability. The results show that the vehicle control group demonstrated an ∼50% degradation of DOCK2 at 8 h post CHX. Conversely, TGF-β stabilized DOCK2 levels for up to 12 h post CHX (Fig. 4, G and H). The data clearly support the concept that TGF-β stabilizes DOCK2 protein in HLFs, in addition to transcriptional regulation.
DOCK2 Is Induced by TGF-β via the Smad3 and ERK Pathways in Primary HLFs
To determine which pathway(s) is involved in TGF-β-induced DOCK2 upregulation, different selective pathway inhibitors were used before the treatment with TGF-β in HLFs. Pretreatment with Smad3 inhibitor SIS3 (10 μM) significantly blocked TGF-β (5 ng/mL, 16 h) induced expression of DOCK2 (Fig. 5, A and B). Similarly, pretreatment with the ERK inhibitor U0126 (10 μM) also significantly attenuated DOCK2 induction by TGF-β (Fig. 5, A and B). By contrast, inhibitors of AKT (Ly294002, 10 μM), JNK (SP600125, 10 μM), and p38 (SB203580, 10 μM) failed to inhibit DOCK2 induction by TGF-β. Further dose-dependent studies using varying concentrations of SIS3 (Fig. 5, C and D) and U0126 (Fig. 5, E and F) confirmed Smad3 and ERK-dependent induction of DOCK2 by TGF-β. Similar findings were also observed in primary IPF HLFs that SIS3 and U0126 attenuate TGF-β-induced DOCK2 expression in a dose-dependent manner (Supplemental Fig. S5). Taken together, these data demonstrate both Smad3-dependent and independent regulation of DOCK2 by TGF-β in primary HLFs.
Figure 5.

TGF-β upregulates DOCK2 expression through Smad3 and Erk pathways. A: primary normal HLFs were pretreated with various inhibitors (10 μmol/L, 30 min) of Smad3 (SIS3), AKT (Ly294002), ERK (U0126), JNK (SP600125), and P38 (SB203580), followed by 5 ng/mL TGF-β treatment for additional 16 h. Whole cell lysates were collected for WB analysis of DOCK2 with GAPDH as a loading control. B is the quantification of A, based on three independent experiments. C and E: primary normal HLFs were pretreated with various concentrations of SIS3 (C) or U0126 (E) for 30 min, followed by treatment with 5 ng/mL TGF-β for additional 16 h. Whole cell lysates were collected for WB analysis of DOCK2 with GAPDH as a loading control. D and F are the quantification of C and E, respectively, based on three independent experiments. ImageJ software was used to quantify the band intensity. *P < 0.05 vs. vehicle control. #P < 0.05 vs. TGF-β alone group, n = 3 independent experiments using the same donor fibroblasts. One-way ANOVA followed by Fisher’s least significant difference test was used for the multiple comparisons. DOCK2, dedicator of cytokinesis 2; HLFs, human lung fibroblasts; TGF-β, transforming growth factor-β; WB, Western blotting.
DOCK2 Expression Is Upregulated in Bleomycin and TGF-β-Induced Pulmonary Fibrosis Models
To determine whether DOCK2 is increased in preclinical pulmonary fibrosis, male wild-type C57BL/6 mice were administered bleomycin via intratracheal injection (IT) canulation once and examined after 28 days, as previously reported (10). Collagen deposition was enhanced in the lungs of mice given bleomycin treatment compared with the saline control group and a significant difference was observed as determined by the Reciprocal intensity method (34, 35) (Fig. 6, A and B). Importantly, IHC staining showed dramatically increased DOCK2 expression in the lungs of bleomycin-treated mice compared with the saline control (Fig. 6C). Similarly, a notable increase in DOCK2 expression was also observed in the lungs of C57BL/6 mice that received intratracheal TGF-β adenovirus as compared with control vector group (Supplemental Fig. S6).
Figure 6.

DOCK2 expression was induced in mice lung tissues from bleomycin-induced pulmonary fibrosis. A: Masson’s trichrome staining showed increased collagen deposition (blue) in the lung tissues of bleomycin (BLEO)-treated male C57BL/6 mice as compared with saline control group, (n = 3 mice/group). B: reciprocal intensity method was used for the quantification of collagen staining as in A from three views per slide/mouse, n = 3 mice/group. *P < 0.05 compared with saline group, as tested by a two-tailed, unpaired Student’s t test. (a.u., arbitrary units). C: representative immunohistochemical (IHC) staining of DOCK2 in the lung tissues from control and BLEO (28 days)-treated male C57BL/6 mice. D: immunofluorescence (IF) staining showed that DOCK2 was induced and colocalized with FMT marker α-SMA (red) in lung tissues of C57BL/6 mice given bleomycin for 28 days. DAPI stains the nuclei (blue). Scale bars (black and white) represent 100 µm, images were taken at the same magnification. Boxes show enlarged views of the areas of interest. α-SMA, smooth muscle α-actin; DOCK2, dedicator of cytokinesis 2; FMT, fibroblast to myofibroblast transition.
One notable feature of the pathogenesis of IPF is the appearance of α-SMA-positive myofibroblasts, which largely derive from FMT. Compared with the saline control group, DOCK2 was dramatically induced in the lung tissues of bleomycin-treated mice (green, Fig. 6D), which was largely located in myofibroblasts as indicated by the colocalization of DOCK2 with α-SMA (red, Fig. 6D). Negative control using IgG and relevant secondary antibodies did not show positive staining (Supplemental Fig. S7). Together, these data suggest that DOCK2 may contribute to pulmonary fibrosis by modulating FMT.
DOCK2 Expression Is Upregulated in the Lung Tissues of Patients with IPF
Finally, we determined whether there is an altered expression of DOCK2 in patients with IPF. Representative images of the lung tissues from patients with IPF showed increased collagen deposition compared with the normal human lung tissues, as determined by Masson’s Trichrome staining (Fig. 7A). While DOCK2 was barely detectable in the normal human lung tissues, its expression was dramatically induced in the lungs of patients with IPF as shown by IF staining (Fig. 7B). Moreover, the mean immunofluorescence intensity (MFI) of DOCK2 was significantly increased in IPF lung specimens compared with that of normal controls (n = 3/group, Fig. 7C). In addition, IHC staining of DOCK2 using lung tissues from control subjects and patients with IPF showed consistent results (Supplemental Fig. S8). These data suggest that aberrant DOCK2 expression is involved in the development of IPF.
Figure 7.

DOCK2 expression was induced in lung tissues from human patients with idiopathic pulmonary fibrosis (IPF). A: representative images of Masson’s trichrome staining showed extensive collagen deposition (blue) in the lung tissues of patients with IPF (n = 3/group). B: representative IF staining showed notably increased DOCK2 expression (red) in the lung tissues from patients with IPF as compared with normal control. DAPI stains the nuclei (blue). C: the quantification and comparison of DOCK2 mean fluorescence intensity (MFI) was based on measurements from 10 representative fields per slide from each of three control and patients with IPF. *P < 0.05 compared with normal controls, as tested by a two-tailed, unpaired Student’s t test. Scale bars (black and white) represent 100 µm, images were taken at the same magnification. DOCK2, dedicator of cytokinesis 2; IF, immunofluorescence.
DOCK2 Deficiency Attenuates FMT and Pulmonary Fibrosis In Vivo
To test the role of DOCK2 in pulmonary fibrosis development in vivo, male wild-type and DOCK2−/− mice were challenged with bleomycin. Lung tissues were collected for examination of collagen deposition by Masson’s Trichrome staining and α-SMA expression by IF staining. Extensive collagen staining was observed in the lungs of WT mice challenged with bleomycin. Conversely, DOCK2−/− mice showed much-decreased collagen staining (Fig. 8A). Significant decrease in collagen deposition was observed in DOCK2−/− group compared with WT group that received bleomycin (Fig. 8B). Consistently, the FMT marker α-SMA expression was notably increased in WT mice but not in the DOCK2−/−mice (Fig. 8C and Supplemental Fig. S9). DOCK2 expression was induced by bleomycin in wild-type mice while DOCK2 knockout mice did not show positive staining (Fig. 8D), suggesting the successful knockout of DOCK2. These results indicate that DOCK2 contributes to FMT and pulmonary fibrosis development in vivo.
Figure 8.

DOCK2 deficiency attenuated bleomycin-induced pulmonary fibrosis and α-SMA expression in the lung tissues. A: representative Masson’s Trichrome staining showed increased collagen deposition (blue) in the lung tissues of bleomycin (BLEO, 21 days)-treated wild-type (WT) C57BL/6 mice as compared with saline control group. DOCK2 knockout (KO) mice received intratracheal BLEO showed less collagen deposition as compared with WT counterparts. B: reciprocal intensity method was used for the quantification of collagen staining as in A from three views per slide/mouse, n = 5 mice/group. *P < 0.05 compared with WT saline group; #P < 0.05, compared with WT BLEO group, tested by one-way ANOVA followed by Fisher’s least significant difference test for the multiple comparisons. (a.u., arbitrary units). C and D: representative immunohistochemical (IHC) staining of α-SMA (C) and DOCK2 (D) expression in the lungs of WT and DOCK2 KO mice received saline or BLEO for 21 days. n = 5 mice/group. Scale bars represent 100 µm, images were taken at the same magnification. Boxes show enlarged views of the areas of interest. E: a diagram showing the role of DOCK in mediating fibroblast to myofibroblast transition and pulmonary fibrosis. TGF-β induces DOCK2 through Smad3 and ERK pathways and at both transcription and posttranslational levels. Increased DOCK2 mediates FMT and contributes to pulmonary fibrosis possibly through affecting the Smad3 signaling and Rac1 activation. Image was created with BioRender.com. α-SMA, smooth muscle α-actin; DOCK2, dedicator of cytokinesis 2; FMT, fibroblast to myofibroblast transition; TGF-β, transforming growth factor-β; WT, wild-type.
The main findings of this study were summarized in Fig. 8E. In HLFs, TGF-β induces DOCK2 expression that is dependent on Smad3 and ERK signaling pathways. Such upregulation occurs at both transcriptional level and posttranslational levels. Increased DOCK2 in turn mediated FMT and is implicated in the development of pulmonary fibrosis. The potential mechanisms might involve the modification of the Smad3 signaling by DOCK2 [as reported in other cell types (27)] and its Rac1 activating function that has been reported to promote pulmonary fibrosis (36, 37). These and our findings warrant further analysis.
DISCUSSION
IPF remains a threatening disease with a poor prognosis, and the mechanisms that govern its progression remain not fully understood. In this study, we sought to characterize the contribution of DOCK2 to the phenotypic transition of fibroblasts to myofibroblasts and thus to the development of pulmonary fibrosis. We revealed a previously undefined role of DOCK2 in promoting myofibroblast transition from fibroblasts. Knockdown of DOCK2 inhibited FMT induced by TGF-β in primary HLFs. In addition, we found that DOCK2 expression was increased in the preclinical models of pulmonary fibrosis and in the lung tissues of patients with IPF. DOCK2 deficiency notably attenuated bleomycin-induced FMT and pulmonary fibrosis in vivo. To our knowledge, this study presents the first evidence that DOCK2 is a critical mediator of FMT, which provides novel insight into the pathogenesis of pulmonary fibrosis.
Recent works from our group and others have demonstrated the contribution of DOCK2 in lung inflammatory injury (22, 38), little if any, however, is known about the role of DOCK2 in interstitial lung disease. We sought to determine the role of DOCK2 in fibroblast activation with a focus on the FMT process. Although DOCK2 has been reported to be one of the most upregulated genes by TGF-β in nonsmall cell lung cancer cells in a screening study using RNA-Sequencing (39), this is the first study to show the induction of DOCK2 by TGF-β in primary HLFs. Our results clearly show that DOCK2 is induced by TGF-β in primary normal and IPF HLFs, along with the induction of FMT markers α-SMA, and Col-1 and FN (Figs. 1 and 2, Supplemental Fig. S1). These findings are consistent with the observation of elevated DOCK2 expression in IPF lungs (Fig. 7), which are reported to promote cytokine production such as TGF-β (40). Next, we found that knockdown of DOCK2 abolished TGF-β induced expression of FMT markers α-SMA, Col-1, and FN through independent experiments in both normal and IPF lung fibroblasts (Fig. 3, Supplemental Fig. S2). The data suggest an indispensable role of DOCK2 in FMT and is in accord with our in vivo observation of the colocalization of DOCK2 with α-SMA in induced pulmonary fibrosis (Fig. 6D). Together, our data demonstrate that DOCK2 is required in FMT induction of primary normal and IPF HLFs.
Currently, there is limited knowledge about the regulation mechanism controlling DOCK2 expression or induction in mammalian cells. Recently, we initially showed the transcriptional regulation of DOCK2 by TGF-β in primary human pleural mesothelial cells (23). This study expands these findings and now delineates the transcriptional regulation of DOCK2 by TGF-β in lung fibroblasts. In this study, we found that TGF-β-induced DOCK2 mRNA expression as early as 4 h after treatment and prolonged up to 24 h (Fig. 4, A and B, Supplemental Fig. S4). Inhibition of transcription with Act D efficiently blocked TGF-β-induced DOCK2 upregulation at both protein and mRNA levels in both normal and IPF HLFs (Fig. 4, C–F). What adds to the complex regulation of DOCK2 by TGF-β is the involvement of a posttranslational mechanism, i.e., TGF-β stabilizes DOCK2 protein (Fig. 4, G and H). The detailed mechanisms underlying stabilization of DOCK2 by TGF-β are under active investigation by our group. Our work demonstrates that regulation of DOCK2 is complex following TGF-β signaling. In the aggregate, these findings strengthen the importance of DOCK2 in mediating TGF-β signaling and suggest that DOCK2 represents a candidate target for interventions to prevent pulmonary fibrosis.
Previous studies have identified multiple noncanonical pathways that are activated by TGF-β and convey its downstream effects including the FMT process. These noncanonical pathways include the PI3K/AKT, ERK, JNK, and p38 pathways, etc. (41, 42). To explore the mechanisms underlying DOCK2 induction by TGF-β, we employed different pathway inhibitors to determine the pathways responsible for DOCK2 induction by TGF-β. The results indicate that the Smad3 inhibitor SIS3 and the ERK inhibitor U0126 suppressed DOCK2 induction caused by TGF-β while the other inhibitors of AKT, p38, and JNK pathways are largely spared (Fig. 5, A and B, Supplemental Fig. S5A). Further dose-dependent experiments confirmed the effect of SIS3 and U0126 in inhibiting TGF-β induced DOCK2 induction in normal (Fig. 5, C–F) and IPF (Supplemental Fig. S5, B and C) HLFs. These data indicate that TGF-β can induce DOCK2 expression through Smad3-dependent and independent pathways. Whether specific pathway-dependent transcriptional and posttranslational regulation of DOCK2 occurs after TGF-β stimulation warrants further study in the future.
Recently, we reported an important role of DOCK2 in high-fat high-fructose-induced pulmonary inflammation with organization (22) and in the development of pleural fibrosis (23). This study extends our earlier work and likewise implicates DOCK2 in the pathogenesis of pulmonary fibrosis, thereby addressing a potentially important knowledge gap. In this study, we observed dramatically increased DOCK2 expression in the lung tissues of patients with IPF compared with normal subjects (Fig. 7 and Supplemental Fig. S8), providing a proof of concept that DOCK2 is involved in the pathogenesis of IPF. In view of the potential variations in human tissues, future cross-sectional study using a large sample size that can adjust for potential variables is warranted to test the association of DOCK2 with IPF.
On the other hand, DOCK2 expression was also upregulated in the bleomycin (Fig. 6C) and TGF-β (Supplemental Fig. S6) induced pulmonary fibrosis model, further supporting the involvement of DOCK2 in the development of parenchymal pulmonary fibrosis. Intriguingly, we found that a large portion of cells positive for DOCK2 staining also expressed α-SMA in the lung tissues of mice that received bleomycin but not saline (Fig. 6D), suggesting that DOCK2 may affect FMT during pulmonary fibrosis progression in vivo. Inflammatory infiltration in the lung resulting from various insults to the alveolar epithelia is usually recognized as an initial response that involves proinflammatory cytokines and chemokines, followed by the resolving phase where TGF-β gradually takes over and drives the profibrotic responses in pathological conditions (4, 43, 44). In diet-associated pulmonary inflammation and fibrosis, DOCK2 was dramatically induced in the lung tissues while DOCK2 deficiency blocks the inflammatory responses and collagen deposition (22). The findings in this study provide novel evidence that DOCK2 is also closely related to the profibrotic changes in the lungs of mice (Fig. 8) apart from playing an important role in promoting early phase inflammation. Further time-course-dependent studies using the bleomycin model might reveal if DOCK2 is also upregulated during the first few days of bleomycin exposure when an inflammatory response dominates (45).
In this study, we provided in vivo evidence that DOCK2 deficiency attenuates pulmonary fibrosis as indicated by collagen deposition as well as the expression of myofibroblast marker α-SMA (Fig. 8, A and C, Supplemental Fig. S9), further supporting an important role of DOCK2 in promoting FMT and pulmonary fibrosis. It needs to be noted that due to its GEF function to activate Rac, DOCK2 has been implicated in diverse diseases by regulating immune responses (13, 14). Recently, DOCK2 was reported to stimulate macrophage proinflammatory activity to regulate endotoxemia-induced acute lung injury (46). DOCK2 deficient macrophages demonstrate defects in chemotaxis and migration (38). Because of the important role of macrophages in IPF pathogenesis (47), it is likely that DOCK2 expression by macrophages may also contribute to pulmonary fibrosis. Future studies are warranted to explore the immune aspect of DOCK2 in the pathogenesis of IPF. On the other hand, it is critical to establish cell-specific DOCK2 knockout mice model and study the cell-type-specific function (e.g., lung fibroblast and macrophage) of DOCK2 in pulmonary fibrosis development. These works specify a future direction of our continued efforts to characterize the role of DOCK2 in IPF development. Based on the findings in this study, a graphic summary is presented in Fig. 8E. DOCK2 is induced by TGF-β through Smad3 and ERK pathways that involve both transcriptional and posttranslational mechanisms. Upregulated DOCK2 in turn mediates FMT process and contributes to pulmonary fibrosis development. The mechanisms may involve Rac activation and potential interaction with the Smad3 signaling. Further mechanistic studies are needed to delineate the potential modes of action of DOCK2 in mediating FMT and promoting pulmonary fibrosis.
In summary, we report a previously unrecognized role of DOCK2 that promotes FMT and implicate DOCK2 in the development of pulmonary fibrosis. These novel findings suggest that DOCK2 may represent a novel preventive or therapeutic target.
SUPPLEMENTAL DATA
Supplemental Table S1 and Supplemental Figs. S1–S9: https://doi.org/10.6084/m9.figshare.19169975.
GRANTS
This study was supported by the University of Texas Health Science Center at Tyler Startup fund (to G.Q. and X.G.) and seed grant (to G.Q.), the National Institutes of Health Grants HL141583 (to X.G.) and R01HL147313 (to S.-Y.C.), and the University of Texas Rising Star Award (to X.G.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
X.G. and G.Q. conceived and designed research; X.G., O.A., C.S., V.M., and A.O. performed experiments; X.G. and G.Q. analyzed data; X.G. and G.Q. interpreted results of experiments; X.G. and G.Q. prepared figures; X.G. and G.Q. drafted manuscript; S.H, S.-Y.C., S.I., and T.A.T. edited and revised manuscript; X.G., G.Q., V.M., S.H., S.-Y.C., S.I., and T.A.T. approved final version of manuscript.
REFERENCES
- 1.Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev 28: 180100, 2019. doi: 10.1183/16000617.0100-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 21: 355–361, 2012. doi: 10.1183/09059180.00002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 198: e44–e68, 2018. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 4.Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res 19: 32, 2018. doi: 10.1186/s12931-018-0730-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 9: 157–179, 2014. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature 587: 555–566, 2020. doi: 10.1038/s41586-020-2938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez IE, Eickelberg O. The impact of TGF-β on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc 9: 111–116, 2012. doi: 10.1513/pats.201203-023AW. [DOI] [PubMed] [Google Scholar]
- 8.Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis 4: 367–388, 2010. doi: 10.1177/1753465810379801. [DOI] [PubMed] [Google Scholar]
- 9.Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J, Sethi T. Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med 185: 537–546, 2012. doi: 10.1164/rccm.201106-0965OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeffers A, Qin W, Owens S, Koenig KB, Komatsu S, Giles FJ, Schmitt DM, Idell S, Tucker TA. Glycogen synthase kinase-3β inhibition with 9-ING-41 attenuates the progression of pulmonary fibrosis. Sci Rep 9: 18925, 2019. doi: 10.1038/s41598-019-55176-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr, Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 282: L585–593, 2002. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 13.Guo X, Chen SY. Dedicator of cytokinesis 2 in cell signaling regulation and disease development. J Cell Physiol 232: 1931–1940, 2017. doi: 10.1002/jcp.25512. [DOI] [PubMed] [Google Scholar]
- 14.Kunimura K, Uruno T, Fukui Y. DOCK family proteins: key players in immune surveillance mechanisms. Int Immunol 32: 5–15, 2020. doi: 10.1093/intimm/dxz067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, Inayoshi A, Noda M, Oike M, Shirai T, Sasazuki T. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature 412: 826–831, 2001. doi: 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka Y, Hamano S, Gotoh K, Murata Y, Kunisaki Y, Nishikimi A, Takii R, Kawaguchi M, Inayoshi A, Masuko S, Himeno K, Sasazuki T, Fukui Y. T helper type 2 differentiation and intracellular trafficking of the interleukin 4 receptor-α subunit controlled by the Rac activator Dock2. Nat Immunol 8: 1067–1075, 2007. doi: 10.1038/ni1506. [DOI] [PubMed] [Google Scholar]
- 17.Jing Y, Kang D, Liu L, Huang H, Chen A, Yang L, Jiang P, Li N, Miller H, Liu Z, Zhu X, Yang J, Wang X, Sun J, Liu Z, Liu W, Zhou X, Liu C. Dedicator of cytokinesis protein 2 couples with lymphoid enhancer-binding factor 1 to regulate expression of CD21 and B-cell differentiation. J Allergy Clin Immunol 144: 1377–1390.e4, 2019. [Erratum in J Allergy Clin Immunol 147: 1528, 2021]. doi: 10.1016/j.jaci.2019.05.041. [DOI] [PubMed] [Google Scholar]
- 18.Ushijima M, Uruno T, Nishikimi A, Sanematsu F, Kamikaseda Y, Kunimura K, Sakata D, Okada T, Fukui Y. The Rac activator DOCK2 mediates plasma cell differentiation and IgG antibody production. Front Immunol 9: 243, 2018. doi: 10.3389/fimmu.2018.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo X, Shi N, Cui XB, Wang JN, Fukui Y, Chen SY. Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ Res 116: e71–e80, 2015. doi: 10.1161/CIRCRESAHA.116.305863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao BJ, Wang XW, Zhu L, Zou RJ, Lu ZQ. Dedicator of cytokinesis 2 silencing therapy inhibits neointima formation and improves blood flow in rat vein grafts. J Mol Cell Cardiol 128: 134–144, 2019. doi: 10.1016/j.yjmcc.2019.01.030. [DOI] [PubMed] [Google Scholar]
- 21.Guo X, Li F, Xu Z, Yin A, Yin H, Li C, Chen SY. DOCK2 deficiency mitigates HFD-induced obesity by reducing adipose tissue inflammation and increasing energy expenditure. J Lipid Res 58: 1777–1784, 2017. doi: 10.1194/jlr.M073049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian G, Adeyanju O, Sunil C, Huang SK, Chen SY, Tucker TA, Idell S, Guo X. Dedicator of cytokinesis 2 (DOCK2) deficiency attenuates lung injury associated with chronic high-fat and high-fructose diet-induced obesity. Am J Pathol 192: 226–238, 2021. doi: 10.1016/j.ajpath.2021.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian G, Adeyanju O, Roy S, Sunil C, Jeffers A, Guo X, Ikebe M, Idell S, Tucker TA. DOCK2 promotes pleural fibrosis by modulating mesothelial to mesenchymal transition. Am J Respir Cell Mol Biol 66: 171–182, 2021. doi: 10.1165/rcmb.2021-0175OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian G, Adeyanju O, Sunil C, Huang SK, Chen SY, Tucker TA, Idell S, Guo X. Dedicator of cytokinesis 2 (DOCK2) deficiency attenuates lung injury associated with chronic high-fat and High-fructose diet-induced obesity. Am J Pathol 192: 226–238, 2022. doi: 10.1016/j.ajpath.2021.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian G, Adeyanju O, Roy S, Sunil C, Jeffers A, Guo X, Ikebe M, Idell S, Tucker TA. DOCK2 Promotes Pleural Fibrosis by Modulating Mesothelial to Mesenchymal Transition. Am J Respir Cell Mol Biol 66: 171–182, 2022. doi: 10.1165/rcmb.2021-0175OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo X, Sunil C, Adeyanju O, Parker A, Huang S, Ikebe M, Tucker TA, Idell S, Qian G. PD-L1 mediates lung fibroblast to myofibroblast transition through Smad3 and β-catenin signaling pathways. Sci Rep 12: 3053, 2022. doi: 10.1038/s41598-022-07044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin W, Jeffers A, Owens S, Chauhan P, Komatsu S, Qian G, Guo X, Ikebe M, Idell S, Tucker TA. NOX1 promotes mesothelial-mesenchymal transition through modulation of ROS-mediated signaling. Am J Respir Cell Mol Biol 64: 492–503, 2021. doi: 10.1165/rcmb.2020-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang S, Wettlaufer SH, Hogaboam C, Aronoff DM, Peters-Golden M. Prostaglandin E2 inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am J Physiol Lung Cell Mol Physiol 292: L405–L413, 2007. doi: 10.1152/ajplung.00232.2006. [DOI] [PubMed] [Google Scholar]
- 29.Qian G, Guo J, Vallega KA, Hu C, Chen Z, Deng Y, Wang Q, Fan S, Ramalingam SS, Owonikoko TK, Wei W, Sun SY. Membrane-associated RING-CH 8 functions as a novel PD-L1 E3 ligase to mediate PD-L1 degradation induced by EGFR inhibitors. Mol Cancer Res 19: 1622–1634, 2021. doi: 10.1158/1541-7786.MCR-21-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian G, Wang D, Magliocca KR, Hu Z, Nannapaneni S, Kim S, Chen Z, Sun SY, Shin DM, Saba NF, Chen ZG. Human papillomavirus oncoprotein E6 upregulates c-Met through p53 downregulation. Eur J Cancer 65: 21–32, 2016. doi: 10.1016/j.ejca.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng L, Qian G, Zhang S, Zheng H, Fan S, Lesinski GB, Owonikoko TK, Ramalingam SS, Sun SY. Inhibition of mTOR complex 1/p70 S6 kinase signaling elevates PD-L1 levels in human cancer cells through enhancing protein stabilization accompanied with enhanced β-TrCP degradation. Oncogene 38: 6270–6282, 2019. doi: 10.1038/s41388-019-0877-4. [DOI] [PubMed] [Google Scholar]
- 32.Qian G, Yao W, Zhang S, Bajpai R, Hall WD, Shanmugam M, Lonial S, Sun SY. Co-inhibition of BET and proteasome enhances ER stress and Bim-dependent apoptosis with augmented cancer therapeutic efficacy. Cancer Lett 435: 44–54, 2018. doi: 10.1016/j.canlet.2018.07.033. [DOI] [PubMed] [Google Scholar]
- 33.Weiss CH, Budinger GR, Mutlu GM, Jain M. Proteasomal regulation of pulmonary fibrosis. Proc Am Thorac Soc 7: 77–83, 2010. doi: 10.1513/pats.200906-055JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen DH, Zhou T, Shu J, Mao JH. Quantifying chromogen intensity in immunohistochemistry via reciprocal intensity. Cancer InCytes 2, 2013. doi: 10.1038/protex.2013.097. [DOI] [Google Scholar]
- 35.Girard RA, Chauhan PS, Tucker TA, Allen T, Kaur J, Jeffers A, Koenig K, Florova G, Komissarov AA, Gaidenko TA, Chamiso MB, Fowler J, Morris DE, Sarva K, Singh KP, Idell S, Idell RD. Increased expression of plasminogen activator inhibitor-1 (PAI-1) is associated with depression and depressive phenotype in C57Bl/6J mice. Exp Brain Res 237: 3419–3430, 2019. doi: 10.1007/s00221-019-05682-0. [DOI] [PubMed] [Google Scholar]
- 36.Osborn-Heaford HL, Ryan AJ, Murthy S, Racila AM, He C, Sieren JC, Spitz DR, Carter AB. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J Biol Chem 287: 3301–3312, 2012. doi: 10.1074/jbc.M111.308387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murthy S, Adamcakova-Dodd A, Perry SS, Tephly LA, Keller RM, Metwali N, Meyerholz DK, Wang Y, Glogauer M, Thorne PS, Carter AB. Modulation of reactive oxygen species by Rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 297: L846–L855, 2009. doi: 10.1152/ajplung.90590.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji L, Chen Y, Xie L, Liu Z. The role of Dock2 on macrophage migration and functions during Citrobacter rodentium infection. Clin Exp Immunol 204: 361–372, 2021. doi: 10.1111/cei.13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Rouhi O, Chen H, Ramirez R, Borgia JA, Deng Y. RNA-Seq and network analysis revealed interacting pathways in TGF-β-treated lung cancer cell lines. Cancer Inform 13: 129–140, 2014. doi: 10.4137/CIN.S14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Broekelmann TJ, Limper AH, Colby TV, McDonald JA. Transforming growth factor β1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci USA 88: 6642–6646, 1991. doi: 10.1073/pnas.88.15.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costanza B, Umelo IA, Bellier J, Castronovo V, Turtoi A. Stromal modulators of TGF-β in cancer. J Clin Med 6: 7, 2017. doi: 10.3390/jcm6010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finnson KW, Almadani Y, Philip A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: mechanisms and targets. Semin Cell Dev Biol 101: 115–122, 2020. doi: 10.1016/j.semcdb.2019.11.013. [DOI] [PubMed] [Google Scholar]
- 43.Betensley A, Sharif R, Karamichos D. A systematic review of the role of dysfunctional wound healing in the pathogenesis and treatment of idiopathic pulmonary fibrosis. J Clin Med 6: 2, 2016. doi: 10.3390/jcm6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo X, Sunil C, Qian G. Obesity and the development of lung fibrosis. Front Pharmacol 12: 812166, 2021. doi: 10.3389/fphar.2021.812166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol 83: 111–119, 2002. doi: 10.1046/j.1365-2613.2002.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X, Su Y, Wu K, Pan F, Wang A. DOCK2 contributes to endotoxemia-induced acute lung injury in mice by activating proinflammatory macrophages. Biochem Pharmacol 184: 114399, 2021. doi: 10.1016/j.bcp.2020.114399. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Wang Y, Wu G, Xiong W, Gu W, Wang CY. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res 19: 170, 2018. doi: 10.1186/s12931-018-0864-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 and Supplemental Figs. S1–S9: https://doi.org/10.6084/m9.figshare.19169975.