

Abstract
mRNA vaccines have recently proved to be highly effective against SARS-CoV-2. Key to their success is the lipid-based nanoparticle (LNP), which enables efficient mRNA expression and endows the vaccine with adjuvant properties that drive potent antibody responses. Effective cancer vaccines require long-lived, qualitative CD8 T cell responses instead of antibody responses. Systemic vaccination appears to be the most effective route, but necessitates adaptation of LNP composition to deliver mRNA to antigen-presenting cells. Using a design-of-experiments methodology, we tailored mRNA-LNP compositions to achieve high-magnitude tumor-specific CD8 T cell responses within a single round of optimization. Optimized LNP compositions resulted in enhanced mRNA uptake by multiple splenic immune cell populations. Type I interferon and phagocytes were found to be essential for the T cell response. Surprisingly, we also discovered a yet unidentified role of B cells in stimulating the vaccine-elicited CD8 T cell response. Optimized LNPs displayed a similar, spleen-centered biodistribution profile in non-human primates and did not trigger histopathological changes in liver and spleen, warranting their further assessment in clinical studies. Taken together, our study clarifies the relationship between nanoparticle composition and their T cell stimulatory capacity and provides novel insights into the underlying mechanisms of effective mRNA-LNP-based antitumor immunotherapy.
Keywords: LNP, mRNA, cancer, immunotherapy, vaccination, design-of-experiments methodology, extrahepatic delivery
Graphical abstract
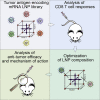
Using design-of experiments methodology, mRNA-lipid-based nanoparticle compositions conferring strong antitumor CD8 T cell responses after systemic administration were identified. Increased immunogenicity depended on mRNA expression by splenic antigen-presenting cells and induction of type I interferon. The study provides novel insights into the underlying mechanisms of effective mRNA-based antitumor immunotherapy.
Introduction
Messenger RNA (mRNA) vaccines are extremely versatile, as mRNA sequences can be easily tailored to encode any antigen of interest, enabling both rapid and mass-scale vaccine development against emerging pathogens as well as personalized vaccine design against cancers. Key to the success of the currently approved SARS-CoV2 mRNA vaccines and of many mRNA-based prophylactic vaccines in clinical development are lipid-based nanoparticles (LNPs), delivery vehicles that mediate efficient mRNA expression in situ and endow the vaccine with intrinsic adjuvant properties.1 LNPs are composed of an ionizable lipid, a phospholipid, cholesterol, and a PEGylated lipid, with the ionizable lipid being considered the most important driver of mRNA expression. In contrast to their predecessors having a cationic charge across a wide variety of pH ranges, ionizable lipids are pH-sensitive lipids that endow the LNPs with a positive, membrane fusion-promoting charge at endosomal pH while having a neutral charge at physiological pH.2 The ionizable lipid is required to encapsulate the mRNA and drives endosomal escape. The PEGylated lipid improves LNP stability and controls the interaction of the LNPs with blood proteins and cells. Cholesterol and the phospholipid contribute to LNP stability and endosomal membrane destabilization.2 Intramuscular3 or subcutaneous4 administration of mRNA-LNPs has been reported to elicit CD8 T cell responses against the mRNA encoded antigen. In these strategies, mRNA is delivered to local antigen-presenting cells (APCs), resulting in low to moderate antigen-specific CD8 T cell levels in the circulation. However, to effectively combat tumors (especially at distant locations) potent, long-lasting, and systemic CD8 T cell responses are required. Intravenous (i.v.) delivery of mRNA vaccines has been described to confer enhanced T cell responses and antitumor immunity,5, 6, 7 which is likely related to its capacity to mobilize the large pools of APCs present in the spleen. Induction of strong CD8 T cell responses has been confirmed clinically, upon i.v. immunization of metastatic melanoma patients with mRNA lipoplexes composed of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and the cationic lipid DOTMA,8 but no LNP-based formulation has yet been used in the clinic for i.v. therapeutic cancer vaccination. LNPs might show even further improved activity compared with lipoplexes given their enhanced intrinsic potential to transfect cells. Nonetheless, standard LNP formulations typically deliver RNA to hepatocytes upon i.v. delivery,9, 10, 11 instead of the APCs needed to instigate CD8 T cells. In this study, we speculated that LNPs could be tailored toward systemic vaccination through optimization of the molar ratio between the four lipids and selection of an appropriate polyethylene glycol (PEG) lipid. These parameters are known to impact LNP features such as size, ζ potential, pharmacokinetics, and biodistribution, which ultimately will determine in vivo immunogenicity.4,9,12, 13, 14, 15, 16 To design and screen a virtually unlimited variety of potential LNP compositions in a time- and cost-effective manner, we adopted a quality-by-design approach based on a statistical design-of-experiment (DOE) coupled to Bayesian regression modeling. This strategy enabled us to identify the key composition parameters that determine immunogenicity and to predict optimal LNP compositions within a single round of in vivo immunogenicity assessment. Optimal LNP compositions (within the constraints of the design space) resulted in strong CD8 T cell responses that could be boosted by repeated administration and conveyed antitumor efficacy in a syngeneic mouse TC-1 tumor model. Mechanistic studies revealed that the T cell response was dependent on initial mRNA expression by various APCs in the spleen and on the induction of type I interferons (IFNs). Surprisingly, we also discovered a previously unrecognized role of B cells in instigating antigen-specific CD8 T cell responses upon intravenous mRNA-LNP vaccination.
Results
DOE-driven optimization of LNP composition maximizes T cell responses
We aimed to maximize the T cell response elicited by antigen-encoding mRNA-LNPs composed of the ionizable lipid Coatsome SS-EC,17 cholesterol, DOPE, and a PEGylated lipid. The human papillomavirus 16 (HPV16) oncoprotein E7 was used as the antigen of choice. HPV16 is a frequent cause of cervical, anal, and oropharyngeal cancer. The oncogenic potential of HPV is driven by persistent expression of the viral oncoproteins E6 and E7, which cause degradation of the tumor-suppressor proteins p53 and Rb.18 Whereas E6 is not immunogenic in standard inbred mice strains,19 E7 contains a well-established class I epitope recognized in C57BL/6 mice, making it a highly relevant and suitable antigen to assess the potency of vaccine platforms in mice. Using DOE methodology, an LNP library was designed to address the impact of lipid molar ratio and PEG-lipid chemistry on the T cell response. Three PEGylated lipids (DMG-PEG2000, DSG-PEG2000, and DSPE-PEG1000) that differ in length and chemistry of the acyl chain and/or the molecular weight of PEG were explored. Per PEG-lipid chemistry, LNPs were formulated at 11 different molar ratios of the four lipid components, yielding a library of 33 LNPs (Table S1). These 11 molar ratios were derived from a Roquemore hybrid design20 in which the molar percentage of ionizable lipid, DOPE, and PEG lipid each had five levels. Cholesterol was used as a “filler” variable that completed the total molar percentage to 100% (Figure 1A). This design is particularly suited for the generation of quadratic response surface models and more economic than traditional central composite or Box-Behnken designs (15 and 13 conditions required, respectively). The composition and properties (size, polydispersity index, percentage of mRNA, encapsulation, and ζ potential) of the LNPs are shown in Table S1. All LNPs remained stable at 4°C for 1 month (Figure S1). LNPs were administered i.v. three times at weekly intervals. Five days after the third injection, the percentage of E7-specific CD8 T cells in blood was determined. Data were subjected to Bayesian statistical modeling to identify the LNP components critical for activity (Figure 1B). Results showed that the magnitude of the CD8 T cell response heavily depends on the LNP composition (Figure 1C). Bayesian effect analysis (supplemental methods) identified PEG-lipid chemistry and molar percentage of PEG lipid as the most critical LNP attributes. In the cases of DSG-PEG2000- and DSPE-PEG1000-based LNPs, the percentage of ionizable lipid also had a critical impact. Interaction effects were found between the percentages of DOPE and the PEG lipids, indicating that LNP optimization required fine-tuning of all lipid components simultaneously. Bayesian regression modeling was applied to predict the immunogenicity of LNP compositions not included in our initial library (Figure 1D). For all PEG-lipid types, the predicted optimal T cell response region corresponded to high percentages of the ionizable lipid SS-EC (>50%) and low percentages of PEG lipid (<0.6%) and DOPE (<10%). To validate the predictive value of the models, we tested six new LNP compositions (LNP34–LNP39, Table S1) in vivo. T cell responses higher than or equal to the best performers of the original library were predicted for mice immunized with LNP34 (DMG-PEG2000), LNP36 (DSG-PEG2000), and LNP38 (DSPE-PEG1000) (optimal LNPs), whereas poor T cell responses were predicted for LNP35 (DMG-PEG2000), LNP37 (DSG-PEG2000), and LNP39 (DSPE-PEG1000) (non-optimal LNPs). The experimental data largely matched the predictions and validated the models in the cases of DMG-PEG2000 and DSG-PEG2000 LNPs (Figures 1E and 1G; supplemental methods). In contrast to the predictions, LNP39 elicited T cell responses nearly equal to those of LNP38 (Figure 1F), meaning that the DSPE-PEG1000 LNP model could not be validated. Because of this unpredictable behavior and visual toxicity in mice (lethargy, pain symptoms) DSPE-PEG1000 LNPs were excluded from further assessment.
Figure 1.

LNP composition was optimized for maximal T cell responses using DOE methodology
(A) Plot showing the uniform distribution of the 11 LNP compositions (with variable lipid ratios) over the experimental domain. An LNP library was prepared covering these 11 lipid ratios for three different PEG lipids (DMG-PEG2000, DSG-PEG2000, and DSPE-PEG1000), yielding a total of 33 LNPs. (B) Schematic representation of LNP optimization approach. (C) Percentage of E7-specific CD8 T cells in blood after three immunizations (weekly intervals, 10 μg of E7 mRNA) with the indicated mRNA-LNP compositions, as determined by MHC class I tetramer stain (n = 3). E7 mRNA was used as antigen of choice encoded by the mRNA. (D) Response surface plots modeled from the data in (C), showing LNP compositions with high (green) and low (red) predicted capacity to elicit CD8 T cell responses. (E and F) Percentage of E7-specific CD8 T cells in blood measured after three immunizations (weekly intervals, 10 μg of E7 mRNA) with predicted optimal and non-optimal DMG-PEG2000, DSG-PEG2000 (E), and DSPE-PEG1000 (F) LNPs. (G) Representative flow cytometry scatterplots of E7-specific CD8 T cells in blood after three immunizations (weekly intervals, 10 μg of E7 mRNA) with LNP34 (optimal) or LNP35 (non-optimal). In (C), (E), and (F), mean ± SD is shown with individual data points in red. In (E) and (F), statistics were assessed by one-way ANOVA with Sidak’s multiple comparison test. ∗∗∗p < 0.001; ns, not significant.
mRNA-LNP vaccines induce qualitative T cell responses and tumor regression
The success of cancer immunotherapy is determined by a multitude of factors, including the T cell phenotype and functionality. We first assessed the quality of the T cell response evoked by the optimal LNPs (LNP34 and LNP36). Therefore, mice received three weekly immunizations followed by a final immunization at day 50. E7 mRNA was supplemented with TriMix, which a mix of three mRNAs encoding murine CD70, murine CD40L, and a constitutively active TLR4, which increases the strength of the T cell response.21 Expression of all four co-formulated mRNAs was confirmed by in vitro transfection (data not shown). For both LNPs, the kinetics and phenotypic traits of the T cell response were similar. Following three immunizations with E7-TriMix LNPs, over 70% of CD8 T cells in blood were E7 specific (Figure 2A). Five weeks after the third immunization (day 49), the percentage of E7-specific CD8 T cells remained highly elevated (approximately 40%, Figure 2A) with the majority displaying a short-lived effector cell phenotype (KLRG1+ CD127−) (Figures 2B and 2C). However, a noteworthy population (∼10%) of memory precursor effector cells (KLRG1− CD127+) was present as well. Functional memory conversion was evidenced by the rapid re-expansion of E7-specific CD8 T cells after the day-50 booster immunization (Figures 2A and 2C). IFN-γ concentrations in serum increased with every immunization (Figure 2D), indicating increased systemic immune stimulation. To assess T cell functionality, we performed an intracellular cytokine staining on splenocytes obtained from mice after three immunizations. Polyfunctional CD8 T cells, which produce more than one cytokine simultaneously, are associated with better control of infectious diseases22,23 and tumors24, 25, 26 and accounted for approximately 11% of CD8 T cells for both optimal LNPs (Figures 2E and 2F). To assess whether the activity of the LNPs also extended to other antigens, mRNA encoding the MC38 colon carcinoma-derived neo-epitope Adpgk or the B16-F10-derived melanocyte differentiation antigen Trp-2 was mixed with TriMix and encapsulated in LNP36. Augmented T cell responses were observed after two immunizations with LNPs containing both types of antigen-encoding mRNAs, especially when combined with TriMix (Figure S2). Next, therapeutic antitumor efficacy was assessed in mice bearing subcutaneous TC-1 tumors, which express HPV16 E7 antigen.27 Treatment was initiated when tumors reached a mean diameter of 52 ± 4 mm3 (Figure 2G). E7-TriMix treatment resulted in profound tumor growth inhibition of the established TC-1 tumors (Figure 2H) and significantly prolonged survival time (Figure 2I), yet tumors relapsed after cessation of treatment. We additionally assessed the capacity of mRNA-LNP-elicited T cells to reach the tumor bed. Two i.v. injections with the mRNA-LNPs led to a strong infiltration of CD8+ tumor-infiltrating lymphocytes (TILs) into the tumor (Figures 2J and 2K), with over 70% being specific for E7 (Figure 2L). Treatment with LNP36 delivering TriMix without E7 or containing an irrelevant mRNA (Firefly luciferase [Fluc]) did not induce regression of tumors, nor did it result in increased percentages of E7-specific CD8 T cells in the tumor (Figures S3A and S3B). To understand why tumors relapse despite the high numbers of vaccine-elicited E7-specific CD8 T cells, we compared tumor E7 expression levels in LNP36 versus Tris-buffered saline (TBS)-treated tumors. Tumors that relapsed after vaccination with E7-TriMix delivered by LNP36 showed a profound decrease in E7 expression levels compared with TBS (Figure S3C), suggesting loss of antigen expression as a major mechanism of immune escape. Moreover, at the time of relapse (day 35) tumors were less infiltrated by E7-specific CD8 TILs compared with 2 days after the second immunization (day 17, regression phase) (Figure S3B). In addition, TILs in regressing tumors showed proliferative potential (Ki67+) and cytolytic capacities (granzyme B+), whereas TILs present in relapsing tumors predominantly had a non-proliferative (Ki67−), non-cytolytic (granzyme B−) phenotype and expressed high levels of the immune-checkpoint PD-1 (Figures S3D and S3E). This “non-reactive” phenotype might be inflicted by immune-suppressive factors present in the tumor microenvironment or driven by the lack of E7 presence. Further immunophenotyping revealed no significant increases in the levels of myeloid-derived suppressor cells, regulatory T cells, or M2 macrophages in relapsing tumors compared with regressing tumors (Figure S3F), arguing against a major role of these cells in the observed tumor escape.
Figure 2.

Optimal mRNA-LNPs induce qualitative T cell responses and strong antitumor efficacy
(A) Mice were immunized with optimal LNPs encapsulating 5 μg of E7 and 5 μg of TriMix mRNA at days 0, 7, 14, and 50. Blood CD8 E7-specific T cell responses were determined at days 5, 20, 49, and 55. (B and C) Differential expression of KLRG1 and CD127 on blood E7-specific (+) and non-specific (−) CD8 T cells at day 49 and day 55. Representative contour plots (B, day 49) and quantification (C, mean ± SD [n = 4]) of KLRG1 and CD127 expression are shown. (D) IFN-γ levels in serum were measured 6 h after each immunization (n = 4–6). (E and F) IFN-γ and TNF-α expression by splenic CD8 T cells after three immunizations with TBS or mRNA-LNPs (weekly intervals, 10 μg of E7-TriMix). Representative contour plots (E) and quantification (F: mean ± SD [n = 3 for TBS, n = 5 for LNPs]) is shown. (G) When TC-1 tumors reached a mean volume of 52 mm3, mice were injected i.v. with 5 μg of E7-TriMix mRNA-LNPs (weekly intervals). (H) TC-1 growth curves in mice. Data are shown as mean ± SEM (n = 6–7). (I) Kaplan-Meier survival curves of TC-1-bearing mice. (J and K) Representative contour plots (J) and graph (K, mean ± SD [n = 4] with individual data points shown in red) quantifying the infiltration of CD8 T cells in TC-1 tumors. (L) E7 specificity of CD8 TILs in TC-1 tumors. Data are shown as mean ± SD with individual data points in red (n = 4). Statistical differences were assessed using a Mantel-Cox log-rank test with Bonferroni multiple testing correction (I), one-way ANOVA with Tukey post hoc test (J), or two-way Student’s t test (L).∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
Early LNP association with splenic APCs correlates with immunogenicity
To address whether the magnitude of the T cell responses after the third immunization can be correlated to the initial biodistribution of mRNA uptake and expression at organ level and cell-type level, we encapsulated a mixture of Cy5-labeled (25% substituted cyanine-5 uridine-5′-triphosphate [Cy5-UTP], 75% substituted 5-methoxy UTP), and unlabeled (and without nucleoside modification) Fluc mRNA in the LNPs screened in the original immunogenicity assessment (Figure 3A). Fluc activity was measured in liver, spleen, lung, heart, and kidney homogenates 4 h after LNP injection. LNP composition had a strong impact on the intensity and organ specificity of mRNA expression (Figure 3B). Across the library, Fluc expression was highest in the liver, followed by the spleen (Figure 3C), but the liver-to-spleen ratio differed strongly between LNPs. The magnitude of the E7-specific CD8 T cell responses positively correlated with absolute (DSG-PEG2000 LNPs) and relative (DMG-PEG2000 LNPs) spleen expression (Table S2). The importance of organ-specific mRNA delivery was further highlighted by the absence of correlation between total expression and T cell responses (Table S2). Significant correlations were also identified between LNP size (in turn strongly dependent on lipid composition) and relative expression in the spleen (Figure S4A) and between LNP size and the E7-specific CD8 T cell response (Figure S4B). Using Cy5-labeled mRNA as a fluorescent reporter molecule, we further assessed whether T cell responses can be linked to early (4 h post administration) mRNA uptake by specific immune cell types in the spleen (Figure 3D). Significant correlations were observed between the T cell response and early mRNA association with splenic monocytes, macrophages, plasmacytoid dendritic cells (pDCs), and B cells (Figure 3E). In contrast, such correlations could not be established between T cell responses and mRNA association with non-parenchymal liver cells (Figures S4C and S4D).
Figure 3.

LNP immunogenicity correlates with splenic immune cell association
(A) A mixture of 5 μg Cy5-labeled (25% substituted Cy5-UTP, 75% substituted 5-methoxy UTP) and 5 μg unlabeled (and without nucleoside modifications) Fluc mRNA in LNPs was injected i.v. (n = 2–4). After 4 h, kidneys, lungs, heart, liver, and spleen were isolated. A part of spleens and livers was kept for immediate flow cytometry analysis to assess Cy5 association with multiple cell types. Remaining tissues were homogenized and assayed for luciferase activity. (B) Luciferase activity per mg of tissue. (C) Luciferase activity (expressed as percent of luciferase activity per mg in all assayed organs) was mainly localized in liver and spleen. (D) Association of Cy5-mRNA with immune cell types in the spleen, expressed as difference in Cy5 mean fluorescence intensity between cells of LNP-injected mice and vehicle-injected mice (ΔCy5 MFI). (E) Correlations of ΔCy5 MFI after first LNP administration with T cell response after three immunizations (Figure 1C) were strongest for monocytes, pDCs, macrophages, and B cells based on Pearson's correlation coefficients. In (B), (C), and (D), mean ± SD is shown. RLU, relative light unit.
Optimal LNP compositions increase mRNA uptake by splenic immune cells and trigger enhanced innate responses
To further validate these findings, we compared the organ-specific and splenic cell distribution of the optimal, highly immunogenic LNPs (LNP34 and LNP36) with the non-optimal, poorly immunogenic LNPs (LNP35 and LNP37) following the same method as for the DOE library LNPs (Figure 3A). Compared with the non-optimal LNPs, LNP34 and LNP36 showed increased splenic mRNA expression (Figure 4A) and mRNA association with splenic macrophages, monocytes, B cells, and dendritic cells (DCs) (Figure 4B). The increased association of optimal LNPs with innate immune cells translated to stronger innate activation, illustrated by enhanced IFN-α and IFN-γ-induced protein 10 (IP-10) levels in blood (Figure 4C) and invigorated expression of the co-stimulatory molecules CD86 (Figures 4D, S5A, and S5B) and CD40 (Figures S5A and S5B) on conventional DC (cDC1 and cDC2) subsets. To fully elucidate which splenic cell types translate the LNP-delivered mRNA, we injected LNPs with Cre recombinase mRNA into ROSA-loxP-STOP-loxP-tdTomato (Ai9) mice (Figure 4E). Of the cell types analyzed, DCs and macrophages showed the highest percentage of transfected cells (Figure 4F). These cell types are potent APCs and hence presumably contribute to the observed T cell responses elicited by antigen mRNA-LNPs. Despite their strong association with Cy5 mRNA, mRNA expression by monocytes and B cells was comparatively low, which might reflect either limited mRNA internalization or poor endosomal release in these cell types.
Figure 4.

Optimal LNPs show increased splenic immune cell transfection and activation compared with non-optimal LNPs
(A) Fluc expression distribution over organs 4 h after injection of 10 μg of Fluc mRNA encapsulated in optimal (LNP34 and LNP36) or non-optimal LNPs (LNP35 and LNP37). Mean ± SD is shown (n = 3). (B) Association of optimal LNPs with various splenic immune cell subsets is stronger compared with non-optimal LNPs. Mean ± SD is shown with individual data points in red (n = 3). (C) Transient increases in IFN-α and IP-10 cytokine levels in serum were observed after injection of optimal, but not suboptimal LNPs (n = 5 for E7 LNPs [10 μg], n = 3 for TBS). (D) CD86 expression on splenic cDC1 and cDC2 is weakly upregulated by non-optimal LNPs and strongly upregulated by optimal LNPs (n = 5 for E7-TriMix LNPs [5 μg], n = 4 for TBS). (E) Cre mRNA (10 μg) in optimal LNPs was injected i.v. into transgenic Cre reporter mice (ROSA-loxP-STOP-loxP-tdTomato). After 48 h, spleens were isolated and assayed for tdTomato fluorescence in immune cell types. (F) In spleen, optimal LNPs mainly transfected DCs and macrophages, as indicated by the difference between the percentage of tdTomato-positive cells in LNP-injected mice and the average of TBS-injected mice for each cell population (ΔtdTomato+ (%)). Mean ± SD is shown (n = 3). In (B), (C), and (F) Statistics were assessed by one-way (B, C) or two-way (F) ANOVA with Sidak’s multiple comparison test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant. RLU, relative light unit. Blue background: DMG-PEG2000-based LNPs, Pink background: DSG-PEG2000-based LNPs.
PEG-lipid chemistry differentially affects LNP biodistribution upon repeated administration
Intravenous administration of mRNA-LNPs28 has been reported to trigger antibody production against the PEG moiety, a response driven by the activation of splenic PEG-specific B cells.29, 30, 31 Presence of anti-PEG antibodies has presumably opposing impacts on the T cell response: it results in decreased expression of the delivered mRNA but also shifts LNP uptake from hepatocytes to phagocytic cells14 that can function as APCs. We therefore determined whether the optimized LNPs we identified—characterized by different PEG-lipid chemistries—induce the generation of anti-PEG antibodies and whether this impacts the intensity and biodistribution of mRNA expression upon repeated administration. I.v. administration of LNP34 and LNP36 provoked a pronounced B cell activation, as evidenced by a strong upregulation of the B cell activation markers CD69 (Figure 5A), CD86, and CD40 (Figure S5C), with the presence of mRNA in the LNPs being a clear prerequisite for B cell activation. Anti-PEG immunoglobulin M (IgM) and IgG became detectable in the serum of mice after respectively the first and second immunization with E7-TriMix LNPs (Figure 5B), and antibodies were still readily detectable 40 days after the last immunization (Figure S6A). Titers were more elevated after treatment with LNP36 compared with LNP34, especially at the later time point (Figure S6A). We observed reduced mRNA expression upon weekly administrations of Fluc mRNA in LNP34 and LNP36, both in liver and spleen (Figure 5C). In the case of DSG-PEG2000-containing LNP36, this reduced expression was accompanied by a relative shift in mRNA expression from liver to spleen, with the spleen/liver ratio increasing from 0.8 at the first injection to 3 after the third injection. The relative increase in splenic expression observed after the third injection was mirrored by an increased association of Cy5-mRNA with splenic macrophages, monocytes, DCs, and B cells (Figure 5D). Despite a drop in the intensity of mRNA expression, the relative distribution of mRNA uptake and expression at the organ and cellular level, in contrast, remained relatively constant in the case of the DMG-PEG2000-stabilized LNP34 (Figures 5C and 5D). We next assessed whether the differential behavior of LNP36 compared with LNP34 could be explained by qualitative changes in the protein corona adsorbed to the LNPs upon repeated administration. LNP34 and LNP36 were incubated with plasma from either naive mice or mice immunized twice with the matching LNPs (“immune plasma”) and subsequently recovered using differential centrifugation. Proteomics analysis identified abundant plasma proteins, i.e., albumin, apolipoprotein E and several immunoglobulins, in all samples (Figure S6B). Differential enrichment analysis showed different intensity of 25 precipitated proteins when LNP36 was incubated with plasma samples from LNP36-immunized mice compared with plasma of naive mice. On the other hand, only one protein was differentially enriched upon incubation of LNP34 with plasma from LNP34-immunized mice versus plasma from naive mice. The enriched proteins after incubation with immune plasma clustered together and were highly distinct from the proteins pelleted after incubation with naive plasma (Figure 5E), although the differences were clearly more pronounced for LNP36 compared with LNP34. Out of the 25 differentially enriched proteins identified for LNP36, 23 were increased in immune plasma compared with naive plasma and all represented immunoglobulins (Figure 5F). In the case of LNP34, Ighv3-6 was the only Ig family member enriched after incubation with immune plasma (Figure 5F). In addition, immunoglobulins in immune plasma make up a higher percentage of the total precipitated proteome for LNP36 compared with LNP34 (Figures S6C and S6D). We hypothesized that both LNP types bind anti-PEG antibodies, but that anti-PEG is released faster from LNP34 than from LNP36 due to the shorter acyl chain of its PEG lipid. Indeed, LNP34 and LNP36 showed a comparable initial opsonization by anti-PEG antibodies in vitro. However, upon incubation with mouse serum, the amount of anti-PEG antibody bound to LNP34 decreased over time, whereas it remained constant in the case of LNP36 (Figure S6E). The gradual loss of anti-PEG from LNP34—presumably due to shedding of the PEG lipid—coincided with an increase in LNP size (Figure S6F). In contrast, LNP36 size was relatively unaltered after 2 h incubation with serum, indicating DSG-PEG2000 was still incorporated in LNP and bound to anti-PEG antibodies. Taken together, these data strongly indicate that LNP36 becomes increasingly opsonized by antibodies upon repeated administration, potentially altering its biodistribution. In contrast, LNP34 shows only short-lived immunoglobulin opsonization.
Figure 5.

PEG-lipid chemistry differentially affects mRNA biodistribution upon repeated administration
(A) mRNA-encapsulating LNPs (5 μg E7-TriMix), but not empty LNPs (equivalent lipids to hypothetical 5 μg mRNA), activate splenic B cells 4 h after a single administration, evidenced by upregulation of CD69. Mean ± SD is shown with individual data points in red (n = 4–5). Statistics were assessed by one-way ANOVA with Tukey’s multiple comparison test. (B) Mice were immunized with 10 μg of E7-TriMix on days 0 and 7. Anti-PEG IgM and IgG titers were determined in plasma collected 6 days after each injection. Both LNPs induce the generation of anti-PEG IgM (left) and IgG (right) upon systemic injection. Mean ± SD is shown (n = 5). (C) Overall luciferase expression decreases with repeated administration of LNP34 and LNP36 encapsulating 10 μg of Fluc mRNA. Mean ± SD is shown (n = 3). Statistics were assessed by one-way ANOVA with Tukey’s multiple comparison test. (D) Splenic cellular Cy5-mRNA association upon repeated administration (5 μg Fluc + 5 μg Cy5-Fluc) remains similar for LNP34, but increases in the case of LNP36. Mean ± SD is shown (n = 3). (E and F) Cluster analysis (E) and volcano plots (F) for differential enrichment (false discovery rate 5%, log2 fold change ≥1.5; padj < 0.05) of proteins in centrifuged pellets after incubation of LNP34 or LNP36 with plasma of naive mice or mice immunized twice with the respective LNP (10 μg E7-TriMix). LLOQ, lower limit of quantification. (G) After two immunizations with a 1:1 mixture of LNPs loaded with either E6 or E7 mRNA combined with TriMix (100 μg mRNA in total) in non-human primates, both mRNAs strongly accumulated in spleen. Mean ± SD is shown with individual data points in red (n = 2). Statistics in (D) were assessed by two-way ANOVA with Tukey’s multiple comparison test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
The spleen-centered biodistribution of mRNA deposition is retained in non-human primates
To exclude species-specific differences in the biodistribution of LNPs that could hamper clinical translation, we assessed the biodistribution of LNP36-delivered mRNA in female and male cynomolgus monkeys. E6- or E7-encoding mRNA mixed with (human) TriMix were formulated in LNP36 and co-administered i.v. two times with a 1-week interval. The LNPs were well tolerated with no histopathological changes observed in major vital organs. Twenty-four hours after the second administration, tissues were collected to quantify mRNA content. Both mRNAs were mostly present in the spleen (based on pg mRNA per g of tissue), followed by the liver, bone marrow, draining lymph node, and lung (Figure 5G), irrespective of the administered dose (Figure S7). E6- and E7-mRNA levels in brain, heart, kidney, and adrenal gland were below quantifiable levels. Taken together, the spleen-centered biodistribution, which is linked to strong T cell responses in rodents, is preserved in non-human primates and warrants further clinical translation.
IFNAR signaling, phagocytes, and B cells are essential for eliciting mRNA-LNP-induced T cell responses
We next sought to determine whether the observed type I IFN response is vital for the T cell response elicited. Type I IFNs are pleiotropic cytokines that are typically induced upon sensing (viral) RNAs. Opposing effects of type I IFNs on the T cell response induced by mRNA vaccines have been reported.5, 6, 7,32, 33, 34 As shown in Figure 4C, intravenous injection of E7 mRNA delivered by LNP34 and LNP36 resulted in a robust type I IFN response. This response was found to be critical for achieving T cell immunity, as blockade of the IFN-α/β receptor (IFNAR) prior to immunization—abrogating type I IFN signaling—had a profound negative impact on the CD8 T cell response (Figures 6A and 6B). In a parallel setup, we depleted pDCs (>80% depletion, Figures S8A and S8B), a cell type capable of producing large amounts of type I IFN in response to RNA sensing.35 Although pDC depletion resulted in reduced yet not fully abrogated IFN-α titers in serum (Figure S8C), the impact of pDC depletion on T cell responses was moderate to non-significant in the case of LNP34 or LNP36, respectively. As our biodistribution studies showed correlations between T cell responses and early LNP association with phagocytic cells and B cells (Figure 3E), we decided to further assess the roles of these immune cells in the LNP-induced T cell response. Clodronate liposomes were used to deplete phagocytes in liver and spleen prior to each immunization.36 We confirmed that this treatment resulted in strong reductions of macrophages in both organs, but also reduced numbers of monocytes and DCs in spleen (Figure S8D). After LNP immunization, strongly reduced E7-specific CD8 T cell responses were measured in the clodronate-pretreated mice (Figure 6D), indicating a vital role for phagocytic cell types in modulating T cell responses to mRNA-LNPs, either by antigen presentation or immunomodulation through cytokine production. However, cytokine levels were similar in clodronate-treated mice compared with wild-type mice after LNP immunization, supporting the former mechanism (Figure S6E). Given the association of mRNA with B cells observed in the biodistribution study, we also assessed the involvement of B cells in the T cell response via comparative immunization studies in wild-type mice versus μMT mice, which lack mature B cells.37 With LNP-binding antibodies resulting in an increased uptake by splenic APCs upon repeated immunization with LNP36 but not LNP34, we speculated that B cells could be major mediators of the T cell response evoked by LNP36, yet not by LNP34. Surprisingly, however, μMT mice showed strongly reduced E7-specific CD8 T cell responses after immunization with both LNPs compared with wild-type mice, indicating a role of B cells in the T cell response that extends beyond antibody opsonization and altering biodistribution upon repeated immunization (Figure 6E). Of note, μMT mice showed preserved innate immune activation after mRNA-LNP injection compared with wild-type mice (Figure S8E), suggesting that the lack of response to mRNA-LNP immunization is not due to complete immunodeficiency.
Figure 6.

CD8 T cell responses are dependent on IFNAR signaling, phagocytes, and B cells
(A) Mice were pretreated with isotype antibodies, anti-CD317 pDC depleting antibody, anti-IFNAR blocking antibody, or clodronate liposomes. μMT mice were used as B cell knockout model. After two or three i.v. immunizations with 10 μg of E7-TriMix mRNA-LNP (weekly intervals), CD8 T cell responses were assessed in blood. (B) IFNAR blocking by i.p. administration of anti-IFNAR antibodies abrogated the E7-specific CD8 T cell response in blood after three immunizations. (C) Depletion of pDCs by i.p. administration of anti-CD317 antibodies had minor negative impact on the percentage of E7-specific CD8 T cells in blood after three immunizations. (D and E) Absence of phagocytes (clodronate-pretreated) (D) or B cells (knockout) (E) strongly reduced the percentage of E7-specific CD8 T cells in blood after two immunizations. In (B), (C), (D), and (E), mean ± SD is shown (n = 4–5). Statistics were assessed by two-way ANOVA with Sidak’s multiple comparison test. ∗p < 0.05, ∗∗∗p < 0.001; ns, not significant.
Discussion
Although LNPs are highly efficient carriers for mRNA delivery, they are not a one-size-fits-all solution, and their composition needs to be tailored toward the route of administration and the desired biological activity.15,16,38, 39, 40 Here, we investigated whether mRNA-LNPs could be designed to induce strong CD8 T cell responses upon intravenous immunization by tailoring lipid ratios and PEG-lipid chemistry. A DOE methodology was used to set up and screen an initial library of 33 LNPs for their capacity to evoke CD8 T cell responses. Maximized T cell responses were predicted and confirmed for LNPs composed of low percentages of PEG lipid, high percentages of SS-EC ionizable lipid, and low percentages of DOPE. An increased T cell response by reduced mol % of PEG lipid is likely caused by the combined effects of reduced shielding and increased size resulting in increased LNP uptake by phagocytes,41 as we observed in the biodistribution studies. Aside of the chemistry of the ionizable lipid used, our initial library was highly similar, in terms of lipid ratios assessed, to the initial library described by Kauffman et al.9 (DOPE: 4%–16%; DMG-PEG2000: 0.5%–2.5%), who optimized erythropoietin (EPO)-encoding mRNA-LNP compositions for maximal hepatic EPO production. Interestingly, the different goals and readouts of both studies led to the identification of different critical LNP attributes, second-order interactions, and predicted optimal LNP compositions. In the study of Kauffman et al., LNPs that were selected as optimal performers (i.e., achieving highest hepatic EPO production) contained high molar percentages of DOPE and PEG lipid and low percentages of ionizable lipid, which is opposite to the optimal mRNA-LNP composition we identified. Hence, DOE enables optimization of LNP compositions for the intended biological activity. Moreover, the design we employed proved to be cost effective, allowing single-step fine-tuning of all lipid components simultaneously in vivo with as little as 11 LNP compositions per PEG lipid. Early biodistribution of LNPs was linked to the magnitude of the T cell response, with mRNA uptake by splenic immune cells, including macrophages, monocytes, and pDCs, correlating positively with the T cell response evoked. By administering Cre mRNA formulated in optimal LNPs to tdTomato reporter mice, we identified splenic DCs and macrophages as the cell types showing the highest percentages of mRNA-expressing cells. Optimal LNPs led to an increased functional maturation of DCs in the spleen and to increased production of T cell stimulatory cytokines and chemokines such as IFN-α and IP-10. Opposing effects of type I IFNs on the T cell response induced by mRNA vaccines have been reported.5, 6, 7,32, 33, 34 Type I IFN can promote DC activation and T cell differentiation but can also interfere with the expression of the mRNA encoded protein by inducing RNase activity and instigating translational arrest. A favorable impact of type I IFN is expected when type I IFN induction and T cell receptor triggering occurs simultaneously, i.e., by systemic immunization.34 In this setting, blocking the type I IFN receptor IFNAR at the time of immunization had a strongly detrimental impact on the magnitude of the antigen-specific CD8 T cell response. Because the biodistribution studies also indicated a positive correlation between early mRNA association with splenic B cells and T cell response, we assessed the involvement of B cells at instigating the antigen-specific CD8 T cell response by conducting vaccination studies in μMT mice, which lack mature B cells. Surprisingly, the CD8 T cell response was nearly fully abrogated in these mice, pinpointing a crucial role of B cells in priming and/or expanding CD8 T cell responses. As B cells are not the typical APCs involved in priming CD8 T cells, we hypothesized that B cells might promote CD8 T cell responses via a more indirect mechanism involving the induction of anti-PEG antibodies, which would lead to LNP opsonization upon repeated administration, as has been postulated for PEGylated liposomes,29,42 and subsequent increased uptake by conventional APCs such as DCs and macrophages. Repeated LNP injection indeed increased anti-PEG IgM and IgG levels in mouse serum, in line with other studies.28 Anti-PEG titers were more elevated after treatment with LNP36 compared with LNP34, likely resulting from the more stable incorporation of DSG-PEG2000 compared with DMG-PEG2000 into the LNPs’ surface, a phenomenon previously described for small interfering RNA LNPs.14 Despite this induction of anti-PEG antibodies in mice, the vaccine-evoked T cell response was easily boostable by repeated immunization, which is of key importance given the increasing prevalence of anti-PEG antibodies in humans43 and a substantial portion of the global population at risk of acquiring anti-PEG antibodies after vaccination with PEGylated LNPs in the context of the COVID-19 pandemic. We also evaluated how the protein corona of LNP34 and LNP36 changed upon repeated administration. For the DSG-PEG2000 stabilized LNP36, we observed a strong enrichment of immunoglobulins in the protein corona after repeated administration, which was absent in case of the DMG-PEG2000 stabilized LNP34. In line with these findings, LNP36 showed a shifted distribution toward the spleen compared with liver upon repeated injection, whereas LNP34 displayed a nearly unaltered biodistribution upon repeated administration, reflecting its minor immunoglobulin opsonization. Nonetheless, compared with wild-type mice, B cell knockout mice (μMT) showed strongly reduced E7-specific CD8 T cell responses after immunization with both LNPs, indicating a role of B cells in the T cell response that extends beyond antibody opsonization and altering biodistribution upon repeated immunization. The role of B cells in promoting antitumoral immune responses is increasingly recognized, with recent studies showing that B cells can act as APCs to T cells.44, 45, 46, 47 Hence B cell responses could enhance T cell responses to mRNA-LNP vaccines by multiple mechanisms, ranging from cytokine production to direct roles in antigen presentation themselves. Of note, as B cells contribute to the generation and maintenance of functional CD4 and CD8 T cells48,49 and help in the organization of lymphoid organ architecture,50 it cannot be ruled out that μMT mice are intrinsically less competent in responding to mRNA-LNP immunization compared with wild-type mice. Further work is needed to unravel the exact mechanism by which B cells contribute to the generation of antigen-specific CD8 T cell responses upon systemic mRNA-LNP vaccination.
In summary, the DOE approach enabled us to identify LNP compositions that induced high-magnitude CD8 T cell responses upon i.v. administration. This increased T cell response coincided with increased mRNA expression in splenic DCs and macrophages alongside augmented DC activation and cytokine release. Type I IFNs, phagocytes, and B cells were vital in instigating the antigen-specific CD8 T cell response. Repeated immunization resulted in high-magnitude and long-lasting tumor antigen-specific CD8 T cells that showed a high degree of multifunctionality, infiltrated subcutaneous antigen-expressing tumors, and provoked high antitumor efficacy. The promising immunogenicity data in mice combined with the beneficial biodistribution/tolerability results in non-human primates warrant further assessment in clinical studies.
Materials and methods
Animals
All experiments with mice were performed with approval from the Utrecht Animal Welfare Body of the UMC Utrecht or by the Animal Ethics Committee of Ghent University. Animal care was according to established guidelines. All mice had unlimited access to water and standard laboratory animal chow. Female C57Bl/6J mice (20–25 g) were obtained from Charles River Laboratories (Germany/France). Female μMT B cell knockout and male and female B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J mice (Ai9) were obtained from The Jackson Laboratory (USA). The study of non-human primates (two male and two female cynomolgus monkeys) was performed at Charles River Laboratories (France) according to local regulations.
Design of experiments and Bayesian-based analysis
A Roquemore hybrid design was used to select the LNP compositions to test in the initial library. The investigation model used to optimize the LNP composition was described by a quadratic response surface model structure. The posterior distributions of the model parameters were computed using the Hamiltonian Monte Carlo algorithm, a generalization of the Metropolis algorithm. After qualification of the model, the latter was used to predict mean CD8 T cell responses for all combinations of the three factors (%ionizable lipid, %PEG lipid, %DOPE) and select the lipid ratio predicted to result in maximal immunogenicity. Details of the statistical model, methods, and analysis can be consulted in supplemental methods.
mRNA synthesis and purification
Codon optimized E7, Adpgk, Trp-2, TriMix, and Firefly luciferase mRNAs were prepared by eTheRNA in vitro transcription (IVT) from eTheRNA plasmids. No nucleoside modifications were incorporated. The E7 mRNA used in the DOE was capped with anti-reverse cap analog. All later experiments were performed using CleanCapped mRNAs. After IVT, double-stranded RNA was removed by cellulose purification. mRNA quality was monitored by capillary gel electrophoresis (Agilent, Belgium). Cy5-labeled Firefly luciferase (Fluc) mRNA and Cre mRNA were purchased from TriLink Biotechnologies. Uridine is fully substituted with 5-methoxyuridine in the Cre mRNA. Cy5-labeled luciferase mRNA is transcribed with cyanine 5-UTP/5-methoxy-UTP at a ratio of 1:3.
LNP production and characterization
For biodistribution and cellular uptake studies, LNPs were loaded with a mixture of Fluc encoding mRNA (eTheRNA Immunotherapies) and CleanCap Cy5-labeled Fluc mRNA (TriLink Biotechnologies) in a 1:1 ratio. For the DOE immunogenicity study, LNPs were loaded with E7 mRNA. All other studies were performed with a mixture of E7 and TriMix (CD40L, CD70, and TLR4 mRNA) in a 3:1:1:1 ratio. The mRNA was diluted in 100 mM sodium acetate buffer (pH 4), and lipids were dissolved and diluted in ethanol. A nitrogen/phosphate ratio of 16 was employed for all LNPs. The mRNA and lipid solutions were mixed using a NanoAssemblr Benchtop microfluidic mixing system (Precision Nanosystems) at a flow rate of 9 mL/min and an aqueous/organic phase flow rate ratio of 2:1, followed by dialysis overnight against TBS (20 mM Tris, 0.9% NaCl, pH 7.4). Amicon Ultra Centrifugal Filters (Merck Millipore, 100 kDa molecular weight cutoff) were used for concentration of LNPs. Size, polydispersity index, and ζ potential were measured with a Zetasizer Nano (Malvern). mRNA encapsulation efficiency was determined via Quant-iT Ribogreen RNA assay (Thermo Fisher) by comparing lysed LNPs in 1% Triton X-100 (all RNA detectable) with LNPs in TE buffer (only non-encapsulated RNA detectable). The assay was performed according to manufacturer’s recommendations. Composition and physicochemical properties of all LNPs are summarized in Table S1.
Biodistribution and cellular uptake
Mice were injected i.v. via the tail vein with 10 μg of mRNA in selected LNP formulations. After 4 h, mice were anesthetized with 250 μL of pentobarbital (6 mg/mL, intraperitoneal [i.p.]). Blood samples were collected in tubes with gel clotting factor (Sarstedt). Subsequently, the chest cavity was opened, the portal vein was cut, and mice were perfused with 7 mL of PBS through the heart’s right ventricle. Organs were excised and snap-frozen in liquid nitrogen. For liver and spleen tissues, a part of the organ was kept in ice-cold PBS for immediate flow cytometry analysis. Non-human primates received two immunizations with a 1-week interval by a 30-min infusion of LNP36 (formulated as a 1:1 mixture of LNP36 containing either E6 or E7 mRNA and TriMix at total dose of 100 μg [Figure 5G] or 40 μg [Figure S7] mRNA). Twenty-four hours after the second LNP administration adrenal gland, bone marrow, brain, heart, kidney, liver, lung, spleen, and one lymph node were collected and immediately frozen in liquid nitrogen until completely frozen and solid, then transferred to −80°C. mRNA quantification in tissues was performed using a QuantiGene Singleplex assay (Thermo Fisher) with a custom-designed probe set hybridizing to E7 mRNA. In addition, tissues were histopathologically inspected by a trained pathologist from Charles River Laboratories (France).
Cellular association
Liver and spleen tissues were placed in Petri dishes with RPMI 1640 medium containing 1 mg/mL collagenase A (Roche) or 20 μg/mL Liberase (Roche), respectively, and 10 μg/mL DNAse I, grade II (Roche). Tissues were minced using surgical blades and incubated for 30 min at 37°C. Subsequently, tissue suspensions were passed through 100-μm nylon cell strainers. Liver suspensions were centrifuged for 3 min at 70 × g to deplete parenchymal cells. Supernatants and spleen suspensions were centrifuged 7 min at 500 × g to pellet cells. Red blood cells were lysed in ACK buffer (Gibco) for 5 min, inactivated with PBS, and subsequently passed through a 100-μm cell strainer. Cells were washed with RPMI 1640 containing 1% fetal bovine serum, mixed with trypan blue, and counted using a Luna-II Automated Cell Counter (Logos Biosystems). 3 × 105 (liver) or 6 × 105 (spleen) live cells were seeded in 96-well plates, pelleted for 5 min at 500 × g, and resuspended in 2% BSA in PBS (2% PBSA) containing 50% Brilliant Stain Buffer (BD Biosciences) and 2 μg/mL TruStain FcX (BioLegend). Cells were incubated for 10 min on ice and mixed 1:1 with 2% PBSA containing applicable antibodies (Table S3) in duplicate. Cells were incubated for 15 min at room temperature on a shaker, washed two times with 2% PBSA, and resuspended in 2% PBSA containing 0.25 μg/mL 7-AAD Viability Stain (BioLegend). Samples were acquired on a 4-laser BD LSRFortessa flow cytometer. Analysis was carried out using FlowJo software. Gating strategies for all analyses are included in supplemental information.
Whole-body distribution
Approximately 50–100 mg of each tissue was dissected, weighed, and placed in 2-mL microtubes with a layer of approximately 5 mm of 1.4-mm ceramic beads (Qiagen). For each mg of tissue, 3 μL of cold Cell Culture Lysis Reagent (Promega) was added, and tissues were homogenized using a Mini-BeadBeater-8 (BioSpec) at full speed for 60 s at 4°C. Homogenates were stored at −80°C, thawed, centrifuged at 10,000 × g for 10 min at 4°C to remove beads and debris, and supernatants were stored again at −80°C. Ten microliters of each lysate was aliquoted in duplicate on a white 96-well plate. Using a SpectraMax iD3 platereader (Molecular Devices) equipped with injector, 50 μL of Luciferase Assay Reagent (Promega) was dispensed in each well while mixing, followed by a delay of 2 s and recording of luciferase emission for 10 s. Luciferase activity was normalized for background signal obtained from organ lysates of mice injected with TBS.
mRNA expression
Ai9 mice were injected intravenously in the tail vein with 10 μg of Cre mRNA encapsulated in selected LNPs. Forty-eight hours after administration, mice were anesthetized with pentobarbital and perfused with PBS as described above. Single-cell suspensions of spleen tissues were prepared and counted as described under “cellular association.” Cells were stained with FcR blocking antibodies and Zombie Aqua viability dye prior to staining with antibodies in 1% PBSA. Subsequently cells were washed two times with 2% PBSA and analyzed using a 4-laser BD LSRFortessa flow cytometer. Analysis was carried out using FlowJo software. Gating strategies are included in supplemental information.
T cell response
Mice were immunized intravenously via the tail vein with 10 μg of mRNA in selected LNPs at weekly intervals. Blood for flow cytometry stainings was collected 5–7 days after immunizations. After lysing of red blood cells, the cells were incubated with FcR block and Zombie Aqua viability dye. After incubation and washing, APC-labeled tetramer/dextramer was added (E7: RAHYNIVTF; Adpgk: ASMTNRELM; or Trp-2: SVYDFFVWL) and incubated at room temperature for 30 min. Excess tetramer was washed away, and an antibody mixture for surface molecules CD3, CD8, KLRG1, and CD127 (Table S3) was added to the cells and incubated for 30 min at 4°C. Samples were acquired on a 3-laser AttuneNxt flow cytometer or a 4-laser BD LSRFortessa flow cytometer. Intracellular cytokine production was determined in spleen 7 days after the third immunization. Single-cell suspensions of splenocytes were prepared by crushing the spleens, lysing the red blood cells, and filtering the samples over a 40-μm cell strainer. Two million cells/well/sample were plated in duplicate in a 96-well plate. Four micrograms of E7 peptide (Genscript) was added for stimulation before cells were incubated at 37°C. After 1 h of peptide stimulation, GolgiPlug (BD Cytofix/Cytoperm kit; BD Biosciences) was added. Cells were incubated for another 4 h. Thereafter, cells were incubated with FcR block and viability dye. After incubation and washing, APC-labeled E7 (RAHYNIVTF)-tetramer was added and incubated at room temperature for 30 min. Excess tetramer was washed away, and an antibody mixture for surface molecules CD3 and CD8 (Table S3) was added to the cells and incubated for 30 min at 4°C. Further steps were according to the manufacturer’s instructions of the BD Cytofix/Cytoperm kit (BD Biosciences). After permeabilization, cells were stained for IFN-γ and tumor necrosis factor α (TNF-α) (Table S3). Samples were acquired on a 4-laser BD LSRFortessa flow cytometer. Analysis was carried out using FlowJo software. Gating strategies are provided in supplemental information.
Immune cell activation
Mice were injected i.v. via the tail vein with 5 μg of E7-TriMix mRNA in selected LNPs. Spleens were harvested 4 h later for flow cytometry staining. Single-cell suspensions of splenocytes were prepared and incubated with digestion buffer (DMEM with DNAse-1 and collagenase-III) for 20 min with regular shaking. Thereafter, samples were incubated with Fc block and viability dye. After incubation and washing, cells were stained with cell lineage markers and activation markers (Table S3). Samples were acquired on a 3-laser AttuneNxt flow cytometer. Analysis was carried out using FlowJo software. Gating strategies are provided in supplemental information.
TC-1 tumor model
TC-1 cells were obtained from Leiden University Medical Center. TC-1 cells (0.5 million) in 50 μL of PBS were injected subcutaneously on the right flank of the mice. Tumor measurements were performed using a caliper. Tumor volume was calculated as (smallest diameter2 × largest diameter)/2. For the TC-1 experiment shown in Figure 2, mice were injected i.v. via the tail vein with 5 μg of E7-TriMix mRNA in LNP34 or LNP36. Intravenous immunizations were started at a mean tumor volume of 52 mm3 and performed weekly for a total of three times. For analysis of TILs, tumors were isolated 3 days after the second mRNA-LNP immunization and placed in a 24-well plate filled with MACS tissue storage buffer (Miltenyi Biotec). Tumors were minced and incubated in digestion buffer for 1 h with regular shaking. Thereafter, red blood cells were lysed and all samples were filtered over a 70-μm cell strainer. Lymphocytes were enriched by Ficoll-Paque density gradient purification before proceeding with staining. First, the cells were incubated with FcR block and viability dye. After incubation and washing, APC-labeled E7 (RAHYNIVTF) tetramer was added and incubated at room temperature for 30 min. Excess tetramer was washed away, and an antibody mixture for surface molecules CD45 and CD8 (Table S3) was added to the cells and incubated for 30 min at 4°C. Samples were acquired on a 3-laser AttuneNxt flow cytometer or a 4-laser BD LSRFortessa flow cytometer. Analysis was carried out using FlowJo software. Gating strategies are provided in supplemental information. For the TC-1 experiment shown in Figure S2, the complete method (flow cytometry and qPCR) is provided in supplemental information.
Cell depletion or receptor blocking
pDC depletion was achieved by i.p. injection of anti-CD317 depleting antibody (BioXcell), starting 1 week before the first immunization (for schedules and depletion efficiency, see Figures S8A and S8B). To deplete phagocytes, mice were injected intravenously via the tail vein with 200 μL of clodronate liposomes (Liposoma) 1 day before the immunization (for depletion efficiency, see Figure S8D). To block IFNAR, mice were injected with 100 μg of IFNAR blocking antibody (BioXcell) 1 h before each immunization.
Anti-PEG ELISA
Blood samples were collected in tubes with EDTA or clotting activator (Sarstedt) 5 days after immunization. Blood samples were centrifuged for 10 min at 1,000 × g to obtain plasma (EDTA tubes) or 5 min at 10,000 × g to obtain serum (clotting activator tubes). Samples were stored at −80°C until analysis. ELISA for anti-PEG IgM (Life Diagnostics, PEGM-1) and anti-PEG IgG (Life Diagnostics, PEGG-1) was performed according to kit protocol. Readout was carried out on a TECAN Infinite 200 Pro plate reader.
Inflammatory cytokines
Blood samples were collected in tubes with gel clotting factor (Sarstedt). Clotted blood samples were centrifuged for 5 min at 10,000 × g to obtain serum. Serum samples were stored at −80°C until analysis. ProcartaPlex multiplex assay (Thermo Fisher) was used to determine concentrations of IFN-γ, IFN-α, and IP-10. Serum samples were diluted three times in assay buffer and incubated with fluorescently labeled beads for 120 min. Further steps were performed according to protocol. Samples were acquired on a MagPix instrument (Luminex). Data were analyzed using ProcartaPlex Analyst software.
LNP protein corona
Mice were immunized two times intravenously via the tail vein with 10 μg of E7-TriMix mRNA in selected LNPs at a weekly interval. Blood samples were collected in tubes with EDTA (Sarstedt) 6 days after the second immunization. In addition, blood was collected from naive (non-immunized) mice. Blood samples were centrifuged for 10 min at 1,000 × g to obtain plasma. LNPs were prepared as described above. Plasma samples and LNPs were shipped to Karolinska Institutet (Sweden) for protein corona analysis. The full method is described in supplemental methods. In brief, 10% plasma in PBS adjusted to 10 mM sodium citrate to prevent coagulation was prepared from naive mice, LNP34--immunized, and LNP36-immunized mice. Next, LNP34 and LNP36 were mixed with 10% immune plasma (from mice immunized with the respective LNP) or naive plasma. The mixture was incubated for 1 h at 37°C. The LNP-corona complexes were spun at 20,000 × g at 4°C for 1 h, the supernatant was discarded, and the pellet was resuspended and washed twice with PBS. Next, proteins were precipitated and digested to peptides. Samples were injected into an Ultimate 3000 RSLCnano system (Dionex) using a C18 trap-column (Dionex) and an in-house packed (3 μm C18 particles, Dr. Maisch) analytical 50 cm × 75 μm internal diameter emitter column (New Objective). Peptides were eluted at 200 nL/min with an 8%–40% B 120 min gradient (buffer B: 80% acetonitrile + 0.1% formic acid; buffer A: 0.1% formic acid) to a Q Exactive Plus (Thermo Fisher Scientific) mass spectrometer. Mass spectrometric raw files were analyzed with the MaxQuant software (version 1.6.15.0). Search was performed against the UniProt Mus musculus reference proteome database. Protein and LFQ ratio count (i.e., number of quantified peptides for reporting a protein intensity) was set to 1. Differential enrichment analysis was performed on the LFQ intensity values by using differential enrichment analysis of proteomics data (DEP version 1.10.0 [3]), R version 4.0.2, and RStudio version 1.3.1073.
Statistical analysis
GraphPad Prism software 8.0 and 9.0 was used for the statistical analyses indicated in figure legends. Significant differences are indicated as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns denotes no significance.
Acknowledgments
This research was supported by the Applied and Engineering Sciences domain of the Dutch Research Council (NWO) HTSM grant (“TORNADO”) no. 16169 (S.A.A.K., R.M.S., J.J.J.M.G.-F., S.D.K.) European Union research and innovation Horizon 2020 grant (“EXPERT”) grant no. 825828 (R.M.S., S.D.K., T.B., L.H.), Flanders Innovation & Entrepreneurship (VLAIO, Baekeland grant) no. 2017.0572 (S.B.), and Estonian Research Council grant PSG226 (T.L.).
Author contributions
S.B., S.A.A.K., and S.D.K. designed the experiments. S.B and S.A.A.K. performed the experiments and analyzed the results. S.S., E.V.d.V., J.J.J.M.G.-F., M.J.W.E., N.C.H.v.K., and M.H.A.M.F. helped with experiment execution. T.B. and L.H. designed the DOE and performed related statistical analysis. H.S. and K.E.A. analyzed LNP protein corona. M.M. and D.V.H. provided non-human primate data. R.M.S., S.D.K., E.M., K.B., S.E.A., T.L., and K.F. supervised the work. S.B., S.A.A.K., and S.D.K. wrote the manuscript. All authors have given approval to the final version of the manuscript.
Declaration of interests
S.B., E.V.d.V., S.S., M.M., D.V.H., and S.D.K. are employees of eTheRNA Immunotherapies NV. S.B., S.A.A.K., R.M.S., and S.D.K. have applied for patents related to this study.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.07.007.
Supplemental information
References
- 1.Alameh M.-G.G., Tombácz I., Bettini E., Lederer K., Sittplangkoon C., Wilmore J.R., Gaudette B.T., Soliman O.Y., Pine M., Hicks P., et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. 2021;54:2877–2892.e7. doi: 10.1016/j.immuni.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichmuth A.M., Oberli M.A., Jeklenec A., Langer R., Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016;7:319–334. doi: 10.4155/tde-2016-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cafri G., Gartner J.J., Zaks T., Hopson K., Levin N., Paria B.C., Parkhurst M.R., Yossef R., Lowery F.J., Jafferji M.S., et al. mRNA vaccine–induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Invest. 2020;130:5976–5988. doi: 10.1172/JCI134915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberli M.A., Reichmuth A.M., Dorkin J.R., Mitchell M.J., Fenton O.S., Jaklenec A., Anderson D.G., Langer R., Blankschtein D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017;17:1326–1335. doi: 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kranz L.M., Diken M., Haas H., Kreiter S., Loquai C., Reuter K.C., Meng M., Fritz D., Vascotto F., Hefesha H., et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 6.Broos K., Van der Jeught K., Puttemans J., Goyvaerts C., Heirman C., Dewitte H., Verbeke R., Lentacker I., Thielemans K., Breckpot K. Particle-mediated intravenous delivery of antigen mRNA results in strong antigen-specific T-cell responses despite the induction of type I interferon. Mol. Ther. Nucleic Acids. 2016;5:e326. doi: 10.1038/mtna.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Der Jeught K., De Koker S., Bialkowski L., Heirman C., Tjok Joe P., Perche F., Maenhout S., Bevers S., Broos K., Deswarte K., et al. Dendritic cell targeting mRNA lipopolyplexes combine strong antitumor T - cell immunity with improved inflammatory safety. ACS Nano. 2018;12:9815–9829. doi: 10.1021/acsnano.8b00966. [DOI] [PubMed] [Google Scholar]
- 8.Sahin U., Oehm P., Derhovanessian E., Jabulowsky R.A., Vormehr M., Gold M., Maurus D., Schwarck-Kokarakis D., Kuhn A.N., Omokoko T., et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–112. doi: 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- 9.Kauffman K.J., Dorkin J.R., Yang J.H., Heartlein M.W., Derosa F., Mir F.F., Fenton O.S., Anderson D.G. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 2015;15:7300–7306. doi: 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- 10.Ramaswamy S., Tonnu N., Tachikawa K., Limphong P., Vega J.B., Karmali P.P., Chivukula P., Verma I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA. 2017;114:E1941–E1950. doi: 10.1073/pnas.1619653114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pardi N., Tuyishime S., Muramatsu H., Kariko K., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S., Tam Y.Y.C., Lin P.J.C., Sung M.M.H., Tam Y.K., Cullis P.R. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control. Release. 2016;235:236–244. doi: 10.1016/j.jconrel.2016.05.059. [DOI] [PubMed] [Google Scholar]
- 13.Chen D., Ganesh S., Wang W., Amiji M. The role of surface chemistry in serum protein corona-mediated cellular delivery and gene silencing with lipid nanoparticles. Nanoscale. 2019;11:8760–8775. doi: 10.1039/c8nr09855g. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki T., Suzuki Y., Hihara T., Kubara K., Kondo K., Hyodo K., Yamazaki K., Ishida T., Ishihara H. PEG shedding-rate-dependent blood clearance of PEGylated lipid nanoparticles in mice: faster PEG shedding attenuates anti-PEG IgM production. Int. J. Pharm. 2020;588:119792. doi: 10.1016/j.ijpharm.2020.119792. [DOI] [PubMed] [Google Scholar]
- 15.Hassett K.J., Benenato K.E., Jacquinet E., Lee A., Woods A., Yuzhakov O., Himansu S., Deterling J., Geilich B.M., Ketova T., et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids. 2019;15:1–11. doi: 10.1016/j.omtn.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassett K.J., Higgins J., Woods A., Levy B., Xia Y., Hsiao C.J., Acosta E., Almarsson Ö., Moore M.J., Brito L.A. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Control. Release. 2021;335:237–246. doi: 10.1016/j.jconrel.2021.05.021. [DOI] [PubMed] [Google Scholar]
- 17.Akita H., Noguchi Y., Hatakeyama H., Sato Y., Tange K., Nakai Y., Harashima H. Molecular tuning of a vitamin E-scaffold pH-sensitive and reductive cleavable lipid-like material for accelerated in vivo hepatic siRNA delivery. ACS Biomater. Sci. Eng. 2015;1:834–844. doi: 10.1021/acsbiomaterials.5b00203. [DOI] [PubMed] [Google Scholar]
- 18.Pal A., Kundu R. Human Papillomavirus E6 and E7: the cervical cancer hallmarks and targets for therapy. Front. Microbiol. 2020;10:3116. doi: 10.3389/fmicb.2019.03116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grunwitz C., Salomon N., Vascotto F., Selmi A., Bukur T., Diken M., Kreiter S., Türeci Ö., Sahin U., Grunwitz C., et al. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology. 2019;8:e1629259. doi: 10.1080/2162402X.2019.1629259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis G.A., Mathieu D., Phan-Tan-Lu R. Marcel Dekker, Inc; 1999. Pharmaceutical Experimental Design. [Google Scholar]
- 21.Bonehill A., Tuyaerts S., Van Nuffel A.M., Heirman C., Bos T.J., Fostier K., Neyns B., Thielemans K. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol. Ther. 2008;16:1170–1180. doi: 10.1038/mt.2008.77. [DOI] [PubMed] [Google Scholar]
- 22.Almeida J.R., Price D.A., Papagno L., Arkoub Z.A., Sauce D., Bornstein E., Asher T.E., Samri A., Schnuriger A., Theodorou I., et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 2007;204:2473–2485. doi: 10.1084/jem.20070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciuffreda D., Comte D., Cavassini M., Giostra E., Bühler L., Perruchoud M., Heim M.H., Battegay M., Genné D., Mulhaupt B., et al. Polyfunctional HCV-specific T-cell responses are associated with effective control of HCV replication. Eur. J. Immunol. 2008;38:2665–2677. doi: 10.1002/eji.200838336. [DOI] [PubMed] [Google Scholar]
- 24.Wimmers F., Aarntzen E.H.J.G., Duiveman-deBoer T., Figdor C.G., Jacobs J.F.M., Tel J., de Vries I.J.M. Long-lasting multifunctional CD8+ T cell responses in end-stage melanoma patients can be induced by dendritic cell vaccination. Oncoimmunology. 2016;5:e1067745. doi: 10.1080/2162402X.2015.1067745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spranger S., Koblish H.K., Horton B., Scherle P.A., Newton R., Gajewski T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Groot R., Van Loenen M.M., Guislain A., Nicolet B.P., Freen-Van Heeren J.J., Verhagen O.J.H.M., Van Den Heuvel M.M., De Jong J., Burger P., Van Der Schoot C.E., et al. Polyfunctional tumor-reactive T cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged T cell profile. Oncoimmunology. 2019;8:e1648170. doi: 10.1080/2162402X.2019.1648170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin K.Y., Guarnieri F.G., Staveley-O’Carroll K.F., Levitsky H.I., August J.T., Pardoll D.M., Wu T.C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 28.Besin G., Milton J., Sabnis S., Howell R., Mihai C., Burke K., Benenato K.E., Stanton M., Smith P., Senn J., et al. Accelerated blood clearance of lipid nanoparticles entails a biphasic humoral response of B-1 followed by B-2 lymphocytes to distinct antigenic moieties. Immunohorizons. 2019;3:282–293. doi: 10.4049/immunohorizons.1900029. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu T., Mima Y., Hashimoto Y., Ukawa M., Ando H., Kiwada H., Ishida T. Anti-PEG IgM and complement system are required for the association of second doses of PEGylated liposomes with splenic marginal zone B cells. Immunobiology. 2015;220:1151–1160. doi: 10.1016/j.imbio.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Kozma G.T., Shimizu T., Ishida T., Szebeni J. Anti-PEG antibodies: properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv. Drug Deliv. Rev. 2020;154–155:163–175. doi: 10.1016/j.addr.2020.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Ishida T., Wang X.Y., Shimizu T., Nawata K., Kiwada H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J. Control. Release. 2007;122:349–355. doi: 10.1016/j.jconrel.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 32.De Beuckelaer A., Pollard C., Van Lint S., Roose K., Van Hoecke L., Naessens T., Udhayakumar V.K., Smet M., Sanders N., Lienenklaus S., et al. Type I interferons interfere with the capacity of mRNA lipoplex vaccines to elicit cytolytic T cell responses. Mol. Ther. 2016;24:2012–2020. doi: 10.1038/mt.2016.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pollard C., Rejman J., De Haes W., Verrier B., Van Gulck E., Naessens T., De Smedt S., Bogaert P., Grooten J., Vanham G., et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013;21:251–259. doi: 10.1038/mt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Hoecke L., Roose K., Ballegeer M., Zhong Z., Sanders N.N., De Koker S., Saelens X., Van Lint S. The opposing effect of type I IFN on the T cell response by non-modified mRNA-lipoplex vaccines is determined by the route of administration. Mol. Ther. Nucleic Acids. 2020;22:373–381. doi: 10.1016/j.omtn.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patricia F.-B., Jihong D., Sukhwinder S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19:3–19. doi: 10.1016/j.cytogfr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Rooijen N., van Nieuwmegen R. Elimination of phagocytic cells in the spleen after intravenous injection of liposome-encapsulated dichloromethylene diphosphonate - an enzyme-histochemical study. Cell Tissue Res. 1984;238:355–358. doi: 10.1007/BF00217308. [DOI] [PubMed] [Google Scholar]
- 37.Kitamura D., Roes J., Kühn R., Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 38.Riley R.S., Kashyap M.V., Billingsley M.M., White B., Alameh M.G., Bose S.K., Zoltick P.W., Li H., Zhang R., Cheng A.Y., et al. Ionizable lipid nanoparticles for in utero mRNA delivery. Sci. Adv. 2021;7:eaba1028. doi: 10.1126/sciadv.aba1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arteta M.Y., Kjellman T., Bartesaghi S., Wallin S., Wu X., Kvist A.J., Dabkowska A., Székely N., Radulescu A., Bergenholtz J., et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. USA. 2018;115:E3351–E3360. doi: 10.1073/pnas.1720542115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lokugamage M.P., Vanover D., Beyersdorf J., Hatit M.Z.C., Rotolo L., Echeverri E.S., Peck H.E., Ni H., Yoon J.K., Kim Y.T., et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat. Biomed. Eng. 2021;5:1059–1068. doi: 10.1038/s41551-021-00786-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar V., Qin J., Jiang Y., Duncan R.G., Brigham B., Fishman S., Nair J.K., Akinc A., Barros S.A., Kasperkovitz P.V. Shielding of lipid nanoparticles for siRNA delivery: impact on physicochemical properties, cytokine induction, and efficacy. Mol. Ther. Nucleic Acids. 2014;3:e210. doi: 10.1038/mtna.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimizu T., Abu Lila A.S., Kawaguchi Y., Shimazaki Y., Watanabe Y., Mima Y., Hashimoto Y., Okuhira K., Storm G., Ishima Y., et al. A novel platform for cancer vaccines: antigen-selective delivery to splenic marginal zone B cells via repeated injections of PEGylated liposomes. J. Immunol. 2018;201:2969–2976. doi: 10.4049/jimmunol.1701351. [DOI] [PubMed] [Google Scholar]
- 43.Hong L., Wang Z., Wei X., Shi J., Li C. Antibodies against polyethylene glycol in human blood: a literature review. J. Pharmacol. Toxicol. Methods. 2020;102:106678. doi: 10.1016/j.vascn.2020.106678. [DOI] [PubMed] [Google Scholar]
- 44.Mathieu M., Cotta-Grand N., Daudelin J.F., Boulet S., Lapointe R., Labrecque N. CD40-activated B cells can efficiently prime antigen-specific naïve CD8 + T cells to generate effector but not memory T cells. PLoS One. 2012;7:1–12. doi: 10.1371/journal.pone.0030139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colluru V.T., McNeel D.G. B lymphocytes as direct antigen-presenting cells for anti-tumor DNA vaccines. Oncotarget. 2016;7:67901–67918. doi: 10.18632/oncotarget.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wennhold K., Shimabukuro-Vornhagen A., Von Bergwelt-Baildon M. B cell-based cancer immunotherapy. Transfus. Med. Hemother. 2019;46:36–46. doi: 10.1159/000496166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sagiv-Barfi I., Czerwinski D.K., Shree T., Lohmeyer J.J.K., Levy R. Intratumoral immunotherapy relies on B and T cell collaboration. Sci. Immunol. 2022;7:eabn5859. doi: 10.1126/sciimmunol.abn5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bergmann C.C., Ramakrishna C., Kornacki M., Stohlman S.A. Impaired T cell immunity in B cell-deficient mice following viral central nervous system infection. J. Immunol. 2001;167:1575–1583. doi: 10.4049/jimmunol.167.3.1575. [DOI] [PubMed] [Google Scholar]
- 49.Homann D., Tishon A., Berger D.P., Weigle W.O., von Herrath M.G., Oldstone M.B.A. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-Cell-Deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from μMT/μMT mice. J. Virol. 1998;72:9208–9216. doi: 10.1128/jvi.72.11.9208-9216.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lykken J.M., DiLillo D.J., Weimer E.T., Roser-Page S., Heise M.T., Grayson J.M., Weitzmann M.N., Tedder T.F. Acute and chronic B cell depletion disrupts CD4 + and CD8 + T cell homeostasis and expansion during acute viral infection in mice. J. Immunol. 2014;193:746–756. doi: 10.4049/jimmunol.1302848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.