

Abstract
As early T-cell precursors transition from multipotentiality to T-lineage commitment, they increase or decrease expression of multiple transcription factors. It is unclear if individual transcription factors directly control choices between T-cell identity and some alternative fate, or whether these factors mostly affect proliferation or survival during the normal commitment process. Here, we unraveled the impacts of deleting individual transcription factors at two stages in early T-cell development, using synchronized in vitro differentiation systems, single-cell RNA-seq with batch indexing, and controlled single-cell gene-disruption strategies. First, using a customized method for single-cell CRISPR disruption, we defined how the early-acting transcription factors Bcl11a, Erg, Spi1 (PU.1), Gata3, and Tcf7 (TCF1) function before commitment. The results revealed a kinetic tug-of-war within individual cells between T-cell factors Tcf7 and Gata3 and progenitor factors Spi1 and Bcl11a, with an unexpected guidance role for Erg. Second, we tested how activation of transcription factor Bcl11b during commitment altered ongoing cellular programs. In knockout cells where Bcl11b could not be upregulated, the cells did not undergo developmental arrest, instead following an alternative path as T-lineage commitment was blocked. A stepwise, time-dependent regulatory cascade began with immediate-early transcription factor activation and E protein inhibition, finally leading Bcl11b-knockout cells toward exit from the T-cell pathway. Finally, gene regulatory networks of transcription factor cross-regulation were extracted from the single-cell transcriptome results, characterizing the specification network operating before T-lineage commitment and revealing its links to both the Bcl11b-knockout alternative network and the network consolidating T-cell identity during commitment.
One sentence summary:
Transcription factor gene knockouts revealed regulatory networks controlling kinetics and pathway choices in early T-cell development.
Introduction
Multipotent precursors acquire a T-cell lineage identity in the thymus through sequential signal-modulated gene network steps(1–5), driven primarily by Notch ligands on the thymic stroma(6–9). Pro-T cells become committed to a T-lineage fate before T-cell receptor expression but multiple cell cycles after entering the thymus(4). Multiple transcription factors are implicated in these events, becoming activated or silenced in a regulated way(10). Single-cell transcriptome analyses have provided details of pro-T cells’ individual developmental gene expression changes in vivo (11–13). Still, it remains unclear how these specific transcription factors control the dynamics of cell-type identity change as the T cell program begins.
This intrathymic T cell development process can be mimicked in cell culture systems(6, 14) that enable controlled timing of signaling environment encounters and regulatory perturbations. However, individual T cell precursors starting from the same developmental stage do not differentiate with uniform kinetics, but show interclonal heterogeneity (15), suggesting that the cells may be caught between opposing regulatory influences. To understand how different transcription factors work in the context of developing pro-T cells, parallel knockouts of such factors at specific stages with scRNA-seq analysis at specific times after knockout were needed. Here, to compare multiple knockouts and timepoints without batch effects, we combined Cell Hashing for sample and batch indexing(16) with either Cre-mediated deletion or a specially designed CRISPR/Cas9 knockout strategy, resembling ‘perturb-seq’(17), that exploited the 10X Chromium V3 chemistry’s direct guide RNA (gRNA) capture capability(18). We investigated two problems in early T cell development.
In early murine pro-T cells, at least 10 multilineage and stem-cell regulatory factors have overlapping expression with T-lineage transcription factors in the stages before lineage commitment (termed here, “Phase 1” stages)(11, 19, 20)[reviewed in (5)]. Because many of these factors are essential in hematopoietic stem and progenitor cells, conventional knockout methods have shed little light on their specifically intrathymic roles. While certain “progenitor” transcription factors antagonize T-lineage progression when overexpressed (21–23), their natural roles in the T-cell program have been unclear. Also in the earliest stages, two important T-lineage regulators, TCF1 (encoded by Tcf7, referred to as “Tcf7” below) and Gata3 (rev. in (5)), become upregulated. However, these factors are so important for pro-T cell viability(24–28) that bulk population knockout studies, skewed by survival artifacts, have only hinted at their specific targets, making many aspects of this gene network that initially guides cells into the T-cell pathway unclear.
After the early thymic progenitor stage, expression of the transcription factor Bcl11b initiates, marking commitment to the T cell lineage(29–32), and crucially for generation of α T cells and most γδ T cells. Populations of pro-T cells lacking Bcl11b abnormally retain some immature or lineage-inappropriate features while converting toward abnormal, natural killer (NK)-like cells(33–37). However, the steps through which alternative pathways emerge are undefined, and it is not clear how they relate to the earlier T-cell development pathway.
Here, using scRNA-seq to analyze the time-resolved effects of knocking out key transcription factors, we resolved the separate roles of these factors in developmental pathway choice, proliferation, and speed of differentiation progression. The results identified latent regulatory circuits for the early Phase 1 progression into the T-cell pathway and for the choice between T and non-T cell choices during commitment. Taken together, these results provided insight into the gene regulatory mechanisms operating during the earliest stages of the T-cell developmental program.
Results
Targeting of transcription factors with early dynamic expression patterns through batch-controlled, dual-gRNA direct-capture perturb-seq
This study focused on two phases of the murine pro-T cell program (Fig. 1A): (1) the early gene regulatory network that guides hematopoietic precursors to initiate the T-cell program in Phase 1, and (2) the choices the cells undergo during commitment, dependent on the transcription factor Bcl11b. These events encompass the stages of pro-T differentiation before T-cell receptor expression, which have been divided by cell surface markers into Early T-cell Precursor or ETP (contained within the DN1 population), DN2a, DN2b and DN3 subsets (Fig. 1A)[reviewed in (5)]. The ETP and early DN2a cells are uncommitted and resemble hematopoietic progenitors, and comprise the Phase 1 stages, while commitment accompanies Bcl11b upregulation in the late DN2a stage(32), initiating Phase 2(5). To reveal mechanisms controlling gene expression changes before T lineage commitment (Fig. 1A), we examined the effects on differentiation speed, transcriptomes, and outcomes when we knocked out (KO) any of seven candidate regulators of these events (Fig. 1B). To determine mechanisms operating during commitment, we compared cohorts of wildtype and Bcl11b KO cells during the first days after Bcl11b would normally be turned on (Fig. 1A).
Fig. 1.
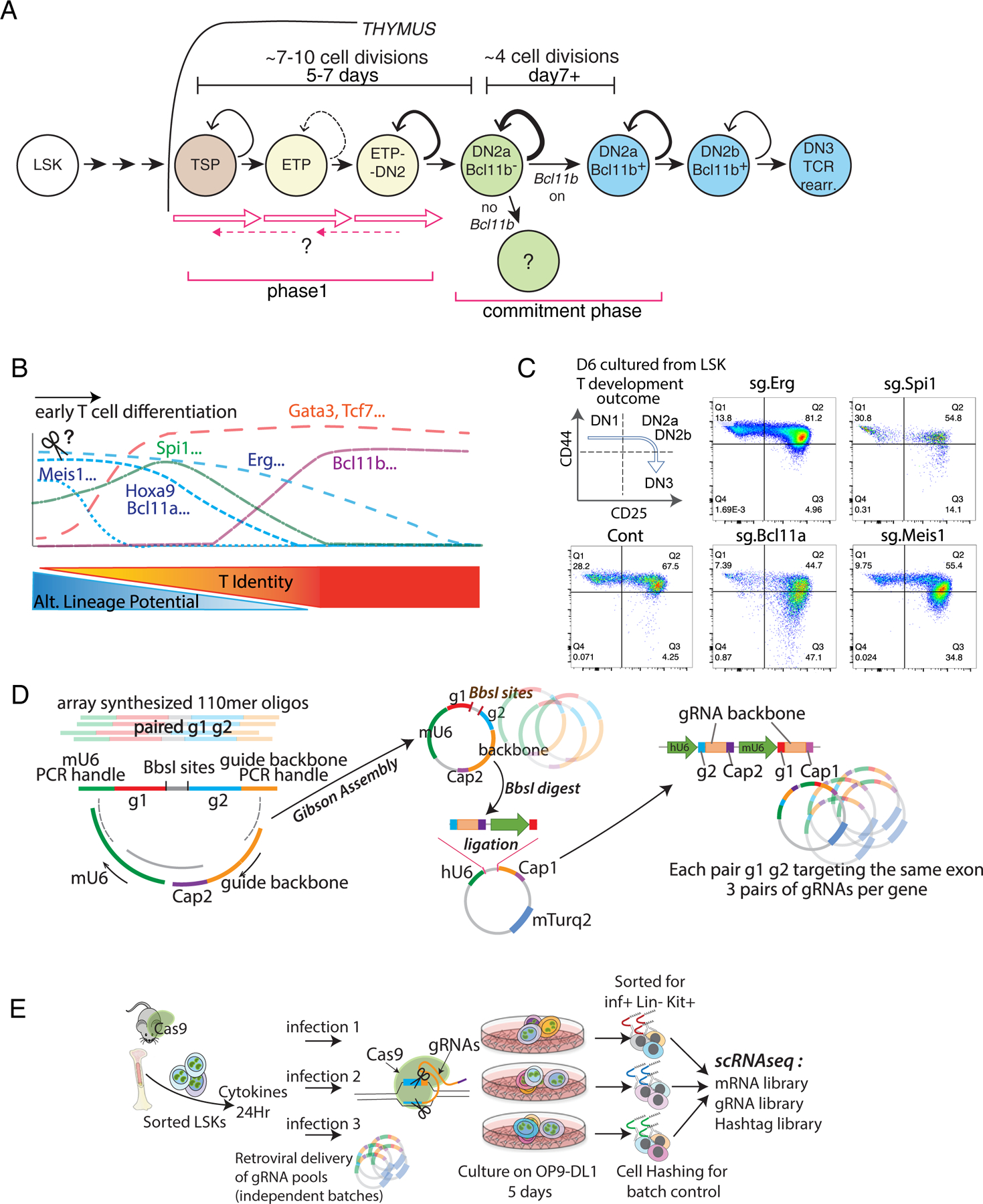
Developmental framework: Pool-based, batch-controlled transcription factor perturbation scRNA-seq to define impacts of timed deletions of developmentally regulated transcription factors. (A) Identification of stages in T cell development in the thymus. All stages indicated are CD4− CD8− TCR− (DN). TSP, thymus-seeding precursor: earliest Flt3+ subset of Early T-cell Precursor (ETP) population; ETP: c-Kithi CD44+ CD25− DN. DN2a: c-Kithi CD44+ CD25+. DN2b: c-Kitint CD44+ CD25+. DN3: c-Kitlo CD44− CD25+, the stage when TCR rearrangement (rearr.) produces complete TCR coding genes. Timing of Bcl11b activation and stages studied here are indicated. (B) Diagram of gene expression patterns of selected transcription factors in relation to measured alternative lineage potentials of the cells at each stage (11). (C) Representative surface expression phenotypes analyzed by flow cytometry. Acute transcription factor deletions were induced in precursors by sgRNA transduction, then cells were cultured for 6 days with OP9-DLL1. Figure shows developmental staging based on surface expression of CD44 and CD25. Stages identified in (A) correspond to the indicated patterns of CD44 and CD25. ETP and TSP cells, but potentially also alternative lineage cells, are included in the “DN1” quadrant. (D) Pool-based dual gRNA cloning strategy used in pool-perturbation experiments in this study. Left: diagram of structure of the paired gRNA oligomers as initially synthesized by array-based oligo synthesis. Middle, each distinct oligomer was joined by Gibson assembly with the invariant Cap2 oligo and mU6 promoter segments in the orientation shown, to generate the paired dual gRNA insert pool. Right, incorporation of the paired dual gRNA inserts into the retroviral vector backbone containing the hU6 promoter and the Cap1 oligo sequence. Note that the assembly of these segments into the indicated site ends up linking the mU6 promoter with g1 and the Cap1 tag, downstream of the hU6 promoter driving g2 and the Cap2 oligo sequence. Retroviral backbones were packaged into the final retroviral vector library. (E) Internal construct- and batch- controlled experimental setup for single-cell pool-perturbation RNA-seq. Two independently packaged stocks of the pool-perturbation viral library were each infected into separate LSK cultures at MOI=1.0 and MOI=0.5, in parallel (plus an extra separate culture for one of the MOI=0.5 samples). These separate transductions of the whole pool were then cultured in parallel as indicated, for 5 days on OP9-DLL1 to initiate T cell development. The five cultures were then harvested, sorting Infected (inf+) Lin− (within the T-cell pathway) Kit+ (uncommitted) cells. Each culture was indexed with different hashtags before cells were pooled for scRNA-seq analysis as described in the text. The cells were processed by 10X Chromium to associate unique barcodes with each cell, and mRNA, gRNA, and hashtag libraries were made. The unique barcodes enabled mRNA sequences from one cell to be correlated with the gRNA and hashtag sequences from the same cell.
To control the time of differentiation experimentally relative to the timing of each knockout, we used two well-established in vitro differentiation systems for these studies, in which the thymus epithelium is replaced by OP9-DLL1 stromal monolayers(6) or by a 3D artificial organoid system (ATO) with MS5-mDLL4 stroma(14)(Supplementary Methods: BM cell differentiation). The OP9-DLL1 system has been widely used, but to validate both in vitro systems for single-cell analysis, we directly compared precursors developing in OP9-DLL1 or in ATO co-culture with corresponding intrathymic (Thy) cells(11)(Fig.S1A). After 5–6 days of in vitro differentiation, DN1, DN2a uncommitted, and DN2a newly committed populations were sorted for RNA-seq analyses as shown (Figs. S1A, S2A; Table S1). Bulk (Fig. S1B) and single-cell analyses (Figs. S2 & S3) confirmed that in vitro and in vivo intrathymic differentiation were highly comparable.
For functional analysis, we focused on Bcl11a, Spi1, Hoxa9, Meis1, and Erg, which encode stem or progenitor-associated factors, and on Tcf7 and Gata3, which encode T lineage-associated factors (5)(Fig. 1B). We knocked out these genes just before initiating T-cell development, using retroviral vectors to introduce single guide RNAs (sgRNAs or gRNAs) against the gene of interest into Cas9-expressing progenitors, Lin− Sca-1+ Kit+(LSK) cells purified from mouse bone marrow (BM). We then transferred the cells to OP9-DLL1 culture to differentiate for 5–6 days before analysis. Although several targeted genes are important for viability of early T cell precursors(25, 28, 39–41), we ensured maximum survival of perturbed cells by using Bcl2-tg;ROSA26-Cas9 input cells which constitutively express antiapoptotic Bcl2. Based on cell surface markers CD44 and CD25, KOs of progenitor-related genes Bcl11a or Meis1 appeared to accelerate the DN2a to DN2b transition, while Erg KO increased the rate of DN1 to DN2 transition (Fig. 1C). Disruption of Spi1, encoding PU.1, a factor important for the myeloid vs lymphoid decision, somewhat accelerated DN2 to DN3 progression (Fig. 1C), in agreement with previous reports where the Bcl2 transgene was not present (39, 40).
In order to understand the precise KO effects on a continuous developmental continuum and to establish their reproducibility while minimizing the potential for technical batch variability, we designed a highly-controlled, multiply-replicated scRNA-seq strategy as follows (Fig. 1D–E). The design was (i) to construct gRNA libraries containing three different pairs of gRNAs to target each gene of interest (Fig. 1D), (ii) to generate them all at similar levels with control constructs within one vector pool to ensure consistency of infection, and (iii) to analyze the effects of all gRNA pairs in the pooled library in multiple parallel replicates of the infections and differentiation cultures, to control for culture variation (Fig. 1E). We used paired gRNAs against the same exon within each construct to optimize target disruption. The three different gRNA pair constructs per target enabled KO effects to be confirmed with independent constructs. By analyzing gRNA sequences from each cell directly, cells infected with individual constructs could be discriminated within the pool. Also, by hashtagging each of the five replicate cultures, the whole set of culture replicates, each containing the whole library of gRNA constructs, could be pooled into a single 10X Genomics Chromium reaction, to eliminate processing variability. Note that this approach was based on generation of a new vector, a new library construction protocol for the pooled samples, and customized methods for assigning reads to specific perturbed single cells within the Chromium samples, as described in Methods and Supplementary Methods (Construct Design and Cloning; CRISPR/Cas9-mediated acute deletion in precursor cell cultures).
Cas9+ Bcl2+ precursor cells (purified LSK) were infected at low multiplicity with the library of vectors, each including paired gRNAs against the same target gene (Fig. 1E). The cells were then cultured in parallel in separate biological replicates on OP9-DL1 stroma for 5 days before sorting for infection-positive populations. The different batches were labeled with barcoded antibody, pooled and then analyzed through scRNA-seq (Fig. 1E; Methods: CRISPR/Cas9-mediated acute deletion in precursor cell cultures). The experimental conditions of 5-day-cultures of LSK with Notch signaling were chosen to monitor loss of function effects in phase 1, before T-lineage commitment. Note that the Bcl2 and Cas9 transgenes in the genetic backgrounds of the developing cells themselves had minimal effects on their developmental pathways or transcriptomes (Fig. S3), as shown by comparing scRNA-seq data from the Bcl2-tg, Cas9-tg, in vitro-differentiated control cells in this study with data from completely wildtype B6 cells freshly taken from mouse thymus (11)(GSE137165). The two populations had nearly indistinguishable transcriptomes (Fig. S3A) from earliest intrathymic ETP stages (Flt3+, Lmo2+) to early commitment (Bcl11b+) (Fig. S3C). Thus, the multiple replicate infections of the Cas9+ Bcl2+ precursor cells, multiple sgRNA pairs against the same target, and pooled sequencing of five separately cultured differentiation replicates in the same 10X Chromium v3 analysis (Fig. 1D,E) yielded a high-quality, internally-controlled, and physiologically relevant data resource in which to measure impacts of deleting specific transcription factors.
Dramatic changes in topology with single transcription factor knockout
In scRNA-seq data, the earliest and latest Phase 1 cells in a low-dimensional transcriptome representation (Fig. 2A–C) were identified by their expression of previously determined landmark genes(11) (Fig. 2B; Fig. S4A, B; top cluster markers listed in Table S2). Some individual transcription factor perturbations resulted in notable pattern changes (Fig. 2C,D). As expected from the asynchronous differentiation observed before(15), control cells spread sparsely across the UMAP plot (Fig. 2D left, gray dashed arrow depicts normal developmental trajectory). In contrast, cells in the same population expressing sg.Tcf7 (Fig. 2D) stalled at the most un-differentiated stages (represented by clusters 7 and 1 in Fig. 2A)(cluster distributions for each gRNA pair given in Fig. S4C), whereas cells expressing sg.Spi1 (Fig. 2D) shifted towards more differentiated stages (clusters 5, 8, 9 in Fig. 2A). Cells expressing sg.Gata3 were more concentrated than controls among the most un-differentiated cells (Fig. 2E), while those expressing sg.Bcl11a mainly showed reductions in their proportions of cells in “ETP-like” clusters 1 and 7 (Fig. 2E). But cells expressing sg.Erg shifted away to form a distinct extended group (Fig. 2D right, upper right of panel, clusters 2–4), parallel to the clusters containing the control trajectory (Fig. 2D). Also, despite matched inputs, more sg.Erg expressing cells (all gRNA pairs) emerged in the pool than controls or cells expressing any other gRNA pairs (Fig. 2F), suggesting that Erg loss may enhance proliferation. In contrast, Tcf7 and Meis1 perturbations significantly reduced cell yields despite the anti-apoptotic Bcl2 transgene (Fig. 2F).
Fig. 2.
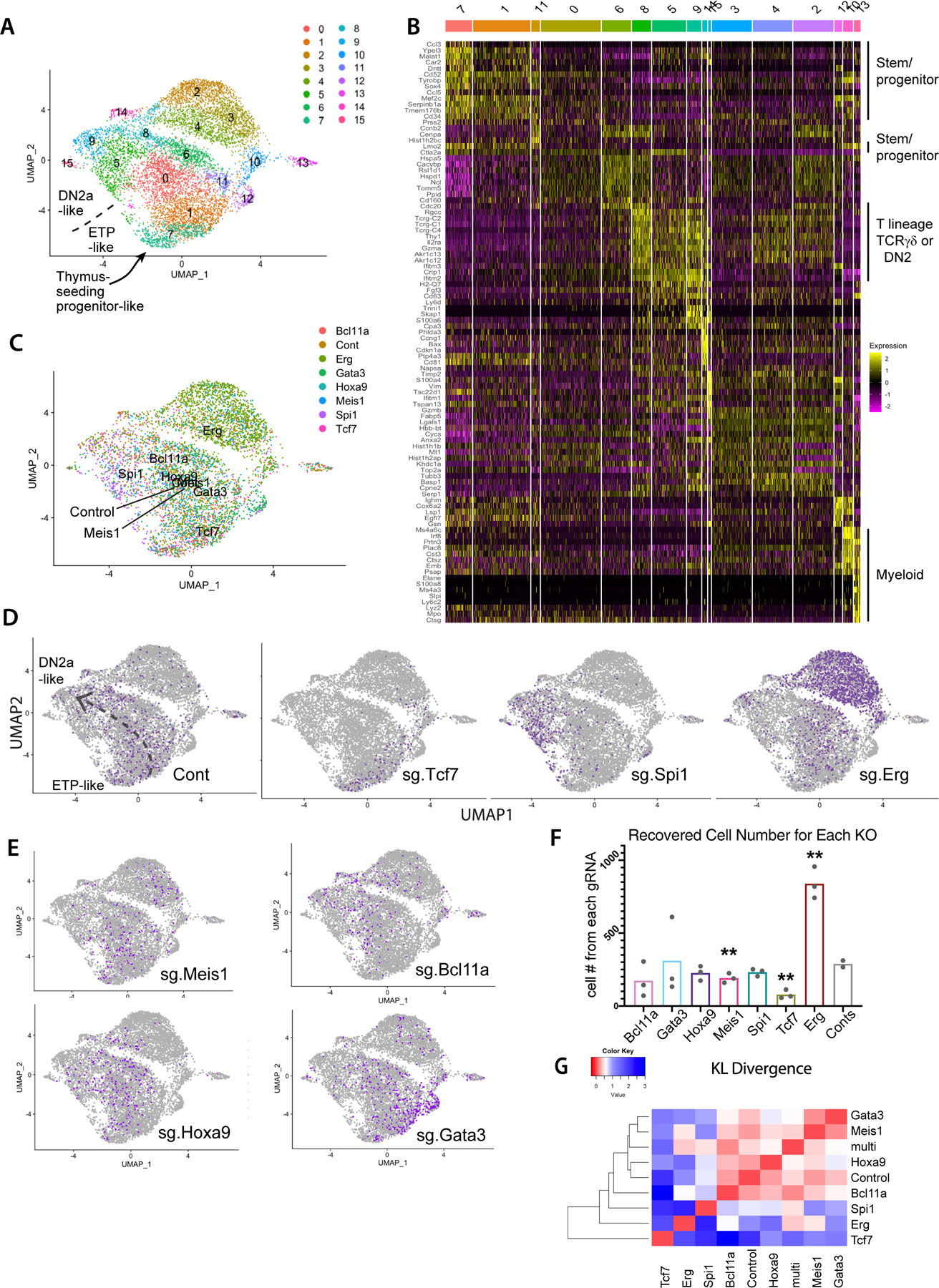
Impacts of transcription factor perturbations on gene expression, cell number, and differentiation trajectories of early pro-T cells. (A) UMAP plot of scRNA-seq data from total set of pool-perturbation samples, here based on PC 1–16. N= 5541 cells for singly assigned gRNA vector, N=8045 for total recovered single cells. The cells are colored by Louvain clustering algorithm. (B) Heatmap displaying the top 10 enriched genes in each sub-cluster, in approximate developmental order. Defined by Seurat 3 pipeline with minimum fraction of expressing cells ≥ 0.25, Wilcoxon rank sum test with avg_logFC threshold of 0.3, full list of genes in Table S2). (C-E) Cells in UMAP display, colored by sgRNA assignment. All cells with any of the 3 pairs of dual sgRNAs against the same gene are colored together. Number of cells of each KO: Controls (Cont.): 793, Bcl11a: 353, Erg: 2101, Gata3: 686, Hoxa9: 503, Meis1: 409, Spi1: 481, Tcf7: 215. (C) Merged representation of results with labels showing the centroids of different KO distributions on a UMAP plot. (D-E) Cells from the indicated KOs (purple dots in each panel) within the total sample (gray dots). The main consensus trajectory for the whole experiment is shown approximately on the “Cont” panel, manually sketched based on marker gene expression shown in panel (B) and Figure S4A. (F) Cell numbers recovered from the scRNA-seq pool. Each dot represents the cell number recovered from one of paired gRNA vectors against the indicated target. Statistical significance calculated in individual pairwise t-tests between Control and each of the KOs. (**: p-val<0.01, *: p-val<0.05). Raw data included in Table S12. (G) Heatmap of KL divergences of cluster distributions (red: high similarity; blue: low similarity) among WT and aggregated KO for each gene. Cluster distribution for each sample used in KL divergence calculation is provided in Table S12.
Pair-wise Kullback-Leibler (KL) divergences among the cluster distributions of each genotype showed that KOs of Tcf7, Erg, and Spi1 each gave dramatically distinct cell distributions across clusters, very different from controls (Fig. 2G). The three Erg KO-specific clusters (clusters 2–4, Fig. 2A, C, D) showed a distinct gene expression signature unlike the ‘early’ clusters (1, 7, 11) or the ‘late clusters (5, 6, 9)(Fig. 2B). However, except for the Erg KO cells, most of the KO and control cells were still found among the same ‘common’ transcriptome clusters as controls, despite these shifting distributions (Fig. 2C; Fig S4B–D). Within the same shared clusters, gene expression patterns in cells from different KOs were nearly indistinguishable from those of the controls (Fig. S4D, S5A). Thus, these KO and wildtype cells were passing through the same states, even if at different speeds. Therefore, these KOs mostly impacted the cluster distributions, which potentially reflect developmental states, rather than resulting in a few genes’ abnormal expression compared to controls within the same clusters.
Shifts in T-cell differentiation pseudotime trajectory caused by KO of individual transcription factors
The impacts of these transcription factor KOs on developmental progression could be related to a common T-lineage differentiation pseudotime metric. Trajectories were calculated from full transcriptome profiles (Monocle3 analysis) in 3D UMAP space (Fig. 3A, pseudotime progressing from blue to yellow). The single dimension of pseudotime showed widely different impacts of different KOs on gross developmental speed (Fig. 3B; red bars, mean). Bcl11a, Spi1 and Erg KOs showed significant pseudotime acceleration relative to controls (Kruskal-Wallis test; significance asterisks in blue), while the Tcf7 KOs had significant deceleration compared to the control (significance asterisks in red). Gata3 KO cells progressed beyond Tcf7 KO cells, but specifically failed to generate cells matching the most advanced controls; Hoxa9 and Meis1 KO effects were less significant. Thus, the natural activities of Spi1, Bcl11a, and Erg appeared to slow early pro-T cell differentiation, while expression of Tcf7 and Gata3 was used (or required) to advance the T lineage program, consistent with previous studies(24, 28, 40).
Fig. 3.
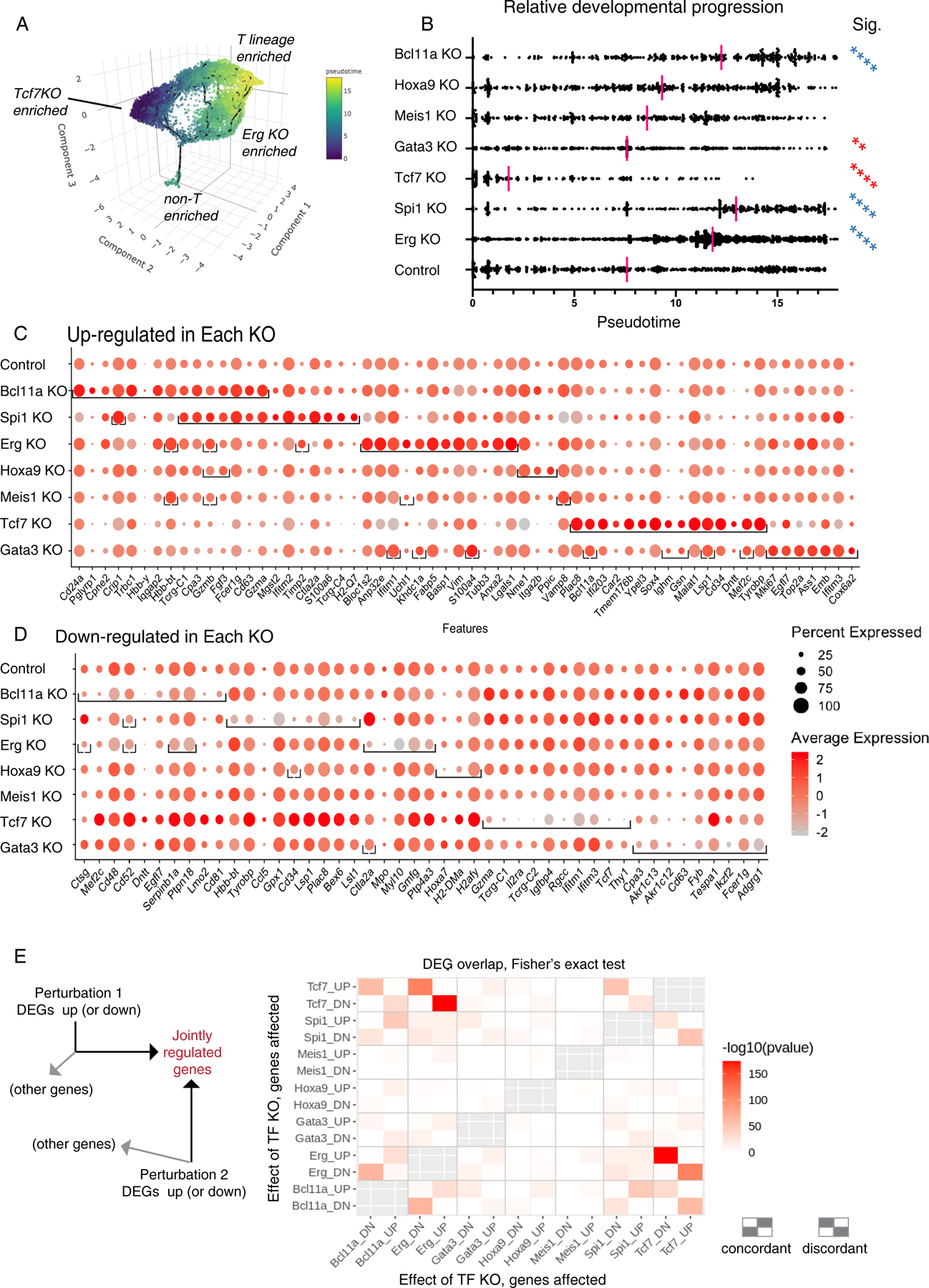
Impact of transcription factor knockouts on gene regulation, pseudotemporal progression, and discrete regulon modules in early pro-T cells. (A) 3D UMAP of all pool-perturbation samples, corresponding to 2D plot shown in Fig. 2A. Here, cells are colored by inferred pseudotime, to relate cells to a common overall pseudotemporal advancement score regardless of trajectories. The pseudotime was calculated with Monocle 3, based on the trajectory inference from 3D UMAP built using the size and cell cycle scaled data as described in Fig. 2A (see Supplementary Methods, Gene and Cell Filtering, Data Alignment, and Clustering Analysis). (B) Pseudotime distributions (x axis) of cells from the indicated KOs (category axis), with medians indicated (red bars). The small “non-T-enriched” population seen in (A) was excluded from the calculation. Statistical significance of comparisons, each KO to control, by Kruskal-Wallis test of multiple comparisons. **, adj.p-val < 1E-02; ****, adj.p-val < 1E-04. Blue asterisks: faster, red asterisks: slower than Control (Cont). (C-D) Genes highly differentially expressed between control and KOs, full gene lists in Table S3. (C) Top 15 up-regulated genes (columns) for each KO (row). In each row (each specific KO), brackets enclose the top differentially expressed genes for that KO. Order of columns: Top 15 genes for Bcl11a KO are followed by top 15 for Spi1 KO if not already listed, followed by top 15 for Erg KO if not already listed, etc. Note that 8 of the top 15 genes in the Spi1 KO are shared with the Bcl11a KO and so add only 7 new genes (columns) to total, whereas 12 of the top 15 genes for Erg KO are not shared with Bcl11a KO or Spi1 KO. Only Meis1 KO and Hoxa9 KO did not define 15 significantly upregulated genes. Top 15 was defined by avg_logFC function on Seurat 3-processed data. Significance determined by Wilcoxon Rank Sum test, minimum expression fraction ≥ 5% cells in either the control or the KO, average “log expression” difference ≥ 0.1 (ln, baseline pseudocount=1), and adjusted p-val < 1E-02. (D) Top 10 down-regulated genes in each KO, with conventions as in panel (C), with average “log expression” difference ≤-0.1, and adjusted p-val < 1E-04. (E) Pairwise comparisons of top differentially expressed genes in each condition, determining to what extent different KOs may affect common target genes either in the same way (concordantly) or in opposite ways (discordantly); pvalues calculated by Fisher’s exact test. Left schematic shows that the chart depicts the intersection between genes increased (UP regulated) or decreased (DOWN regulated) by the perturbation shown on the y axis, and genes UP or DOWN regulated by the perturbation on the x axis. The top left rectangle, e.g., shows the enrichment of genes that are both increased in expression by the Tcf7 KO and decreased in expression by the Bcl11a KO. Other genes affected by an individual KO but not included in the intersection are not depicted. Right schematic: patterns of concordant or discordant regulation.
Factor-specific effects of different KOs at target gene level
All the KOs except Hoxa9 and Meis1 significantly changed expression of many genes (top hits shown as dots within brackets in the row for that KO in Fig. 3C, D; extended data in Table S3). The Bcl11a KO down-regulated progenitor-associated genes (e.g. Lmo2, Mef2c, Egfl7)(11), modestly upregulating T-specification genes Gata3 and Tcf7 and also NK and ILC-associated genes (Zfp105, Zbtb16, Fcer1g, Gzma, Gzmb). The Spi1 KO shared many of the effects of Bcl11a KO, increasing expression of T-cell receptor-associated genes (Tcrg-C1, Tcrg-C4, Trdc, Trat1, Cd247). Erg KO cells also downregulated progenitor genes, but upregulated distinctive cytoskeleton- and growth/signaling-related genes. Tcf7 KO cells lost expression of T-cell genes but showed enrichment for stem/progenitor-cell related genes(11) (Sox4, Cd34, Lmo2, Bcl11a, Spi1, Mef2c). While Gata3 KO cells also enriched expression of some progenitor genes, they showed a different pattern from the Tcf7 KO cells as they upregulated a mosaic of genes enriched in different non-T cell types and cell cycle-related genes.
We related the top genes affected by each KO quantitatively to the ‘normal’ maturation-related gene-expression shift in the control samples between early DN1 to late DN2b, i.e., genes changing expression between early clusters 1, 7, and 11 and late clusters 0, 5, 6, 8, 9, and 14 in Control samples only (Fig. 2B; Fig. S5B). Volcano plots (Fig. S5C–F) showed that the Tcf7 KO effect indeed resembled a simple developmental block (Fig. S5C) relative to genes normally up-regulated (magenta) or down-regulated (cyan) in T-lineage development. Spi1 or Bcl11a KOs caused effects roughly opposite to Tcf7 KO, mostly enhancing expression of genes normally upregulated in development. However, Gata3 KOs affected maturation-related genes irregularly, and both Gata3 and Spi1 KOs affected genes that could not be related purely to the normal T-lineage trajectory (Fig. S5E–F, dark brown dots). Pairwise comparisons showed that when Spi1 KO and Bcl11a KO affected the same genes, they acted concordantly nearly all the time, whereas when Tcf7 KO affected the same genes as Bcl11a KO or Spi1 KO, it nearly always affected them in the opposite direction (Fig. 3E). However, Erg KO effects were distinct. Thus, these regulatory effects were KO-specific, not simply consequences of developmental advancement.
SCENIC analysis-based identification of intermediate transcription factor activities mediating specific subsets of KO effects
The KO effects on gene expression shown in Figures 2 and 3 and Table S3 might have been direct or indirect effects of the targeted transcription factors, as multiple KOs affected expression of other regulators. For example, among the KO factors themselves, the Seurat 3-defined DEGs for each KO (Table S3) implied that Bcl11a repressed Gata3 while Gata3 repressed Bcl11a, that Spi1 activated Bcl11a while Tcf7 repressed it, that Tcf7 activated Gata3, and that Spi1 repressed Tcf7. However, it was not clear whether these effects were direct or how much of any knockout phenotype may have been mediated by these secondarily affected factors. For global insight, we used the SCENIC algorithm(38) to identify groups of genes potentially co-regulated directly by other regulators downstream of the seven targeted transcription factors. SCENIC uses scRNA-seq data to identify possible targets of expressed transcription factors independent of perturbation, based on enrichment of a known transcription factor target motif in genomic regions near the promoters of genes that are expressed co-varying with the candidate factor. Sets of such potential positively-regulated, direct target genes are defined as the factor’s ‘regulons’.
Here, although SCENIC failed to define regulons for some factors of interest in our dataset (e.g. Gata3), SCENIC did identify many regulons with sharply varying activity patterns across the dataset (Fig. 4A) and differential representation in the different KOs (Fig. S6A–F). Activities of representative regulons are shown in Fig. 4A, where calculated regulon activities (based on groups of putative target genes for the indicated factors) were plotted on the same UMAP manifold as shown in Figure 2. These regulon activities were generally similar to reported expression of the corresponding transcription factors in ETP-DN2b stages (Fig. 4A; compare (11, 42)). However, note that this correlation was not essential: transcription factors related to the SCENIC-named regulator could be contributing to regulon control in fact. Moreover, post-translational signaling changes could increase or decrease activity even if the gene encoding a factor did not change expression. Thus, SCENIC may have indicated, in part, when signaling pathways controlling a factor’s activity post-transcriptionally were activated. To check how well a regulon’s gene list truly predicted functional control, we exploited high-sensitivity previous identifications of functionally responding Spi1 (PU.1) target genes (DEGs) that were validated in acute gain and loss of function experiments across the DN2a-DN3 interval(43). Spi1 regulon members, whether defined in the Phase 1 cells here or in later stages (Phase 2) were indeed highly enriched for PU.1-activated DEGs, but had minimal overlap with PU.1-repressed DEGs (Fig. S6G,H; Table S9A), in accord with other ChIP-seq evidence(43).
Fig. 4.
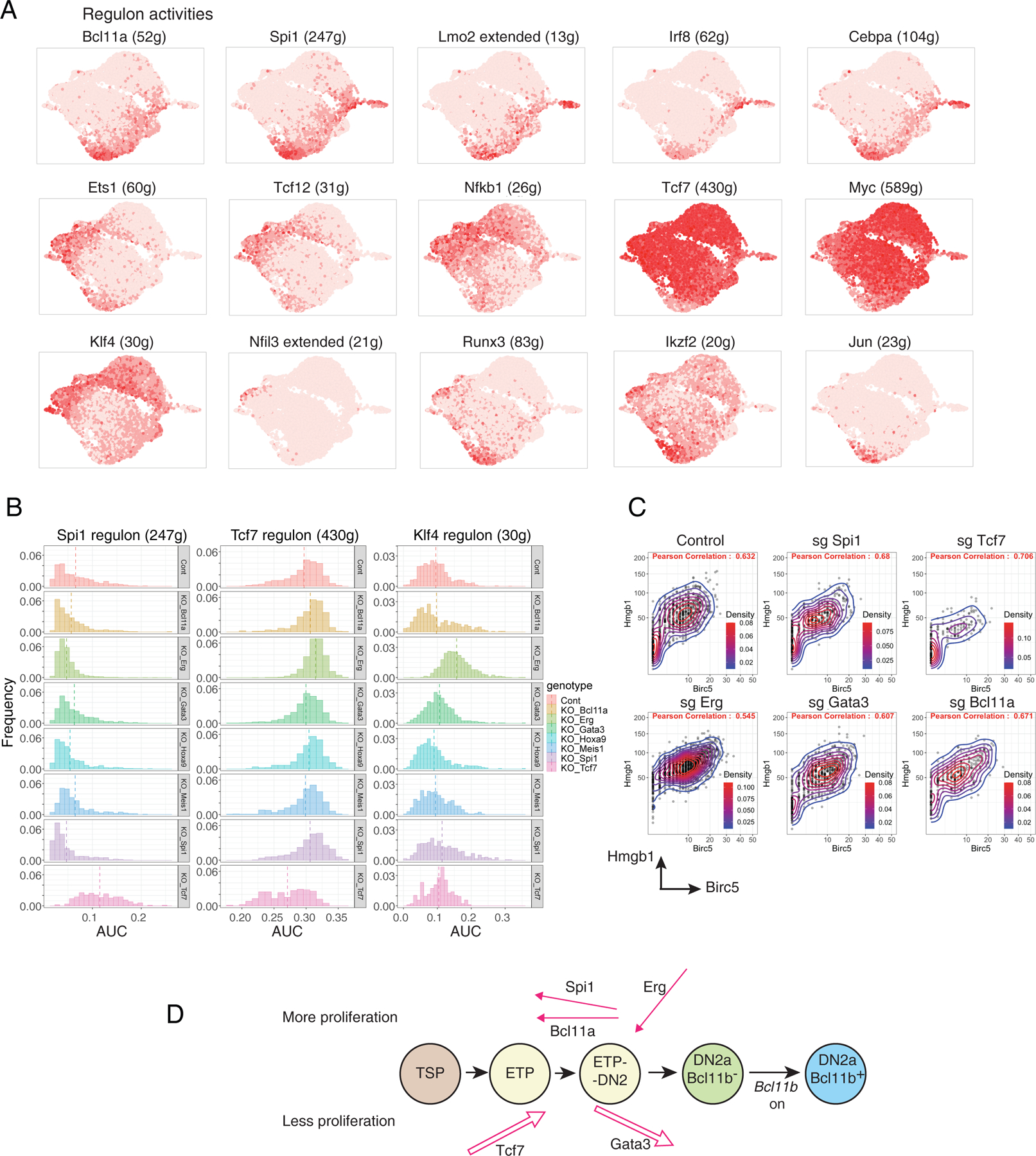
SCENIC analysis of major regulons in early pro-T cell pool-perturbation dataset and impacts of transcription factor knockouts (A) Activities of inferred regulons for different endogenous transcription factors across the whole pool-perturb early pro-T cell dataset. Color intensity in UMAP indicates expression of genes constituting the regulons predicted to be activated by the indicated transcription factors (gene lists in Table S4B). Landmark regulons: in top row, Bcl11a, Spi1, and Lmo2(extended) are progenitor-associated; in middle row, Ets1, Tcf12, and Tcf7 are T lineage progression-associated; in bottom row, Klf4 and Nfil3 are innate-immune cell associated. (B) Histograms of activities of the Spi1, Tcf7, and Klf4 regulons among cells from the 7 KOs and control (“genotypes”). Plots show frequency (y axis) vs. area under the curve (AUC, x axis) for each regulon within the indicated subset. Vertical broken line indicates median AUC score. Number of cells in each genotype: Cont = 793, Bcl11a KO = 353, Erg KO = 1397, Gata3 KO = 686, Hoxa9 KO = 503, Meis1 KO = 409, Spi1 KO= 481, Tcf7 KO = 215. Kolmogorov–Smirnov tests performed comparing each KO to control were included in Table S4. (C) Pairwise scatterplots of the Hmgb1 and Birc5, both cell cycle-associated gene transcripts, showing different enrichments of cycling cells among the different KOs (G2/M phase cells express Birc5). Density scales are contours with color scales automatically adjusted for the distribution of cells in each sample. Density scale for Control, sg Gata3, and sgBcl11a: 0.02–0.08; for sg Spi1, 0.025–0.075; for sg Tcf7, 0.06-approx 0.12; for sg.Erg, 0.025–0.100. Pearson correlations were similar, ranging from 0.54 to 0.71. (D) Summary schematic of inferred physiological roles of Tcf7, Gata3, Spi1, Bcl11a, and Erg in developmental progression (arrow direction left to right for promoting differentiation, right to left for restraining differentiation) and proliferation control (arrow pointing up for enhanced proliferation vs. pointing down for restraining proliferation) in normal early pro-T cell development, as described in the main text. T lineage progression promoting activities shown as outlines, progenitor state-maintaining activities shown as lines with filled arrowheads.
Regulons for Spi1 and Tcf7 transcription factors, calculated from the whole dataset, showed specifically reduced activity in the Spi1 and Tcf7 KOs, respectively, as expected (Fig. 4B, Table S4; stars in Fig. S6A). However, in accord with a global developmental progression block, Tcf7 KO cells were enriched for expression of all progenitor-associated regulons, including the Spi1, Bcl11a, Irf8 and Cebpa regulons (Fig. 4B, Fig. S6A, E, Table S4). In contrast, Spi1 KO increased activity of the predicted Tcf12, Ets1, and Nfkb1 regulons, suggesting that PU.1 encoded by Spi1 normally restrained these factors’ activities (Fig. S6C, Table S4). Most notably, the Erg KO caused specific upregulation of the regulons for Rxra and Klf4 (Fig. 4A, bottom; Fig. 4B; Fig. S6B, D; Table S4), normally myeloid- or ILC-linked factors(42), but reducing activity of the Tcf12-extended regulon (Table S4). Thus, in addition to direct targets, SCENIC gave substantial evidence that the targeted transcription factors functionally controlled the activities of other factors in distinct patterns, and that these impacted intermediate factors contributed to the responses.
Independence of cell cycle and RNA content regulation from regulation of developmental progression speed
Normal T-lineage output depends on proliferation control as well as on gene expression change. RNA content distributions (Fig. S7A) and cell-cycle S phase and G2/M enrichment scores (Fig. S7B; Seurat 3) differed greatly among the various KO samples. The Erg KO had the highest RNA content and cell cycle scores, whereas the Tcf7 KO had the lowest RNA content and cell cycle scores, consistent with their opposing effects on total cell recoveries (compare Fig. 2F). Also, SCENIC identified a Ybx1-extended regulon co-enriched with cell cycle in all samples, comprising S/G2 cell cycle markers, e.g. multiple histone genes and cyclins, Top2a and Hmgb1 (gene list in Table S4B). The Ybx1-extended regulon was also predicted to include Myc, encoding a transcription factor with potent metabolic effects and its own large regulon (Fig. S6F). The Erg KO cells were shifted to a more cycling state, with high Ybx1 expression (Fig. S7B,D) and slightly increased activity of the Myc regulon (Fig. 4C, Figs. S6F, S7D), consistent with the high cell yield, while the Tcf7 KO cells showed the opposite. Gata3 KO cells resembled Erg KO more than Tcf7 KO cells in upregulation of cell-cycle genes Mki67, Top2a, Birc5 and Hmgb1 (Figs. 3C, 4C; Tables S3, S4). Notably, despite their opposite effects on developmental progression, the Spi1 and Tcf7 KOs both had reduced cell-cycle indices (Fig. S7B) and reduced expression of the Myc regulon (Fig. S6F) (40, 44).
Thus, the transcription factor KOs studied here affected T-lineage differentiation progression and cell cycle separately. Besides the dichotomy between factors with developmental progression-promoting and progression-opposing actions, there were also factor-specific effects on cell cycle that cut across this dichotomy. While these results implied that Tcf7 and Gata3 both pushed the cells forward in the T-cell program (outline right-pointing arrows in Fig. 4D), Tcf7 enhanced proliferation (arrow slanted upward) much more than Gata3. Spi1 and Bcl11a pulled them backwards in differentiation (solid left-pointing arrows, Fig. 4D), whereas Erg prevented them from advancing along a variant pathway, restricting their proliferation (down-angled arrow in Fig. 4D). This indicated that early pro-T cells, which individually co-express most or all of the factors studied here (11), were normally subject to distinct, competing forces affecting developmental speed and proliferation (Fig. 4D), contributing to their asynchronous, stochastically progressing differentiation (see Fig. 2D; and (15)).
Routes and kinetics of lineage divergence of Bcl11b KO pro-T cells
A watershed in early pro-T cell development is lineage commitment, occurring as Bcl11b is finally activated(11, 32) (Fig. 1A). Bcl11b acts to preserve lineage identity(30, 37, 45, 46). However, the aberrant cells that emerge when Bcl11b is deleted have obscure origins, emerging from unknown pathway(s) with unclear relationship to pre-commitment stages. Bcl11b KO pro-T cells express some early ‘immature’ genes normally downregulated during commitment, but also abnormally turn on genes that were silent in normal precursors before commitment, i.e. before Bcl11b was there to repress them (34, 37). We therefore investigated the trajectories and regulatory components through which Bcl11b loss causes cells to activate such alternative differentiation pathway(s).
To compare WT and Bcl11b KO precursor differentiation at single-cell resolution around the time of T-lineage commitment, we exploited the longer-term differentiation fidelity of the ATO system (Fig. S1, S2). Because Bcl11b−/− pro-T cells have no survival defect in vitro(28, 33), we used conditional knockout Bcl11bflx/flx;Vav1-iCre mice(37). These delete exon 4 in Bcl11b before T cell development begins; but deletion should only affect cells after ~D7 in the precursors in ATO culture, when Bcl11b expression is normally activated. Multiple parallel ATOs were seeded with LSK precursors purified from different individual wildtype (WT) or Bcl11b KO mice. A first experiment consisted of samples all collected at D10. A second independent experiment generated both D10 samples and D13 samples from aliquots of the same donor cells, which were seeded three days apart so that the D10 and D13 samples could be harvested on the same day, as shown in Figure 5A. In each experiment, Lin− CD45+ CD25+ cells were sorted, separately, from these single-donor replicates (Fig. 5A). The surface staining phenotypes recapitulated previously reported phenotypes for Bcl11b KO pro-T cells(33–37): while WT cells progressively downregulated c-Kit, Bcl11b KO cells sustained c-Kit expression (Figs. 5B, S8A). Samples from different individual animals were separately tagged with cell hashing antibody conjugates to associate different barcodes with each donor(16), then pooled for scRNA-seq and analyzed in a single 10X v3 run. All results were combined in the same reference UMAP1–2 plot. As shown in Figure S8B–D, samples from different donors across both experiments showed excellent reproducibility. In the following, the same UMAP1–2 plot from the combined dataset was used to display all the results highlighted through all parts of Figures 5–7 and Fig. S8.
Fig. 5.
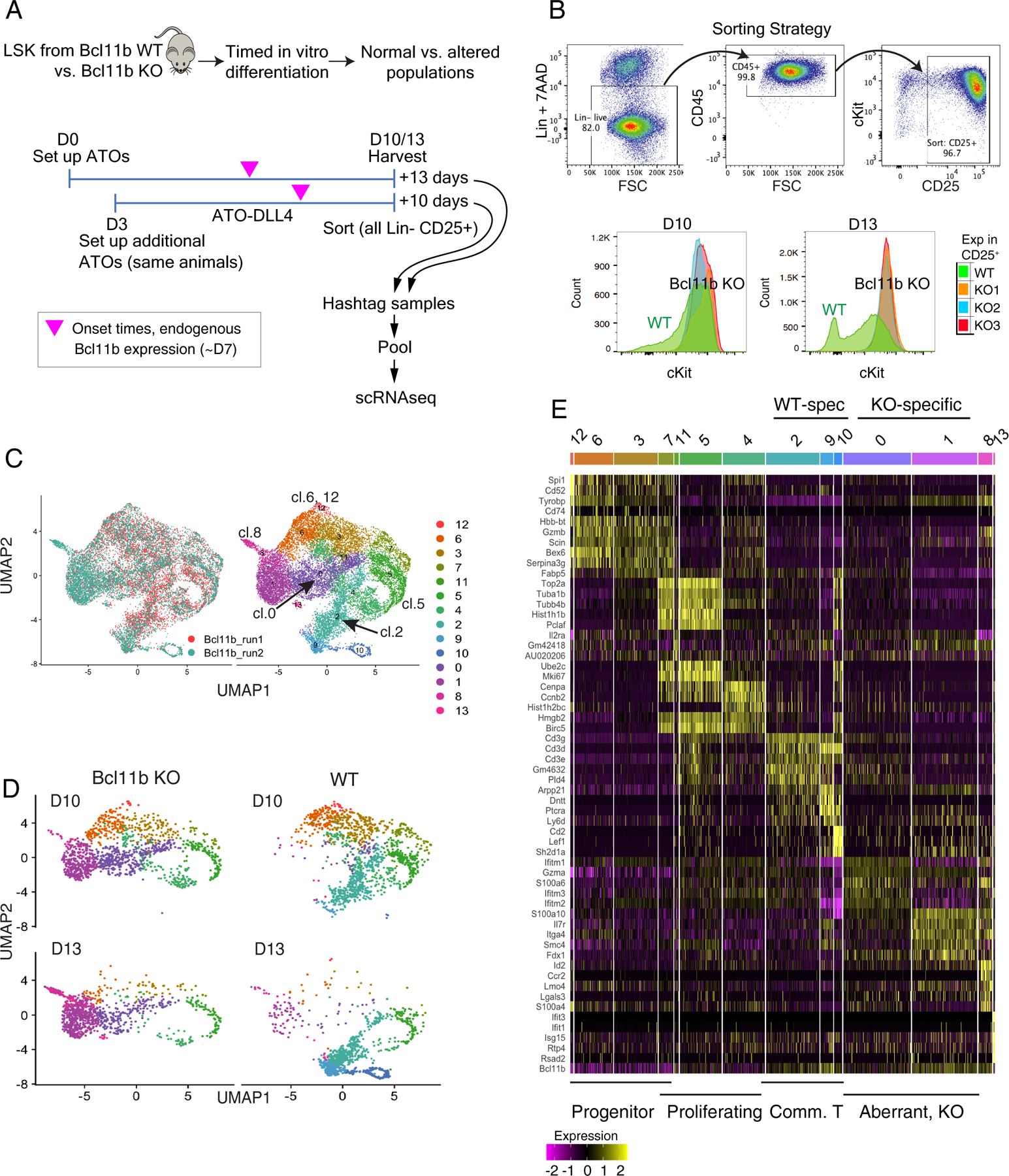
Time-dependent progressive divergence of Bcl11b knockout from wild type gene expression trajectories at late pro-T cell stages. (A) Schematic of experiments to determine pathway of response to loss of Bcl11b. Staggered setup of cultures from aliquots of frozen BM progenitors enables samples from the same donors to be collected after 10 or 13 days of differentiation, simultaneously. Magenta arrowheads indicate that endogenous Bcl11b normally turns on at approximately D7 of differentiation. (B) Top: FACS purification strategy for the Bcl11b scRNA-seq experiments. Bottom: surface staining phenotypes of progenitor-marker c-Kit levels in WT (green) and Bcl11b KOs (replicates in blue, red, orange, also see Fig.S8A). (C) Left: alignment of two experiments of Bcl11b KO scRNA-seq profiles after CCA scaling, as ‘Bcl11b_run1’ and ‘Bcl11b_run2’, in UMAP1–2. N=7451 cells from ‘Bcl11b_run1’, and N=8558 cells from ‘Bcl11b_run2’, total 16009 cells. Note that ‘Bcl11b_run1’ samples were only collected in D10. Right: Louvain clustering of the integrated samples from all timepoints and both genotypes. (D) Samples subsetted according to the hashtag demultiplexed genotype and time of harvest, and displayed in UMAP1–2, colored by the same clustering annotation as in panel C, right (individual mouse samples shown in Fig. S8B). Note that these samples were not subjected to cell cycle regression before UMAP plotting, as this helped to distinguish genotypes. Proliferating cells had higher UMAP1 values (right). (E) Heatmap displaying the top 5 enriched genes in each sub-cluster ordered by approximate developmental trajectories of WT and Bcl11b KO, based on gene expression and connectivity in UMAP displays. (Seurat 3 pipeline, minimum fraction of expressing cells ≥ 0.25, Wilcoxon rank sum test, avg_logFC ≥ 0.3). Full details of cluster-defining marker genes are provided in Table S5.
Fig. 7.
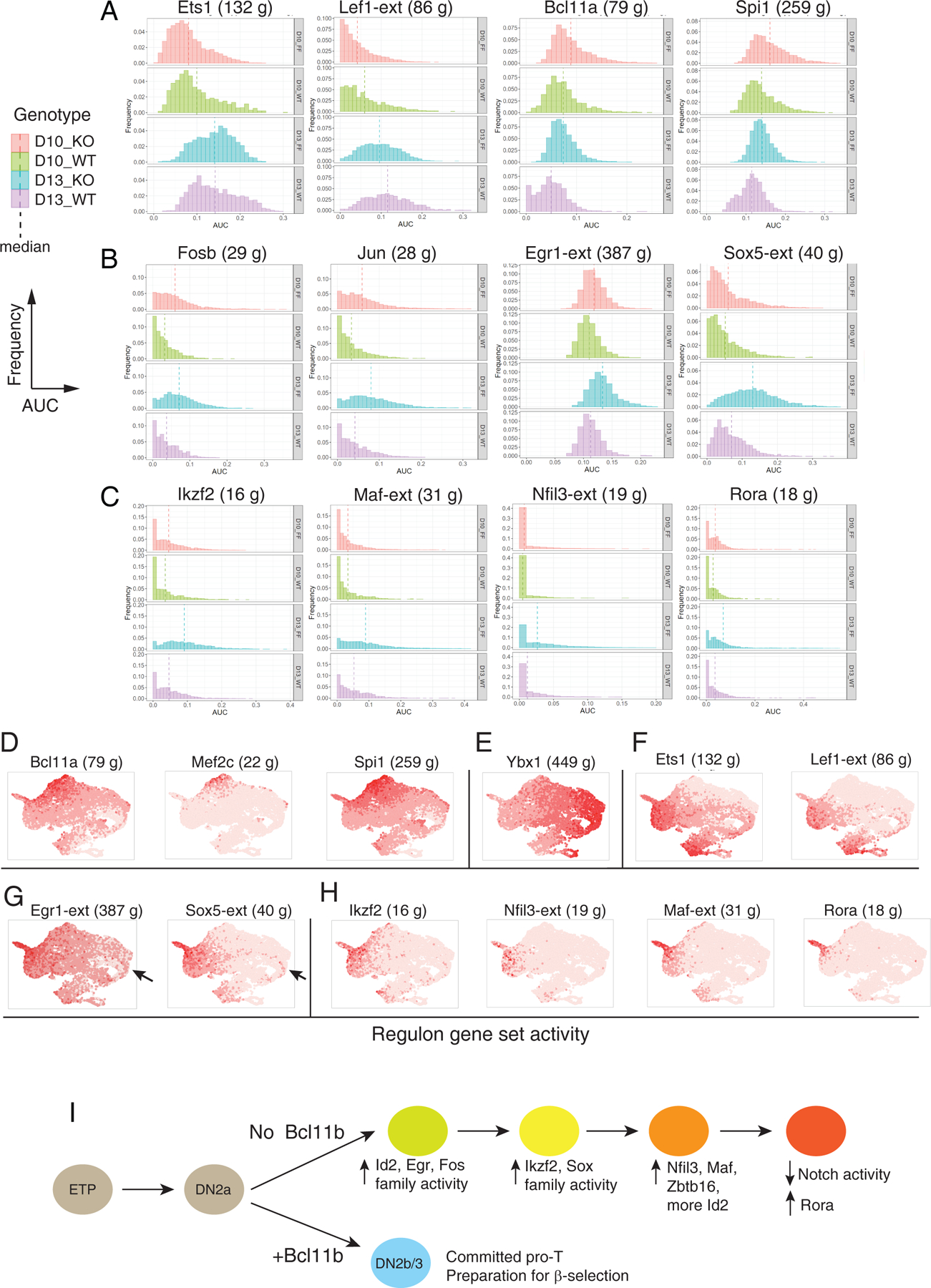
SCENIC analysis of Bcl11b KO impact on gene expression (A-C) Histograms of expression enrichments for indicated regulons, comparing cells from Bcl11b KO (FF) and WT controls (WT) at D10 and D13. Frequency (Y axis) plotted vs. regulon activity (AUC, X axis), means shown by vertical dashed lines. N cells for each sample: FF10 = 3236, FF13 = 3100, WT10 = 1075, WT13 = 1147, all from second experiment to avoid integration introduced data scaling. Full regulon data of individual sample, regulon gene lists and Kolmogorov–Smirnov tests are given in Table S8. (A) General developmental progression indicators: Ets1 and Lef1 activities increasing, Bcl11a and Spi1 activities decreasing with development. (B) AP-1-associated (FosB and Jun), Egr1 (immediate-early response) and Sox5 (TCRγδ-associated) regulons. (C) Ikzf2, Maf, Nfil3, and Rora regulons, showing increasing activity in Bcl11b KO after D10 and/ or concentrated in candidate terminal state. (D-H) Developmental distribution of regulon activation states, shown in UMAP plots of integrated WT and Bcl11b KO cells, colored with intensities showing relative activities of the indicated regulons. (D) Stem/ progenitor regulons (cf. Fig. 4A, Fig. S6A). (E) Cell cycle-associated regulon Ybx1 (compare Figs. S6, S7). (F) Developmental progression-associated regulons (compare panel A). (G) Egr1 and Sox5 regulons (compare panel B). Arrows indicate early-appearing bias of these regulons to Bcl11b KO subset of cluster 5 cells. (H) Other Bcl11b KO-induced regulons (compare panel C). (I) Proposed pathway of transcriptional regulatory cascade leading Bcl11b KO cells to alternative developmental endpoint, inferred from Figs. 6, 7 and Fig. S10. The earliest changes detectable in Bcl11b KO cells at D10 include increases in Fos, Jun, and Egr1 regulon activity and Id2 expression. Upregulation of Ikzf2, Sox5, Zbtb16, Nfil3, and Maf begin later, based on regulon activity and gene expression. A hypothetical order of activation is shown, based on D10 to D13 changes in activities of these regulons and on the positions of the expressing cells in the UMAP plots, along the right-to-left trajectories for Bcl11b KO cells shown in Fig. 6E and Fig. S10A, B. Id2 gene expression is seen initially in the D10 cluster 5 bifurcation between Bcl11b KO and WT cells (right side of UMAP plot, see Fig. 6B and F) but then begins to increase again from D10 to D13 as KO cells move further to the left of the UMAP plots, reaching a maximum in the terminus of the Bcl11b KO pathway (cluster 8, ‘tip of exit’). At this terminus of the Bcl11b KO pathway, at D13 the cells finally upregulate Rora and shut off Notch signaling, thus exiting from the T-cell pathway.
Characteristically, WT-enriched and Bcl11b KO-enriched genes were identified, in agreement with previous bulk measurement reports(34, 37), at both timepoints (Fig. S9A; fold changes D10 to D13 compared in S9B; Table S6). There were also some genes that changed expression over time in opposite directions in the two genotypes (Fig. S9B, genes in red). Single-cell transcriptomes of Bcl11b KO and WT cells (Fig. 5C) showed partial overlap between Bcl11b KO and WT cell clustering patterns at D10, but gave strikingly different patterns at D13 (Fig. 5D, Fig. S8B). Gene expression patterns (Figs. 5E, Table S5 (11)) identified the main clusters in Fig. 5C. Immature, pre-commitment cells were located toward the top of each UMAP plot (Fig. 5C,D), the bottom of the distribution (clusters 2, 9, 10) consisted exclusively of WT cells, whereas the middle left side of the distribution (clusters 0, 1, 8) consisted only of KO cells. As shown in Fig. 5D, at D10, cultures of both genotypes still included many cells in immature, pre-commitment states (clusters 12, 6, 3 from Fig. 5C) as well as intermediate cells expressing cell cycle genes (clusters 7, 5). By D13, the immature clusters were depleted in both genotypes. However, as WT cells increasingly shifted “down” to more mature committed (‘Comm. T’) stages in clusters 4, 2, 9, and 10, the Bcl11b KO cells shifted “left” to alternative ‘aberrant KO’ clusters 0, 1, and 8 instead (Fig. 5D). These assignments were confirmed by the UMAP expression distributions of characteristic stage marker genes (Fig. 6A): Mef2c and Spi1 marking the immature cells in both WT and Bcl11b KO samples, and Lef1, marking cells in later stages of development in both WT and Bcl11b KO samples. Transcripts from non-deleted Bcl11b locus exons were also detectable in WT and Bcl11b KO alike, distinguishing the stages after commitment should normally have occurred (Fig. 6A). The Bcl11b-dependent target Cd3g was turned on during commitment in WT but not in Bcl11b KO cells (Fig. 6B). Genes previously shown to be upregulated specifically in Bcl11b KO fell into two patterns on these plots (Fig. 6B): genes including Zbtb16 and Tyrobp were normally expressed before commitment but abnormally sustained and upregulated in the KO-specific cell states; while genes like Rora, Sox5, Il2rb, and Id2 were highly specific for the KO cells. Cells in the cluster 8 region (see key in Fig. 6B) expressed the most highly Bcl11b KO-specific genes (Fig. 6B). We quantified the cluster distribution differences among all the samples in the experiment, using pair-wise KL divergence (Fig. 6C; see legend for sample details). This confirmed the agreements between the replicates, the contrast between WT and Bcl11b KO samples, and the increasing divergence between WT and Bcl11b KO patterns from D10 to D13.
Fig. 6.
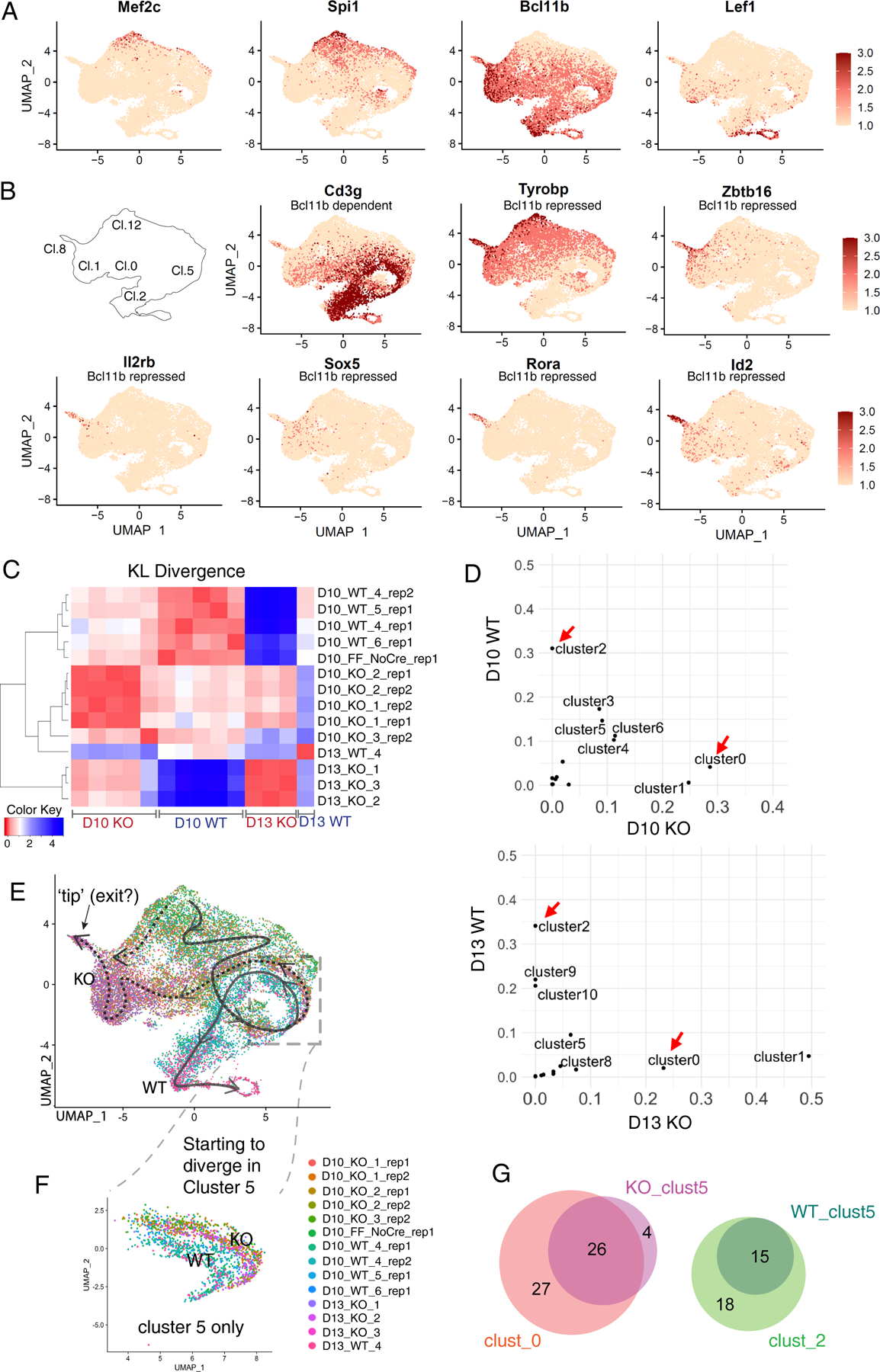
Single-cell analysis of differences between Bcl11b KO and WT: WT and Bcl11b KO trajectories separate immediately after the normal onset of Bcl11b expression. (A, B) Expression patterns of indicative genes in UMAP 1–2 plots of whole WT and Bcl11b KO data ensemble. (A) Genes indicating degrees of progression in the normal pro-T cell pathway. Note that transcripts from Bcl11b locus are detected even from mutant allele. (B) Genes distinctively regulated in Bcl11b KO as opposed to WT cells, with reference to cluster locations on UMAP plot. Cd3g: a representative Bcl11b-dependent gene. Tyrobp, Zbtb16: representative Bcl11b-repressed genes with some normal expression before commitment. Il2rb, Sox5, Rora, Id2: genes with minimal normal expression before commitment, activated selectively in Bcl11b KO. (C) Heatmap showing the KL divergences among all of the integrated samples, calculated based on correlations between cluster distributions, as shown in Fig.5C (Table S6B). Between the two experiments, there were 5 WT control samples (Bcl11b+/+ Vav1-iCre+), 8 KO samples (Bcl11bf/f Vav1-iCre+), and one Cre-negative control (Bcl11bf/f without Cre; “FF_NoCre”). (D) Pair-wise cluster distribution scatterplots comparing WT and Bcl11b KO. Data were from Bcl11b_run2, where the two timepoints were collected from each of the same donor inputs (see comparison, Fig. S8C–F). Pearson correlation r= −0.04 for D10, r= −0.18 for D13. Red arrows indicate the most dramatic and consistent differences between the two genotypes in both time points, cluster 0 and 2. (Raw cluster distributions for each sample in Table S6b). (E-F) UMAP 1–2 colored by individual demultiplexed samples. (E) Schematic, overview of inferred trajectories of differentiation of WT and Bcl11b KO cells around stages of commitment. Solid arrow, trajectory of wildtype cells. Dotted arrows, trajectories followed by Bcl11b KO cells after branching off from main trajectory. See panel (B), key, for cluster numbers. Dashed box, region of UMAP containing proliferating cells (cluster 5 in Fig. 5C) where most divergence appears to occur. Most Bcl11b KO cells follow the horizontal right-to-left path from cluster 5 through clusters 0 and 1, but our data leave open the possibility that an additional minor pathway may exist for some Bcl11b KO cells (dotted downward arrow along the left-hand edge of UMAP plot). (F) Zoom-in view of subset of cells only within cluster 5 from Fig. 6E, with the color of cells showing the slight separation between genotypes. This separation is also indicated in panel (B) by comparing patterns of expression of Cd3g (Bc11b dependent) and Tyrobp (Bcl11b repressed) in this part of the UMAP plot. N=1630 cells. (G) Proportional Venn diagrams comparing genes showing differential expression between WT and Bcl11b KO within cluster 5 (WT_clust5, KO_clust5)(heatmap comparison in Fig. S9D), with the genes showing differential expression between the strongly divergent clusters 0 (KO-specific) and 2 (WT-specific; heatmap comparison in Fig. S9C).
Differences in gene expression between WT and Bcl11b KO at the earlier time point, D10, could shed light on the origin of the divergence, as Bcl11b KO cells were already entering unique transcriptome states on the UMAP manifold. Clusters 2 and 0 were early indices of divergence: at both D10 and D13, cluster 2 was always enriched in the WT cells and cluster 0 was always enriched in the KO, while other differences only emerged at D13 (Fig. 6D; Fig. S8C–F). Cluster 2 cells preferentially expressed T lineage-associated genes compared to cluster 0 (Fig. S9C; full list in Table S7A), while cluster 0 cells expressed Bcl11b KO-specific genes (34, 37). Most genes distinguishing between clusters 0 and 2 continued to be differentially expressed between WT and Bcl11b KO with further differentiation (to clusters 9, 10 for WT and clusters 1, 8 for Bcl11b KO, respectively; Fig. S9C).
Taken together, these gene expression analyses suggested the likely differentiation trajectories shown in Fig. 6E for WT cells (solid arrow, top to bottom) and Bcl11b KO cells (dotted arrows, moving to left). Closer comparison of the individual-sample profiles (Fig. 5D, Fig. S8B) further suggested that although cluster 5 (box in Fig. 6E) included cells of both genotypes, it was not homogeneous. Within cluster 5, the WT and Bcl11b KO cells formed parallel but slightly separate loops (Fig. 6F). Although cluster 5 cells in general shared a cell-cycle-associated signature (Fig. 5E), comparison between the WT and Bcl11b KO cells within this cluster (Fig. S9D, Table S7B) showed numerous genes already significantly differentially expressed before the cells emerged into clusters 0 or 2. The split indicated in Figure 6F is also visible for Cd3g-expressing vs. Id2- and Tyrobp-expressing cells in the cluster 5 region of Fig. 6B. Of the KO-specific and WT-specific genes that became differentially expressed between cluster 0 and cluster 2 (Fig. 6G, clust_0 and clust_2, respectively), nearly half were already differentially expressed between the KO and WT cells within cluster 5 (KO_clust5 and WT_clust5, proportional Venn diagrams in Fig. 6G). Within cluster 5, the WT to KO difference also increased from D10 to D13 (Fig. S9D, genes indicated by red dots). Thus, the initial transcriptome distinction between WT and Bcl11b KO emerged during a shared proliferative phase represented by cluster 5, around the time when the Bcl11b locus was normally turned on (Fig. 6A), before the cells diverged to discrete transcriptome states in UMAP1–2 space.
Defining a unique terminus of the Bcl11b KO pathway
Bcl11b KO pro-T populations abnormally express B-, NK-, other innate-, and some progenitor-associated genes as well as increased γδ-associated genes(34, 37). In contrast to those genes that were abnormally sustained from earlier progenitors in the KO, those that were activated de novo appeared to emerge late in the Bcl11b KO pathway, in cells with the lowest UMAP1 values (Fig. 6B). Alternative cell-type genes specifically upregulated in Bcl11b KO cells nearly all reached their highest levels in the same ‘tip of exit’ population with the lowest UMAP1 value (Fig. 6B), corresponding to transcriptome cluster 8 (Fig. 5C). Thus, most changes activated in Bcl11b KO cells appeared to be compatible with convergence toward a unique regulatory state. This was visualized most clearly when Bcl11b KO cells only were selected (through sample hashtags) and displayed on new Bcl11b-KO-specific UMAP1,3 axes, with early clusters computationally masked (Fig S10A–E). The UMAP3 dimension distinguished cells in this ‘tip of exit’ unambiguously at D13. At D10, Bcl11b KO cells were proliferating (Fig. S10A,B, right), but at D13 they had stopped proliferating (Fig. S10A,B, left) while they upregulated Ikzf2, Cd163l1, and Zbtb16 (Fig S10E). Notably, only cells reaching the ‘tip of exit’ (Fig. S10A–E, see label on UMAP in Fig. S10A) also downregulated Notch target genes Il2ra, Ptcra, and Nrarp (Fig. S10C,D). This terminal inhibition of the Notch pathway and Rag1 silencing could result from the strong secondary upregulation of Id2 also occurring in these cells (Fig. S10E; Fig. 6B), as Id2 is known to antagonize transcription factors needed to support Rag1 and Notch1 activity (47, 48). Thus, the end-stage of the Bcl11b KO pathway may include the effects of an intrinsically downregulated Notch response.
SCENIC evidence for a cascade of drivers in the Bcl11b KO pathway
Continued expression of progenitor-associated genes could simply reflect lack of Bcl11b-mediated repression(37, 49) in Bcl11b KO cells. However, it is unclear what positive regulator triggers Bcl11b KO pro-T cells to upregulate the signature genes that are normally silent, even in pro-T cells before Bcl11b is present to repress them(34). Although the known innate-cell regulator, Id2, was upregulated early in the Bcl11b KO pathway (in cluster 5; Fig. 6B; arrow, Fig. S9D) as well as in the terminus of the pathway (Fig. S10E), Id2 encodes a non-DNA binding antagonist and thus cannot directly mediate this novel positive regulation. To search for other cryptic transcriptional drivers for this process, we used SCENIC (Table S8A–D). SCENIC did not predict direct targets of Bcl11b itself in the datasets of Figs. 5–7, potentially failing to define a Bcl11b regulon because the Bcl11b KO does not eliminate transcription across the locus(37). However, SCENIC shed light on the first regulatory activities that were unleashed when Bcl11b was absent, initiating the KO-specific alternative pathway.
Bcl11b KO cells might be undergoing a mixture of continuing progression (Bcl11b-independent events), failure to complete exit from phase 1 (abnormal progenitor regulator persistence), or de novo activation of alternative programs (new driver activation). Comparison of the WT and Bcl11b KO samples at D10 and D13 showed regulons that exemplified each of these behaviors (Fig. 7A–C). First, Ets1 and Lef1 both normally increase expression at commitment, starting immediately after Bcl11b(11, 19, 42). Their regulons underwent similar increasing activation from D10 to D13 in WT and Bcl11b KO samples alike (Fig. 7A, left). By this criterion, both genotypes progressed. Second, the Bcl11a and Spi1 regulons originally operating before commitment lost activity in both WT and KO samples, but decreased faster in the WT than in KO samples (Fig. 7A, right). This difference suggests weaker silencing of progenitor-specific factors in the KO than in the WT, possibly creating a shortcut pathway to the KO endstate from the progenitor state (see upper left dashed arrow in Fig. 6E); however, Fig. 6A shows that Spi1 expression was extinguished in most cells that were starting the Bcl11b KO-specific pathway. Third, multiple regulons showed specifically increased activity only in the KO, some starting from D10 (Fig. 7B) and others only evident at D13 (Fig. 7C). These KO-associated regulons were most likely to shed light on the identities of effective positive drivers of the Bcl11b KO-specific pathway.
In Bcl11b KO cells, regulons associated with Fosb, Jun, and Junb bZIP family members, i.e. components of the classic signal-activated AP-1 factor (Fig. 7B) were already prominently active by D10 and increased activity at D13. The regulon associated with immediate-early activation factor Egr1 (Egr1-extended, Egr1-ext) was also significantly upregulated in Bcl11b KO cells by D10 and further at D13. Ikzf2 and Sox5 regulons were also upregulated slightly in Bcl11b KO cells at D10, but strongly by D13 (Fig. 7B,C). Among normal T lineage cells, Sox5 expression is rare and limited to γδ cells, similar to Sox13 (42), but Sox5 itself was upregulated in many Bcl11b KO cells (Fig. 6B), and the regulon evidence (Fig. 7B) suggested that Sox5 itself or a related factor had a strong functional impact (Sox5-ext regulon). Between D10 and D13, some Bcl11b KO cells also markedly activated regulons for Nfil3, Rora, and the bZIP factor Maf (Fig. 7C). Fig. 7D–H shows that these regulons were differentially activated across the cells in the UMAP1–2 space. Different parts of the population were marked by activities of progenitor-associated regulons (Fig. 7D), the cell cycle-associated regulon Ybx1 (Fig. 7E), genotype-unbiased regulons associated with developmental progression (Fig. 7F), regulons activated early in Bcl11b KO cells (Fig. 7G; arrow indicates activation occurring within cluster 5), and later-activated Bcl11b KO regulons (Fig. 7H). This time-dependent activation of new regulons associated with distinct transcriptome phenotypes suggested an ordered cascade of transcription factors that could be unleashed in pro-T cells when Bcl11b was absent (as summarized in Fig. 7I).
To test which regulon transcription factors might be functionally responsible for the specific differentiation changes seen in Bcl11b KO cells, we inspected their SCENIC-predicted target lists (Table S8B) for overlaps with a high-confidence list of Bcl11b-responsive DEGs, previously well-validated in multiple knockout conditions (37)(Fig. S10A–C)(Table S9A). The Sox5, Egr1(extended, -Xt), Ikzf2, and Klf2-extended regulon members showed highly significant, specific overlaps with known Bcl11b-repressed targets (Fig. S10F). Three progenitor factor-associated regulons could still be defined in D10 populations, showing significant overlap with subsets of Bcl11b-regulated DEGs, both Bcl11b-activated and -repressed (Fig. S10G). While Jun family regulon members showed low overlap (Fig. S10H), Fos-FosB regulons included 40% of the genes initially induced by Bcl11b loss in cluster 5 (Fig. S10I). Thus, Sox5-, Egr1-, Ikzf2- and Klf-family, and transiently also Fos-family factors, appeared to be strong candidates for activators functionally important in the Bcl11b KO regulatory cascade.
A three-part regulatory relay in Bcl11b KO cells
To assess how cells could be driven progressively along the alternative pathway in Bcl11b KO cells, we investigated relationships between different potential inputs to individual genes within the alternative pathway itself. First, we selected the regulons that were most significantly Bcl11b KO-affected at D10, using the statistical criteria shown in Fig. S11A, and focused on the subsets of genes in those regulons that were also functionally validated as Bcl11b-repressed in the high-sensitivity Bcl11b KO DEG lists (Table S9B). Then, Bcl11b-repressed DEGs within the 11 regulons that were most enhanced in the Bcl11b KO were analyzed to determine whether groups of regulators might be driving expression of the same genes, as a measure of likely coordinate action (Table S9B,C). Of the 65 individual genes in this set, about half appeared in more than one regulon, and about 1/3 had predicted inputs from at least three (Fig. 8A). Furthermore, the pattern of shared regulon memberships predicting inputs into joint target genes appeared to split the regulons themselves into three clusters. Regulons we call “progenitor-type” included Bcl11a, Spi1 (PU.1), and Irf5-Irf8, Phase 1 antagonists of T-cell specification. Another group was “alternative lineage-type” regulons (Ikzf2, Sox5, Rora and Klf family)(Fig. 8A). Between these, a “pivot-type” group included Egr1, Fos family, and Jun family immediate-early activators.
Fig. 8.
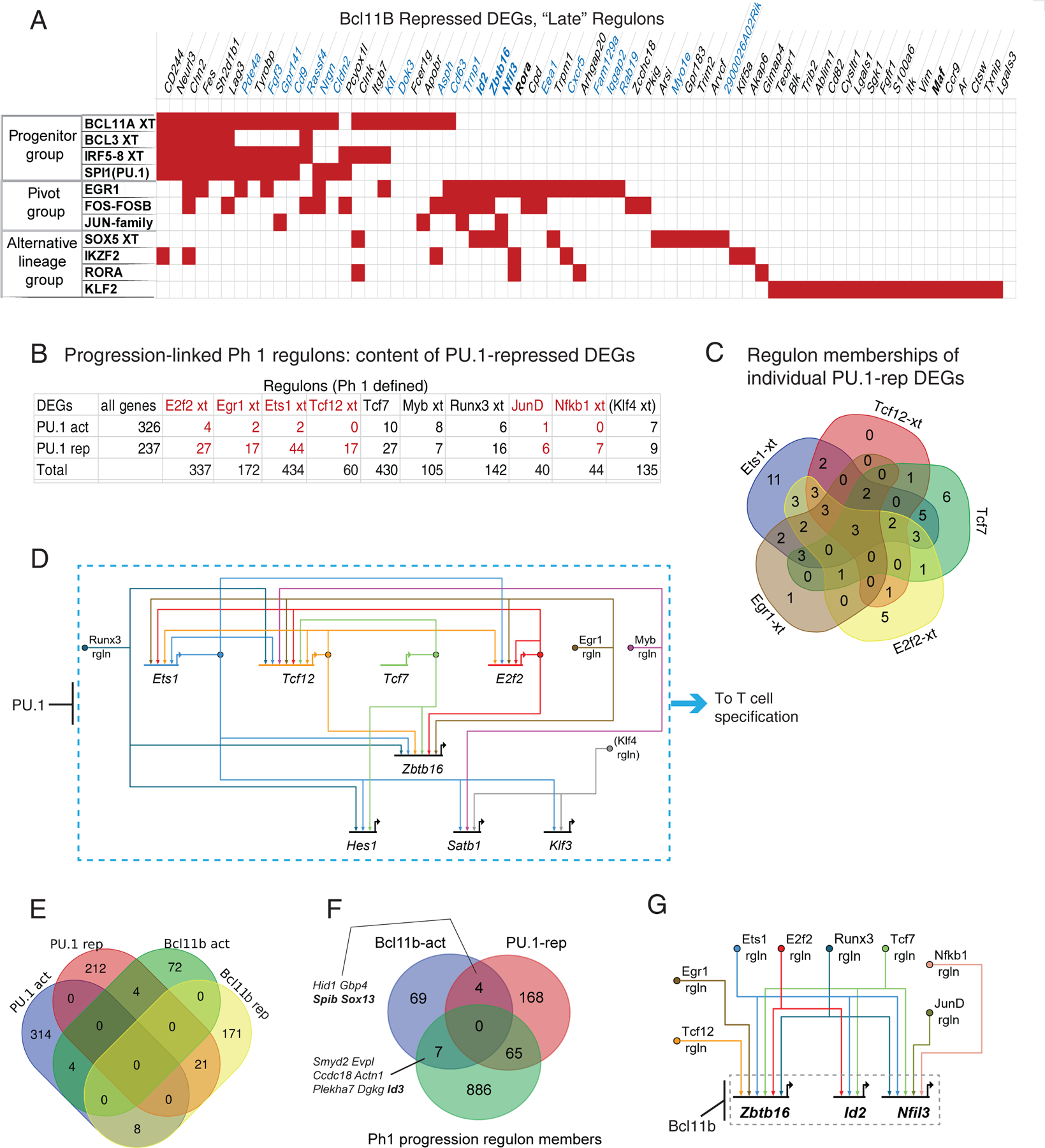
Inference of underlying network architectures for Bcl11b KO response and its relationship with an initial Phase 1 T progression circuit (A) Input-target relationship matrix for validated Bcl11b-repressed DEGs within the Bcl11b KO-upregulated regulons. Red cells in matrix indicate that a given gene listed (columns) is a member of a given regulon (rows). Flow chart of data analysis logic and threshold criteria is shown in Fig. S11A, from full data in Table S8. Panel shows that individual Bcl11b-repressed DEGs(37) often can be members of more than one indicated regulon, and that the cases where the same gene is a member of more than one regulon fall roughly into three pattern groups. (B) Identification of an early T-lineage progression-associated regulon ensemble among Phase 1-defined regulons (Table S4). Klf4 regulon, identifiable from data in the pooled-perturbation experiment, is in parentheses here and below because Klf4 is minimal or absent in normal pro-T cells. Flow chart of selection criteria is shown in Fig. S11B, using data from Table S4. Panel shows number of individual genes in each Spi1 KO-enriched Phase 1 regulon that overlap with validated PU.1-activated or PU.1-repressed DEGs(43). Red: highly significant bias favoring overlap with PU.1-repressed rather than PU.1-activated DEGs. Fisher’s exact test p values are given in Table S9D. (C) Extensive overlap in Phase 1 regulon memberships among PU.1-repressed DEGs. Full gene lists are given in Table S9E. A live interactive model of predicted gene network relationships across all the regulon-DEG intersection genes in this analysis (http://bioinfoweb.caltech.edu/tf_targets/) is shown in Fig. S11. (D) Highly-interconnected transcription factor network associated with progress through T-lineage specification is predicted by shared regulon memberships of genes that encode major PU.1-repressed Phase 1 transcription factors themselves. Network model uses the Biotapestry graphic framework (64), which displays oriented gene networks in which regulator is identified as distinct from target, with arrows pointing from regulator to target gene’s control sequences. Shown are the relationships between regulons enriched in Spi1 (PU.1) KO samples that also have PU.1-repressed transcription factor genes among their members, based on Table S3, Table S11, and (43). PU.1 is depicted as a repressor (line with blocked end) of all the genes and regulons (rgln) shown in the dashed box. Gene models, horizontal lines with bent arrows, here represent transcription factor coding genes seen to be transcriptionally repressed by PU.1. Filled circles represent transcription factor protein activities inferred from activities of their regulons, as shown in panel (B). Arrows represent mapping of regulon transcription factors to putative gene targets, based on their regulon memberships. Different regulons are depicted with different colored lines to help trace each putative regulator (circle) to its various targets. Transcription factor coding genes in this circuit are given the same color as the circles representing the factors they encode and the arrows connecting their activities to target genes in their regulons. Regulon membership is depicted as positive regulation here (pointed arrows) but could include negative regulation in some cases. Klf4 rgln: seen here to be upregulated in Spi1 KO, but shown in parentheses because not normally expressed in early pro-T cells. (E) Venn diagram of relationship between validated Bcl11b KO DEGs(37) and validated Spi1 (PU.1) KO DEGs(39): no preferential association seen between Bcl11b-dependent “commitment” genes and PU.1-repressed “T lineage progression” genes. (F) Genes that are both Bcl11b-dependent DEGs and PU.1-repressed DEGs are separate from Phase 1 progression-associated (specification-associated) regulon membership. Phase 1 regulon member pool here includes whole regulon memberships of Ets1-ext, Tcf12-ext, E2f2-ext, Egr1-ext, Tcf7, Runx3-ext, JunD, Myb-ext, and Nfkb1-ext. (G) Biotapestry gene network model of predicted inputs from Phase 1 progression-associated transcription factors into transcription factor genes that are Bcl11b-repressed, based on memberships of the Phase 1 regulons. Relationships are depicted as in (D), with the same color code for the same factors encoded by the genes shown in (D). For gene lists see Table S9G.
Bcl11b-repressed DEGs in the Bcl11a-extended (Bcl11a-xt), Bcl3-xt, Irf5–8-xt, and Spi1 regulons often had multiple predicted regulators within this group but were mostly separated from the other Bcl11b-repressed DEGs (Fig. 8A, left), consistent with expression of these transcription factors limited to progenitor-type cells (e.g. Spi1; see Fig. 6A). Further, at this stage of development, these “progenitor-group” regulons included no transcription factor coding genes. A second group of genes (Fig. 8A, middle) shared membership in multiple regulons of pivot-type factors, often with membership in alternative lineage-type factor regulons as well. Strikingly, this group included the genes encoding the four key alternative-lineage factors, Zbtb16, Id2, Nfil3, and Rora themselves. A final group of genes appeared only to receive single, alternative lineage-type inputs; these included only one transcription factor coding gene (Maf)(Fig. 8A, right). Thus, as a source of regulatory inputs to turn on genes coding for the alternative-lineage factors themselves, the “pivot”-type regulon factors appeared to most likely be the initiators, whether or not other inputs were present.
Taken together, the results in Figs. 7, 8A, and S10 indicate that pro-T cells reaching the threshold of commitment, when they would normally upregulate Bcl11b, responded to the lack of functional Bcl11b by initiating an activation-like response and progressively upregulating ‘abnormal’ genes in an alternative developmental pathway. As summarized in Fig. 7I, the earliest detectable events in this cascade involved an immediate-early type activation response, together with Id2 activation. Ikzf2 and Sox5/ Sox family regulators appeared to be activated next, through undefined drivers. A third set of alternative regulators was next activated, with SCENIC analysis suggesting that the initial “pivot” regulators could have direct input into the genes encoding Zbtb16 and Nfil3 among them, as well as into Rora and Id2. Finally, the late increase of Id2 could directly cause the loss of Notch signaling at the terminus of the program (47, 48).
Relation of early T-cell specification circuit to lineage choice at commitment, from augmented SCENIC analysis
Results from the augmented SCENIC analyses shown in Fig. 8A (Fig. S11A) and Fig. S10F–I suggested that a similar method could also shed light on earlier connections between genes most important for establishing competence for commitment, during the Phase 1 stage of T cell development. Returning to the Phase 1 pool-perturbation data (Figs. 1–4), we therefore refined the SCENIC analysis of dynamically affected regulons by identifying the genes within them that were most closely correlated with faster differentiation progression speed. As Spi1 (encoding PU.1) was found to be the most powerful brake on earlier T-lineage progression (Fig. 3B), the various regulons it inhibited were presumably enriched for genes promoting a T specification program. As shown above (Fig. S6G, H), the Spi1 regulon itself contained PU.1-activated but not PU.1-repressed DEGs, consistent with ChIP-seq evidence that it represses indirectly(43); thus, other factors must be the positive drivers of genes that Spi1 repressed. We therefore identified 10 regulons in the phase 1 pool perturbation samples most significantly increased in activity in the Spi1 KO (Table S4D), and specifically identified the PU.1-repressed DEGs these regulons contained (68 of 237 total PU.1-repressed DEGs), using the criteria diagrammed in Fig. S11B.
The extended Ets1(Ets1(ext)), Tcf12(ext), E2f2(ext), and Egr1(ext) regulons especially contained PU.1-repressed rather than PU.1-activated DEGs (Fig. 8B; Table S9D). Together with the Tcf7 regulons, these five regulons covered 60 of the total PU.1-repressed DEGs (Fig. 8C; Table S9D). Ets1, Tcf12, E2f2, and Tcf7 were themselves downregulated by PU.1 in pro-T cells, and here the statistical prominence of these regulons (Fig. 8B; Table S9D) underlined their potential impact on development. In addition, the Ets1(ext), Tcf12(ext), Egr1(ext), and Runx3(ext) regulons all were also upregulated by Bcl11a KO and Spi1 KO, and reduced in activity by Tcf7 KO and Gata3 KO (Table S9D), supporting their identification as central to early T-lineage developmental progression in general and not simply repression targets of one transcription factor. Multiple regulons contained the same PU.1-repressed DEGs, suggesting parallel inputs (Fig. 8C, Table S9E; Fig. S11C, D). Notably, the PU.1-repressed DEGs within these specification-associated regulons also frequently included transcription factor-coding genes, especially Ets1, Tcf12, and E2f2 themselves, which were predicted targets in multiple regulons. The combined DEG and SCENIC analyses indicated that these transcription factor coding genes could not only be common targets of PU.1 inhibition, but could also cross-regulate each other, in a network with the relationships shown in Fig. 8D (regulon memberships highlighted in Table S9D). A live interactive model (Fig. S11C, D) is presented at http://bioinfoweb.caltech.edu/tf_targets/. Thus, the developmental progression program that Spi1 (PU.1) restrains was likely based on a densely interconnected gene regulatory network, incorporating differentiation drivers including ETS factors, Tcf12, E2f2, and Egr1, that potentially cross-activate.
The T-lineage commitment program promoted by Bcl11b in DN2b stage was now seen to be substantially disconnected from the early specification-associated network. Bcl11b DEGs and PU.1 DEGs had minimal overlaps (Fig. 8E). Furthermore, Bcl11b-activated (Bcl11b-dependent) DEGs also had minimal overlap with the core Phase1-defined specification-associated regulons (Fig. 8F). This argued against a model in which Bcl11b simply sustained T-cell specification through the same mechanisms set in place earlier. Instead, specification-associated regulons were mostly biased to overlap with Bcl11b-repressed DEGs. Multiple genes upregulated in the Bcl11b KO (Fig. 8A, blue font) were predicted to receive input from Phase 1 specification transcription factors. Notably, the regulon analysis suggested that Bcl11b KO-associated regulatory genes Zbtb16 (Fig. 8D), Id2 and Nfil3 all could receive inputs from multiple specification-associated Phase 1 factors besides pivot factor Egr1 (Table S9G; Fig. 8G). This analysis thus indicated candidate linkages that could connect the early drivers of T-cell program entry to an alternative pathway restrained later by Bcl11b, while Bcl11b-dependent targets appeared to create discontinuity.
Discussion
Cell fate commitment represents a milestone in the developmental process, an irreversible transition for multipotent cells. However, for a stem cell-derived population, approaching lineage commitment is a developmental continuum guided by a gene regulatory network. In early T cell development there is marked variation among clones in differentiation speeds leading to commitment(15). The process involves multiple transcription factors (5, 10, 19, 20, 34, 50–52), and previous studies tested perturbations of individual transcription factors through bulk assays, often at later stages(24, 25, 28, 34, 37, 40, 41, 53). Here, using single-cell approaches, we elucidated the gene regulatory network leading up to commitment and controlling the fate choice that cells undergo at commitment.Most individual ETP cells in vivo still co-express progenitor-associated genes even after activating the Notch-induced T-cell regulatory genes, Gata3 and Tcf7 (11). The lack of single-cell resolution in such perturbation studies has hindered the full definition of the roles of individual factors leading up to T-cell lineage commitment. Here, we used scRNA-seq coupled with specifically designed and optimized perturbations to dissect the cooperative and antagonistic relationships among key regulators within single early T cell precursors. We leveraged a controlled in vitro differentiation system to score knockout effects at specific timepoints in the development of cohorts of cells from defined primary-cell precursors. Thus, we clarified the stage-specific roles of Tcf7, Spi1, Gata3, Bcl11a, and Erg leading up to lineage commitment, computationally defined a core network circuit involved in pre-commitment T-lineage specification opposed by Spi1, and characterized a separate alternative-lineage cascade of poised factors that were later restrained by Bcl11b as it directed and enforced lineage commitment.
The ETP population, the starting point for T cell development, has been especially problematic to interpret because the bulk population shows so much similarly with non-T hematopoietic precursors in the bone marrow and fetal liver(13, 42, 54). Progenitor-associated transcription factors in the bulk ETP population, like Spi1, Bcl11a, and others (42) could in principle be working in completely different cells than those that go on to give rise to T cells(11). Thus, it was notable that during ETP to DN2a progression, disruption of Spi1 or Bcl11a shifted the cells further along the same T cell developmental trajectory with the same intermediate transcriptome states as the control cells, indicating that normally these factors work to control speed along this canonical pathway. Effects of Hoxa9 and Meis1 KOs were not resolved here. However, Erg KO also accelerated the cells, but caused the population to shift to an aberrant, highly proliferative altered-DN2 state. Erg activity is a distinguishing feature of T-cell precursors before TCR expression(55), with vital roles in hematopoietic stem cell maintenance, differentiation restriction, and self-renewal (56, 57), but its functional activity in T-lineage cells has previously been associated only with malignancy(58). The Erg KO altered pathway was marked by high Klf4 activity, with E protein activity (Tcf12 regulon) distinctively inhibited, potentially via Id1 upregulation. The elevated cell cycling, Klf4, and Myc activities resembled a self-renewal phenotype induced in macrophages(59) as well as roles of these factors in pluripotency(60). Our results thus indicated a role for Erg, at normal endogenous levels, in keeping T-cell precursors on the mainstream developmental trajectory.
SCENIC analysis(38) further showed that the effects of different Phase 1 transcription factors were individually distinct as they controlled proliferation independently of their effects on T-lineage progression. SCENIC analysis also identified a tightly-interlinked core circuit of transcription factor activities that appeared to underlie the progression of T-lineage specification in the Phase 1 cells. This circuit was promoted by Tcf7 and/or Gata3 and restrained by Spi1, and included regulatory genes beyond those that we perturbed experimentally here. Taken together, the results confirmed that the progenitor-associated factors as well as the “T-lineage” factors were actively regulating T-lineage developmental progression speed, with Bcl11a and Spi1 slowing progression and Erg partially steering it, while Gata3 and Tcf7 accelerated it within the same cells. The network instability implied by these results could help explain why the absolute timing with which individual cells progress through the ETP and DN2a stages is so variable (15).
As cells progress through lineage commitment, Bcl11b is through an incompletely understood mechanism. Bcl11b KO populations in vivo and in vitro appear to be developmentally arrested at an abnormal DN2/DN3 stage (29, 33, 35–37). Eventually they produce NK-like CD25− CD44+ cells(33, 36), outside of the T-cell pathway(34), but the precursors of these emerging cells are not clear. Our results showed that despite the retention of DN2-like cell surface phenotype, loss of Bcl11b rapidly caused a switch in pathway choice across the majority of the population, rather than arresting developmental progression. Based on SCENIC, a transient activation of immediate-early response factors of Egr and Fos families appeared to lead to activation of the regulators that were specifically upregulated in the Bcl11b KO-specific pathway, including Ikzf2, Sox family factors, Nfil3, Zbtb16 and Maf factors. Despite dysregulation of genes normally associated with separate NK, ILC, and TCRγδ pathways(36, 37), our results further showed that all the regulatory changes induced by Bcl11b loss appeared to overlap in the same cell clusters and later converge, reaching maximum expression at a single common endpoint at which Notch signaling response genes were finally turned off. The terminal upregulation of Rora and Id2 with loss of Notch input, together with high Gata3, resembles the gene network circuit recently described for natural ILC2 differentiation from fetal thymocytes (61).
Cells proliferating around the time of commitment appeared primed to enter one of two divergent developmental trajectories, depending on whether Bcl11b was activated; yet both represented a clear departure from the pre-commitment states of ETP-DN2 stage cells. SCENIC analysis supported the model that some of the same factors that earlier promote T-cell specification in opposition to Spi1 could themselves be direct regulators of Zbtb16, Nfil3, and Id2, consistent with the genetic requirements for T-cell-associated factors in ILC(62). Further studies will be required to interrogate fully the impacts of each of the predicted intermediate regulators and their predicted network connections. However, these single-cell transcriptome analyses have indicated how six transcription factors interact to control speed and correct pathway choice in early T cells, provided evidence for poised gene network circuitry that may connect their targets into coherent pathways, and resolved their additive and stage-specific contributions to programs for T cell and innate lymphoid development.
MATERIALS AND METHODS
Study design
This study was designed to dissect the roles of transcription factors Tcf7, Spi1, Gata3, Bcl11a, Erg, Hoxa9, Meis1, and Bcl11b in the gene networks leading to and implementing T-cell lineage commitment. The goal was to clarify how differentiation speed and cell fate outcomes were controlled by these transcription factors in early T cell development. We therefore acutely perturbed expression of these transcription factors in primary pro-T cells through synchronized in vitro differentiation systems and controlled gene-disruption strategies. The impacts of perturbation were analyzed by scRNA-seq with batch indexing, 3–6 days after perturbation or after onset of the gene’s normal expression.
Mouse models
Cells were from young adult mice with a C57BL/6 (B6) background. B6.Cg-Tg(BCL2)25Wehi/J (Bcl2-tg), B6N.Cg-Commd10Tg(Vav1–icre)A2Kio/J (Vav1-iCre), B6.Gt(ROSA)26Sortm1.1(CAG-cas9*,- EGFP)Fezh/J (Cas9), and B6.Bcl11byfp/yfp mice (32) were bred as reported previously(37). Bcl11b+/+ and Bcl11bfl/fl ROSA26R-YFP mice both with Vav1-iCre (37) are denoted here as WT and Bcl11b KO. For care and details see Supplementary Methods.
Cell lines
OP9-DLL1 co-cultures(6) and mATO cultures(14) were used for in vitro differentiation, as described in detail in Supplementary Methods.
Cell purification and culture
For in vitro (ex-vivo) differentiation of pro-T cells, bone marrow hematopoietic progenitors were used for input. Bone marrow (BM) was removed from the femurs and tibiae of 10–12 week-old mice, and cells were depleted with magnetic beads to remove Lineage marker-positive (Lin+) cells, and the resulting Lin− cells were further FACS sorted to purify live (7AADnegative), CD45+ “LSK” cells (Linnegative Scalhigh c-Kithigh), as a source of multipotent hematopoietic stem and progenitor cells. Purified cells were stored frozen in liquid N2. Methods including antibodies are spelled out in Supplementary Materials and Methods (Cell Purification and Culture: Primary Cell Purification). Flow Cytometry conditions are detailed in Supplementary Methods (Cell Purification and Culture: Flow Cytometry and Cell Sorting). Flowjo analysis software was used to analyze flow cytometric data. For bulk RNA-seq and scRNA-seq samples, gating strategies are included in Fig.S1, S2, and 5B.
CRISPR/Cas9-mediated acute deletion in precursor cell cultures
Stored Lin− or LSK cells were thawed and recovered in cytokines (details in Supplementary Methods: Cell Purification and Culture, CRISPR/Cas9-mediated Acute Deletion in Precursor Cell Cultures). Then the cells were transduced with retroviral vectors encoding reporters (CFP) and the indicated guide RNAs (sgRNAs), and then seeded to OP9-DLL1 culture as described previously(39). For infecting LSK precursors for scRNA-seq (CRISPR), pre-titered batches of virus (Fig. S12B) were delivered to target cells at a precise multiplicity of infection (MOI) of 0.5 or 1, to achieve maximum single-vector infected cells. The whole library was prepared in two independent packaging reactions which were each separately infected into BM cells in at least two parallel, separate replicate cultures (Fig. 1E; Perturbseq pools 1 & 2, Fig. S12B). After 5 d incubation, these replicates were harvested and each labeled with distinct antibody-conjugated “cell-hashing” oligonucleotides(16). This enabled pooling for scRNA-seq analysis in a single Chromium reaction and library preparation, to eliminate the need to correct for batch effects or depth normalization between replicates.
Construct design and cloning
The retroviral vector cloning used for sgRNA expression cloning was based on previously published E42-dTet(39) with the modifications for dual sgRNA cloning compatibility and incorporation of capture sequences for 3’ direct capture of sgRNA for single cell sequencing (details in Supplementary Methods: Construct Design and Cloning and Table S10). The pool-based dual gRNA cloning was performed similarly to the protocol described in (63) (workflow shown in Fig. 1D). Briefly, we designed and pool-synthesized 3 pairs of sgRNAs per gene (together with sgRNA for non-expressing and non-targeting controls), cloned them into a pool of sgRNA expressing plasmids, and then packaged them into the retroviral sgRNA library. The paired sgRNAs in each vector were designed to be compatible with direct capture of both sgRNA sequences in scRNA-seq using 10X Chromium V3 chemistry (‘Cap1’ and ‘Cap2’ in Fig. 1D). We verified equal representation of the guides in the pool (Fig. S12A), the titer of each packaging of the pool (Fig. S12B), and improvement of knockout efficiency using two guides per vector rather than one (Fig. S12C, D), as described in Supplementary Methods (RNA-SEQ AND SINGLE-CELL RNA-SEQ METHODS: Dual gRNA validation).
Bulk RNA-seq
To compare in vitro vs. in vivo development (Fig. S1), Lin− BM cells were harvested from B6.Bcl11byfp/yfp animals, and cultured in ATO or OP9-DLL1 differentiation conditions as indicated. Cells were sorted into CD25low for DN1 (ETP-enriched), Bcl11b-YFPneg CD25hi for DN2a uncommitted, and Bcl11b-YFPposCD25hi for DN2a/b newly committed fractions (32), followed by RNA purification and cDNA preparation. Details are given in Supplementary Methods (Bulk RNAseq Analysis for in vivo vs. in vitro reference analysis).
Single-cell mRNA sequencing
Full details of procedures for each experiment are given in Supplementary Methods as individual subsections under major subheading RNA-SEQ AND SINGLE-CELL RNA-SEQ METHODS. In each experiment, biological replicates for scRNA-seq were FACS sorted on the same day, then antibody hashtagged (TotalSeq A hashtag antibodies, Biolegend), and then finally combined to target equal cell numbers from each of the hashtagged samples for a pooled scRNA-seq analysis. cDNA was prepared following the instruction manual of Chromium v2 or v3 (10X Genomics), while the hashtag library was prepared following the Biolegend TotalseqA guide.
After sequencing libraries of the cDNA, sgRNA, and hashtags from the same experiment, the resulting FASTQ files were aligned using CellRanger3 (for cDNA) (details in Supplementary Methods: Data Analysis: Mapping of scRNA-seq Sequences, Hashtag and gRNA Identification). The single-cell hashtags, as well as the dual guide RNA sequencing data in CRISPR-pool perturbation experiment, were aligned and quantified using in-house tools (hashtag_tool and guiderna_tool) (https://github.com/gaofan83/single_cell_perturb_seq/releases/tag/v.1.0.0), processing from raw fastq data and generate count tables (Fig.S12E, F).
Data analyses
For bulk RNA-seq, base calls were performed with RTA (1.13.48.0) followed by conversion to FASTQ with bcl2fastq 1.8.4. RNA-seq reads were mapped onto the mouse genome build GRCm38/mm10 using STAR (v2.4.0) and were post-processed with RSEM (v1.2.25), and differential expression analysis was performed with EdgeR (v3.6.8). Seurat 3, Monocle 3, and SCENIC were used for transcriptome analyses. The scRNA-seq downstream analysis were performed mainly in R (version 4.0.2) with the following packages: ggplot2(v3.3.2), dplyr(v1.0.2), cowplot(1.1.0), Seurat(v3.2.2), AUCell(v1.10.0), RcisTarget(v1.8.0), GENIE3(v1.10.0), SCENIC(v1.2.2), monocle3(v0.2.3.0), ggraph(v2.0.4), igraph(v1.2.6), philentropy (v 0.4.0), gplots (v 3.1.0). Details of each separate scRNA-seq experiment and its data analysis are described in Supplementary Methods (individual subsections of section Data Analysis). SCENIC analysis using AUCell was carried out as detailed in Supplementary Methods (Data Analysis: SCENIC Analysis and Visualization Graphics).
Statistics
Statistical analyses were conducted using Seurat 3, Monocle 3, SCENIC, R scripts, or GraphPad Prism. Seurat 3 was used for differential gene expression analysis with default settings (Supplementary Methods: Data Analysis: Differential Gene Expression Analysis); however, one alternative analysis of the pool-perturbation differential expression results (Table S3) using modified Seurat settings is shown in Table S11. Note that in the pool-perturbation experiments, most noted effects were seen with at least 2/3 of the sg.RNA pairs for a given gene, although there were some construct-specific differences (Fig. S5B). We combined data from all sg.RNA pairs against the same gene to compare with controls for our estimates of the ‘KO’ impact. Kullback-Leibler divergence tests were described in Supplementary Methods (Data Analysis: Kullback-Leibler (KL) Divergence Calculation). Fisher’s exact test calculations assumed that the maximum number of expressed genes potentially detectable by 10X Chromium was 7000. All SCENIC analysis p-values are shown in Tables S4A–D and S8A–D, and p-values for enrichments of SCENIC regulon/ DEG overlaps are given in Table S9A, B, and D. Statistical tests used, p values and significance levels are reported in respective figure legends and/or tabulated for all data sets and comparisons in Supplementary Tables S2–S9 and S11.
Supplementary Material
Supplemental Material
Supplemental Text: Supplemental Materials and Methods
Fig. S1. Bulk RNA-seq analysis of in vivo and ex-vivo derived early pro-T cells.
Fig. S2. Gene expression profiles of BM-derived early pro-T cells in vitro recapitulate those of in vivo thymic early pro-T cells, on single-cell level.
Fig. S3. Alignment of Cas9;Bcl2-transgenic control cells developing in vitro with wildtype non-transgenic B6 thymic pro-T cells developing in vivo.
Fig. S4. Knockout effects on cluster distributions of cells relative to normal developmental trajectory.
Fig. S5. Relationship of knockout effects to gene expression changes occurring naturally with T-cell lineage developmental progression.
Fig. S6. Inferred TF activities and regulatory connections by SCENIC: histograms of individual representative regulons in the 8 CRISPR pool-perturbation genotypes.
Fig. S7. Transcription factor knockout impacts on cell biological parameters of developing pro-T cells.
Fig. S8. Surface marker and transcriptome profiles of WT and Bcl11b KO single-cell samples.
Fig. S9. Time-dependent gene expression deviations between Bcl11b KO and WT late pro-T cells.
Fig. S10. Fine-grained examination of the Bcl11b KO trajectory.
Fig. S11. Gene network analysis of Phase 1 specification and the Bcl11b knockout substructure.
Fig. S12. Technical and analytical details of dual gRNA perturb-seq.
Table S1. In vivo vs. in vitro T cell development comparison.
Table S2. Genes enriched in each sub-cluster of early pro-T pool-perturbation scRNA-seq.
Table S3. Differential Expression analysis comparing Control (Cont) and seven TF KOs in early pro-T cell pool-perturbation scRNA-seq.
Table S4. SCENIC analysis of pool-perturbation results in Phase 1 cells.
Table S5. Characterization of gene expression subclusters in WT and Bcl11b KO samples.
Table S6. Differential gene expression analysis between D10 and D13 timepoints in WT and Bcl11b KO.
Table S7. Differentially expressed genes between specific Clusters in WT and Bcl11b KO scRNA-seq analysis.
Table S8. SCENIC analysis of Bcl11b KO effects.
Table S9. Gene lists and statistics for gene network predictions based on the regulon-DEG intersection genes.
Table S10. Designed gRNA list (A) and pool oligo list (B) used for generating pool-perturbation dual gRNA vectors.
Table S11. Differential Expression analysis in early pro-T cell pool-perturbation scRNA-seq using an alternative method to increase sensitivity to low levels of expression.
Table S12. Table of raw data
ACKNOWLEDGMENTS
We thank Jeff Park and Sisi Chen from the Caltech Single Cell Profiling and Engineering Center for providing support for processing 10X Chromium samples, Boyoung Shin for helpful discussions and additional data visualization, Rochelle Diamond and members of the Caltech Flow Cytometry and Cell Sorting facility for sorting, Ingrid Soto for mouse care, Maria Quiloan for mouse genotyping and supervision, and Igor Antoshechkin and Vijaya Kumar of the Caltech Jacobs Genomics Facility for bulk RNA sequencing. We also thank Gay Crooks and Amélie Montel-Hagen (UCLA) for sharing the mATO system with us.
FUNDING:
Support for this project came from USPHS grants (R01AI135200, R01HL119102, and R01HD100039) to E.V.R., The Beckman Institute at Caltech for support of all the Caltech facilities, the Biology and Biological Engineering Division Bowes Leadership Chair Fund, the Louis A. Garfinkle Memorial Laboratory Fund, and the Al Sherman Foundation. E.V.R. gratefully acknowledges past support from the Albert Billings Ruddock Professorship.
Footnotes
COMPETING INTERESTS:
WZ is employed by BillionToOne, Inc. and has been employed by 10X Genomics. EVR is a member of the Scientific Advisory Board for Century Therapeutics and has advised Kite Pharma and A2 Biotherapeutics.
DATA AND MATERIALS AVAILABILITY:
Custom-made software for analysis of single-cell transcriptome perturbations using gRNA and batch hashtags has been released, and is now available at https://github.com/gaofan83/single_cell_perturb_seq/releases/tag/v.1.0.0. All genomic sequencing data have been deposited in Gene Expression Omnibus under accession numbers GSE165835 and GSE183026. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and notes
- 1.Porritt HE, Gordon K, Petrie HT, Kinetics of steady-state differentiation and mapping of intrathymic-signaling environments by stem cell transplantation in nonirradiated mice. J. Exp. Med 198, 957–962 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothenberg EV, Moore JE, Yui MA, Launching the T-cell-lineage developmental programme. Nat. Rev. Immunol 8, 9–21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taghon TN, David ES, Zúñiga-Pflücker JC, Rothenberg EV, Delayed, asynchronous, and reversible T-lineage specification induced by Notch/Delta signaling. Genes Dev 19, 965–78 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yui MA, Feng N, Rothenberg EV, Fine-Scale Staging of T Cell Lineage Commitment in Adult Mouse Thymus. J. Immunol 185, 284–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yui MA, Rothenberg EV, Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol 14, 529–45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitt TM, Zúñiga-Pflücker JC, Induction of T cell development from hematopoietic progenitor cells by Delta-like-1 in vitro. Immunity 17, 749–756 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Peydro M, de Yebenes VG, Toribio ML, Sustained Notch1 signaling instructs the earliest human intrathymic precursors to adopt a gd T cell fate in fetal thymus organ culture. Blood 102, 2444–2451 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Radtke F, Wilson A, Mancini SJ, MacDonald HR, Notch regulation of lymphocyte development and function. Nat Immunol 5, 247–253 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Mohtashami M, Shah DK, Nakase H, Kianizad K, Petrie HT, Zúñiga-Pflücker JC, Direct comparison of Dll1- and Dll4-mediated Notch activation levels shows differential lymphomyeloid lineage commitment outcomes. J Immunol 185, 867–876 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Hosokawa H, Rothenberg EV, How transcription factors drive choice of the T cell fate. Nat. Rev. Immunol 21, 162–176 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou W, Yui MA, Williams BA, Yun J, Wold BJ, Cai L, Rothenberg EV, Single-Cell Analysis Reveals Regulatory Gene Expression Dynamics Leading to Lineage Commitment in Early T Cell Development. Cell Syst 9, 321–337.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le J, Park JE, Ha VL, Luong A, Branciamore S, Rodin AS, Gogoshin G, Li F, Loh Y-HE, Camacho V, Patel SB, Welner RS, Parekh C, Single-Cell RNA-Seq Mapping of Human Thymopoiesis Reveals Lineage Specification Trajectories and a Commitment Spectrum in T Cell Development. Immunity 52, 1105–1118.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavaert M, Liang KL, Vandamme N, Park JE, Roels J, Kowalczyk MS, Li B, Ashenberg O, Tabaka M, Dionne D, Tickle TL, Slyper M, Rozenblatt-Rosen O, Vandekerckhove B, Leclercq G, Regev A, Van Vlierberghe P, Guilliams M, Teichmann SA, Saeys Y, Taghon T, Integrated scRNA-Seq Identifies Human Postnatal Thymus Seeding Progenitors and Regulatory Dynamics of Differentiating Immature Thymocytes. Immunity 52, 1088–1104.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Montel-Hagen A, Sun V, Casero D, Tsai S, Zampieri A, Jackson N, Li S, Lopez S, Zhu Y, Chick B, He C, de Barros SC, Seet CS, Crooks GM, In Vitro Recapitulation of Murine Thymopoiesis from Single Hematopoietic Stem Cells. Cell Rep 33, 108320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olariu V, Yui MA, Krupinski P, Zhou W, Deichmann J, Andersson E, Rothenberg EV, Peterson C, Multi-scale Dynamical Modeling of T Cell Development from an Early Thymic Progenitor State to Lineage Commitment. Cell Rep 34, 108622 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, Smibert P, Satija R, Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, Adamson B, Norman TM, Lander ES, Weissman JS, Friedman N, Regev A, Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Replogle JM, Norman TM, Xu A, Hussmann JA, Chen J, Cogan JZ, Meer EJ, Terry JM, Riordan DP, Srinivas N, Fiddes IT, Arthur JG, Alvarado LJ, Pfeiffer KA, Mikkelsen TS, Weissman JS, Adamson B, Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol 38, 954–961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV, Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell 149, 467–482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mingueneau M, Kreslavsky T, Gray D, Heng T, Cruse R, Ericson J, Bendall S, Spitzer MH, Nolan GP, Kobayashi K, von Boehmer H, Mathis D, Benoist C, Immunological Genome Consortium, Best AJ, Knell J, Goldrath A, Joic V, Koller D, Shay T, Regev A, Cohen N, Brennan P, Brenner M, Kim F, Nageswara Rao T, Wagers A, Heng T, Ericson J, Rothamel K, Ortiz-Lopez A, Mathis D, Benoist C, Bezman NA, Sun JC, Min-Oo G, Kim CC, Lanier LL, Miller J, Brown B, Merad M, Gautier EL, Jakubzick C, Randolph GJ, Monach P, Blair DA, Dustin ML, Shinton SA, Hardy RR, Laidlaw D, Collins J, Gazit R, Rossi DJ, Malhotra N, Sylvia K, Kang J, Kreslavsky T, Fletcher A, Elpek K, Bellemare-Pelletier A, Malhotra D, Turley S, The transcriptional landscape of αβ T cell differentiation. Nat. Immunol 14, 619–632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothenberg EV, Hosokawa H, Ungerback J, Mechanisms of Action of Hematopoietic Transcription Factor PU.1 in Initiation of T-Cell Development. Front Immunol 10, 228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zohren F, Souroullas GP, Luo M, Gerdemann U, Imperato MR, Wilson NK, Göttgens B, Lukov GL, Goodell MA, The transcription factor Lyl-1 regulates lymphoid specification and the maintenance of early T lineage progenitors. Nat Immunol 13, 761–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cleveland SM, Smith S, Tripathi R, Mathias EM, Goodings C, Elliott N, Peng D, El-Rifai W, Yi D, Chen X, Li L, Mullighan C, Downing JR, Love P, Dave UP, Lmo2 induces hematopoietic stem cell-like features in T-cell progenitor cells prior to leukemia. Stem Cells 31, 882–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber BN, Chi AW-S, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, Bhandoola A, A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476, 63–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, Arnett KL, Blacklow SC, Aifantis I, Aster JC, Gounari F, T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc. Natl. Acad. Sci. U. S. A 108, 20060–20065 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hattori N, Kawamoto H, Fujimoto S, Kuno K, Katsura Y, Involvement of transcription factors TCF-1 and GATA-3 in the initiation of the earliest step of T cell development in the thymus. J. Exp. Med 184, 1137–1147 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hosoya T, Kuroha T, Moriguchi T, Cummings D, Maillard I, Lim K-C, Engel JD, GATA-3 is required for early T lineage progenitor development. J. Exp. Med 206, 2987–3000 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scripture-Adams DD, Damle SS, Li L, Elihu KJ, Qin S, Arias AM, Butler RR, Champhekar A, Zhang JA, Rothenberg EV, GATA-3 dose-dependent checkpoints in early T cell commitment. J Immunol 193, 3470–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, Hitomi J, Yamamoto T, Utsuyama M, Niwa O, Aizawa S, Kominami R, Bcl11b is required for differentiation and survival of αβ T lymphocytes. Nat. Immunol 4, 533–539 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Liu P, Li P, Burke S, Critical roles of Bcl11b in T-cell development and maintenance of T-cell identity: Bcl11b has essential functions in T cells. Immunol. Rev 238, 138–149 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Shibata K, Yamada H, Nakamura M, Hatano S, Katsuragi Y, Kominami R, Yoshikai Y, IFN-γ–Producing and IL-17–Producing γδ T Cells Differentiate at Distinct Developmental Stages in Murine Fetal Thymus. J. Immunol 192, 2210–2218 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Kueh HY, Yui MA, Ng KK, Pease SS, Zhang JA, Damle SS, Freedman G, Siu S, Bernstein ID, Elowitz MB, Rothenberg EV, Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol 17, 956–65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Leid M, Rothenberg EV, An Early T Cell Lineage Commitment Checkpoint Dependent on the Transcription Factor Bcl11b. Science 329, 89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longabaugh WJR, Zeng W, Zhang JA, Hosokawa H, Jansen CS, Li L, Romero-Wolf M, Liu P, Kueh HY, Mortazavi A, Rothenberg EV, Bcl11b and combinatorial resolution of cell fate in the T-cell gene regulatory network. Proc. Natl. Acad. Sci 114, 5800–5807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, Kominami R, Katsura Y, Kawamoto H, An Essential Developmental Checkpoint for Production of the T Cell Lineage. Science 329, 93–96 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Li P, Burke S, Wang J, Chen X, Ortiz M, Lee S-C, Lu D, Campos L, Goulding D, Ng BL, Dougan G, Huntly B, Gottgens B, Jenkins NA, Copeland NG, Colucci F, Liu P, Reprogramming of T Cells to Natural Killer-Like Cells upon Bcl11b Deletion. Science 329, 85–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hosokawa H, Romero-Wolf M, Yui MA, Ungerbäck J, Quiloan MLG, Matsumoto M, Nakayama KI, Tanaka T, Rothenberg EV, Bcl11b sets pro-T cell fate by site-specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat. Immunol 19, 1427–1440 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S, SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hosokawa H, Ungerbäck J, Wang X, Matsumoto M, Nakayama KI, Cohen SM, Tanaka T, Rothenberg EV, Transcription Factor PU.1 Represses and Activates Gene Expression in Early T Cells by Redirecting Partner Transcription Factor Binding. Immunity 49, 782 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Champhekar A, Damle SS, Freedman G, Carotta S, Nutt SL, Rothenberg EV, Regulation of early T-lineage gene expression and developmental progression by the progenitor cell transcription factor PU.1. Genes Dev 29, 832–848 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S, Liu P, Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med 209, 2467–2483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, Tellier J, Kim E, Dwyer D, Shinton S, Nabekura T, Qi Y, Yu B, Robinette M, Kim KW, Wagers A, Rhoads A, Nutt SL, Brown BD, Mostafavi S, Buenrostro JD, Benoist C, Immunological Genome P, The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912 e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ungerbäck J, Hosokawa H, Wang X, Strid T, Williams BA, Sigvardsson M, Rothenberg EV, Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Res 28, 1508–1519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schilham MW, Wilson A, Moerer P, Benaissa-Trouw BJ, Cumano A, Clevers HC, Critical involvement of Tcf-1 in expansion of thymocytes. J Immunol 161, 3984–3991 (1998). [PubMed] [Google Scholar]
- 45.Avram D, Califano D, The multifaceted roles of Bcl11b in thymic and peripheral T cells: impact on immune diseases. J Immunol 193, 2059–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kojo S, Tanaka H, Endo TA, Muroi S, Liu Y, Seo W, Tenno M, Kakugawa K, Naoe Y, Nair K, Moro K, Katsuragi Y, Kanai A, Inaba T, Egawa T, Venkatesh B, Minoda A, Kominami R, Taniuchi I, Priming of lineage-specifying genes by Bcl11b is required for lineage choice in post-selection thymocytes. Nat Commun 8, 702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Del Real MM, Rothenberg EV, Architecture of a lymphomyeloid developmental switch controlled by PU.1, Notch and Gata3. Development 140, 1207–19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yashiro-Ohtani Y, He Y, Ohtani T, Jones ME, Shestova O, Xu L, Fang TC, Chiang MY, Intlekofer AM, Blacklow SC, Zhuang Y, Pear WS, Pre-TCR signaling inactivates Notch1 transcription by antagonizing E2A. Genes Dev 23, 1665–76 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cismasiu VB, Adamo K, Gecewicz J, Duque J, Lin Q, Avram D, BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 24, 6753–6764 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Naito T, Tanaka H, Naoe Y, Taniuchi I, Transcriptional control of T-cell development. Int. Immunol 23, 661–668 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Thompson PK, Zúñiga-Pflücker JC, On becoming a T cell, a convergence of factors kick it up a Notch along the way. Semin. Immunol 23, 350–359 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Maslova A, Ramirez RN, Ma K, Schmutz H, Wang C, Fox C, Ng B, Benoist C, Mostafavi S, Immunological Genome Project, Deep learning of immune cell differentiation. Proc. Natl. Acad. Sci. U. S. A 117, 25655–25666 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.García-Ojeda ME, Klein Wolterink RGJ, Lemaître F, Richard-Le Goff O, Hasan M, Hendriks RW, Cumano A, Di Santo JP, GATA-3 promotes T-cell specification by repressing B-cell potential in pro-T cells in mice. Blood 121, 1749–1759 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Zeng Y, Liu C, Gong Y, Bai Z, Hou S, He J, Bian Z, Li Z, Ni Y, Yan J, Huang T, Shi H, Ma C, Chen X, Wang J, Bian L, Lan Y, Liu B, Hu H, Single-cell RNA sequencing resolves spatiotemporal development of pre-thymic lymphoid progenitors and thymus organogenesis in human embryos. Immunity 51, 930–948.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Anderson MK, Hernandez-Hoyos G, Diamond RA, Rothenberg EV, Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development 126, 3131–3148 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Loughran SJ, Kruse EA, Hacking DF, de Graaf CA, Hyland CD, Willson TA, Henley KJ, Ellis S, Voss AK, Metcalf D, Hilton DJ, Alexander WS, Kile BT, The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat.Immunol 9, 810–819 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Knudsen KJ, Rehn M, Hasemann MS, Rapin N, Bagger FO, Ohlsson E, Willer A, Frank AK, Sondergaard E, Jendholm J, Thoren L, Lee J, Rak J, Theilgaard-Monch K, Porse BT, ERG promotes the maintenance of hematopoietic stem cells by restricting their differentiation. Genes Dev 29, 1915–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thoms JAI, Birger Y, Foster S, Knezevic K, Kirschenbaum Y, Chandrakanthan V, Jonquieres G, Spensberger D, Wong JW, Oram SH, Kinston SJ, Groner Y, Lock R, MacKenzie KL, Göttgens B, Izraeli S, Pimanda JE, ERG promotes T-acute lymphoblastic leukemia and is transcriptionally regulated in leukemic cells by a stem cell enhancer. Blood 117, 7079–89 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Aziz A, Soucie E, Sarrazin S, Sieweke MH, MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science 326, 867–871 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Takahashi K, Yamanaka S, A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 17, 183–93 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Ferreira ACF, Szeto ACH, Heycock MWD, Clark PA, Walker JA, Crisp A, Barlow JL, Kitching S, Lim A, Gogoi M, Berks R, Daly M, Jolin HE, McKenzie ANJ, RORα is a critical checkpoint for T cell and ILC2 commitment in the embryonic thymus. Nat. Immunol 22, 166–178 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Obaldia ME, Bhandoola A, Transcriptional regulation of innate and adaptive lymphocyte lineages. Annu Rev Immunol 33, 607–42 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Vidigal JA, Ventura A, Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun 6, 8083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Longabaugh WJR, Davidson EH, Bolouri H, Computational representation of developmental genetic regulatory networks. Dev.Biol 283, 1–16 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Franco CB, Scripture-Adams DD, Proekt I, Taghon T, Weiss AH, Yui MA, Adams SL, Diamond RA, Rothenberg EV, Notch/Delta signaling constrains reengineering of pro-T cells by PU.1. Proc Natl Acad Sci U A 103, 11993–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taghon T, Yui MA, Rothenberg EV, Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat Immunol 8, 845–55 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE, Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, Doench JG, Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun 9, 5416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meier JA, Zhang F, Sanjana NE, GUIDES: sgRNA design for loss-of-function screens. Nat. Methods 14, 831–832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P, Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 5, e12776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material
Supplemental Text: Supplemental Materials and Methods
Fig. S1. Bulk RNA-seq analysis of in vivo and ex-vivo derived early pro-T cells.
Fig. S2. Gene expression profiles of BM-derived early pro-T cells in vitro recapitulate those of in vivo thymic early pro-T cells, on single-cell level.
Fig. S3. Alignment of Cas9;Bcl2-transgenic control cells developing in vitro with wildtype non-transgenic B6 thymic pro-T cells developing in vivo.
Fig. S4. Knockout effects on cluster distributions of cells relative to normal developmental trajectory.
Fig. S5. Relationship of knockout effects to gene expression changes occurring naturally with T-cell lineage developmental progression.
Fig. S6. Inferred TF activities and regulatory connections by SCENIC: histograms of individual representative regulons in the 8 CRISPR pool-perturbation genotypes.
Fig. S7. Transcription factor knockout impacts on cell biological parameters of developing pro-T cells.
Fig. S8. Surface marker and transcriptome profiles of WT and Bcl11b KO single-cell samples.
Fig. S9. Time-dependent gene expression deviations between Bcl11b KO and WT late pro-T cells.
Fig. S10. Fine-grained examination of the Bcl11b KO trajectory.
Fig. S11. Gene network analysis of Phase 1 specification and the Bcl11b knockout substructure.
Fig. S12. Technical and analytical details of dual gRNA perturb-seq.
Table S1. In vivo vs. in vitro T cell development comparison.
Table S2. Genes enriched in each sub-cluster of early pro-T pool-perturbation scRNA-seq.
Table S3. Differential Expression analysis comparing Control (Cont) and seven TF KOs in early pro-T cell pool-perturbation scRNA-seq.
Table S4. SCENIC analysis of pool-perturbation results in Phase 1 cells.
Table S5. Characterization of gene expression subclusters in WT and Bcl11b KO samples.
Table S6. Differential gene expression analysis between D10 and D13 timepoints in WT and Bcl11b KO.
Table S7. Differentially expressed genes between specific Clusters in WT and Bcl11b KO scRNA-seq analysis.
Table S8. SCENIC analysis of Bcl11b KO effects.
Table S9. Gene lists and statistics for gene network predictions based on the regulon-DEG intersection genes.
Table S10. Designed gRNA list (A) and pool oligo list (B) used for generating pool-perturbation dual gRNA vectors.
Table S11. Differential Expression analysis in early pro-T cell pool-perturbation scRNA-seq using an alternative method to increase sensitivity to low levels of expression.
Table S12. Table of raw data