

PURPOSE
The National Cancer Institute–Children's Oncology Group Pediatric MATCH trial aimed to facilitate evaluation of molecular-targeted therapies in biomarker-selected cohorts of childhood and young adult patients with cancer by screening tumors for actionable alterations.
PATIENTS AND METHODS
Tumors from patients age 1-21 years with refractory solid tumors, lymphomas, or histiocytic disorders were subjected to cancer gene panel sequencing and limited immunohistochemistry to identify actionable alterations for assignment to phase II treatment arms. The rates of treatment arm assignment and enrollment were compared between clinical and demographic groups.
RESULTS
Testing was completed for 94.7% of tumors submitted. Actionable alterations were detected in 31.5% of the first 1,000 tumors screened, with treatment arm assignment and enrollment occurring in 28.4% and 13.1% of patients, respectively. Assignment rates varied by tumor histology and were higher for patients with CNS tumors or enrolled at Pediatric Early Phase Clinical Trials Network sites. A reported history of prior clinical molecular testing was associated with higher assignment and enrollment rates. Actionable alterations in the mitogen-activated protein kinase signaling pathway were most frequent (11.2%). The most common reasons provided for not enrolling on treatment arms were patients receiving other treatment or poor clinical status.
CONCLUSION
The Pediatric MATCH trial has proven the feasibility of a nationwide screening Protocol for identification of actionable genetic alterations and assignment of pediatric and young adult patients with refractory cancers to trials of molecularly targeted therapies. These data support the early use of tumor molecular screening for childhood patients with cancer whose tumors have not responded to standard treatments.
INTRODUCTION
Although outcomes for children diagnosed with cancer have improved, effective treatment options for most recurrent cancers are limited and novel approaches are needed. Significant research effort has focused on interrogation of the genomic landscapes of pediatric cancers, providing insight into tumor biology and revealing potentially targetable genetic alterations in key signaling pathways.1,2 Clinical trials have demonstrated the benefit of such therapies for selected pediatric patients whose cancers harbor relevant (actionable) genetic alterations.3-5
CONTEXT
Key Objective
Cancer therapies targeting genetic alterations in key signaling pathways offer benefit for selected patients, but the utility of a molecular screening approach for children with refractory cancers is unknown. National Cancer Institute–Children's Oncology Group Pediatric MATCH created a nationwide Protocol to facilitate enrollment in phase II trials of molecularly targeted therapies using next-generation sequencing to identify tumors harboring actionable genetic alterations. This analysis examined the frequency and spectrum of actionable tumor alterations in 1,000 patients and the resulting rates of treatment arm assignment and enrollment.
Knowledge Generated
Actionable genetic alterations in diverse genes were identified in 31% of tumors sequenced. Assignments to treatment arms were made for 28% of patients screened and 13% of patients enrolled in those studies.
Relevance
Next-generation tumor sequencing was effective at identifying actionable genetic alterations for investigational targeted therapies in pediatric and young adult patients with refractory cancers, supporting the use of molecular screening in this population.
These alterations each only occur in a fraction of pediatric tumors, however, and often in multiple disease histologies,1,2 complicating efforts to evaluate novel molecularly targeted agents. Access to investigational molecularly targeted therapies in children has also presented a limitation to the performance of biomarker-selected trials.6 Moreover, data are limited regarding the frequency of actionable tumor alterations in recurrent pediatric cancers, and the utility of a molecular screening approach for pediatric patients is not known. The National Cancer Institute–Children's Oncology Group Pediatric Molecular Analysis for Therapy Choice (NCI-COG Pediatric MATCH) trial was therefore developed to provide a national framework for trials of investigational molecularly targeted therapies in biomarker-selected populations.7
The NCI-COG Pediatric MATCH screening Protocol (online only) allows enrollment of patients from COG institutions across the United States. Centralized tumor sequencing was performed, and patients with actionable tumor alterations were then eligible for phase II treatment arms of targeted therapies. We describe here the frequency and spectrum of alterations detected in the first 1,000 patients screened and the experience with treatment arm assignment and enrollment for children and young adults with advanced cancers enrolled in this national trial.
PATIENTS AND METHODS
Patient Selection and Clinical Data Collection
Patients age 1-21 years with treatment-refractory or recurrent solid tumors, non-Hodgkin lymphomas, or histiocytic disorders treated at US-based COG sites were eligible for the NCI-COG Pediatric MATCH screening trial (NCT03155620). Patients enrolled in the screening Protocol were required to have a tumor sample available for study testing (see the Biopsy Specimens and Tumor Testing section) or be planned to have a clinically indicated procedure to obtain such a sample. The trial was approved by the NCI Central Institutional Review Board, which serves as the Institutional Review Board of record for COG institutions. A written informed consent document was signed by all patients, and assent was obtained as appropriate. Patient demographic and clinical data were reported by study sites.
Selection of Study Drugs
Drugs studied in Pediatric MATCH (Fig 1 and Data Supplement, online only) were used as single agents. A Target and Agent Prioritization study committee (Data Supplement) longitudinally reviewed potential classes of agents and individual drugs and recommended agents for inclusion in the trial.7 The study included 13 treatment arms with agents inhibiting a wide range of molecular targets (Fig 1); seven of these were activated with the screening Protocol, and six at later time points. Multiple arms with agents targeting mitogen-activated protein kinase (MAPK) signaling were included, collectively referred to as MAPK pathway arms: Arm E, selumetinib; Arm G, vemurafenib; Arm J, ulixertinib; and Arm M, tipifarnib.
FIG 1.
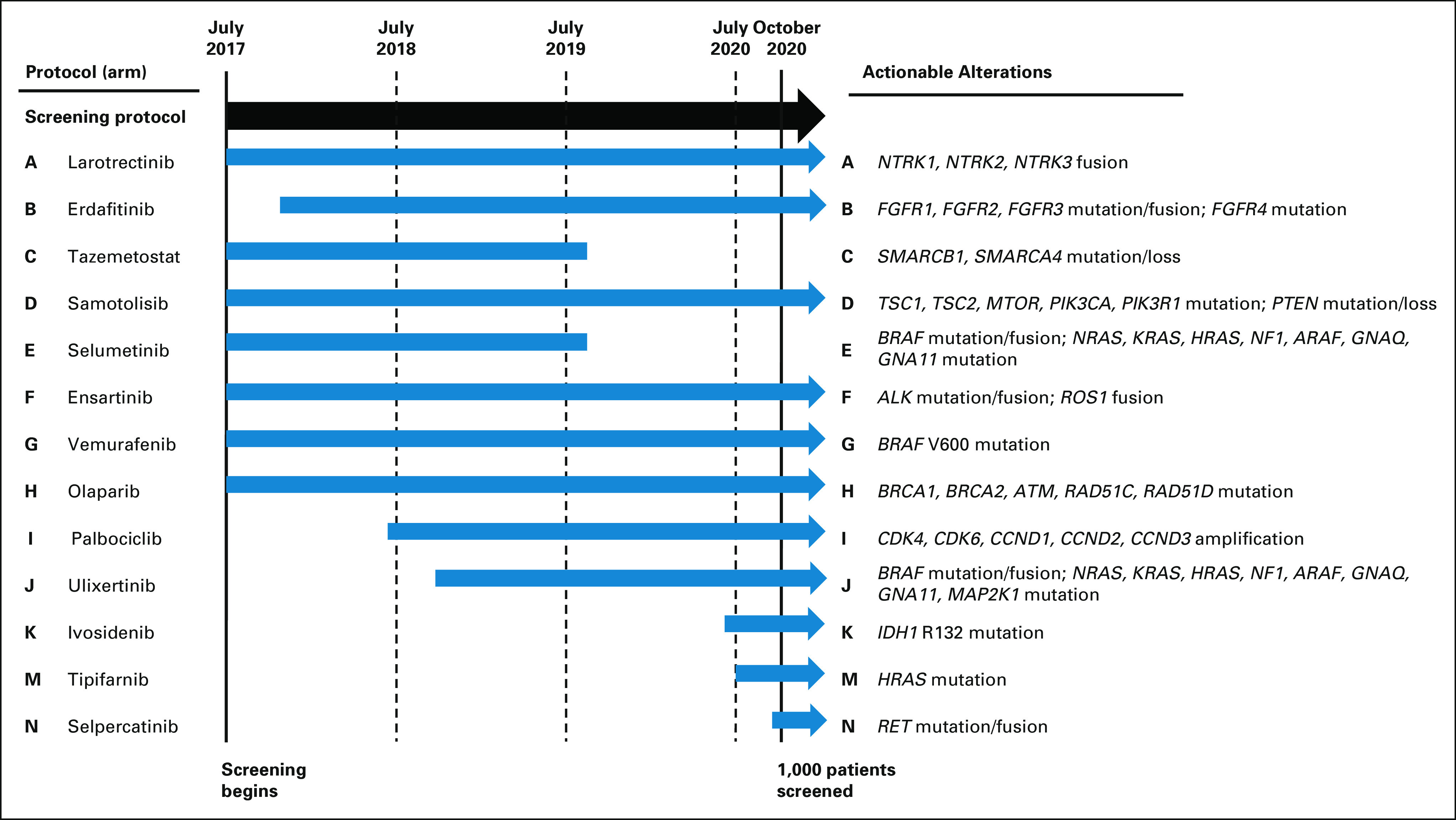
Pediatric MATCH trial arms and actionable cancer genes. The screening protocol and the seven treatment arms (A, C, D, E, F, G, and H) were activated in July 2017. Six additional treatment arms (B, I, J, K, M, and N) had been activated by the time the 1,000th patient screened was enrolled in October 2020. Arms C and E were closed after completion of study accrual.
Definition of Targetable Alterations
Lists of actionable molecular alterations were generated for each treatment arm on the basis of predefined levels of clinical and preclinical evidence (Fig 1 and Data Supplement). Actionable alteration lists for treatment arms were nonoverlapping except for the MAPK pathway arms. For patients with tumors harboring actionable alterations for more than one treatment arm, the arm with the highest level of evidence linking the alteration to the agent was prioritized. All treatment arms were histology-agnostic except Arms E (selumetinib) and G (vemurafenib) for which patients with low-grade glioma were ineligible because previous clinical trials of those agents had already established activity.8,9
Biopsy Specimens and Tumor Testing
Formalin-fixed paraffin-embedded (FFPE) tumors submitted for Pediatric MATCH screening were required to have been obtained at any point after initial tumor recurrence or progression. A tumor sample from a diagnostic (pretreatment) procedure was only acceptable for use in screening for patients with high-grade gliomas of the brainstem or thalamus. The study Protocol advised that tumor samples that had been subjected to acid-based decalcification were generally not suitable for testing. All specimens were processed in the COG Biopathology Core laboratory at Nationwide Children's Hospital. Isolated DNA and RNA for next-generation sequencing and slides for immunohistochemistry (IHC) were then sent to Pediatric MATCH laboratories (Data Supplement) for testing.
Next-Generation Sequencing and IHC Assays
DNA and RNA extracted from the FFPE tumor samples were sequenced using an Oncomine AmpliSeq cancer gene panel (Thermo Fisher Scientific, Waltham, MA) designed for detection of single-nucleotide variants (SNVs), insertions and deletions, amplifications, and selected fusions in 159 cancer genes (Data Supplement).10 A variant allele fraction of 0.05 was required for reporting of SNVs and 0.10 for indels, with amplification defined as ≥ 7 gene copies. Copy number losses and consensus splice site mutations were not analyzed. Loss of protein expression for selected tumor suppressor genes (SMARCB1, SMARCA4, and PTEN) was evaluated by IHC; reflex testing for RB1 expression was performed only in tumors with an actionable alteration for Arm I (palbociclib). A software application (MATCHBOX) to facilitate processing and analysis of sequence data, treatment arm assignment, and patient tracking on the basis of that used for the adult NCI-MATCH trial was developed for the current study.10,11
Study Definitions and Statistical Analysis
The study planned to screen 200-300 patients per year (17-25 per month), of which 10% were expected to have an actionable alteration of interest for which a treatment Protocol was available. For the match rate feasibility analysis, the match rate along with the 95% score CI was assessed among patients with confirmed variant reports. The actionable alteration rate was defined as the fraction of tumors that had an actionable alteration detected for any study arm (regardless of slot availability). The treatment arm assignment (match) rate and enrollment rate were defined as the fraction of patients screened who were assigned to a treatment Protocol and the fraction of assigned patients who enrolled in a treatment arm, respectively. The 95% score type of CI was reported. Chi-square tests (or Fisher's exact tests as appropriate) were used to compare treatment arm matching and enrollment rates between groups. Statistical significance was set at P-values < .05. Data were analyzed using SAS version 9.4 and R version 4.0.2. Data as of December 31, 2020 were used in this article.
RESULTS
Patient Characteristics
The Pediatric MATCH screening Protocol was activated in July 2017. Tumor profiling was completed by December 2020 for 1,000 patients enrolled at 138 COG study sites in the United States between July 2017 and October 2020 (Appendix Fig A1, online only). The median screening Protocol enrollment rate was 28 patients per month over this period (range, 2-52; Appendix Fig A2, online only). Patient characteristics are summarized in Table 1. Patients ranged in age from 1 to 21 years (median 13 years) with a slight predominance of males (55.6%). Adolescent and young adult patients (age 15-21 years) comprised 40.6% of the cohort. Most patients were White (69.0%) and of non-Hispanic or Latino ethnicity (72.7%). The median performance status score (Lansky or Karnofsky) was 90, with a score < 80 reported for 15.1% of patients. Fewer than half of the screened patients (45.2%) were enrolled at one of the 37 COG study sites in the United States that are members of the Pediatric Early Phase Clinical Trials Network (PEP-CTN).12
TABLE 1.
NCI-COG Pediatric MATCH Screening Trial Patient Characteristics

Solid tumors occurring outside the CNS comprised 72.3% of patient diagnoses, with CNS tumors in 24.6% and lymphomas/histiocytoses in 3.1% (Tables 1 and 2). Osteosarcoma (18.4%), rhabdomyosarcoma (12.0%), and Ewing sarcoma (10.4%) were most common; sarcomas as a group comprised 50.5% of the cohort. High-grade glioma (6.9%), neuroblastoma (6.0%), Wilms tumor (5.6%), and low-grade glioma (5.4%) were the only other tumor types observed in more than 5% of patients (Table 2). The remaining 353 patients (35.3%) had less frequent tumor types. In total, 80 different diagnoses were included in the study cohort (Data Supplement), with 61 of these found in fewer than 1% of patients.
TABLE 2.
Diagnoses and Matching Results of NCI-COG Pediatric MATCH Screening Trial Patients

Feasibility of Molecular Profiling
Tumors were submitted for 1,056 of 1,111 (95.0%) patients enrolled between July 2017 and October 2020 (Fig 2). Tumor screening was completed for 1,000 of these 1,056 patients (94.7%): the cohort being analyzed in this report. The RNA panel testing component of the assay was unsuccessful in 11.9% of these patients. The median and 75th percentile times from the receipt of the tumor sample to completion of tumor testing were 12 days and 16 days, respectively; the median time from screening Protocol enrollment to completion of tumor testing was 24 days (range 9-570 days). Grade 3 or higher biopsy-related adverse events were reported in 2 of 256 (0.8%) patients who had a clinically indicated procedure performed after study enrollment: one patient with headache (grade 3, probable attribution) and the other with surgical site infection and meningitis (grade 3, probable).
FIG 2.

Patient flow diagram for Pediatric MATCH trial. Reasons provided for matched patients not enrolling in treatment protocols are described further in the Data Supplement.
Treatment Protocol Assignment and Enrollment
At least one actionable tumor alteration for a study treatment arm was identified in 315 of 1,000 (31.5%) patients screened. Open treatment arm slots were not available for 31 patients with actionable tumor mutations; therefore, treatment arm assignments were made for 284 (28.4%) patients (95% CI, 25.7 to 31.3; Table 2 and Fig 2). A total of 131 patients, 13.1% of those screened (95% CI, 11.1%-15.3%), were enrolled in a treatment arm, yielding an enrollment rate of 46.1% among those patients for whom a treatment assignment was made. The most common reasons provided for not enrolling in a treatment arm (Data Supplement) were patient receiving other treatment (32% of responses), poor clinical status (15%), lack of measurable disease (11%), or ineligible diagnosis (10%).
Factors Related to Treatment Assignment and Enrollment
No significant differences in the treatment arm match rate or enrollment rate were observed for patient sex, age, race, or ethnicity (Data Supplement). A reported history of prior molecular testing (38.2% v 21.8%, P < .001) and enrollment at a PEP-CTN site (33.2% v 24.5%, P = .002) were associated with significantly higher match rates. Higher enrollment rates were observed for patients with a reported history of prior molecular testing (53.3% v 38.4%, P = .039).
Patients with CNS tumors were nearly twice as likely as those with non-CNS solid tumors to be matched to treatment Protocols (44.3% v 22.8%, P < .001; Data Supplement); however, no difference in enrollment rate was observed (P = .253). Marked variability in match rates was observed across individual tumor types (Table 2). Of the common non-CNS solid tumors, match rates were highest in rhabdomyosarcoma (50 of 120, 41.7%) and neuroblastoma (25 of 60, 41.7%). Matches were much less frequent in other sarcomas, including osteosarcoma (28 of 184, 15.2%) and Ewing sarcoma (10 of 105, 9.6%). Treatment arm match rates for common CNS tumors were highest in low-grade glioma (37 of 54, 68.5%) and high-grade glioma (45 of 69, 65.2%).
Genomic Tumor Landscape
A total of 343 actionable alterations were detected in 315 tumors (Fig 3A and Data Supplement). DNA panel testing identified actionable mutations (SNVs or indels) in 206 tumors (20.6%) and amplifications in 48 tumors (4.8%), RNA testing revealed actionable fusions in 35 tumors (3.5%), and tumor IHC detected actionable tumor suppressor gene loss in 45 tumors (4.5%). Mutations or fusions in BRAF were most frequent (4.2%), followed by SMARCB1 loss/mutation (3.9%), CDK4 amplification (3.1%), FGFR1 mutation/fusion (2.9%), and NF1 mutation (2.4%; Fig 3B and Data Supplement). Actionable alterations in 11 genes were detected in more than 1% of patients.
FIG 3.
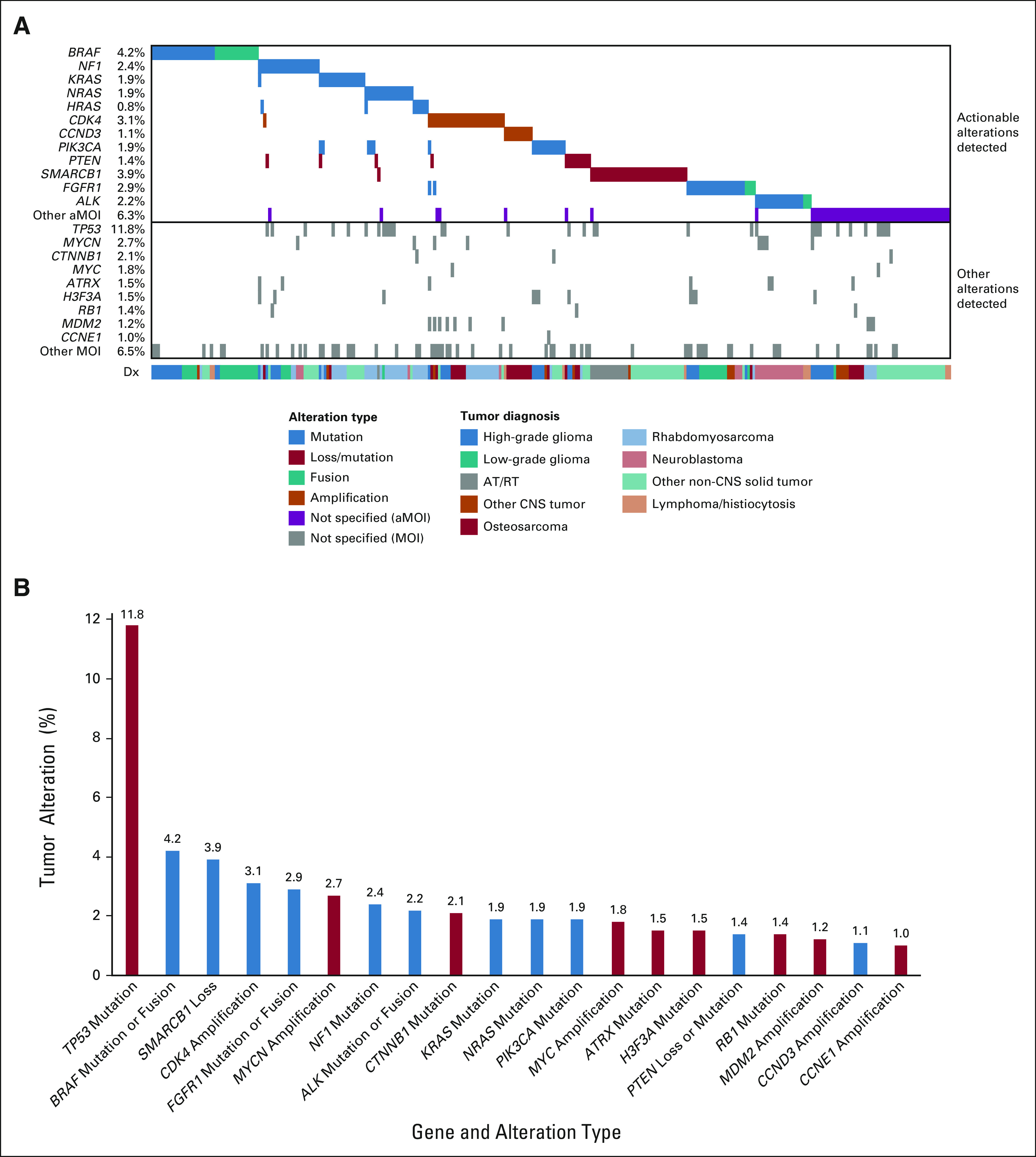
Genomic landscape of tumors with actionable alterations in the Pediatric MATCH trial (n = 315). A genomic waterfall plot (A) displaying the genes with most frequent actionable alterations at the top of the figure, categorized by variant type (mutation, loss/mutation, fusion, and amplification). Each tumor is represented by a column. The variant types for the less common actionable genes (shaded in purple) are not specified. Concurrent alterations in nonactionable genes occurring at a frequency of 1% or greater in the full study cohort are presented at the bottom of the figure. The lower bar categorizes the tumors on the basis of Dx. For the purposes of this figure, negative expression of SMARCB1, SMARCA4, or PTEN by IHC is assigned the loss/mutation variant type. (B) Twenty genes with tumor alterations detected in at least 1% of Pediatric MATCH patients screened. Actionable genes for Pediatric MATCH study arms are marked in blue. Genes with nonactionable alterations detected in patient tumors are marked in red. aMOI, actionable mutation; AT/RT, tumor histology of atypical teratoid rhaboid tumor; Dx, diagnosis; IHC, immunohistochemistry.
The frequency of actionable alterations by treatment arm for the diagnoses occurring in more than 5% of patients is presented in Figure A3, online only and the Data Supplement. Alterations for MAPK pathway study arms (11.2%), Arm I (palbociclib, 4.8%), Arm C (tazemetostat, 4.6%), and Arm D (samotolisib, 4.2%) were most frequent. Actionable alterations for Arm B (erdafitinib) were common in CNS tumors (9.3%). Only 21 tumors (2.1%) harbored actionable alterations for more than one treatment arm (Data Supplement). Of these, eight were tumors with actionable alterations in both the MAPK and phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/MTOR) pathways. Since MAPK mutations are known to confer resistance to PI3K inhibitors, these patients were not eligible for assignment to the PI3K/MTOR inhibitor samotolisib (Arm D).
Actionable mutations for MAPK pathway arms were detected in 25 tumor types (Fig 4A). Rhabdomyosarcoma (n = 32), low-grade glioma (n = 26), and high-grade glioma (n = 23) collectively comprised 72.3% of MAPK-altered tumors; the remaining 22 tumor types had four or fewer mutated tumors. The specific pattern of MAPK pathway alterations varied markedly by tumor type (Figs 4B-4G and Data Supplement); for example, RAS gene mutations predominated in rhabdomyosarcoma (Fig 4E), whereas BRAF V600E and NF1 mutations were most frequent in high-grade glioma (Fig 4F), and BRAF fusions accounted for more than half of the alterations in low-grade glioma (Fig 4G).
FIG 4.
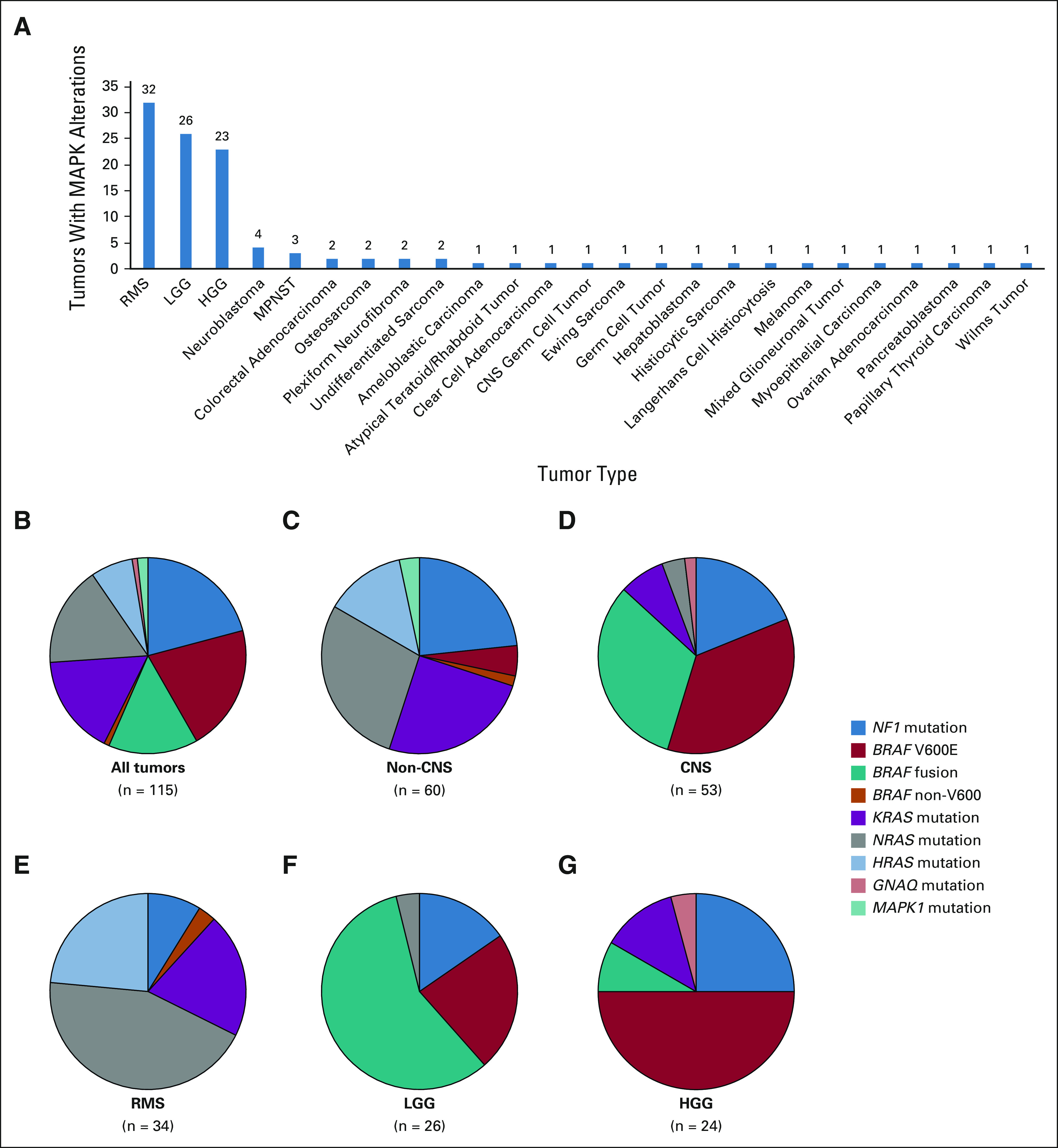
Actionable MAPK pathway alterations by tumor type. A total of 115 alterations were detected in 112 tumors. (A) Tumor diagnoses with actionable MAPK pathway alterations identified. (B-G) Specific MAPK alterations detected in selected diagnostic groups and tumor types. The number of alterations in each group is shown. (B) All tumors, (C) non-CNS solid tumors, (D) CNS solid tumors, (E) RMS, (F) LGG, and (G) HGG. HGG, high-grade glioma; LGG, low-glade glioma; MAPK, mitogen-activated protein kinase; MPNST, malignant peripheral nerve sheath tumor; RMS, rhabdomyosarcoma.
Cancer gene alterations that were not actionable for Pediatric MATCH treatment arms were detected in 347 tumors (Fig 3B and Data Supplement). Tumors with more than one nonactionable mutation were uncommon (9.8%). The nine genes with alterations occurring at a frequency of 1% or greater are shown in Figure 3B. Mutation of TP53 was by far the most common (11.8%), followed by MYCN amplification (2.7%), CTNNB mutation (2.1%), and MYC amplification (1.8%).
DISCUSSION
The NCI-COG Pediatric MATCH trial has demonstrated the feasibility of a nationwide screening and centralized testing Protocol to select patients with tumors harboring actionable alterations for enrollment in clinical trials of molecularly targeted therapies. Study accrual exceeded projections, and tumor profiling was completed for the majority of patients, using predominantly archived FFPE samples. Although tumor biopsy was not part of the Pediatric MATCH Protocol, collection of adverse event data after study enrollment revealed few procedure-related adverse events. Most importantly, the screening Protocol has successfully matched patients to investigational therapies, with 28% of patients assigned to treatment arms and 13% enrolled in those trials.
The trial has provided access to clinical molecular screening and early-phase trials of molecularly targeted therapies for pediatric and young adult patients with cancer across the United States. The study cohort was geographically diverse (Appendix Fig A1), with patients screened and enrolled at 138 and 61 COG institutions, respectively, and representative of the race and ethnicity of the pediatric cancer population in the United States (Data Supplement). Nearly half of patients were enrolled in treatment arms at sites that are not members of the PEP-CTN (Table 1). Despite the known challenges of adolescent and young adult participation in clinical trials,13,14 patients age 15-21 years comprised more than 40% of patients screened. These data suggest that the study was broadly accessible to patients including those at COG institutions with less extensive access to investigational therapies.
Multiple factors might have affected the diagnoses and demographics of patients enrolled in the study, including technical challenges of sequencing FFPE tumor samples from bone biopsies, an inability to take pills for younger patients, and differential availability of competing trials for different tumor types. Despite this, Pediatric MATCH has provided a useful snapshot of the spectrum of refractory solid tumor histologies (and their key molecular features) in pediatric and young adult patients, highlighting opportunities for further research. Although the tumor diagnoses were diverse (n=80), more than half of patients had sarcomas (Data Supplement). Except for rhabdomyosarcoma, a relatively low frequency of actionable alterations (16.4%) was observed in sarcomas (Table 2), demonstrating the need for alternative therapeutic strategies for these patients. Across the full cohort, the most frequently detected actionable genes (BRAF, SMARCB1, and CDK4) and pathways/functions (MAPK, CDK4/6, and MTOR/PI3K signaling) were consistent with previous genomic studies of pediatric cancers (Fig 3, Appendix Fig A3, and Data Supplement).1,2,15-19 Alterations in MAPK signaling genes (Fig 4) were by far the most common. Continuous drug development and incorporation of pediatric patients into innovative clinical trials targeting this pathway, including combination therapies, remains a high priority.
Comparison of data from Pediatric MATCH data and the adult NCI-MATCH trial, which used the same testing platform and a similar study design (albeit different treatment arms),11 allows several observations regarding the feasibility of precision oncology strategies for childhood patients with cancer. First, analysis of archived FFPE tumor samples in Pediatric MATCH resulted in a similarly high profiling success rate (> 94%) as seen in the adult NCI-MATCH trial, which primarily used FFPE tumor samples that were collected from mandatory tumor biopsy procedures to obtain fresh tumor.11 Second, despite the largely distinct cancer diagnoses in the two trials, the rates of actionable genetic alterations (31.5% pediatric v 37.6% adult) and treatment arm enrollment (13.1% pediatric v 12.4% adult) were also similar. Third, co-occurring resistance mutations that excluded patients from treatment arms were markedly less common in Pediatric MATCH as compared with the adult trial (2.1% v 34.7%).
Patients with a reported history of prior clinical molecular testing were significantly more likely to both match to and enroll in treatment arms, suggesting preferential enrollment of patients with tumors known to harbor actionable alterations. Most reasons provided for matched patients not enrolling on treatment arms (Data Supplement) could be considered issues of timing, with patients screened too late to receive treatment in the study (ie, after initiating alternative treatment for their refractory tumor or when poor clinical status precluded trial enrollment). Given the frequency of actionable alterations detected by Pediatric MATCH testing across most tumor types (Table 2 and Appendix Fig A3) and the increasing potential of those results to guide selection of both US Food and Drug Administration–approved and investigational therapies, early molecular testing for children and young adults with refractory cancers seems a reasonable strategy to inform treatment options in these patients.
Our trial design had several limitations that suggest opportunities to increase the rate of actionable alteration detection and trial enrollment. First, a more comprehensive tumor testing strategy could be used. Alternative approaches would be necessary to more comprehensively identify inactivating events (deletions and splice site mutations) in tumor suppressor genes (eg, SMARCB1, NF1, PTEN, TSC1, TSC2, BRCA1, and BRCA2) and fusion events between oncogenes (eg, BRAF and NTRK genes) and novel or uncommon partners. More comprehensive analysis of study tumors (including exome, whole genome, and transcriptome sequencing) will provide further insight into the additional yield of those approaches for identification of molecular targets.20-22 Second, determination of alteration actionability could be made in real time following trial rules (Data Supplement) instead of using static variant lists. This would also facilitate the use of molecular profiling results from outside laboratories, increasing trial access for patients without available tumor for testing: an approach that will be implemented in Pediatric MATCH beginning in early 2022.
In a minority of cases, alterations were classified as nonactionable in Pediatric MATCH that could be considered actionable for agents not included in the trial (Data Supplement), including biomarkers for US Food and Drug Administration–approved therapies (eg, PTCH1 or SMO mutations for vismodegib) and molecular targets of investigational agents (eg, PDGFRA and KIT amplifications).23,24 Finally, a review of the most frequently identified nonactionable alterations detected (Figs 3B and Data Supplement) serves as a stark reminder that many of the most frequent genomic events in pediatric solid tumors (TP53 and CTNNB1 mutations, MYC and MYCN amplifications, and EWSR-FLI1 fusions) remain therapeutic challenges. Novel strategies for targeting these genes and related pathways will be necessary to dramatically increase the potential clinical benefit of precision oncology strategies for childhood patients with cancer.
APPENDIX
FIG A1.
Geographic distribution of COG sites at which Pediatric MATCH patients were enrolled. COG, Children's Oncology Group.
FIG A2.

Projected and actual Pediatric MATCH screening protocol accrual.
FIG A3.

Actionable alteration rates for Pediatric MATCH study arms by tumor type. Study arms: A (larotrectinib), B (erdafitinib), C (tazemetostat), and D (samotolisib); MAPK arms (E, selumetinib; G, vemurafenib; J, ulixertinib; and M, tipifarnib), F (ensartinib), H (olaparib), K (ivosidenib), and N (selpercatinib). (A) All tumors, (B) non-CNS solid tumors, (C) CNS solid tumors, (D) osteosarcoma, (E) rhabdomyosarcoma, (F) Ewing sarcoma, (G) high-grade glioma, (H) neuroblastoma, and (I) low-grade glioma. MAPK, mitogen-activated protein kinase.
D. Williams Parsons
Patents, Royalties, Other Intellectual Property: Coinventor on current and pending patents related to cancer genes discovered through sequencing of several cancer types. Participants in royalty sharing related to those patents
Katherine A. Janeway
Honoraria: Foundation Medicine, Takeda
Consulting or Advisory Role: Bayer, Ipsen
Travel, Accommodations, Expenses: Bayer
Brent Coffey
Stock and Other Ownership Interests: Pfizer, AbbVie
P. Mickey Williams
Research Funding: Illumina (Inst)
Patents, Royalties, Other Intellectual Property: I was a coinventor of the DLBCL cell of origin patent recently filed by the NIH
Gregory J. Tsongalis
Stock and Other Ownership Interests: ChromaCode
Stacey L. Berg
Other Relationship: I am a member of the Children's Oncology Group Developmental Therapeutics Steering Committee. Some clinical trials may be partially industry funded. My institution may receive some funding for these trials, Pediatric Early Phase Clinical Trials Network
Douglas S. Hawkins
Research Funding: Loxo (Inst), Bristol Myers Squibb (Inst), Merck Sharp & Dohme (Inst), Bayer (Inst), Lilly (Inst), Eisai (Inst), Amgen (Inst), Seattle Genetics (Inst), Incyte (Inst), Jazz Pharmaceuticals (Inst), Pfizer (Inst)
Travel, Accommodations, Expenses: AstraZeneca
No potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the 2021 ASCO Annual Meeting, Chicago IL, June 5, 2021.
SUPPORT
CLINICAL TRIAL INFORMATION
AUTHOR CONTRIBUTIONS
Conception and design: D. Williams Parsons, Katherine A. Janeway, David R. Patton, Brent Coffey, P. Mickey Williams, Todd A. Alonzo, Stacey L. Berg, Elizabeth Fox, Jeffrey S. Abrams, Naoko Takebe, James V. Tricoli, Nita L. Seibel
Administrative support: David R. Patton
Provision of study materials or patients: Katherine A. Janeway, David R. Patton, Sinchita Roy-Chowdhuri, Nilsa C. Ramirez
Collection and assembly of data: D. Williams Parsons, Katherine A. Janeway, David R. Patton, Cynthia L. Winter, Brent Coffey, Mark Routbort, Nilsa C. Ramirez, Lauren Saguilig, Stacey L. Berg, Elizabeth Fox, Margaret Mooney, Naoko Takebe, Nita L. Seibel
Data analysis and interpretation: D. Williams Parsons, Katherine A. Janeway, David R. Patton, Brent Coffey, Sinchita Roy-Chowdhuri, Gregory J. Tsongalis, Lauren Saguilig, Jin Piao, Todd A. Alonzo, Stacey L. Berg, Elizabeth Fox, Douglas S. Hawkins, Naoko Takebe, Nita L. Seibel
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients with Refractory Cancers in the NCI–COG Pediatric MATCH Trial
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
D. Williams Parsons
Patents, Royalties, Other Intellectual Property: Coinventor on current and pending patents related to cancer genes discovered through sequencing of several cancer types. Participants in royalty sharing related to those patents
Katherine A. Janeway
Honoraria: Foundation Medicine, Takeda
Consulting or Advisory Role: Bayer, Ipsen
Travel, Accommodations, Expenses: Bayer
Brent Coffey
Stock and Other Ownership Interests: Pfizer, AbbVie
P. Mickey Williams
Research Funding: Illumina (Inst)
Patents, Royalties, Other Intellectual Property: I was a coinventor of the DLBCL cell of origin patent recently filed by the NIH
Gregory J. Tsongalis
Stock and Other Ownership Interests: ChromaCode
Stacey L. Berg
Other Relationship: I am a member of the Children's Oncology Group Developmental Therapeutics Steering Committee. Some clinical trials may be partially industry funded. My institution may receive some funding for these trials, Pediatric Early Phase Clinical Trials Network
Douglas S. Hawkins
Research Funding: Loxo (Inst), Bristol Myers Squibb (Inst), Merck Sharp & Dohme (Inst), Bayer (Inst), Lilly (Inst), Eisai (Inst), Amgen (Inst), Seattle Genetics (Inst), Incyte (Inst), Jazz Pharmaceuticals (Inst), Pfizer (Inst)
Travel, Accommodations, Expenses: AstraZeneca
No potential conflicts of interest were reported.
REFERENCES
- 1.Gröbner SN, Worst BC, Weischenfeldt J, et al. : The landscape of genomic alterations across childhood cancers. Nature 555:321-327, 2018 [DOI] [PubMed] [Google Scholar]
- 2.Ma X, Liu Y, Liu Y, et al. : Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555:371-376, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laetsch TW, DuBois SG, Mascarenhas L, et al. : Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 19:705-714, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross AM, Wolters PL, Dombi E, et al. : Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med 382:1430-1442, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mossé YP, Voss SD, Lim MS, et al. : Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A Children's Oncology Group study. J Clin Oncol 35:3215-3221, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang TJ, Orenstein L, DuBois SG, et al. : Pediatric trials for cancer therapies with targets potentially relevant to pediatric cancers. J Natl Cancer Inst 112:224-228, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen CE, Laetsch TW, Mody R, et al. : Target and agent prioritization for the Children's Oncology Group-National Cancer Institute pediatric MATCH trial. J Natl Cancer Inst 109:djw274, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fangusaro J, Onar-Thomas A, Young Poussaint T, et al. : Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol 20:1011-1022, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fangusaro J, Onar-Thomas A, Poussaint TY, et al. : A phase 2 trial of selumetinib in children with recurrent optic pathway and hypothalamic low-grade glioma without NF1: A pediatric brain tumor consortium study. Neuro Oncol 23:1777-1788, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lih CJ, Harrington RD, Sims DJ, et al. : Analytical validation of the next-generation sequencing assay for a nationwide signal-finding clinical trial: Molecular Analysis for Therapy Choice clinical trial. J Mol Diagn 19:313-327, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flaherty KT, Gray RJ, Chen AP, et al. : Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J Clin Oncol 38:3883-3894, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.National Cancer Institute: Pediatric Early Phase Clinical Trials Network (PEP-CTN). https://ctep.cancer.gov/initiativesPrograms/pep-ctn.htm
- 13.Faulk KE, Anderson-Mellies A, Cockburn M, et al. : Assessment of enrollment characteristics for Children’s Oncology Group (COG) upfront therapeutic clinical trials 2004-2015. PLoS One 15:e0230824, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siembida EJ, Loomans-Kropp HA, Tami-Maury I, et al. : Barriers and facilitators to adolescent and young adult cancer trial enrollment: NCORP site perspectives. JNCI Cancer Spectr 5:pkab027, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackay A, Burford A, Carvalho D, et al. : Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32:520-537, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryall S, Zapotocky M, Fukuoka K, et al. : Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37:569-583, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee RS, Stewart C, Carter SL, et al. : A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122:2983-2988, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pajtler KW, Witt H, Sill M, et al. : Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728-743, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shern JF, Selfe J, Izquierdo E, et al. : Genomic classification and clinical outcome in rhabdomyosarcoma: A report from an international consortium. J Clin Oncol 39:2859-2871, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong M, Mayoh C, Lau LMS, et al. : Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med 26:1742-1753, 2020 [DOI] [PubMed] [Google Scholar]
- 21.van Tilburg CM, Pfaff E, Pajtler KW, et al. : The pediatric precision oncology INFORM registry: Clinical outcome and benefit for patients with very high-evidence targets. Cancer Discov 11:2764-2779, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman S, Nakitandwe J, Kesserwan CA, et al. : Genomes for kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov 11:3008-3027, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cortes JE, Gutzmer R, Kieran MW, et al. : Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat Rev 76:41-50, 2019 [DOI] [PubMed] [Google Scholar]
- 24.A study of avapritinib in pediatric patients with solid tumors dependent on KIT or PDGFRA signaling. https://clinicaltrials.gov/ct2/show/NCT04773782