

Abstract
Skin manifestations are well‐recognized non‐motor symptoms of Parkinson's disease (PD) and other hypokinetic and hyperkinetic movement disorders. Skin conditions are usually well visible during routine clinical examination and their recognition may play a major role in diagnostic work‐up. In this educational review we: (1) briefly outline skin conditions related to Parkinson's disease, including therapy‐related skin complications and their management; (2) discuss the role of skin biopsies in early diagnosis of PD and differential diagnosis of parkinsonian syndromes; and focus more on areas which have not been reviewed in the literature before, including (3) skin conditions related to atypical parkinsonism, and (4) skin conditions related to hyperkinetic movement disorders. In case of rare hyperkinetic movement disorders, specific dermatological manifestations, like presence of angiokeratomas, telangiectasias, Mongolian spots, lipomas, ichthyosis, progeroid skin changes and others may point to a very specific group of disorders and help guide further investigations.
Keywords: skin, movement disorders, skin biopsy, atypical parkinsonism, hyperkinetic
Introduction
Skin disorders are a well‐recognized, although often overlooked, symptom associated with Parkinson's disease (PD) and other movement disorders. Multiple skin conditions have been previously well‐described and comprehensively reviewed in the context of PD. 1 , 2 Skin changes may be present also in atypical parkinsonism, although the body of literature in this regard is rather limited and related almost exclusively to autonomic skin changes and skin biopsies in the differential diagnosis of parkinsonian syndromes. In addition to hypokinetic disorders, several hyperkinetic movement disorders may be related to prominent dermatological manifestations, such as presence of lipomas related to mitochondrial disorders, 3 angiokeratomas related to lysosomal storage disorders, 4 or presence of progeroid skin changes, skin hypersensitivity and tumors associated with defects in DNA repair mechanisms. 5 In this educational review we provide a brief overview of non‐iatrogenic and iatrogenic therapy‐related skin conditions related to PD, including the role of skin biopsies in the diagnosis and differential diagnosis of parkinsonian syndromes (for a more in‐depth overview see e.g. the reviews of Skorvanek et Bhatia 1 or Niemann et al. 2 ) and we concentrate more on review of skin conditions associated with atypical parkinsonism and hyperkinetic movement disorders, which have not been comprehensively reviewed in the literature before.
Methods
We searched articles published in English using Pubmed without restriction on dates. We used a combination of medical subject headings (MeSH): “Skin” and/or “cutaneous,” “dermat*,” “melanoma,” “basal cell carcinoma,” “squamous cell carcinoma,” “granuloma,” “lipoma*,” “seborhoeic dermatitis,” “rosacea,” “sweating,” “bullous pemphigoid,” “erythema*,” “pigmentary changes,” “progeroid,” “alopecia,” “hypertrichosis,” “hair,” “self‐injurious behavior,” “self‐mutilations,” “parkinson*,” “progressive supranuclear palsy”, “multiple system atrophy,” “corticobasal,” “Dementia with Lewy bodies”, “dystonia,” “chorea,” “myoclonus,” “tremor,” “ataxia,” “tics.” We also included additional references from relevant research and review articles.
Skin Conditions Related to Parkinson's Disease
Seborrheic dermatitis
The relationship between seborrheic dermatitis (SD) and PD has been known since 1927. 6 It is a chronic, relapsing dermatitis characterized by erythematous lesions with greasy scales, affecting sebum‐rich areas of the scalp, face, hairline, nasolabial folds, ears and upper chest (Fig. 1A). SD is primarily seen in infants, in adults over the age 50 and more in men. 7 In the study of Tanner et al. 8 SD was associated with increased risk of PD. Moreover, in 4% of their PD cases the diagnosis of SD was made prior to the diagnosis of PD, suggesting a potential prodromal marker of the disease. 8
FIG 1.
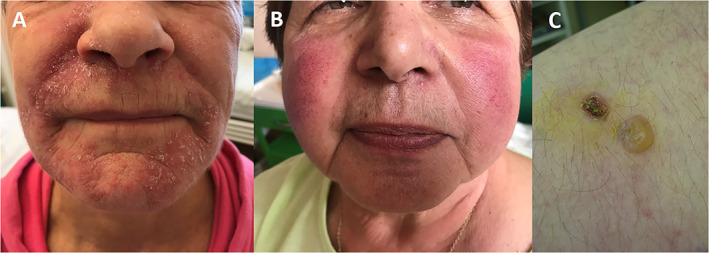
Skin changes in Parkinson's disease. (A) Seborrheic dermatitis, (B) Rosacea, (C) Bullous pemphigoid. 1
The exact mechanisms underlying the association of PD and SD are not fully clear. Increased prevalence of SD in PD may be influenced by Malassezia, a yeast which is present in lipid‐rich skin areas, 9 and gene polymorphisms in GBA, LRRK2, PINK1 or SNCA, playing a role in lipid regulation and coating of lipid droplets. 7
Some of the contradictory causes of SD include autonomic dysfunction, 10 although this was not confirmed in other studies;11 the role of androgens or testosterone relating to higher incidence of SD in male populations; 11 , 12 and the systemic effect of melanocyte‐stimulating hormone. 13
Appropriate anti‐fungal treatment, such as ketoconazole can be useful for PD patients to reduce Malassezia growth and enzyme production. Anti‐inflammatory agents like topical steroids or topical calcineurin inhibitors can also be used in the treatment. 14
Rosacea
Rosacea is a chronic inflammatory disorder classified to 4 subtypes: erythematotelangiectatic, papulopustular, phymatous and ocular. 15 Skin manifestations include centrofacial erythema, telangiectasia, papules, pustules, skin thickening of the nose, rarely beard or ear regions due to fibrosis and glandular hypertrophy (Fig. 1B). The pathogenesis of rosacea is not fully understood, but genetics, immune and environmental factors, neurovascular dysregulation, and microorganisms, may play a role in the pathogenesis. 2
Association of rosacea with PD was shown in two large population‐based studies with an almost 2‐fold increase in the risk of PD among patients with rosacea, mostly the ocular type. 16 , 17 Possible mechanisms linking PD and rosacea include: (1) up‐regulation of matrix metalloproteinase (MMP) enzymes, notably MMP‐1 and MMP‐3 (which plays a role in tissue breakdown and repair in rosacea 18 and contributes to nigrostriatal dopaminergic neuronal loss and neuroinflammation in animal models of PD 19 ), (2) microbiome gut‐brain axis, 16 and (3) Neurokinin B which often accompanies the onset of rosacea. 20
Treatment of rosacea includes avoidance of triggering factors, topical treatment (metronidazole, azelaic acid, antibiotics, topical alfa‐agonists, ivermectin, retinoids), systemic medications (doxycycline, beta blockers, hydroxychloroquine, in case of severe rosacea fulminans retinoids and corticosteroids), systemic treatment with anti IL17 injections, intradermal botulinum toxin injections, and surgical care (electrosurgery, 585‐nm pulsed dye laser, mechanical dermabrasion, carbon dioxide laser peel, and surgical shave techniques). 21
Sweating disturbances
Sweating disturbances are common and distressing non‐motor symptoms of PD that are related mainly to autonomic dysfunction, off periods, and dyskinesias. 22 They occur mainly on the head, neck and trunk and may be asymmetric. Several reports indicate that sweating disturbance may occur in 30% to 60% of PD patients, with hyperhidrosis being more common (9–100%, average 38%) compared to hypohidrosis (9–46%, average 15%). 1 , 2
Bullous pemphigoid
Several studies and case‐series confirmed a significant association between bullous pemphigoid (BP) and several neurological diseases. 23 BP is an autoimmune bullous disease which commonly affects elderly people and is characterized by the generation of autoantibodies directed in particular against BP180/collagen XVII and BP230/dystonin. BP clinically presents with itchy erythematous patches, on which tense bullae with clear or hemorrhagic content later occur (Fig. 1C). After rupture, bullae leave erosions and crusts. Mucosal involvement is observed in 10%–30% cases. 24
BP has a significant association with PD, and the risk ratio of BP in patients with PD has been reported between 2.2 and 9.0. Based on current evidence it is not possible to differentiate whether the elevated risk of BP is driven by the neurological disorder itself or its treatment. 23
Systemic corticosteroid therapy represents the treatment of choice for severe forms. Adjunctive therapy includes combination with azathioprine, mycophenolate mofetil, tetracyclines plus nicotinamide, methotrexate, and dapsone. Bacterial superinfection of erosions should be treated with local antiseptics. 23
Malignant melanoma
The risk of dying from cancer is lower in PD patients compared to the general population. On the other hand, PD patients have a significantly higher risk of developing melanomas, which does not seem to be related to dopaminergic therapy. 25 , 26 , 27 , 28 , 29
Cutaneous melanoma (CM) causes 90% of skin cancer mortality. It is a malignant tumor that arises from melanocytes, primarily involves the skin, and can occur from a preexisting pigmented mole. In this case skin signs of CM are often based on ABCDE criteria (Asymmetry, Border irregularity, Color variety, Diameter, Evolution). 30 CM can occur also de novo and in that case the lesion usually looks like an ”ugly duckling,“ being different from the others.
Clinical manifestations of CM subtypes include: (1) superficial spreading melanoma (macule or patch of irregular shape and different brown to black pigmentation), (2) nodular melanoma (nodular, exophytic, often bleeding tumor with brown‐black pigmentation), (3) lentigo malignant melanoma (located on sun‐damaged areas mainly face of elderly individuals), (4) acral lentiginous melanoma (subungual and palmoplantar location), (5) amelanotic melanoma (lesion with little or no pigmentation). 31
Alterations in melanin and melanin‐synthesizing enzymes, genetic factors, and abnormal autophagy, along with the fact that both melanocytes and neurons share their embryological origins from the neural crest, have all been proposed to underlie the increased risk of melanoma in PD. 1 , 32
No evidence‐based recommendations can be made regarding the need for periodic dermatological screening in PD specifically. Nevertheless, in general any changing or otherwise concerning melanocytic nevus should be checked by a dermatologist, otherwise preventive dermatological screening for CM or nonmelanoma skin cancer should be done every year.
Therapy‐related skin disorders in PD
Iatrogenic skin complications of PD therapy were summarized in detail in several previously published reviews, with recommendations for both prevention and treatment. 1 , 2 Apart from possible allergic reactions, oral pharmacotherapy‐associated skin disorders were generally mild, with resolution after drug discontinuation in most cases. Lower limb oedema (associated with levodopa, dopamine agonists), livedo reticularis (amantadine), alopecia, vitiligo, hyperpigmentation (levodopa) can be listed as the most common ones. 2 In case of other administration routes such as transdermal patch (rotigotine), subcutaneous infusion (apomorphine, levodopa) or via percutaneous endoscopic gastrostojejunostomy (levodopa‐carbidopa intestinal gel), skin complications were mostly procedure and maintenance related—wound infections, application/infusion site reactions, subcutaneous nodules etc., manageable by standard prevention and treatment measures. 2 As for deep brain stimulation, hardware‐related infections and subsequent skin erosion were described. 33 The overview of skin complications and their management is outlined in Table 1.
TABLE 1.
Therapy‐related skin complications and their management in Parkinson's disease, adapted from Niemann et al. 2
| Therapy | Possible complications | Recommendations | |
|---|---|---|---|
| Pharmacotherapy | Route of administration | ||
| Levodopa/carbidopa | Oral | Lower extremity oedema, allergic cutaneous reactions, alopecia, vitiligo, skin hyperpigmentation, Laugier–Hunziker syndrome, Henoch–Schönlein syndrome, pseudobullous morphea, scleroderma‐like illness | Drug discontinuation (resolution of most of the complications) |
| Subcutaneous infusion | Subcutaneous nodules | Prevention: rotation of infusion sites | |
| Other infusion site reaction (hematoma, infections, pain) | As in other subcutaneous delivery therapies; under investigation | ||
| Via PEG‐J (LCIG) | Wound infections, erythema at stoma site | Prevention: remove dressing before cleaning, clean with soap and water (avoid alcohol, iodine), mobilize PEG‐J 2–3 cm in and out of stoma one healed; treatment: systemic antibiotics, possible removal of PEG‐J | |
| Excessive granulation tissue | Topical silver nitrate, surgical removal | ||
| Stoma leakage | Avoid topical creams/ointments, apply dressing to keep stoma dry | ||
| Amantadine | Oral | Livedo reticularis | Resolution 2–4 weeks after drug discontinuation |
| Dopamine agonists:ropinirole, pramipexole (non‐ergoline) | Oral | Lower limb oedema | Resolution with drug discontinuation |
| Dopamine agonists: rotigotine (non‐ergoline) | Transdermal patch | Localized skin reactions (erythema, oedema, pruritus) | Prevention: rotation of application sites, apply patch to clean and dry skin, clean area with soap and water after patch removal; treatment: topical steroids |
| Dopamine agonists: bromocriptine, cabergoline, pergolide, lisuride (ergoline) | Oral | Erythromelalgia‐like skin eruption ‐ painful erythema and oedema of the feet and ankles | Resolution with drug discontinuation |
| Apomorphine | Subcutaneous infusion | Subcutaneous nodules, infusion site erythema (panniculitis) | Prevention: rotation of infusion sites, use Teflon needles, apply needle at 45–90° delivery angle, use lower concentration of apomorphine solution; treatment: massage, silicone dressing, ultrasound management |
| Sublingual | Oropharyngeal erythema | Resolution with drug discontinuation | |
| COMT inhibitors: entacapone | Oral | Bullous eruptions | Case report – i.v. antibiotic and corticosteroid treatment |
| Surgery | |||
| DBS | Subcutaneous placement of hardware | Hardware infections | Prevention: use curved scalp incisions during placement, avoid externalization of hardware following surgery; treatment: wound debridement, hardware removal, i.v. antibiotics |
| Skin erosions | Prevention: use of smaller hardware with lower profile, recessing hardware into drilled bone, using curved scalp incisions, implanting hardware under muscle fascia when possible; treatment: surgical revision, wound debridement, hardware removal or re‐implantation | ||
Abbreviations: PEG‐J, percutaneous endoscopic gastrostojejunostomy; LCIG, Levodopa‐carbidopa intestinal gel; COMT, catechol‐O‐methyltransferase; i.v., intravenous; DBS, deep brain stimulation.
Skin conditions related to atypical Parkinsonism
Multiple system atrophy
Multiple system atrophy (MSA) is a progressive neurodegenerative disorder, caused by underlying α‐synuclein (α‐syn) pathology and characterized by a combination of parkinsonism, ataxia, corticospinal dysfunction and autonomic failure. 34 Although formerly considered as a pure CNS degeneration, a peripheral component was also proposed recently as phosphorylated α‐syn (p‐α‐syn) was detected in cutaneous small fiber nerves in MSA patients. 35
Anhidrosis occurs in majority of MSA subjects and tends to be progressive and widespread, compared to non‐progressive asymptomatic hypohidrosis restricted to hands and feet in PD. 36 Pathogenetic mechanisms probably involve both postganglionic and preganglionic sudomotor dysfunction. 37 Several electrophysiological tests can detect autonomic skin abnormities and distinguish between various neurodegenerative disorders. Mental or physiological stimuli normally lead to a transient rise of sweat secretion (sympathetic sweat response, SSwR) and transient reduction of skin blood flow in the palm/sole (skin vasomotor reflex, SkVR). Pure autonomic failure (PAF) also presents a condition of dysautonomia, but without extrapyramidal symptomatology. Diminished SSwR and preserved SkVR was observed in MSA, while attenuation of both seems to be characteristic of PAF, which can help to differentiate MSA and PAF, especially in the early stages. 38 Sympathetic skin response (SSR) tends to be abnormal in MSA (69–100%) much more frequently than in PD (0–7.7%). 39 , 40 The Odds Ratio (OR) for having MSA‐parkinsonism (MSA‐P) vs. PD based on reduced sweating function (assessed by electrochemical skin conductance for hands and feet) is 4.94; in combination with orthostatic hypotension and reduced heart rate variations (HRV) to deep breathing the OR can be further increased to 11.68. 41
Another autonomic cutaneous manifestation in MSA is disturbed neurovascular thermoregulation of distal extremities—the so‐called cold hand sign. 42 Palm skin temperature is significantly lower in MSA patients than in controls; the temperature <28°C can even distinguish MSA from PD, although with low sensitivity. 43
Dementia with Lewy bodies
Dementia with Lewy bodies (DLB) is a synucleinopathy characterized by dementia and varying combinations of the core clinical features of parkinsonism, REM sleep behavior disorder (RBD), fluctuating cognition/alertness and visual hallucinations. 44 Impaired sudomotor function can be detected by electrophysiological testing—typical findings are severely reduced SSwR 45 and absent or reduced SSR. 46 Skin vasomotor dysfunction is represented by reduced SkVR amplitudes. 45 Combined cutaneous and cardiovascular features of dysautonomia (abnormal SSR, reduced HRV) may distinguish DLB from non‐synucleinopathy causes of dementia such as Alzheimer's disease. 47
Progressive supranuclear palsy
Progressive supranuclear palsy (PSP) is a neurodegenerative disease characterized pathologically by 4 repeat tau deposition in various cell types and anatomical regions. Richardson's syndrome (RS) is the initially described and one of the clinical phenotypes associated with PSP pathology, characterized by vertical supranuclear gaze palsy, postural instability with early falls and subcortical frontal dementia. PSP can manifest as several other clinical phenotypes, including PSP‐parkinsonism, pure akinesia with gait freezing, frontotemporal dementia, corticobasal syndrome, speech/language impairment. 48 In a previous study of Kikkawa et al., 49 SSwR was severely diminished, whereas SVR was maintained and cardiovascular function was well preserved compared to PD. Abnormal results of electrophysiological tests (abnormal SSR, HRV) without clinical evidence of dysautonomia is more suggestive of PSP, while in MSA a significant correlation between the intensity of clinical symptoms of dysautonomia and electrophysiological tests results can be found. 50
Skin biopsies and α‐Synuclein in Parkinsonian syndromes
Skin biopsy is a promising in vivo diagnostic tool for synucleinopathies, relevant for confirmation of the diagnosis, prodromal diagnostics and as a possible biomarker in future clinical trials. 51 α‐syn deposition is most prominent in sympathetic adrenergic nerve fibers innervating the arrector pili muscles, but also in sympathetic cholinergic nerve fibers. 52
In general, specificity of α‐syn detection in PD is high (even up to 100%); multiple studies yielded various results especially for sensitivity (30–100%), depending on methodological differences e.g. biopsy site and size, number of samples, type of fixative and time duration, thickness of skin sections, technique—conventional immunohistochemistry (using different antibodies) or novel techniques assessing the seeding activity of α‐syn. 51
Apart from α‐syn detection, peripheral neuropathy (reduced intraepidermal nerve fiber density, IENFD) was documented in skin samples of PD patients, affecting predominantly distal small fibers and autonomic fibers. 53 , 54 , 55 Different underlying mechanisms were proposed, including levodopa intake or B vitamins deficiency. However, IENFD was reduced also in idiopathic RBD (iRBD) patients with normal vitamin levels and without levodopa medication, suggesting that small fiber neuropathy is intrinsic to the disease itself, 56 although the length‐dependent reduction in IENFD and the α‐syn‐associated autonomic neuropathy are suggested to have different pathogenetic mechanisms. 52
P‐α‐syn is detected in PD skin samples from very early stages of the disease 52 and even in patients with iRBD. 57 Skin biopsies therefore represent a promising biomarker of prodromal PD, as well as a helpful differential diagnostic tool, given that patients with secondary parkinsonism (vascular) without underlying α‐syn pathology stain negative. 58 Regarding various genetic forms of PD, patients with SNCA, DJ‐1, LRRK2 or GBA mutations had substantial α‐syn deposition in cutaneous sympathetic noradrenergic nerves, whereas those with biallelic PRKN mutations did not. 59
Localization and load differences of α‐syn aggregates may potentially help distinguish different synucleinopathies. In PD, DLB and PAF the α‐syn deposition was in autonomic fibers mainly at proximal sites, with PAF and DLB showing the highest α‐syn load and a widespread involvement of autonomic skin nerve fibers, whereas in MSA‐P the deposits were in somatic fibers, mainly at distal sites. MSA‐cerebellar type displayed no skin deposits in the majority of analyzed patients. 60 In another study, p‐α‐syn was detected in all DLB subjects and no non‐synucleinopathy dementia patients, proposing skin biopsy as a useful differential diagnostic marker. 61 Through skin biopsy, PAF was also differentiated from acquired autonomic neuropathies. 62 Concerning IENFD, somatosensory fibers are more affected in MSA compared to PD with more dominant involvement of autonomic fibers. 35
In atypical parkinsonism caused by tauopathies (PSP, CBS), cutaneous α‐syn was either not detected by conventional immunohistochemistry at all 63 or in just negligible minority with clinical features suggesting atypical synucleinopathy or a mixed pathology;64 RT‐QuIC and PMCA analyses of seeding activity of α‐syn were also negative. 65 Neither of the tau‐pathologies is spread widely in the periphery—with only a minority of skin samples of CBS/PSP patients staining positive for total tau and none for phosphorylated tau (p‐tau) 66 ; another study showed p‐tau positivity in PSP patients and mixed p‐tau/α‐syn pathology in PD. 67
Skin Conditions in Hyperkinetic Movement Disorders
Movement disorders related to skin hypersensitivity, pigmentary changes, skin tumors and progeroid syndromes
Disorders related to skin hypersensitivity, skin tumors (i.e. malignant melanoma, squamous cell or basal cell carcinoma), pigmentary changes and progeroid syndromes are often associated with defects in the nuclear envelope and DNA repair mechanisms, 5 such as Xeroderma pigmentosum and Ataxia telangiectasia (Table 2). Skin hypersensitivity may present with visible or non‐visible subjective symptoms such as dry skin, irritation, eczema, redness, desquamation, burning, itching or stinging. Progeroid syndromes are a heterogeneous group of very rare disorders resembling features of accelerated aging, i.e. hair loss, short stature, skin atrophy with loss of cutaneous elasticity, dysfunction of cutaneous appendices, increased susceptibility for malignant tumors, cardiovascular diseases and osteoporosis. 68 , 69 , 70
TABLE 2.
Specific skin manifestations associated with hyperkinetic movement disorders
| Skin manifestations | Disease | Movement disorder | Specification of skin manifestation |
|---|---|---|---|
| Photosensitivity, freckles | Hartnup disease | Ataxia, dystonia | |
| Xeroderma pigmentosum | Chorea, ataxia | ||
| Pigmentary changes | Xeroderma pigmentosum | Chorea, ataxia | Progressive freckle‐like pigmentary changes |
| Ataxia telangiectasia | Ataxia, dystonia, chorea, myoclonus | Vitiligo‐like hypopigmented macules, Café‐au‐lait macules | |
| Incontinentia pigmenti | Ataxia | ||
| Hypomelanosis of Ito | Ataxia, hyperkinesias | ||
| Waardenburg‐Shah syndrome | Ataxia | Skin and hair hypopigmentation | |
| ADAR1‐related disease | Dystonia, ataxia, tremor, chorea | Dyschromatosis symmetrica hereditaria | |
| GM1 gangliosidosis | Ataxia, dystonia, parkinsonism | Mongolian spots | |
| Chediak‐Higashi syndrome | Ataxia, parkinsonism | Oculocutaneous albinism | |
| GM3 Synthase deficiency | Choreoathetosis, stereotypies | Salt and pepper pigmentary changes, atopic dermatitis, ichthyosis, SIB | |
| Ichthyosis | SCA34 | Ataxia | Ichthyosis, erythrokeratoderma |
| Refsum disease | Ataxia | Ichthyosis | |
| Type 2 Gaucher disease | Striatal toes, myoclonus, parkinsonism | Ichthyosis | |
| GM3 Synthase deficiency | Choreoathetosis, stereotypies | Salt and pepper pigmentary changes, atopic dermatitis, ichthyosis, SIB | |
| Telangiectasias | Ataxia telangiectasia | Ataxia, dystonia, chorea, myoclonus | |
| GM1 gangliosidosis | Ataxia, dystonia, parkinsonism | ||
| Progeric skin changes | Xeroderma pigmentosum | Chorea, ataxia | |
| Ataxia telangiectasia | Ataxia, dystonia, chorea, myoclonus | ||
| POLR3A‐related disease | Ataxia, dystonia, tremor | ||
| Malignant tumors | Xeroderma pigmentosum | Chorea, ataxia | |
| Ataxia telangiectasia | Chorea, dystonia, myoclonus, ataxia | ||
| Benign tumors | Mitochondrial disorders (especially MERRF) | Myoclonus, ataxia | Multiple systemic lipomatosis |
| POLR3A‐related disease | Ataxia, dystonia | Lipomas | |
| Ataxia telangiectasia | Ataxia, dystonia, chorea, myoclonus | Cutaneous granulomatosis | |
| Cerebrotendinous xantomatosis | Ataxia, parkinsonism, dystonia, myoclonus, postural tremor | Tendinous xantomas | |
| Erythema | Sydenham's chorea | Chorea, tics | Erythema marginatum |
| Antiphospholipid syndrome | Chorea | ||
| Systemic lupus erythematosus | Chorea | Malar rash | |
| Biotinidase deficiency | Myoclonus, dystonia, ataxia | Erythematous dermatitis | |
| Phenylketonuria | Stereotypies, tics, tremor | Eczematous dermatitis, Acrodermatitis dysmetabolica | |
| GM3 Synthase deficiency | Choreoathetosis, stereotypies | Salt and pepper pigmentary changes, atopic dermatitis, ichtyosis, SIB | |
| Pellagra | Ataxia, myoclonus, parkinsonism, tremor | Dermatitis, hyperpigmentation, hyperkeratosis, vesicles and bullae in “wet pellagra” | |
| Angiokeratomas | Galactosialidosis | Myoclonus | |
| Fucosidosis | Dystonia | ||
| GM1 gangliosidosis | Ataxia, dystonia, parkinsonism | ||
| Fabry disease | Ataxia, parkinsonism | ||
| Skin adnexes | KMT2B‐related disease | Dystonia | Hypertrichosis |
| SURF1‐Leigh syndrome | Dystonia, ataxia, chorea | Hypertrichosis | |
| Ataxia telangiectasia | Ataxia, dystonia, chorea, myoclonus | Hypertrichosis | |
| Mucopolysaccharidoses (MPS‐II, MPS‐III, MPS‐VII) | Choreoathetosis, ataxia | Hypertrichosis | |
| Biotinidase deficiency | Myoclonus, dystonia, ataxia | Alopecia | |
| Woodhouse‐Sakati syndrome | Dystonia | Alopecia | |
| Systemic lupus erythematosus | Chorea | Alopecia | |
| Satoyosi syndrome | Pseudodystonia (muscle spasms) | Alopecia | |
| X‐linked adrenoleukodystrophy | Ataxia | Alopecia, skin hyperpigmentations | |
| Gomez‐López‐Hernandez syndrome | Ataxia | Partial Alopecia | |
| Klinefelter syndrome | Tremor | Insufficient facial, axillary and pubic hair | |
| Giant axonal neuropathy | Ataxia | Curly/frizzly hair | |
| FA2H neurodegeneration | Ataxia, dystonia | Bristle‐like appearance of hair | |
| Nail‐patella syndrome | Dystonia | Small, poorly developed nails | |
| Self‐injurious behavior | Tics/Tourette syndrome | Chorea | |
| Neuroacanthocytosis | Chorea, dystonia, tics | ||
| Lesch–Nyhan syndrome | Dystonia, chorea, stereotypies | ||
| NMDAR encephalitis | Chorea, dystonia, stereotypies | ||
| SCA17 | Ataxia, chorea, dystonia | ||
| Rett syndrome | Dystonia, stereotypies, tremor | ||
| Phenylketonuria | Stereotypies, tics, tremor | ||
| 6‐Pyruvoyl‐Tetrahydropterin Synthase deficiency | Dystonia, chorea | ||
| GM3 Synthase deficiency | Choreoathetosis, stereotypies | Salt and pepper pigmentary changes, atopic dermatitis, ichtyosis, SIB | |
| Other specific skin conditions | Ehlers‐Danlos syndrome | Dystonia, tremor, chorea, myoclonus, tic disorders | Abnormal skin fragility |
| Complex regional pain syndrome | Fixed dystonia | Color changes | |
| Aicardi‐Goutières syndromes | Dystonia | Chilblain lesions | |
| GNB1 encephalopathy | Dystonia, tics, ataxia, chorea | Mastocytosis and others | |
| Coeliac disease | Ataxia, myoclonus | Dermatitis herpetiformis | |
| WDR73‐associated disease (CAMOS syndrome) | Dystonia, ataxia | Abnormal osmiophilic pattern of skin vessels | |
| Erdheim‐Chester disease | Ataxia | Periorbital xanthelasma, xanthoma‐like papules | |
| Behçet's disease | Chorea, ataxia, myoclonus | Genital, oral and skin ulcers, folliculitis‐like lesions | |
| NAXE and NAXD mutations | Ataxia, hypotonia | Leyll‐like bullous skin lesions |
Xeroderma pigmentosum (XP) is an autosomal recessive disorder with 100% penetrance, characterized by enzymatic defect in DNA repair pathway known as nucleotide excision repair (NER) and a combination of cutaneous, ophthalmological and neurological symptoms. Depending on the involved genes, complementation groups from XP‐A to XP‐G and XP‐V have been described. 71 Patients with XP typically show extreme hypersensitivity to UV exposure with sunburns often occurring from early infancy (60% of infant cases after minimal UV exposure), skin xerosis (dryness), progressive freckle‐like pigmentary changes (often prominent already around 2 years of age), progeroid skin changes and an increased incidence of skin malignancies (Fig. 2A, B). 71 Skin cancer usually occurs in the face/head/neck area and compared to healthy population may be increased 2000‐fold for cutaneous melanoma and up to 10,000‐fold for squamous cell and basal cell carcinomas. 72 In addition to cutaneous manifestation, ophthalmological symptoms may include ocular surface pathology, eyelid damage, and ophthalmic malignancies. Approximately 20–30% of cases (esp. XP‐A and XP‐B) may present with neurological symptoms, which typically start with decreased tendon reflexes and sensorineural hearing loss and later may progress to severe ataxia, swallowing difficulties and progressive cognitive impairment. 73 In addition, patients with XP‐E complementation group may present with a Huntington's disease‐like syndrome including prominent chorea, ataxia, progressive cognitive decline and neuropsychiatric symptoms. 74 Classic dermatological and oncological features of XP need to be investigated in choreic patients with negative genetic tests for Huntington's disease.
FIG 2.
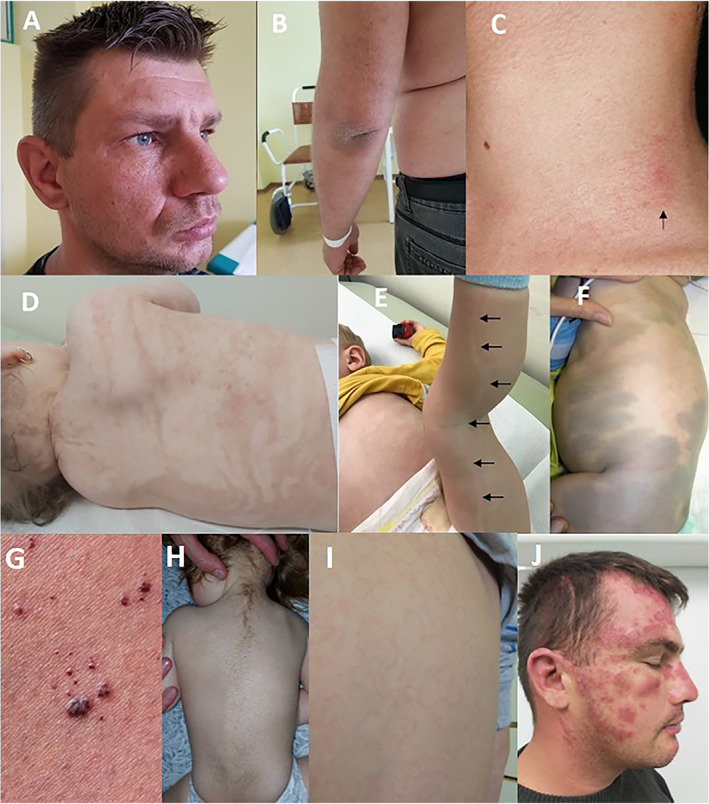
Skin changes in hyperkinetic movement disorders. (A) Freckle‐like pigmentary changes and (B) Skin xerosis on left elbow in a patient with biallelic ERCC4 variants causing Xeroderma pigmentosum type F presenting also with generalized chorea, ataxia and mild cognitive problems, (C) Cutaneous telangiectasias (arrow) in a patient with biallelic ATM variants, (D) Incontinentio pigmenti, (E) Hypomelanosis of Ito (hypomelanotic streak on the left leg—black arrows), (F) Mongolian spots in a child with GM1‐gangliosidosis, (G) Angiokeratoma (adapted with permissions from Cuestas et al. 2019 97 ), (H) Hypertrichosis in a child with biallelic SURF1 variants, (I) Erythema marginatum, (J) Malar rash in lupus erythematosus.
Ataxia telangiectasia (AT) is an autosomal recessive disorder caused by biallelic variants in the ATM gene, which is involved in cell cycle progression, nuclear and mitochondrial DNA repair, protection of chromosome ends and apoptosis. 75 , 76 The dysfunctional ATM protein in addition induces expression of hypoxia inducible factor (HIF‐1) and subsequently vascular endothelial growth factor (VEGF), leading to the development of telangiectasia. 77 It is a multisystem disease characterized by progressive neurologic decline, oculocutaneous telangiectasias, immunodeficiency, susceptibility to sinopulmonary infections, autoimmune or other chronic inflammatory diseases, radiation sensitivity, and malignancies. In typical cases, progressive ataxia starts in the first years of life, leads to a wheelchair‐bound state around the second decade and is variably accompanied by other movement disorders like chorea, dystonia, or myoclonus. 75 Nevertheless, increasing evidence expands the spectrum of AT to atypical or variant forms with a later age of onset, prolonged survival, and absence of ataxia or telangiectasias in the disease course. 75 , 78 While ocular telangiectasia may be present in up to 97% of AT cases, telangiectasia on other body parts may be less frequent (Fig. 2C). Other skin manifestations include progeric changes of the skin and hair (premature hair graying since childhood), cutaneous atrophy with decreased subcutaneous tissue, pigmentary anomalies, including café‐au‐lait macules and vitiligo‐like hypopigmented macules, melanocytic nevi, facial papulosquamous rash and hypertrichosis. 75 , 76 Cutaneous granulomatosis presenting as skin nodules and ulcerated erythematous plaques disseminated on the face, and on trauma‐prone areas of upper and lower extremities was described as well. 79 In addition, approximately 24.7% of AT cases develop malignancies over their lifetime, mostly hematological, and although the incidence of melanoma has not been characterized well, previous reports emphasize the importance of performing periodic longitudinal complete skin examinations with a focus on changing nevi in AT population. 80
In addition, progeroid changes and movement disorders may be a prominent feature of biallelic POLR3A variants, related to the Wiedemann‐Rautenstrauch syndrome, a rare neonatal progeroid syndrome with growth and developmental retardation, lipoatrophy, a distinctive face, sparse scalp hair, prominent scalp veins, and dental anomalies. Ataxia, tremor and dystonia may be a common manifestation in these individuals. 81 , 82
Hartnup disease is an autosomal recessive disorder caused by SLC6A19 mutations leading to impaired functioning of the sodium dependent B0 AT1 neutral amino acid transporter and impaired intestinal uptake and tubular reabsorption of all neutral amino acids. The disorder typically manifests with cutaneous and neurological symptoms, including a typical pellagra‐like rash, light‐sensitive dermatitis, intermittent cerebellar ataxia and psychiatric symptoms. 83 In addition, phenotypes presenting with intermittent dystonia and hereditary spastic paraplegia have been described. 84 , 85
Other movement disorders related to pigmentary changes
Incontinentio pigmenti is a rare X‐linked dominant neurocutaneous disorder caused by NEMO gene variants, affecting ectodermal tissues—the skin, skin adnexes, eyes, central nervous system and teeth. 86 Pigmentary skin changes are very typical (Fig. 2D) and commonly occur with a distribution on Blaschko lines, which present lines of normal cell development in the skin. They usually follow a V shape over the back, S shaped whirls over the chest and sides and wavy shades on the head. Skin manifestations commonly occur in 4 stages, which may overlap or not occur at all—erythema, then vesicles and pustules (Stage 1, first weeks of life); verrucous lesions (Stage 2, first months of life); linear hyperpigmentation (Stage 3, around 6 months of life); and depigmentation (Stage 4, mostly described in adults). 87 Neurological features occur in about 20–30% of cases and typically present with cerebellar ataxia, seizures (most common), mental retardation, pyramidal involvement and microcephaly. 88
Hypomelanosis of Ito is a rare neurocutaneous disorder related to reduced melanin in the epidermis, which is characterized by presence of hypopigmented lesions that can also follow the Blaschko lines or have a block‐like configuration (Fig. 2E). 87 Hypopigmentations may fade in later life. Neurological manifestations include especially mental retardation, epilepsy, motor system dysfunction, including cerebellar ataxia and psychiatric symptoms including autism and cortical visual impairment. 87 , 89
Chediak‐Higashi syndrome is a rare autosomal recessive disorder characterized by oculocutaneous albinism and immune deficiency with an increased susceptibility to infections. Neurological symptoms usually occur in early adulthood and may include cerebellar deficits, polyneuropathies, spasticity, cognitive decline and parkinsonism. 90
Waardenburg‐Shah syndrome due to dominant SOX10 mutations combines Waardenburg syndrome (congenital hearing loss and eye, skin and hair hypopigmentation) and demyelinating neuropathy, central dysmyelination and Hirschsprung disease. Ataxia, spasticity and developmental delay are common. 91
Variants in ADAR1 gene are responsible for rare neurological and dermatological syndromes including Aicardi‐Goutieres syndrome type 6 (severe infantile encephalopathy with intracranial calcifications), bilateral striatal necrosis and dyschromatosis symmetrica hereditaria, which is characterized by a mixture of hypopigmented and hyperpigmented macules on the dorsa of both hands and feet, and freckle‐like macules on the face. 92 In a recent review of 57 subjects with ADAR1 variants, movement disorders were present in 60% of these cases, with dystonia being the most common (39%, in 2 cases presenting with a severe status dystonicus), followed by spasticity (19%), tremor (7%) and rigidity, chorea and ataxia (all 5%). 93
GM1 gangliosidosis is an autosomal recessive lysosomal storage disorder with a spectrum of phenotypes ranging from severe infantile to mild chronic/adult forms. 94 Types II and III may present with progressive ataxia or generalized dystonia followed by later development of akinetic‐rigid parkinsonism and cognitive decline. Skin manifestations typically include Mongolian spots, generally presenting as large, non‐blanching, hyperpigmented, blueish to slate‐gray macules or patches over the lower back, buttocks, and occasionally flanks or shoulders (Fig. 2F), as well as angiokeratomas (discussed in more details below), eczematoid facial rash, truncal macular rash, ecchymosis and generalized telangiectasias. 95
Pigmentary changes described as “salt and pepper” may be present in about 70% of cases with GM3 Synthase deficiency caused by recessive ST3GAL5 mutations. All of the individuals have profound intellectual disability, choreoathetosis, and hearing loss. In addition, severe atopic dermatitis, ichthyosis and self‐injurious behavior have been described in this condition. 96
Movement disorders related to angiokeratomas
Angiokeratomas (AKs) (Fig. 2G) 97 are benign capillary malformations that can form dark red to black papules, nodules, or plaques with associated overlying epidermal hyperplasia (acanthosis or hyperkeratosis). 97 The diffuse form—angiokeratoma corporis diffusum (ACD) is characterized by multiple AKs typically located between the naval and upper thigh and is related to lysosomal storage disorders (LSDs). Several LSDs, including GM1 gangliosidosis described in more details above, have been previously associated with presence of AKs and hyperkinetic movement disorders. Fucosidosis is an autosomal recessive neurodegenerative disorder presenting with coarse facial features, growth retardation, recurrent upper respiratory tract infections, dysostosis multiplex and AKs. Focal dystonia has been previously reported in 12% of fucosidosis cases and generalized dystonia has been also reported. 98 , 99 Galactosialidosis is an autosomal recessive disorder caused by a primary defect of protective protein/cathepsin A and/or a secondary defect of components of the lysosomal multienzyme complex (LMC), which includes galactosidase and neuraminidase‐1. 100 Patients with juvenile/adult type of Galactosialidosis present with a spectrum of coarse facies, vertebral changes, cherry‐red spots and neurological symptoms including cognitive impairment, epilepsy, ataxia and myoclonus. 101 In addition to LSDs related to hyperkinetic disorders, Fabry disease has been previously linked to presence of Parkinson's disease. In a report of Wise et al. 102 2.2% of Fabry patients and 7.4% of their first line relatives fit the criteria for a conservative diagnosis of PD. In addition to angiokeratomas, patients with Fabry disease may also present with other skin abnormalities including scant body hair and hypo/hyper/anhidrosis. 4
Movement disorders related to lipomas
Lipomas are a rare disorder of the adipose tissue. 103 While the etiology is still unclear, a potential link with mitochondrial dysfunction has been hypothesized 3 best known in the association of Myoclonic epilepsy with ragged red fibers (MERRF) and presence of cervical lipomas. A recent study on mitochondrial disorders in Italy identified 22 (1.7%) patients with lipomas among the 1300 enrolled mitochondrial patients. 104 The most common presentation of lipomas in this study was in form of multiple systemic lipomatosis affecting symmetrically the cervical‐cranial‐thoracic region, only 2 patients had lipomas localized in a single anatomical site. Eighty‐six percent had mutations in mtDNA coding for tRNA lysine (MERRF). In addition to mitochondrial disorders, presence of lipomas and movement disorders has been reported in POLR3A‐related disease, 82 discussed in more details above.
Movement disorders related to skin adnexes pathology
The skin appendages (or adnexes) are epidermal and dermal‐derived components of the skin and include hair, nails, sweat glands, and sebaceous glands. 105 In the context of hyperkinetic movement disorders, abnormalities of the hair are the most common. Alopecia, or hair loss, is associated with several following conditions. In Woodhouse‐Sakati syndrome, a rare autosomal recessive disease caused by DCAF17 mutations, alopecia occurs along with hypogonadism, diabetes mellitus, hypothyroidism, intellectual disability, sensorineural hearing loss and movement disorders (mainly dystonia and chorea). Childhood‐onset hair thinning often progresses to alopecia totalis in adulthood. Eyebrows and facial hair are sparse or absent. 106 , 107 In biotinidase deficiency, caused by BTD mutations, various neurological symptoms such as epilepsy, deafness, optic atrophy, spastic paralysis, ataxia or dystonia, combine with skin manifestations, including alopecia, dermatitis, dry skin, yellow hair, keratitis and others. Biotin replacement therapy is usually effective. 108 The combination of alopecia totalis, painful spasms and diarrhea suggests Satoyosi syndrome. 109 Other differential diagnoses of alopecia with movement disorders include X‐ALD presenting additionaly with ataxia, pyramidal signs, leukodystrophy and skin hyperpigmentations;110 and a very rare Gomez‐López‐Hernandez syndrome presenting with a classic triad of rhomboencephalosynapsis, partial non‐scarring alopecia and trigeminal anesthesia with additional ataxia in a proportion of cases. 111 Recently, RHOBTB2 gene variants, usually presenting with epileptic encephalopathy, intellectual disability, microcephaly, facial dysmorphia, choreatic and/or dystonic dyskinesias (including paroxysmal), were anecdotally reported also with aplasia cutis congenita. 112 In Klinefelter syndrome (mostly 47, XXY), sparse body and facial hair are often present besides the cardinal features (hypogonadism, cryptorchism, infertility, gynecomastia, decreased testosterone levels and learning problems) and various movement disorders (ET‐like tremor, myoclonus and even parkinsonism). 113 , 114 A condition seen in children is giant axonal neuropathy. Disease typically starts in childhood with sensory‐motor axonal neuropathy and ataxia with curly/frizzy hair found in nearly all patients. MRI is also characteristic, with T2/FLAIR hyperintensity in the white matter surrounding the dentate nucleus. 115 In addition, bristle‐like appearance of hair is commonly present in Fatty acid 2‐hydroxylase (FA2H) neurodegeneration, which can present with ataxia, pyramidal signs and frequent dystonia (50%) in childhood. Leukodystrophy, cerebellar and pontine atrophy, thin corpus callosum and T2‐hypointensity of globus pallidus are typical imaging clues. 116
Hypertrichosis may be a clinical clue to SURF1‐associated Leigh syndrome (Fig. 2H), which typically presents also with feeding difficulties, developmental delay, hypotonia, ataxia, choreoathetosis, dystonia and symmetrical T2 hyperintense lesions in the brainstem and/or basal ganglia. 117 In addition, hypertrichosis may present also in mucopolysaccharidoses (MPS‐II, MPS‐III, MPS‐VII), which may manifest also with choreoathetosis and ataxia. 118
Unusual paroxysmal cranial dystonic dyskinesias triggered by wind, touch and water, have been recently described in a large multigenerational Danish family as a novel feature of nail‐patella syndrome. This autosomal dominantly inherited disorder is otherwise characterized by morphological changes of nails, joints, bones, renal disease, polyneuropathy and behavioral and psychiatric symptoms. 119
Movement disorders related to erythema and dermatitis
In addition to polyarthritis, carditis, and Sydenham chorea, another major feature of rheumatic fever (RF), an autoimmune response to a preceding Streptococcus group A infection, is erythema marginatum (Fig. 2I). It refers to erythematous, peripherally spreading macules developing patchy areas, and typically occurring several weeks after Streptococcal infection. Lesions are typically fluctuant, migrating, fading and reappearing within hours. However, although typical, it is recognized only in <6% of patients. Another cutaneous manifestation of RF are subcutaneous nodules. 120 , 121 Cutaneous symptoms are frequent in systemic lupus erythematosus (SLE) and antiphospholipid syndrome (APS), both manifesting sometimes with movement disorders (with chorea being the most common in both, and ataxia and parkinsonism less often in SLE). 122 The skin is involved in up to 85% of SLE cases. Some of the numerous skin manifestations of SLE include malar rash (a butterfly‐shaped erythema over the cheeks and bridge of the nose, Fig. 2J), macular or diffuse erythematous rash in sun‐exposed areas, discoid rash, alopecia, and cutaneous vasculitis. 123 Cutaneous manifestations of APS include livedo reticularis, necrotizing vasculitis, thrombophlebitis, erythematous macules, purpura, and others. 124
Another skin symptom, called dermatitis herpetiformis, may appear along with ataxia, both in the context of gluten sensitivity. 125 A number of nutritional deficiencies can present with skin changes. Myoclonus, ataxia, tremor and parkinsonism have been reported in pellagra, caused by nicotinic acid (vitamin B 3) deficiency, which is otherwise characterized by a combination of 3Ds—dermatitis, diarrhea and dementia—and is possibly fatal if left untreated (death as the 4th D). 126 , 127 Although being predominantly found in developing regions such as India, Africa, and China in context of unbalanced diet, pellagra occurs sporadically also in developed countries, particularly in patients with alcohol abuse, malabsorption syndromes, HIV infection, specific medication (immunosuppressants and antituberculotics), carcinoid syndrome, Hartnup disease, food faddism and eating disorders such as anorexia nervosa, and in homeless people. 128 Pellagra dermatitis is a photosensitive eruption, typically bilateral, symmetrical, limited to sun‐exposed sites with a sharp line of demarcation from unexposed skin (dorsal hands and feet, face, neck/upper chest—collar‐like lesion on the neck is called “Casal's necklace“); reddish at the beginning and dry, dark, scaly and cracked later on (hyperpigmented, hyperkeratotic plaques, Fig. 3A);129 with vesicles or bullae in severe form termed wet pellagra. 130 Pellagra typically responds well to niacin or nicotinamide administered either orally or intravenously (300 mg/day divided into 3–4 doses, 3–4 weeks) and a diet enriched in niacin, as well as to supplementation of other group B vitamins (multivitamin deficiency is seen in majority of patients, especially alcohol abusers). 131
FIG 3.
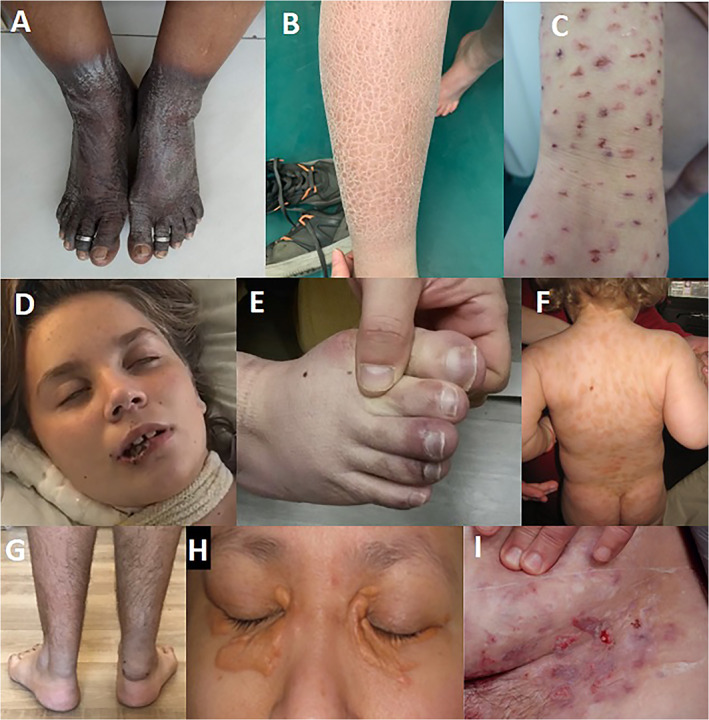
Skin changes in hyperkinetic movement disorders. (A) Classical pellagra dermatitis over sun‐exposed area of the lower limb with a clear cut‐off at the margins of clothing (adapted with permissions from Madhyastha et al. 2020), 129 (B) Ichthyosis, (C) self‐injurious behavior—artificial skin changes, (D) Self‐injurious behavior—lip biting in antiNMDAR encephalitis, (E) Chilblain lesions in Aicardi‐Gutieres syndrome (adapted with premissions from Videira et al. 2020) 149 (F) Mastocytosis, (G) Tendon xanthomas in Cerebrotendinous xanthomatosis (courtesy of Dr. Paldaufova, Dr. Serdahely, Dept. of Neurology, Skalica, Slovakia), (H) Periorbital xanthelasma in Erdheim‐Chester disease (adapted with permissions from Chasset et al. 2016), 154 (I) Abdominal skin ulcerations in Behçet disease (adapted with permissions from Scherrer et al. 2017). 155
Movement disorders related to ichthyosis
Ichthyosis is characterized by dry, thickened, scaly skin (Fig. 3B). While it may be associated with a number of genetic conditions, several present also with movement disorders or ataxia, especially ELOVL4 (SCA34), Refsum disease, type 2 Gaucher disease and GM3 Synthase deficiency. In addition to ichthyosis, patients with ELOVL4‐related ataxia (SCA34) present with adult‐onset ataxia and frequent erythrokeratoderma (40%). Also, 40–50% of SCA34 subjects may present with a hot cross bun sign on brain MRI mimicking MSA. 132 , 133 , 134 , 135 Refsum disease is associated with elevated plasma phytanic acid levels, late childhood‐onset (or later) retinitis pigmentosa, and variable combinations of ichthyosis, anosmia, polyneuropathy, deafness, ataxia, and potentially life‐threatening cardiac complications including arrythmias and heart failure caused by cardiomyopathy. Dietary restriction of phytanic acid intake helps resolve ichthyosis, sensory neuropathy, and ataxia. 136 Ichthyosis may be associated also with early‐onset forms of Gaucher disease, including the perinatal lethal form or type 2 Gaucher disease, which affects infants and typically leads to death before the age of 2–3 years. 137 , 138 Neurological features may include among others oculomotor apraxia, head retroflexion, striatal toes, myoclonus and hypertonus with or without hypokinesia. 139
Movement disorders related to self‐injurious behaviors
Some cutaneous or mucosal changes may appear as part of self‐injurious behavior (SIB), presenting also in several hyperkinetic movement disorders. By definition, SIB encompasses deliberate, repetitive and persistent behaviors directed towards the body (e.g. hitting, biting, scratching), leading to significant physical injury, in the absence of suicidal intent or sexual arousal (Fig. 3C, D). Management of SIB includes wearing protective restrains (e.g. helmets, bite splints), behavioral therapy, pharmacotherapy and surgical therapy (tooth extraction, deep brain stimulation). 140 , 141 The most common and representative hyperkinetic disorders with SIB are Tourette syndrome (TS), Lesch–Nyhan syndrome (LNS) and chorea‐acanthocytosis (CHAc). SIB in TS occurs in up to 60% of individuals and correlates with impulsivity and tic severity. 142 LNS is a rare X‐linked recessive neurometabolic disorder caused by severe inborn deficiency of hypoxanthine‐guanine phosphoribosyl transferase (HPRT) enzyme. Consequently, patients with LNS have increased uric acid in both serum and urine. Clinical symptoms include mental retardation, dystonia, choreoathetosis, spastic paresis, and finally, aggressive self‐mutilating behavior, typically in perioral region (especially lower lip). Skin and tissues of the fingers and hands are frequently seriously damaged. 143 CHAc, a progressive autosomal recessive disorder, typically presents in early adulthood with chorea, orolingual (tongue protrusion) dystonia, epilepsy, peripheral neuropathy, head drops, parkinsonism, tics, obsessive–compulsive behavior, and SIB (tongue‐, lip‐, cheek‐ or finger biting and others), where the latter are caused by psychiatric features rather than movement disorder. 144
SIB has been described also in other disorders with hyperkinetic movement disorders, like Rett syndrome, anti‐NMDAR encephalitis (Fig. 3D), spinocerebelar ataxia type 17, 141 , 145 6‐Pyruvoyl‐Tetrahydropterin Synthase deficiency, 146 or phenylketonuria which may present also with skin conditions including eczematous dermatitis, acrodermatitis dysmetabolica and movement disorders typically including stereotypies, tics and tremor. 147
Movement disorders related to other skin manifestations
The list of skin manifestations associated with hyperkinetic movement disorders is certainly broader than the list presented here. Many of them do not fit the above‐mentioned skin manifestation categories, for instance skin color changes in complex regional pain syndrome (often presenting with dystonia);148 chilblain lesions in Aicardi‐Goutières syndrome (Fig. 3E);149 cutaneous mastocytosis (Fig. 3F) in GNB1 encephalopathy;150 tendon xantomas in cerebrotendinous xantomatosis (Fig. 3G) typically associated also with ataxia, dystonia, diarrhea and cataracts;151 abnormal osmiophyllic pattern of skin vessels in WDR73‐associated disease labeled also as Cerebellar ataxia with Mental Retardation, Optic Atrophy and Skin Abnormalities (CAMOS) syndrome;152 periorbital xanthelasma and xanthoma‐like papules in Erdheim‐Chester disease which may present also with ataxia (Fig. 3H)153, 154 genital, oral and skin ulcers (Fig. 3I) in Behçet's disease presenting also with chorea, ataxia and myoclonus. 155 , 156 Leyll‐like bullous skin lesions may be present in potentially treatable NAXE and NAXD mutations, which can lead in addition to infantile fever‐related subacute encephalopathy, with hypotonia, ataxia and frequent respiratory failure. There are some reports of vitamin B3 stabilizing this disease. 157 , 158
Conclusions
Skin conditions are usually easily visible during routine clinical examination and their recognition may play a major role in diagnostic work‐up. Skin changes may precede onset of motor symptoms in parkinsonian disorders and presence of α‐syn in skin biopsies seems to be a promising biomarker for prodromal PD or differentiation of neurodegenerative parkinsonian syndromes. Literature on skin disorders in atypical parkinsonism is very limited. It is not clear whether skin manifestations typically associated with PD are disease‐specific or their presence extends also to other neurodegenerative parkinsonian disorders and further studies in this regard are warranted. In case of hyperkinetic movement disorders, specific dermatological manifestations, like presence of angiokeratomas, telangiectasias, Mongolian spots, lipomas, progeroid skin changes, etc. may point to a very specific group of disorders and help guide further investigations. While several of the non‐iatrogenic and iatrogenic therapy‐related skin conditions associated with movement disorders may be very bothersome and decrease patient quality of life, 22 their active screening and management as well as involvement of a dermatologist in movement disorders multidisciplinary teams may significantly improve diagnostic and therapeutic outcomes in movement disorder patients.
Author Roles
1. Research project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
K.K.: 1B, 1C, 3A, 3B.
J.B.: 1C, 3A, 3B.
J.N.: 1C, 3A, 3B.
M.S.: 1A, 1B, 1C, 3A, 3B.
Disclosures
Funding Sources and Conflicts of Interest
This work was supported by the Operational Programme Integrated Infrastructure, funded by the ERDF under No. ITMS2014+:313011 V455 for KK, JB and MS.
The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months
KK, JB and JN declare no financial disclosures. MS received speaker honoraria and consultation fees from Abbvie, Krka, Medtronic, Sanofi Genzyme, Stada and UCB.
Ethical Compliance Statement
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. All patients providing their photographs for the manuscript singed a written informed consent which meets the journal requirements, and an institutional review board approval was not required for this work.
References
- 1. Skorvanek M, Bhatia KP. The skin and Parkinson's Disease: Review of clinical, diagnostic, and therapeutic issues. Mov Disord Clin Pract 2016;4:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Niemann N, Billnitzer A, Jankovic J. Parkinson's disease and skin. Parkinsonism Relat Disord 2021;82:61–76. [DOI] [PubMed] [Google Scholar]
- 3. Berkovic SF, Andermann F, Shoubridge EA, et al. Mitochondrial dysfunction in multiple symmetrical lipomatosis. Ann Neurol 1991;29:566–569. [DOI] [PubMed] [Google Scholar]
- 4. Mohrenschlager M, Braun‐Falco M, Ring J, Abeck D. Fabry Disease: Recognition and management of cutaneous manifestations. Am J Clin Dermatol 2003;4:189–196. [DOI] [PubMed] [Google Scholar]
- 5. Gordon LB, Rothman FG, López‐Otín C, Misteli T. Progeria: A paradigm for translational medicine. Cell 2014;156:400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krestin D. The seborrheic facies as a manifestation of post‐encephalitic parkinsonism and allied disorders. Q J Med 1927;21:177–186. [Google Scholar]
- 7. Rietcheck HR, Maghfour J, Rundle CW, et al. A review of the current evidence connecting Seborrheic dermatitis and Parkinson's disease and the potential role of oral cannabinoids. Dermatology 2021;237:872–877. [DOI] [PubMed] [Google Scholar]
- 8. Tanner C, Albers K, Goldman S, Fross R, Leimpeter A, Klingman J, van den Eeden S. Seborrheic dermatitis and risk of future Parkinson's Disease. Neurology 2012;78:2012. [Google Scholar]
- 9. Rosso JQ. Adult seborrheic dermatitis. A status report on practical topical management. J Clin Aesthet Dermatol 2011;4:32–38. [PMC free article] [PubMed] [Google Scholar]
- 10. Sariahmetoglu H, Soysal A, Sen A, Yuksel B, Celiker S, Ciftci‐Kavaklioglu B, Arpaci B. Forehead sympathetic skin responses in determining autonomic involvement in Parkinson's disease. Clin Neurophysiol 2014;125:2436–2440. [DOI] [PubMed] [Google Scholar]
- 11. Martignoni E, Godi L, Pacchetti C, et al. Is seborrhea a sign of autonomic impairment in Parkinson's disease? J Neural Transm 1997;104:1295–1304. [DOI] [PubMed] [Google Scholar]
- 12. Pochi PE, Strauss JS, Mescon H. Sebum production and fractional 17‐ketosteroid excretion in parkinsonism. J Invest Dermatol 1962;38:45–51. [DOI] [PubMed] [Google Scholar]
- 13. Shuster S, Thody AJ, Goolamali SK, Burton JL, Plummer N, Bates D. Melanocyte‐stimulating hormone and parkinsonism. Lancet 1973;3:463–464. [DOI] [PubMed] [Google Scholar]
- 14. Ravn AH, Thyssen JP, Egeberg A. Skin disorders in Parkinson's disease: Potential biomarkers and risk factors. Clin Cosmet Investig Dermatol 2017;10:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan J, Berg M, Gallo RL, Del Rosso JQ. Applying the phenotype approach for rosacea to practice and research. Br J Dermatol 2018;179:741–746. [DOI] [PubMed] [Google Scholar]
- 16. Egeberg A, Hansen PR, Gislason GH, Thyssen JP. Exploring the association between rosacea and Parkinson Disease: A Danish Nationwide cohort study. JAMA Neurol 2016;73:529–534. [DOI] [PubMed] [Google Scholar]
- 17. Mathieu RJ, Guido N, Ibler E, et al. Rosacea and subsequent diagnosis for Parkinson's disease: A large, urban, single center, US patient population retrospective study. J Eur Acad Dermatol Venereol 2018;32:e141–e144. [DOI] [PubMed] [Google Scholar]
- 18. Jang YH, Sim JH, Kang HY, Kim YC, Lee ES. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non‐granulomatous rosacea. J Eur Acad Dermatol Venereol 2011;25:544–548. [DOI] [PubMed] [Google Scholar]
- 19. Chung YC, Kim YS, Bok E, Yune TY, Maeng S, Jin BK. MMP‐3 contributes to nigrostriatal dopaminergic neuronal loss, BBB damage, and neuroinflammation in an MPTP mouse model of Parkinson's disease. Mediators Inflamm 2013;2013:370526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karpouzis A, Avgeridis P, Tripsianis G, Gatzidou E, Kourmouli N, Veletza S. Assessment of Tachykinin receptor 3' gene polymorphism rs3733631 in rosacea. Int Sch Res Notices 2015;2015:469402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang H, Tang K, Wang Y, Fang R, Sun Q. Rosacea treatment: Review and update. Dermatol Ther (Heidelb) 2021;11:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swinn L, Schrag A, Viswanathan R, Bloem BR, Lees A, Quinn N. Sweating dysfunction in Parkinson's disease. Mov Disord 2003;18:1459–1463. [DOI] [PubMed] [Google Scholar]
- 23. Försti AK, Huilaja L, Schmidt E, Tasanen K. Neurological and psychiatric associations in bullous pemphigoid‐more than skin deep? Exp Dermatol 2017;26:1228–1234. [DOI] [PubMed] [Google Scholar]
- 24. Di Lernia V, Casanova DM, Goldust M, Ricci C. Pemphigus vulgaris and bullous Pemphigoid: Update on diagnosis and treatment. Dermatol Pract Concept 2020;10:e2020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bose A, Petsko GA, Eliezer D. Parkinson's Disease and melanoma: Co‐occurrence and mechanisms. J Parkinsons Dis 2018;8:385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olsen JH, Tangerud K, Wermuth L, Frederiksen K, Friis S. Treatment with levodopa and risk for malignant melanoma. Mov Disord 2007;22:1252–1257. [DOI] [PubMed] [Google Scholar]
- 27. Bertoni JM, Arlette JP, Fernandez HH, et al. Increased melanoma risk in Parkinson disease: A prospective clinicopathological study. Arch Neurol 2010;67:347–352. [DOI] [PubMed] [Google Scholar]
- 28. Liu R, Gao X, Lu Y, Chen H. Meta‐analysis of the relationship between Parkinson disease and melanoma. Neurology 2011;76:2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang P, Yang XD, Chen SD, Xiao Q. The association between Parkinson's disease and melanoma: A systematic review and meta‐analysis. Transl Neurodegener 2015;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. American Academy of Dermatology Ad Hoc Task Force for the ABCDEs of Melanoma , Tsao H, Olazagasti JM, et al. Early detection of melanoma: Reviewing the ABCDEs. J Am Acad Dermatol 2015;72:717–723. [DOI] [PubMed] [Google Scholar]
- 31. Garbe C, Amaral T, Peris K, et al. European consensus‐based interdisciplinary guideline for melanoma. Part 1: Diagnostic—update 2019. Eur J Cancer 2020;126:141–158. [DOI] [PubMed] [Google Scholar]
- 32. Pan T, Li X, Jankovic J. The association between Parkinson's disease and melanoma. In J Cancer 2011;128:2251–2260. [DOI] [PubMed] [Google Scholar]
- 33. Jitkritsadakul O, Bhidayasiri R, Kalia SK, Hodaie M, Lozano AM, Fasano A. Systematic review of hardware‐related complications of deep brain stimulation: Do new indications pose an increased risk? Brain Stimul 2017;10:967–976. [DOI] [PubMed] [Google Scholar]
- 34. Fanciulli A, Stankovic I, Krismer F, et al. Multiple system atrophy. Int Rev Neurobiol 2019;149:137–192. [DOI] [PubMed] [Google Scholar]
- 35. Doppler K, Weis J, Karl K, et al. Distinctive distribution of phospho‐alpha‐synuclein in dermal nerves in multiple system atrophy. Mov Disord 2015;30:1688–16892. [DOI] [PubMed] [Google Scholar]
- 36. Iodice V, Lipp A, Ahlskog JE, et al. Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests. J Neurol Neurosurg Psychiatry 2012;83:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Donadio V, Nolano M, Elam M, et al. Anhidrosis in multiple system atrophy: A preganglionic sudomotor dysfunction? Mov Disord 2008;23:885–888. [DOI] [PubMed] [Google Scholar]
- 38. Asahina M, Akaogi Y, Yamanaka Y, Koyama Y, Hattori T. Differences in skin sympathetic involvements between two chronic autonomic disorders: Multiple system atrophy and pure autonomic failure. Parkinsonism Relat Disord 2009;15:347–350. [DOI] [PubMed] [Google Scholar]
- 39. Bordet R, Benhadjali J, Destee A, Hurtevent JF, Bourriez JL, Guieu JD. Sympathetic skin response and R‐R interval variability in multiple system atrophy and idiopathic Parkinson's disease. Mov Disord 1996;11:268–272. [DOI] [PubMed] [Google Scholar]
- 40. De Marinis M, Stocchi F, Gregori B, Accornero N. Sympathetic skin response and cardiovascular autonomic function tests in Parkinson's disease and multiple system atrophy with autonomic failure. Mov Disord 2000;15:1215–1220. [DOI] [PubMed] [Google Scholar]
- 41. Pavy‐LeTraon A, Brefel‐Courbon C, Dupouy J, Ory‐Magne F, Rascol O, Senard JM. Combined cardiovascular and sweating autonomic testing to differentiate multiple system atrophy from Parkinson's disease. Neurophysiol Clin 2018;48:103–110. [DOI] [PubMed] [Google Scholar]
- 42. Pietzarka K, Reimann M, Schmidt C, et al. The cold hand sign in multiple system atrophy: Skin perfusion revisited. J Neural Transm (Vienna) 2010;117:475–479. [DOI] [PubMed] [Google Scholar]
- 43. Asahina M, Low DA, Mathias CJ, et al. Skin temperature of the hand in multiple system atrophy and Parkinson's disease. Parkinsonism Relat Disord 2013;19:560–562. [DOI] [PubMed] [Google Scholar]
- 44. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB consortium. Neurology 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Akaogi Y, Asahina M, Yamanaka Y, Koyama Y, Hattori T. Sudomotor, skin vasomotor, and cardiovascular reflexes in 3 clinical forms of Lewy body disease. Neurology 2009;73:59–65. [DOI] [PubMed] [Google Scholar]
- 46. Toru S, Kanouchi T, Yokota T, Yagi Y, Machida A, Kobayashi T. Utility of autonomic function tests to differentiate dementia with Lewy bodies and Parkinson Disease with dementia from Alzheimer Disease. Eur Neurol 2018;79:27–32. [DOI] [PubMed] [Google Scholar]
- 47. Negami M, Maruta T, Takeda C, Adachi Y, Yoshikawa H. Sympathetic skin response and heart rate variability as diagnostic tools for the differential diagnosis of Lewy body dementia and Alzheimer's disease: A diagnostic test study. BMJ Open 2013;3:e001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Giagkou N, Höglinger GU, Stamelou M. Progressive supranuclear palsy. Int Rev Neurobiol 2019;149:49–86. [DOI] [PubMed] [Google Scholar]
- 49. Kikkawa Y, Asahina M, Suzuki A, Hattori T. Cutaneous sympathetic function and cardiovascular function in patients with progressive supranuclear palsy and Parkinson's disease. Parkinsonism Relat Disord 2003;10:101–106. [DOI] [PubMed] [Google Scholar]
- 50. Nojszewska M, Potulska‐Chromik A, Jamrozik Z, Janik P, Zakrzewska‐Pniewska B. Electrophysiological and clinical assessment of dysautonomia in multiple system atrophy (MSA) and progressive supranuclear palsy (PSP): A comparative study. Neurol Neurochir Pol 2019;53:26–33. [DOI] [PubMed] [Google Scholar]
- 51. Donadio V. Skin nerve α‐synuclein deposits in Parkinson's disease and other synucleinopathies: A review. Clin Auton Res 2019;29:577–585. [DOI] [PubMed] [Google Scholar]
- 52. Gibbons CH, Garcia J, Wang N, Shih LC, Freeman R. The diagnostic discrimination of cutaneous α‐synuclein deposition in Parkinson disease. Neurology 2016;87:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nolano M, Provitera V, Estraneo A, et al. Sensory deficit in Parkinson's disease: Evidence of a cutaneous denervation. Brain 2008;131:1903–1911. [DOI] [PubMed] [Google Scholar]
- 54. Wang N, Gibbons CH, Lafo J, Freeman R. α‐Synuclein in cutaneous autonomic nerves. Neurology 2013;81:1604–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Navarro‐Otano J, Casanova‐Mollà J, Morales M, Valls‐Solé J, Tolosa E. Cutaneous autonomic denervation in Parkinson's disease. J Neural Transm (Vienna) 2015;122:1149–1155. [DOI] [PubMed] [Google Scholar]
- 56. Schrempf W, Katona I, Dogan I, et al. Reduced intraepidermal nerve fiber density in patients with REM sleep behavior disorder. Parkinsonism Relat Disord 2016;29:10–16. [DOI] [PubMed] [Google Scholar]
- 57. Doppler K, Jentschke HM, Schulmeyer L, et al. Dermal phospho‐alpha‐synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson's disease. Acta Neuropathol 2017;133:535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Donadio V, Incensi A, Leta V, et al. Skin nerve α‐synuclein deposits: A biomarker for idiopathic Parkinson disease. Neurology 2014;82:1362–1369. [DOI] [PubMed] [Google Scholar]
- 59. Isonaka R, Goldstein DS, Zhu W, et al. α‐Synuclein deposition in sympathetic nerve fibers in genetic forms of Parkinson's Disease. Mov Disord 2021;36:2346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Donadio V, Incensi A, El‐Agnaf O, et al. Skin α‐synuclein deposits differ in clinical variants of synucleinopathy: An in vivo study. Sci Rep 2018;8:14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Donadio V, Incensi A, Rizzo G, et al. A new potential biomarker for dementia with Lewy bodies: Skin nerve α‐synuclein deposits. Neurology 2017;89:318–326. [DOI] [PubMed] [Google Scholar]
- 62. Donadio V, Incensi A, Cortelli P, Giannoccaro MP, Jaber MA, Baruzzi A, Liguori R. Skin sympathetic fiber α‐synuclein deposits: A potential biomarker for pure autonomic failure. Neurology 2013;80:725–732. [DOI] [PubMed] [Google Scholar]
- 63. Ikemura M, Saito Y, Sengoku R, et al. Lewy body pathology involves cutaneous nerves. J Neuropathol Exp Neurol 2008;67:945–953. [DOI] [PubMed] [Google Scholar]
- 64. Giannoccaro MP, Avoni P, Rizzo G, Incensi A, Infante R, Donadio V, Liguori R. Presence of skin α‐Synuclein deposits discriminates Parkinson's Disease from progressive Supranuclear palsy and Corticobasal syndrome. J Parkinsons Dis 2021;12:585–591. 10.3233/JPD-212904. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Z, Becker K, Donadio V, et al. Skin α‐Synuclein aggregation seeding activity as a novel biomarker for Parkinson Disease. JAMA Neurol 2020;78:1–11. Erratum in: JAMA Neurol. 2021; 78: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dugger BN, Hoffman BR, Scroggins A, et al. Tau immunoreactivity in peripheral tissues of human aging and select tauopathies. Neurosci Lett 2019;696:132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rodríguez‐Leyva I, Chi‐Ahumada EG, Carrizales J, et al. Parkinson disease and progressive supranuclear palsy: Protein expression in skin. Ann Clin Transl Neurol 2016;3:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kamenisch Y, Berneburg M. Progeroid syndromes and UV‐induced oxidative DNA damage. J Investig Dermatol Symp Proc 2009;14:8–14. [DOI] [PubMed] [Google Scholar]
- 69. Ahmad A, Enzlin JH, Bhagwat NR, et al. Mislocalization of XPF‐ERCC1 nuclease contributes to reduced DNA repair in XP‐F patients. PLoS Genet 2010;6:e1000871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mori T, Yousefzadeh MJ, Faridounnia M, et al. ERCC4 variants identified in a cohort of patients with segmental progeroid syndromes. Hum Mutat 2018;39:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Piccione M, Belloni Fortina A, Ferri G, et al. Xeroderma Pigmentosum: General aspects and management. J Pers Med 2021;11:1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kraemer KH, Lee M, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer‐the Xeroderma Pigmentosum paradigm. Arch Dermatol 1994;130:1018–1021. [PubMed] [Google Scholar]
- 73. Krasikova Y, Rechkunova N, Lavrik O. Nucleotide excision repair: From molecular defects to neurological abnormalities. Int J Mol Sci 2021;22:6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Garcia‐Moreno H, Fassihi H, Sarkany RPE, Phukan J, Warner T, Lehmann AR, Giunti P. Xeroderma pigmentosum is a definite cause of Huntington's disease‐like syndrome. Ann Clin Transl Neurol 2017;5:102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Levy A, Lang AE. Ataxia‐telangiectasia: A review of movement disorders, clinical features, and genotype correlations. Mov Disord 2018;33:1238–1247. [DOI] [PubMed] [Google Scholar]
- 76. Greenberger S, Berkun Y, Ben‐Zeev B, Levi YB, Barziliai A, Nissenkorn A. Dermatologic manifestations of ataxia‐telangiectasia syndrome. J Am Acad Dermatol 2013;68:932–936. [DOI] [PubMed] [Google Scholar]
- 77. Ganguly J, Bernaola MT, Goobie S, Prasad A, Jog M. Myoclonus‐dystonia presentation of ATM gene mutation in a Canadian Mennonite. Mov Disord Clin Pract 2021;9:264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Necpál J, Zech M, Škorvánek M, Havránková P, Fečíková A, Winkelmann J, Jech R. Ataxia telangiectasia gene mutation in isolated segmental dystonia without ataxia and telangiectasia. Mov Disord Clin Pract 2017;5:89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Szczawińska‐Popłonyk A, Olejniczak K, Tąpolska‐Jóźwiak K, et al. Cutaneous and systemic granulomatosis in ataxia‐telangiectasia: A clinico‐pathological study. Postepy Dermatol Alergol 2020;37:760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Oska S, Zarbo A, Yeager D, Friedman BJ, Shwayder T. Melanoma arising in a patient with ataxia‐telangiectasia: A call for full skin examinations in this patient population. Pediatr Dermatol 2020;37:767–768. [DOI] [PubMed] [Google Scholar]
- 81. Majethia P, Girisha KM. Wiedemann‐Rautenstrauch syndrome in an Indian patient with biallelic pathogenic variants in POLR3A. Am J Med Genet A 2021;185:1602–1605. [DOI] [PubMed] [Google Scholar]
- 82. Di Donato I, Gallo A, Ricca I, et al. POLR3A variants in hereditary spastic paraparesis and ataxia: Clinical, genetic, and neuroradiological findings in a cohort of Italian patients. Neurol Sci 2021;23. [Epub ahead of print]:1071–1077. 10.1007/s10072-021-05462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hashmi MS, V G. Hartnup Disease. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021. [Google Scholar]
- 84. Darras BT, Ampola MG, Dietz WH, Gilmore HE. Intermittent dystonia in Hartnup disease. Pediatr Neurol 1989;5:118–120. [DOI] [PubMed] [Google Scholar]
- 85. Wang X, Li XY, Piao Y, et al. Hartnup disease presenting as hereditary spastic paraplegia and severe peripheral neuropathy. Am J Med Genet A 2022;188:237–242. [DOI] [PubMed] [Google Scholar]
- 86. Migeon BR, Axelman J, de Beur SJ, et al. Selection against lethal alleles in females heterozygous for incontinentia pigmenti. Am J Hum Genet 1989;44:100–106. [PMC free article] [PubMed] [Google Scholar]
- 87. Bodemer C. Incontinentia pigmenti and hypomelanosis of Ito. Handb Clin Neurol 2013;111:341–347. [DOI] [PubMed] [Google Scholar]
- 88. Hadj‐Rabia S, Froidevaux D, Bodack N, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol 2003;139:1163–1170. [DOI] [PubMed] [Google Scholar]
- 89. Pini G, Faulkner LB. Cerebellar involvement in hypomelanosis of Ito. Neuropediatrics 1995;26:208–210. [DOI] [PubMed] [Google Scholar]
- 90. Introne WJ, Westbroek W, Groden CA, et al. Neurologic involvement in patients with atypical Chediak‐Higashi disease. Neurology 2017;88:e57–e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pingault V, Zerad L, Bertani‐Torres W, Bondurand N. SOX10: 20 years of phenotypic plurality and current understanding of its developmental function. J Med Genet 2022;59:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, Tomita Y. Mutations of the RNA‐specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003;73:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Di Lazzaro G, Graziola F, Sancesario A, et al. Movement disorders in ADAR1 disease: Insights from a comprehensive cohort. Parkinsonism Relat Disord 2020;79:100–104. [DOI] [PubMed] [Google Scholar]
- 94. Regier DS, Tifft CJ, Rothermel CE. GLB1‐Related Disorders. 2013 Oct 17 [Updated 2021 Apr 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993. ‐2021. [PubMed] [Google Scholar]
- 95. Dweikat I, Libdeh BA, Murrar H, Khalil S, Maraqa N. GM1 gangliosidosis associated with neonatal‐onset of diffuse ecchymoses and mongolian spots. Indian J Dermatol 2011;56:98–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gordon‐Lipkin E, Cohen JS, Srivastava S, Soares BP, Levey E, Fatemi A. ST3GAL5‐related disorders: A deficiency in Ganglioside metabolism and a genetic cause of intellectual disability and Choreoathetosis. J Child Neurol 2018;33:825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cuestas D, Perafan A, Forero Y, et al. Angiokeratomas, not everything is Fabry disease. Int J Dermatol 2019;58:713–721. [DOI] [PubMed] [Google Scholar]
- 98. Wali G, Wali GM, Sue CM, Kumar KR. A novel homozygous mutation in the FUCA1 gene highlighting fucosidosis as a cause of dystonia: Case report and literature review. Neuropediatrics 2019;50:248–252. [DOI] [PubMed] [Google Scholar]
- 99. Stepien KM, Ciara E, Jezela‐Stanek A. Fucosidosis—Clinical manifestation, long‐term outcomes, and genetic profile—Review and case series. Genes 2020;11:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. D'Azzo A, Hoogeveen A, Reuser AJ, Robinson D, Galjaard H. Molecular defect in combined beta‐galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci U S A 1982;79:4535–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nakajima H, Ueno M, Adachi K, et al. A new heterozygous compound mutation in the CTSA gene in galactosialidosis. Hum Genome Va 2019;6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wise AH, Yang A, Naik H, et al. Parkinson's disease prevalence in Fabry disease: A survey study. Mol Genet Metab Rep 2017;14:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Madelung OW. Ueber den fetthals (diffuses lipom des halses). Arch Klin Chir 1888;37:106–130. [Google Scholar]
- 104. Musumeci O, Barca E, Lamperti C, et al. Lipomatosis incidence and characteristics in an Italian cohort of mitochondrial patients. Front Neurol 2019;10:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yousef H, Miao JH, Badri AM. Histology, Skin Appendages. [Internet]. Treasure Island (FL): StatPearls Publishing; 2021. [PubMed] [Google Scholar]
- 106. Woodhouse NJ, Sakati NA. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet 1983;20:216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Agopiantz M, Corbonnois P, Sorlin A, et al. Endocrine disorders in Woodhouse‐Sakati syndrome: A systematic review of the literature. J Endocrinol Invest 2014;37:1–7. [DOI] [PubMed] [Google Scholar]
- 108. Yang Y, Yang JY, Chen XJ. Biotinidase deficiency characterized by skin and hair findings. Clin Dermatol 2020;38:477–483. [DOI] [PubMed] [Google Scholar]
- 109. Solís‐García Del Pozo J, de Cabo C, Solera J. Treatment of Satoyoshi syndrome: A systematic review. Orphanet J Rare Dis 2019;14:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Raymond GV, Moser AB, Fatemi A. X‐Linked Adrenoleukodystrophy. 1999 [Updated 2018]. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993. –2022. [PubMed] [Google Scholar]
- 111. Perrone E, Perez ABA, D'Almeida V, et al. Clinical and molecular evaluation of 13 Brazilian patients with Gomez‐López‐Hernández syndrome. Am J Med Genet A 2021;185:1047–1058. [DOI] [PubMed] [Google Scholar]
- 112. Necpál J, Zech M, Valachová A, et al. Severe paroxysmal dyskinesias without epilepsy in a RHOBTB2 mutation carrier. Parkinsonism Relat Disord 2020;77:87–88. [DOI] [PubMed] [Google Scholar]
- 113. Bonomi M, Rochira V, Pasquali D, et al. Klinefelter syndrome (KS): Genetics, clinical phenotype and hypogonadism. J Endocrinol Invest 2017;40:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fabbri M, Zibetti M, Martone T, Lopiano L. Expanding the spectrum of movement disorders in Klinefelter syndrome. Neurol Sci 2018;39:1303–1304. [DOI] [PubMed] [Google Scholar]
- 115. Echaniz‐Laguna A, Cuisset JM, Guyant‐Marechal L, et al. Giant axonal neuropathy: A multicenter retrospective study with genotypic spectrum expansion. Neurogenetics 2020;21:29–37. [DOI] [PubMed] [Google Scholar]
- 116. Rattay TW, Lindig T, Baets J, et al. FAHN/SPG35: A narrow phenotypic spectrum across disease classifications. Brain 2019;142:1561–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ostergaard E, Bradinova I, Ravn SH, et al. Hypertrichosis in patients with SURF1 mutations. Am J Med Genet A 2005;138:384–388. [DOI] [PubMed] [Google Scholar]
- 118. Ebrahimi‐Fakhari D, Hildebrandt C, Davis PE, Rodan LH, Anselm I, Bodamer O. The Spectrum of movement disorders in childhood‐onset Lysosomal storage diseases. Mov Disord Clin Pract 2018;5:149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bech S, Løkkegaard A, Nielsen TT, et al. Paroxysmal cranial dyskinesia and nail‐patella syndrome caused by a novel variant in the LMX1B gene. Mov Disord 2020;35:2343–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tani LY, Veasy LG, Minich LL, Shaddy RE. Rheumatic fever in children younger than 5 years: Is the presentation different? Pediatrics 2003;112:1065–1068. [DOI] [PubMed] [Google Scholar]
- 121. Heard MA, Green MC, Royer M. Acute rheumatic fever: A review of essential cutaneous and histological findings. Cureus 2021;13:e12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Baizabal‐Carvallo JF, Bonnet C, Jankovic J. Movement disorders in systemic lupus erythematosus and the antiphospholipid syndrome. J Neural Transm (Vienna) 2013;120:1579–1589. [DOI] [PubMed] [Google Scholar]
- 123. Uva L, Miguel D, Pinheiro C, Freitas JP, Marques Gomes M, Filipe P. Cutaneous manifestations of systemic lupus erythematosus. Autoimmune Dis 2012;2012:834291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997;36:970–982. [DOI] [PubMed] [Google Scholar]
- 125. Helsing P, Frøen H. Dermatitis herpetiformis presenting as ataxia in a child. Acta Derm Venereol 2007;87:163–165. [DOI] [PubMed] [Google Scholar]
- 126. Cavanna AE, Nani A, Williams AC. Parkinsonian features in a case of pellagra: A historical report. J Parkinsons Dis 2013;3:539–545. [DOI] [PubMed] [Google Scholar]
- 127. Park K, Oeda T, Sawada H. A case of alcoholic pellagra encephalopathy presenting with spinal myoclonus. Neurol Clin Pract 2015;5:472–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Delgado‐Sanchez L, Godkar D, Niranjan S. Pellagra: rekindling of an old flame. Am J Ther 2008;15:173–175. [DOI] [PubMed] [Google Scholar]
- 129. Madhyastha SP, Shetty GV, Shetty VM, Reddy CT. The classic pellagra dermatitis. BMJ Case Rep 2020;13:e239741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wong CY, Chu DH. Cutaneous signs of nutritional disorders. Int J Womens Dermatol 2021;7:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hołubiec P, Leończyk M, Staszewski F, Łazarczyk A, Jaworek AK, Rojas‐Pelc A. Pathophysiology and clinical management of pellagra–A review. Folia Med Cracov 2021;61:125–137. [DOI] [PubMed] [Google Scholar]
- 132. Cadieux‐Dion M, Turcotte‐Gauthier M, Noreau A, et al. Expanding the clinical phenotype associated with ELOVL4 mutation: Study of a large French‐Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 2014;71:470–475. [DOI] [PubMed] [Google Scholar]
- 133. Ozaki K, Doi H, Mitsui J, et al. A novel mutation in ELOVL4 leading to Spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking Erythrokeratodermia: A broadened Spectrum of SCA34. JAMA Neurol 2015;72:797–805. [DOI] [PubMed] [Google Scholar]
- 134. Haeri G, Hajiakhoundi F, Alavi A, Ghiasi M, Munhoz RP, Rohani M. Congenital Ichthyosis in a case of Spinocerebellar ataxia type 34: A novel presentation for a known mutation. Mov Disord Clin Pract 2021;8:275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bourque PR, Warman‐Chardon J, Lelli DA, et al. Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34). Neurol Genet 2018;4:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Waterham HR, Wanders RJA, Leroy BP. Adult Refsum Disease. 2006 [Updated 2021]. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993. –2022. [PubMed] [Google Scholar]
- 137. Mignot C. Aspects cliniques des forms neurologiques précoces de la maladie de Gaucher [Clinical aspects of early‐stage neurological forms of Gaucher disease]. Rev Med Interne 2006;27(Suppl 1):S14–S147. [DOI] [PubMed] [Google Scholar]
- 138. Haverkaemper S, Marquardt T, Hausser I, Timme K, Kuehn T, Hertzberg C, Rossi R. Congenital ichthyosis in severe type II Gaucher disease with a homozygous null mutation. Neonatology 2011;100:194–197. [DOI] [PubMed] [Google Scholar]
- 139. Chetrit EB, Alcalay RN, Steiner‐Birmanns B, Altarescu G, Phillips M, Elstein D, Zimran A. Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells Mol Dis 2013;50:218–221. [DOI] [PubMed] [Google Scholar]
- 140. Huisman S, Mulder P, Kuijk J, et al. Self‐injurious behavior. Neurosci Biobehav Rev 2018;84:483–491. [DOI] [PubMed] [Google Scholar]
- 141. Fischer JF, Mainka T, Worbe Y, Pringsheim T, Bhatia K, Ganos C. Self‐injurious behaviour in movement disorders: Systematic review. J Neurol Neurosurg Psychiatry 2020;91:712–719. [DOI] [PubMed] [Google Scholar]
- 142. Mathews CA, Waller J, Glidden D, et al. Self injurious behaviour in Tourette syndrome: Correlates with impulsivity and impulse control. J Neurol Neurosurg Psychiatry 2004;75:1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Jathar P, Panse AM, Jathar M, Gawali PN. Lesch‐Nyhan syndrome: Disorder of self‐mutilating behavior. Int J Clin Pediatr Dent 2016;9:139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Walker RH. Untangling the thorns: Advances in the Neuroacanthocytosis syndromes. J Mov Disord 2015;8:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bonomo R, Latorre A, Bhatia KP. Self‐injurious behaviour in SCA17: A new clinical observation. Tremor Other Hyperkinet Mov (N Y) 2019;9:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Latorre A, Salgado P, Salari M, Jesuthasan A, Bhatia KP. Combined dystonia with self‐mutilation in 6‐Pyruvoyl‐Tetrahydropterin synthase (PTPS) deficiency: A case report. Mov Disord Clin Pract 2018;6:81–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Mainka T, Fischer JF, Huebl J, et al. The neurological and neuropsychiatric spectrum of adults with late‐treated phenylketonuria. Parkinsonism Relat Disord 2021;89:167–175. [DOI] [PubMed] [Google Scholar]
- 148. Taylor SS, Noor N, Urits I, et al. Complex regional pain syndrome: A comprehensive review. Pain Ther 2021;10:875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Videira G, Malaquias MJ, Laranjinha I, Martins R, Taipa R, Magalhães M. Diagnosis of Aicardi‐Goutières syndrome in adults: A case series. Mov Disord Clin Pract 2020;7:303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Szczałuba K, Biernacka A, Szymańska K, Gasperowicz P, Kosińska J, Rydzanicz M, Płoski R. Novel GNB1 de novo mutation in a patient with neurodevelopmental disorder and cutaneous mastocytosis: Clinical report and literature review. Eur J Med Genet 2018;61:157–160. [DOI] [PubMed] [Google Scholar]
- 151. Stelten BML, van de Warrenburg BPC, Wevers RA, Verrips A. Movement disorders in cerebrotendinous xanthomatosis. Parkinsonism Relat Disord 2019;58:12–16. [DOI] [PubMed] [Google Scholar]
- 152. Vodopiutz J, Seidl R, Prayer D, et al. WDR73 mutations cause infantile Neurodegeneration and variable glomerular kidney Disease. Hum Mutat 2015;36:1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Cohen Aubart F, Idbaih A, Galanaud D, et al. Central nervous system involvement in Erdheim‐Chester disease: An observational cohort study. Neurology 2020;95:e2746–e2754. [DOI] [PubMed] [Google Scholar]
- 154. Chasset F, Barete S, Charlotte F, et al. Cutaneous manifestations of Erdheim‐Chester disease (ECD): Clinical, pathological, and molecular features in a monocentric series of 40 patients. J Am Acad Dermatol 2016;74:513–520. [DOI] [PubMed] [Google Scholar]
- 155. Scherrer MAR, Rocha VB, Garcia LC. Behçet' disease: Review with emphasis on dermatological aspects. An Bras Dermatol 2017;92:452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Menozzi E, Mulroy E, Akbarian‐Tefaghi L, Bhatia KP, Balint B. Movement disorders in systemic autoimmune diseases: Clinical spectrum, ancillary investigations, pathophysiological considerations. Parkinsonism Relat Disord 2021;88:116–128. [DOI] [PubMed] [Google Scholar]
- 157. Zhou J, Li J, Stenton SL, et al. NAD(P)HX dehydratase (NAXD) deficiency: A novel neurodegenerative disorder exacerbated by febrile illnesses. Brain 2020. Erratum in: Brain 2020 143:e15;143:e8. [DOI] [PubMed] [Google Scholar]
- 158. Trinh J, Imhoff S, et al. NAXE variants as a cause for neurometabolic disorder: Implications for treatment. J Neurol 2020;267:770–782. [DOI] [PubMed] [Google Scholar]