

Abstract
Although it is widely accepted that protein function is largely dependent on its structure, intrinsically disordered proteins (IDPs) lack defined structure but are essential in proper cellular processes. Mammalian high mobility group proteins (HMGA) are one such example of IDPs that perform a number of crucial nuclear activities and have been highly studied due to their involvement in the proliferation of a variety of disease and cancers. Traditional structural characterization methods have had limited success in understanding HMGA proteins and their ability to coordinate to DNA. Ion mobility spectrometry and mass spectrometry provide insights into the diversity and heterogeneity of structures adopted by IDPs and are employed here to interrogate HMGA2 in its unbound states and bound to two DNA hairpins. The broad distribution of collision cross sections observed for the apo-protein are restricted when HMGA2 is bound to DNA, suggesting that increased protein organization is promoted in the holo-form. Ultraviolet photodissociation was utilized to probe the changes in structures for the compact and elongated structures of HMGA2 by analyzing backbone cleavage propensities and solvent accessibility based on charge-site analysis, which revealed a spectrum of conformational possibilities. Namely, preferential binding of the DNA hairpins with the second of three AT-hooks of HMGA2 is suggested based on the suppression of backbone fragmentation and distribution of DNA-containing protein fragments.
Graphical Abstract
INTRODUCTION
A central theme in the understanding of a protein’s function has historically been focused on understanding its structure. A class of proteins exists, however, in which a protein or a region therein lacks any higher order structural characteristics. These intrinsically disordered proteins (IDPs) or protein regions (IDPRs) are nevertheless crucial for proper function in their respective biological pathways, which can be as varied as the structures the IDPs adopt.1,2 The mammalian high mobility group proteins (HMGA) are one such family of IDPs that have been implicated in many disease states, both benign and malignant.3–5 HMGA2 is a small (11.6 kDa) protein that contains three highly basic DNA-binding regions and an acidic C-terminus. Each DNA-binding region, or AT-hook, possesses a conserved Arg-Gly-Arg-Pro motif surrounded by additional basic residues. These AT-hooks can bind to the minor groove of AT-rich DNA5 (or major groove if the minor groove is otherwise occupied6) with possible multivalent binding of one protein to multiple AT tracts,7,8 contributing to its functions involving DNA replication and repair as well as gene transcription and regulation.3,4 The absence of HMGA2 has been linked to dwarfism,9 and overexpression has been attributed to lung cancer,10,11 prostate cancer,12,13 breast cancer,14 and obesity.15 The important functions and specificity of HMGA proteins make them key biomarkers and drug targets, yet their intrinsically disordered regions have impeded their biophysical characterization. Interestingly, although it is considered an IDP, a truncated form of HMGA has been crystallized and observed to adopt an ordered structure when bound to DNA.16,17 The adaptability and flexibility of the protein is thought to contribute to the many roles it plays in the nucleus.4
X-ray crystallography is traditionally employed to elucidate protein structure, but owing to ineffective X-ray scattering by IDPs or simply their resistance to crystallization, other methods are employed to uncover their structures.18 For example, the Stoke’s radius can be determined from small-angle X-ray scattering or size-exclusion chromatography to provide a size estimation of the IDPs, while circular dichroism spectroscopy offers insight into the secondary structure composition.18 Limited proteolysis followed by chromatographic separation of the constituent peptides and mass-spectrometric analysis (i.e., bottom-up mass spectrometry) has also been used to determine the relative flexibility and unfolding state of protein regions.18 NMR has likewise contributed greatly to the understanding of IDPs and IDPRs, particularly for realizing dynamics interactions with their biological partners.1,2,18 Results from these analyses provide constraints for various models and molecular dynamics simulations to assist in visualization in IDP and IDPR structures.1,2
Several more specialized mass spectrometric methods have proved valuable in determining the conformational diversity of IDPs and IDPRs. The exchange of amide hydrogens in the protein backbone in deuterated water, for which rates of exchange correspond with the solvent accessibility and rigidity of the protein, can be determined by hydrogen−deuterium exchange (HDX) mass spectrometry.19 HDX and other MS-based surface labeling methods have been helpful in modeling the structural changes that proteins undergo upon binding ligands, including DNA, lipids, and other proteins.20–22 However, all of the analyses mentioned thus far, including spectroscopy and spectrometry, do not differentiate between the multitude of structures adopted by disordered proteins and instead report on the overall features of the many compact and extended states that undoubtably exist. To that end, ion mobility spectrometry (IMS) can be employed to separate protein ions based on their size and charge.23
Intact proteins can be analyzed via mass spectrometry in native-like states using nanoelectrospray ionization (nESI). Typical native MS experiments result in a narrow conformational distribution of compact (folded) proteins that carry relatively few charges. The nESI mass spectra of IDPs, however, frequently exhibit bimodal charge-state distributions characteristic of a mixed population of more extended, unfolded conformers that accept more charges during the nESI process (high charge states) in addition to a more compact population in low charge states.24 IMS together with native MS can be used to determine the collision cross section (CCS) of each charge state to determine the size and shape of the full array of gas-phase conformations of proteins that are indicative of folding in solution.25,26 The ability of IMS to separate conformations of a protein allows interrogation of individual structures and can contribute to existing solution studies described above that work toward understanding the broad structural landscapes of IDPs that are inadequately described by a single conformation.
Tandem mass spectrometry (MS/MS) can be employed to further probe the structure of protein conformers transferred to the gas phase from native-like solution conditions. Some MS/MS methods have been successfully implemented to interrogate the higher order structures of gas-phase proteins, namely electron transfer (ETD) and electron capture dissociation (ECD) as well as ultraviolet photodissociation (UVPD). ETD and ECD induce covalent cleavage at surface-exposed regions of proteins providing insight into overall fold.27,28 UVPD induces greater covalent cleavages and is sensitive to the noncovalent interactions that dictate protein conformations.29,30 Moreover, changes in the backbone cleavage efficiencies of apo- (unbound) and holo- (ligand-bound) states of a protein upon UVPD have been used to localize active sites and other regions of the protein that are perturbed due to structural rearrangement upon ligand binding.31,32
Here, native MS methods including IMS-MS and UVPD-MS are applied to investigate the various structures of the protein HMGA AT-hook 2 (HMGA2) alone and in complex with a 22-nucleotide (DNA22) or 50-nucleotide (DNA50) AT-rich DNA hairpin. A modular, ambient drift tube provides information on the broad conformational landscape that HMGA2 adopts. CCS values calculated from transient decay profiles in an Orbitrap mass analyzer suggest that the solution structures are maintained in the gas phase, allowing structural characterization using UVPD. While HMGA2 may bind to the AT-rich hairpins in any of its three AT-hooks, suppression of photofragmentation along with results from charge site analysis using UVPD supports preferential binding of DNA22 and DNA50 at AT-hook 2 over AT-hooks 1 and 3.
MATERIALS AND METHODS
The methods for HMGA2 expression and purification have been previously described.8 The sequence of HMGA2 is shown in Figure 1A with the acidic C-terminal tail shown in blue and the AT-hook regions shown in red with the conserved Arg-Gly-Arg-Pro core underlined. The two AT-rich deoxy-oliogonucleotides, DNA22 (6.6 kDa) and DNA50 (15.3 kDa) (Figures 1B,C, respectively) were purchased from Eurofins Scientific (Luxembourg City, Luxembourg) and used as received. These two putative DNA hairpins are each composed of a base pair stem and an AT-rich region in the middle of the stem.
Figure 1.
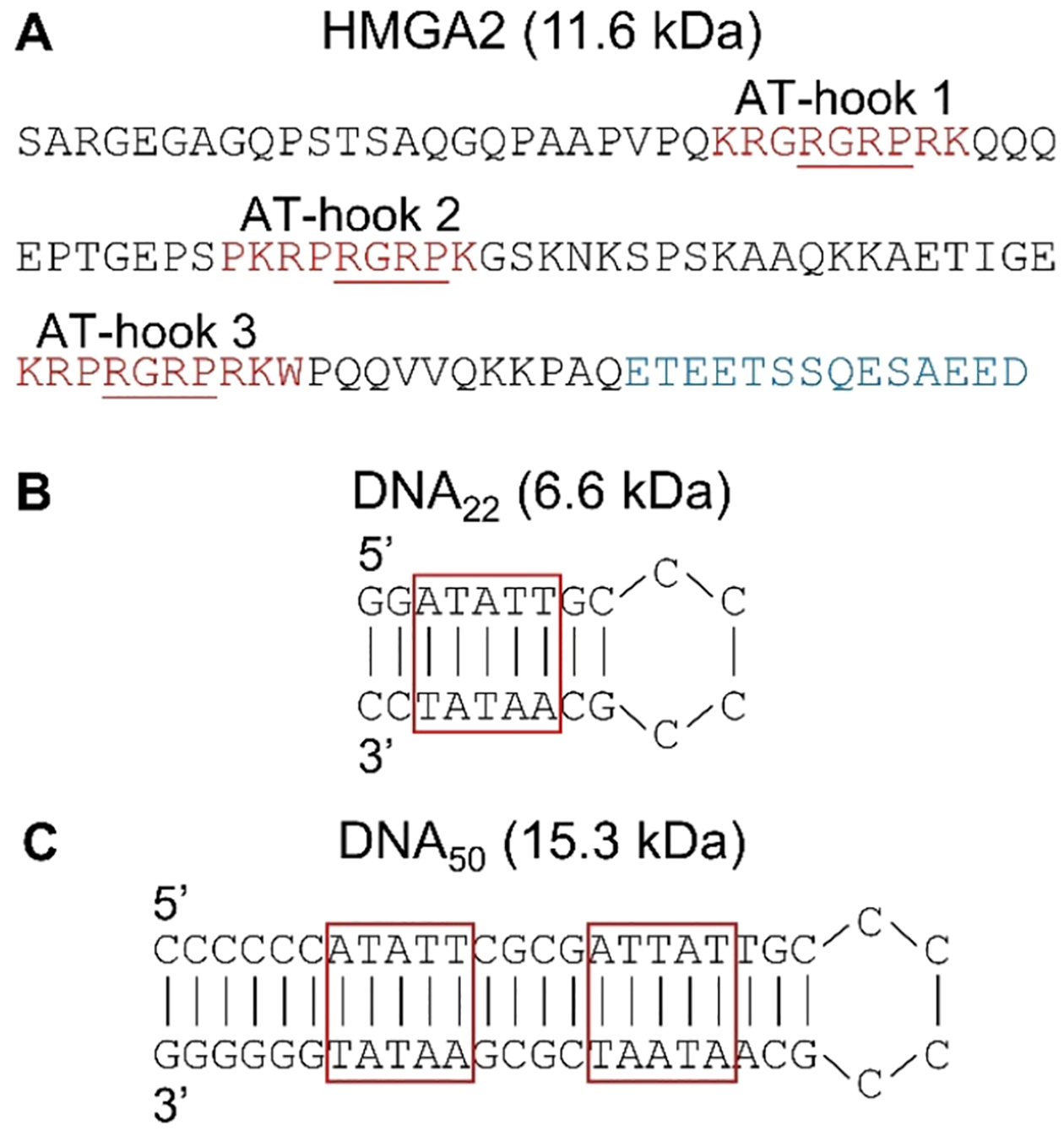
Sequences of (A) HMGA2 and the DNA hairpins, (B) DNA22, and (C) DNA50. HMGA2 sequence shows the negatively charge C-terminus (blue) and three AT-hook DNA binding regions (red). The underlined residues represent the conserved core of each AT-hook. The AT-rich domains of the DNA hairpins are outlined in red.
HMGA2 and the DNA hairpins were exchanged into 10 mM ammonium acetate solutions using Micro Bio-Spin P6 size-exclusion columns per manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA). All samples were analyzed at concentrations of approximately 5 μM for UVPD-MS and Orbitrap CCS measurements and at 10 μM for DTIMS−MS experiments. Protein−DNA complexes were analyzed in equimolar ratios. Solutions were introduced using a NanoFlex ESI source with borosilicate capillaries that were pulled in-house using a Sutter Instruments P-2000 (Novato, CA) and coated with a Au/Pd alloy. Electrospray voltages between 900 and 1200 V were optimized for each protein solution. All experiments were carried out on a Thermo Fisher Scientific Orbitrap Elite mass spectrometer (Bremen, Germany) equipped with a 193 nm Coherent Excistar XS excimer laser (Santa Clara, CA) as displayed in Figure S1A. The temperature of the MS inlet was set to 200 °C for all analyses to promote desolvation of the large protein−DNA complexes. Instrument modifications have been previously described.33 The Orbitrap Elite mass spectrometer is also equipped with electron transfer dissociation (ETD) performed in the linear ion trap. Further discussion of ETD for HMGA2 is provided in the Supporting Information.
Drift tube ion mobility spectrometry−mass spectrometry (DTIMS−MS) was performed using a modular ambient-pressure drift tube constructed from printed circuit boards with a 10 cm drift region.34,35 The drift tube is equipped with two gates operated simultaneously in a Fourier transform mode prior to ion entrance into the mass spectrometer.36–38 Further details are available in the Supporting Information. Briefly, a square waveform was used to open and close gates 1 and 2 with a frequency sweeping from 5 to 7005 Hz over a period of 8 min. Arrival times were converted to collision cross section (CCS) using the Mason−Schamp equation.23
All collision cross sections (CCSs) measured using the drift tube were also evaluated transient decay analysis (TDA) in the Orbitrap mass analyzer, as described previously.39 For these measurements, low transfer potentials were used to maintain solution conformations of ions in the gas phase. Additional details are available in the Supporting Information. All experiments were replicated at least three times.
UVPD was used to investigate the structures of HMGA2 alone and in complex with DNA hairpins. Mass-selected charge states of HMGA2 or HMGA2·DNA complexes were subjected to photoirradiation in the HCD cell using a single laser pulse of 3 mJ. UVPD mass spectra were composed of 100 transient averages that were acquired in the Orbitrap analyzer using a resolving power of 480000 (at m/z 200) and deconvoluted using the Xtract algorithm in QualBrowser or the THRASH algorithm in ProSight PC 4.1 with an S/N ratio of 3 in each case. Fragment ions generated by UVPD were identified from the deconvoluted spectra using UV-POSIT,40 a software developed in-house and available publicly, with a tolerance of 6 ppm. Product ions of the DNA-containing portions of the protein, of the protein-containing portions of the DNA, or of portions of the protein-containing portions of the DNA were not included in the search due to the high probability of false positive matches. The average fractional abundance and standard deviation of at least three replicates were calculated. Significant differences in the abundances of a- and x-type were considered with a p-value of less than 0.02 using the ttest2 function in Matlab (R2020a). Protonated residues were localized using the charge-state analysis tool in UV-POSIT as described previously.41
RESULTS AND DISCUSSION
nESI and CCS Measurements of HMGA2 and Complexes.
The nESI mass spectra of HMGA2 sprayed from aqueous solutions containing 10 mM ammonium acetate are displayed in Figure 2A. A bimodal distribution is observed centered around the 7+ and 12+ charge states. Such distributions are commonly observed for intrinsically disordered proteins that have broad conformational landscapes spanning from compact structures at low charge states (here 6+ to 8+) to intermediate and extended structures at moderate to high charge states (9+ to 15+). ESI of the solutions containing HMGA2 and DNA result in 1:1 protein·DNA complexes as shown in Figure 2B,C. For the solution containing HMGA2 and DNA22, an array of high charge states (10+ to 16+) of low abundance and a series of low charge states (7+ to 9+) of greater abundance are observed. This bimodal distribution of HMGA2·DNA22 complexes suggests that the extended structures of HMGA2 bind less tightly to the hairpin than the compact structures that result in lower charge states (7+ to 9+). The abundances of the HMGA2·DNA50 complexes are low, and only a single distribution of low charge states is observed, suggesting that the complex adopts primarily compact conformations.
Figure 2.
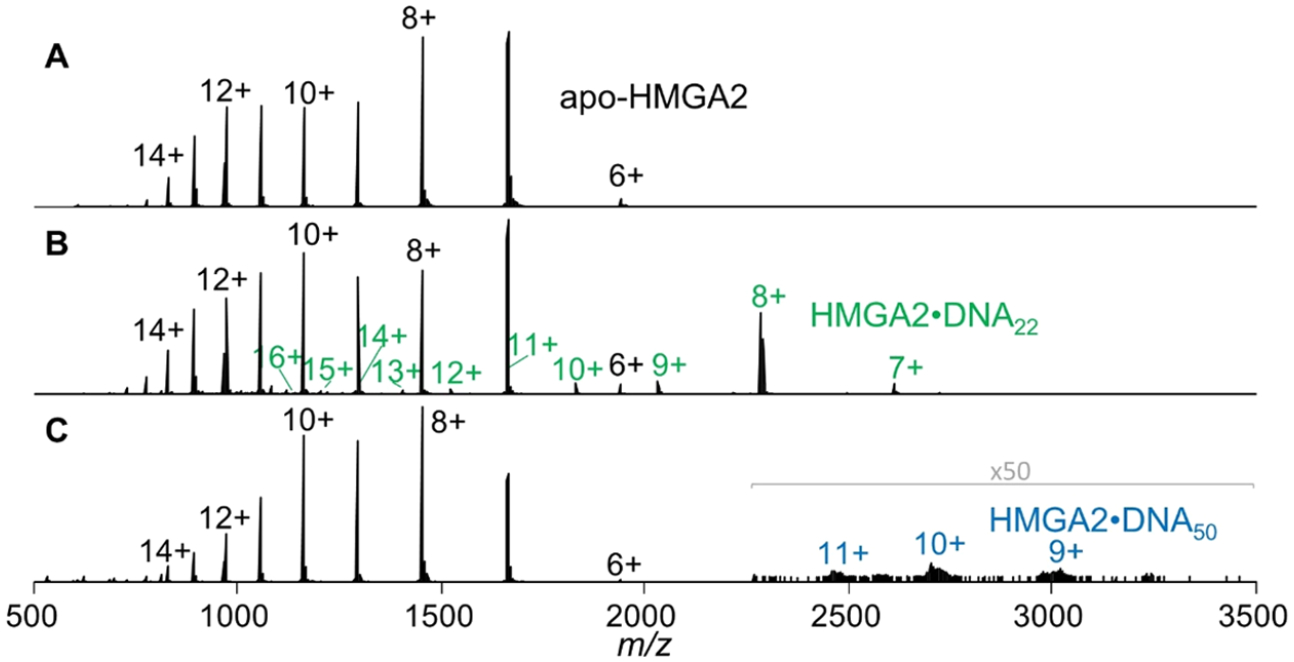
Native ESI mass spectra generated from 10 mM ammonium acetate solutions containing (A) HMGA2, (B) HMGA2·DNA22 (green), and (C) HMGA2·DNA50 (blue).
The vast array of conformations adopted by apo-HMGA2 is demonstrated by the DTIMS−MS results depicted in Figure 3A, with collision cross sections spanning from 1400 to 3000 Å2 depending on charge state. Narrow distributions are observed for the lowest 6+ and 7+ charge states, indicative of a homogeneous population of similar compact structures. The intermediate and high charge states from 9+ to 13+ exhibit broad CCS distributions, consistent with heterogeneous populations of many elongated structures adopted by HMGA2. A relatively narrow CCS distribution is observed again for the 14+ charge state, implying a population of closely related elongated structures.
Figure 3.
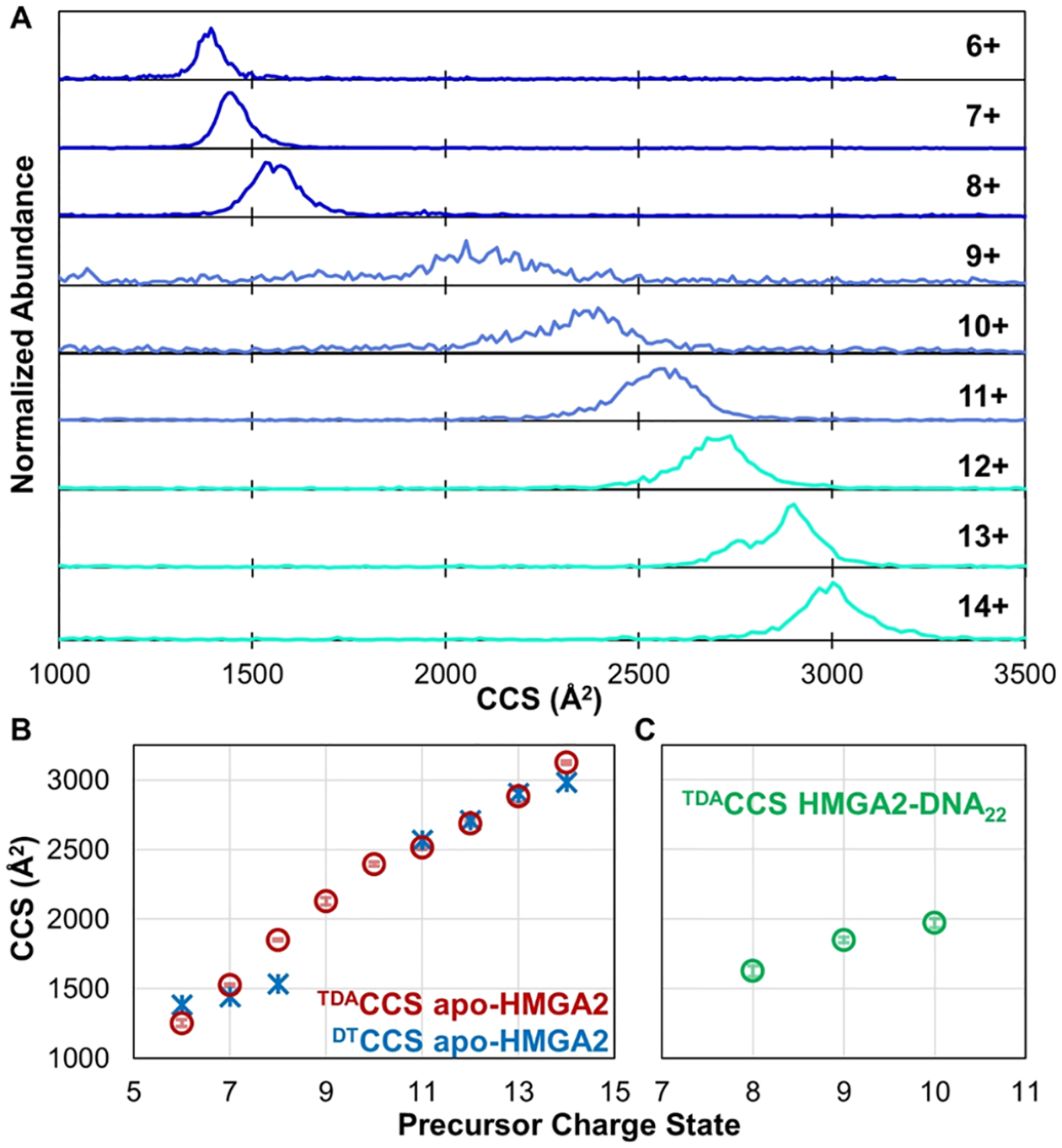
(A) CCS distributions obtained from an ambient drift tube operated in a FT mode prior to entrance into the Orbitrap mass spectrometer. (B) Peak DTCCSs; values for the 9+ and 10+ charge states of apo-HMGA2 are omitted because broad distributions in (A) are not adequately represented by single values. CCS values based on transient decay measurements in the Orbitrap mass analyzer (TDACCS) are shown in (B) HMGA2 and (C) HMGA2·DNA22.
The drift profiles of the HMGA2·DNA complexes were also examined using the ambient drift tube to characterize their CCS values. However, there was insufficient transmission of the complexes through the drift tube due to the low abundance of the complexes and decreased transmission of large, slow ions through the ion gates using high gating frequencies, thus impeding measurement of CCS of the complexes.42–44 Owing to the inability to determine CCS distributions of the protein· DNA complexes using the drift tube, an alternative method was used. Transient decay analysis (TDA) was employed to estimate TDACCS based on the decay rates of time-domain transient signals in the Orbitrap mass analyzer, as described previously.39 The results using TDA for apo-HMGA2 ions are displayed in Figure 3B, and the DTCCS results are overlaid for comparison. With the exception of the 8+ charge state (discussed further in the Supporting Information), the CCS values determined by the transient decay method for apo-HMGA are in good agreement with those measured using the drift tube.
TDA of HMGA2·DNA complexes was also undertaken to estimate CCS values, with results for the 8+ through 10+ charge states of HMGA2·DNA22 displayed in Figure 3C. Interestingly, the CCS values of the HMGA2·DNA22 complexes are lower than the TDACCS values of the apo-HMGA2 species of the same charge state, indicating that the complexes are more compact than the protein alone. This important outcome suggests that the DNA22 hairpin is itself quite compact and that binding to HGMA2 organizes the protein and creates a more ordered structure, an outcome which has been observed previously.16,17 HMGA2 has also been shown to induce DNA bending,17,45 which may contribute to the compact structure observed for the HMGA2·DNA22 complexes. Accurate TDACCS measurements of HMGA2·DNA50 were hindered by the large size (27 kDa) and the presumed increased order of the complexes, leading to potential deviation from the assumption that a single collision is sufficient to cause ion decay in the Orbitrap (an essential prerequisite of the transient decay analysis as discussed in the Supporting Information) as the complex has more avenues to disperse the internal energy gained from a collision.39 It should be noted that this same reasoning could also mean that the TDACCS values of the HMGA2·DNA22 complexes underestimate the true sizes. However, CCS measurements using trapped ion mobility (TIMS) on a Bruker time-of-flight platform yielded values between 1700 and 2000 Å2 for the 8+ through 10+ charge states of the HMGA2·DNA22 complex46 and are in reasonably good agreement with the CCS values between 1625 and 1970 Å2 reported herein using TDA (Figure 3C).
Folding of HMGA2 Analyzed by nESI-UVPD-MS.
UVPD of proteins and protein complexes using 193 nm photons has demonstrated a correlation between the efficiency of backbone cleavages and structural features of proteins. For example, the propensity for backbone fragmentation decreases in regions that are more structured owing to stabilizing, noncovalent interactions which impede the separation and release of fragment ions.29,32,47 Moreover, cleavages of covalent backbone bonds may occur upon UVPD without disruption of noncovalent interactions with a ligand, thus allowing localization of binding sites.31,32 For these reasons, UVPD-MS was employed to localize DNA binding sites and reveal regions of enhanced protein flexibility or rigidity based on monitoring changes in the fragmentation patterns of apo-HMGA2 versus the corresponding HMGA2·DNA complexes. The 7+ through 12+ charge states of apo-HMGA2, the 7+ through 10+, 12+, and 13+ charge states of HMGA2·DNA22, and the 10+ charge state of HMGA2·DNA50 were individually isolated and activated with a single, 3 mJ laser pulse. The 11+ charge state of HMGA2·DNA22 was not analyzed owing to m/z overlap with the 7+ charge state of apo-HMGA2. Other charge states (6+, 13+, 14+ of apo-HMGA2, all charge states greater than 13+ of HMGA2·DNA22, and 9+ and 11+ of HMGA2·DNA50) were omitted owing to low precursor abundance and/or unsatisfactory signal-to-noise of the resulting UVPD mass spectra (data not shown). Representative UVPD mass spectra are displayed in Figures S2 and S3 for apo-HMGA2 and HMGA2·DNA complexes, respectively.
UVPD of all charge states of the apo-HMGA2 resulted in an average of 89% sequence coverage compared to an average of 55% sequence coverage obtained using higher energy collisional dissociation (HCD). The latter demonstrated decreased sequence coverage with increasing charge state (Figure S4A,B) attributed to an enhancement in charge-directed preferential cleavages and possible secondary dissociation for highly charged proteins. Because UVPD performance (i.e., sequence coverage) remains consistent across a range of charge states for many proteins,33,48 it provides an effective way to broadly profile changes in fragmentation based on ligand binding or conformational states.
The distribution and abundance of sequence ions resulting from cleavages at each position along the backbone of apo-HMGA2 are depicted in Figure 4A. The absence of sequence ions originating from cleavages of certain backbone positions, such as in the case of the C-terminal region of HMGA2, is typically ascribed to the presence of noncovalent interactions that impede separation and release of the complementary C- and N-terminal fragments after the backbone is cleaved. These noncovalent interactions may include hydrogen bonds and salt bridges between polar, acidic, and basic residues as well as secondary structural features (hydrogen bonding in α-helices or β-sheets). For HMGA2, the suppressed fragmentation of the C-terminus may be caused by salt bridges between the acidic tail of the protein with one of the basic AT-hook regions that span residues 25 through 33, 45 through 52, and 73 through 82. Intermolecular coordination of the negatively charge C-terminus with the positively charge AT-hooks has been observed in the self-associating homodimers of HMGA2 and similar intramolecular interactions are presumed likely.25 Alternatively, the low number of possible protonation sites in the 25 residues following AT-hook 3 (only two basic residues in the C-terminal region) also decreases the probability of detecting charged ion fragments covering this region. In contrast, the relatively high abundance and large array of fragment ions observed throughout the N-terminus and first two AT-hooks suggests greater protein flexibility and facile release of fragment ions not tethered by extensive networks of noncovalent interactions, consistent with an intrinsically disordered protein.
Figure 4.
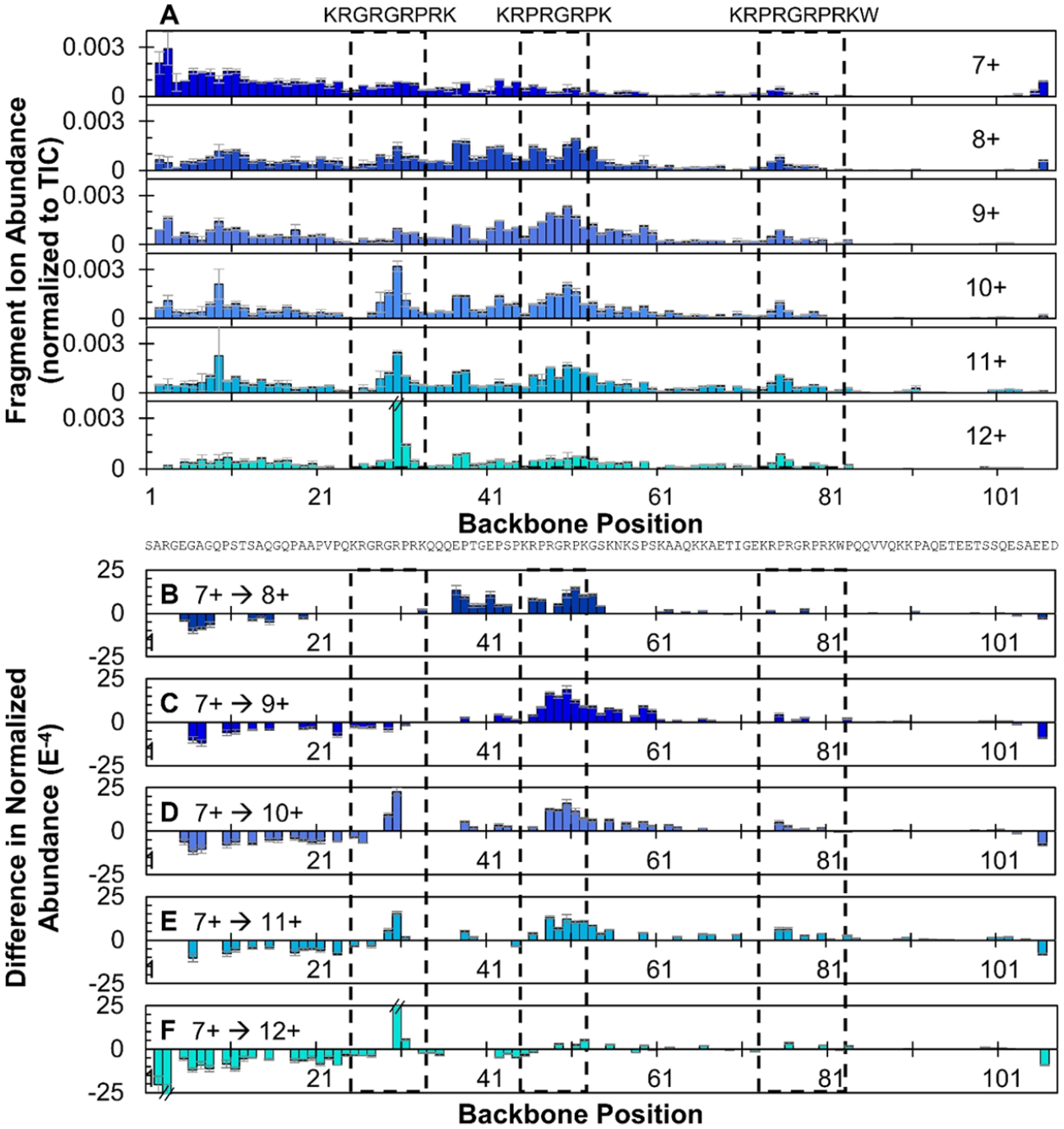
(A) Distribution of backbone cleavages at each residue in the primary sequence of HMGA2 for the 7+ through 12+ charge states based on the summation of N- and C-terminal ions generated from UVPD using a single 3 mJ laser pulse. (B−F) Difference in normalized abundances of sequence ions arising from backbone cleavages generated upon UVPD of HMGA2, 8+ through 12+, relative to the 7+ charge state. Positive values indicate regions of enhanced backbone cleavages for the higher charge state of HMGA2 relative to the 7+ charge state; negative values indicate a suppression of backbone cleavages relative to the 7+ charge state. Residues encompassed in the dashed black boxes denote the three AT-hook regions. Error bars are equivalent to the standard deviation of normalized sequence ion abundances across replicates. Only differences that are statistically different at a 98% confidence level are shown and insignificant difference are omitted, represented as “zero” values.
Changes in UVPD patterns observed for different precursor charge states (7+ to 12+) may suggest structural transitions from ordered (low charge states) to disordered conformations (high charge states). Variations of UVPD across each charge state of HMGA2 relative to the 7+ charge state are shown as difference plots in Figure 4B–F, allowing direct comparison of fragmentation trends without influence of the primary sequence. With respect to the AT-hook regions, the extent of fragmentation is generally lowest for the two lowest charge states (7+ and 8+). The increase in fragment ion abundance in the AT-hook regions for the higher charge states (9+, 10+, 11+) coincides with their more extended conformations (Figure 3) and lower degree of structural organization. The low abundance or absence of sequence ions originating from the C-terminus limits structural insights from UVPD. Similar analysis using conventional collision-activated dissociation (CAD) methods, such as collision-induced dissociation (CID) and its higher energy analogue (HCD), is not feasible given their significant dependence on charge state and availability of mobile protons (Figure S4).49,50
The addition of just one proton going from the 7+ to 8+ charge state of HMGA2 affords a 21% increase in the TDACCS (1526 ± 9 Å2 to 1851 ± 8 Å2 in Figure 3) and results in significant changes in backbone cleavage propensities at 38 sites upon UVPD (Figure 4B). Notably, sequence ion abundances are decreased in the N-terminal region of the 8+ charge state relative to the 7+ charge state and increased throughout residues 37−54, including the second AT-hook. The enhanced fragmentation in the midsection of the protein suggests greater flexibility and elongation of the protein. The suppression of fragmentation at the N-terminus for the 8+ charge state would typically suggest increased protein rigidity (e.g., higher degree of noncovalent interactions) relative to the 7+ charge state; however, in the case of HMGA2, this behavior may alternatively point to a shift in how energy is distributed throughout the protein prior to dissociation. Following 193 nm photon absorption and excitation to an excited electronic state, a protein may undergo direct dissociation from the excited state (typically resulting in a/x-type fragment ions) or internal conversion and intramolecular vibrational energy redistribution (IVR) preceding fragmentation from a ground electronic state.51 In the latter process, the resulting fragment ions are similar to those generated from slow-heating activation methods, such as CAD or infrared multiphoton dissociation (i.e., primarily b/y ions).51 For the 7+ charge state, the fragment ions covering the flexible N-terminus region (residues 1−21) are mainly a + 1 ions (Figure S5), the abundances of which diminish as the charge state of HMGA2 increases. As the charge state increases and HMGA2 unfolds, fragmentation following IVR becomes more probable across the entire length of the protein, as evidenced by the increasing portion of IVR-type fragments (b/y ions) upon UVPD of HMGA2 in higher charge states, as displayed in Figure S6A.
UVPD backbone cleavage propensity of the 9+ charge state of HMGA2 is similar to that of the 8+ charge state (Figure 4A), suggesting that the addition of a second proton to the compact HMGA2 conformer (7+) has relatively little impact on the structure of this more extended conformer (8+ and 9+). This result is also demonstrated by the similarity of the difference plots in Figure 4B,C, which shows a suppression of fragmentation at the N-terminus and enhancement in the midsection, extending further into the primary sequence of the 9+ charge state (Figure 4C), indicative of additional protein unfolding. The 10+ and 11+ charge states also show similar trends with suppression of fragmentation to residue 26 in the first AT-hook region and enhanced fragmentation extending deeper in the primary sequence to residue 90 in the case of the 11+ charge state (Figures 4D,E), consistent with greater unfolding of the protein. Backbone cleavage N-terminal to Pro31, a known preferential pathway of slow-heating activation methods, is prominent for the 10+ to 12+ charge states. With exception of this single prominent Pro cleavage, the 12+ charge state exhibits a relatively flat fragmentation profile (Figure 4A). This flat distribution suggests that the likelihood of a backbone cleavage is roughly equivalent across the length of the protein and that any structural features arising from noncovalent interactions that typically prohibit the release of sequence ions have been lost.
Charge State Analysis Suggests Regions of Solvent Exposure of HMGA2.
While the variation in the backbone cleavages of HMGA2 provides insight into the rigidity or flexibility of the protein as mediated by the presence of noncovalent interactions, localizing protonation sites offers feedback about how charge sites shift for different protein conformations.41 Basic residues on the surface of a protein are more likely to sequester and retain protons during the ESI process, whereas buried residues of proteins in more compact, folded conformations will be less likely to capture and retain protons and are more likely to be engaged in noncovalent electrostatic interactions with other residues. The high sequence coverage afforded by UVPD provides a means to track charge sites of HMGA2, in many cases with single-residue specificity, termed charge-site analysis. In essence, the transitions in the charge states of fragment ions are used to bracket the locations of each additional proton. The charge state analysis tool in UV-POSIT identifies sequence ions (namely a, a + 1, x, and x + 1 ions) and their charge states from deconvoluted UVPD mass spectra using ProSight PC 4.1. Only these a/x-type ions are used for this analysis because they are unique to UVPD and not modulated by the mobile proton model prominent upon CAD.50,52 Although a-ions can be derived from secondary dissociation of b-ions,49 they have low prevalence (<2% abundance) upon HCD of HMGA2 ions (Figure S6B). This suggests that a-ions are more uniquely generated from UVPD for HMGA2, not from processes involving vibrational excitation akin to CAD. Figure S7 displays the distributions of charge states of a/x-type ions generated upon UVPD of HMGA2 (7+ through 12+ precursors). Larger fragment ions appear in higher charge states, as expected, and in many cases, there are stepwise progressions from one charge state to the next as the backbone is cleaved across the protein. The locations of the backbone cleavages that result in the a/x ions are shown along the primary sequence of HMGA2 in Figure S8. The transitions in charge states of fragment ions are used to assign the approximate locations of each added proton as the sizes (i.e. number of residues) of the fragment ions increase. For example, consider the formation of x104 and x105 ions which are markers for backbone cleavage between residues Arg3 and Gly4 and residues Ala2 and Arg3, respectively (Figure S7B). The step increase in charge state going from x104 to x105 for nearly every precursor charge state suggests the additional proton for x105 is localized at Arg3. The an ions in Figure S7A are complementary to the xn ions with no a-type ions detected that are smaller than a3 (containing residues SAR, 1+). This result is consistent with sequestration of a proton on the basic Arg3 residue. Protonation of Arg3 is unsurprising given the high gas-phase basicity of arginine and the flexibility of the N-terminus suggested by the enhanced fragmentation of this region upon UVPD as described earlier.
The more gradual changes in charge-state distributions of sequence ions that appear as “tailing” of the transitions in Figure S7 suggest the presence of two or more protonated forms. This tailing is most apparent for the 7+, 8+, and 9+ charge states of HMGA, which likely possess a mixture of compact and partially unfolded conformations. In contrast, the 10+, 11+, and 12+ charge states, which have larger CCSs characteristic of substantially elongated conformations, exhibit more discrete step changes in the charge states of the fragment ions. The more significant variability in proton locations for the compact conformations of HMGA2 (in lower charge states) may provide further support for the greater flexibility of the N-terminal region of HMGA2 relative to the C-terminus. Indeed, there is evidence that four of the seven protons of the 7+ charge state of HMGA2 are localized in the N-terminus and extending to first AT-hook regions (residues 1−33), with the other three protons dispersed over the remaining ~70% of the protein. This finding supports that the acidic C-terminus is folded and engaged in salt bridges with one of the AT-hooks. The presumed high flexibility (based on enhanced fragmentation) of the first AT-hook makes it a less likely binding partner to the C-terminus.
Based on the charge-state analysis of fragment ions, predicted protonated residues are shown in Figure S9 in red font with gray brackets indicating regions in which a proton may be mobile or indicative of multiple proton configurations. In essence, Figure S9 provides visualization of the locations of each added proton as the charge state of HMGA2 increases. Going from the 7+ to 8+ charge states, for example, there appears to be some shuffling of protonation sites, and a lone proton at Arg50 is “replaced” by charges at Arg46 and Lys61 to accommodate the additional proton. This change is consistent with greater exposure of the amino acids in the center of the protein and parallels the enhanced UVPD fragmentation in that region (Figure 4B). Additional protonation occurs in the third AT-hook, suggesting it too becomes more exposed as HMGA2 unfolds in higher charge states. As the protein becomes even more extended and dynamic, as suggested by the larger CCS and enhanced fragmentation of the 12+ charge state, the protons are relatively evenly spaced throughout the primary sequence, with particular affinity for the highly basic AT-hooks. One proposed unfolding scheme of HMGA2 is displayed in Figure S10, which depicts HMGA2 in a compact conformation and low charge state (top) with one of the AT-hooks (AT-hook 2, although interaction with AT-hook 3 is also likely) in contact with the C-terminal region. The conformation extends and CCS increases as the N-terminus becomes more exposed in the intermediate state (middle) until the tertiary interactions involving the C-terminus are lost, resulting in a fully unfolded structure (bottom).
ETD is another activation method that is known to provide structural information based on cleavage of primarily surface-exposed regions of the protein backbone. However, the charge dependence of ETD often limits its utility for native proteins that carry fewer charges than denatured ones. ETD was used in the present study to characterize HMGA2 and the HMGA2·DNA complexes. Details about the experimental parameters and the resulting spectra and sequence maps are provided in the Supporting Information and in Figures S11−S14. Briefly, ETD resulted in moderate sequence coverage (34−52%) of apo-HMGA2 species; however, ETD of the HMGA2·DNA complexes resulted in few backbone cleavages, limiting structural characterization. Only three holo-fragments from the HMGA2·DNA22 complex were identified (shown in Figure S14B), which support the UVPD findings. Owing to the insufficient insight offered for the HMGA2·DNA complexes, ETD was not pursued further.
Increasing Noncovalent Interactions upon Binding to DNA Suppresses UVPD Fragmentation of HMGA2.
UVPD was employed to probe the structures of HMGA2· DNA complexes as done for apo-HMGA2. The vast change in size and degrees of freedom of apo-protein relative to the protein−DNA complexes warranted consideration of normalization of laser energy, as discussed in the Supporting Information. Ultimately, the same laser pulse energy (3 mJ) was used for analysis of HMGA2 as for the HMGA2·DNA complexes because as the size and number of chromophores of the complex increases, so does its photoabsorptivity. As shown in Figure S15, DNA itself absorbs 193 nm photons and undergoes UVPD, although not quite as efficiently as proteins based on the precursor ion depletion measurements shown in Figure S16. It is unknown how much this precursor depletion is affected by the higher order structure of the hairpins in addition to photoabsorptivity of the ion. Therefore, normalization of laser energy was not undertaken.
The UVPD mass spectra of HMGA2·DNA complexes in Figure S3 were used to generate maps of backbone cleavages that result in apo-fragments (sequence ions that do not retain DNA, Figure S17) or holo-fragment ions (ones that retain DNA, Figure 5). Overall, the abundances of apo fragment ions are greater than those of holo fragment ions for the HMGA2· DNA complexes. Evident in Figure S17 and Figure 5 is that UVPD of the HMGA2·DNA complexes exhibits more prominent backbone cleavage throughout the N-terminus of HMGA2 than throughout the rest of the protein. Suppression of backbone cleavages throughout the midsection is distinct (Figures 5 and S17), presumably owing to networks of noncovalent interactions with DNA that stabilize the complexes and curb separation and release of fragment ions. Finally, some notable fragmentation of the C-terminal region is observed for the HMGA2·DNA complexes, a feature not noted upon UVPD of apo-HMGA.
Figure 5.
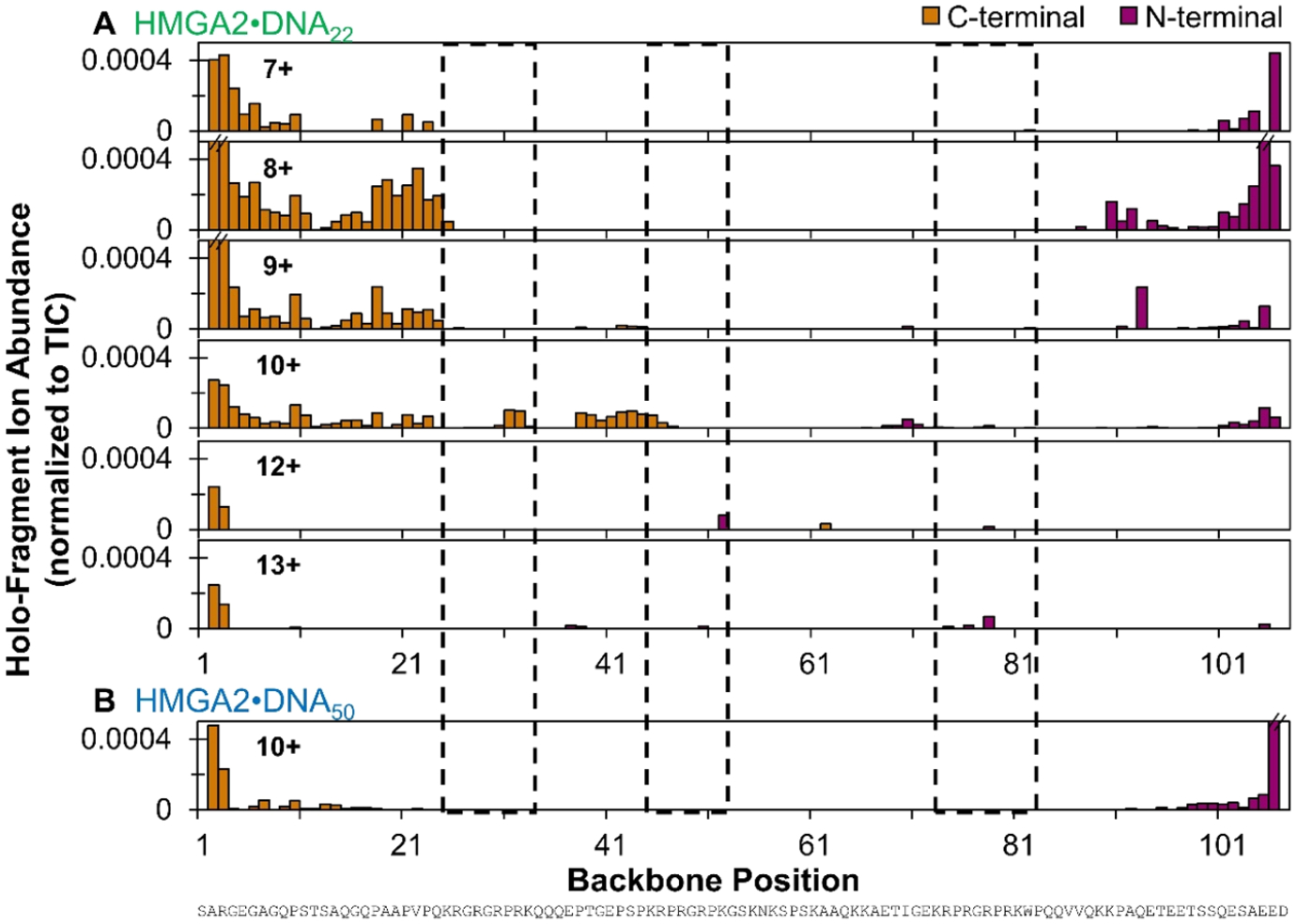
Distribution of backbone cleavages at each residue in the primary sequence of (A) HMGA2·DNA22 (7+ through 13+ charge states) and (B) the HMGA2·DNA50 complex (10+ charge state) based on holo (DNA-containing) fragment ions generated by UVPD using a single 3 mJ laser pulse. Residues encompassed in dashed black boxes denote the three AT-hook regions. Error bars are equivalent to the standard deviation of replicate measurements. Only holo-fragments containing the entire DNA are considered. N-terminal and C-terminal fragments include a/b/c and x/y/z ions, respectively. The corresponding distribution of backbone cleavages resulting in apo fragment ions (ones that do not retain the DNA) is shown in Figure S17.
Direct comparisons of the fragmentation patterns of apo-HMGA2 (8+) relative to the summed total of apo- and holo-fragments from HMGA2·DNA22 (8+) and HMGA2·DNA50 (10+) are shown as difference plots in Figure 6A,B, respectively. Comparison of these selected charge states was considered appropriate because they are consistently the most abundant and thus represent the greatest population of conformations of compact structures present in solution. The 7+ apo-HMGA2 species is also dominant, and the fragmentation patterns of this charge state are also compared to HMGA2·DNA22 (8+) and HMGA2·DNA50 (10+) in Figure S18, revealing trends similar to those in Figure 6. An overall suppression in the abundance of sequence ions is observed for the HMGA2·DNA complexes relative to HMGA2, occurring primarily between residues 21 and 73 (Figure 6), and is shown as negative values beginning at the first AT-hook, spanning the second AT-hook, and terminating at the third AT-hook. This is consistent with previous reports that all three AT-hooks are capable of binding to AT-rich DNA.5,6 As illustrated in Figure S17, the overall fragmentation efficiency of the third AT-hook region is similar. The greater suppression of fragmentation of the first and second AT-hooks relative to the third suggests preferential binding of DNA to these regions of the HMGA2· DNA complexes, which is supported by a previous study in which AT-hooks 1 and 2 induced greater thermal stability of DNA22 (referred to as FL876) than did AT-hook 3.6 This does not discount any DNA binding to AT-hook 3, and it is likely that all three binding modes for both DNA hairpins exist in equilibrium in solution. That is, DNA22 may be bound to either AT-hook 1, 2, or 3 and DNA50 may be bound to AT-hooks 1 and 2, 1 and 3, or 2 and 3.
Figure 6.
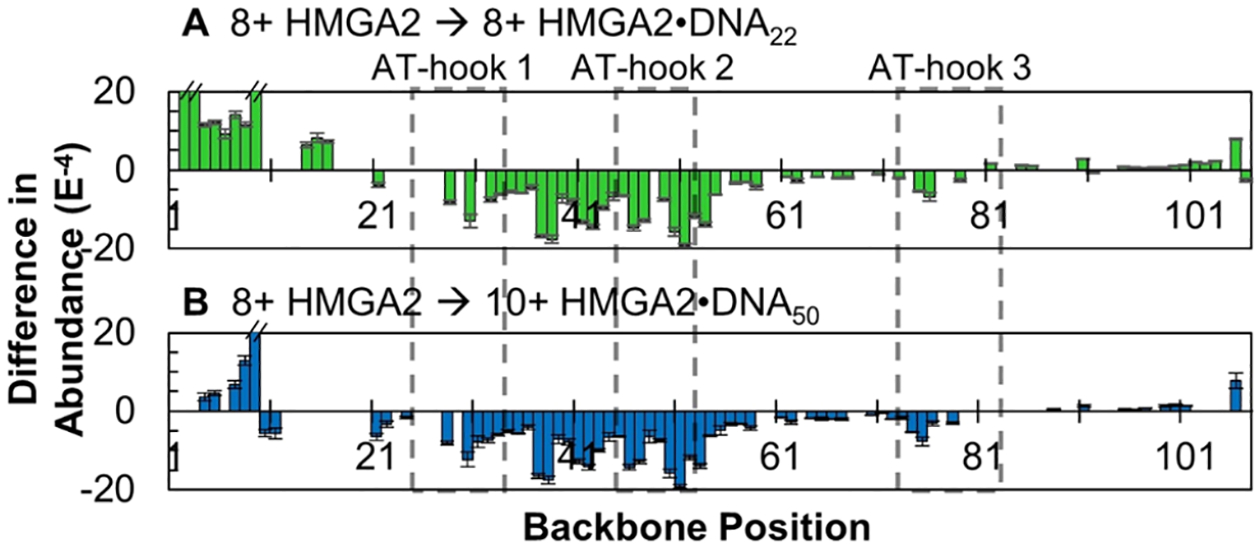
Differences in normalized abundances of sequence ions arising from backbone cleavages generated upon UVPD of HMGA2 (8+) relative to all UVPD sequence ions (i.e., the sum of apo- and holo-fragments) of (A) HMGA2·DNA22 (8+) and (B) HMGA2·DNA50 (10+). Positive values indicate regions of enhanced backbone cleavages for the HMGA2·DNA complex relative to the apoprotein; negative values indicate a suppression of backbone cleavages. Residues encompassed in dashed black boxes denote the three AT-hook regions. Only differences that are statistically different at a 98% confidence level are shown, and insignificant difference are omitted, represented as “zero” values.
Fragmentation of the C-terminus region, though generally sparse, is in fact more prominent for the HMGA2·DNA complexes than for apo-HMGA2, demonstrated by positive values between residues 88 and 101 in Figure 6 (and notable in Figure S17). The more efficient release and detection of these C-terminal sequence ions suggests a decrease in noncovalent interactions in that region of the protein. This interesting result supports two complementary postulates: (1) the acidic C-terminus interacts with one or more of the AT-hooks when they are unoccupied in apo-HMGA2 and (2) the DNA hairpin binds to one or more AT-hooks, mitigating their interactions with the C-terminus. The N-terminus of HMGA2 also exhibited increased photofragmentation, suggesting it too is flexible when bound to DNA.
Holo-Sequence Ion and Charge Site Analysis Aids Localization of Binding Sites.
The ability of a protein to maintain noncovalent interactions with ligands after activation and cleavage of covalent bonds is a recognized hallmark of UVPD.31,32 Maps of the backbone cleavages that lead to identified holo-sequence ions of HMGA2 are shown in Figure 5. The abundances of these holo-fragments, however, are approximately 10 times lower than the apo-fragments (Figure S17), an outcome that is unsurprising considering that many holo-ions may disassemble, releasing the DNA ligand to generate apo-ions during IVR if not produced promptly from precursors in excited electronic states. All of the holo-fragments correspond to very large ions typically containing 70 or more amino acids, meaning they span nearly the entire protein sequence and thus maintain sufficient structure and noncovalent interactions to retain the DNA and be successfully detected. A segment of identified holo-sequence ions is displayed in the inset of Figure S3A. C-Terminal holo-fragments extending from residues 34 to 25 and N-terminal holo-fragments from residues 45 to 72 suggest preferential binding of DNA22 to AT-hook 2 (Figure 5, most notably 9+ and 10+) and that DNA binding to the AT-hooks 1 and 3 is less prevalent. These results support the observation of DNA binding in the center of the protein as noted based on the greater suppression of fragmentation observed in Figure 6.
Charge-site analysis was performed for HMGA2·DNA complexes with results shown in Figures S19 and S20. The limited sequence coverage of the HMGA2·DNA complexes leads to incomplete assignment of protonation sites but can still provide insight into the exposure of basic residues in HMGA2. The majority of protons on the HMGA2·DNA22 complex are concentrated at the N-terminal half of the protein, suggesting it maintains some degree of exposure of basic residues although it is generally more structured relative to the apoprotein (Figure 6). As summarized schematically in Figure S20, the second and third AT-hook regions each possess just one proton for the 8+ and 9+ charge states of HMGA2·DNA22, which is relatively deficient in charge considering the highly basic nature of the AT-hooks. The deficiency of charge in AT-hook 3 was determined based on the lack of any charge increase for x202+ to x292+ ions in the AT-hook3 region for different charge states of HMGA2·DNA50, suggesting that AT-hook 3 does not sequester more than a single proton. As the charge of the complex increases, AT-hook 3 is eventually protonated, followed by AT-hook 2. These data taken into consideration with the results presented in Figure 6A suggest that the second AT-hook is most likely involved in binding DNA22 and, therefore, less available to capture and retain charge during ESI. The two AT-rich regions of DNA50 allow it to bind to the second AT-hook of HMGA2 in addition to either the first AT-hook, supported by suppressed UVPD fragmentation (Figure 6B), or the third AT-hook, accounting for the sparsity of protons between residues 45 and 81 (Figures S19C,D and S20B). These results are consistent with previously published solution studies showing that two AT-hooks can simultaneously bind to the AT-rich DNA substrate.7,8 Moreover, salt-dependent studies showed that electrostatic interactions are the dominant force for the binding dynamics of HMGA2 and the AT-rich DNA substrate.8 Thus, binding dynamics may change in different salt conditions than carried out in this experiment. It is of note that some binding isomers (i.e., DNA22 bound to either AT-hook 1, 2, or 3) may exist in different charge states due to differences in conformations and subsequent protonation sites, contributing to different fragmentation patterns. Additionally, existence of the various isomers in the same charge state convolutes interpretation of the data.
CONCLUSION
The structural characterization of mammalian high mobility group protein AT-hook 2 using traditional methods is elusive due to its categorization as an intrinsically disordered protein. However, utilization of native mass spectrometry coupled to ion mobility spectrometry and UVPD has provided insights into the broad conformational landscape adopted by HMGA2. The range of compact to extended structures was observed using ambient drift tube IMS and maintained in the gas phase as confirmed using transient decay analysis. These structures were characterized using UVPD, the results of which support the presence of higher order interactions between the acidic C-terminal region of HMGA2 with the highly basic AT-hooks (most likely AT-hook 2) of the compact conformations, leaving the N-terminus relatively more exposed and flexible. These salt bridges are not maintained in the extended structures of apo-HMGA2 as suggested by the increase in the number of protonated residues and fragmentation throughout the AT-hook regions. The structural heterogeneity of HMGA2 decreased upon binding a DNA hairpin, which acted to stabilize HMGA2 and enhance organization to more compact structures as suggested by the relatively small TDACCS values observed for the HMGA2·DNA22 complex and the suppressed UVPD of both protein·DNA complexes. The hairpins displaced the acidic C-terminus by binding preferentially to AT-hook 2 as supported by an increase in fragmentation at the C-terminal tail of HMGA2, a concurrent decrease in fragmentation throughout the middle of the protein sequence, and the presence of DNA-containing holo-fragment ions extending to AT-hook 2. AT-hooks 1 and 3 may also be involved in binding DNA50 according to suppressed fragmentation and protonation of those regions, respectively, corresponding with previous reports demonstrating that all three AT-hooks are capable of binding DNA and that DNA50 is capable of occupying more than one AT-hook at a time. The two mass spectrometry methods used in the present study, IMS-MS and UVPD-MS, have demonstrated the ability to uncover structural features difficult to discern for intrinsically disordered proteins.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the financial support from the National Science Foundation Division of Chemistry, under CAREER award CHE-1654274, with cofunding from the Division of Molecular and Cellular Biosciences to F.F.-L. Funding from the National Institutes of Health (R35GM139658) and the Robert A. Welch Foundation (F-1155) is acknowledged by J.S.B.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jasms.2c00083
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.2c00083.
Description of transient decay analysis, ETD analysis of proteins and protein·DNA complexes, and UVPD of DNA; schematic of instrumentation; UVPD spectra for proteins, DNA, and protein·DNA complexes; fragmentation maps, sequence coverage information, and ion-type distributions from MS/MS spectra; and charge-site analysis of proteins and protein·DNA complexes (PDF)
The authors declare no competing financial interest.
Contributor Information
Sarah N. Sipe, Department of Chemistry, University of Texas, Austin, Texas 78712, United States.
Kevin Jeanne Dit Fouque, Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, United States.
Alyssa Garabedian, Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, United States.
Fenfei Leng, Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, United States; Biomolecular Sciences Institute, Florida International University, Miami, Florida 33199, United States.
Francisco Fernandez-Lima, Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, United States; Biomolecular Sciences Institute, Florida International University, Miami, Florida 33199, United States.
Jennifer S. Brodbelt, Department of Chemistry, University of Texas, Austin, Texas 78712, United States.
REFERENCES
- (1).Oldfield CJ; Dunker AK Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem 2014, 83 (1), 553–584. [DOI] [PubMed] [Google Scholar]
- (2).Wright PE; Dyson HJ Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol 2015, 16 (1), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Reeves R HMGA Proteins: Flexibility Finds a Nuclear Niche? Biochem. Cell Biol 2003, 81 (3), 185–195. [DOI] [PubMed] [Google Scholar]
- (4).Reeves R High Mobility Group (HMG) Proteins: Modulators of Chromatin Structure and DNA Repair in Mammalian Cells. DNA Repair 2015, 36, 122–136. [DOI] [PubMed] [Google Scholar]
- (5).Cui T; Leng F Specific Recognition of AT-Rich DNA Sequences by the Mammalian High Mobility Group Protein AT-Hook 2: A SELEX Study. Biochemistry 2007, 46 (45), 13059–13066. [DOI] [PubMed] [Google Scholar]
- (6).Garabedian A; Jeanne Dit Fouque K; Chapagain PP; Leng F; Fernandez-Lima F AT-Hook Peptides Bind the Major and Minor Groove of AT-Rich DNA Duplexes. Nucleic Acids Res 2022, gkac115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Maher JF; Nathans D Multivalent DNA-Binding Properties of the HMG-1 Proteins. Proc. Natl. Acad. Sci 1996, 93 (13), 6716–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cui T; Wei S; Brew K; Leng F Energetics of Binding the Mammalian High Mobility Group Protein HMGA2 to Poly(DA-DT)2 and Poly(DA)-Poly(DT). J. Mol. Biol 2005, 352 (3), 629–645. [DOI] [PubMed] [Google Scholar]
- (9).Zhou X; Benson KF; Ashar HR; Chada K Mutation Responsible for the Mouse Pygmy Phenotype in the Developmentally Regulated Factor HMGI-C. Nature 1995, 376 (6543), 771–774. [DOI] [PubMed] [Google Scholar]
- (10).Rogalla P; Drechsler K; Schröder-Babo W; Eberhardt K; Bullerdiek J HMGIC Expression Patterns in Non-Small Lung Cancer and Surrounding Tissue. Anticancer Res 1998, 18 (5A), 3327–3330. [PubMed] [Google Scholar]
- (11).Winslow MM; Dayton TL; Verhaak RGW; Kim-Kiselak C; Snyder EL; Feldser DM; Hubbard DD; DuPage MJ; Whittaker CA; Hoersch S; Yoon S; Crowley D; Bronson RT; Chiang DY; Meyerson M; Jacks T Suppression of Lung Adenocarcinoma Progression by Nkx2-1. Nature 2011, 473 (7345), 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Müller MH; Drieschner N; Focken T; Bartnitzke S; Winter N; Klemke M; Bullerdiek J HMGA2 Expression in the PC-3 Prostate Cancer Cell Line Is Autonomous of Growth Factor Stimulation. Anticancer Res 2013, 33 (8), 3069–3078. [PubMed] [Google Scholar]
- (13).Bussemakers MJG; Ven WJM; van de Debruyne FMJ; Schalken JA Identification of High Mobility Group Protein I(Y) as Potential Progression Marker for Prostate Cancer by Differential Hybridization Analysis. Cancer Res 1991, 51 (2), 606–611. [PubMed] [Google Scholar]
- (14).Sun M; Song C-X; Huang H; Frankenberger CA; Sankarasharma D; Gomes S; Chen P; Chen J; Chada KK; He C; Rosner MR HMGA2/TET1/HOXA9 Signaling Pathway Regulates Breast Cancer Growth and Metastasis. Proc. Natl. Acad. Sci 2013, 110 (24), 9920–9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Anand A; Chada K In Vivo Modulation of Hmgic Reduces Obesity. Nat. Genet 2000, 24 (4), 377–380. [DOI] [PubMed] [Google Scholar]
- (16).Huth JR; Bewley CA; Nissen MS; Evans JNS; Reeves R; Gronenborn AM; Clore GM The Solution Structure of an HMG-I(Y)–DNA Complex Defines a New Architectural Minor Groove Binding Motif. Nat. Struct. Mol. Biol 1997, 4 (8), 657–665. [DOI] [PubMed] [Google Scholar]
- (17).Fonfría-Subirós E; Acosta-Reyes F; Saperas N; Pous J; Subirana JA; Campos JL Crystal Structure of a Complex of DNA with One AT-Hook of HMGA1. PLoS One 2012, 7 (5), No. e37120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dunker AK; Lawson JD; Brown CJ; Williams RM; Romero P; Oh JS; Oldfield CJ; Campen AM; Ratliff CM; Hipps KW; Ausio J; Nissen MS; Reeves R; Kang C; Kissinger CR; Bailey RW; Griswold MD; Chiu W; Garner EC; Obradovic Z Intrinsically Disordered Protein. J. of Mol. Graphics Modell 2001, 19 (1), 26–59. [DOI] [PubMed] [Google Scholar]
- (19).Keppel TR; Howard BA; Weis DD Mapping Unstructured Regions and Synergistic Folding in Intrinsically Disordered Proteins with Amide H/D Exchange Mass Spectrometry. Biochemistry 2011, 50 (40), 8722–8732. [DOI] [PubMed] [Google Scholar]
- (20).Russel D; Lasker K; Webb B; Velázquez-Muriel J; Tjioe E; Schneidman-Duhovny D; Peterson B; Sali A Putting the Pieces Together: Integrative Modeling Platform Software for Structure Determination of Macromolecular Assemblies. PLOS Biology 2012, 10 (1), No. e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Leman JK; Weitzner BD; Lewis SM; Adolf-Bryfogle J; Alam N; Alford RF; Aprahamian M; Baker D; Barlow KA; Barth P; Basanta B; Bender BJ; Blacklock K; Bonet J; Boyken SE; Bradley P; Bystroff C; Conway P; Cooper S; Correia BE; Coventry B; Das R; De Jong RM; DiMaio F; Dsilva L; Dunbrack R; Ford AS; Frenz B; Fu DY; Geniesse C; Goldschmidt L; Gowthaman R; Gray JJ; Gront D; Guffy S; Horowitz S; Huang P-S; Huber T; Jacobs TM; Jeliazkov JR; Johnson DK; Kappel K; Karanicolas J; Khakzad H; Khar KR; Khare SD; Khatib F; Khramushin A; King IC; Kleffner R; Koepnick B; Kortemme T; Kuenze G; Kuhlman B; Kuroda D; Labonte JW; Lai JK; Lapidoth G; Leaver-Fay A; Lindert S; Linsky T; London N; Lubin JH; Lyskov S; Maguire J; Malmström L; Marcos E; Marcu O; Marze NA; Meiler J; Moretti R; Mulligan VK; Nerli S; Norn C;Ó’Conchúir S; Ollikainen N; Ovchinnikov S; Pacella MS; Pan X; Park H; Pavlovicz RE; Pethe M; Pierce BG; Pilla KB; Raveh B; Renfrew PD; Burman SSR; Rubenstein A; Sauer MF; Scheck A; Schief W; Schueler-Furman O; Sedan Y; Sevy AM; Sgourakis NG; Shi L; Siegel JB; Silva D-A; Smith S; Song Y; Stein A; Szegedy M; Teets FD; Thyme SB; Wang RY-R; Watkins A; Zimmerman L; Bonneau R Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Methods 2020, 17 (7), 665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Biehn SE; Lindert S Accurate Protein Structure Prediction with Hydroxyl Radical Protein Footprinting Data. Nat. Commun 2021, 12 (1), 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Revercomb HE; Mason EA Theory of Plasma Chromatography/Gaseous Electrophoresis. Review. Anal. Chem 1975, 47 (7), 970–983. [Google Scholar]
- (24).Testa L; Brocca S; Santambrogio C; D’Urzo A; Habchi J; Longhi S; Uversky VN; Grandori R Extracting Structural Information from Charge-State Distributions of Intrinsically Disordered Proteins by Non-Denaturing Electrospray-Ionization Mass Spectrometry. Intrinsically Disord. Proteins 2013, 1 (1), No. e25068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Frost L; Baez MAM; Harrilal C; Garabedian A; Fernandez-Lima F; Leng F The Dimerization State of the Mammalian High Mobility Group Protein AT-Hook 2 (HMGA2). PLoS One 2015, 10 (6), No. e0130478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Stuchfield D; Barran P Unique Insights to Intrinsically Disordered Proteins Provided by Ion Mobility Mass Spectrometry. Curr. Opin. Chem. Biol 2018, 42, 177–185. [DOI] [PubMed] [Google Scholar]
- (27).Geels RBJ; van der Vies SM; Heck AJR; Heeren RMA Electron Capture Dissociation as Structural Probe for Noncovalent Gas-Phase Protein Assemblies. Anal. Chem 2006, 78 (20), 7191–7196. [DOI] [PubMed] [Google Scholar]
- (28).Li H; Nguyen HH; Ogorzalek Loo RR; Campuzano IDG; Loo JA An Integrated Native Mass Spectrometry and Top-down Proteomics Method That Connects Sequence to Structure and Function of Macromolecular Complexes. Nat. Chem 2018, 10 (2), 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Warnke S; von Helden G; Pagel K Analyzing the Higher Order Structure of Proteins with Conformer-Selective Ultraviolet Photodissociation. Proteomics 2015, 15 (16), 2804–2812. [DOI] [PubMed] [Google Scholar]
- (30).Theisen A; Black R; Corinti D; Brown JM; Bellina B; Barran PE Initial Protein Unfolding Events in Ubiquitin, Cytochrome c and Myoglobin Are Revealed with the Use of 213 Nm UVPD Coupled to IM-MS. J. Am. Soc. Mass Spectrom 2019, 30 (1), 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).O’Brien JP; Li W; Zhang Y; Brodbelt JS Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2014, 136 (37), 12920–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cammarata MB; Brodbelt JS Structural Characterization of Holo- and Apo-Myoglobin in the Gas Phase by Ultraviolet Photodissociation Mass Spectrometry. Chem. Sci 2015, 6 (2), 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc 2013, 135 (34), 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Reinecke T; Clowers BH Implementation of a Flexible, Open-Source Platform for Ion Mobility Spectrometry. HardwareX 2018, 4, No. e00030. [Google Scholar]
- (35).Sanders JD; Butalewicz JP; Clowers BH; Brodbelt JS Absorption Mode Fourier Transform Ion Mobility Mass Spectrometry Multiplexing Combined with Half-Window Apodization Windows Improves Resolution and Shortens Acquisition Times. Anal. Chem 2021, 93 (27), 9513–9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Knorr FJ; Eatherton RL; Siems WF; Hill HH Fourier Transform Ion Mobility Spectrometry. Anal. Chem 1985, 57 (2), 402–406. [DOI] [PubMed] [Google Scholar]
- (37).Davis AL; Reinecke T; Morrison KA; Clowers BH Optimized Reconstruction Techniques for Multiplexed Dual-Gate Ion Mobility Mass Spectrometry Experiments. Anal. Chem 2019, 91 (2), 1432–1440. [DOI] [PubMed] [Google Scholar]
- (38).Poltash ML; McCabe JW; Shirzadeh M; Laganowsky A; Clowers BH; Russell DH Fourier Transform-Ion Mobility-Orbitrap Mass Spectrometer: A Next-Generation Instrument for Native Mass Spectrometry. Anal. Chem 2018, 90 (17), 10472–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sanders JD; Grinfeld D; Aizikov K; Makarov A; Holden DD; Brodbelt JS Determination of Collision Cross-Sections of Protein Ions in an Orbitrap Mass Analyzer. Anal. Chem 2018, 90 (9), 5896–5902. [DOI] [PubMed] [Google Scholar]
- (40).Rosenberg J; Parker WR; Cammarata MB; Brodbelt JS UV-POSIT: Web-Based Tools for Rapid and Facile Structural Interpretation of Ultraviolet Photodissociation (UVPD) Mass Spectra. J. Am. Soc. Mass Spectrom 2018, 29 (6), 1323–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Morrison LJ; Brodbelt JS Charge Site Assignment in Native Proteins by Ultraviolet Photodissociation (UVPD) Mass Spectrometry. Analyst 2016, 141 (1), 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kwantwi-Barima P; Reinecke T; Clowers BH Increased Ion Throughput Using Tristate Ion-Gate Multiplexing. Analyst 2019, 144 (22), 6660–6670. [DOI] [PubMed] [Google Scholar]
- (43).Kirk AT; Grube D; Kobelt T; Wendt C; Zimmermann S High-Resolution High Kinetic Energy Ion Mobility Spectrometer Based on a Low-Discrimination Tristate Ion Shutter. Anal. Chem 2018, 90 (9), 5603–5611. [DOI] [PubMed] [Google Scholar]
- (44).Reinecke T; Davis AL; Clowers BH Determination of Gas-Phase Ion Mobility Coefficients Using Voltage Sweep Multiplexing J. Am. Soc. Mass Spectrom 2019. DOI: 10.1007/s13361-019-02182-x. [DOI] [PubMed] [Google Scholar]
- (45).Chen B; Young J; Leng F DNA Bending by the Mammalian High-Mobility Group Protein AT Hook 2. Biochemistry 2010, 49 (8), 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jeanne Dit Fouque K; Sipe SN; Garabedian A; Mejia G; Su L; Md Lockman Hossen; Chapagain PP; Leng F; Brodbelt JS; Fernandez-Lima F Exploring the Conformational and Binding Dynamics of HMGA2 DNA Complexes Using Trapped Ion Mobility Spectrometry − Mass Spectrometry. J. Am. Soc. Mass Spectrom 2022, DOI: 10.1021/jasms.2c00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Theisen A; Yan B; Brown JM; Morris M; Bellina B; Barran PE Use of Ultraviolet Photodissociation Coupled with Ion Mobility Mass Spectrometry To Determine Structure and Sequence from Drift Time Selected Peptides and Proteins. Anal. Chem 2016, 88 (20), 9964–9971. [DOI] [PubMed] [Google Scholar]
- (48).Sipe SN; Brodbelt JS Impact of Charge State on 193 Nm Ultraviolet Photodissociation of Protein Complexes. Phys. Chem. Chem. Phys 2019, 21 (18), 9265–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wysocki VH; Tsaprailis G; Smith LL; Breci LA Mobile and Localized Protons: A Framework for Understanding Peptide Dissociation. J.l of Mass Spectrom 2000, 35 (12), 1399–1406. [DOI] [PubMed] [Google Scholar]
- (50).Chanthamontri C; Liu J; McLuckey SA Charge State Dependent Fragmentation of Gaseous α-Synuclein Cations via Ion Trap and Beam-Type Collisional Activation. Int. J. Mass Spectrom 2009, 283 (1–3), 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Julian RR The Mechanism Behind Top-Down UVPD Experiments: Making Sense of Apparent Contradictions. J. Am. Soc. Mass Spectrom 2017, 28 (9), 1823–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mistarz UH; Bellina B; Jensen PF; Brown JM; Barran PE; Rand KD UV Photodissociation Mass Spectrometry Accurately Localize Sites of Backbone Deuteration in Peptides. Anal. Chem 2018, 90 (2), 1077–1080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.