

Abstract
Enhancer of zeste homolog 2 (EZH2), a catalytic subunit of polycomb repressive complex 2 (PRC2), is overexpressed in triple-negative breast cancer (TNBC), correlating with poor prognosis. However, EZH2 catalytic inhibitors are ineffective in suppressing the growth of TNBC cells that are dependent on EZH2. Knockdown of EZH2 inhibits the proliferation of these cells, suggesting that EZH2 protein overexpression but not its catalytic activity is critical for driving TNBC progression. Several proteolysis targeting chimera (PROTAC) degraders of EZH2, including the von Hippel–Lindau (VHL)-recruiting PROTAC YM281, have been reported. However, the effects of these EZH2 PROTACs in TNBC cells were not investigated. Here, we report the discovery and characterization of a novel, potent, and selective EZH2 PROTAC degrader, MS8815 (compound 16), which induced robust EZH2 degradation in a concentration-, time-, and proteasome-dependent manner in TNBC cells. Importantly, 16 effectively suppressed the cell growth in multiple TNBC cell lines and primary patient TNBC cells.
Keywords: triple-negative breast cancer, PROTAC, EZH2, epigenetics, cancer treatment
Epigenetic regulation of gene expression plays a fundamental role in disease progression, especially relating to cancers.1 Targeting the regulatory proteins that mediate these epigenetic changes has been considered a promising therapeutic avenue to advance cancer treatment.2 Enhancer of zeste homolog 2 (EZH2) is the main catalytic subunit of the polycomb repressive complex 2 (PRC2), which is responsible for epigenetically silencing transcription by catalyzing trimethylation of lysine 27 on histone H3 (H3K27me3).3−5 Two additional components in PRC2, embryonic ectoderm development (EED) and suppressor of zeste 12 (SUZ12), are required for EZH2-mediated H3K27me3, which is the primary methylation mark associated with PRC2- and EZH2-dependent diseases.6−8 As the main catalytic subunit of PRC2, EZH2’s role in oncogenesis has been evaluated in a variety of cancers, including diffuse large B-cell lymphomas (DLBCL),9 gastric cancer,10 ovarian cancer,11 prostate cancer,12 and breast cancer.12 For example, DLBCL typically displays a dependence on the canonical methyltransferase activity of PRC2. EZH2 frequently harbors somatic gain-of-function mutations in its catalytic domain that lead to the enhanced methyltransferase activity of PRC2 in DLBCL, resulting in suppressed gene expression.13 Another example of canonical PRC2 regulation is observed with the tumor suppressor p21. EZH2 binds the p21 promoter region, depositing the repressive H3K27me3 mark and resulting in p21 transcriptional silencing, thus promoting the growth of gastric cancer cells.14
EZH2 drives oncogenesis not only in a canonical H3K27me3-dependent manner but also through non-canonical H3K27me3-independent functions.15,16 For example, EZH2 has been shown to methylate non-histone proteins such as GATA4,17 STAT3,18 and β-catenin,19 among others. EZH2 can also act as a transcriptional activator by direct binding to a promoter region of several targets, such as NOTCH120 and TRIM28.21 Recently, we have reported that EZH2 drives tumorigenesis in acute leukemias through a catalytically independent non-canonical function in activating oncogenes by binding cMyc and p300 via its hidden transactivation domain (TAD).16 EZH2 has also been found to possess several non-canonical roles in solid tumors, particularly breast cancer.15 When phosphorylated at Thr367 by p38, EZH2 can dissociate from PRC2, enter the cytoplasm, and drive migration and invasion of metastasis in PR-, ER-, and triple-negative breast cancers in a PRC2-independent manner.22
Several EZH2 catalytic inhibitors, such as GSK126,9 CPI-1205,23 PF-06821497,24 SHR2554,25 DS-3201,26 EPZ-643827,28 (also known as Tazemetostat), UNC1999,29 and C24,30 have been developed. Six of them (CPI-1205, GSK126, EPZ-6438, SHR2554, DS-3201, and PF-06821497) have advanced into clinical trials for investigating their effects in a variety of H3K27me3-dependent tumors, such as DLBCL5,31,32 (Figure 1). Among them, EPZ-6438 is the first FDA-approved EZH2 inhibitor for the treatment of patients with metastatic or locally advanced epithelioid sarcoma and follicular lymphoma,33 ushering in a new wave of potential for pharmacological targeting of epigenetic proteins in cancer treatment. However, EZH2 inhibition in DLBCL can result in resistance to EZH2 inhibitors by upregulating IGF-1R, MEK, or PI3K pathways to circumvent EZH2 inhibition.34 Furthermore, it has been shown that acquired resistance mutations (such as C663Y and Y726F) in EZH2 render EZH2 inhibitors GSK126 and EPZ-6438 ineffective.34
Figure 1.

Chemical structures of representative EZH2 catalytic inhibitors.
A significant challenge that prevents further utilization of EZH2 inhibitors is that many EZH2-dependent cancers, especially solid tumors, are not dependent on the catalytic activity of EZH2 for tumor progression.5,15,22,35−37 Hence, targeting the non-catalytic functions of EZH2 through targeted protein degradation (TPD) has been investigated. Several EZH2 protein degraders have been developed to date. We previously reported MS1943, a hydrophobic tag (HyT)-mediated EZH2 degrader, which effectively induced EZH2 degradation and suppressed the growth of triple-negative breast cancer (TNBC) cells both in vitro and in vivo.38 However, the mechanism of HyT-mediated degradation has yet to be fully elucidated. Proteolysis targeting chimeras (PROTACs) are a class of heterobifunctional small molecules which link a binder of the protein of interest (POI) to an E3 ligase ligand, leading to the selective polyubiquitination and subsequent degradation of the POI at the 26S proteasome.39 Recently, MS17716 and E7,40 two cereblon (CRBN)-recruiting EZH2 PROTACs, and YM281, a von Hippel–Lindau (VHL)-recruiting EZH2 PROTAC,41 have been reported. While these compounds induced EZH2 degradation and inhibited cell growth in lymphoma and leukemia cell lines, the anti-proliferative effects of these compounds in solid tumors such as breast cancer were not investigated, even though EZH2 degradation was achieved in prostate40,41 and lung cancer cell lines.41 Herein, we report the discovery of a novel, potent, and selective VHL-recruiting EZH2 PROTAC degrader, MS8815 (compound 16), through a structure–activity relationship (SAR) study in TNBC cells. We also developed MS8815N (compound 17), which is incapable of recruiting the VHL E3 ligase but retains the same EZH2 binding moiety and linker, as a negative control for degrader 16. Compound 16 but not 17 effectively induced EZH2 degradation in a time- and concentration-dependent manner through the ubiquitin–proteasome system (UPS) in multiple TNBC cell lines. Degrader 16 was more effective in inducing EZH2 degradation and suppressing the proliferation in multiple TNBC cell lines than the previously reported VHL-recruiting EZH2 PROTAC YM281, while the parental inhibitor EPZ-6438 displayed no effect on EZH2 degradation and cell growth inhibition. Furthermore, degrader 16, but not compound 17 or EPZ-6438, effectively inhibited the growth of primary patient TNBC cells. Moreover, compound 16 is bioavailable in mice, with sufficient plasma exposure levels achieved after intraperitoneal (IP) injection. Collectively, these results demonstrate that 16 is a highly effective VHL-recruiting EZH2 PROTAC degrader. Our work also provides further support that pharmacological degradation of EZH2 is a promising therapeutic strategy for the treatment of TNBC.
Results and Discussion
Design and Evaluation of Putative EZH2 Degraders in TNBC Cells
In the design of our EZH2 degraders, we selected EPZ-6438 as the EZH2 binder because EPZ-6438 is the first and only FDA-approved EZH2 inhibitor with high potency (Ki = 2.5 ± 0.5 nM) and selectivity. We reported the first EZH2-selective degrader MS1943, which is a HyT-based EZH2 degrader, and employed C24 as the EZH2 binding moiety with the piperazine motif identified as the solvent-exposed position.38 EPZ-6438 is structurally similar to C24, and we reasoned that the morpholinyl group of EPZ-6438 was potentially exposed to the solvent, providing a suitable position to attach a linker with an E3 ligase ligand without interfering with EZH2 binding. Furthermore, the molecular docking model of EPZ-6438 also confirmed our hypothesis that the morpholine moiety extended into solvent without interactions with other PRC2 subunits.41 We therefore replaced this morpholinyl group with the piperazine moiety as the exit vector to generate precursor 1 (Figure 2). We then generated a series of EZH2 putative degraders, 2-16, by connecting precursor 1 to VHL-142,43 (KD = 188 nM), a well-known ligand of the VHL E3 ligase,44 through various linkers of different composition and length (Figure 2).
Figure 2.

Chemical structures of precursor 1, VHL E3 ligase ligand VHL-1, and designed putative EZH2 degraders.
We first assessed the effect of these compounds on reducing the EZH2 protein level by Western blotting in MDA-MB-453 cells, a TNBC cell line (Figure 3A). Compounds 2–4, which contain a short PEG linker, induced little or no EZH2 degradation, while compounds 5–7, bearing a medium length PEG linker, were somewhat effective in reducing the EZH2 protein level at 3 μM. However, compounds 8–10, with a long PEG linker (4–5 PEGs), had no effect on degrading EZH2. Interestingly, among compounds with an alkylene linker (compounds 11–16), compounds 11–14, which contain a short linker (2–5 methylene units), were generally less effective in reducing the EZH2 protein level than compounds 15 and 16, which contain a longer linker (6–7 methylene units). Compounds 15 and 16 induced nearly complete degradation of EZH2, with compound 16 exhibiting the most robust degradation at 0.3 μM. In addition, we also evaluated the effect of these compounds on degrading EZH2 in BT549 cells, another TNBC cell line (Figure 3B). Compounds with a short PEG linker (compounds 2–6) were generally ineffective in reducing the EZH2 protein level in BT549 cells. Consistent with the results in MDA-MB-453 cells, compound 7 with a three-PEG unit linker effectively degraded EZH2 at 3 μM in BT549 cells, and compounds with a long PEG linker (compounds 8–10) were inactive in inducing EZH2 degradation. Among the compounds with an alkylene linker (compounds 11–16), compounds 15 and 16 with a six- and seven-methylene linker, respectively, exhibited the most effective EZH2 degradation in BT549 cells. Overall, the SAR results observed in MDA-MB-453 cells are generally in agreement with that observed in BT549 cells. Through these SAR studies, we identified compound 16 as the most effective EZH2 degrader when compared to 15 in both MDA-MB-453 and BT549 cells. We therefore selected compound 16 for further evaluation and characterization.
Figure 3.

Effect of compounds 2–16 on reducing the EZH2 protein level in TNBC cells. MDA-MB-453 (A) and BT549 (B) cells were treated with DMSO or the indicated compound at 0.3 and 3 μM for 48 h. The cell lysates were analyzed by Western blotting to examine the protein level of EZH2. β-Actin, H3, or both were used as the loading controls. Results shown are representative of at least two independent experiments.
To facilitate our understanding of the mechanism of action (MOA) and phenotypic effects mediated by EZH2 degradation induced by compound 16, we developed a structurally similar negative control, compound 17 (Figure 4). Compound 17 features a benzyl-protected hydroxyl proline group (VHL-1-Bn)45 at the VHL binding region while keeping the same EZH2 binding moiety and linker as compound 16 (Figure 2). This modification was designed to disrupt the key interactions between the hydroxyl group and VHL,46 as recently demonstrated and confirmed by isothermal titration calorimetry (ITC).45 Moreover, we also synthesized the previously reported VHL-recruiting EZH2 PROTAC YM281 as a positive control (Figure 4).
Figure 4.

Chemical structures of the negative control (compound 17) and previously reported VHL-recruiting EZH2 PROTAC YM281.
Compound 16 Is Selective for EZH2 over Other Protein Methyltransferases
We next assessed the potency of the parental inhibitor EPZ-6438, degrader 16, and its negative control 17 in inhibiting the EZH2 enzymatic activity by using a biochemical assay (Figure 5A). In comparison with EPZ-6438 (IC50 = 3.3 ± 0.1 nM), degrader 16 (IC50 = 8.6 ± 0.9 nM) displayed 2.6-fold lower inhibitory potency but still maintained high potency in inhibiting the EZH2 methyltransferase activity. As expected, the negative control 17 that was designed to bind EZH2, but not VHL, retained a comparable potency in EZH2 inhibition (IC50 = 13 ± 7.6 nM). These results confirmed our design hypothesis that introducing a linker attached to the VHL-1 ligand at the piperazine motif of precursor 1 had no significant detrimental effect on EZH2 binding. Moreover, degrader 16 (IC50 = 62 ± 10 nM) also showed significant potency at inhibiting EZH1, which is another catalytic subunit of PRC2 and is highly homologous to EZH2 (Figure 5B). This result is consistent with previous reports that the parental compound EPZ-6438 inhibited EZH1 in addition to EZH2.28 Interestingly, we found that compound 16 did not induce significant EZH1 degradation in MDA-MB-453 cells (Figure 5C) even though it potently inhibited EZH1 in a biochemical assay (Figure 5B). We also evaluated the EZH1 protein level in SUM159 cells treated with compound 16 and found that 16 did not degrade EZH1 in SUM159 cells (Figure S1). It has been reported that PROTACs can induce isoform/subtype-selective degradation, even though the POI ligands utilized in the PROTACs are not isoform/subtype-selective.47−50 To further assess the selectivity of degrader 16, we performed inhibition assays with a panel of protein methyltransferases (Figure 5D). About 50% inhibition of MLL1 at 10 μM was observered, and no appreciable inhibition (>50% at 10 μM) was detected across the panel of other 19 protein methyltransferases, demonstrating that compound 16 is generally selective for EZH2 over other protein methyltransferases.
Figure 5.

Compound 16 is selective for EZH2 over other protein methyltransferases. (A) Effects of EPZ-6438, 16, and 17 on inhibiting the EZH2 catalytic activity in an EZH2 methyltransferase assay (3H-labeled S-adenosyl methionine [SAM] was used as the methyl donor). IC50 results shown are the mean values ± SD from duplicate experiments. (B) Effect of 16 on inhibiting the EZH1 catalytic activity in an EZH1 methyltransferase assay (3H-labeled SAM was used as the methyl donor). IC50 results shown are the mean values ± SD from duplicate experiments. (C) Effect of 16 on reducing the EZH1 protein level in MDA-MB-453 cells. MDA-MB-453 cells were treated with DMSO or 16 at the indicated concentration for 48 h. The cell lysates were analyzed by Western blotting to examine the EZH1 protein level. β-Actin and H3 were used as the loading controls. Results shown are representative of at least two independent experiments. (D) Selectivity of compound 16 (at 10 μM) against a panel of 20 protein methyltransferases in radioactive methyltransferase assays. Error bars represent ± SD in duplicate experiments.
Compound 16 Induced Potent EZH2 Degradation in a Time-, Concentration-, and UPS-Dependent Manner
We next further evaluated the EZH2 degradation activity of compound 16 in MDA-MB-453 cells (Figure 6). Noticeable EZH2 degradation and concomitant reduction in the protein levels of other PRC2 core components EED and SUZ12 were observed at as early as 6 h, and maximal degradation was achieved at 48 h, suggesting a relatively slow kinetics of the EZH2 degradation induced by compound 16 (Figure 6A,B). We also found that 16 degraded EZH2, EED, and SUZ12 in a concentration-dependent manner, and the DC50 value was 140 ± 56 nM for EZH2 degradation in MDA-MB-453 cells (Figure 6C–E). Interestingly, compound 16 significantly reduced the repressive H3K27me3 mark at 48–72 h post-treatment (Figure 6A). As expected, EPZ-6438 effectively reduced the H3K27me3 mark in MDA-MB-453 cells (Figure S2).
Figure 6.

Compound 16 induces EZH2 degradation in a time- and concentration-dependent manner. (A) Western blots of EZH2, SUZ12, EED, and H3K27me3 post-treatment of MDA-MB-453 cells with DMSO (48 h) or compound 16 (1 μM) at the indicated time points. β-Actin and H3 were used as the loading controls. (B) Quantification of EZH2 blots in (A) and their replicates. (C) Western blots of EZH2, SUZ12, and EED post-treatment of MDA-MB-453 cells with DMSO or compound 16 at the indicated concentrations for 48 h. β-Actin was used as the loading control. (D) Quantification of EZH2 blots in (C) and their replicate. (E) DC50 curve of 16, generated using the data in (D).
To investigate the MOA of the EZH2 degradation induced by compound 16, we conducted a series of competition experiments (Figure 7). The EZH2 degradation induced by compound 16 (1 μM) was completely rescued when co-treated with an excess amount of either the EZH2 inhibitor EPZ-6438 (5 μM) or VHL ligand VHL-1 (5 μM) in MDA-MB-453 cells for 24 h. As shown in Figure 7A, treatment with EPZ-6438 or VHL-1 ligand at 5 μM alone did not reduce the EZH2 protein level, confirming that 16 is competing with the parental inhibitor EPZ-6438 and VHL-1 for the same binding sites in EZH2 and VHL, respectively. Next, we assessed whether 16-mediated EZH2 degradation was proteasome-dependent. We pre-treated MDA-MB-453 cells with the proteasome inhibitor MG132 at 0.5 μM for 30 min and found that EZH2 degradation induced by 16 was fully recovered. Similarly, treatment with MG132 alone did not result in any EZH2 degradation, providing strong evidence that 16-induced degradation is proteasome-dependent (Figure 7B). Lastly, we also found that the EZH2 degradation induced by 16 can be partially rescued by pre-treatment with the NEDD8-activating E1 enzyme (NAE) inhibitor MLN4924, suggesting that an active cullin RING family E3 ubiquitin ligase (CRL) complex was required for 16-induced EZH2 degradation (Figure 7C and Figure S3). We next evaluated the ability of compound 17 to degrade EZH2 in BT549 and MDA-MB-453 cells. Compound 17 treatment (48 h, 3 μM) did not lead to EZH2 degradation in both BT549 (Figure 7D) and MDA-MB-453 cells (Figure 7E), confirming compound 17 as a negative control and further suggesting that binding to the E3 ligase VHL is required for 16-induced EZH2 degradation. Collectively, these results clearly demonstrate that compound 16 is a bona fide EZH2 PROTAC degrader.
Figure 7.

Compound 16 induces EZH2 degradation through the UPS. (A) MDA-MB-453 cells were co-incubated with compound 16 (1 μM) and VHL-1 (5 μM) or EPZ-6438 (5 μM) for 24 h. (B) MDA-MB-453 cells were pre-treated with MG132 (0.5 μM) for 30 min and then co-incubated with compound 16 (1 μM) for 24 h. (C) MDA-MB-453 cells were pre-treated with MLN4924 (0.5 μM) for 30 min and then co-incubated with compound 16 (1 μM) for 24 h. (D) BT549 cells were treated with DMSO, compound 16 (3 μM), or compound 17 (3 μM) for 48 h. (E) MDA-MB-453 cells were treated with DMSO, compound 16 (3 μM), or compound 17 (3 μM) for 48 h. The EZH2 protein level was determined by Western blot analysis. β-Actin, H3, or both were used as the loading controls. Results shown in panels A–E are representative of three independent experiments.
Degrader 16 Displayed Superior Anti-proliferative Activity in TNBC Cells Compared to the EZH2 Inhibitor EPZ-6438 and Compound 17
We next assessed the growth inhibition activity of degrader 16 together with the negative control 17, as well as the parental inhibitor EPZ-6438, and reported VHL-recruiting EZH2 PROTAC YM281 in a panel of EZH2-sensitive TNBC cell lines (BT549, MDA-MB-468, MDA-MB-453, and SUM159). In all four cell lines, degrader 16 effectively suppressed the cell proliferation with GI50 values ranging from 1.7 to 2.3 μM, while EPZ-6438 or compound 17 had no effect on cell growth inhibition (Figure 8). Furthermore, EPZ-6438 effectively inhibited H3K27me3 in MDA-MB-453 cells (Figure S2), suggesting that EZH2 degradation, but not EZH2 inhibition, is a major contributor to compound 16’s anti-proliferation effect in TNBC cells. YM281 was also effective in inhibiting the cell growth in these TNBC cell lines but exhibited slightly weaker inhibitory potency (GI50 = 2.9–3.3 μM) than degrader 16 in BT549, MDA-MB-468, and SUM159 cells. Overall, these results indicate that EZH2 degraders 16 and YM281 have anti-proliferative activities much superior to those of EZH2 inhibitors EPZ-6438 and 17 in TNBC cells, and degrader 16 is slightly more potent than YM281.
Figure 8.

Compound 16 shows superior cellular growth inhibition activity in a panel of TNBC cells compared to the EZH2 inhibitor EPZ-6438 and compound 17. BT549 (A), MDA-MB-468 (B), SUM159 (C), and MDA-MB-453 (D) cells were treated with serial dilution (starting from 10 μM, with 2-fold dilution) of 16, EPZ-6438, YM281, or negative control 17 for 5 days. Cell viability was determined using a WST-8 assay. GI50 results shown are the mean values ± SD from three independent experiments.
We next conducted the side-by-side comparison of VHL-recruiting EZH2 PROTAC degraders 16 and YM281 to probe their capability in inducing EZH2 degradation in these four TNBC cell lines (BT549, MDA-MB-468, SUM159, and MDA-MB-453). We found that compound 16 was slightly more effective in degrading EZH2 than YM281 in BT549, MDA-MB-468, and SUM159 cells (Figure 9A–C). In MDA-MB-453 cells, compound 16 and YM281 displayed similar effectiveness in degrading EZH2 (Figure 9D). The EZH2 degradation results of these two compounds are consistent with their cell viability data (Figure 8). Taken together, the head-to-head comparisons of these two VHL-recruiting PROTACs in their EZH2 degradation and cell anti-proliferation activities suggest that compound 16 is a slightly better EZH2 degrader than YM281. In addition, we compared the cell anti-proliferation effect of 16 to our previously reported CRBN-recruiting EZH2 degrader MS17716 in BT549 cells. As indicated in Figure S5A, 16 was more effective than MS177 in suppressing the growth in BT549 cells. Furthermore, we also found that 16 induced EZH2 degradation more effectively than MS177 in BT549 cells, which was in line with the cell viability data (Figure S5B,C).
Figure 9.

Compound 16 displays superior EZH2 degradation compared to YM281 in TNBC cells. BT549 (A), MDA-MB-468 (B), SUM159 (C), and MDA-MB-453 (D) cells were treated with DMSO, 16, or YM281 at indicated concentrations for 48 h. Cell lysates were analyzed by Western blotting to examine the EZH2 protein level with β-actin or H3 used as the loading control. Results shown are representative of at least two independent experiments.
In order to evaluate 16 in a more clinically relevant model, we utilized primary patient TNBC ductal adenocarcinoma cells (515a) to evaluate the growth inhibitory effects of 16. Compound 16 exerted potent growth inhibition (GI50 = 1.4 ± 0.05 μM) in the primary patient TNBC cells, while EZH2 inhibitors EPZ-6438 and compound 17 had no effect (Figure 10A), recapitulating the effect observed in TNBC cell line models (Figure 8). We next assessed the effect of 16 on degrading EZH2 in 515a cells. We found that compound 16 effectively degraded EZH2 and achieved nearly complete degradation of EZH2 at a concentration as low as 100 nM (Figure 10B). These results suggest a potential translational aspect of EZH2 PROTAC degraders in targeting TNBC.
Figure 10.
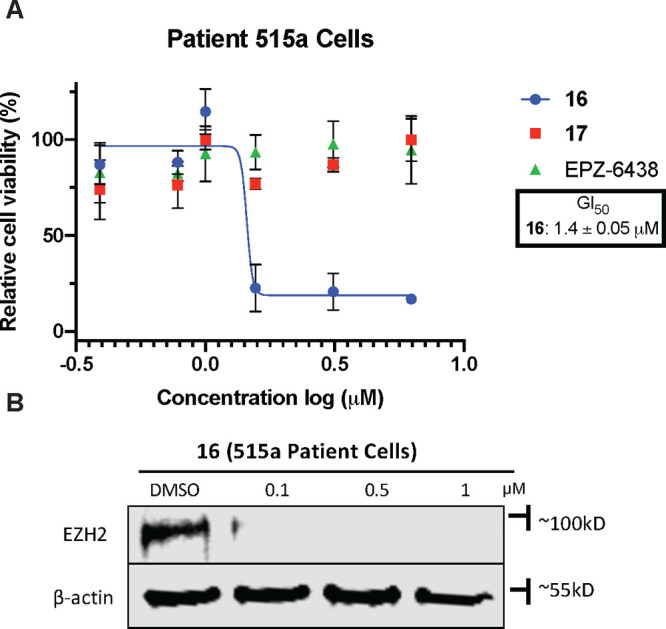
Compound 16 displays potent cell growth inhibition and EZH2 degradation in primary TNBC patient cells (515a ductal TNBC). (A) 515a cells were treated with serial dilution of 16 (starting from 10 μM, with 2-fold dilution) for 5 days. Cell viability was determined using a WST-8 assay. GI50 results shown are the mean values ± SD from three independent experiments. (B) 515a cells were treated with DMSO and 16 at concentrations of 0.1, 0.5, and 1 μM for 48 h, and the cell lysate was analyzed with Western blotting to assess the EZH2 protein level. β-Actin was used as the loading control. Results shown are representative of two independent experiments.
Compounds 16 Is Bioavailable in Mice
Lastly, we assessed the pharmacokinetic (PK) properties of compound 16 in mice (Figure 11). The maximum plasma concentration (Cmax) of 3.7 μM was achieved at 1 h after a single-dose IP administration of 16 at 50 mg/kg. The plasma concentrations remained at approximately 3 μM over 4 h and above 250 nM over 12 h post-injection. This high plasma exposure suggested that degrader 16 could be potentially suitable for future in vivo efficacy studies. Moreover, it should be noted that the mice treated with compound 16 in the PK study did not display obvious clinical signs of toxicity, suggesting that degrader 16 was well tolerated at the tested dose.
Figure 11.
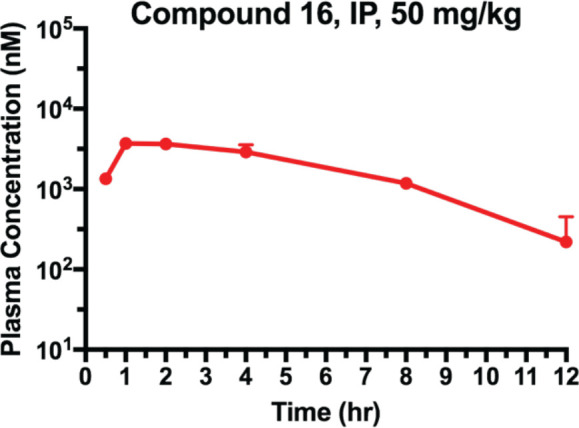
EZH2 degrader 16 is bioavailable in mice. Plasma concentrations of 16 following IP administration in male Swiss Albino mice at a single dose of 50 mg/kg over 12 h. Experiments were performed in biological triplicate per time point, with the values representing the mean concentrations ± SEM.
Chemical Syntheses
The precursor 1(51) and compounds 18–3252 were prepared following the previously reported procedures. As shown in Scheme 1, EPZ-6438-based VHL-recruiting EZH2 degraders 2–16 were prepared by amide coupling of intermediate 1 and various VHL-1-derived linkers 18–32.
Scheme 1. Syntheses of Compounds 2–16.

Reaction conditions: (a) 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI), 1-hydroxy-7-azabenzo-triazole (HOAt), N-methylmorpholine (NMM), DMSO, rt, 12 h.
The synthetic route for preparing the negative control compound 17 is described in Scheme 2. Intermediate 35 was prepared from commercially available compounds 33 and 34 through an amide coupling reaction followed by the removal of the Boc-group. Amide coupling between 35 and commercially available (S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (36) and subsequent Boc-deprotection afforded intermediate 37, which was then coupled with nonanedioic acid (38) to furnish 39. Negative control compound 17 was synthesized from intermediates 1 and 39 by amide coupling.
Scheme 2. Synthesis of Negative Control Compound 17.

Reaction conditions: (a) EDCI, HOAt, NMM, DMSO, rt, 12 h; (b) TFA, DCM, rt, 30 min.
Conclusions
We previously developed MS1943, the first EZH2-selective degrader, which is a hydrophobic tag-based degrader and effectively induced EZH2 degradation and suppressed the cell growth in TNBC cells.38 Several EZH2 PROTAC degraders, including E740 and MS177,16 which recruit the E3 ligase CRBN, and YM281,41 which recruits the E3 ligase VHL, have been reported very recently. However, the effects of these EZH2 PROTAC degraders in TNBC cells were not investigated. Through an SAR study, we discovered a novel EPZ-6438-based VHL-recruiting EZH2 PROTAC degrader, compound 16, which effectively reduced the EZH2 protein level in a concentration-, time-, and UPS-dependent manner in a variety of TNBC cells. Compound 16 was also able to degrade other PRC2 core components, EED and SUZ12, and significantly decreased the H3K27me3 mark. Compound 16 was highly selective for EZH2 over other protein methyltransferases except EZH1, and interestingly, it did not degrade EZH1 even though it potently inhibited the EZH1 methyltransferase activity biochemically. We also developed a structurally similar compound, 17, which was designed to be unable to recruit the VHL E3 ligase but retain the same EZH2 binding moiety and linker, as a negative control of 16. As expected, compound 17 maintained similar EZH2 inhibitory potency but was incapable of inducing EZH2 degradation. Phenotypically, EZH2 PROTAC degraders 16 and YM281 displayed anti-proliferative activities much superior to those of EZH2 inhibitors EPZ-6438 and compound 17 in multiple TNBC cell lines. We also determined that degrader 16 was slightly more effective than YM281 in degrading EZH2 and suppressing the proliferation in TNBC cells. Furthermore, degrader 16 also effectively degraded EZH2 and suppressed cell growth in primary TNBC patient cells. Lastly, degrader 16 is bioavailable via IP administration in a mouse PK study and could potentially be used for in vivo efficacy studies. Collectively, our results demonstrate that compound 16 is a highly effective EZH2 PROTAC degrader and suggest that it could serve as a potentially useful chemical tool to explore the therapeutic utility of EZH2 degradation in solid tumors such as TNBC. This work also provides further support for pharmacological degradation of EZH2 as a potential therapeutic strategy for treating TNBC.
Experimental Section
Chemistry General Procedures
All solvents and reagents purchased from chemical vendors were used directly without further purification. Normal and reverse-phase flash chromatography were carried out through a Teledyne ISCO CombiFlash instrument and HP C18 RediSep Rf reverse-phase silica columns, respectively. All final compounds 2–17 for biological evaluation were purified with preparative high-performance liquid chromatography (HPLC) on an Agilent Prep 1200 series with the UV detector set to 254 or 220 nm, with solvent A (0.1% of TFA in water) and solvent B (acetonitrile) as eluents at a flow rate of 40 mL/min at rt. Purities of the final compounds were assessed by HPLC and were determined to be greater than 95%. The conditions of HPLC to determine the purity are described as follows: Agilent 1200 series system, 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column; 1–99% gradient of 0.1% trifluoroacetic acid in water, and 0.1% trifluoroacetic acid in acetonitrile as eluents; flow rate, 0.4 mL/min; acquisition time, 8 min. High-resolution mass spectra (HRMS) data were acquired on an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Nuclear magnetic resonance spectra were acquired on either a Bruker DXI 800 MHz or an AVANCE NEO 600 MHz NMR spectrometer. Proton and carbon nuclear magnetic resonance (1H NMR and 13C NMR) spectra are reported in parts per million (ppm) on the δ scale in the following format: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant, and integration.
(2S,4R)-1-((S)-2-(2-(2-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-2-oxoethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (2)
To a solution of intermediate 1(51) (11.4 mg, 0.02 mmol) in DMSO (1 mL) were added intermediate 18 (10.9 mg, 0.02 mmol, 1.0 equiv), EDCI (5.8 mg, 0.03 mmol, 1.5 equiv), HOAt (4.1 mg, 0.03 mmol, 1.5 equiv), and NMM (6.1 mg, 0.06 mmol, 3.0 equiv). After stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10%–100% methanol/0.1% TFA in H2O) to afford compound 2 as a white solid (15.5 mg, yield 70%). 1H NMR (800 MHz, CD3OD) δ 8.97 (s, 1H), 7.84 (d, J = 8.1 Hz, 3H), 7.70 (s, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.51–7.36 (m, 4H), 6.16 (s, 1H), 4.71 (s, 1H), 4.60 (t, J = 8.0 Hz, 1H), 4.53 (s, 4H), 4.49 (d, J = 15.4 Hz, 1H), 4.47–4.37 (m, 6H), 4.18 (d, J = 14.9 Hz, 1H), 4.12 (d, J = 15.2 Hz, 1H), 4.01 (d, J = 11.6 Hz, 3H), 3.93 (d, J = 11.0 Hz, 1H), 3.84 (dd, J = 11.0, 4.0 Hz, 1H), 3.69–3.58 (m, 8H), 3.40 (t, J = 11.9 Hz, 3H), 2.48 (s, 3H), 2.45 (s, 3H), 2.42 (s, 3H), 2.28–2.25 (m, 4H), 2.14–2.08 (m, 1H), 1.84–1.75 (m, 3H), 1.09 (s, 9H), 1.06–1.01 (m, 3H). HRMS (m/z) for C60H78N9O9S+ [M+H]+: calcd 1100.5638, found 1100.5644.
(2S,4R)-1-((S)-2-(3-(3-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-3-oxopropoxy)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (3)
Compound 3 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 19 (11.5 mg, 0.02 mmol, 1.0 equiv). Compound 3 was obtained as a white solid (18.6 mg, yield 82%). 1H NMR (800 MHz, CD3OD) δ 8.96 (s, 1H), 7.91–7.85 (m, 1H), 7.85–7.79 (m, 2H), 7.70 (s, 1H), 7.64 (t, J = 9.0 Hz, 2H), 7.48 (d, J = 7.9 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 6.16 (s, 1H), 4.69 (s, 1H), 4.64–4.57 (m, 1H), 4.56–4.50 (m, 5H), 4.49–4.36 (m, 3H), 4.06–3.97 (m, 3H), 3.92 (d, J = 11.1 Hz, 1H), 3.83 (dd, J = 11.0, 3.8 Hz, 1H), 3.79–3.61 (m, 5H), 3.40 (t, J = 11.8 Hz, 3H), 2.77–2.64 (m, 3H), 2.57–2.50 (m, 3H), 2.46 (s, 6H), 2.42 (s, 9H), 2.27 (s, 4H), 2.14–2.08 (m, 1H), 1.84–1.80 (m, 3H), 1.06 (s, 9H), 1.04–1.00 (m, 3H). HRMS (m/z) for C62H82N9O9S+ [M+H]+: calcd 1128.5951, found 1128.5936.
(2S,4R)-1-((S)-2-(2-(2-(2-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-2-oxoethoxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (4)
Compound 4 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 20 (11.8 mg, 0.02 mmol, 1.0 equiv). Compound 4 was obtained as a white solid (18.4 mg, yield 80%). 1H NMR (800 MHz, CD3OD) δ 8.98 (s, 1H), 7.90 (s, 1H), 7.84 (d, J = 7.9 Hz, 2H), 7.74 (s, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.48–7.38 (m, 4H), 6.16 (s, 1H), 4.72 (s, 1H), 4.59–4.47 (m, 6H), 4.47–4.33 (m, 4H), 4.28 (s, 1H), 4.09–4.04 (m, 2H), 4.04–3.98 (m, 2H), 3.90–3.80 (m, 2H), 3.80–3.63 (m, 13H), 3.40 (t, J = 12.3 Hz, 3H), 2.49 (s, 2H), 2.47 (s, 5H), 2.42 (s, 3H), 2.27 (s, 4H), 2.15–2.08 (m, 1H), 1.82 (s, 3H), 1.05 (d, J = 4.2 Hz, 9H), 1.04–0.99 (m, 3H). HRMS (m/z) for C62H82N9O10S+ [M+H]+: calcd 1144.5900, found 1144.5908.
(2S,4R)-1-((S)-2-(3-(2-(3-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-3-oxopropoxy)ethoxy)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (5)
Compound 5 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 21 (12.3 mg, 0.02 mmol, 1.0 equiv). Compound 5 was obtained as a white solid (17.6 mg, yield 75%). 1H NMR (800 MHz, CD3OD) δ 8.97 (s, 1H), 7.88 (s, 1H), 7.85 (d, J = 7.9 Hz, 2H), 7.71 (s, 1H), 7.66 (d, J = 7.7 Hz, 2H), 7.49 (d, J = 7.7 Hz, 2H), 7.43 (d, J = 7.6 Hz, 2H), 6.16 (s, 1H), 4.68 (s, 1H), 4.61–4.49 (m, 6H), 4.46 (s, 2H), 4.39 (d, J = 15.4 Hz, 1H), 4.05–3.97 (m, 3H), 3.90 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 10.8, 3.8 Hz, 2H), 3.79–3.72 (m, 5H), 3.62 (s, 7H), 3.40 (t, J = 11.9 Hz, 3H), 2.77–2.63 (m, 3H), 2.62–2.53 (m, 2H), 2.50 (d, J = 16.4 Hz, 5H), 2.46 (s, 3H), 2.42 (s, 3H), 2.28–2.23 (m, 5H), 2.14–2.07 (m, 1H), 1.87–1.77 (m, 3H), 1.06 (s, 9H), 1.04 (s, 3H). HRMS (m/z) for C64H86N9O10S+ [M+H]+: calcd 1172.6213, found 1172.6227.
(2S,4R)-1-((S)-2-(tert-Butyl)-14-(4-((3′-(((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-4,14-dioxo-6,9,12-trioxa-3-azatetradecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (6)
Compound 6 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 22 (12.6 mg, 0.02 mmol, 1.0 equiv). Compound 6 was obtained as a white solid (18.3 mg, yield 77%). 1H NMR (800 MHz, CD3OD) δ 8.97 (s, 1H), 7.89 (s, 1H), 7.84 (d, J = 7.9 Hz, 2H), 7.73 (s, 1H), 7.63 (d, J = 8.0 Hz, 2H), 7.51–7.40 (m, 4H), 6.16 (s, 1H), 4.70 (s, 1H), 4.60 (t, J = 8.4 Hz, 1H), 4.57–4.50 (m, 5H), 4.49–4.36 (m, 4H), 4.32–4.25 (m, 1H), 4.24–4.16 (m, 1H), 4.05–3.96 (m, 3H), 3.94–3.85 (m, 2H), 3.85–3.81 (m, 1H), 3.78–3.58 (m, 16H), 3.40 (t, J = 11.8 Hz, 3H), 2.50 (s, 3H), 2.46 (d, J = 5.2 Hz, 3H), 2.42 (s, 3H), 2.27 (s, 5H), 2.15–2.10 (m, 1H), 1.86–1.77 (m, 3H), 1.08–0.97 (m, 12H). HRMS (m/z) for C64H86N9O11S+ [M+H]+: calcd 1188.6162, found 1188.6174.
(2S,4R)-1-((S)-2-(tert-Butyl)-16-(4-((3′-(((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-4,16-dioxo-7,10,13-trioxa-3-azahexadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (7)
Compound 7 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 23 (13.2 mg, 0.02 mmol, 1.0 equiv). Compound 7 was obtained as a white solid (18.5 mg, yield 76%). 1H NMR (800 MHz, CD3OD) δ 9.00 (s, 1H), 7.89 (s, 1H), 7.85 (d, J = 7.8 Hz, 2H), 7.72 (s, 1H), 7.66 (d, J = 7.8 Hz, 2H), 7.49 (d, J = 7.9 Hz, 2H), 7.44 (d, J = 7.7 Hz, 2H), 6.16 (s, 1H), 4.67 (s, 1H), 4.59 (t, J = 8.3 Hz, 1H), 4.57–4.49 (m, 5H), 4.46 (s, 2H), 4.39 (d, J = 15.4 Hz, 1H), 4.05–3.98 (m, 2H), 3.91 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 4.0 Hz, 1H), 3.77 (t, J = 6.1 Hz, 2H), 3.76–3.68 (m, 2H), 3.68–3.55 (m, 18H), 3.40 (t, J = 11.6 Hz, 3H), 2.70 (d, J = 22.1 Hz, 2H), 2.60–2.55 (m, 1H), 2.49 (s, 4H), 2.46 (s, 3H), 2.42 (s, 3H), 2.27 (s, 4H), 2.14–2.07 (m, 1H), 1.86–1.76 (m, 3H), 1.05 (d, J = 12.3 Hz, 12H). HRMS (m/z) for C66H90N9O11S+ [M+H]+: calcd 1216.6475, found 1216.6466.
(2S,4R)-1-((S)-2-(tert-Butyl)-19-(4-((3′-(((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-4,19-dioxo-7,10,13,16-tetraoxa-3-azanonadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (8)
Compound 8 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 24 (14.2 mg, 0.02 mmol, 1.0 equiv). Compound 8 was obtained as a white solid (18.2 mg, yield 72%). 1H NMR (800 MHz, CD3OD) δ 8.99 (s, 1H), 7.89 (s, 1H), 7.85 (d, J = 7.9 Hz, 2H), 7.72 (s, 1H), 7.66 (d, J = 7.7 Hz, 2H), 7.49 (d, J = 7.7 Hz, 2H), 7.44 (d, J = 7.8 Hz, 2H), 6.16 (s, 1H), 4.66 (s, 1H), 4.62–4.49 (m, 6H), 4.49–4.41 (m, 2H), 4.39 (d, J = 15.4 Hz, 1H), 4.01 (d, J = 11.7 Hz, 2H), 3.89 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 4.0 Hz, 1H), 3.79–3.68 (m, 6H), 3.68–3.53 (m, 20H), 3.40 (t, J = 11.8 Hz, 3H), 2.81–2.62 (m, 2H), 2.60–2.53 (m, 1H), 2.50 (s, 3H), 2.46 (s, 3H), 2.42 (s, 3H), 2.28–2.22 (m, 5H), 2.14–2.05 (m, 1H), 1.87–1.74 (m, 3H), 1.05 (d, J = 11.0 Hz, 12H). HRMS (m/z) for C68H94N9O12S+ [M+H]+: calcd 1260.6737, found 1260.6730.
(2S,4R)-1-((S)-2-(tert-Butyl)-20-(4-((3′-(((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-4,20-dioxo-6,9,12,15,18-pentaoxa-3-azaicosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9)
Compound 9 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 25 (14.4 mg, 0.02 mmol, 1.0 equiv). Compound 9 was obtained as a white solid (14.4 mg, yield 56%). 1H NMR (800 MHz, CD3OD) δ 8.98 (s, 1H), 7.84 (d, J = 8.0 Hz, 3H), 7.70 (s, 1H), 7.65 (d, J = 7.9 Hz, 2H), 7.48 (d, J = 7.9 Hz, 2H), 7.46–7.38 (m, 2H), 6.16 (s, 1H), 4.64 (s, 1H), 4.57 (dd, J = 16.1, 7.5 Hz, 1H), 4.55–4.42 (m, 6H), 4.39 (d, J = 15.4 Hz, 1H), 4.29 (s, 2H), 4.14–4.04 (m, 2H), 4.01 (d, J = 11.1 Hz, 3H), 3.86–3.77 (m, 2H), 3.75–3.55 (m, 25H), 3.40 (t, J = 12.0 Hz, 3H), 2.50 (s, 3H), 2.45 (s, 3H), 2.42 (s, 3H), 2.29–2.21 (m, 5H), 2.13–2.07 (m, 1H), 1.86–1.76 (m, 3H), 1.06 (s, 9H), 1.04–0.98 (m, 3H). HRMS (m/z) for C68H94N9O13S+ [M+H]+: calcd 1276.6686, found 1276.6695.
(2S,4R)-1-((S)-2-(tert-Butyl)-22-(4-((3′-(((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-4,22-dioxo-7,10,13,16,19-pentaoxa-3-azadocosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (10)
Compound 10 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 26 (15.0 mg, 0.02 mmol, 1.0 equiv). Compound 10 was obtained as a white solid (18.5 mg, yield 71%). 1H NMR (800 MHz, CD3OD) δ 8.97 (s, 1H), 7.85 (d, J = 8.1 Hz, 3H), 7.66 (d, J = 8.0 Hz, 3H), 7.54–7.37 (m, 4H), 6.15 (s, 1H), 4.66 (s, 1H), 4.62–4.49 (m, 5H), 4.49–4.42 (m, 2H), 4.38 (d, J = 15.4 Hz, 1H), 4.01 (d, J = 11.8 Hz, 2H), 3.89 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 3.9 Hz, 1H), 3.78 (t, J = 5.9 Hz, 2H), 3.75–3.68 (m, 1H), 3.68–3.53 (m, 28H), 3.40 (t, J = 12.0 Hz, 3H), 2.79–2.65 (m, 2H), 2.59 (dd, J = 14.5, 6.4 Hz, 1H), 2.50 (s, 3H), 2.45 (s, 3H), 2.42 (s, 3H), 2.30–2.21 (m, 5H), 2.14–2.06 (m, 1H), 1.81 (s, 3H), 1.06 (s, 9H), 1.05–0.98 (m, 3H). HRMS (m/z) for C70H98N9O13S+ [M+H]+: calcd 1304.6999, found 1304.7006.
(2S,4R)-1-((S)-2-(4-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-4-oxobutanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (11)
Compound 11 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 27 (10.6 mg, 0.02 mmol, 1.0 equiv). Compound 11 was obtained as a white solid (5.2 mg, yield 21%). 1H NMR (800 MHz, CD3OD) δ 8.98 (s, 1H), 7.84 (d, J = 7.7 Hz, 3H), 7.65 (d, J = 8.1 Hz, 3H), 7.52–7.40 (m, 4H), 6.16 (s, 1H), 4.62 (s, 1H), 4.61–4.50 (m, 5H), 4.46 (s, 2H), 4.39 (d, J = 15.4 Hz, 1H), 4.01 (s, 2H), 3.91 (d, J = 10.9 Hz, 1H), 3.83 (dd, J = 10.8, 4.0 Hz, 1H), 3.70–3.52 (m, 10H), 3.40 (t, J = 11.7 Hz, 3H), 2.80–2.56 (m, 5H), 2.50 (s, 3H), 2.44 (s, 3H), 2.42 (s, 3H), 2.30–2.22 (m, 4H), 2.15–2.08 (m, 1H), 1.85–1.71 (m, 3H), 1.06 (s, 9H), 1.05–0.98 (m, 3H). HRMS (m/z) for C60H78N9O8S+ [M+H]+: calcd 1084.5689, found 1084.5698.
(2S,4R)-1-((S)-2-(5-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-5-oxopentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (12)
Compound 12 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 28 (10.9 mg, 0.02 mmol, 1.0 equiv). Compound 12 was obtained as a white solid (15.5 mg, yield 71%). 1H NMR (800 MHz, CD3OD) δ 8.96 (d, J = 10.2 Hz, 1H), 7.89–7.75 (m, 3H), 7.75–7.56 (m, 3H), 7.52–7.37 (m, 4H), 6.16 (s, 1H), 4.63 (d, J = 3.8 Hz, 1H), 4.57 (t, J = 8.3 Hz, 1H), 4.56–4.50 (m, 4H), 4.44 (s, 2H), 4.41–4.37 (m, 1H), 4.03–3.98 (m, 2H), 3.95 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 3.9 Hz, 1H), 3.69–3.57 (m, 10H), 3.40 (t, J = 12.0 Hz, 3H), 2.52–2.43 (m, 9H), 2.42 (s, 3H), 2.39–2.35 (m, 2H), 2.27 (s, 4H), 2.14–2.08 (m, 1H), 1.95–1.88 (m, 2H), 1.84–1.75 (m, 3H), 1.07 (s, 9H), 1.05–0.98 (m, 3H). HRMS (m/z) for C61H80N9O8S+ [M+H]+: calcd 1098.5845, found 1098.5836.
(2S,4R)-1-((S)-2-(6-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-6-oxohexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (13)
Compound 13 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 29 (11.2 mg, 0.02 mmol, 1.0 equiv). Compound 13 was obtained as a white solid (20.5 mg, yield 92%). 1H NMR (800 MHz, CD3OD) δ 8.97 (s, 1H), 7.84 (d, J = 7.9 Hz, 3H), 7.65 (d, J = 7.7 Hz, 3H), 7.49 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 7.8 Hz, 2H), 6.16 (s, 1H), 4.66 (s, 1H), 4.59 (t, J = 8.3 Hz, 1H), 4.56–4.48 (m, 4H), 4.46–4.41 (m, 2H), 4.41–4.37 (m, 1H), 4.01 (d, J = 11.9 Hz, 2H), 3.93 (d, J = 10.9 Hz, 1H), 3.83 (dd, J = 10.9, 3.9 Hz, 1H), 3.73–3.57 (m, 10H), 3.40 (t, J = 12.1 Hz, 3H), 2.52–2.44 (m, 9H), 2.42 (s, 3H), 2.38–2.29 (m, 2H), 2.27 (s, 4H), 2.14–2.08 (m, 1H), 1.86–1.76 (m, 3H), 1.70–1.62 (m, 4H), 1.06 (s, 9H), 1.05–1.00 (m, 3H). HRMS (m/z) for C62H82N9O8S+ [M+H]+: calcd 1112.6002, found 1112.6012.
(2S,4R)-1-((S)-2-(7-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-7-oxoheptanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (14)
Compound 14 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 30 (11.4 mg, 0.02 mmol, 1.0 equiv). Compound 14 was obtained as a white solid (15.6 mg, yield 69%). 1H NMR (800 MHz, CD3OD) δ 8.98 (s, 1H), 7.84 (d, J = 7.9 Hz, 3H), 7.73–7.60 (m, 3H), 7.49 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 7.9 Hz, 2H), 6.16 (s, 1H), 4.66 (s, 1H), 4.59 (t, J = 8.4 Hz, 1H), 4.56–4.50 (m, 4H), 4.45 (s, 2H), 4.39 (d, J = 15.6 Hz, 1H), 4.01 (d, J = 11.7 Hz, 2H), 3.92 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 4.0 Hz, 1H), 3.72–3.52 (m, 10H), 3.40 (t, J = 11.8 Hz, 3H), 2.50 (s, 3H), 2.45 (s, 8H), 2.42 (s, 3H), 2.28–2.22 (m, 4H), 2.14–2.07 (m, 1H), 1.97–1.76 (m, 3H), 1.70–1.61 (m, 4H), 1.43–1.36 (m, 2H), 1.06 (s, 9H), 1.05–1.00 (m, 3H). HRMS (m/z) for C63H84N9O8S+ [M+H]+: calcd 1126.6158, found 1126.6152.
(2S,4R)-1-((S)-2-(8-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-8-oxooctanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (15)
Compound 15 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 31 (11.7 mg, 0.02 mmol, 1.0 equiv). Compound 15 was obtained as a white solid (16.6 mg, yield 73%). 1H NMR (800 MHz, CD3OD) δ 8.98 (s, 1H), 7.85 (d, J = 8.1 Hz, 3H), 7.74–7.62 (m, 3H), 7.49 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 7.9 Hz, 2H), 6.16 (s, 1H), 4.66 (s, 1H), 4.59 (t, J = 8.3 Hz, 1H), 4.57–4.49 (m, 4H), 4.45 (d, J = 10.2 Hz, 2H), 4.39 (d, J = 15.4 Hz, 1H), 4.01 (d, J = 11.7 Hz, 2H), 3.93 (d, J = 10.8 Hz, 1H), 3.83 (dd, J = 11.0, 3.9 Hz, 1H), 3.74–3.58 (m, 10H), 3.40 (t, J = 11.9 Hz, 3H), 2.50 (d, J = 3.4 Hz, 3H), 2.45 (s, 6H), 2.42 (s, 5H), 2.29–2.22 (m, 4H), 2.15–2.08 (m, 1H), 1.86–1.76 (m, 3H), 1.68–1.59 (m, 4H), 1.44–1.35 (m, 4H), 1.06 (s, 9H), 1.05–1.01 (m, 3H). HRMS (m/z) for C64H86N9O8S+ [M+H]+: calcd 1140.6315, found 1140.6322.
(2S,4R)-1-((S)-2-(9-(4-((3′-(((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-9-oxononanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (16)
Compound 16 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (11.4 mg, 0.02 mmol) and 32 (12.0 mg, 0.02 mmol, 1.0 equiv). Compound 16 was obtained as a white solid (16.5 mg, yield 71%). 1H NMR (800 MHz, CD3OD) δ 8.99 (s, 1H), 7.95–7.80 (m, 3H), 7.76–7.60 (m, 3H), 7.54–7.37 (m, 4H), 6.16 (s, 1H), 4.66 (s, 1H), 4.60 (t, J = 8.4 Hz, 1H), 4.57–4.51 (m, 4H), 4.45 (s, 2H), 4.39 (d, J = 15.4 Hz, 1H), 4.04–3.99 (m, 2H), 3.93 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 3.9 Hz, 1H), 3.76–3.63 (m, 10H), 3.41 (t, J = 11.9 Hz, 3H), 2.50 (s, 3H), 2.48–2.43 (m, 6H), 2.42 (s, 3H), 2.35–2.29 (m, 1H), 2.29–2.22 (m, 5H), 2.14–2.08 (m, 1H), 1.83 (s, 3H), 1.67–1.58 (m, 4H), 1.40–1.33 (m, 6H), 1.05 (d, J = 10.1 Hz, 12H). 13C NMR (151 MHz, CD3OD) δ 174.64, 173.11, 172.78, 170.96, 169.55, 163.94, 159.96, 152.81, 152.22, 146.27, 143.95, 140.45, 140.04, 139.36, 131.85 (2C), 128.97 (2C), 128.76, 127.69 (2C), 127.63 (2C), 126.30, 121.49, 120.83, 116.97, 115.59, 115.06, 113.70, 110.15, 69.68, 65.85, 59.62, 59.46, 57.57, 56.63, 51.20, 50.99, 48.46, 42.28, 42.11, 38.14, 37.55, 35.42, 35.20, 35.15, 32.18, 28.87, 28.77 (2C), 28.74 (2C), 28.70 (2C), 25.65 (3C), 25.59, 25.52, 24.67, 18.32, 17.21, 13.72, 9.47. HRMS (m/z) for C65H88N9O8S+ [M+H]+: calcd 1154.6471, found 1154.6478.
(2S,4R)-4-(Benzyloxy)-1-((S)-2-(9-(4-((3′-(((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[1,1′-biphenyl]-4-yl)methyl)piperazin-1-yl)-9-oxononanamido)-3,3-dimethylbutanoyl)-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (17)
To a solution of commercially available (2S,4R)-4-(benzyloxy)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (33, 1 mmol, 321 mg) in DMSO (5 mL) were added (4-(4-methylthiazol-5-yl)phenyl)methanamine (34, 1 mmol, 204 mg), EDCI (288 mg, 1.5 mmol), HOAt (204 mg, 1.5 mmol), and NMM (303 mg, 3 mmol). After being stirred for 2 h at rt, the resulting mixture was purified by reverse phase ISCO (methanol/0.1% TFA in H2O) to afford an intermediate, which was dissolved in TFA (5 mL) and DCM (5 mL). After the resulting mixture was stirred for 30 min, solvents were removed. The resulting residue was purified by reverse phase ISCO (methanol/0.1% TFA in H2O) to afford (2S,4R)-4-(benzyloxy)-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (35, 276 mg, yield 68%). To a solution of 35 (84 mg, 0.2 mmol) in DMSO (1 mL) was added commercially available (S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (36, 48.5 mg, 0.2 mmol), EDCI (11.6 mg, 0.06 mmol), HOAt (8.2 mg, 0.06 mmol), and NMM (12.7 mg, 0.12 mmol), the reaction was stirred at rt for 3 h, and the mixture was purified by preparative HPLC (10% −100% acetonitrile/0.1% TFA in H2O) to afford the intermediate. This intermediate was dissolved in DCM (1 mL) and TFA (1 mL), and the resulting mixture was stirred at rt for 30 min. The solvent was evaporated, and the resulting mixture was purified by preparative HPLC (10% −100% acetonitrile/0.1% TFA in H2O) to afford the (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-(benzyloxy)-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (37, 39.5 mg, yield 38% for two steps) as a white solid. ESI (m/z) [M+H+]: 521.3. To a solution of 37 (15.6 mg, 0.03 mmol) in DMSO (1 mL) were added nonanedioic acid (38, 11.3 mg, 0.06 mmol), EDCI (8.6 mg, 0.045 mmol), HOAt (6.1 mg, 0.045 mmol), and NMM (9.1 mg, 0.09 mmol). After being stirred for 1 h at rt, the resulting mixture was purified by preparative HPLC (10% −100% acetonitrile/0.1% TFA in H2O) to afford the title compound 39 as a white solid (16.4 mg, yield 79%). 1H NMR (600 MHz, CD3OD) δ 9.05 (s, 1H), 7.55–7.39 (m, 4H), 7.39–7.20 (m, 5H), 4.73 (s, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.59–4.49 (m, 3H), 4.39 (d, J = 15.4 Hz, 1H), 4.35–4.29 (m, 2H), 3.76 (dd, J = 11.3, 3.6 Hz, 1H), 2.51 (s, 3H), 2.46–2.39 (m, 1H), 2.34–2.20 (m, 4H), 2.15–2.09 (m, 1H), 1.63–1.55 (m, 4H), 1.34–1.29 (m, 6H), 1.06 (s, 9H). HRMS (m/z) for C38H51N4O6S+ [M+H]+: calcd 691.3524, found 691.3536. Then, compound 17 was synthesized following the standard procedure for preparing compound 2 from intermediates 1 (13.6 mg, 0.024 mmol) and 39 (16.4 mg, 0.024 mmol, 1.0 equiv). Compound 17 was obtained as a white solid (24.3 mg, yield 81%). 1H NMR (600 MHz, CD3OD) δ 8.99 (s, 1H), 7.87 (s, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.69 (s, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 7.27–7.19 (m, 4H), 7.19–7.13 (m, 1H), 6.07 (s, 1H), 4.62 (s, 1H), 4.51 (d, J = 11.6 Hz, 1H), 4.49–4.38 (m, 5H), 4.34 (s, 2H), 4.30 (d, J = 15.5 Hz, 1H), 4.24–4.17 (m, 2H), 3.97–3.87 (m, 3H), 3.75–3.62 (m, 10H), 3.30 (t, J = 11.9 Hz, 3H), 2.40 (s, 3H), 2.38 (s, 3H), 2.34–2.27 (m, 7H), 2.23–2.11 (m, 5H), 2.05–1.98 (m, 1H), 1.80–1.70 (m, 3H), 1.53–1.47 (m, 4H), 1.26–1.19 (m, 6H), 0.99–0.93 (m, 12H). 13C NMR (151 MHz, CD3OD) δ 174.69, 172.99, 172.74, 171.04, 169.60, 164.01, 157.69, 157.41, 152.63, 152.06, 143.86, 140.42, 139.23, 138.05, 131.82 (2C), 129.60, 129.12, 128.98 (2C), 128.67, 128.05, 127.97 (2C), 127.66 (2C), 127.62 (2C), 127.43, 127.31 (2C), 120.89, 116.85, 115.59, 114.94, 113.70, 109.97, 77.15, 70.26, 65.88, 59.63, 59.45, 57.43, 53.11, 51.21, 50.97, 42.27, 42.08, 38.11, 36.08, 35.43, 35.36, 35.19, 34.96, 32.18, 28.99, 28.80 (2C), 28.72 (2C), 28.70 (2C), 25.61 (3C), 25.56, 24.64, 18.29, 17.18, 14.00, 13.67, 9.57. HRMS (m/z) for C72H94N9O8S+ [M+H]+: calcd 1244.6941, found 1244.6954.
Cell Culture
All of the cell lines used were purchased from ATCC (American Type Culture Collection, Manassas, VA), with the exception of patient primary cell line 515a, which was obtained from the Cancer Cell Organoid Core at Mount Sinai. SUM159, MDA-MB-468, MDA-MB-453, and BT549 were grown in Dulbecco’s modified Eagle’s medium (DMEM) or Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin under 5% CO2 in humidified conditions at 37 °C. Cells were passed every 2–3 days depending on confluency. For 515a cells, Complete F-media (50% fresh and 50% conditioned F-media) was utilized and prepared as described by Schlegel et al.53 F-media contained 50% DMEM high glucose, 50% F-12 medium, 5% FBS, 8.4 ng/mL cholera toxin, 5 μg/mL insulin, 10 μg/mL epithelial growth factor, 5 μM Y-27632, and 0.4 μg/mL hydrocortisone. 515a cells were then grown in Complete F-media with 5% FBS and 1% penicillin–streptomycin under 5% CO2 in humidified conditions at 37 °C and passaged every 5–7 days, with media replacements every 2–3 days.
Western Blotting Analysis
Approximately 1 million cells were collected after treatment and lysed in 200–300 μL of radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitor cocktail. After lysis, the concentration of protein was quantified by a bicinchoninic acid (BCA) protein assay kit. Equal amounts of total protein were loaded onto 4–15% SDS–PAGE and separated. The gel was transferred to polyvinylidene difluoride membranes. Then, membranes were blocked for 1 h in PBS or TBS blocking solution (LI-COR) and probed at 4 °C for 12 h with the following primary antibodies: EZH2 (Cell Signaling Technology), SUZ12 (Cell Signaling Technology), EZH1 (Abcam ab176115), EED (Cell Signaling Technology), H3K27me3 (Cell Signaling Technology), H3 (Cell Signaling Technology), and β-actin (Cell Signaling Technology). Membranes were then incubated with the specific rabbit IRDye 800CW secondary antibody (LI-COR) for 1 h at room temperature. After the membrane was washed for 5 min two times with PBS 0.01% Tween20, the blot was detected using an Odyssey CLx Imaging system (LI-COR). Data analysis was performed using Image Studio (LI-COR).
Cell Viability Assays
Cells (10 000 cells/well) were seeded into a 96-well plate in triplicate in 200 μL of media. After 16 h, cells were treated with DMSO or compound through serial dilution and incubated for 5 days. The viability of the cells was assessed by CCK-8 (Cell Counting Kit-8, WST-8). A 1× solution of CCK-8 (Dojindo, CK04) was added (20 μL) to each well and re-incubated at 37 °C for approximately 2–4 h until control DMSO group absorbance at 450 nm read above or equal to 1.0. The absorbance of 690 nm was used as reference using an Infinite F PLEX plate reader (TECAN). GI50 values were analyzed using GraphPad Prism 6. Error bars represent ± SD for three biological independent experiments.
EZH2/EZH1Methyltransferase Inhibition and Selectivity Assays
The methyltransferase assay involves monitoring the transfer of a tritium(3H)-labeled methyl group from the cofactor S-adenosyl methionine (SAM) to histone substrates. In this biochemical assay, 5 nM of the recombinant five-component PRC2 complex (comprising EZH2 (2–746), EED (2–441), SUZ12 (2–739), RbAp48 (2–425), and AEBP2 (2–517); all full-length) or 20 nM of the recombinant five-component PRC2 complex (comprising EZH1 (2–746), EED (2–441), SUZ12 (2–739), RbAp48 (2–425), and AEBP2 (2–517); all full-length) was used as the enzyme, 0.05 μM of core histones as the substrate, and 1 μM of SAM as methyl donor, respectively. The assay was performed in duplicate (carried out by Reaction Biology Corp). Assays for compound selectivity against a panel of methyltransferases (performed by Reaction Biology Corp.) used the same 3H-labeled SAM assay format as described above. Here, the selectivity assays against 20 other methyltransferases were performed in duplicate at the concentration of 10 μM.
Mouse Pharmacokinetic Study
Compound 16 in HCl salt form was dissolved in a formulation vehicle of 5% v/v NMP, 5% v/v Solutol HS-15, and 90% v/v normal saline. In total, six male Swiss Albino mice were administered intraperitoneally with a solution formulation of compound 16 at a single dose of 50 mg/kg. All samples were stored below −70 ± 10 °C until bioanalysis. Approximately 60 μL plasma samples were collected from three mice at each of the six time points (0.5, 1, 2, 4, 8, and 12 h). The plasma concentration–time data of compound 16 were used for the PK analysis, which was performed using the NCA module of Phoenix WinNonlin (Version 8.0), and plasma concentrations were quantified by a fit-for-purpose LC-MS/MS method (LLOQ: 1.03 ng/mL). Compound concentrations in plasma at each time point are mean values from three tested mice. Error bars represent ±SEM. Peak plasma concentration (Cmax) and time to peak plasma concentration (Tmax) were the observed values. Experiments involving mice were performed according to the Institutional Animal Care and Use Committee (IACUC)-approved protocol.
Acknowledgments
This research was supported in part by the grants R01CA230854 (to J.J.), R01CA218600 (to J.J.), and R01CA268519 (to J.J.) from the National Cancer Institute (NCI) at the U.S. National Institutes of Health. J.J. acknowledges the support by an endowed professorship by the Icahn School of Medicine at Mount Sinai. This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504. B.D. acknowledges support from the Medical Scientist Training Program (training grant T32GM007280) at the Icahn School of Medicine at Mount Sinai and the grant (3R01CA230854S1) from the U.S. National Institutes of Health. We thank Dr. Pamela Cheung at the Cancer Cell Organoid Core (CCOC) at Mount Sinai for providing patient primary cell line 515a.
Glossary
Abbreviations Used
- CRBN
cereblon
- CRL
cullin RING family E3 ubiquitin ligase
- DLBCL
diffuse large B-cell lymphomas
- DMSO
dimethyl sulfoxide
- EED
embryonic ectoderm development
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- EZH2
enhancer of zeste homolog 2
- HOAt
1-hydroxy-7-azabenzo-triazole
- Hyt
hydrophobic tag
- IP
intraperitoneal
- MOA
mechanism of action
- NAE
NEDD8-activating E1 enzyme
- PDX
patient cells-derived xenograft
- PEG
polyethylene glycol
- PK
pharmacokinetic
- POI
proteins of interest
- PRC2
polycomb repressive complex 2
- PROTAC
proteolysis targeting chimera
- SAR
structure–activity relationship
- SUZ12
suppressor of zeste 12
- TAD
transactivation domain
- TNBC
triple-negative breast cancer
- TPD
targeted protein degradation
- UPS
ubiquitin–proteasome system
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00100.
Effect of 16 on reducing the EZH1 protein level in SUM159 cells; EPZ-6438 reduces the H3K27me3 mark in MDA-MB-453 cells; the EZH2 degradation induced by compound 16 is rescued by the NEDD8-activating E1 enzyme (NAE) inhibitor MLN4924; compound 16 inhibits the growth of MDA-MB-453 cells; compound 16 displays growth inhibition and EZH2 degradation superior to that of MS177 in BT549 cells; 1H NMR, 13C NMR, and HPLC spectra of compounds 16 and 17 (PDF)
Molecular formula strings for all compounds (XLSX)
Author Contributions
# B.D. and C.A. contributed equally to this work.
The authors declare the following competing financial interest(s): J.J. is a cofounder and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc., and Cullinan Oncology, Inc.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- Sharma S.; Kelly T. K.; Jones P. A. Epigenetics in Cancer. Carcinogenesis 2010, 31 (1), 27–36. 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150 (1), 12–27. 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Margueron R.; Reinberg D. The Polycomb Complex PRC2 and Its Mark in Life. Nature 2011, 469 (7330), 343–349. 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L.; Helin K. Transcriptional Regulation by Polycomb Group Proteins. Nat. Struct. Mol. Biol. 2013, 20 (10), 1147–1155. 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- Kim K. H.; Roberts C. W. M. Targeting EZH2 in Cancer. Nat. Med. 2016, 22 (2), 128–134. 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Garcia E.; Licht J. D. Deregulation of H3K27 Methylation in Cancer. Nat. Genet. 2010, 42 (2), 100–101. 10.1038/ng0210-100. [DOI] [PubMed] [Google Scholar]
- Ezponda T.; Licht J. D. Molecular Pathways: Deregulation of Histone H3 Lysine 27 Methylation in Cancer - Different Paths, Same Destination. Clin. Cancer Res. 2014, 20 (19), 5001–5008. 10.1158/1078-0432.CCR-13-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe B. R.; Maxham L. A.; Hamey J. J.; Wilkins M. R.; Partridge J. F. Histone H3Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers (Basel) 2019, 11 (5), 660. 10.3390/cancers11050660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe M. T.; Ott H. M.; Ganji G.; Korenchuk S.; Thompson C.; Van Aller G. S.; Liu Y.; Graves A. P.; Della Pietra A. III; Diaz E.; LaFrance L. V.; Mellinger M.; Duquenne C.; Tian X.; Kruger R. G.; McHugh C. F.; Brandt M.; Miller W. H.; Dhanak D.; Verma S. K.; Tummino P. J.; Creasy C. L. EZH2 Inhibition as a Therapeutic Strategy for Lymphoma with EZH2-Activating Mutations. Nature 2012, 492 (7427), 108–112. 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Gan L.; Xu M.; Hua R.; Tan C.; Zhang J.; Gong Y.; Wu Z.; Weng W.; Sheng W.; Guo W. The Polycomb Group Protein EZH2 Induces Epithelial-Mesenchymal Transition and Pluripotent Phenotype of Gastric Cancer Cells by Binding to PTEN Promoter. J. Hematol. Oncol. 2018, 11 (1), 1–12. 10.1186/s13045-017-0547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. A.; Varambally S.; Arend R. C. Histone Methyltransferase EZH2: A Therapeutic Target for Ovarian Cancer. Mol. Cancer Ther. 2018, 17 (3), 591–602. 10.1158/1535-7163.MCT-17-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann I. M.; Halvorsen O. J.; Collett K.; Stefansson I. M.; Straume O.; Haukaas S. A.; Salvesen H. B.; Otte A. P.; Akslen L. A. EZH2 Expression Is Associated with High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. J. Clin. Oncol. 2006, 24 (2), 268–273. 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- Yap D. B.; Chu J.; Berg T.; Schapira M.; Cheng S. W. G.; Moradian A.; Morin R. D.; Mungall A. J.; Meissner B.; Boyle M.; Marquez V. E.; Marra M. A.; Gascoyne R. D.; Humphries R. K.; Arrowsmith C. H.; Morin G. B.; Aparicio S. A. J. R. Somatic Mutations at EZH2 Y641 Act Dominantly through a Mechanism of Selectively Altered PRC2 Catalytic Activity, to Increase H3K27 Trimethylation. Blood 2011, 117 (8), 2451–2459. 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Wang Z.; Lu W.; Jiang H.; Lu J.; Qiu J.; Ye G. EZH2 Promotes Gastric Cancer Cells Proliferation by Repressing P21 Expression. Pathol. Res. Pract. 2019, 215 (6), 152374. 10.1016/j.prp.2019.03.003. [DOI] [PubMed] [Google Scholar]
- Anwar T.; Gonzalez M. E.; Kleer C. G. Noncanonical Functions of the Polycomb Group Protein EZH2 in Breast Cancer. Am. J. Pathol. 2021, 191 (5), 774–783. 10.1016/j.ajpath.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Yu X.; Gong W.; Liu X.; Park K.-S.; Ma A.; Tsai Y.-H.; Shen Y.; Onikubo T.; Pi W.-C.; Allison D. F.; Liu J.; Chen W.-Y.; Cai L.; Roeder R. G.; Jin J.; Wang G. G. EZH2 Noncanonically Binds CMyc and P300 through a Cryptic Transactivation Domain to Mediate Gene Activation and Promote Oncogenesis. Nat. Cell Biol. 2022, 24, 384. 10.1038/s41556-022-00850-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He A.; Shen X.; Ma Q.; Cao J.; von Gise A.; Zhou P.; Wang G.; Marquez V. E.; Orkin S. H.; Pu W. T. PRC2 Directly Methylates GATA4 and Represses Its Transcriptional Activity. Genes Dev. 2012, 26 (1), 37–42. 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.; Kim M.; Woo D. H.; Shin Y.; Shin J.; Chang N.; Oh Y. T.; Kim H.; Rheey J.; Nakano I.; Lee C.; Joo K. M.; Rich J. N.; Nam D. H.; Lee J. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell 2013, 23 (6), 839–852. 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeyer K.; Junghans D.; Kanzler B.; Kemler R. Trimethylation and Acetylation of β-Catenin at Lysine 49 Represent Key Elements in ESC Pluripotency. Cell Rep. 2017, 18 (12), 2815–2824. 10.1016/j.celrep.2017.02.076. [DOI] [PubMed] [Google Scholar]
- Shi B.; Liang J.; Yang X.; Wang Y.; Zhao Y.; Wu H.; Sun L.; Zhang Y.; Chen Y.; Li R.; Zhang Y.; Hong M.; Shang Y. Integration of Estrogen and Wnt Signaling Circuits by the Polycomb Group Protein EZH2 in Breast Cancer Cells. Mol. Cell. Biol. 2007, 27 (14), 5105–5119. 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Xi Y.; Li W.; McCarthy R. L.; Stratton S. A.; Zou W.; Li W.; Dent S. Y.; Jain A. K.; Barton M. C. TRIM28 Interacts with EZH2 and SWI/SNF to Activate Genes That Promote Mammosphere Formation. Oncogene 2017, 36 (21), 2991–3001. 10.1038/onc.2016.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar T.; Arellano-Garcia C.; Ropa J.; Chen Y.-C.; Kim H. S.; Yoon E.; Grigsby S.; Basrur V.; Nesvizhskii A. I.; Muntean A.; Gonzalez M. E.; Kidwell K. M.; Nikolovska-Coleska Z.; Kleer C. G. P38-Mediated Phosphorylation at T367 Induces EZH2 Cytoplasmic Localization to Promote Breast Cancer Metastasis. Nat. Commun. 2018, 9 (1), 2801. 10.1038/s41467-018-05078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani R. G.; Gehling V. S.; Dakin L. A.; Cook A. S.; Nasveschuk C. G.; Duplessis M.; Iyer P.; Balasubramanian S.; Zhao F.; Good A. C.; Campbell R.; Lee C.; Cantone N.; Cummings R. T.; Normant E.; Bellon S. F.; Albrecht B. K.; Harmange J. C.; Trojer P.; Audia J. E.; Zhang Y.; Justin N.; Chen S.; Wilson J. R.; Gamblin S. J. Identification of (R)-N-((4-Methoxy-6-Methyl-2-Oxo-1,2-Dihydropyridin-3-Yl)Methyl)-2-Methyl-1-(1-(1-(2,2,2-Trifluoroethyl)Piperidin-4-Yl)Ethyl)-1H-Indole-3-Carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitabl. J. Med. Chem. 2016, 59 (21), 9928–9941. 10.1021/acs.jmedchem.6b01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung P. P.; Bingham P.; Brooun A.; Collins M.; Deng Y. L.; Dinh D.; Fan C.; Gajiwala K. S.; Grantner R.; Gukasyan H. J.; Hu W.; Huang B.; Kania R.; Kephart S. E.; Krivacic C.; Kumpf R. A.; Khamphavong P.; Kraus M.; Liu W.; Maegley K. A.; Nguyen L.; Ren S.; Richter D.; Rollins R. A.; Sach N.; Sharma S.; Sherrill J.; Spangler J.; Stewart A. E.; Sutton S.; Uryu S.; Verhelle D.; Wang H.; Wang S.; Wythes M.; Xin S.; Yamazaki S.; Zhu H.; Zhu J.; Zehnder L.; Edwards M. Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(Methoxy(Oxetan-3-Yl)Methyl)-2-((4-Methoxy-6-Methyl-2-Oxo-1,2-D. J. Med. Chem. 2018, 61 (3), 650–665. 10.1021/acs.jmedchem.7b01375. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang D.; Ding N.; Mi L.; Yu H.; Wu M.; Feng F.; Hu L.; Zhang Y.; Zhong C.; Ye Y.; Li J.; Fang W.; Shi Y.; Deng L.; Ying Z.; Song Y.; Zhu J. The Synergistic Anti-Tumor Activity of EZH2 Inhibitor SHR2554 and HDAC Inhibitor Chidamide through ORC1 Reduction of DNA Replication Process in Diffuse Large B Cell Lymphoma. Cancers (Basel). 2021, 13 (17), 4249. 10.3390/cancers13174249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma D.; Nosaka E.; Shiroishi M.; Takata Y.; Hama Y.; Yamamoto Y.; Adachi N.; Maruyama D.; Tobinai K.; Ishida T.; Kusumoto S.; Ishitsuka K.; Imaizumi Y.; Takeuchi S.; Tsukasaki K.; Fujioka T.; Watanabe J.; Kanno O.; Kumazawa E.; Fujitani S.; Araki K.; Fujiwara K. DS-3201, a Potent EZH1/2 Dual Inhibitor, Demonstrates Antitumor Activity Against Non-Hodgkin Lymphoma (NHL) Regardless of EZH2 Mutation. Blood 2018, 132 (Supplement 1), 2217. 10.1182/blood-2018-99-113379. [DOI] [Google Scholar]
- Knutson S. K.; Wigle T. J.; Warholic N. M.; Sneeringer C. J.; Allain C. J.; Klaus C. R.; Sacks J. D.; Raimondi A.; Majer C. R.; Song J.; Scott M. P.; Jin L.; Smith J. J.; Olhava E. J.; Chesworth R.; Moyer M. P.; Richon V. M.; Copeland R. A.; Keilhack H.; Pollock R. M.; Kuntz K. W. A Selective Inhibitor of EZH2 Blocks H3K27 Methylation and Kills Mutant Lymphoma Cells. Nat. Chem. Biol. 2012, 8 (11), 890–896. 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Porter Scott M.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M.; Kuntz K. W.; Keilhack H. Durable Tumor Regression in Genetically Altered Malignant Rhabdoid Tumors by Inhibition of Methyltransferase EZH2. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (19), 7922–7927. 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D.; Ma A.; Li F.; Barsyte-Lovejoy D.; Parton T.; MacNevin C. J.; Liu F.; Gao C.; Huang X.-P.; Kuznetsova E.; Rougie M.; Jiang A.; Pattenden S. G.; Norris J. L.; James L. I.; Roth B. L.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Hahn K. M.; Wang G. G.; Vedadi M.; Jin J. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8 (6), 1324–1334. 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Li F.; Konze K. D.; Meslamani J.; Ma A.; Brown P. J.; Zhou M. M.; Arrowsmith C. H.; Kaniskan H. Ü.; Vedadi M.; Jin J. Structure-Activity Relationship Studies for Enhancer of Zeste Homologue 2 (EZH2) and Enhancer of Zeste Homologue 1 (EZH1) Inhibitors. J. Med. Chem. 2016, 59 (16), 7617–7633. 10.1021/acs.jmedchem.6b00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Martini M. L.; Jin J. Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev. 2018, 118 (3), 989–1068. 10.1021/acs.chemrev.6b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat K. P.; Kaniskan H. Ü.; Jin J.; Gozani O. Epigenetics and beyond: Targeting Writers of Protein Lysine Methylation to Treat Disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. 10.1038/s41573-020-00108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy S. M. Tazemetostat: First Approval. Drugs 2020, 80 (5), 513–521. 10.1007/s40265-020-01288-x. [DOI] [PubMed] [Google Scholar]
- Bisserier M.; Wajapeyee N. Mechanisms of Resistance to Ezh2 Inhibitors in Diffuse Large B-Cell Lymphomas. Blood 2018, 131 (19), 2125–2137. 10.1182/blood-2017-08-804344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C. L.; Baldwin A. S. Non-Canonical EZH2 Transcriptionally Activates RelB in Triple Negative Breast Cancer. PLoS One 2016, 11 (10), e0165005 10.1371/journal.pone.0165005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Gou H.; Yao J.; Yi K.; Jin Z.; Matsuoka M.; Zhao T. The Noncanonical Role of EZH2 in Cancer. Cancer Sci. 2021, 112 (4), 1376–1382. 10.1111/cas.14840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B.; Sun R.; Wang D.; Sheng H.; Wei T.; Wang L.; Zhang J.; Ho T. H.; Yang L.; Wei Q.; Huang H. ZMYND8 Preferentially Binds Phosphorylated EZH2 to Promote a PRC2-Dependent to -Independent Function Switch in Hypoxia-Inducible Factor–Activated Cancer. Proc. Natl. Acad. Sci. U.S.A. 2021, 118 (8), 1–11. 10.1073/pnas.2019052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A.; Stratikopoulos E.; Park K.-S.; Wei J.; Martin T. C.; Yang X.; Schwarz M.; Leshchenko V.; Rialdi A.; Dale B.; Lagana A.; Guccione E.; Parekh S.; Parsons R.; Jin J. Discovery of a First-in-Class EZH2 Selective Degrader. Nat. Chem. Biol. 2020, 16 (2), 214–222. 10.1038/s41589-019-0421-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale B.; Cheng M.; Park K.-S.; Kaniskan H. Ü.; Xiong Y.; Jin J. Advancing Targeted Protein Degradation for Cancer Therapy. Nat. Rev. Cancer 2021, 21 (10), 638–654. 10.1038/s41568-021-00365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Hu X.; Wang Q.; Wu X.; Zhang Q.; Wei W.; Su X.; He H.; Zhou S.; Hu R.; Ye T.; Zhu Y.; Wang N.; Yu L. Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2. J. Med. Chem. 2021, 64 (5), 2829–2848. 10.1021/acs.jmedchem.0c02234. [DOI] [PubMed] [Google Scholar]
- Tu Y.; Sun Y.; Qiao S.; Luo Y.; Liu P.; Jiang Z.; Hu Y.; Wang Z.; Huang P.; Wen S. Design, Synthesis, and Evaluation of VHL-Based EZH2 Degraders to Enhance Therapeutic Activity against Lymphoma. J. Med. Chem. 2021, 64 (14), 10167–10184. 10.1021/acs.jmedchem.1c00460. [DOI] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. U.S.A. 2016, 113 (26), 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; Den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Xu J.; Shen Y.; Cahuzac K. M.; Park K.; Dale B.; Liu J.; Parsons R. E.; Jin J. Discovery of Potent, Selective, and In Vivo Efficacious AKT Kinase Protein Degraders via Structure–Activity Relationship Studies. J. Med. Chem. 2022, 65 (4), 3644–3666. 10.1021/acs.jmedchem.1c02165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein-Protein Interaction between the von Hippel-Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57 (20), 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G. M.; Smith B. E.; Lai A. C.; Jaime-Figueroa S.; McQuaid D. C.; Bondeson D. P.; Toure M.; Dong H.; Qian Y.; Wang J.; Crew A. P.; Hines J.; Crews C. M. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25 (1), 67–77. 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B.; Wang E. S.; Donovan K. A.; Liang Y.; Fischer E. S.; Zhang T.; Gray N. S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem., Int. Ed. 2019, 58 (19), 6321–6326. 10.1002/anie.201901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L.; Zhou H.; Xu R.; Zhao Y.; Chinnaswamy K.; McEachern D.; Chen J.; Yang C. Y.; Liu Z.; Wang M.; Liu L.; Jiang H.; Wen B.; Kumar P.; Meagher J. L.; Sun D.; Stuckey J. A.; Wang S. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36 (5), 498–511. 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S.; Zhang X.; Lv D.; Zhang Q.; He Y.; Zhang P.; Liu X.; Thummuri D.; Yuan Y.; Wiegand J. S.; Pei J.; Zhang W.; Sharma A.; McCurdy C. R.; Kuruvilla V. M.; Baran N.; Ferrando A. A.; Kim Y.-m.; Rogojina A.; Houghton P. J.; Huang G.; Hromas R.; Konopleva M.; Zheng G.; Zhou D. A Selective BCL-XL PROTAC Degrader Achieves Safe and Potent Antitumor Activity. Nat. Med. 2019, 25 (12), 1938–1947. 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Despotovic N.; Kovacs M. S.; Pin C. L.; Luyt L. G. 18 F-Labeled PET Probe Targeting Enhancer of Zeste Homologue 2 (EZH2) for Cancer Imaging. ACS Med. Chem. Lett. 2019, 10 (3), 334–340. 10.1021/acsmedchemlett.8b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.; Yang X.; Liu J.; Xiong Y.; Poulikakos P.; Karoulia Z.; Wu X.; Ahmed T.. Compositions and Methods for Treating CDK4/6-Mediated Cancer. WO 2018106870, 2018.
- Liu X.; Krawczyk E.; Suprynowicz F. A.; Palechor-Ceron N.; Yuan H.; Dakic A.; Simic V.; Zheng Y. L.; Sripadhan P.; Chen C.; Lu J.; Hou T. W.; Choudhury S.; Kallakury B.; Tang D.; Darling T.; Thangapazham R.; Timofeeva O.; Dritschilo A.; Randell S. H.; Albanese C.; Agarwal S.; Schlegel R. Conditional Reprogramming and Long-Term Expansion of Normal and Tumor Cells from Human Biospecimens. Nat. Protoc. 2017, 12 (2), 439–451. 10.1038/nprot.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.