

Abstract
Stiff person syndrome spectrum disorders (SPSD) are a group of rare neuroimmunological disorders that often include painful spasms and rigidity. However, patients have highly heterogeneous signs and symptoms which may reflect different mechanistic disease processes. Understanding subsets of patients based on clinical phenotype may be important for prognosis and guiding treatment. The goal of this review is to provide updates on SPSD and its expanding clinical spectrum, prognostic markers, and treatment considerations. Further, we describe the current understanding in immunopathogenesis and highlight gaps in our knowledge appropriate for future research directions. Examples of revised diagnostic criteria for SPSD based on phenotype are also presented.
Keywords: Stiff person spectrum disorders, Stiff person syndrome, glutamic acid decarboxylase, GAD65, autoimmunity
1. Introduction
Stiff person syndrome (SPS) is a rare neuroimmunological disorder that is associated with multiple symptoms and varying levels of disability. SPS is highly heterogeneous and a broad spectrum of signs and symptoms are captured under the umbrella of SPS spectrum disorders (SPSD). Indeed, multiple phenotypes of SPSD exist, which may have different immune underpinnings. Importantly, a general lack of awareness and familiarity with the various clinical phenotypes results in people with SPS being misdiagnosed early on in their disease course. Moreover, increasing the diagnostic challenge is that many patients may not exhibit objective findings on examination early in their disease course, which may result in incomplete work ups and delayed diagnoses. This is important, as delayed diagnoses can negatively impact an individual’s quality of life and may impact the development of future disability. This review article will describe important updates in SPSD including the expanding clinical spectrum of SPSD with considerations around updating diagnostic criteria by SPS phenotype, prognostic markers, suspected immunopathogenesis, treatment considerations, and proposes future research directions.
2. Expanding clinical spectrum of SPSD
The original descriptions of SPS date back to 1956 and are attributed to Moersch and Woltman. They identified 14 cases who were seen at the Mayo Clinic over a 30-year time span. The clinical features of these patients included painful spasms, rigidity, and hyperlordosis. The body regions involved in these patients fit best under the classic phenotype of SPS 1. Since the original description, additional phenotypes have been identified including partial SPS, in which symptoms are limited to extremities and often only one limb (stiff limb syndrome) or to the torso, SPS-plus, in which classic SPS symptoms exist in combination with cerebellar and/or brainstem findings, pure cerebellar ataxia (CA), in which musculoskeletal symptoms and signs are lacking, and progressive encephalomyelitis with rigidity and myoclonus (PERM) 2–4. Currently, there are varying opinions on how some phenotypes should be described or designated. For example, some experts do not consider PERM as a separate phenotype and include it under the SPS-plus phenotype. Also, some physicians do not include the pure cerebellar phenotype under SPSD and instead consider it as a separate autoantibody associated condition. Moreover, there are some patients who do not fit perfectly into the individual phenotypes and are considered to have an overlapping syndrome, for example patients with classic SPS with epilepsy or limbic encephalitis 5. Regardless of the nuances between the varying conditions and phenotypes, they are often associated with similar autoantigens (e.g., antibodies to the glutamic acid decarboxylase 65-kilodalton isoform (GAD65)- see Immune Specificities in SPS section below) and are treated with a combination of non-pharmacological and pharmacological interventions.
In clinical practice, the classic phenotype is the most commonly encountered, and accounts for approximately 70% of patients, followed by SPS-plus, which accounts for between 12–30% of patients 2,5,6. The majority (~95%) of patients with SPSD have non-paraneoplastic disease etiology. However, a variety of malignancies have been associated with SPSD including breast cancer, small cell lung cancer, lymphoma, and thymoma 6,7. It is therefore important to consider a paraneoplastic process in individuals that present within five years from symptom onset and who are of older age 8.
Overall, the majority of people affected with SPSD are middle-aged Caucasian women. However, similar to other immune related conditions, SPSD does occur in patients with diverse backgrounds and can occur across the age spectrum 2,5–7,9–12. Multiple studies, including those conducted by The National Organization for Rare Diseases and a large case series, report that due to the rarity of this disease and its varied presentations most patients wait several years for a diagnosis. Moreover, the delay in diagnosis occurs for both pediatric and adult onset SPSD 6,9. Obtaining a definitive diagnosis of SPSD remains challenging and relies on multiple factors since there is no gold standard test or sole clinical marker. This is especially true when we consider the various phenotypes within SPSD and conditions that may mimic SPSD 3,4,13. As highlighted above, there are a significant portion of people with SPSD who present with symptoms or signs on exam outside of the musculoskeletal system that localize to the cerebellum, brainstem, spinal cord, or cortices. Indeed, the symptoms experienced by the patient depend on the clinical phenotype which might provide prognostic markers for future disability (see Prognostic Markers section below). Additionally, most patients will co-present with or develop a systemic co-morbidity such as thyroid disorders, diabetes mellitus, and pernicious anemia. Further, patients often have co-existing psychiatric conditions. For example, anxiety appears intrinsic to SPSD 2,6,7,12. Hence, it is important for clinicians to be aware of these associations in order to periodically monitor for the development of these medical co-morbidities and to treat mood related conditions that are impacting quality of life.
A few recent studies underscore the expanding spectrum of SPSD. A case series of eight, primarily older male patients, demonstrate that early prominent vestibular and ocular motor (VOM) dysfunction can occur in SPSD. The patients in this study initially presented to multiple non-neurology subspecialists with dizziness and diplopia. All patients were ultimately deemed to have an SPS-plus or CA phenotype, which was diagnosed approximately six years after the onset of their initial symptom(s). The majority of the cohort had extremely high serum anti-GAD65 antibody titers and almost two-thirds had the presence of anti-GAD65 antibodies within their cerebrospinal fluid (CSF). Interestingly, common clinical exam features suggestive of early cerebellar or brainstem involvement included spontaneous down beat nystagmus, with or without fixation, and saccadic smooth pursuit. The patients experienced both symptomatic and functional improvement after starting a combination of immune and symptomatic therapies 14. In a different study, it was shown that the anterior visual system could be affected by SPSD. The authors were interested in assessing this region of the body since the retina is an area that is highly enriched with γ-aminobutyric acid (GABA)-ergic (GABAergic) neurons and clinically some SPSD patients report severe photosensitivity, which is thought to be a symptom that localizes to the retina. In this study, optical coherence tomography (OCT) was used to assess for differences in retinal layer thicknesses between healthy controls (HCs) and patients with SPSD. Further, in a subgroup of these participants, visual acuity measures were obtained to assess functional visual outcomes. Interestingly, SPSD patients did have thinning of their retinal layers along with impaired visual acuity when compared to HCs. Moreover, ganglion cell-inner plexiform layer thickness correlated with number of body regions involved. A marked decrease of 1.25 μm (95% confidence interval, −2.2 to −0.3 μm; p = 0.008) per additional body region affected was detected in patients with SPSD even when adjusting for age, sex, diabetes history, disease duration, and history of immunomodulatory therapy 15. Another study in a large cohort (>200) of patients with SPSD demonstrated that approximately a quarter of patients experience gastrointestinal (GI) dysfunction, most commonly including dysphagia and constipation. However, greater than 50% of patients who underwent motility testing demonstrated objective evidence of upper, lower, or diffuse GI dysmotility 16. Additionally, in a recent study, Chan and colleagues described detailed cognitive and mood profiles in a subgroup of people with SPSD. Sixty-six out 205 patients with SPSD reported cognitive symptoms (32%) in their cohort, of which 20 underwent detailed cognitive testing 17. The most common cognitive domains affected in these individuals included verbal fluency/recall, processing speed, and attention. The cognitive dysfunction was felt to be multifactorial since many of these patients had co-existing mood disorders and/or were on medications that could impact cognitive function. However, despite these confounding factors, the authors suggested that cognitive deficits and mood changes could be intrinsic to SPSD since reduced GABA levels have been associated with cognitive dysfunction, anxiety, and depression in other diseases.
The diffuse regions of involvement with SPSD is not entirely unexpected when one considers the ubiquitous presence of GABAergic neurons throughout the nervous system. However, the complexity of this syndrome has resulted in challenges in recognizing the full spectrum of symptoms and signs on exam, especially when physicians are less familiar or unaware of SPSD. The different presentations and phenotypes could be further delineated within diagnostic criteria which may simplify diagnosis. This approach has been successful with other diseases such as multiple sclerosis, neuromyelitis optica spectrum disorder, and myelin oligodendrocyte glycoprotein antibody disorders. Delineating these phenotypes quickly is important for patients since there is emerging evidence showing that disease burden, treatment response, and future prognosis may differ between SPSD phenotypes 12,18. Examples of revised diagnostic criteria based on SPS phenotype are represented in Table 1. Prior criteria focused on the classic SPS phenotype and now need updating since there are multiple phenotypes within SPSD that have unique features and possibly different long-term outcomes. The clinical and paraclinical markers (e.g., high-titer anti-GAD65 antibody) within the individual criteria are being used in clinical practice for diagnosis. However, it is unclear what marker(s) are the most sensitive and/or specific for helping aid in the diagnosis of each phenotype. Moreover, these data would help experts develop consensus guidelines around diagnostic criteria and are currently not available and beyond the scope of this review.
Table 1.
Examples of Expanded Diagnostic Criteria for Stiff Person Syndrome Spectrum Disorders
| Phenotype | Classic | Partial | SPS-plus | Pure Cerebellar Ataxia | Progressive encephalomyelitis with rigidity and myoclonus (PERM) |
|---|---|---|---|---|---|
| Major criteria | • Clinical presentation including typical body regions involved (torso and lower extremities > upper extremities) • Hallmark triggers for spasms/increased rigidity* • Hallmark exam findings: hyperlordosis, rigidity of torso and/or extremity, paravertebral/abdominal muscle spasm/tightness, spasticity in extremity and/or gait, hyperreflexia with lower extremities > upper extremities • Presence of serum autoantibody to GAD65 (high titer), glycine receptor, or amphiphysin • Exclusion of alternative diagnoses and no better explanation for syndrome |
• Clinical presentation including typical body regions involved (isolated to one extremity or torso) • Hallmark triggers for spasms/increased rigidity* • Hallmark exam findings: hyperlordosis, rigidity of torso or extremity, paravertebral/abdominal muscle spasm/tightness, spasticity and/or hyperreflexia in affected extremity • Presence of serum autoantibody to GAD65 (high titer), glycine receptor, or amphiphysin • Exclusion of alternative diagnoses and no better explanation for syndrome |
• Clinical presentation including typical body regions involved (classic phenotype regions plus brainstem and/or cerebellar symptoms) • Hallmark triggers for spasms/increased rigidity* • Hallmark exam findings: classic phenotype exam findings plus brainstem (e.g., ocular motor dysfunction, dysarthria, dysphagia) and/or cerebellar signs (e.g., central nystagmus, appendicular or gait ataxia) • Presence of serum autoantibody to GAD65 (high titer), glycine receptor, or amphiphysin • Exclusion of alternative diagnoses and no better explanation for syndrome |
• Clinical presentation including typical body regions involved (e.g., central vertigo, unsteady ambulation, incoordination, poor manual dexterity, etc.) • Exam findings: scanning speech, ocular motor dysfunction (vertigo nystagmus, sustained gaze evoked nystagmus, overshooting), appendicular dysmetria, and/or gait ataxia • Presence of serum autoantibody to GAD65 (high titer), glycine receptor, or amphiphysin • Exclusion of alternative diagnoses and no better explanation for syndrome |
• Clinical presentation including typical body regions involved (neck, torso, extremities, brainstem and/or cerebellar symptoms) • Hallmark triggers for spasms/increased rigidity* • Exam findings: admixture of other phenotypes findings plus encephalopathy and severe torso rigidity and/or myoclonus (multifocal or generalized) • Presence of serum autoantibody to GAD65 (high titer), glycine receptor, or amphiphysin • EEG (generalized slowing and/or epileptic discharges) • Exclusion of alternative diagnoses and no better explanation for syndrome |
| Minor criteria | • Presence of CSF autoantibody to GAD65, glycine receptor, or amphiphysin • CSF-restricted OCB • Electromyography demonstrating cocontraction of agonist and antagonist muscles and/or continuous motor unit activity in affected muscles (paraspinal/abdominal musculature and/or legs>arms) • Robust response to muscle relaxers early on; especially GABAergic agonists (e.g., diazepam) |
• Presence of CSF autoantibody to GAD65, glycine receptor, or amphiphysin • CSF-restricted OCB • Electromyography demonstrating cocontraction of agonist and antagonist muscles and/or continuous motor unit activity in affected muscles (paraspinal/abdominal musculature or legs/arms) • Robust response to muscle relaxers early on; especially GABAergic agonists (e.g., diazepam) |
• Presence of CSF autoantibody to GAD65, glycine receptor, or amphiphysin • CSF-restricted OCB • Electromyography demonstrating cocontraction of agonist and antagonist muscles and/or continuous motor unit activity in affected muscles (paraspinal/abdominal musculature and/or legs/arms) • Robust response to muscle relaxers early on; especially GABAergic agonists (e.g., diazepam) |
• Presence of CSF autoantibody to GAD65, glycine receptor, or amphiphysin • CSF-restricted OCB • Brain MRI demonstrates cerebellar volume loss/atrophy • Brain FDG-PET demonstrates hyper-or- • hypometabolism of the cerebellum |
• Autonomic dysfunction • Presence of CSF autoantibody to GAD65, glycine receptor, or amphiphysin • CSF pleocytosis • CSF-restricted OCB • Electromyography demonstrating cocontraction of agonist and antagonist muscles and/or continuous motor unit activity in affected muscles (paraspinal/abdominal musculature and/or legs/arms) • Brain MRI demonstrates T2/contrast-enhancing lesion(s) in brainstem • Brain FDG-PET demonstrates hyper-or-hypometabolism within cortices |
Abbreviations: OCB, oligoclonal bands; FDG-PET, fluorodeoxyglucose (FDG)-positron emission tomography; GAD65, glutamic acid decarboxylase 65-kilodalton isoform; EEG, electroencephalography; MRI, magnetic resonance imaging; CSF, cerebrospinal fluid.
abrupt loud noises, cold weather, open spaces, emotional stress (good and bad), and/or tactile stimuli
if paraneoplastic, typical cancer seen in SPSD found within 5 years of symptom onset.
Primarily upper torso/neck involvement is typical for amphiphysin associated paraneoplastic SPSD.
Less typical: involvement of face and neck; epilepsy; normal musculoskeletal and/or neurological exam over time; seronegative; pediatric onset
All Phenotypes (definitive, probable, or possible diagnosis)
Meets all major and minor criteria= Definitive diagnosis
Meets all major criteria and no minor criteria= Definitive diagnosis
Meets 4 major criteria (must include serum autoantibody and exclusion of alternative diagnoses and no better explanation for syndrome) and at least 2 minor criteria= Definitive diagnosis
Meets 3 major criteria (must include serum autoantibody and exclusion of alternative diagnoses and no better explanation for syndrome) and at least 2 minor criteria= Definitive diagnosis
Meets 3 major criteria (must include exclusion of alternative diagnoses and no better explanation for syndrome) and at least 2 minor criteria= Probable
Meets 3 major criteria (must include exclusion of alternative diagnoses and no better explanation for syndrome) and less than 2 minor criteria= Possible
Meets 2 major criteria (must include exclusion of alternative diagnoses and no better explanation for syndrome) and at least 2 minor criteria= Possible
3. Prognostic markers in SPS
There is a lack of established clinical or paraclinical markers for SPSD that correlate with disease burden or long-term prognosis. Traditionally, the presence of a high-titer autoantibodies targeting GAD65 or amphiphysin, has helped aid in the diagnosis of SPS. However, previous studies demonstrated that there was no correlation between antibody titers and disease severity 19. Further, due to the rarity of SPSD and its heterogeneous clinical presentations, it remains unclear if specific phenotypes, the presence of certain symptoms or signs, or the presence of particular immune markers are associated with greater disability or have any predictive value.
A few recent studies that have included larger cohorts of patients with SPSD are providing important insights into which factors may account for a greater disease burden and worse outcomes. In a cohort of 212 patients with high-titer anti-GAD65 associated disorders, Budrahm and colleagues showed that the presence of either cerebellar ataxia, an initial visit disease burden with an modified Rankin Scale (mRS) score over two, or serum GAD65 antibody titers >500 nmol/L were independent predictors of a poorer outcome over time 12. Similarly, Mukaresh and colleagues reported in approximately 200 patients with SPSD that brainstem and/or cerebellar symptoms and those with SPS-plus/pure cerebellar phenotypes have greater disability at presentation compared to those with classic or stiff limb phenotypes. The authors further concluded that the early involvement of these regions and that these specific disease phenotypes are clinical markers of higher disease burden and may warrant starting an immune therapy earlier in the disease course 18. In addition to the insights into disease phenotype, immune specificities may also be prognostic. In a study of 121 patients with SPSD Martinez-Hernandez and colleagues found that that patients with GAD65 antibodies had worse outcomes compared to those with glycine receptor antibodies 2.
4. Update on treatment considerations in SPSD
A detailed review of all symptomatic and immune therapies is beyond the scope of this review; hence, this section will mainly focus on general therapeutic considerations and recent studies of interest. The treatment of SPSD is multifaceted and usually requires a combination of pharmacological (symptomatic and immune therapies) and non-pharmacological interventions. The number and type of therapies depends on the individual; however, most patients will require GABA-ergic agonist symptomatic treatments and many will need immune based treatments.
Benzodiazepines have been the cornerstone of symptomatic therapies in SPSD given their main mechanism of action, enhancing GABAergic pathways, and observed positive treatment response. The first report of diazepam being used in SPSD dates back to 1963 and it continues to be one of the more common GABA-ergic agonists prescribed 20,21. In clinical practice, patients with SPSD often require at least 20 to 30 mgs of diazepam monotherapy or in combination with other symptomatic therapies (clonazepam, baclofen, tizanidine, botulinum toxin, etc.) 4,22. Non-pharmacological interventions are also key to the multipronged approach to treating SPS and could include selective physical therapy (stretching, ultrasound, gait, and balance training), heat therapy, aqua therapy, deep tissue massage/myofascial techniques, osteopathic/chiropractic manipulation, acupuncture, acupressure, etc 2. Similar to pharmacotherapies, these therapies vary based on patient needs, SPS phenotype, treatment response and tolerability to the intervention(s).
There are certain classes of symptomatic medications that are not recommended for patients with SPSD including opioids and any medication that has norepinephrine reuptake inhibitors (NRIs) within their mechanism of action (MOA). The combination of opioids and any centrally-acting muscle relaxer (e.g., benzodiazepines, baclofen, tizanidine, methocarbamol, etc.) may result in severe respiratory depression. Tricyclic antidepressants and duloxetine, both of which have NRI as part of their MOA, were observed to have temporal worsening in SPS symptoms when starting the therapy and/or upon medication dosage increase 23,24. Baclofen pumps have been used in patients with SPSD with refractory spasticity, although, malfunctioning pumps have been associated with severe drug withdrawal and death 25. Therefore, it is recommended to avoid baclofen pumps.
If a patient is experiencing increased symptoms and burden of disease despite symptomatic interventions, then immune therapies should be considered (Table 2). There is limited data on which subgroups of patients would benefit from immune modulatory therapies earlier in diseases course and the optimal timing to initiate these therapies also remains unclear. These data are urgently needed in order to help identify the most appropriate candidate(s) for immune based therapies. A recent study attempted to address this issue by assessing if the burden of disease increased early in disease course in patients with SPSD, primarily including those with the classic phenotype 26. In this study, 32 patients that were immune treatment naïve were examined every six months over a two-year time period. As compared to their baseline, patients were found to have an increased number of body regions that become stiff (4.15 vs. 3.25; p < 0.0001), along with increased fall frequency, and impaired ability to ambulate. In addition, the majority of patients experienced limitations with their ability to work by the end of the study time period. The authors suggests that SPS could be a progressive disease and that starting immune therapies as early as possible may be beneficial to patients 26. Further studies are needed to confirm that early immune interventions may alter disease progression.
Table 2.
Escalation Treatment Approach in Stiff Person Syndrome Spectrum Disorders
| First-line Therapies | Second-line Therapies | Third-line Therapies | Fourth-line Therapies |
|---|---|---|---|
| Intravenous immunoglobulin | Plasma exchange* | Combination of therapies# | Stem Cell Therapies |
| Subcutaneous immunoglobulin | Rituximab | Cyclophosphamide | |
| Plasma exchange* | Mycophenolate mofetil | ||
| Corticosteroids** | Azathioprine | ||
| Combination of therapies# |
Therapeutic plasma exchange can be used in setting of acute exacerbations, as bridge therapy to other immune treatments, and/or as part of maintenance immune treatment regimen.
Try to avoid long-term use of steroids given increased risk of patients with stiff person syndrome spectrum disorders developing diabetes
Some patients with stiff person syndrome spectrum disorders require multiple immune therapies. For example, intravenous immunoglobulin and rituximab or mycophenolate mofetil.
Once it is determined that a patient requires an immune therapy, intravenous immunoglobulin (IVIG) is often the initial treatment. IVIG was shown in a randomized, placebo controlled cross over study to be effective in treating SPS 27. A total of 16 patients with SPS were enrolled in this eight-month study and during the months of receiving high dose IVIG treatment (2 grams/kilogram over two consecutive days), participants were noted to have improvements in their stiffness, spasms, and sensitivity to stimuli (e.g., noise-induced spasms, stress-induced spasms, etc.). Moreover, participants had increased mobility and decreased falls while on IVIG and importantly, improved ability to perform activities of daily living. The durability of the IVIG treatment effect varied from several weeks to up to a year 27. In clinical practice, there are different IVIG treatment protocols used, which are based on medication tolerability, treatment response, and perceived loss of effect. In line with the SPS IVIG treatment trial, we have observed that high dose IVIG appears to provide a more robust treatment response than lower dose treatment protocols. Hence, patients may benefit by being maintained on high dose IVIG (total collective dose of 2 grams/kilogram) throughout the duration of their IVIG treatment. However, IVIG dosing and frequency of administration should be tailored over time based on patient response and safety related concerns (e.g., infusion reactions, risk of thrombosis, renal dysfunction, and aseptic meningitis).
Subcutaneous immunoglobulin (SCIG) is another potential treatment option for SPSD. A recent case series demonstrated that patients with SPS who did not tolerate IVIG, successfully transitioned to SCIG 28. Importantly, patients’ symptoms remained stable during medication transition and beyond. Most patients in this case series tolerated SCIG well, but one patient discontinued SCIG due to worsening respiratory distress with ongoing treatment 28. However, this patient had a long-standing history of reactive airway disease and a suspected bronchospasm episode with IVIG, therefore this response may not be specific to SCIG. Other limiting factors for SCIG could include injection site reactions and difficulty with achieving the equivalent IVIG dose.
Therapeutic plasma exchange (TPE) has been used to treat a multitude of neuroimmunological disorders including SPS. While there are no randomized studies with TPE, it has shown to be effective for some patients with SPSD 29,30. TPE is often reserved for treating patients in acute crises and when patients are having subtherapeutic treatment responses to first-line therapies (e.g., symptomatic and IVIG). TPE catheter associated complications, along with hemodynamic effects and logistical issues, have limited enthusiasm for more regular TPE treatments. However, a small proportion of patients will use TPE as a maintenance treatment in parallel with other therapies. This could become more common with the successful implementation of outpatient administration of TPE.
If patients do not respond favorably to the above interventions, it is appropriate to consider escalating to a stronger immunotherapy. Rituximab has been used in this setting since B-cells are thought to play some role in the pathogenesis and/or propagation of SPS 31. Dalakas and colleagues recently published the results of a randomized, double-blinded, placebo-controlled trial assessing rituximab in SPS 32. Twenty-four patients with SPS enrolled in this six-month trial and were randomized to placebo or rituximab in a 1:1 fashion. The rituximab treated group received one treatment course at baseline with no further drug administered throughout the study. This study did not meet its primary endpoint of improvements in stiffness index score, nor did it demonstrate statistically significant differences in other outcome measures assessed between groups 32. However, there were a subset of patients with a more severe burden of disease that appeared to have meaningful clinical improvements due to rituximab. This suggests that patients who have experienced a subtherapeutic response to other treatments (e.g., IVIG and GABAergic agonists) could potentially benefit from rituximab 32. This preliminary study had important limitations including a small sample size, a strong placebo effect, and the noted heterogeneous response to treatment. Moreover, the duration of the study was only six months and study participants received only one treatment course of rituximab. Most chronic neuroimmunological conditions require longer treatment durations to see the maximum benefit of immune therapies. An independent study confirms that some patients with SPSD will benefit from rituximab 33. In this study, patients were given at least two years of consecutive treatment) and wereconsidered to have improvement if they experienced subjective or objective improvement in any of the cardinal signs or symptoms of SPS (e.g., spasms, rigidity, gait function) or improvement in walking speed (>20% change in the timed 25-foot walk test). Again, the clinical outcomes were heterogeneous and some patients continued to worsen clinically despite receiving rituximab. The patients who had the most robust response to rituximab were younger in age, had shorter disease duration, and were able to walk independently at baseline 33. However, conclusions are limited as this study was not a randomized, placebo-controlled trial. Collectively, these studies suggest that some patients do respond to rituximab, although predicting which patients will is not currently possible. Further, it remains unclear if non-responders to rituximab may benefit from a broader B-cell depletion or therapies directed towards different parts of the immune system, such as T-cells.
Autologous hematopoietic stem cell transplantation (auto-HSCT) has also been attempted in SPS. Auto-HSCT has been used in other autoimmune conditions such as multiple sclerosis, neuromyelitis optica, myasthenia gravis, and systemic sclerosis with varying successes 34. Hence, it has been postulated that auto-HSCT might help patients with SPS. Individual case reports and case series have documented that auto-HSCT can be well-tolerated and helpful for some patients with refractory SPS 35,36. A recently published open-label study including 23 participants with SPS demonstrated that a subgroup of patients with mostly intermittent symptoms responded to auto-HSCT. A major inclusion criteria for this study was being dependent or intolerant of benzodiazepines and IVIG. The patients who had intermittent spasms, absence of limb rigidity, lack of hyperreflexia, presence of GAD65 antibodies in the CSF, and unremarkable EMGs seemed to respond to auto-HSCT. Less than half of these responders reportedly stayed in remission for more than a few years and those participants who were deemed responders continued to have symptoms (e.g., stiffness) and some continued to require GABAergic agonist therapies. As the aim of this treatment is to restore immunologic tolerance through intense lymphodepleting conditioning, patients pre-HSCT transplant regimens included immunosuppressant therapies that are used to treat people with SPS. Therefore, it is difficult to attribute the clinical improvement to the auto-HSCT and not to the immune suppressive therapies themselves. One patient in these reports died a year after their transplant although the death was reported to be secondary to SPS disease related progression 37. Nonetheless, caution is urged with this treatment intervention for patients with SPSD because of the lack of persistent and definitive clinical benefit and the risk of development of serious adverse immunological events 38–40.
While immunomodulatory therapies are beneficial to patients with SPSD, the optimal timing of initiation of these therapies remains unclear. Studies are needed to determine if a proactive treatment approach, initiated shortly after initial symptom onset, results in better outcomes as compared to a reactive treatment approach, in which immune therapies are initiated only after a worsening of symptoms and disease burden despite attempted symptomatic interventions. In other neuroimmunological conditions, for example MS, starting immune therapies as early as possible appears to help prevent future disability 41. A preliminary study by Reyes-Mantilla and colleagues, suggest that there may be subgroups of patients with SPSD that benefit from commencing immunotherapy earlier in their disease course. In this study, 159 patients with SPSD received some type of immunotherapy of which 99 patients were followed for more than 18 months, with a median follow-up of 44 months 42. Over 95% of patients in this cohort received IVIG as their first-line immune therapy. The patients who started an immune therapy later than 60 months after symptom onset appeared to have a higher disease burden as compared to those who initiated therapy earlier in disease course 42. However, this study was not able to elucidate if early immune intervention would benefit all patients with SPSD or if early intervention should be tailored to certain subgroups of patients, such as those with SPS-plus or a pure cerebellar phenotype. What is clear is that many people with SPSD will require an immune therapy at some point and more studies are needed to better understand the optimal treatment approaches in SPSD.
5. Immune specificities associated with SPSD
Several autoantigens are associated with SPSD (Table 3). The majority of patients have antibodies to GAD65, but other autoantigens have also been described in subsets of patients. Some of these immune specificities associate with a distinctive phenotype or other disease process. For example, antibodies to the glycine receptor are often found in patients with the PERM phenotype 43,44 whereas antibodies to GAD65 are typically associated with the classic SPS phenotype. Some autoantibodies present in patients with SPS are strongly associated with cancers, such as antibodies to amphiphysin 45,46. 47. The different immune specificities may indicate distinct underlying disease mechanisms and underpinnings which could account for some of the variations in clinical phenotype. Conceptually, it is thought that immune responses in SPSD target inhibitory interneurons and their pathways, albeit through different mechanisms (Figure 1).
Table 3.
Autoantigens associated with Stiff Person Syndrome Spectrum Disorders
| Antigen | Patients with antibody (%) | Reference |
|---|---|---|
| GAD65 * | ~70–85% | 2,47,55 |
| Glycine receptor * | Unknown | 43,44 |
| Amphiphysin * | <5% | 78,108 |
| Zic4 | Case reports | 85 |
| DPPX | ~3% | 76,109 |
| Gephyrin | Case reports | 110 |
| GABAA receptor | ~3% | 65 |
| GABARAP | ~70% | 67 |
These autoantibodies are thought to be the most important of those identified in Stiff Person Syndrome Spectrum Disorders and testing for them is commercially available.
Figure 1. Potential mechanisms of immunopathology in Stiff Person Syndrome Spectrum Disorders.
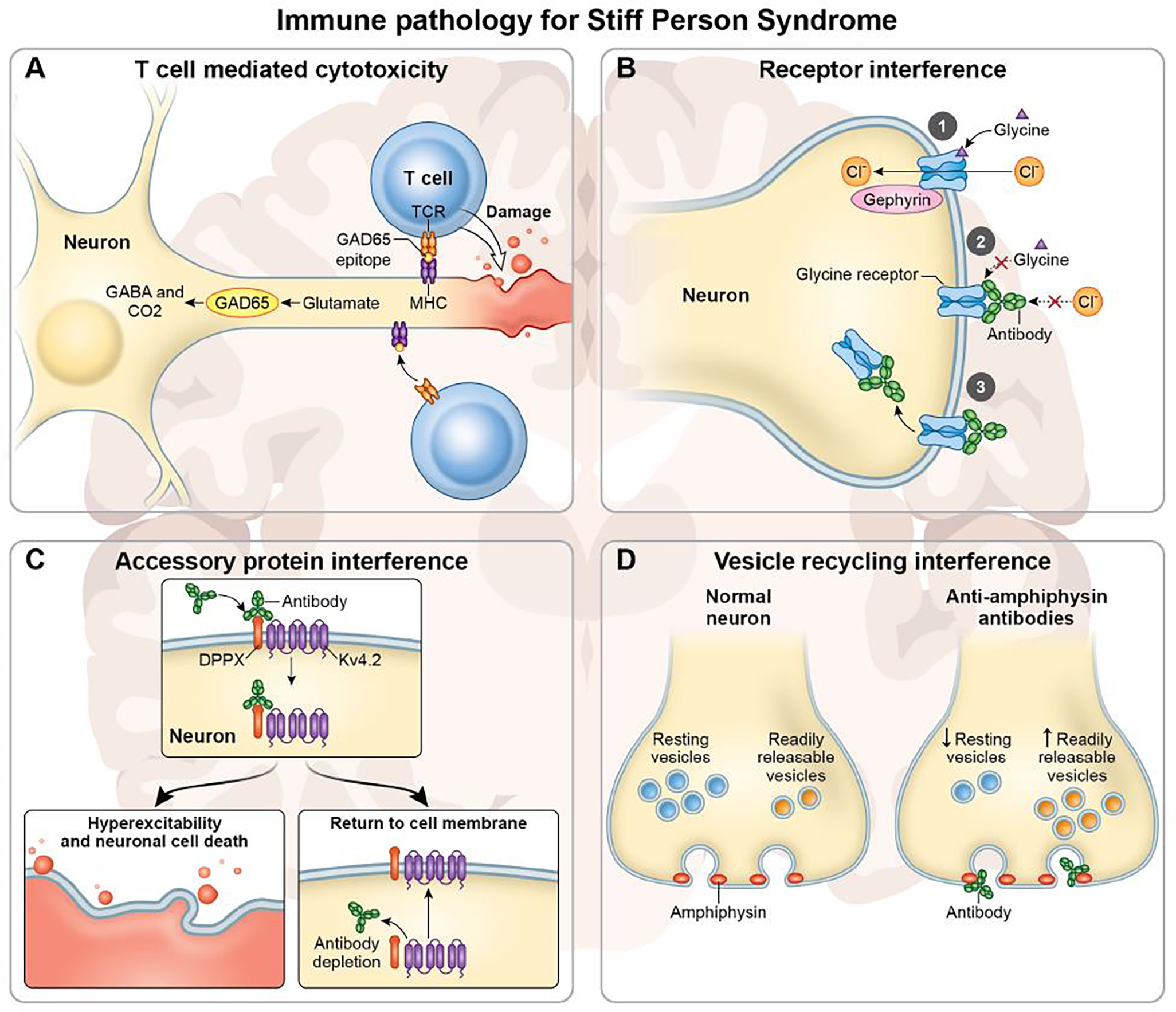
(A) T cell mediated cytotoxicity. GAD65 expressing neurons synthesize the neurotransmitter GABA from glutamate. GAD65 epitopes are processed and presented in MHC molecules which are recognized by autoreactive T cells. T cells that recognized GAD65 epitopes can initiate cytotoxic immune responses including the release of perforin and granzyme B. (B) Receptors are impaired by antibody binding. 1) The normal function of glycine receptors. Upon binding by glycine, glycine receptors open and allow for chloride ions to flow through into the cell. 2) Antibody mediated inhibition of receptor functioning. Antibodies to the glycine receptor may cause the channel to remain closed despite the presence of glycine. 3) Antibody mediated internalization of receptors. Upon antibody binding, glycine receptors are internalized by the cell thereby limiting the number of active and available receptors for glycine binding and ion flux. (C) Antibodies to accessory proteins can also result in channel internalization. DPPX and Kv4.2 are both expressed on neuronal membranes. Antibodies specific to DPPX initiate internalization of both DPPX and KV4.2 (top), resulting in hyperexcitability, and potentially death, of the neuron (bottom left). Both DPPX and Kv4.2 can re-establish on the membrane surface upon antibody removal (bottom right). (D) Antibodies can interfere with vesicle recycling and neuronal signaling. Amphiphysin is an important regulator of clathrin-coated synaptic vesicles. In a healthy neuron (left) there is continuous recycling of vesicles. Antibodies to amphiphysin are internalized and interfere with vesicle recycling (right). This results in an accumulation of readily releasable vesicles (yellow) and a decrease in resting vesicles (blue), resulting in impairments of neuronal signaling.
The main inhibitory signals in the CNS are γ-aminobutyric acid (GABA) and glycine (recently reviewed in 48). These neurotransmitters are released by inhibitory interneurons, which are a diverse group of neurons that collectively represent approximately 10–20% of neurons in the CNS (recently reviewed in 49). These nerve cells are important in shaping and modulating neural circuits and alterations in inhibitory interneuron functions have been implicated in epilepsy 50, Alzheimer’s disease 51, and in developmental disorders such as autism 52. Glycine and GABA activate their respective ion channels, the glycine receptor and GABAA receptors, which allows for an influx of chloride ions into the cell thereby inducing hyperpolarization of the postsynaptic neurons and raising the threshold for firing of an action potential 48,53. This inhibition is critical for appropriate CNS development and overall function 49,54.
The immune specificities defined in SPSD demonstrate that these immune responses are targeting inhibitory interneurons (Figure 1). GAD65, which is the most common immune specificity associated with SPSD 2,4,6,47,55, is the rate limiting enzyme in the synthesis of GABA. GAD65 is expressed in the CNS as well as in the β-cells of the pancreas and GAD65 is also an autoantigen in patients with type 1 diabetes mellitus 56, which often co-occurs with SPSD. Although patients have antibodies to GAD65, these antibodies do not appear to be directly pathogenic. Antibodies to GAD65 are not internalized by neurons, and therefore do not engage with the intracellular antigen 57. Further, antibody titers to GAD65 have traditionally not correlated with clinical disease 26, although recent data may suggest some association with burden of disease 12,15. However, GAD65 specific antibodies do recognize linear epitopes 57 and therefore suggest a robust T cell response to GAD65 (Figure 1A). Memory T cells recognizing GAD65 peptides could therefore enter the CNS and mount effector responses against GAD65 expressing neurons 58. Supporting this, GAD65 specific T cells have been isolated from the CSF of patients with SPS 59. Further, post-mortem studies in patients with SPS and GAD65 immune specificities have revealed decreases in GABA expressing interneurons in the cerebellum 60 as well as infiltrating CD8+ T cells 59. It is important to note that GAD65 autoimmune responses can occur with other immune specificities 55 and that multiple mechanisms of immune-mediated damage, driven by multiple immune specificities, can exist within the same patient.
Pathogenic autoantibodies do exist in association with other immune specificities found in patients with SPSD. These antibodies result in disruption of inhibitory interneuron functioning through diverse mechanisms including internalization of proteins from the membrane, inducing alterations in receptor functioning, and modulating synaptic vesicle trafficking (Figure 1). For example, antibodies against the glycine receptor from patients with SPS disrupt receptor trafficking and functioning. Binding of these antibodies results in the internalization of the glycine receptor in vitro 61,62 and in vivo 63, although continued expression of the glycine receptor on the membrane surface was detected despite internalization of the antibody-associated receptor. Additional in vivo studies utilizing zebra fish revealed altered glycine receptor functioning in the presence of glycine receptor antibodies from patients despite membrane expression of the receptor 64.
GABAA receptors are also an immunologic target in SPS 65 and antibodies to GABAA share a similar pathogenic mechanism as glycine receptor antibodies. Autoantibodies to subunits of GABAA receptors from patients with encephalitis cause decrease membrane expression of GABAA receptor 66 which in turn decreases inhibitory neuronal signaling resulting in increased neuronal excitation. Other proteins associated with GABAA receptors are also targets of immune responses in SPS. These immune specificities include GABAA receptor-associated protein (GABARAP) 67, which helps to guide the intracellular trafficking of GABAA receptors 68, and gephyrin which interacts with both GABAA receptors and glycine receptors 69. Although the pathogenic mechanism of gephyrin antibodies has not been elucidated, antibodies against GABARAP from patients with SPS decrease membrane expression of GABAA receptors in vitro 67.
Autoantibodies against dipeptidyl-peptidase-like protein-6 (DPPX) also result in decreased expression of DPPX and the Kv4.2 potassium channel, of which DPPX is a cell surface auxiliary subunit of 70, on neuronal membranes 71. The Kv4.2 potassium channel is an A-type channel 72, and the decrease of the Kv4.2 potassium channel results in hyperexcitability of neurons 71,73 which may lead to neurotoxicity and loss of neurons in patients (Figure 1C). Although associated with SPS in the literature the clinical syndrome has differences as compared to SPSD. A case report of a patient with DPPX-antibody associated encephalitis revealed infiltrating T cells and neuronal loss in the CA4 and CA3 regions of the hippocampus 74. However, in patients with DPPX antibodies, immune modulatory therapies do improve outcomes and membrane expression of DPPX and Kv4.2 reestablish after removal of DPPX antibodies 75,76. More data are required to determine if this immune response is associated with SPSD.
Amphiphysin, a member of the Bin/Amphiphysin/Rvs (BAR)-domain containing proteins, is an important regulator of clathrin-coated synaptic vesicles 77 and is also an immune target in patients with SPS 78. Loss of amphiphysin in murine models results in reductions in synaptic vesicle recycling 79 and antibodies to amphiphysin from patients with SPS cause SPS like symptoms in a rat model 80. Amphiphysin antibodies are internalized in an antigen specific manner and cause a decrease in the release of GABA 80. Further investigations into the mechanism behind this loss of GABA secretion revealed that antibodies to amphiphysin caused alterations in the composition of synaptic vesicles 81. Amphiphysin antibodies caused an increase in synaptobrevin-2, found on readily releasable vesicles, and a decrease in synaptobrevin-7, found on resting pool vesicles 81. Collectively these data indicate an impairment in vesicle trafficking induced by amphiphysin antibodies which may lead to dysfunctional synapses resulting in the clinical observed in SPSD (Figure 1D).
Zic4 is a neuronal autoantigen that is primarily associated with paraneoplastic disease. Autoantibodies to Zic4 are enriched in patients with small-cell lung cancer 45, however with the advent of checkpoint inhibitor therapy Zic4 antibodies are emerging in association with other cancers 82. The majority of patients with Zic4 autoantibodies have cerebellar degeneration 83,84, but recently these antibodies were identified in a patient with seronegative SPS without obvious cerebellar involvement 85. The co-occurrence of cerebellar ataxia and SPS is uncommon and may be an epiphenomenon 86–88 however cerebellar involvement in SPSD does occur and can be considered a distinct clinical phenotype (e.g., SPS-plus, see Table 1).
A subgroup of patients with SPSD test negative for antibodies to known autoantigens 2. Defining additional immune specificities associated with SPSD is a critical area of research and represents an unmet need. Studies suggest that responses to immune therapy may be more likely beneficial to patients with certain immune specificities, such as patients with glycine receptor autoantibodies readily responding to immune modulation 61,62 whereas patients with GAD65 show mixed responses to immune modulation 7 which may be dependent upon antibody titer 89. Additionally, some autoantibodies may confer protection rather than be directly pathogenic 90 by participating in sequestering or clearing over abundant or altered proteins. Further characterizing immune specificities in SPSD may be beneficial for understanding disease mechanism as well as for prognosis, diagnosis, and guiding therapeutic interventions. Importantly, potentially new immune specificities are being described 85 and likely more are yet to be discovered.
6. Immunopathogenesis of SPS
Although several mechanisms of immune-mediated loss of neuronal inhibitory pathways have been elucidated, work is still needed to understand the factors that contribute to the loss of tolerance and the subsequent development of autoimmunity in patients with SPSD.
Autoimmune diseases have been described as occurring in four phases: susceptibility, initiation, propagation, and regulation 91. The first phase is susceptibility, which describes the time before disease onset but where conditions exist for future disease initiation. These conditions include factors such as altered signaling thresholds, impairment of central tolerance, or inhibition in apoptosis or clearance pathways (reviewed in 91) which can all result in the loss of immune tolerance. The second phase is initiation, which is before disease symptoms appear, but during which there is presentation of epitopes to T cells that result in an activating immune response. It is during this time that loss of immune tolerance unfolds, and disease processes start. Although the susceptibility conditions and initiation events of SPSD remain undefined, emerging data, described below, suggest that both genetic and acquired risk factors may exist that contribute to the development and propagation of SPSD.
6.1. Lymphocyte signaling thresholds
Evidence of altered susceptibility factors defined in other autoimmune diseases appear to also occur in patients with SPSD, however robust data are lacking. For example, immune signaling thresholds are important for determining if lymphocyte signaling ultimately results in T cell activation or inhibition. One important negative regulator of T cell activation is cytotoxic T lymphocyte antigen-4 (CTLA-4) and mutations in this gene are known to confer susceptibility in autoimmunity 92. Four patients with SPS were included in a recent study investigating potential genetic variants that may be linked to GAD65 autoimmune associated neurologic disease 93. All four patients with SPS had a known CTLA-4 variant that is associated with type 1 diabetes mellitus 94, suggesting alterations in T cell signaling thresholds, mediated by CTLA-4, may contribute to the development of SPS. However, future studies will need to confirm these findings in larger cohorts of patients and assess if only subgroups of SPSD have these alterations.
6.2. Cryptic antigens
As in other autoimmune diseases, associations with human leukocyte antigen (HLA) haplotypes and SPS are recognized. Certain HLA-DR and DQ alleles are associated with SPS, including DQB1*0201 95 and DRB1*0301 26,47. These have been identified in cohort studies which found these HLA alleles highly enriched in patients with SPS as compared to the general population. Recent work in type I diabetes mellitus indicates that certain HLA haplotypes increase risk of autoimmune diseases because the HLA forms autoantigen-HLA complexes with low stability 96, thereby these autoantigen epitopes may not be stably expressed in the thymus during T cell development. T cell receptors that are specific for these epitopes may therefore not encounter these epitopes resulting in a lack of negative selection during central tolerance selection processes. These epitopes, known as cryptic epitopes 97, when expressed in the periphery, are capable of activating these autoreactive T cells. However, the factors that would allow for increased stability of antigen presentation by these specific HLA molecules in the periphery are not yet identified. Some evidence suggests that high affinity binding of GAD65 by autoantibodies increases GAD65 antigen presentation 98, similar to what has been described for other autoantigens 99.
Another important loss of tolerance can occur when neoantigens are produced from somatic mutations. These neoantigens are recognized by T cells as foreign and allow for immune responses to neoplasms 100. However, antibody responses against the wild-type version of the mutated protein can be generated via linked recognition, resulting in the development of autoimmune disease as has been demonstrated in scleroderma 101. This process may explain how patients with SPSD lose tolerance to certain antigens known to be altered in associated cancers and develop autoantibodies to wild-type proteins.
Aside from lack of thymic expression or mutations, other factors, including epitope degradation of CNS antigens by proteases expressed in the thymus 102,103, post-translational modifications 104, cleavage of autoantigens during inflammatory processes 105, inhibition of apoptosis and clearance 91, and netosis 106 contribute to the loss of tolerance to autoantigens in other autoimmune diseases. These mechanisms may be relevant to SPSD pathogenesis. Infections and inflammation due to major life stressors may also initiate autoimmune diseases and both these mechanisms are suggested to occur in SPSD. Additional investigations into forces driving loss of peripheral tolerance in SPSD are greatly needed. Examining antigen presentation and autoantigen modifications, determined directly from antigen presenting cells from patients 107 with SPSD, may be particularly informative.
7. Outlook
There are several great unmet needs in SPSD, which span across the clinical and basic science arenas. Clinically, more refined diagnostic criteria are needed especially if this will help clinicians make earlier and accurate diagnoses. It often takes several years from symptom onset to diagnosis for people with SPSD, which in part is due to lack of awareness of these conditions and outdated diagnostic criteria. In addition, further studies are needed to determine if specific (or all) SPSD phenotypes require immune based therapies shortly after symptom onset versus starting with only symptomatic interventions and adding immune therapies if and when people experience increasing disease burden. There is a precedent set with other autoimmune disorders (e.g., MS, NMOSD, MG, etc.) for starting immune therapies early on in order to help prevent future disability and has become the standard of care treatment approach. The uncertainty with what may be the best treatment approach in SPSD stems from a lack of clinical and paraclinical biomarkers that correlate with future disability and treatment response. Hence, identifying such biomarkers would help push the field forward and help improve long-term outcomes for our patients.
Basic science studies and further investigations into the immunopathogenesis of SPSD are also required. In particular, studies that further our understanding of T cell responses in SPSD, and in particular identifying pathogenic T cell clones, is a critical area of research. Not only will this help inform disease mechanisms but may also provide a unique target for therapeutics. Recent advances have demonstrated that elimination of pathogenic T cell clones via bi-specific single chain variable fragment antibodies without globally impairing T cell function is possible 101. However, the application of this novel therapeutic approach to autoimmune disease requires the identification of pathogenic T cell clones. Future investigations into SPSD pathogenesis should focus on identification and confirmation of T cell clones that could be drivers of disease.
Highlights.
Stiff person syndrome spectrum disorders (SPSD) are immune mediated and impact different regions of the nervous system.
SPSD have heterogenous presentations and are under-recognized.
A combination of pharmacological and non-pharmacological interventions can help mitigate the burden of disease in SPSD.
Advances in understanding of pathogenic mechanisms are important for guiding therapy development.
Acknowledgements
The authors would like to acknowledge Ethan Tyler for assistance in creating Figure 1.
Abbreviations:
- SPS
Stiff person syndrome (SPS)
- SPSD
SPS spectrum disorders (SPSD)
- CA
cerebellar ataxia (CA)
- PERM
progressive encephalomyelitis with rigidity and myoclonus
- GAD65
glutamic acid decarboxylase 65-kilodalton isoform
- VOM
vestibular and ocular motor
- CSF
cerebrospinal fluid
- GABA
γ-aminobutyric acid
- GABAergic
γ-aminobutyric acid (GABA)-ergic
- GI
gastrointestinal
- mRS
modified Rankin Scale
- NRIs
norepinephrine reuptake inhibitors
- MOA
mechanism of action
- IVIG
intravenous immunoglobulin
- SCIG
subcutaneous immunoglobulin
- TPE
therapeutic plasma exchange
- auto-HSCT
Autologous hematopoietic stem cell transplantation
- MS
multiple sclerosis
- GABARAP
GABAA receptor-associated protein
- DPPX
dipeptidyl-peptidase-like protein-6
- BAR
Bin/Amphiphysin/Rvs
- CTLA-4
cytotoxic T lymphocyte antigen-4
- HLA
human leukocyte antigen
- NETs
neutrophil extracellular traps
Footnotes
Conflict of interest: Dr. Johnson does not have any conflicts of interest to declare. Dr. Scott Newsome has received consultant fees for scientific advisory boards from Biogen, Genentech, Bristol Myers Squibb, EMD Serono, Greenwich Biosciences, Novartis, Horizon Therapeutics, is an advisor for BioIncept and Autobahn, is the study lead PI for a Roche clinical trial, was a clinical adjudication committee member for a medDay Pharmaceuticals clinical trial, and has received research funding (paid directly to institution) from Biogen, Roche, Genentech, National Multiple Sclerosis Society, Department of Defense, and Patient Centered Outcomes Institute.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome); report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin 1956; 31(15): 421–7. [PubMed] [Google Scholar]
- 2.Martinez-Hernandez E, Arino H, McKeon A, et al. Clinical and Immunologic Investigations in Patients With Stiff-Person Spectrum Disorder. JAMA Neurol 2016; 73(6): 714–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol 2011; 11(5): 272–82. [DOI] [PubMed] [Google Scholar]
- 4.Johnston MV, Adams HP, Fatemi A. Neurobiology of disease. Second edition. ed. Oxford; New York: Oxford University Press; 2016. [Google Scholar]
- 5.Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008; 131(Pt 10): 2553–63. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Tian F, Aljarallah S, Fitzgerald K, Newsome SD. A Clinical Characterization of Stiff Person Syndrome Spectrum Disorders (2547). Neurology 2020; 94(15 Supplement): 2547. [Google Scholar]
- 7.McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol 2012; 69(2): 230–8. [DOI] [PubMed] [Google Scholar]
- 8.Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004; 75(8): 1135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeshokumar AK, Sun LR, Newsome SD. Defining the Expanding Clinical Spectrum of Pediatric-Onset Stiff Person Syndrome. Pediatr Neurol 2021; 114: 11–5. [DOI] [PubMed] [Google Scholar]
- 10.Schreck KC, Orthmann-Murphy JL, Newsome SD. Clinical Reasoning: A 70-year-old woman with acute-onset weakness and progressive hemiataxia. Neurology 2016; 87(22): e264–e8. [DOI] [PubMed] [Google Scholar]
- 11.Clardy SL, Lennon VA, Dalmau J, et al. Childhood onset of stiff-man syndrome. JAMA Neurol 2013; 70(12): 1531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Budhram A, Sechi E, Flanagan EP, et al. Clinical spectrum of high-titre GAD65 antibodies. J Neurol Neurosurg Psychiatry 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabatino JJ, Jr., Newsome SD. Stiff person syndrome masquerading as multiple sclerosis. J Neurol Sci 2017; 372: 297–9. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Tourkevich R, Bosley J, Gold DR, Newsome SD. Ocular Motor and Vestibular Characteristics of Antiglutamic Acid Decarboxylase-Associated Neurologic Disorders. J Neuroophthalmol 2020. [DOI] [PubMed] [Google Scholar]
- 15.Lambe J, Rothman A, Prince J, Saidha S, Calabresi PA, Newsome SD. Retinal pathology occurs in stiff-person syndrome. Neurology 2020; 94(20): e2126–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koshorek J, Maldonado DP, Reyes-Mantilla M, Obando D, Wang Y, Newsome S. The Evolving Spectrum of Gastrointestinal Dysfunction in Stiff Person Syndrome (4200). Neurology 2021; 96(15 Supplement): 4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan CK, Maldonado DAP, Wang Y, Obando D, Hughes AJ, Newsome SD. Cognitive and mood profiles among patients with stiff person syndrome spectrum disorders. Frontiers in Neurology 2022; 13:865462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukharesh L, Fitzgerald K, Wang Y, Newsome S. Can Certain Baseline Characteristics in Patients with Stiff Person Syndrome (SPS) Predict Disease Burden and Functional Outcomes? (5277). Neurology 2020; 94(15 Supplement): 5277. [Google Scholar]
- 19.Rakocevic G, Raju R, Dalakas MC. Anti-glutamic acid decarboxylase antibodies in the serum and cerebrospinal fluid of patients with stiff-person syndrome: correlation with clinical severity. Arch Neurol 2004; 61(6): 902–4. [DOI] [PubMed] [Google Scholar]
- 20.Olafson RA, Mulder DW, Howard FM. “Stiff-Man” Syndrome: A Review of the Literature, Report of Three Additional Cases and Discussion of Pathophysiology and Therapy. Mayo Clin Proc 1964; 39: 131–44. [PubMed] [Google Scholar]
- 21.Howard FM Jr. A new and effective drug in the treatment of the stiff-man syndrome: preliminary report. Proc Staff Meet Mayo Clin 1963; 38: 203–12. [PubMed] [Google Scholar]
- 22.Murinson BB. Stiff-person syndrome. Neurologist 2004; 10(3): 131–7. [DOI] [PubMed] [Google Scholar]
- 23.Benavides DR, Newsome SD. Serotonin-norepinephrine reuptake inhibitors may exacerbate stiff-person syndrome. Neurol Neuroimmunol Neuroinflamm 2016; 3(5): e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meinck HM, Ricker K, Conrad B. The stiff-man syndrome: new pathophysiological aspects from abnormal exteroceptive reflexes and the response to clomipramine, clonidine, and tizanidine. J Neurol Neurosurg Psychiatry 1984; 47(3): 280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meinck HM, Tronnier V, Rieke K, Wirtz CR, Flugel D, Schwab S. Intrathecal baclofen treatment for stiff-man syndrome: pump failure may be fatal. Neurology 1994; 44(11): 2209–10. [DOI] [PubMed] [Google Scholar]
- 26.Rakocevic G, Alexopoulos H, Dalakas MC. Quantitative clinical and autoimmune assessments in stiff person syndrome: evidence for a progressive disorder. BMC Neurol 2019; 19(1): 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dalakas MC, Fujii M, Li M, Lutfi B, Kyhos J, McElroy B. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med 2001; 345(26): 1870–6. [DOI] [PubMed] [Google Scholar]
- 28.Aljarallah S, Newsome SD. Use of subcutaneous immunoglobulin in stiff person syndrome: Case series. Medicine (Baltimore) 2021; 100(12): e25260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albahra S, Yates SG, Joseph D, De Simone N, Burner JD, Sarode R. Role of plasma exchange in stiff person syndrome. Transfus Apher Sci 2019; 58(3): 310–2. [DOI] [PubMed] [Google Scholar]
- 30.Pagano MB, Murinson BB, Tobian AA, King KE. Efficacy of therapeutic plasma exchange for treatment of stiff-person syndrome. Transfusion 2014; 54(7): 1851–6. [DOI] [PubMed] [Google Scholar]
- 31.Rizzi M, Knoth R, Hampe CS, et al. Long-lived plasma cells and memory B cells produce pathogenic anti-GAD65 autoantibodies in Stiff Person Syndrome. PLoS One 2010; 5(5): e10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol 2017; 82(2): 271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aljarallah S, Wang Y, Shoemaker T, Newsome S. Long-term Rituximab Use Benefits Patients with Stiff Person Syndrome (P2.2–033). Neurology 2019; 92(15 Supplement): P2.–033. [Google Scholar]
- 34.Sharrack B, Saccardi R, Alexander T, et al. Autologous haematopoietic stem cell transplantation and other cellular therapy in multiple sclerosis and immune-mediated neurological diseases: updated guidelines and recommendations from the EBMT Autoimmune Diseases Working Party (ADWP) and the Joint Accreditation Committee of EBMT and ISCT (JACIE). Bone Marrow Transplant 2020; 55(2): 283–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kass-Iliyya L, Snowden JA, Thorpe A, et al. Autologous haematopoietic stem cell transplantation for refractory stiff-person syndrome: the UK experience. J Neurol 2021; 268(1): 265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanders S, Bredeson C, Pringle CE, et al. Autologous stem cell transplantation for stiff person syndrome: two cases from the Ottawa blood and marrow transplant program. JAMA Neurol 2014; 71(10): 1296–9. [DOI] [PubMed] [Google Scholar]
- 37.Burt RK, Balabanov R, Han X, et al. Autologous Hematopoietic Stem Cell Transplantation for Stiff-Person Spectrum Disorder: A Clinical Trial. Neurology 2021; 96(6): e817–e30. [DOI] [PubMed] [Google Scholar]
- 38.Strunz PP, Froehlich M, Gernert M, et al. Immunological Adverse Events After Autologous Hematopoietic Stem Cell Transplantation in Systemic Sclerosis Patients. Front Immunol 2021; 12: 723349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grant A, Chapman LRM, Mitchell R, O’Brien TA. Engraftment syndrome following hematopoietic stem cell transplant: A review of the literature. Clin Transplant 2020; 34(6): e13875. [DOI] [PubMed] [Google Scholar]
- 40.Cornell RF, Hari P, Drobyski WR. Engraftment Syndrome after Autologous Stem Cell Transplantation: An Update Unifying the Definition and Management Approach. Biol Blood Marrow Transplant 2015; 21(12): 2061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown JWL, Coles A, Horakova D, et al. Association of Initial Disease-Modifying Therapy With Later Conversion to Secondary Progressive Multiple Sclerosis. JAMA 2019; 321(2): 175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reyes M, Wang Y, kalaitzidis G, et al. Immunotherapy in Stiff Person Syndrome, is Timing Really Important? (5176). Neurology 2021; 96(15 Supplement): 5176. [Google Scholar]
- 43.Crisp SJ, Balint B, Vincent A. Redefining progressive encephalomyelitis with rigidity and myoclonus after the discovery of antibodies to glycine receptors. Curr Opin Neurol 2017; 30(3): 310–6. [DOI] [PubMed] [Google Scholar]
- 44.Hutchinson M, Waters P, McHugh J, et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology 2008; 71(16): 1291–2. [DOI] [PubMed] [Google Scholar]
- 45.Bataller L, Wade DF, Graus F, Stacey HD, Rosenfeld MR, Dalmau J. Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer. Neurology 2004; 62(5): 778–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antoine JC, Absi L, Honnorat J, et al. Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol 1999; 56(2): 172–7. [DOI] [PubMed] [Google Scholar]
- 47.Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology 2000; 55(10): 1531–5. [DOI] [PubMed] [Google Scholar]
- 48.Kasaragod VB, Schindelin H. Structure-Function Relationships of Glycine and GABAA Receptors and Their Interplay With the Scaffolding Protein Gephyrin. Front Mol Neurosci 2018; 11: 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swanson OK, Maffei A. From Hiring to Firing: Activation of Inhibitory Neurons and Their Recruitment in Behavior. Front Mol Neurosci 2019; 12: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Magloire V, Mercier MS, Kullmann DM, Pavlov I. GABAergic Interneurons in Seizures: Investigating Causality With Optogenetics. Neuroscientist 2019; 25(4): 344–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Y, Zhao M, Han Y, Zhang H. GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment. Front Neurosci 2020; 14: 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiebe S, Nagpal A, Truong VT, et al. Inhibitory interneurons mediate autism-associated behaviors via 4E-BP2. Proc Natl Acad Sci U S A 2019; 116(36): 18060–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 2011; 71(1): 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J, Yang X, Tang K. Interneuron development and dysfunction. FEBS J 2021. [DOI] [PubMed] [Google Scholar]
- 55.Chang T, Alexopoulos H, McMenamin M, et al. Neuronal surface and glutamic acid decarboxylase autoantibodies in Nonparaneoplastic stiff person syndrome. JAMA Neurol 2013; 70(9): 1140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Towns R, Pietropaolo M. GAD65 autoantibodies and its role as biomarker of Type 1 diabetes and Latent Autoimmune Diabetes in Adults (LADA). Drugs Future 2011; 36(11): 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gresa-Arribas N, Arino H, Martinez-Hernandez E, et al. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS One 2015; 10(3): e0121364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol 2017; 17(3): 179–94. [DOI] [PubMed] [Google Scholar]
- 59.Holmoy T, Skorstad G, Roste LS, Scheie D, Alvik K. Stiff person syndrome associated with lower motor neuron disease and infiltration of cytotoxic T cells in the spinal cord. Clin Neurol Neurosurg 2009; 111(8): 708–12. [DOI] [PubMed] [Google Scholar]
- 60.Warich-Kirches M, Von Bossanyi P, Treuheit T, et al. Stiff-man syndrome: possible autoimmune etiology targeted against GABA-ergic cells. Clin Neuropathol 1997; 16(4): 214–9. [PubMed] [Google Scholar]
- 61.Hinson SR, Lopez-Chiriboga AS, Bower JH, et al. Glycine receptor modulating antibody predicting treatable stiff-person spectrum disorders. Neurol Neuroimmunol Neuroinflamm 2018; 5(2): e438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carvajal-Gonzalez A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 2014; 137(Pt 8): 2178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carvajal-Gonzalez A, Jacobson L, Clover L, et al. Systemic delivery of human GlyR IgG antibody induces GlyR internalization into motor neurons of brainstem and spinal cord with motor dysfunction in mice. Neuropathol Appl Neurobiol 2021; 47(2): 316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rauschenberger V, von Wardenburg N, Schaefer N, et al. Glycine Receptor Autoantibodies Impair Receptor Function and Induce Motor Dysfunction. Ann Neurol 2020; 88(3): 544–61. [DOI] [PubMed] [Google Scholar]
- 65.Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014; 13(3): 276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohkawa T, Satake S, Yokoi N, et al. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci 2014; 34(24): 8151–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raju R, Rakocevic G, Chen Z, et al. Autoimmunity to GABAA-receptor-associated protein in stiff-person syndrome. Brain 2006; 129(Pt 12): 3270–6. [DOI] [PubMed] [Google Scholar]
- 68.Chen ZW, Chang CS, Leil TA, Olsen RW. C-terminal modification is required for GABARAP-mediated GABA(A) receptor trafficking. J Neurosci 2007; 27(25): 6655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tretter V, Mukherjee J, Maric HM, Schindelin H, Sieghart W, Moss SJ. Gephyrin, the enigmatic organizer at GABAergic synapses. Front Cell Neurosci 2012; 6: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nadal MS, Ozaita A, Amarillo Y, et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 2003; 37(3): 449–61. [DOI] [PubMed] [Google Scholar]
- 71.Piepgras J, Holtje M, Michel K, et al. Anti-DPPX encephalitis: pathogenic effects of antibodies on gut and brain neurons. Neurology 2015; 85(10): 890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol 2005; 569(Pt 1): 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kiselycznyk C, Hoffman DA, Holmes A. Effects of genetic deletion of the Kv4.2 voltage-gated potassium channel on murine anxiety-, fear- and stress-related behaviors. Biol Mood Anxiety Disord 2012; 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stokin GB, Popovic M, Gelpi E, Kogoj A, Dalmau J, Graus F. Neuropathologic features of anti-dipeptidyl-peptidase-like protein-6 antibody encephalitis. Neurology 2015; 84(4): 430–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hara M, Arino H, Petit-Pedrol M, et al. DPPX antibody-associated encephalitis: Main syndrome and antibody effects. Neurology 2017; 88(14): 1340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boronat A, Gelfand JM, Gresa-Arribas N, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol 2013; 73(1): 120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.David C, McPherson PS, Mundigl O, de Camilli P. A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Proc Natl Acad Sci U S A 1996; 93(1): 331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Camilli P, Thomas A, Cofiell R, et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of Stiff-Man syndrome with breast cancer. J Exp Med 1993; 178(6): 2219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Di Paolo G, Sankaranarayanan S, Wenk MR, et al. Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron 2002; 33(5): 789–804. [DOI] [PubMed] [Google Scholar]
- 80.Geis C, Weishaupt A, Hallermann S, et al. Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain 2010; 133(11): 3166–80. [DOI] [PubMed] [Google Scholar]
- 81.Werner C, Pauli M, Doose S, et al. Human autoantibodies to amphiphysin induce defective presynaptic vesicle dynamics and composition. Brain 2016; 139(Pt 2): 365–79. [DOI] [PubMed] [Google Scholar]
- 82.Iyer SG, Khakoo NS, Aitcheson G, Perez C. Case of anti-Zic4 antibody-mediated cerebellar toxicity induced by dual checkpoint inhibition in head and neck squamous cell carcinoma. BMJ Case Rep 2020; 13(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sabater L, Bataller L, Suarez-Calvet M, Saiz A, Dalmau J, Graus F. ZIC antibodies in paraneoplastic cerebellar degeneration and small cell lung cancer. J Neuroimmunol 2008; 201–202: 163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bataller L, Wade DF, Fuller GN, Rosenfeld MR, Dalmau J. Cerebellar degeneration and autoimmunity to zinc-finger proteins of the cerebellum. Neurology 2002; 59(12): 1985–7. [DOI] [PubMed] [Google Scholar]
- 85.Bernardo F, Rebordao L, Rego A, et al. Stiff person spectrum disorders: An illustrative case series of their phenotypic and antibody diversity. J Neuroimmunol 2020; 341: 577192. [DOI] [PubMed] [Google Scholar]
- 86.Seneviratne SO, Buzzard KA, Cruse B, Monif M. Cerebellar Ataxia Followed by Stiff Person Syndrome in a Patient with Anti-GAD Antibodies. Case Reports Immunol 2020; 2020: 8454532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rakocevic G, Raju R, Semino-Mora C, Dalakas MC. Stiff person syndrome with cerebellar disease and high-titer anti-GAD antibodies. Neurology 2006; 67(6): 1068–70. [DOI] [PubMed] [Google Scholar]
- 88.Kono S, Miyajima H, Sugimoto M, Suzuki Y, Takahashi Y, Hishida A. Stiff-person syndrome associated with cerebellar ataxia and high glutamic acid decarboxylase antibody titer. Intern Med 2001; 40(9): 968–71. [DOI] [PubMed] [Google Scholar]
- 89.Munoz-Lopetegi A, de Bruijn M, Boukhrissi S, et al. Neurologic syndromes related to anti-GAD65: Clinical and serologic response to treatment. Neurol Neuroimmunol Neuroinflamm 2020; 7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Darrah E, Giles JT, Davis RL, et al. Autoantibodies to Peptidylarginine Deiminase 2 Are Associated With Less Severe Disease in Rheumatoid Arthritis. Front Immunol 2018; 9: 2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Antiochos B, Rosen A. Mechanisms of Autoimmunity. 2019: 677–84.e1. [Google Scholar]
- 92.Rai E, Wakeland EK. Genetic predisposition to autoimmunity--what have we learned? Semin Immunol 2011; 23(2): 67–83. [DOI] [PubMed] [Google Scholar]
- 93.Thaler FS, Bangol B, Biljecki M, Havla J, Schumacher AM, Kumpfel T. Possible link of genetic variants to autoimmunity in GAD-antibody-associated neurological disorders. J Neurol Sci 2020; 413: 116860. [DOI] [PubMed] [Google Scholar]
- 94.Marron MP, Raffel LJ, Garchon HJ, et al. Insulin-dependent diabetes mellitus (IDDM) is associated with CTLA4 polymorphisms in multiple ethnic groups. Hum Mol Genet 1997; 6(8): 1275–82. [DOI] [PubMed] [Google Scholar]
- 95.Pugliese A, Solimena M, Awdeh ZL, et al. Association of HLA-DQB1*0201 with stiff-man syndrome. J Clin Endocrinol Metab 1993; 77(6): 1550–3. [DOI] [PubMed] [Google Scholar]
- 96.Miyadera H, Tokunaga K. Associations of human leukocyte antigens with autoimmune diseases: challenges in identifying the mechanism. J Hum Genet 2015; 60(11): 697–702. [DOI] [PubMed] [Google Scholar]
- 97.Sercarz EE, Lehmann PV, Ametani A, Benichou G, Miller A, Moudgil K. Dominance and crypticity of T cell antigenic determinants. Annu Rev Immunol 1993; 11: 729–66. [DOI] [PubMed] [Google Scholar]
- 98.Reijonen H, Daniels TL, Lernmark A, Nepom GT. GAD65-specific autoantibodies enhance the presentation of an immunodominant T-cell epitope from GAD65. Diabetes 2000; 49(10): 1621–6. [DOI] [PubMed] [Google Scholar]
- 99.Lanzavecchia A How can cryptic epitopes trigger autoimmunity? J Exp Med 1995; 181(6): 1945–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Engelhorn ME, Guevara-Patino JA, Noffz G, et al. Autoimmunity and tumor immunity induced by immune responses to mutations in self. Nat Med 2006; 12(2): 198–206. [DOI] [PubMed] [Google Scholar]
- 101.Joseph CG, Darrah E, Shah AA, et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science 2014; 343(6167): 152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Manoury B, Mazzeo D, Fugger L, et al. Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol 2002; 3(2): 169–74. [DOI] [PubMed] [Google Scholar]
- 103.Serre L, Girard M, Ramadan A, et al. Thymic-Specific Serine Protease Limits Central Tolerance and Exacerbates Experimental Autoimmune Encephalomyelitis. J Immunol 2017; 199(11): 3748–56. [DOI] [PubMed] [Google Scholar]
- 104.Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Ann N Y Acad Sci 2005; 1050: 1–9. [DOI] [PubMed] [Google Scholar]
- 105.Darrah E, Kim A, Zhang X, et al. Proteolysis by Granzyme B Enhances Presentation of Autoantigenic Peptidylarginine Deiminase 4 Epitopes in Rheumatoid Arthritis. J Proteome Res 2017; 16(1): 355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303(5663): 1532–5. [DOI] [PubMed] [Google Scholar]
- 107.Tiniakou E, Fava A, McMahan ZH, et al. Definition of Naturally Processed Peptides Reveals Convergent Presentation of Autoantigenic Topoisomerase I Epitopes in Scleroderma. Arthritis Rheumatol 2020; 72(8): 1375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murinson BB, Guarnaccia JB. Stiff-person syndrome with amphiphysin antibodies: distinctive features of a rare disease. Neurology 2008; 71(24): 1955–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Balint B, Jarius S, Nagel S, et al. Progressive encephalomyelitis with rigidity and myoclonus: a new variant with DPPX antibodies. Neurology 2014; 82(17): 1521–8. [DOI] [PubMed] [Google Scholar]
- 110.Butler MH, Hayashi A, Ohkoshi N, et al. Autoimmunity to gephyrin in Stiff-Man syndrome. Neuron 2000; 26(2): 307–12. [DOI] [PubMed] [Google Scholar]