

Abstract
Adoptive transfer of suppressive thymic CD4+CD25+ Tregulatory cells (tTreg) can control auto- and allo-immune responses, but typically require in vitro expansion to reach the target cell number for efficacy. Although the adoptive transfer of expanded tTreg purified from umbilical cord blood ameliorated GVHD in patients receiving hematopoietic stem cell transplantation for lympho-hematopoietic malignancy, individual Treg products of 100×106 cells/kg were manufactured over an extended 19 day time period using a process that yielded variable products and was both laborious and costly. These limitations could be overcome with the availability of ‘off the shelf’ Treg. Previously, we reported a repetitive restimulation expansion protocol that maintains Treg phenotype (CD4+25++127-Foxp3+), potentially providing hundreds to thousands of patient infusions. However, repetitive stimulation of Teffectors induces a well-defined program of exhaustion that leads to reduced T cell survival and function. Unexpectedly, we find that multiply stimulated human tTreg do not develop an exhaustion signature and instead maintain their Treg gene expression pattern. We also find that tTreg expanded with 1 or 2 rounds of stimulation, and tTreg expanded with 3 or 5 round of stimulation, preferentially express distinct subsets of a group of five transcription factors that lock-in Treg Foxp3 expression, Treg stability and suppressor function. Multiply restimulated Treg also had increased transcripts characteristic of Tfollicular regulatory cells, a Treg subset. These data demonstrate that repetitively expanded human tTreg have a Treg locking transcription factor with stable FoxP3 and without classical T cell exhaustion gene expression profile, desirable properties that support the possibility of off-the-shelf Treg therapeutics.
Keywords: regulatory T cell, Treg, Cell therapy, cGMP production, graft versus host disease
Introduction
Regulatory T-cells (Treg) are CD4+ suppressor cells that co-express the high affinity IL-2Ralpha chain, CD25, and the master transcription factor, Foxp3. Treg are essential immune system controllers, limiting self-reactive T-cells responsible for autoimmunity, and alloreactive T-cells in transplant recipients that destroy foreign antigen-bearing tissues resulting in tissue inflammation and injury1. Treg generated in response to commensal bacteria or their products facilitate tissue and organism homeostasis2–4. Treg can be classified by their site of differentiation in the thymus (thymic Treg, or tTreg)5–7 or induced in the periphery (pTreg) or in vitro (iTreg)8,9. Under specific inflammatory conditions, Treg can become unstable and convert to Teffectors (Teffs).
Allogeneic hematopoietic stem cell transplantation (HSCT) can be complicated by graft-versus-host disease (GVHD)10–12 caused by donor T-cells that attack host tissues; GVHD occurs in up to 70% of transplant recipients, leading to high rates of morbidity and mortality13,14. The adoptive transfer of high Treg numbers can prevent murine GVHD15–19. Donor and third-party Tregs were similarly effective in suppressing murine GVHD20. Circulating human Treg are present at low frequency (1–2%), and as such may require in vitro expansion to achieve favorable Treg: Teff ratios needed to control adverse Teff responses. Because short-course expansion of Treg is often inadequate to obtain sufficient Treg yields16, we developed an in vitro expansion protocol that preferentially supports Treg over Teffs. Robust expansion was achieved by repetitively stimulating Treg after return to resting size. Integral to this process is the immunosuppressant rapamycin, which selectively expands murine and human CD4+25+Foxp3+ Treg vs. Teffector/memory cells21–23. In the clinic, a cord blood tTreg expansion protocol that incorporated a re-stimulation step allowed high donor Treg:Teff ratios of ~7:1; the infusion of these in vitro expanded, third-party HLA 3–6/6 tTregs resulted in virtually no acute GVHD24.
During the course of preclinical optimization of re-stimulation cultures, we tested an expansion platform consisting of flow cytometer-sorted peripheral blood (PB) human tTreg expanded on GMP-grade artificial antigen-presenting cells (aAPC) for a total of 5 stimulations with rapamycin present in the culture to suppresses Teff expansion and enhance Foxp3 and tTreg stability22,25,26. Treg expansion of ≥10,000,000-fold was attained, while retaining Foxp3 expression and in vitro and in vivo xenogeneic GVHD suppressor function15,16,27. Using a computational network inference approach, Fu identified 5 redundant transcription factors (TF) (Eos, Lef1, Satb1, IRF4 and GATA1) that individually synergize with Foxp3 to stabilize Foxp3 expression and “lock in” the Treg transcriptional signature28. Recent studies characterized additional TF that maintain Foxp3 expression and Treg stability29–32. Expression of two of these TF (TCF-1 and LEF1) defines three tTreg subsets as resting (TCF-1+LEF1+), activated (TCF-1+LEF1+/lo), and effector (TCF-1-LEF1-). TCF-1 and LEF1 were required for increased expression of specific Treg signature genes, in vivo Treg competitive fitness and Tfollicular regulatory (Tfr) cell development but were dispensable for in vitro suppressive function31,32.
Chronic Teff stimulation can drive cells into a state of exhaustion resulting in Teff dysfunction33–35. The TF and gene sets enforcing exhaustion have been documented and include TOX, associated with exhausted murine and human CD8 T-cells, and TCF-1 associated with exhausted T-cell precursors36–39. One of the dominant T-cell exhaustion characteristics is the upregulation of multiple inhibitory receptors, including PD-1, Lag-3, Tim-3, CD160 and CTLA440.
Herein, we show that naïve PB Treg either stimulated 2 times without rapamycin (resulting in ~10,000-fold expansion) or Treg stimulated 5 times in rapamycin (resulting in ~10,000,000-fold expansion) preferentially express different sets of previously defined Treg locking factors (Eos, Lef1, Satb1, IRF4, GATA1), TCF1 and Bach2 that promotes the differentiation of Treg and homeostasis. Importantly, Treg cultured in rapamycin did not increase exhaustion gene expression even after 5 stimulations, in contrast to Treg stimulated 2 times without rapamycin that upregulated genes found in previously annotated exhaustion gene sets. The lack of observed signs of exhaustion of Tregs stimulated at resting size in rapamycin suggests that highly expanded Treg could become a clinically applicable off-the-shelf cellular therapeutic.
Methods
Thymic Treg and CD4 T-cell purification and validation
Non-mobilized peripheral blood (PB) leukapheresis products were purchased from Memorial Blood Center (St. Paul, MN). Naïve human PB tTreg (CD4+25+127−45RA+) were sort-purified from PB mononuclear cells (PBMNCs) (Ficoll-Hypaque, Amersham Biosciences, city, state) in a two-step procedure in which CD25+ cells were initially enriched from PBMNCs by AutoMACS (PosselD2) with GMP grade anti-CD25 microbeads (Miltenyi Biotec, Auburn, CA). CD25high cells were stained with CD4, CD25, CD127 and CD45RA and sorted via FACSAria as CD4+, CD25high, and CD127− CD45RA using a fluorochome-conjugated anti-CD25 antibody that recognizes a different epitope from bead-bound anti-CD25. Naïve human CD4 T-cells were sort-purified from the CD25- fraction as CD4+25–127+45RA+ (Figure S1). To determine the methylation status of the TSDR in the Foxp3 gene, DNA was purified from sort-purified tTregs expanded. Samples were submitted to EpigenDx Inc. (Worcester, MA 01606) for bisulfite modification and sequencing of 11 CpG motifs in the TSDR.
In vitro tTreg and CD4 T-cell expansion
Purified naïve tTreg and CD4 T-cells were stimulated with a human myelogenous leukemia cell line, K562, engineered to express CD86 and CD64, the high-affinity Fc receptor (KT64/86) (2:1 tTreg:KT64/86), which had been irradiated with 10,000 cGy and loaded with anti-CD3 mAb (Miltenyi Biotec, Auburn, CA)16,24. tTreg and CD4+ Teff were re-stimulated up to 4 times as shown in Figure S2. tTreg and CD4+ Teff cell size was monitored 3x weekly, and re-stimulation was performed after they had returned to a resting size (8.5μm and 9.2μm, respectively), which we have shown maximizes expansion16. In some experiments, tTregs that had been stimulated with KT64/86 cells that were preloaded, irradiated, and frozen were used at a 1:1 tTreg:KT64/86. Naive tTreg and CD4 T-cells were cultured in X-Vivo-15 (BioWhittaker, Walkersville, MD) media supplemented with 10% human AB serum (Valley Biomedical, Winchester, VA). Recombinant IL-2 (300 IU/mL for Treg, 50 IU/mL for CD4+ T-cells; Chiron, Emeryville, CA) was added on day 2 and maintained for culture duration. Cultures were refed at least every Monday, Wednesday and Friday, and plated at 0.25 × 106 to 0.5 × 106 viable nucleated cells/ml. Where indicated, rapamycin (Rapamune, Wyeth-Ayerst, Princeton, NJ) at 109 nM was added on day 0 and with subsequent media supplementations. At the termination of each culture (7–10 days following the last stimulation), cells were aliquoted and frozen. To avoid experiment variability, analyses were conducted concurrently with all samples after thawing.
Suppression assays
The in vitro–suppressive capacity of expanded tTreg was assessed with a carboxyfluorescein succinimidyl ester inhibition assay as published41. Briefly, PBMNCs were purified, labeled with the intracellular dye CFSE (Invitrogen) that dilutes with each division, and stimulated with anti-CD3 mAb-coated beads (Dynal, InVitrogen) ± cultured tTreg (1:2–1:32 tTreg/PBMNCs). On day 4, cells were stained with antibodies to CD4 and CD8 and suppression was determined from Division Index (FlowJo). tTreg comparably suppressed CD4 and CD8 T cell responses; therefore, only CD8 data are presented.
Flow cytometry and antibodies
Human-specific antibodies were used for flow cytometry and included CD4 (clone RPA-T4), CD8 (RPA-T8), CD14 (MSE2), CD19 (HIB19), CD25 (M-A251), CD45RA (HI100), PD-1 (eBioJ105) and Lag-3 (3DS223H) were purchased from BD Pharmingen or eBioscience (San Diego, CA). Anti-Foxp3 (clone 249D) was from BioLegend (San Diego, CA) and stains were performed with the Foxp3 staining kit (BioLegend, 421403) per manufacturer recommendations. Acquisition was performed using a FACScalibur or LSRII (BD Bioscience, San Jose, CA) and data were analyzed using FlowJo software (TreeStar Inc., Ashland, OR).
Microarray analysis
Cell pellets (1–2×106 cells) were frozen in RNAlater at harvest. 100 ng of total RNA extracted using the RNeasy Mini Plus Kit with on-column DNase digestion (Qiagen) was amplified using Ambion WT expression kit (Life Technologies) according to manufacturer’s instructions. mRNA was reverse transcribed into cDNA and a second strand cDNA was generated, then used as a template for in vitro transcription for cRNA synthesis. Following purification, the cRNA was converted into cDNA and excess RNA was removed by RNase H treatment. cDNA was fragmented by restriction digestion and end terminal labeled with GeneChip WT Terminal Label kit (Affymetrix). Samples were hybridized to Human Gene ST 1.0 Arrays (Affymetrix) in a GeneChip Hybridization Oven 640 >16 hours, 45°C, 60 rpm, stained on a GeneChip Fluidics Station 450, and scanned by GeneChip Scanner (Affymetrix).
Bioinformatic Analysis
Fluorescent intensity data (CEL files) were imported and Robust Multi-array Average (‘RMA’) normalized using the Bioconductor ‘oligo’ package. Probesets were annotated using the Affymetrix annotation file and the Bioconductor ‘biomart’ package. Normalized fluorescent intensities were filtered using a variance cutoff of 0.3 with the Bioconductor ‘genefilter’ package. Differential gene expression thresholds included a log2 fold-change cutoff of 0.5 and significance threshold of 0.05 using a moderated T-statistic corrected for multiple hypothesis testing (Bioconductor ‘limma’ package). Gene modules were identified using the WGCNA package.
Gene-set enrichment analysis (GSEA) and its single-sample variant were performed using gene sets from the MSigDB. Single sample GSEA to assess correlation of in vitro expanded naïve tTreg and CD4 Teff with ImmSig gene sets comparing Treg and conventional CD4 T cells isolated from: mouse adipose tissue (GEO accession code 7852), a composite of mouse adipose/spleen/lymph node (GEO accession code 7852), or human fetus (GEO accession code 25087). Single sample GSEA to assess correlation of in vitro expanded naïve tTreg and CD4 Teff with ImmSig gene sets comparing acute and chronically stimulated T cells, including: CD4+ T cells isolated from mice infected with the clone 13 vs Armstrong variant of LCMV (GEO accession code 30431), a composite of mouse and human transcriptomic-derived exhaustion-specific genes (GEO accession code GSE96578) or human CD8+ T cells isolated from patients with a progressing HIV infection versus those with controlled HIV infection (GEO accession code 25087).
Statistical analyses
Data were analyzed by one-way analysis of variance (ANOVA) or Student t test. Probability (P) values less than or equal to 0.05 were considered statistically significant.
Results
Sort-purified naïve tTreg maintain Foxp3 and suppressive function after 5 repetitive stimulations over 50-day time period.
Chronic activation of human or murine T cells leads to exhaustion associated with hypofunction and shortened longevity33,35,42. To assess culture expansion effects on human naïve tTreg and non-Treg CD4 T cell populations, naïve tTreg (CD4+25++127–45RA+) and control naïve CD4 T cells (CD4+25–127+45RA+) were sort-purified from the same donors and expanded in parallel (see Figure S1 for details). Naïve tTreg yield from PBMC following sorting was 0.08±0.05% tTreg can be distinguished from iTreg or pTreg by hypo-methylation of the conserved non-coding sequence 2 located in the first intron of the Foxp3 gene, known as the Treg-specific demethylated region (TSDR)43,44. Since Foxp3 is on the X-chromosome, male donors were selected for TSDR analysis. Foxp3 expression in the sorted population (85±7%) correlated with TSDR demethylation (77±5%) (Figures S1D, E).
Sort-purified naive CD4 T cells and naïve tTreg were separately expanded using KT64/86 cells27,41. tTreg without rapamycin (tTreg-No Rapa) were re-stimulated after returning to resting size (day 12), when Treg are highly receptive to TCR signals and hence expansion16 (Figure S2). CD4+25–127+45RA+ T-cells (referred to as CD4 Teff) increased 3.9±0.2×104-fold after 2 stimulations, while tTreg expanded ~3.5-fold less, at 1.1±0.7×104-fold under the same conditions (Figure 1A). Such tTreg maintained high Foxp3 (75±2% and 69±10% for 1 and 2 stimulations, respectively) and concomitant TSDR demethylation (Figure 1B and C, respectively). We have previously shown that tTreg cultures stimulated a third time in the absence of rapamycin have greatly decreased %Foxp3+16. Because a decrease in %Foxp3+ cells was observed in naïve tTreg cultures stimulated a third time in the absence of rapamycin (Figure S2C), this condition was excluded from further analyses.
Figure 1: Sort-purified naïve tTreg maintain Foxp3 and suppressive function after multiple stimulations. In vitro expansion of naïve human tTreg and naive CD4 T cells.
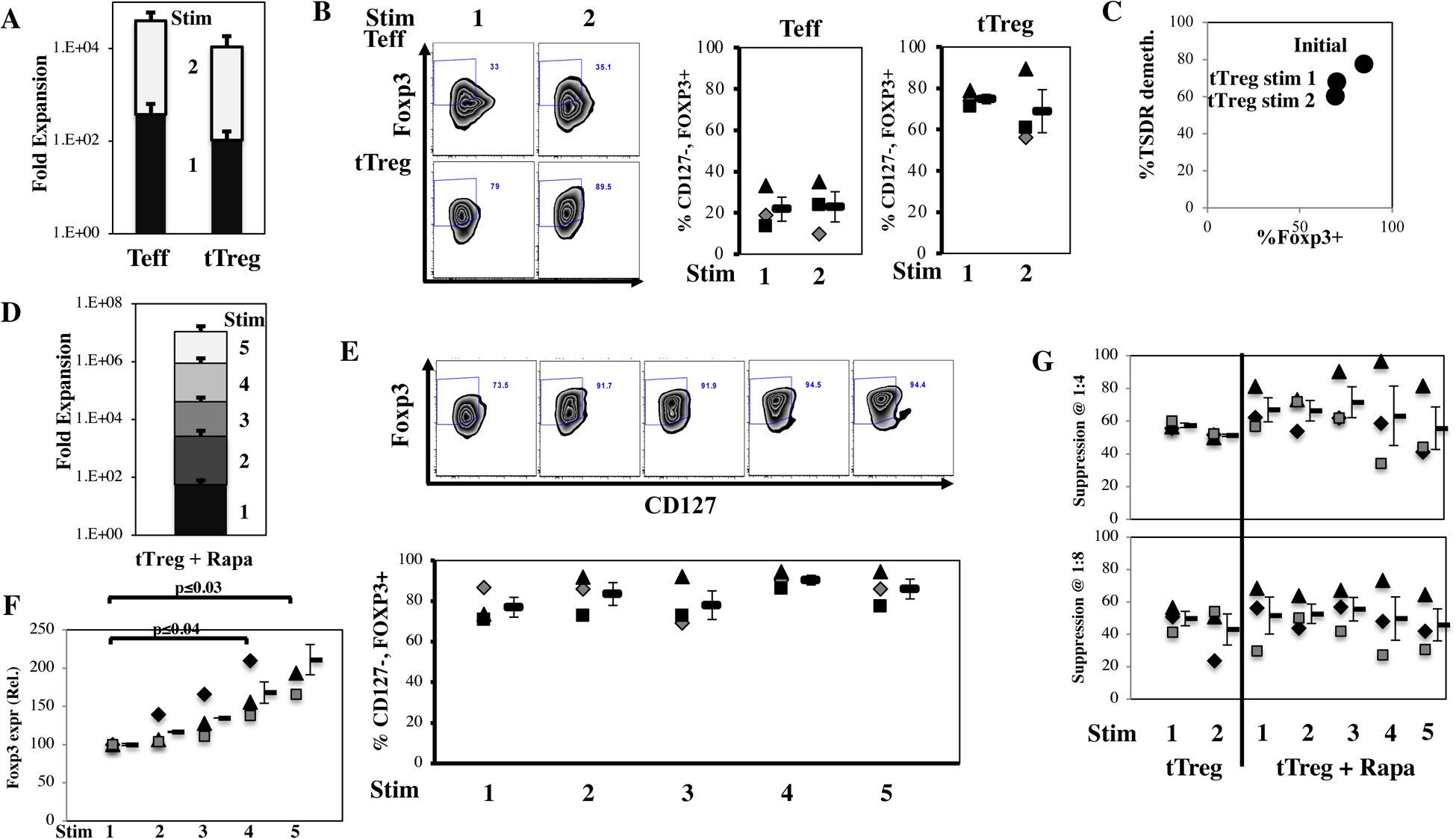
Naïve PB CD4+ T cells and tTreg were sort-purified (CD4+25–127+45RA+ and CD4+25hi127−45RA+, respectively) and expanded with anti-CD3 mAb-loaded KT64/86 using two rounds of stimulation. tTreg were also stimulated 5 times in the presence of rapamycin. (A) Average expansion for CD4 T cells (Teff) and tTreg-No Rapa. (B) Representative example (left) and average percent CD127−Foxp3+ (CD4-gated). (C) Average (n=3) %TSDR demethylation of naïve tTreg stimulated once or twice in the absence of rapamycin compared with Foxp3 expression. (D) Average expansion for naïve tTreg stimulated 1–5 times in the presence of rapamycin. (E) Representative example (above) and individual data points along with average percent CD127−Foxp3+ (CD4-gated) for tTreg+Rapa stimulated 1–5 times as indicated. Mean ± 1 SEM is shown. (F) Average relative Foxp3 expression in naïve tTreg expanded in rapamycin with the indicated number of stimulations normalized to 1 stimulation. (G) Percent suppression of in vitro CD8 T cell proliferation tTreg/PBMNC ratios of 1:2, 1:4 and 1:8 for tTreg cultures stimulated the indicated number of times in the presence or absence of rapamycin. Data shown are mean ± SEM. No significant differences were observed in suppression. All results are from 3–4 independent experiments.
To determine whether a far greater expansion of tTreg would retain the phenotype observed with 2 stimulations as used in our clinical trials24,27, a portion of the naïve tTreg sorted from the same donor was stimulated a total of 5 times (Figure S2D). In these longer-term cultures, the addition of rapamycin suppressed the proliferation of Teffs and favored tTreg expansion and stability16. Despite known rapamycin anti-proliferative properties, naive tTreg cultured in rapamycin and stimulated for a total of 5 times expanded 1.0±0.6×107-fold (Figure 1D and S2E), >1,000-fold more than twice-stimulated tTreg without rapamycin. All cultures remained ≥69% CD127-Foxp3+, and averaged 86±5% CD127-Foxp3+ (Figure 1E). Compared to one stimulation, Foxp3 mean fluorescent intensity progressively increased in tTreg+Rapa (relative Foxp3 expression stimulated 4 or 5 times was 168% or 211%, respectively) (Figure 1F). To compare suppressive function of all culture conditions without potential assay variations, frozen Treg aliquots were thawed and concurrently assayed in a CFSE-based, anti-CD3 mAb-driven CD8+ T cell proliferation assay at tTreg:PBMNC ratios from 1:2–1:8. All cultures had ≥ 42% suppressor function at 1:4 (Treg:PBMNC). While the average suppression of Treg stimulated 5 times in the presence of rapamycin was lower, it was not statistically significant, and these cultures were still highly suppressive (Figure 1G).
tTreg expanded >10-million-fold maintained tTreg-associated gene expression.
To ascertain the impact of expansion on the expression of Treg-associated genes after multiple stimulations, microarray profiling was conducted on RNA purified from CD4 Teff or tTregs (tTreg-No Rapa) expanded 1 or 2 times and tTreg expanded with 1, 2, or 5 times in rapamycin (tTreg+Rapa) (n=3 donors for each group).
Principal component analysis (PCA) showed significant clustering of gene expression patterns by cell type and by number of stimulations (Fig. 2A; see also Table 1), indicating individual groups of cells propagated under these discrete conditions were distinct. To discern the transcripts most closely associated with each sample type, we employed single-sample gene-set enrichment analysis (ssGSEA)45 with special attention to the extent to which each population maintained tTreg-associated gene expression. Gene expression differences in expanded cell populations were analyzed with previously curated sets of Treg-associated genes expressed in Tregs purified from three different sources, PB46, cord blood47,48, and adipose tissues46. To interrogate genes directly regulated by Foxp3, we also performed ssGSEA against a published murine CHIPseq dataset that identifies genes directly bound by FOXP349. In vitro expanded tTreg vs Teff showed enrichment in each of these four canonical tTreg gene sets (Figure 2B).
Figure 2: Thymic Treg expanded >10-million fold maintain expression of tTreg-associated genes.
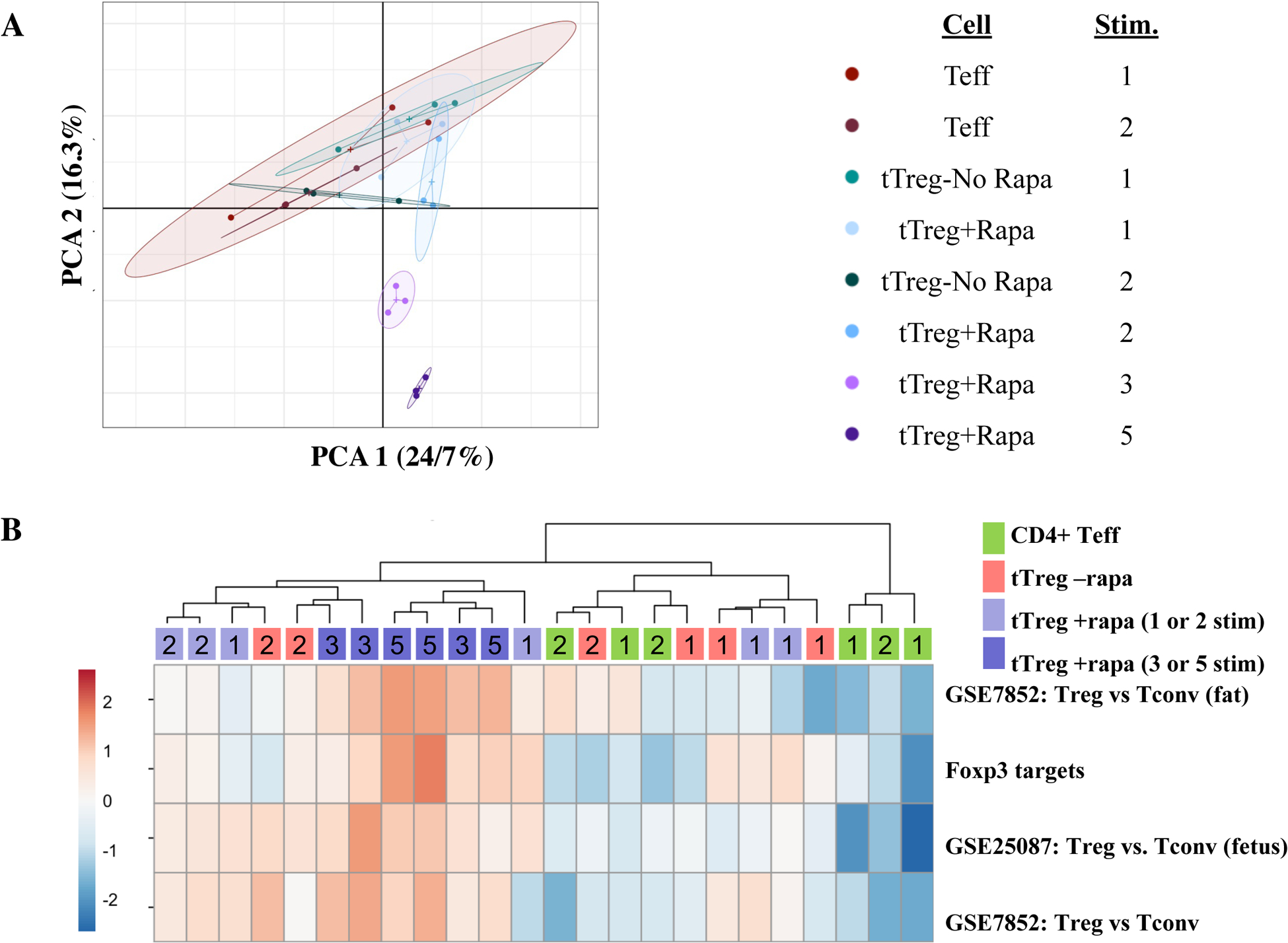
Naïve PB CD4+ T cells and tTreg were sort-purified (CD4+25–127+45RA+ and CD4+25hi127−45RA+, respectively) and expanded with anti-CD3 mAb-loaded KT64/86 using up to 5 rounds of stimulation, and microarray profiling performed. (A) Principle-component analysis (PCA) comparing CD4 Teff, tTreg-No Rapa, and tTreg-Rapa expanded with low (1 or 2) and high (3 or 5) number of stimulations. PCA1, PCA2: principle components 1 and 2. (B) Heatmap of Treg-related gene sets enriched between CD4 Teff, tTreg-No Rapa and tTreg+Rapa stimulated the indicated number of times using ssGSEA. Gene sets were comparisons of Treg vs. conventional T cells isolated from adipose tissue (GSE25087), fetal tissue (GSE25087) and peripheral blood (GSE7852). Foxp3 targets is a gene set identified by Foxp3 ChIP assay.
Table I:
Global summary of CD4 Teff and tTreg Data
| Cell type | Stim# | Rapa | Fold Exp. (Approx.) | Hierarchical clustering | TF WGCNA | Overall conclusions | ||
|---|---|---|---|---|---|---|---|---|
| Module | Representative genes | Module | Representative TF | |||||
| CD4 Teff | 1 or 2 | NA | 400–40,000 | Green | IL-7RA, Themis, TBX21, ID2, CD40L Eomes, TOX; multiple inhibitory receptors and cytokines | Green | Tbx21, Eomes, BATF |
|
| tTreg | 1 or 2 | − | 100–1000 | Red | Foxp3, Helios, UTS2, SLC14A1, CDKN2B, Eos, PLCL1, SOCS2, CD80, GPR55, LSR | Violet | Foxp3, Helios, Eos, Satbl.Lefl, Bach2, TCF1 |
|
| Lilac | RTKN2, CASK, SELP, MY05C, CD27, DGKA, and CCR6 | |||||||
| tTreg | 1 or 2 | + | 50–2500 | Red | Foxp3, Helios, UTS2, SLC14A1, CDKN2B, Eos, PLCL1, SOCS2, CD80, GPR55, LSR | Violet | Foxp3, Helios, Eos, Satbl, Lefl, Bach2, TCF1 |
|
| Lilac | RTKN2, CASK, SELP, MY05C, CD27, DGKA, and CCR6 | |||||||
| tTreg | 3 or 5 | + | 40,000–10,000,000 | Red | Foxp3, Helios, UTS2, SLC14A1, CDKN2B, Eos, PLCL1, SOCS2, CD80, GPR55, LSR | Yellow | IRF4, GATA1, PRDM1 |
|
| Purple | RTKN2, CASK, SELP, MY05C, CD27, DGKA, and CCR6 | |||||||
Unlike CD4 Teff, tTreg repetitively stimulated in rapamycin do not upregulate genes associated with exhaustion.
Exhaustion was initially described as a condition in which T cells chronically activated by virus displayed decrease proliferation and effector function35. Gene expression profiling of acute vs. chronically activated T cells has now identified a core group of exhaustion-related genes40,42,50. Multiply re-stimulated tTreg maintained both proliferative and suppressive function (Figure 1D and G, respectively), providing important functional evidence against tTreg exhaustion. To further evaluate exhaustion, we performed ssGSEA to compare our transcriptome data to three published gene sets. The first gene set was identified as distinguishing exhausted CD4 and CD8 gene expression patterns from mice infected with lymphochoriomeningitis virus (LCMV) clone 13 that causes chronic viral T cell stimulation and exhaustion vs LCMV Armstrong that causes acute infection and does not result in T cell exhausion40. The other two gene sets were identified by comparing human CD8 T cell expression patterns from HIV+ patients able to control their infection (“controllers”, putatively non-exhausted) in contrast to “progressors” non-controllers who developed AIDS (putatively exhausted)42,50 (Figure 3). Genes differentially expressed in CD4 Teffs stimulated once versus twice, corresponding to an increase in expansion from ~400-fold to ~40,000-fold, respectively, showed significant enrichment for genes associated with exhausted murine CD4 and CD8 T cells and human CD8 T-cells from patients who progressed to AIDS (Figure S3A; false discovery rate (FDR)=0.001). Similarly, gene expression changes associated with tTreg stimulated once or twice without rapamycin (tTreg-No Rapa) showed enrichment for gene sets associated with murine CD4 and CD8, and human CD8 T-cell exhaustion (Figure S3B; FDR=0.001). In contrast, no significant enrichment in exhaustion-related genes) was observed in tTreg+Rapa, even though expansion with 5 rounds of stimulation was 100,000-fold higher than one stimulation (Figure S3C; FDR=0.18). This lack of an exhaustion signature suggests that tTreg highly expanded in rapamycin may have greater in vivo therapeutic advantages than tTreg stimulated without rapamycin.
Figure 3: Thymic Treg expanded >10-million do not upregulate genes associated with exhaustion.
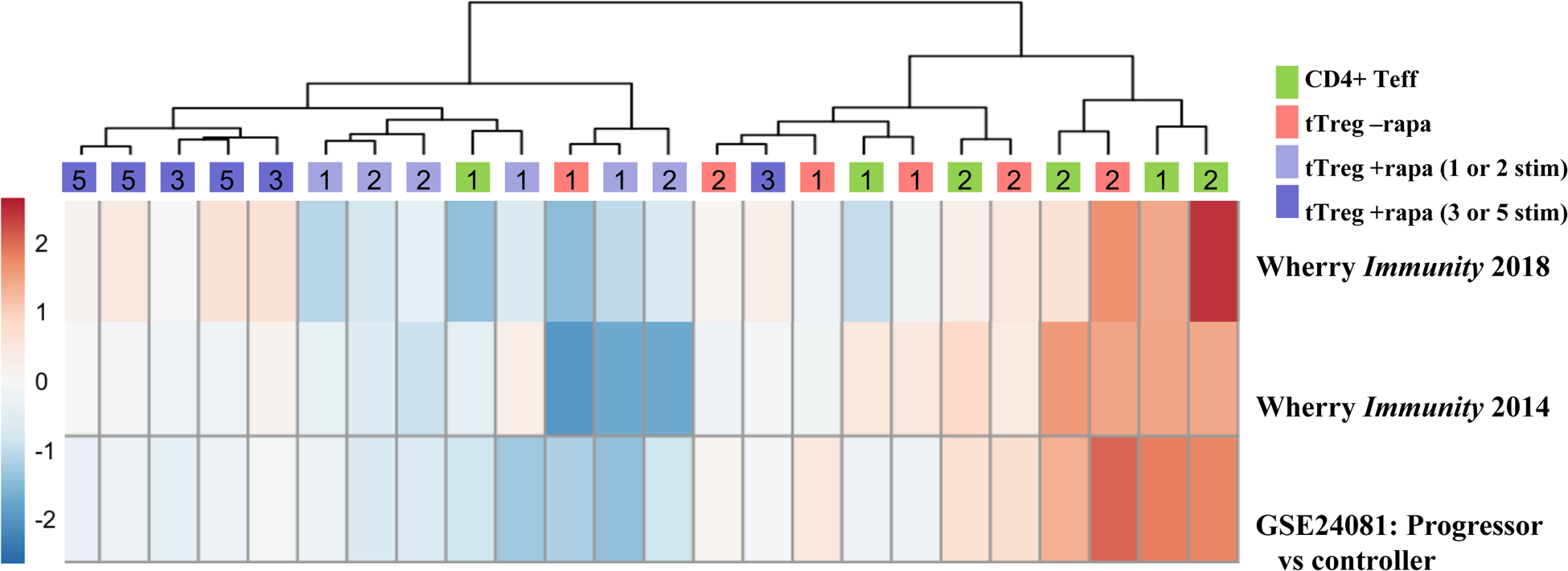
Naïve PB CD4+ T cells and tTreg were sort-purified (CD4+25–127+45RA+ and CD4+25hi127−45RA+, respectively) and expanded with anti-CD3 mAb-loaded KT64/86 using up to 5 rounds of stimulation, and microarray profiling performed. Heatmap of exhaustion-related gene sets enriched between CD4 Teff, tTreg-No Rapa and tTreg+Rapa stimulated the indicated number of times using ssGSEA. Gene sets were murine genes associated with exhaustion in both murine CD4 and CD8 T cells (Wherry Immunity 2014), and two gene sets from human CD8 T cell isolated from HIV+ patients able to control their infection (controllers) vs. those who developed AIDS (progressors) (Wherry Immunity 2018 and GSE24081).
tTreg+Rapa repetitively stimulated 3 or 5 times develop a gene signature distinct from tTreg cultured under other conditions.
Supervised hierarchical clustering and expression analyses were performed comparing naïve CD4 T cells (expanded with 1 or 2 stimulations), to naïve tTreg (expanded with 1 or 2 stimulations without rapamycin) and to naïve tTreg (expanded with 1, 2, 3 or 5 stimulations in rapamycin). A total of 4359 differentially expressed genes (FDR p<0.05) were identified, distributing into 7 independent clusters (Figure 4A). The cluster color-coded ‘red’ identified genes enriched in all tTreg groups, and included canonical Treg genes such as Foxp3 and Helios, and other genes shown to be differentially expressed in expanded human tTreg (e.g. UTS2, SLC14A1, CDKN2B, Eos, PLCL1, SOCS2, CD80, GPR55, LSR)51. The cluster color-coded ‘lilac’ was associated with tTreg undergoing one or two rounds of stimulation ± rapamycin, and included a set of previously reported tTreg-related genes (e.g. RTKN2, CASK, SELP, MYO5C, CD27, DGKA, and CCR6)51.
Figure 4: tTreg+Rapa stimulated 3 or 5 times develop a gene signature distinct from tTreg stimulated 1 or 2 times.
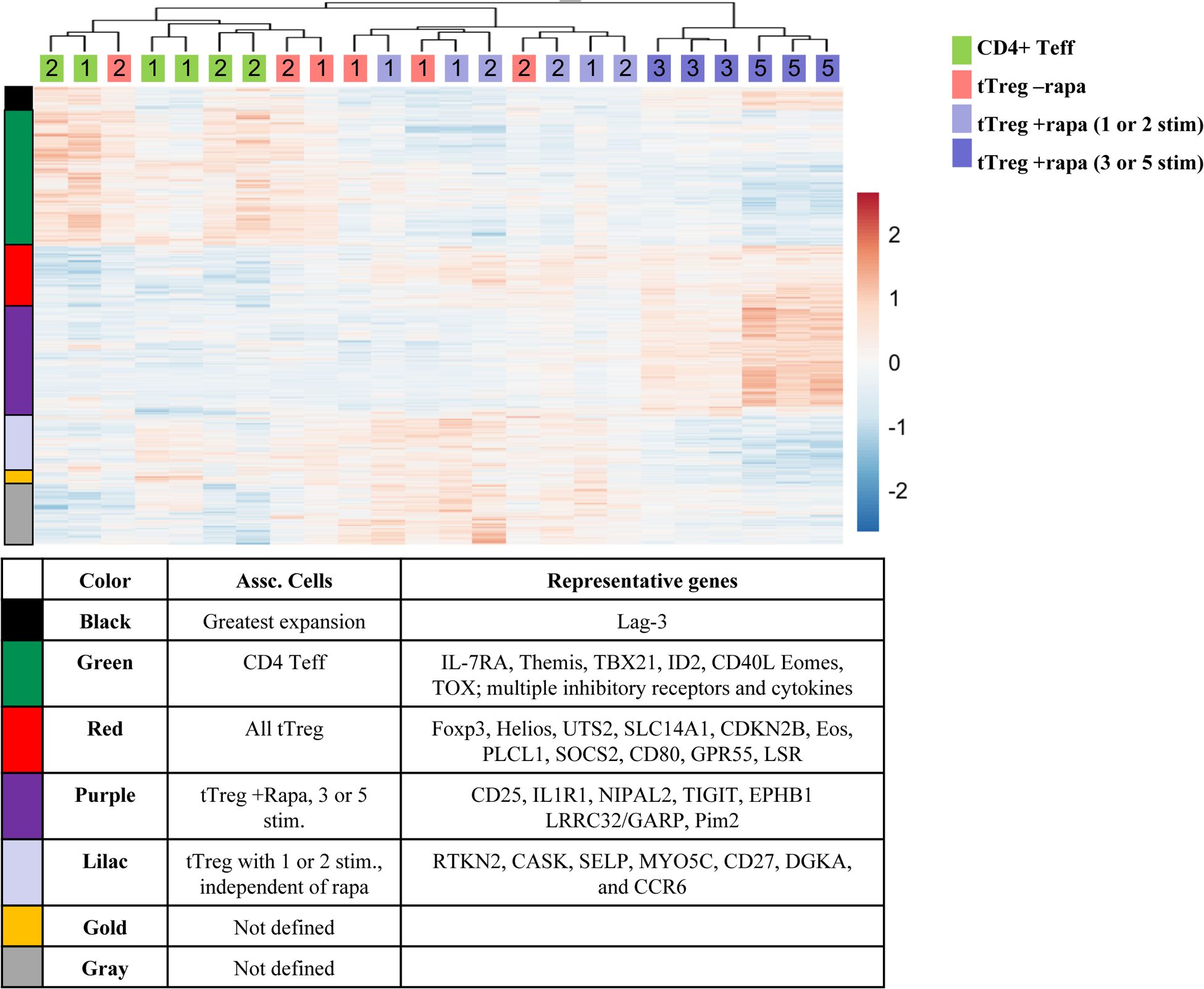
To assess in vitro expansion-related changes in global gene expression between CD4 Teff, tTreg-No Rapa and tTreg+Rapa, RNA was purified from cultured cells and microarray analysis performed. Heatmap of genes differentially expressed between CD4+ Teff and tTreg-No Rapa stimulated once or twice, and tTreg+Rapa stimulated 1, 2, 3 or 5 times (FDR-adjusted P value, <0.05).
The cluster color-coded ‘purple’ associated with tTreg expanded with 3 or 5 rounds of stimulation in rapamycin, and included a separate set of tTreg-related genes (including IL1R1, NIPAL2, IL2RA)51,52. Furthermore, several genes in the purple cluster are known to be differentially expressed in follicular regulatory T cells (Tfr) including LRRC32 (GARP), CTLA4, IRF4, and TIGIT53,54, and this cluster also included two additional genes (EPHB1, INHA) that may encode potential Tfr effector molecules55,56. EPHB1 inhibits Tfollicular helper recruitment and germinal center retention56, while INHA antagonizes INHBA-supported Tfollicular helper differentiation55. In agreement with flow phenotyping, the purple cluster was enriched in IL2Rα. Aptly, Pim-2, a functional Akt homolog which confers rapamycin resistance in Treg25, was associated with the purple cluster. While the purple cluster could simply represent genes regulated by rapamycin, tTreg expanded in rapamycin receiving more limited stimulation (1–2 stimulations) did not associate with the purple cluster. Moreover, ssGSEA showed that tTreg+Rapa expanded with 3 or 5 stimulations did not preferentially associate with several defined rapamycin-dependent gene sets (Figure S4). The gold and gray clusters were not defined by specific cell subsets.
Expanded CD4 Teff have increased expression of multiple inhibitory co-receptors compared to tTreg expanded with or without rapamycin.
The cluster color-coded ‘green’, associated with CD4 Teff populations, and was comprised of a large set of genes enriched in murine and human CD4 (non-Treg) cells including: IL-7Rα (CD127) and Themis51,57, as well as several genes involved in effector (e.g. TBX21, ID2, CD40L) and exhaustion (Eomes, TOX) differentiation40,42,50. Co-expression of multiple inhibitory receptors which regulate TCR signaling represent a molecular hallmark of exhausted CD4 and CD8 T-cells40,42. Studies indicate that the degree of exhaustion was not linked to expression of any single inhibitory receptor, but to the aggregate number of individual receptors co-expressed on a given cell, typically including PD-1, Lag-3, Tim-3, CD160 and CTLA440. More recently, TIGIT and KLRG1 were shown to be upregulated in exhausted human CD8 T cells42. In agreement with ssGSEA, the green cluster contained several inhibitory receptors previously correlated with exhaustion (Lilrb3, PD-L1, PD-L2, Tim-3, CD274, KLRG1, CD96 and LAIR1)58 as well as several Killer Inhibitory Receptors (KIR) genes, both inhibitory (KIR2DL2, KIR2DL3, KIR2DL4, KIR3DL3, KIR2DL5B) and activating (KIR2DS2, KIR2DS3, KIR2DS4), associated with a late memory CD4 T-cell phenotype59. Two inhibitory receptors (CTLA4, TIGIT), which are typically expressed on tTreg, were associated with the purple cluster which defined tTreg stimulated 3–5 times in rapamycin.
To determine inhibitory receptor protein expression in CD4 Teff and tTreg cultures, aliquots were concurrently thawed and stained with antibodies to CTLA4, TIGIT, PD-1 and Lag-3. As expected, ≥70% of tTreg+Rapa cultures expressed CTLA4 and TIGIT after 1 stimulation (Figure 5A and B). CTLA4 did not further increase with up to 5 stimulations. CTLA4 and TIGIT expression on naïve and memory CD4+ T cells (CD45RA+ and CD45RA−, respectively) was compared to tTreg directly analyzed ex vivo. CTLA4 expression was higher in both naïve and memory tTreg than in CD4+ T cells (Figure 5A). Consistent with activation-induced expression of inhibitory receptors, a higher % of memory CD4+ T cells expressed TIGIT compared to naïve CD4+ T cells (Figure 5B). Compared to CD4+ T cell subsets, a higher % of naïve and memory tTreg expressed TIGIT and memory tTreg had greater TIGIT expression than naïve tTreg.
Figure 5: Expanded CD4 Teff have increased expression of multiple inhibitory co-receptors genes compared to tTreg expanded with or without rapamycin.
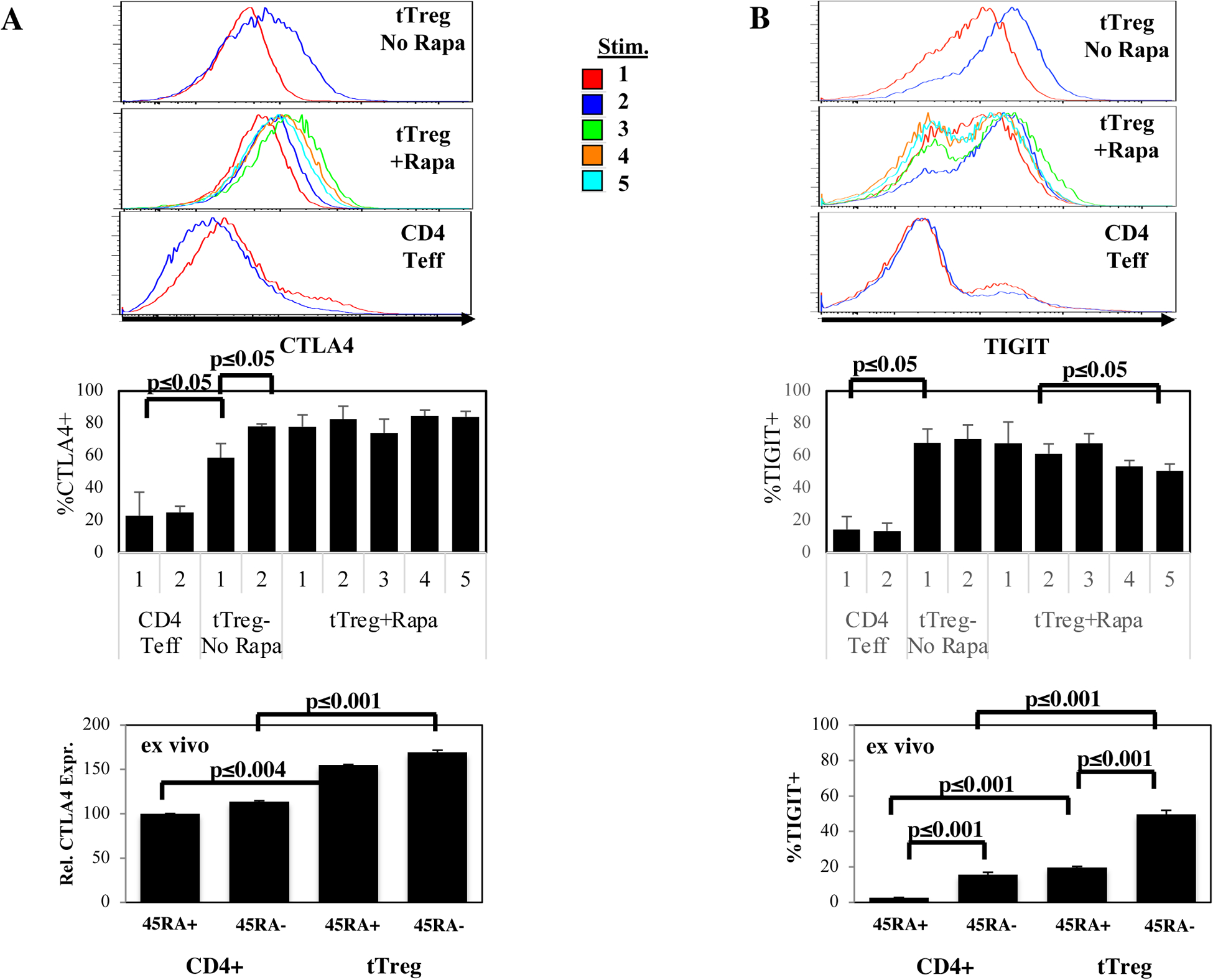
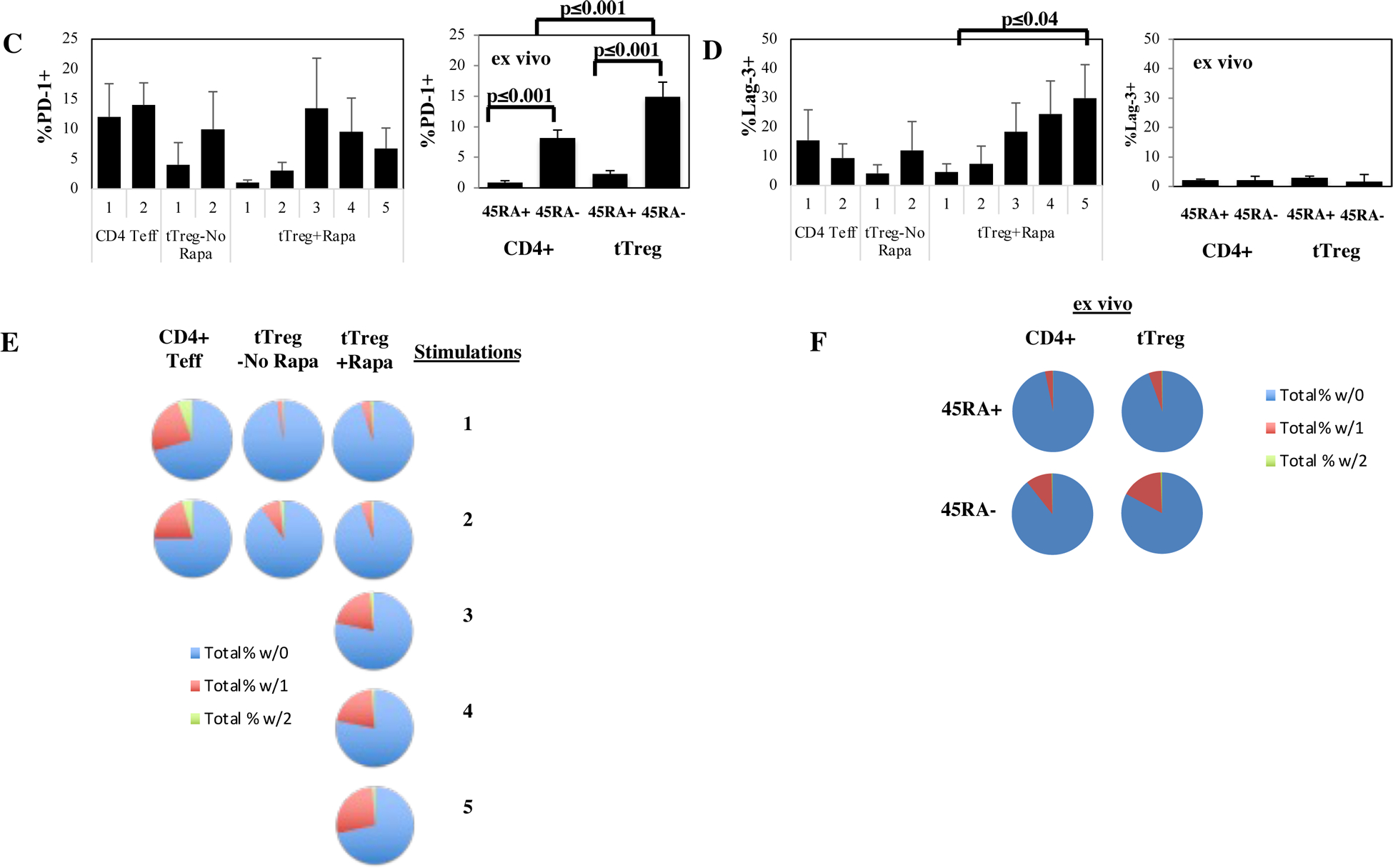
To assess protein expression of specific inhibitory receptors, aliquots of CD4 Teff and tTreg cultured with or without rapamycin undergoing the indicated number of stimulations were concurrently thawed and stained with antibodies to CTLA4, TIGIT, PD-1 and Lag-3. To determine how this expression compares to naïve and memory CD4 T cells and tTreg directly ex vivo, PBMC were also stained for these markers. Representative example and average % of CD4+ Teff, tTreg-No Rapa or tTreg+Rapa that express CTLA4 (A) or TIGIT (B). Summary of PD-1 (C) and Lag-3 expression (D) in CD4+ Teff, tTreg-No Rapa or tTreg+Rapa after expansion or directly ex vivo. (E, F) Pie chart showing Boolean gating for PD-1 and Lag-3 co-expression in CD4+ Teff, tTreg-No Rapa or tTreg+Rapa after expansion (E) or directly ex vivo (F) from a representative experiment. Results from expanded cells are n=3 independent donors and ex vivo data is from n=12 donors.
PD-1 was not identified in the cluster color-coded ‘black’ in Figure 4 and was expressed in only ~5% of tTreg+Rapa stimulated 5 times (Figure 5C). Memory CD4 T cells and tTreg expressed higher PD-1 levels than naïve counterparts and, unlike what was seen following in vitro expansion, a higher % of memory tTreg expressed PD-1 than memory CD4 T cells. Lag-3 was present in the black cluster and associated with cultures undergoing the greatest fold-expansion, increasing to ~30% Lag3+ cells in tTreg+Rapa after 5 stimulations (Figure 5D). While Lag-3 has been shown to inhibit Teff cells, Treg with an activated phenotype have been shown to express this inhibitory receptor, and Lag-3 expressing Treg specifically inhibit IL-1ß and IL-23 expression by CX3CR1+ macrophages60,61. Flow cytometry showed decreasing TIGIT expression in tTreg+Rapa cells stimulated 2–5 times (61±4% to 51±3%; p<0.05) and a paucity (≤4%) of tTreg+Rapa stimulated 5 times co-expressed PD-1 and Lag-3 (Figure 5E and F). As tTregs stimulated 5 times had potent suppressor function (Figure 1G) and no evidence of an exhaustion signature (Figure 3), in aggregate. these data are consistent with tTreg not becoming exhausted despite substantial expansion rates.
Transcription factor WGCNA identifies differentially expressed ‘tTreg locking’ transcription factors in tTreg undergoing 1 or 2 vs. 3 or 5 stimulations.
To interrogate the transcriptional programs driving Treg specification and rapamycin effects using an unsupervised statistical approach, we applied weighted gene correlation network analysis (WGCNA)62 to construct a gene co-expression network63,64. To identify TF that might drive expression of target genes associated with differentially cultured Treg, we focused the network analysis on the top-1000 most variant TFs identified by gene array. To improve statistical power for network analysis, we grouped our samples into CD4 Teff (1 and 2 stimulations), tTreg-No Rapa (1 and 2 stimulations), tTreg+Rapa (1 and 2 stimulations), and tTreg+Rapa (3 and 5 stimulations). WGCNA for TFs identified 3 discrete self-assembling modules, illustrated by a new cluster color schema (with modules color-coded ‘green’; ‘violet’; ‘yellow’) (Figure 6A). CD4 Teff cultures were associated with the green module (Figure 6B) enriched for Teff TF including Tbet (Tbx21), Eomes, and BATF. This module also includes 52 separate histone genes, shown to increase greatly during S-phase in CD4 T-cells65. Correspondingly, flow phenotyping showed that CD4 Teff had significantly higher expression of Ki-67, a proliferation marker most highly expressed during S-phase, than tTreg grown in the presence or absence of rapamycin (not shown).
Figure 6: Transcription factor WGCNA shows tTreg stimulated 1– 2 times express different ‘tTreg locking’ transcription factors than tTreg stimulated 3–5 times.
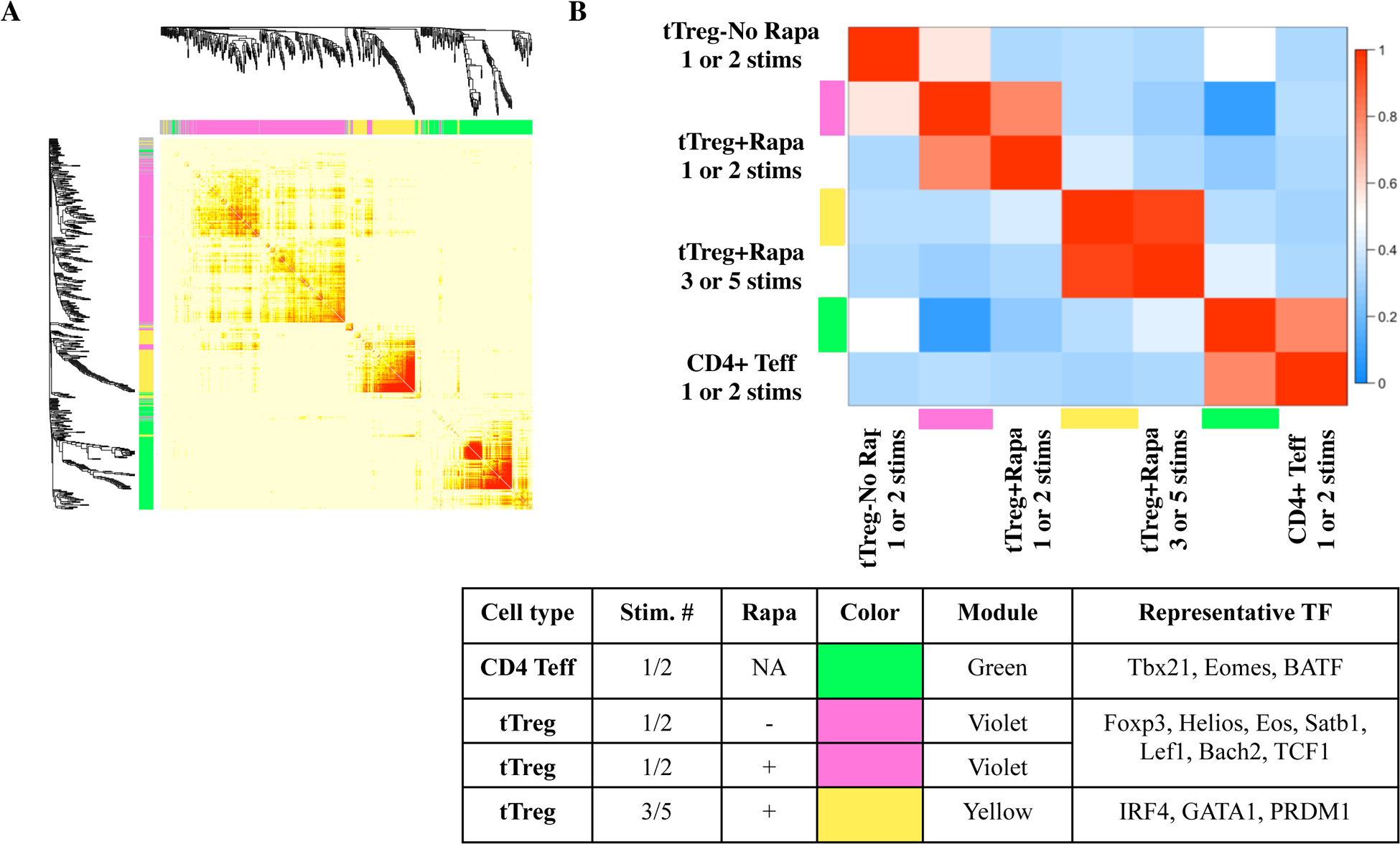
Naïve PB CD4+ T cells and tTreg were sort-purified (CD4+25– 127+45RA+ and CD4+25hi127−45RA+, respectively) and expanded with anti-CD3 mAb-loaded KT64/86 using up to 5 rounds of stimulation, and microarray profiling performed. (A) Topological overlap matrix plot with hierarchical clustering tree and the resulting gene modules from a weighted network of total CD4 Teff, tTreg-No Rapa and tTreg+Rapa transcripts. (B) Eigengene adjacency heatmap showing module eigengene similarity to CD4 Teff, tTreg-No Rapa and tTreg+Rapa.
tTreg-No Rapa and tTreg+Rapa (1 and 2 stimulations each) cultured cells associated with the violet module contained many canonical Treg-associated TF, including Foxp3. Importantly, while Foxp3 gene expression was more enriched in the violet-associated tTreg-No Rapa and tTreg+Rapa (1 and 2 stimulations) populations, flow cytometry phenotyping demonstrated that Foxp3 protein was highest in the tTreg+Rapa (3 and 5 stimulations) cultures, suggesting post-transcriptional regulation of Foxp3 is also likely involved (Figure 1C,S5A). Helios, an Ikaros-family transcription factor and Foxp3 co-factor that is required to maintain Foxp3 expression in a chronic inflammatory microenvironment66,67, was also present in the violet module, consistent with flow cytometry data showing increased protein expression in tTreg cultures with 1 or 2 stimulations (Figure S5B). Eos, another Ikaros-family TF that binds to and maintains Foxp3 expression, preventing Treg reprogramming into Teff68, was also significantly enhanced in the violet TF module.
Using a computational algorithm [the context likelihood of relatedness (CLR)69] to predict TF that contribute to the Treg signature, a recent study identified five TFs (Eos, IRF4, Satb1, Lef1, GATA-1), in addition to Helios and Xbp1, that act in synergy with Foxp3 in a redundant manner to ‘lock in’ the Treg cell signature28,70. The violet and yellow modules were enriched in specific subsets of these ‘Treg locking factors. The violet module contained three of the ‘Treg locking factors’, (Eos, Lef1 and Satb1)71. We previously showed that Bach2 has a similar ‘Treg-locking’ function and maintains Treg stability by repressing effector gene expression30,72. Bach2 was present in the violet transcription factor module and associated with tTreg groups stimulated 1–2 times.
tTreg stimulated 3 or 5 times in rapamycin was associated with the yellow module (Figure 5B), enriched in IRF4 and GATA1. IRF4 is a positive regulator of Treg homeostasis and, by upregulating PRDM1 expression (also present in the yellow module) and IL-10 production, controls differentiation of effector Treg which undergo stimulus-specific differentiation to Treg helper types73,74. IRF4 expression is enhanced by TCR stimulation and IRF4 mRNA expression was further increased when Treg were activated in the presence of the mTOR inhibitors Torin 1 or PP24275. IRF4 increases Pim-2, the rapamycin-independent Akt functional homolog25. One of the top Treg connections for GATA1 identified by CLR analysis was CD2528, consistent with tTreg+Rapa (3 and 5 stimulations) having the highest CD25 expression (Figure 1B). GATA1 also potently down-regulates IFNγ expression76, consistent with the lack of enrichment for this effector cytokine in tTreg cultures in rapamycin, providing supporting evidence for preservation of a tTreg signature.
Discussion
Our goals were two-fold: to determine whether naive tTreg maintain expression of tTreg-associated genes after multiple rounds of in vitro expansion, and whether such a technique would result in an exhausted phenotype as can be seen in human Teff cells expanded for cancer immunotherapy71. Using a bioinformatics approach, we report that in vitro expanded tTreg maintained a tTreg signature, even after ten million-fold expansion. While a single re-stimulation of CD4 Teff resulted in a significant increase in the expression of mRNAs present in exhaustion-related gene sets, no increase was observed in tTreg stimulated 5 times in the presence of rapamycin. One of the hallmarks of exhaustion is the accumulated expression of a group of inhibitory receptors. Hierarchical clustering found a group of 13 inhibitory receptor genes that were preferentially associated with CD4+ Teff cultures but not in the highly expanded tTreg. The 3 inhibitory receptors (CTLA4, TIGIT and Lag-3) with increased expression on expanded Treg compared to CD4 Teff have each been shown to mark Treg with enhanced suppressive function77. The ability of this stimulation protocol to generate such robust expansion without significant signs of exhaustion or activation induced cell death are likely multifactorial. These may include the conditions and use of a cell-based artificial antigen presenting cell (aAPC) for stimulation, choice of aAPCs expressing co-stimulatory molecules (including 4.1BBL and TRAIL), surface expression of CD58 that binds to CD2 on T cells to strengthen cell adhesion, and the timing of stimulation. With respect to the latter, we have previously reported that allowing Tregs to return to a resting size prior to additional stimulation increases their expansion which we ascribe to avoiding restimulation during a period of relative TCR signaling refractoriness.
This bioinformatic approach also found that, while all expanded tTreg express a core group of genes (that includes Foxp3 and Helios), one key difference between tTreg expanded with 1 or 2 rounds versus 3 or 5 rounds of stimulation was the differential expression level of 5 previously defined ‘Treg locking genes’ (Eos, Lef1, Satb1, IRF4, GATA1). tTreg expanded with 1 or 2 stimulations had increased expression of Eos, Lef1 and Satb1 (as well as the more recently defined Bach2 and TCF1), whereas tTreg+Rapa cells expanded 3 or 5 times had increased expression of 2 of the 5 Treg locking signature genes, IRF4 and GATA1, associated with effector tTreg74. To our knowledge, this is the first example of an expansion-based, differential expression of these TF for human Tregs, suggesting an ordered approach to the transcriptional control of Treg stability and function.
tTreg receiving 3 or 5 rounds of stimulation in rapamycin also developed an independent gene expression pattern that did not overlap with genes previously demonstrated to be regulated by rapamycin suggesting that tTreg had intrinsic properties that favored a locking Treg signature and disfavored the acquisition of an exhaustion signature. The link between this cluster of genes and Tfr-associated genes is intriguing and is reminiscent, especially in the context of differential Treg-locking gene expression, of the recently described connection between murine resting Treg and Tfr associated with the activated Treg subset32. Expression of Lef1 and TCF1 can be used to divide Treg into resting (TCF1+LEF1+), activated (TCF1+LEF1+/lo), and effector (TCF1-LEF1-) subsets32. Similar to our data, this report found, in addition to Lef1 and Tcf1, resting Treg had higher expression of Satb1 and Bach2. Additionally, Tfr cells were almost exclusively derived from the activated Treg pool that uniquely, amongst the three subsets, expressed both GATA1 and IRF4.
In conclusion, this study has shown that, in addition to maintaining Foxp3 expression and suppressive function, large-scale in vitro expanded tTreg maintained a canonical tTreg transcriptional profile and did not develop a signature consistent with exhaustion. The presence of increased inhibitory receptor expression on memory tTreg analyzed directly ex vivo suggests clinical products would best be started with naïve tTreg, which also maintain higher Foxp3+ expression after expansion than memory tTreg78, even though this would reduce initial yield. In regards to potential clinical banking, a typical apheresis unit of 12×109 cells would yield ~10×106 purified naïve tTreg which, after 10×106-fold expansion from 5 rounds of stimulation in the presence of rapamycin, would generate a bank of ~1×1014 cells. Since CD45RA expression on CD4 T cells decreases with age79, and our starting tTreg population was only 14±3% CD45RA+, bank size could be further increased by restricting donor age or by using UCB tTreg, which are uniformly CD45RA+80. While, admittedly, many additional factors will play into how many patients a bank of this size could treat, it is interesting to note that, at the 100×106/kg dose our recent clinical trial with UCB tTreg showed almost completely ameliorated GVHD, the upper limit would be 10,000 patients receiving a single tTreg infusion. These data pave the way for consideration of generating tTreg banks of multiply re-stimulated cells to provide an off-the-shelf product, especially given that our prior data24,81and that of others82 indicate that related or unrelated donor Treg need not be more than HLA 3/6 matched with the possibility that HLA-matching requirements would be limited if at all. These studies provide relevant insights on the mechanisms of Treg-mediated protection from GVHD and support for the use of third-party Treg in clinical trials.
Supplementary Material
Acknowledgements
The authors wish to acknowledge Drs. Bruce Levine, James Riley and Carl June (University of Pennsylvania Abramson Family Cancer Center Research Institute) for providing the aAPC (KT64/86) used to expand the CD4 Teff and tTreg cells. This work was supported in part by research grants from the Children’s Cancer Research Fund (K.L.H.), Leukemia and Lymphoma Translational Research Grant R6029–07 (B.R.B.), NIH grants R01 HL114512-01 (M.L.M, K.L.H.), R37 AI34495, R01 HL11879, P01 AI056299 (B.R.B.), NHLBI N01HB037164 (J.E.W., J.S.M., K.L.H.), and NCI P01 CA067493 (B.R.B., J.E.W., J.S.M.). LSK was supported by NIH 2U19 AI051731, NIH 2R01 HL095791. This work was supported in part by an NIH Clinical and Translational Science Award to the University of Minnesota (8UL1TR000114) and an NIH P30 CA77598 using the shared resource Flow Cytometry Core from the Masonic Cancer Center, University of Minnesota.
Disclosure of Conflicts of Interest statement
BRB is a founder of Tmunity Therapeutics, serves as an advisor for and receives research support from BlueRock Therapeutics, and, along with KLH holds patents for the production and use of Treg for clinical trials.
LSK is on the scientific advisory board for HiFiBio. She reports research funding from Bristol Myers Squibb, Kymab Limited, Magenta Therapeutics, BlueBird Bio, and Regeneron Pharmaceuticals. She reports consulting fees from Equillium, FortySeven Inc, Novartis Inc, EMD Serono, Gillead Sciences and Takeda Pharmaceuticals. She has a patent “Method to prevent relapse after transplant” which is pending, and a patent “Method to prevent GVHD after transplant” with royalties paid.
Footnotes
Supplemental Information
Supplemental Information includes Supplemental figure legends along with five figures.
References
- 1.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30(5):626–635. [DOI] [PubMed] [Google Scholar]
- 2.Hegazy AN, Powrie F. MICROBIOME. Microbiota RORgulates intestinal suppressor T cells. Science. 2015;349(6251):929–930. [DOI] [PubMed] [Google Scholar]
- 3.Khoruts A, Hippen KL, Lemire AM, Holtan SG, Knights D, Young JH. Toward revision of antimicrobial therapies in hematopoietic stem cell transplantation: target the pathogens, but protect the indigenous microbiota. Transl Res. 2017;179:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staffas A, Burgos da Silva M, van den Brink MR. The intestinal microbiota in allogeneic hematopoietic cell transplant and graft-versus-host disease. Blood. 2017;129(8):927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nature reviews Immunology. 2010;10(7):490–500. [DOI] [PubMed] [Google Scholar]
- 6.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30(5):636–645. [DOI] [PubMed] [Google Scholar]
- 7.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nature reviews Immunology. 2008;8(7):523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 2008;29(9):429–435. [DOI] [PubMed] [Google Scholar]
- 9.Roncarolo MG, Gregori S, Lucarelli B, Ciceri F, Bacchetta R. Clinical tolerance in allogeneic hematopoietic stem cell transplantation. Immunological reviews. 2011;241(1):145–163. [DOI] [PubMed] [Google Scholar]
- 10.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012;12(6):443–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeiser R, Blazar BR. Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med. 2017;377(22):2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeiser R, Blazar BR. Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets. N Engl J Med. 2017;377(26):2565–2579. [DOI] [PubMed] [Google Scholar]
- 13.Holtan SG, Pasquini M, Weisdorf DJ. Acute graft-versus-host disease: a bench-to-bedside update. Blood. 2014;124(3):363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald-Hyman C, Turka LA, Blazar BR. Advances and challenges in immunotherapy for solid organ and hematopoietic stem cell transplantation. Sci Transl Med. 2015;7(280):280rv282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hippen KL, Merkel SC, Schirm DK, et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11(6):1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hippen KL, Merkel SC, Schirm DK, et al. Massive ex vivo expansion of human natural regulatory T cells (Tregs) with minimal loss of in vivo functional activity. Science Translational Medicine. 2011;3(78):78ra33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hippen KL, Watkins B, Tkachev V, et al. Preclinical Testing of Antihuman CD28 Fab’ Antibody in a Novel Nonhuman Primate Small Animal Rodent Model of Xenogenic Graft-Versus-Host Disease. Transplantation. 2016;100(12):2630–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99(10):3493–3499. [DOI] [PubMed] [Google Scholar]
- 19.Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. 2009;30(5):656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pierini A, Colonna L, Alvarez M, et al. Donor Requirements for Regulatory T Cell Suppression of Murine Graft-versus-Host Disease. J Immunol. 2015;195(1):347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177(12):8338–8347. [DOI] [PubMed] [Google Scholar]
- 22.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105(12):4743–4748. [DOI] [PubMed] [Google Scholar]
- 23.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178(1):320–329. [DOI] [PubMed] [Google Scholar]
- 24.Brunstein CG, Miller JS, McKenna DH, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127(8):1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basu S, Golovina T, Mikheeva T, June CH, Riley JL. Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol. 2008;180(9):5794–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeiser R, Leveson-Gower DB, Zambricki EA, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111(1):453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKenna DH Jr., Sumstad D, Kadidlo DM, et al. Optimization of cGMP purification and expansion of umbilical cord blood-derived T-regulatory cells in support of first-in-human clinical trials. Cytotherapy. 2017;19(2):250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu W, Ergun A, Lu T, et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol. 2012;13(10):972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grant FM, Yang J, Nasrallah R, et al. BACH2 drives quiescence and maintenance of resting Treg cells to promote homeostasis and cancer immunosuppression. J Exp Med. 2020;217(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roychoudhuri R, Hirahara K, Mousavi K, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature. 2013;498(7455):506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xing S, Gai K, Li X, et al. Tcf1 and Lef1 are required for the immunosuppressive function of regulatory T cells. J Exp Med. 2019;216(4):847–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang BH, Wang K, Wan S, et al. TCF1 and LEF1 Control Treg Competitive Survival and Tfr Development to Prevent Autoimmune Diseases. Cell Rep. 2019;27(12):3629–3645 e3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35(2):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alfei F, Kanev K, Hofmann M, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571(7764):265–269. [DOI] [PubMed] [Google Scholar]
- 37.Scott AC, Dundar F, Zumbo P, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571(7764):270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekine T, Perez-Potti A, Nguyen S, et al. TOX is expressed by exhausted and polyfunctional human effector memory CD8(+) T cells. Sci Immunol. 2020;5(49). [DOI] [PubMed] [Google Scholar]
- 39.Seo H, Chen J, Gonzalez-Avalos E, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A. 2019;116(25):12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crawford A, Angelosanto JM, Kao C, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity. 2014;40(2):289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hippen KL, Harker-Murray P, Porter SB, et al. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4–1BB expressed on artificial antigen-presenting cells. Blood. 2008;112(7):2847–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bengsch B, Ohtani T, Khan O, et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity. 2018;48(5):1029–1045 e1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. 2009;9(2):83–89. [DOI] [PubMed] [Google Scholar]
- 44.Polansky JK, Kretschmer K, Freyer J, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38(6):1654–1663. [DOI] [PubMed] [Google Scholar]
- 45.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6):899–911. [DOI] [PubMed] [Google Scholar]
- 48.Mold JE, Venkatasubrahmanyam S, Burt TD, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010;330(6011):1695–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marson A, Kretschmer K, Frampton GM, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445(7130):931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quigley M, Pereyra F, Nilsson B, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16(10):1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhairavabhotla R, Kim YC, Glass DD, et al. Transcriptome profiling of human FoxP3+ regulatory T cells. Hum Immunol. 2016;77(2):201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cuadrado E, van den Biggelaar M, de Kivit S, et al. Proteomic Analyses of Human Regulatory T Cells Reveal Adaptations in Signaling Pathways that Protect Cellular Identity. Immunity. 2018;48(5):1046–1059 e1046. [DOI] [PubMed] [Google Scholar]
- 53.Wu H, Chen Y, Liu H, et al. Follicular regulatory T cells repress cytokine production by follicular helper T cells and optimize IgG responses in mice. Eur J Immunol. 2016;46(5):1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wing JB, Kitagawa Y, Locci M, et al. A distinct subpopulation of CD25(−) T-follicular regulatory cells localizes in the germinal centers. Proc Natl Acad Sci U S A. 2017;114(31):E6400–E6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Locci M, Wu JE, Arumemi F, et al. Activin A programs the differentiation of human TFH cells. Nat Immunol. 2016;17(8):976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu P, Shih C, Qi H. Ephrin B1-mediated repulsion and signaling control germinal center T cell territoriality and function. Science. 2017;356(6339). [DOI] [PubMed] [Google Scholar]
- 57.Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. 2018;19(3):291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity. 2016;44(5):1052–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Bergen J, Kooy-Winkelaar EM, van Dongen H, et al. Functional killer Ig-like receptors on human memory CD4+ T cells specific for cytomegalovirus. J Immunol. 2009;182(7):4175–4182. [DOI] [PubMed] [Google Scholar]
- 60.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44(5):989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bauche D, Joyce-Shaikh B, Jain R, et al. LAG3(+) Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1(+) Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity. 2018;49(2):342–352 e345. [DOI] [PubMed] [Google Scholar]
- 62.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. [DOI] [PubMed] [Google Scholar]
- 63.Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xue Z, Huang K, Cai C, et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500(7464):593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao J Coordination of DNA synthesis and histone gene expression during normal cell cycle progression and after DNA damage. Cell Cycle. 2004;3(6):695–697. [PubMed] [Google Scholar]
- 66.Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim HJ. Instability of Heliosdeficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci U S A. 2016;113(22):6248–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yates K, Bi K, Haining WN, Cantor H, Kim HJ. Comparative transcriptome analysis reveals distinct genetic modules associated with Helios expression in intratumoral regulatory T cells. Proc Natl Acad Sci U S A. 2018;115(9):2162–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma MD, Huang L, Choi JH, et al. An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity. 2013;38(5):998–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Faith JJ, Hayete B, Thaden JT, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akimova T, Zhang T, Negorev D, et al. Human lung tumor FOXP3+ Tregs upregulate four “Treg-locking” transcription factors. JCI Insight. 2017;2(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. 2018;18(5):340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roychoudhuri R, Clever D, Li P, et al. BACH2 regulates CD8(+) T cell differentiation by controlling access of AP-1 factors to enhancers. Nat Immunol. 2016;17(7):851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng Y, Chaudhry A, Kas A, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458(7236):351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cretney E, Xin A, Shi W, et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. 2011;12(4):304–311. [DOI] [PubMed] [Google Scholar]
- 75.Chapman NM, Zeng H, Nguyen TM, et al. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat Commun. 2018;9(1):2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sundrud MS, Vancompernolle SE, Eger KA, et al. Transcription factor GATA-1 potently represses the expression of the HIV-1 coreceptor CCR5 in human T cells and dendritic cells. Blood. 2005;106(10):3440–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity. 2019;50(2):302–316. [DOI] [PubMed] [Google Scholar]
- 78.Hoffmann P, Eder R, Boeld TJ, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. 2006;108(13):4260–4267. [DOI] [PubMed] [Google Scholar]
- 79.Pilarski LM, Yacyshyn BR, Jensen GS, Pruski E, Pabst HF. Beta 1 integrin (CD29) expression on human postnatal T cell subsets defined by selective CD45 isoform expression. J Immunol. 1991;147(3):830–837. [PubMed] [Google Scholar]
- 80.Seay HR, Putnam AL, Cserny J, et al. Expansion of Human Tregs from Cryopreserved Umbilical Cord Blood for GMP-Compliant Autologous Adoptive Cell Transfer Therapy. Mol Ther Methods Clin Dev. 2017;4:178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brunstein CG, Miller JS, Cao Q, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Di Ianni M, Falzetti F, Carotti A, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117(14):3921–3928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.