

Abstract
Nucleic acids are now considered as one of the most potent therapeutic modalities, as their roles go beyond storing genetic information and chemical energy or as signal transducer. Attenuation or expression of desired genes through nucleic acids have profound implications in gene therapy, gene editing and even in vaccine development for immunomodulation. Although nucleic acid therapeutics bring in overwhelming possibilities towards the development of molecular medicines, there are significant loopholes in designing and effective translation of these drugs into the clinic. One of the major pitfalls lies in the traditional design concepts for nucleic acid drug carriers, viz. cationic charge induced cytotoxicity in delivery pathway. Targeting this bottleneck, several pioneering research efforts have been devoted to design innovative carriers through charge-conversion approaches, whereby built-in functionalities convert from cationic to neutral or anionic, or even from anionic to cationic enabling the carrier to overcome several critical barriers for therapeutics delivery, such as serum deactivation, instability in circulation, low transfection and poor endosomal escape. This review will critically analyze various molecular designs of charge-converting nanocarriers in a classified approach for the successful delivery of nucleic acids. Accompanied by the narrative on recent clinical nucleic acid candidates, the review concludes with a discussion on the pitfalls and scope of these interesting approaches.
Keywords: Nucleic Acid, Drug Delivery, Charge Conversion, Non-cationic, Endosomal Escape
Graphical Abstract
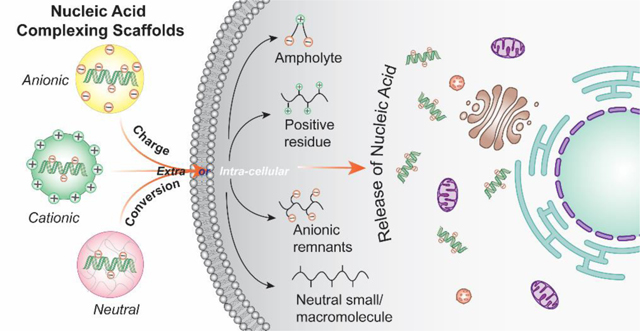
1. Introduction: nucleic acid as ‘old-good’ therapeutics
Advanced genetic understanding of disease pathogenesis has opened up a plethora of avenues for the development of therapeutics and prophylactic drugs for hard-to-cure diseases and preventive therapies.[1–2] Nucleic acids are biologics with many promising candidates in clinical trials and in pharma developmental pipeline.[3–5] As attenuation or amplification of gene expressions influence specific disease progression, it is now practically possible to design nucleic acids that can alter the therapeutic outcome for almost any disease including personalized medicines.[6–7] Several promising nucleic acid candidates are being investigated as therapeutic molecules, e.g., antisense oligonucleotide (ASO), small interfering RNA (siRNA), micro RNA (miRNA), messenger RNA (mRNA), CpG oligodeoxynucleotide, plasmid DNA (pDNA) and aptamers.[8–11] The common work-place for all these biomacromolecules is intracellular milieu, either nucleus (DNA-based) or cytoplasm (RNA-based). As all these candidates possess high negative charge with bulky polyelectrolyte-like behavior, this makes them impermeable through the lipid bilayer of plasma membrane. Also, systemic instability of bare nucleic acids in presence of serum nucleases followed by fast renal clearance include other biological barriers.[12–13] Apart from these common hurdles, different nucleic acid cargoes might demand customized delivery strategies based on their size and functional group chemistry.[14–15] Due to the presence of anionic phosphodiester backbone, nucleic acids possess high residual negative charge which allows electrostatic interactions with cationic delivery agents. Smaller candidates like siRNA, miRNA and ASO (~20–25 bp) might face less electrostatic interactions owing to lower number of negatively charged groups in comparison to larger polycations like pDNA and mRNAs (~2000–10,000 bp), which could essentially compromise condensation efficiency and hamper stability of the complex.[14–16] When a design of experiment screening was performed with variation of molar mass and charge density on copolymers of poly(2-ethyl-2-oxazoline) and PEI, it was found that a limited correlation could be possible between the nucleic acid size and the polymer size or charge densities; however the scope of this correlation might depend on specific polymer chemistry and composition.[16] Despite similarities in the challenges, developing an efficient and safe nucleic acid delivery approach still remains a grand challenge as there seems to be no possible ‘one-size-fits-all’ carrier design strategy.
To address these critical pitfalls, several approaches have been proposed with either chemical modifications of the nucleic acids and/or innovative carrier designs for nucleic acid cargoes.[17–20] Chemical modifications enhance systemic stability with nuclease resistance (e.g. backbone and 5’-phosphate, base, sugar) and increase biological half-life (PEGylation, hydrophobic modifications with cholesterol, bile or fatty acid).[21–23] Although base modifications impart higher stability of the nucleic acid cargo under in vivo conditions, structure-activity studies are essential to optimize binding affinity, loss of potency of the nucleic acid cargo and elevation of systemic toxicity.[23–25] On the other hand, potent nucleic acid carriers like viral vectors impart high transfection efficacy; albeit instigate elevated risk of immunogenicity and undesired mutagenesis.[26–28] In this scenario, non-viral delivery vehicles like cationic lipids, peptides and polymers provide rather clean and straight-forward choices, wherein electrostatic forces between cationic delivery agents and negatively charged nucleic acids result in a condensed complex.[29–37] The stability of such complexes in biological environment is essentially proportional to the electrostatic attractions between the nucleic acid and the binder, which is dictated by (i) the density of cationic charge in play and also by (ii) the ability of these molecular systems to optimize the interactions (e.g. controlled by factors such as molecular flexibility). Typically, a high positive charge, often dictated by the high N/P ratio (molar ratio of cationic nitrogen in delivery agent to anionic phosphate groups in nucleic acid), is found to be critical for complete complexation and protection.[38] Complexes with high cationic charge, under in vivo conditions, pose increased toxicity and adverse side effects such as plasma membrane damage, release of cytochrome C from mitochondria and alteration in membrane potential.[38–39] As a remedy, reduction of positive charge at lower N/P ratio in the complex has been investigated to attenuate toxicity.[40] However, this approach might compromise the complex stability under in vivo conditions and does not address the inherent problem of cationic charge driven toxicity.[41] PEGylation of cationic vectors, another promising strategic solution to this problem, enhances steric crowding in the system diminishing the binding efficacy with cationic vectors and retards efficient cellular uptake of the nucleic acid carriers.[42–46] In essence, cationic charge acts in favor of increasing binding affinity and stability, but jeopardizes safety features of the delivery vector.
As a remedy, charge-reversal approaches, wherein overall charge of the complexes are either masked or altered, have potential implications in toxicity profiles and gene silencing. Whereas charge masking strategy has less deleterious effect on the plasma membrane, the exposed cationicity of the carrier after cellular internalization can be utilized for endosomal escape. Similarly, ionizable units which are neutral at physiological pH and changes to cationic at lower pH have been used to impart efficient endosomal escape feature in the delivery vector. However, a much desired logically safe strategy would be the permanent decationization of the delivery agent to potentially minimize cytotoxicity in sub-cellular organelles.[18] In this review, we will discuss the structural aspects of various charge-altering mechanisms built-in the nucleic acid delivery materials (Figure 1), whereby the original charge of the delivery aid is altered to reduce toxicity, fasten release kinetics of the nucleic acid cargo, help in endosomal escape or any of their combinations. In the field of nucleic acid delivery materials, charge-conversion strategies are comparatively newer and thus are still in the early developmental stages. Clinical translation of such methods would require considerable research investigations involving in vivo studies of toxicity, efficacy and immunogenicity with appropriate disease models. In addition to nucleic acid-based gene therapies and RNAi technologies, efficacious delivery systems can have major impact on the translation of more advanced gene editing technologies.[47–48] Moreover, acknowledging the recent optimistic research results, focus is currently shifting from conventional cationic charge-based complexation approaches to alternative charge-converting molecular designs accommodating demands of complex human diseases. With a discussion on major ongoing nucleic acid clinical drugs, this review will critically analyze unmet needs, pitfalls, and future opportunities. Overall, we will focus on the recent trends in charge-alteration based approaches discussing its potential to address the evolving clinical requirements to create smarter and robust delivery platforms.
Figure 1.
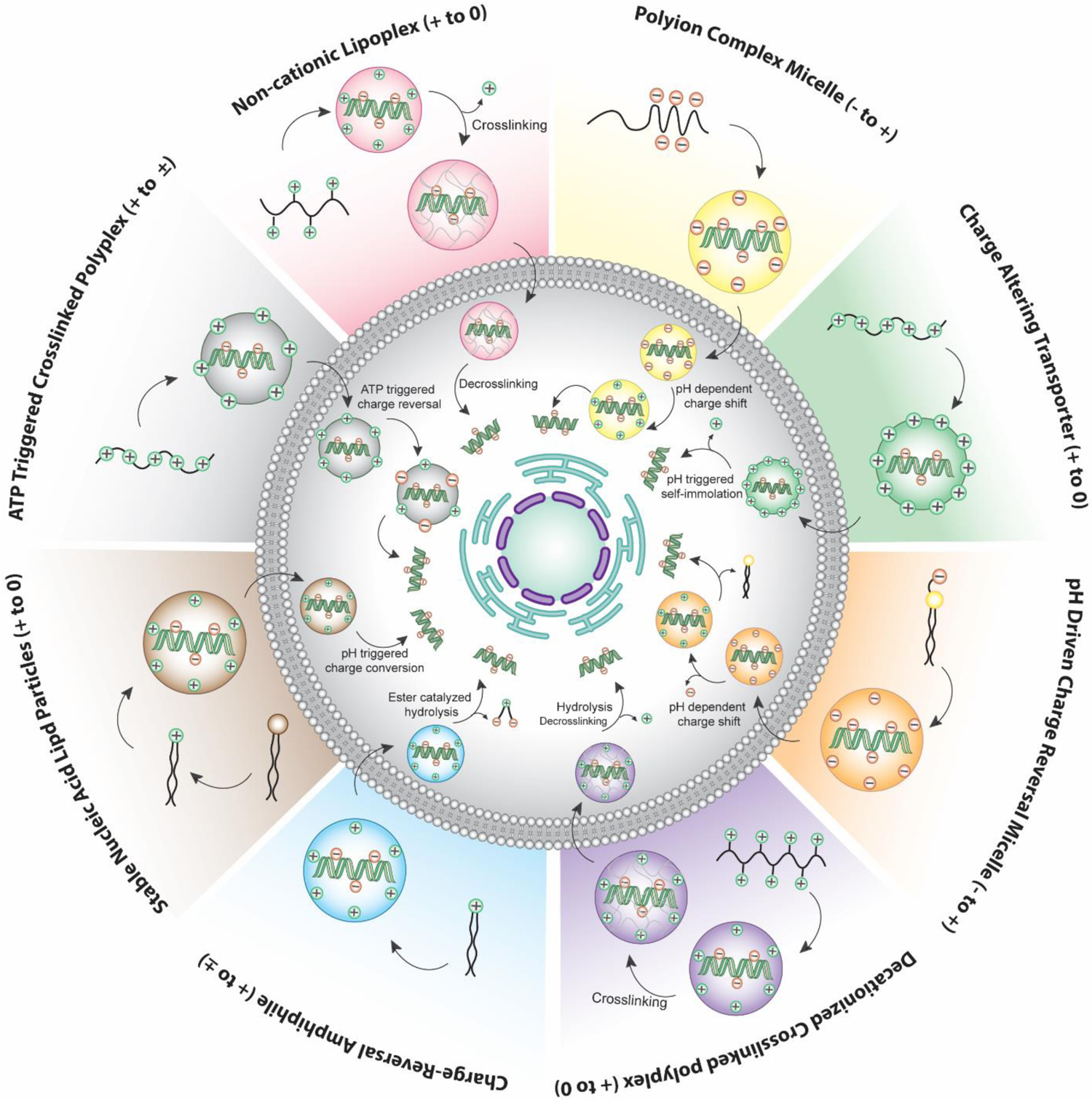
Schematic diagram of various classified charge-conversion approaches for nucleic acid delivery
2. Structural designs of charge-converting complexation scaffolds
Encapsulation of negatively charged nucleic acids with cationic vectors is a straight-forward strategy practiced widely. However, given the limitations owing to cytotoxicity concerns discussed above, it is necessary to develop approaches that can address the associated challenges. Possible solutions to this could be designing delivery agents that can either bypass electrostatic compensation needed for complexation or can utilize built-in mechanisms in the systems to cloak in nucleic acids and convert on demand to reveal opposite charges. The following sections are devoted to discuss several exciting approaches classified under conversion of charge which would potentially be applicable to address the unmet need for clinical translation of several nucleic acid candidates.
2.1. Cationic charge neutralizing strategies
2.1.1. Installation of permanent cationic charge and its removal via crosslinking
Neutralizing cationic charge after complexation of nucleic acids will serve as a promising methodology, albeit associated with the risk of leakage of the cargo due to lack of interactive forces to maintain the integrity of the complex. To address this, we have reported a ‘bait-and-switch’ strategy to incarcerate dsRNA stably encapsulated in a neutral complex (Figure 2a).[49] Methacrylate-based random copolymers were synthesized incorporated with cationic methylated pyridyl disulfide (MPDS) and polyethylene glycol (PEG) moieties in the side chains (Figure 2a). The ratio of the cationic MPDS and PEG groups (0.88:0.22) was optimized to enhance complexation with a dsRNA for Tuba1a gene (dsTuba1a). The strategy utilized a molecular mechanism to shed-off the cationic charge of the MPDS groups by a crosslinking reaction with dithiothreitol (DTT). Agarose gel electrophoresis study showed that the encapsulated dsRNA, even after removing the positive charge via crosslinking, remained stably encapsulated in the neutral nanocomplex. Evaluation of cytotoxicity studies in both human cancer and mouse embryo fibroblast cells revealed the non-toxic nature of the nanocomplex. Moreover, utilization of polymer-dsTuba1a nanocomplex resulted in decrease of corresponding mRNA levels, which ultimately stopped the generation of the blastocyst stage during mammalian embryo development. However, the development of embryo remained unaffected with control dsRNA or nano-complex encapsulated with negative control dsRNA. Inspired by this, a follow up study investigated a post-functionalization strategy for RNA interference (RNAi).[50] By tuning the molecular weight of the polymer and crosslinking degree, successful encapsulation of smaller GFP siRNA (siGFP) was achieved with tunable siRNA release kinetics. The strategy also demonstrated GFP silencing without any prominent toxicity.
Figure 2.
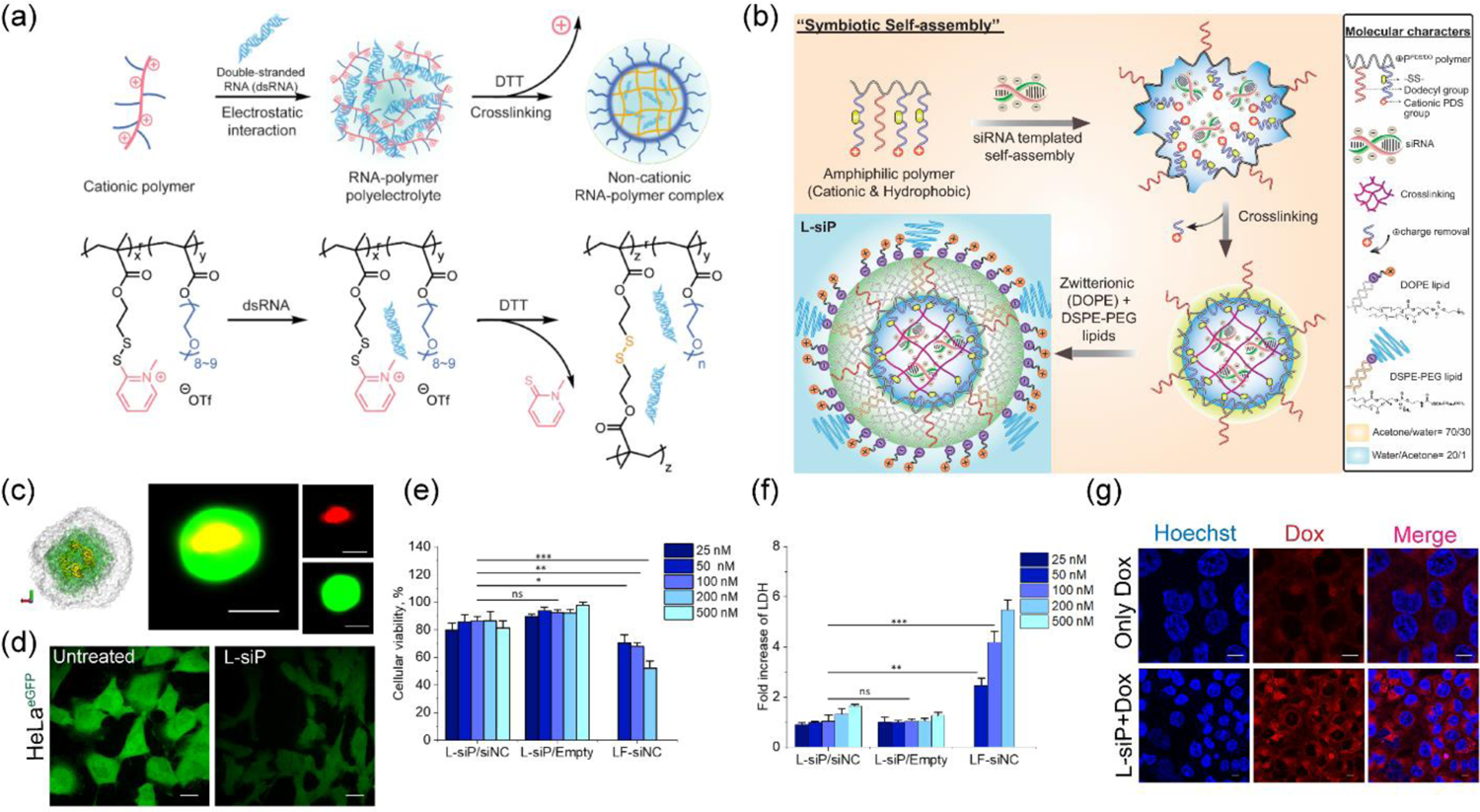
(a) Non-cationic polymer-RNA complex formation in aqueous media. The decationized complex was prepared via a crosslinking reaction that removed the positive charge (reproduced with permission from reference 49, Copyright 2019 American Chemical Society); (b) Tri-component self-assembly of a cationic polymer, an anionic siRNA and zwitterionic lipids via solvophobic forces and physical incarcerations with crosslinking to form L-siP nanoassembly; (c) left: Lipid-coated L-siP nanoassembly constructed with CG–MD simulation, green-polymer, yellow-oligonucleotide, transparent gray-lipid later; right: N-STORM confocal microscopy image of an individual L-siP nanoassembly, red- cy3-siRNA, green- carboxyfluorescein-labeled DSPE-PEG lipid, scale: 100 nm; (d) Silencing of eGFP gene in HeLa cells with L-siP nanoassembly showing reduction of green fluorescence, scale bar: 20 μm; (e) Cytotoxicity measurement via cell viability in HeLa cells with Lipofectamine-negative control siRNA complex (LF-siNC), empty L-siP and L-siP loaded with negative control siRNA (L-siP/siNC); (f) Cell membrane damage assessment with LDH-assay in HeLa cells showing no apparent membrane damage upon treatment with L-siP nanoparticles; (g) Uptake of doxorubicin increased in L-siP/siMDR1 treated NCI-ADR/RES cells, resistant to doxorubicin, scale: 10 μm (reproduced with permission from reference 53, Copyright 2019 American Chemical Society).
The mechanism of viral infections, caused by rapid ejection of genomes from capsid after binding to host surface, is believed to be guided by ‘electrostatic pressure’ of the tightly-packed charged nucleic acids inside the capsid core, which are due for charge compensation.[51–52] This, in combination with the fact that polyelectrolyte complexation is enhanced in low dielectric media and is entropically driven, inspired us to design a three-component self-assembly strategy consisting of an amphiphilic polymer with a cationic and a hydrophobic tail, siRNA and zwitterionic lipids (Figure 2b).[53] The electrostatic complexation between the polymer and siRNA was enhanced in a low dielectric mixed-solvent system followed by a crosslinking reaction to get rid of the cationic charge. Interestingly, the retention of caged siRNA was dictated by a combination of physical crosslinking and solvophobic forces originating from insolubility of highly negatively charged siRNA, and not merely by the conventional electrostatic interactions. The symbiotic process was completed by the self-assembly of zwitterionic lipids sheathing around the hydrophobic alkyl tail of the polymer. The prepared nanoassemblies showed efficient silencing of GFP, PLK1 and MDR1 genes (Figure 2c) with negligible cytotoxicity compared to Lipofectamine, a commercial transfection agent composed of cationic liposomes (Figure 2d).
2.1.2. Ionizable cationic charge and its removal through hydrolysis
Wherein a crosslinking reaction was utilized above to chop-off the positive charge from the delivery vector, another interesting strategy explored a hydrolysis reaction to pursue a similar goal. In an elegant approach, polyplexes composed of a cationic block copolymer poly(hydroxypropylmethacrylamide-dimethylaminoethyl-co-pyridyldithioethylamine-methacryl amide)-b-polyethylene glycol) (p(HPMA-DMAE-co-PDTEMA)-b-PEG or pHDP-PEG in short) and pDNA (pCMV-luc or pCMV-EGFP) were prepared by electrostatic interactions (Figure 3a–b).[54–55] Interestingly, the cationic charge of the polymer was generated by an ionizable amine moiety instead of a permanent quaternary ammonium group. This was followed by an interchain crosslinking reaction via a thiol-disulfide exchange reaction. To remove the ionizable cationic charge from the dimethylaminoethyl group, hydrolysis of the carbonate ester was performed at pH 8.5 leaving a disulfide crosslinked polyplex encapsulated with pDNA. The pHDP-PEG polyplexes were able to release the pDNA cargo only in response to a thiol (dithiothreitol, DTT), whereas no release was observed for the control cationic variants owing to the prevailing electrostatic interactions present therein. The decationized polyplexes were found to be non-toxic via XTT assay (for measuring cell viability) and LDH release assay (to check membrane integrity) with gene (luciferase) expression efficacy.[54] To overcome the issues with high non-specific uptake of cationic nanoparticles, this strategy was further improved via incorporating a targeting ligand.[56] Studies showed that the folate ligand incorporated polyplexes (pHDP-PEG-FA) were proficient in cellular uptake only for folate receptor overexpressing cells, HeLa and OVCAR-3, whereas transfection was minimal for A549, a cell line with low folate receptor concentration. To evaluate systemic toxicity of the polyplexes, a zebrafish embryo model was employed wherein fish eggs were incubated with different concentrations of polymer (Figure 3c–d, 3c: pre-test in HUVEC cells, 3d: with fish embryos).[55] Unlike cationic polyplexes which showed high teratogenicity at concentrations as low as 0.1 mg/mL, the neutral decationized ones did not show any developmental defects and mortality issues till 1 mg/mL concentrations. Assessments of systemic stability and biodistribution are important to understand nanoparticle’s integrity while in circulation and for the eventual localization in tumor.[55] Fluorescence single particle tracking (FSPT) studies of the polyplexes in human plasma showed a stable particle size distribution for 48 h without any aggregation. Finally, systemic administration of decationized cy7-labelled polyplexes into A431 tumor bearing mice showed efficient tumor localization measured through noninvasive optical imaging in comparison to cationic versions. Further, ex vivo analysis with fluorescence reflectance imaging (FRI), histological staining and fluorescence microscopy validated high tumor accumulation and expression of a polyplex delivered transgene (eGFP).
Figure 3.
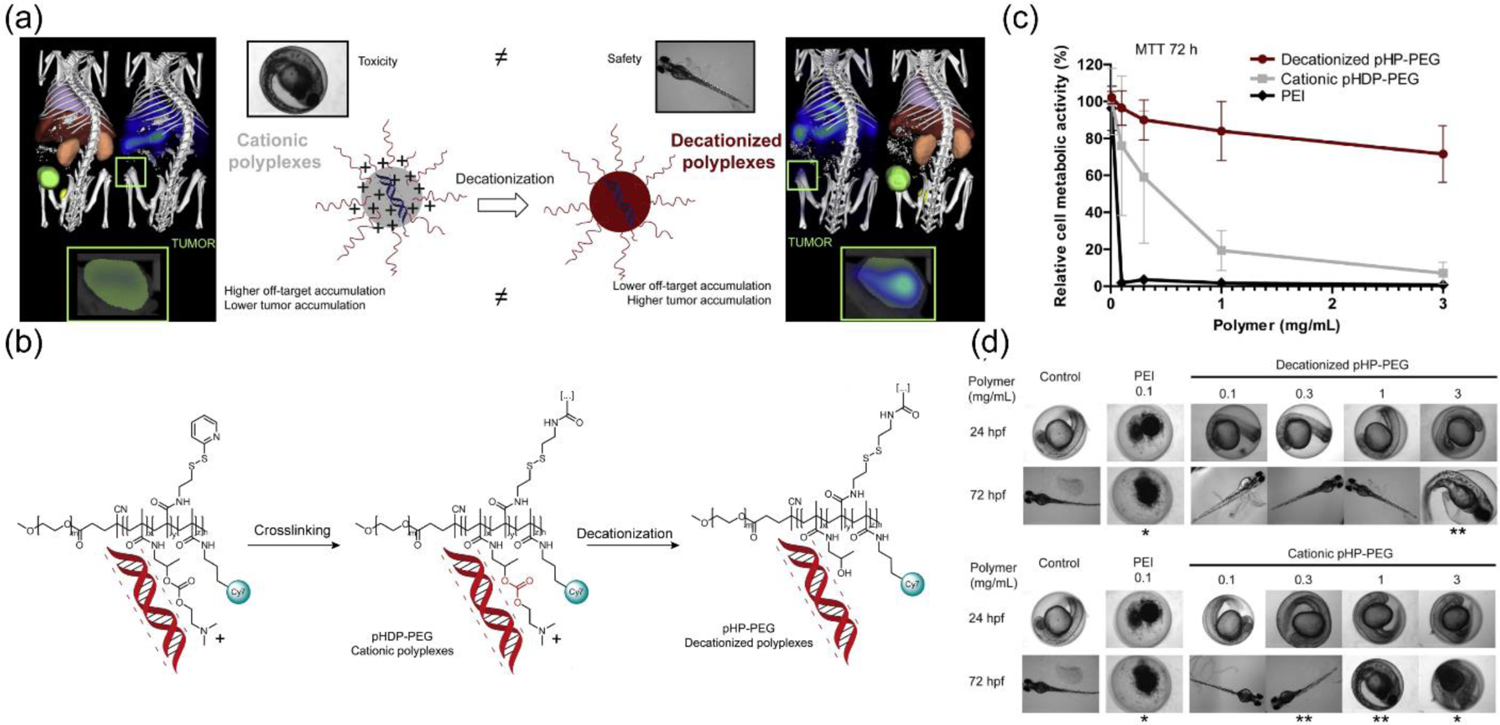
(a) Concept of decationized polyplexes for improved biodistribution and safety profile; (b) pDNA encapsulation in crosslinked pHDP-PEG polyplexes that can be decationized via hydrolysis reaction; (c) Safety evaluation by cell viability measurement in HUVEC cells upon treatment with non-cationic pHDP-PEG polyplex, cationic control pHP-PEG and polyethylene imine (PEI) where decationized polyplex shows better safety profile compared to cationic controls; (d) Images of zebrafish embryos upon treatment with increasing dosages of decationized pHP-PEG and its cationic control revealing elevated levels of toxicity only for cationic control and PEI in comparison to decationized pHP-PEG polyplex, * = significant mortality, ** = significant developmental defect (reproduced with permission from reference 55, Copyright 2014 Elsevier).
Encouraged by the safety profiles and gene transfection studies on pDNA, folate conjugated polyplexes (pHDP-PEG-FA) were extended to small interfering RNA (siRNA).[57] Owing to much smaller size of siRNA (~13.3 kDa, 21 bp) compared to pDNA (3000–5000 bp),[57] the polyplexes were optimized to tune monomer ratio in the polymer, N/P ratio during complexation (ratio of cationic to anionic groups) and crosslinked density. Also, fluorescence correlation spectroscopy (FCS) studies in human plasma showed that the polyplexes could successfully hold ~40% of siRNA for 2 h. The pHP-PEG-FA decationized polyplexes were also found to be non-toxic in cell viability assay with even a very high siRNA dosage (1000 nM, in 0.05 mg/mL pHDP-PEG-FA). Further, the utility of the system for silencing specific gene activity was demonstrated via folate targeted silencing of luciferase gene in Skov3 cell line, that was stably expressing luciferase. However, to be effective in delivery of nucleic acids at lower polymer concentrations, these strategies need further evaluation to overcome the endosomal entrapment of the polyplexes.
2.1.3. Ionizable cationic charge removal via self-immolation
Recent development of synthetic oligo(carbonate-b-α-amino ester)s, a new class of materials adding to the charge neutralizing strategies, has truly broadened the scope of this field by stepping into the domain of other nucleic acid types, like mRNA. While mRNA is long known for serving as the key component in cellular protein synthesis machinery, its application as a therapeutic candidate is considered to be of immense importance, as evident from recent research articles and ongoing clinical trials, including vaccine candidates for COVID-19.[58–59] To this end, polycationic oligo(carbonate-b-α-amino ester), synthesized via organocatalytic ring-opening polymerization (OROP), served as a delivery vector to electrostatically complex mRNA, protect and unwrap afterwards via a self-immolative mechanism to generate a neutral byproduct (Figure 4a–b).[60] As expected with charge neutralizing approaches, this CART (charge-altering releasable transporter) system (cation: anion = 10:1) showed no alarming toxicity when assessed the cellular viability of the polyplexes and the small molecule diketopiperazine byproduct separately (Figure 4c). CART system with ~13 lipid and ~11 cationic units in the oligomer backbone demonstrated >90% transfection efficacy and EGFP expression in various cell lines, including the hard-to-transfect ones (Figure 4d). In vivo bioluminescence studies for quantification of firefly luciferase mRNA delivery with CART polyplexes demonstrated observable presence of luciferase activity even after 48 h of intramuscular and tail vein injections.
Figure 4.
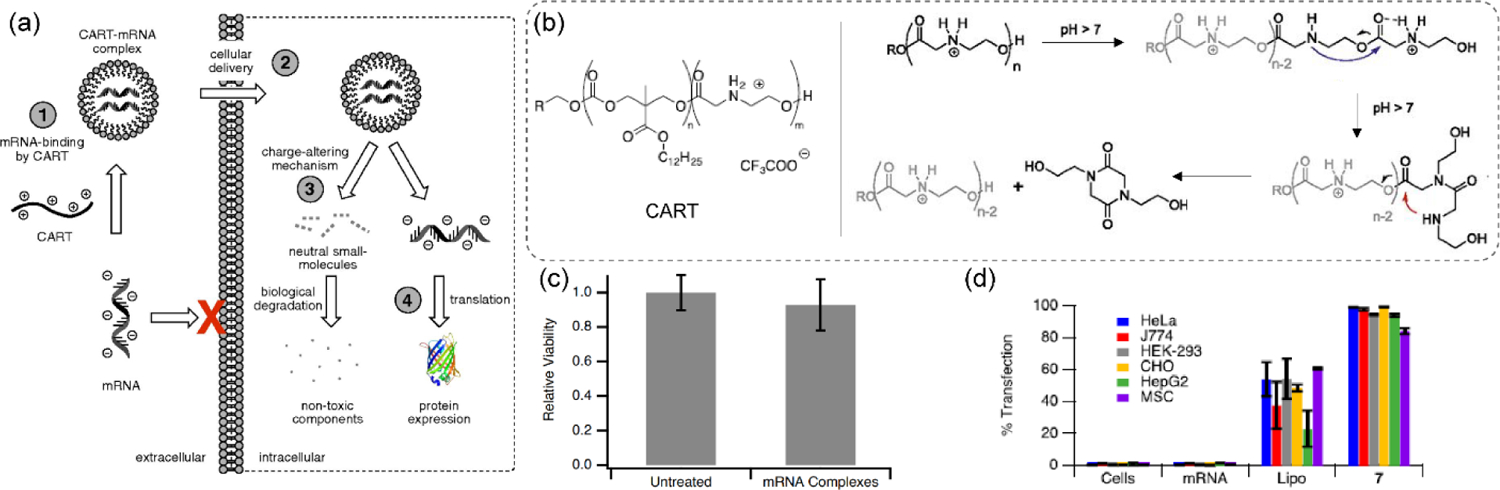
(a) mRNA delivery strategy with CART systems, incorporated with charge-altering mechanism, to induce intracellular protein expression; (b) Proposed cationic charge-alteration mechanism for oligo(α-amino ester)s involving two sequential 5- and 6-membered transition states; (c) MTT cell viability assay over 72 h in HeLa cells showing low toxicity of CART/mRNA complexes; (d) eGFP mRNA transfection efficacy in different cell lines with a preferred CART system (denoted as 7: with 13 lipid and 11 cationic chains, where n=13, m=11, R=3) (reproduced with permission from reference 60, Copyright 2017 The National Academy of Sciences of the USA).
To explore the strategy for the delivery of pDNA, CART oligomers were modified with a series of lipid tail, including noneyl, dodecyl, steryl, oleyl, linoleyl and cholesterol moieties.[61] A structure-activity study revealed oleyl- and linoleyl-based CART systems performed ~2–4x better in expressing GFP on successful delivery of the corresponding pDNA of a fusion protein, GFP-protein kinase C δ (GFP-pPKCδ), while maintaining a high cellular viability. Moreover, with the help of bryostatin 1, a PKC modulator, it was shown that the delivery of pDNA not only helped express a properly folded GFP-pPKCδ in the cytosol, but the fused protein was active enough to be able to translocate to the plasma membrane highlighting it with green fluorescence (Figure 5a). Hydrophobicity of the nanoparticle is known to impart significant influence in transfection efficacy.[62–63] Inspired by this, it was hypothesized that the CART platform might outperform in transfection efficacy for lymphocytes, including T cells, when incorporated with a mixture of lipid side chains. To test this, a combinatorial library approach was taken to identify the best performing lipid-CART binary mixture from 64 candidates (Figure 5b).[64] Oleyl- and nonenyl-CARTs at 1:1 mixture complexed with corresponding mRNA provided highest luciferase expression (Figure 5c). Finally, a single CART system incorporated with both lipid components was tested to find 6–9 fold higher transfection efficacy of mRNA into T and B lymphocytes compared to individual CART systems or the commercial transfection agent Lipofectamine 2000. Translation to in vivo mice model resulted in similar observations of high delivery efficacy of mRNA and expression of luciferase gene in splenocytes of mice. Moreover, transfection efficacy was found to increase across CD4 (~1–1.6%) and CD8 (~1.5%) T cells and B cells (11%) which was higher than the conventional mRNA delivery systems (Figure 5d).
Figure 5.
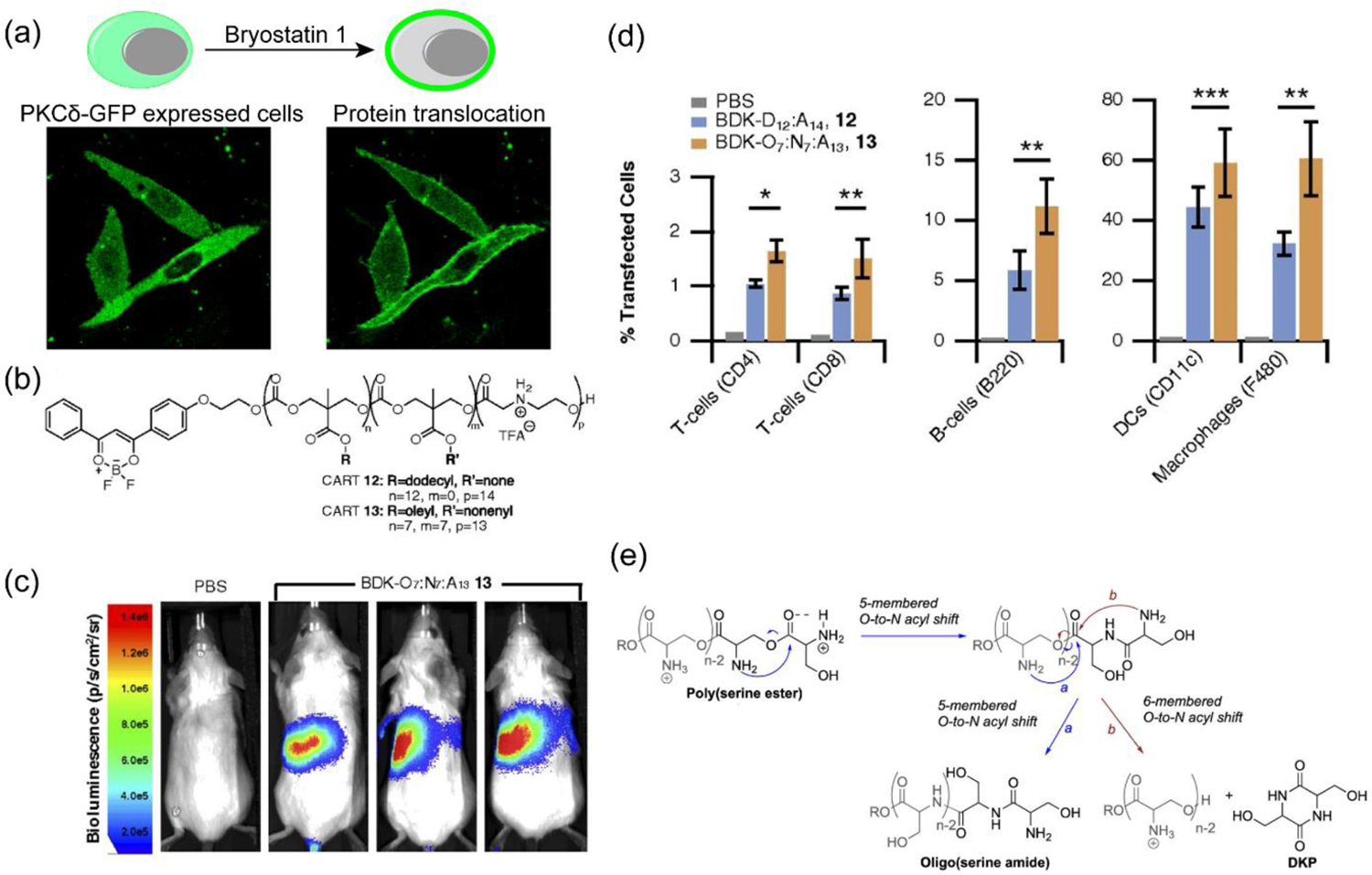
(a) CART-pPKCδ-GFP transfected CHO-K1 cells expressing pPKCδ-GFP fused protein in cytosol. The fused protein was able to translocate to cell membrane upon treatment with bryostatin 1, attesting the structure and activity of expressed fused protein (reproduced with permission from reference 61, Copyright 2018 American Chemical Society); (b) Fluorescently labeled single- and mixed-lipid CART systems; (c) Bioluminescence studies for the in vivo delivery of luciferase mRNA in BALB/c mice with mixed lipid CART system (CART 13, shown in Figure 5b) showing high levels of gene expression in spleen, considered as a favorable indication of lymphocyte transfection; (d) Transfection of immune cells for single- vs. mixed-lipid CART systems in spleen (reproduced with permission from reference 64, Copyright 2018 The National Academy of Sciences of the USA); (e) Proposed charge-altering rearrangement mechanism for poly(serine ester) CART system (reproduced with permission from reference 65, Copyright 2019 American Chemical Society).
As the charge-altering mechanism is directed by the activation of α-amino ester moiety towards rearrangement at higher pH, an oligo(serine ester) based CART (ser-CART) system was investigated owing to its favorable activating influence through O-to-N acyl shift (Figure 5e).[65] When complexed with eGFP mRNA, the ser-CAR system was found to show >95% transfection efficacy in multiple cell lines with high eGFP expression. The system could also be utilized under in vivo conditions to express luciferase gene expression via both intramuscular and intravenous injections. Importantly, like its other counterparts, the ser-CART system was also found to have improved cellular survival (~78–87%) for 10:1 (cationic: anionic) system.
2.2. Charge-reversing strategies from cationic-to-anionic species
Charge-reversing materials, designed to possess positively charged functionalities, capture negatively charged nucleic acids through conventional electrostatic interactions. However, through structural reorganization influenced by environmental cues, cationic functionalities can be reverted to anionic groups that render dissociation of the polyelectrolyte-type complexes. Undoubtably, these strategies are also considered for encapsulation and delivery of nucleic acids, while minimizing cytotoxicity owing to eventual transformation to negatively charged species. Note that anionic nanomaterials are generally considered as safe compared to cationic species.[39] The idea of complete reversal of charge is beneficial as it introduces an ejection mechanism of nucleic acids from the complex through Coulombic repulsive forces, and thereby eliminates the possibility of incomplete cargo release due to left over cationicity of the polyplex. Also, this approach provides an opportunity to temporally control the polyplex degradability affecting release kinetics of the encapsulated anionic cargo. Inspired by this, significant research effort has been devoted to identify suitable structural parameters integrated into the materials design, as discussed below.
Ideally, condensed polyplexes of nucleic acids are made to be thermodynamically stable with a high dose of cationic complexing agent to resist the inherent possibility of dissociation while in systemic circulation. A charge-reversal mechanism for unpacking complexed nucleic acid triggered by endogenous stimuli would be an effective strategy having minimal interference with other intracellular machinery. Demonstrating the practicality of the concept, a reactive oxygen species (ROS) responsive charge-reversing cationic polymeric system, poly[(2-acryloyl)ethyl(p-boronic acid benzyl)diethylammonium bromide] (B-PDEAEA), had been utilized for the delivery of a therapeutic DNA (pTRAIL) selectively to A549 tumor bearing mice (Figure 6a).[66] When exposed to elevated ROS level (e.g., H2O2) inside tumor cells, the boronic acid moiety installed in the polymer backbone was oxidized releasing a quinone methide from the adjacent quaternary ammonium moiety, converting it to a tertiary ammonium center (Figure 6a–b). This subsequently generated negatively charged poly(acrylic acid) after a self-catalyzed hydrolysis reaction of the ester group in the polymer. As evident with the phenyl boronic acid-based systems, the rate of charge conversion was found to increase with a decrease in pH. Polyplexes of B-PDEAEA showed enhanced gene (luciferase, GFP) transfection efficacy and anti-tumor activity (with TRAIL plasmid) in cancer cells. To impart an esterase responsive version of the above polyplex system, the phenyl boronic acid group was replaced with a p-acetyloxyphenyl moiety, that showed enhanced apoptosis upon delivery of TRAIL plasmid in HeLa i.p. tumor model (Figure 6c).[67]
Figure 6.
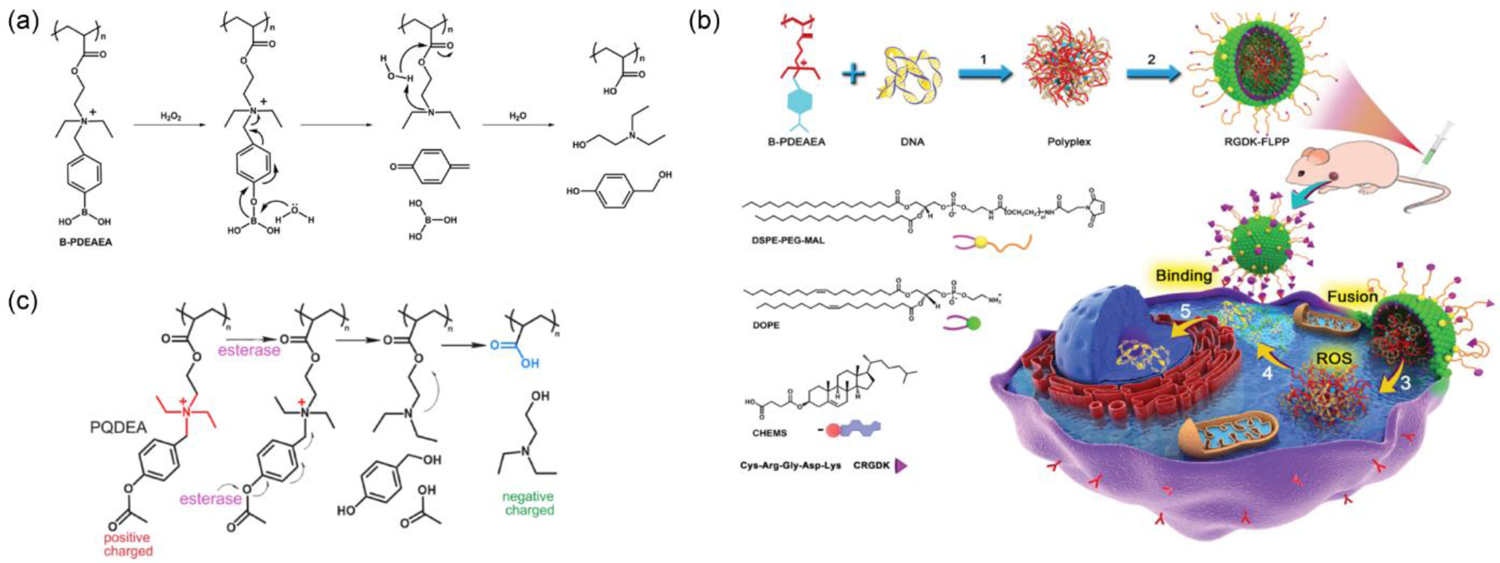
(a) ROS-responsive cationic-to-anionic charge conversion for B-PDEAEA polymer to generate anionic polyacrylic acid; (b) Complexation of pDNA with B-PDEAEA polymer and intracellular gene delivery: 1. complexation, 2. targeting lipid coating, 3. membrane fusion, 4. ROS-responsive release of DNA, 5. localization of DNA in nucleus (reproduced with permission from reference 66, Copyright 2016 Wiley-VCH Verlag GmbH & Co); (c) Structural modifications to introduce esterase-responsiveness in PQDEA polymer and the proposed charge-reversal mechanism (reproduced with permission from reference 67, Copyright 2018 American Chemical Society).
2.3. Charge-reversing from cationic to zwitterionic species
2.3.1. Conversion from cationic species to polyampholytes
While previous studies showed charge conversion from positive to negative in response to a stimulus of interest, another simple yet attractive approach could be dynamic introduction of anionic charge in the cationic polyplex to generate polyampholyte species. Similar concept had been tested with a cationic polyamine with an ester functionality that was gradually hydrolyzed to expose anionic carboxylate groups.[68] Dynamic variation in cation to anion ratios had been shown to control release of pDNA from self-assembled structures. In another approach, a multicompartmental polyplex comprised of a triblock copolymer, polybutadiene-b-poly(methacrylic acid)-b-poly(2-(dimethylamino)ethyl methacrylate) (BMAAD), was employed for pDNA complexation, wherein the overall charge was dictated by the pH-dependent ionization and reorganization of methacrylate and dimethylamino groups around the polyplex surface (Figure 7a).[69] Compared to polyethyleneimine (PEI) or poly(2-(dimethylamino)ethyl methacrylate (PDMAEMA) alone, BMAAD showed high cellular viability and eGFP transfection. Another interesting strategy relied on a combination of permanent and convertible cationic charges installed in a polymer scaffold. A poly[3-aminopropylmethacrylamide-co-N,N(dimethylamino) ethylacrylate] (p[APM-co-DMAEMA]: PAD) copolymer was designed that was able to produce a high cationic charge from aminopropyl and dimethylamino groups to complex pDNA, and then shifted to a ‘less-cationic’ variant via ester-hydrolysis to generate anionic acrylate moieties in the polymer backbone (Figure 7b).[70–71] Studies substantiated that PAD copolymers containing higher DMAEMA content (80 mol%) showed high cellular transfection efficiency for cy3-labelled pDNA (92%) and uncompromised cellular viability even at higher concentrations (N/P ratio = 5:1). Importantly, HeLa cells treated with polyplexes richer in APM showed more apoptotic cells than the polyplexes richer in DMAEA, which was reasoned to be due to the higher cationic and cytotoxic APM and low DMAEA units that were able to undergo charge-shifting hydrolysis producing anionic acrylates to increase zwitterionic nature of the complex.[70] To deliver pDNA in tumor without activating growth and metastasis promoter fibroblasts, the B-PDEAEA polymer system, reported in previous section, was modified to include an esterase sensitive 4-acetoxybenzyl ester moiety (Figure 7c).[72] Due to high esterase activity in cancer cells compared to fibroblasts, hydrolysis of polymer reversed the charge from cationic to zwitterionic liberating pDNA for TRAIL to induce apoptosis in a cell-selective manner.
Figure 7.
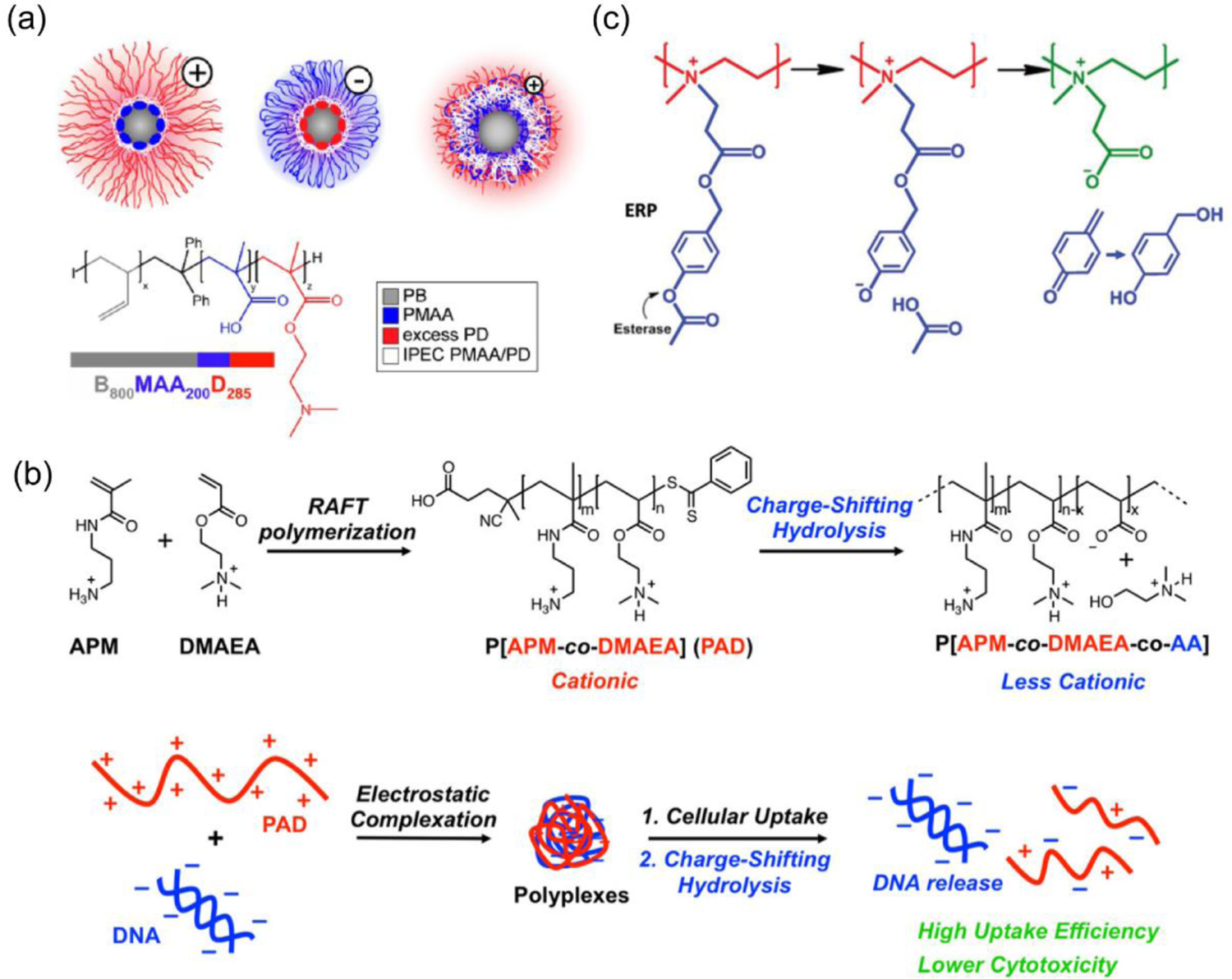
(a) Chemical structure of the BMAAD polymer and Cryo-TEM images of the formed micelles with charge-conversion at different pH (reproduced with permission from reference 69, Copyright 2013 American Chemical Society); (b) Synthesis of PAD polymer and charge-shifting mechanism through hydrolysis that facilitated the release of nucleic acid (reproduced with permission from reference 70, Copyright 2020 American Chemical Society); (c) Propionic 4-acetoxybenzyl ester modified PEI reversed cationic charge via esterase-mediated hydrolysis to form a zwitterionic species (reproduced with permission from reference 72, Copyright 2016 Wiley-VCH Verlag GmbH & Co).
Utilization of an endogenous stimulus to unload nucleic acid cargo triggered by charge conversion in polyplexes could serve as a fail-safe approach for nucleic acid delivery at target site. To this end, cationic polyplexes of CrossPPA/siRNA (crosslinked polyethylene imine-phenylboronic acid-alginate) were prepared comprising phenyl boronic acid (PBA) linker covalently crosslinked with alginate.[73] The overall cationic charge on the polyplex can be reversed to anionic after preferential binding between PBA and 1,2-diol from adenosine triphosphate (ATP),[74] an endogenous stimulus present at elevated concentration in cytosol. A sharp decrease in zeta potential, from positive to negative, was observed with CrossPPA polyplexes in response to ATP. The Bcl2-siRNA loaded polyplexes were able to downregulate the anti-apoptosis factor initiating apoptosis in malignant 4T1 tumor bearing mice. The polyplexes were also shown to impose minimal cytotoxicity when tested in 4T1 cells. Similar cationic to polyampholytic conversions were reported with FPBA grafted polyetheleneimine (PEI), which was assembled with cholesterol conjugated dopamine (via interactions with diol) and the ternary complex PFCD (PEI-FPBA/Cholesterol-Dopamine) was used for ATP responsive siRNA delivery.[75] To improve serum stability, cationic PEI-PBA/siRNA complexes were covalently cross-linked with ATP grafted hyaluronic acid to impart negatively charged polyplex, which was then successfully targeted to lower the expressions of eGFP, luciferase and BCl2 genes.[76]
2.3.2. Supramolecular assembly comprising lipids
Owing to the membrane compatibility, lipids are long considered as biocompatible material, and thus have been heavily explored for gene delivery purposes. Conventional cationic lipids, like DOTAP, do possess complexing ability with nucleic acids, but suffer from gene delivery efficacy due to the absence of any release mechanism of complexed nucleic acid cargo.[77] To address this, a supramolecular complex was prepared via interactions between DNA and a synthetic charge-reversing lipid (Figure 8a).[78] The amphiphilic lipid consists of a quaternary ammonium ion with ester moieties associated in the two lipid tails (Figure 8b). A supramolecular structure with bilayered-amphiphiles and DNA were formed initially through electrostatic charge complementary interactions. Upon hydrolysis in presence of esterase enzymes inside cell, the anionic carboxylate functionalities were exposed, and this released DNA through electrostatic repulsion caused by the increase in anionicity of the lipid. Understandably, the control anionic lipid did not show efficient binding affinity with DNA and lipid with non-hydrolyzable amide functionality was not able to release the nucleic acid from the assembly (Figure 8b–c). The DNA transfection efficacy was found to be significantly higher than the conventional lipid DOTAP. Further, investigations had been conducted to elucidate the influence of spacer group, introduced between the quaternary ammonium headgroup and the hydrophobic alkyl tail of the lipid, towards gene delivery.[79] Based upon previous benzyl ester lipid tail construct, different spacer groups with varying chain length (2, 5 and 7 atoms), rigidity, hydrophobicity or H-bonding ability were introduced in the amphiphiles. High transfection efficacy with DNA and efficient gene knockdown for siRNA were observed with two glycine spacer containing lipid complex.
Figure 8.
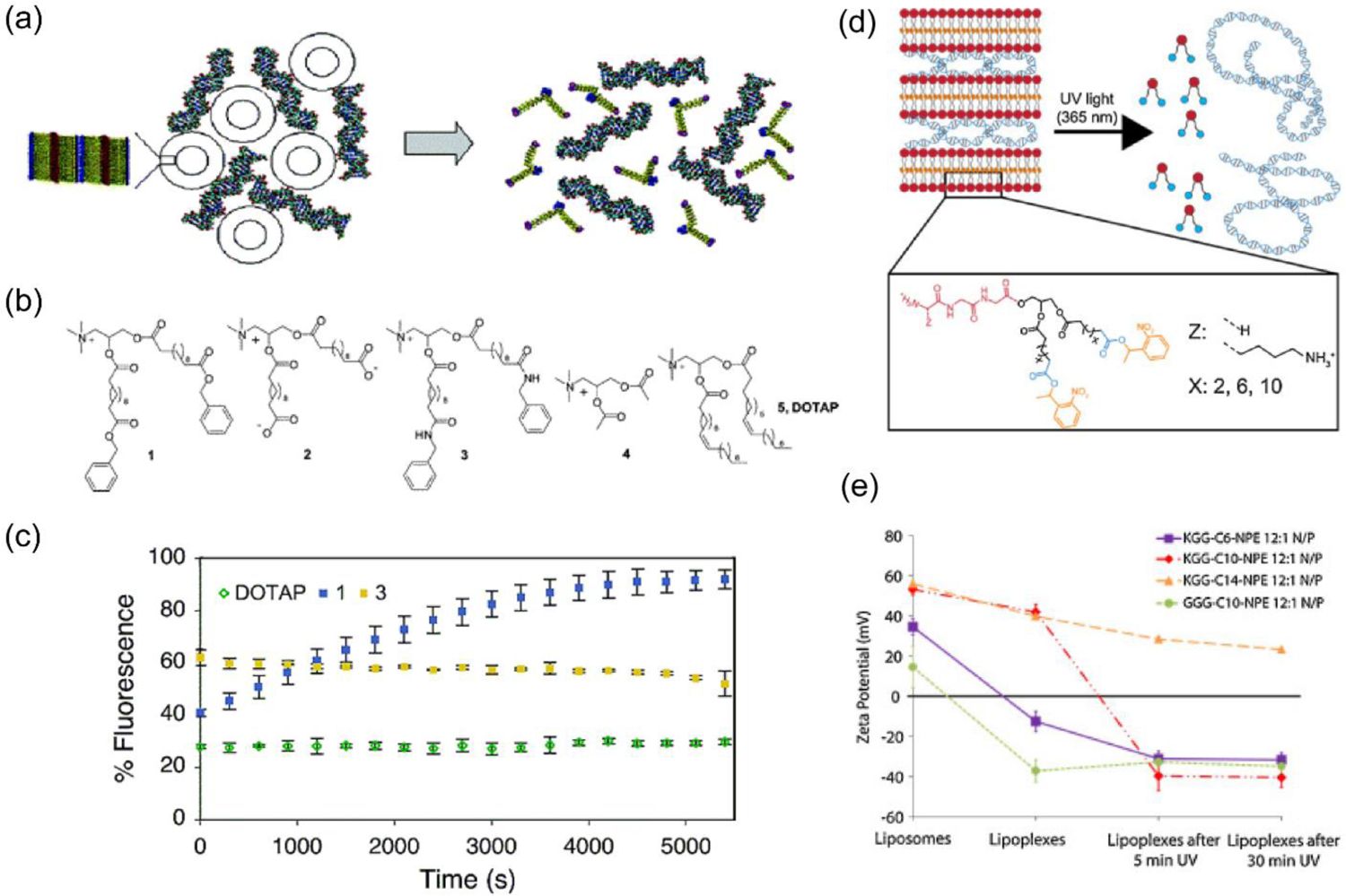
(a) Supramolecular assembly between DNA and multi-walled vesicles of a charge-reversing lipid amphiphile, and the release mechanism of nucleic acid via hydrolysis of ester moieties present in the lipid to form anionic species; (b) Structures of charge-reversing (1) and control lipids (2–5), wherein the favored prototype 1 was equipped with quaternary ammonium headgroup (for nucleic acid complexation), hydrophobic acyl chain (for bilayer formation) and terminal benzyl ester (for hydrolysis driven anionic group generation); (c) Kinetic measurements of fluorescence from ethidium bromide displacement assay to show the release of DNA from the assembly in response to porcine liver esterase (reproduced with permission from reference 78, Copyright 2004 American Chemical Society); (d) A supramolecular assembly of cation lipid with tripeptide headgroup and anionic nucleic acid, and light-triggered disassembly to release nucleic acids via charge conversion to generate anionic carboxylate groups; (e) Zeta potential studies showing charge-reversal of different tripeptide lipid-pDNA assemblies upon exposure with UV light (reproduced with permission from reference 81, Copyright 2016 Elsevier).
From steric accessibility and chemical reactivity perspectives, it is understandable that the charge-switching capability and kinetics of these amphiphiles will depend on the position and nature of the ester moiety in the alkyl lipid tail. To optimize this factor, different lipid amphiphiles were synthesized with varying tail length and chemical functionality (ester tail variations: -R-CO-R’ where R’= acetyl, ethyl, butyl benzyl and R= -(CH2)8, 10 or 14).[80] Benzyl ester amphiphile with C-10 chain showed fastest esterase-response to release DNA and highest transfection efficacy in CHO, HEK293 and K562 cells. Note that no apparent cytotoxic effect was observed with any of these lipid complexes. This stimuli-triggerable platform was further developed to include a photo-responsive unit that can spatiotemporally control the release of encapsulated nucleic acid. To this end, light-sensitive nitrophenylethyl (NPE) ester containing lipid amphiphiles were synthesized and their charge reversal was confirmed with UV (365 nm) light irradiation for 30 min (Figure 8d–e).[81] Studies demonstrated successful encapsulation and release of DNA and siRNA from these supramolecular assemblies.
2.4. Reversal of charge from anionic to neutral or cationic
In contrary to the previously discussed polycations that can reverse their surface charge from cationic to anionic, another appealing strategy to minimize nonspecific interactions with negatively charged serum components and plasma membrane is to utilize a negatively charged polyplex system. The negative charge can be reversed to neutral or cationic while inside endosome to impart efficient escape through “proton-sponge” effect.[82]
2.4.1. Polyion complex (PIC) micelles
In a noteworthy approach pAsp-(DET) (poly(N-[N’-(2-aminoethyl)-2-aminoethyl]aspartamide)), possessing an endosome-disrupting property, was utilized to form polyplexes with plasmid DNA (Figure 9a).[83] pAsp-(DET) polyplexes showed positive surface zeta potential of +40 mV (N/P ratio 4–8). However, the cationic polyplexes were coated with a charge-conversion polymer pAsp-(DET-Aco) (poly(N-(N’-[(N”-cis-aconityl)- 2-aminoethyl]-2-aminoethyl)aspartamide)) to form an overall anionic ternary polyion complex (PIC) micelle (zeta potential ≈ −40 mV, Figure 9b–c). Due to the carboxylic acid groups of citraconic amide in the pAsp-(DET-Aco), the micelles were negatively charged at neutral or basic pH, but eventually became positive after degradation to cationic primary amines at acidic pH 5,[84] an analogous environment as in endosome. Transfection with ternary PIC micelles containing luciferase pDNA on HUVEC cells (hard to transfect and sensitive to toxicity) offered more than ten times efficiency compared to a commercial transfection agent ExGen500 with linear PEI, and with high cellular viability. However, there are concerns regarding thermodynamic stability of the PIC micelles, specifically when loaded with short chain length nucleic acids, e.g., siRNA, under relevant physiological conditions. To address this issue, cationic polymer PEG-b-P(Lys/FPBA), wherein some of the lysine residues were post-functionalized with 3-fluoro-4-carboxyphenylboronic acid (FPBA), was employed to deliver siRNA (Figure 9d).[85] The rationale to use the PBA functionality is to from reversible covalent esters with the 1,2- or 1,3-cis-diols on the ribose rings present at the 3’ end of the siRNA along with possible intermolecular crosslinking events improving equilibrium binding.[74] Moreover, encapsulated siRNAs could be released from the FPBA modified PICs triggered by intracellular ATP through competitive binding with PBA. PEG-b-P(Lys/FPBA) polymers were optimized for siRNA delivery targeting PLK1 gene by varying the mol% of FPBA, where 23% and 35% of FPBA modification were identified for high serum stability and low cytotoxicity.[86]
Figure 9.
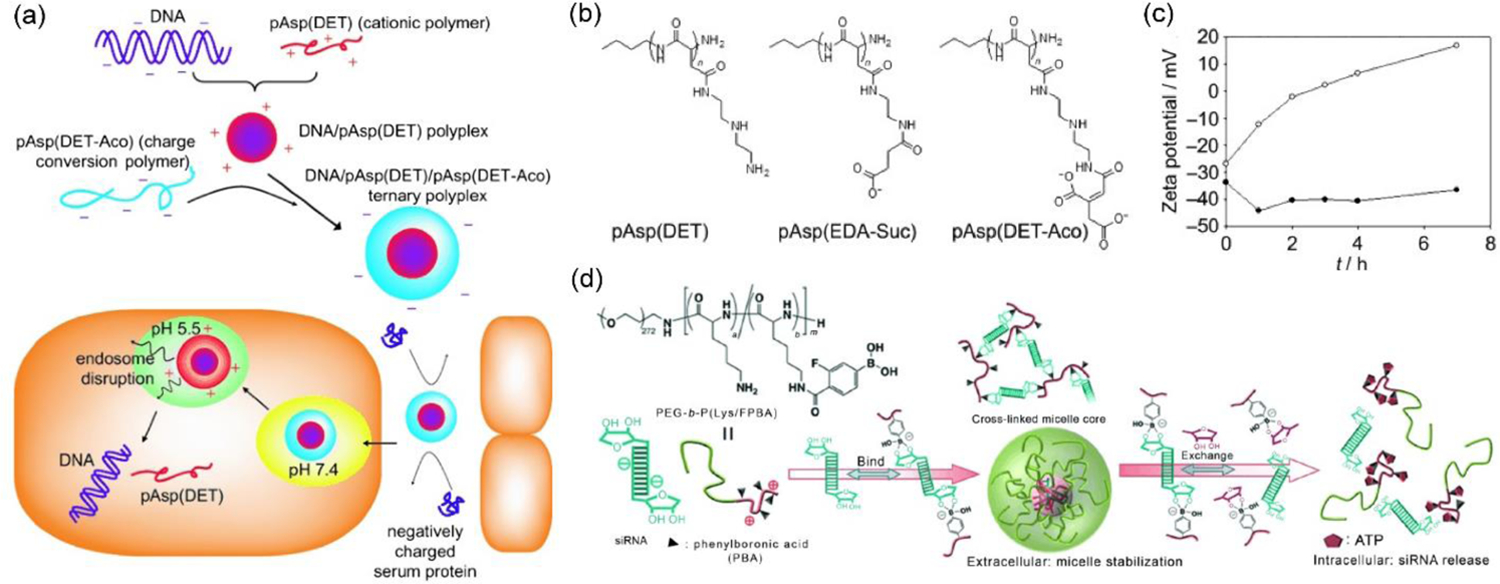
(a) Schematic for charge-reversing PIC micelle strategy for pDNA delivery, wherein the anionic ternary polyplex reverses to cationic under acidic pH condition in endosome causing endosomal disruption; (b) Structure of cationic pAsp(DET), charge-reversing pAsp(DET-Aco), and non-charge reversing control pAsp(EDA-Suc) polymers; (c) Zeta potential measurements to observe charge conversion in pDNA/pAsp(DET)/pAsp(DET-Aco), solid sphere: at pH 7.4, dark sphere: at pH 5.5 (reproduced with permission from reference 83, Copyright 2008 Wiley-VCH Verlag GmbH & Co.); (d) Utilization of phenylboronic acid-diol interaction for siRNA encapsulation in PIC micelles and its ATP-responsive release (reproduced with permission from reference 85, Copyright 2012 Wiley-VCH Verlag GmbH & Co.).
2.4.2. pH sensitive diblock polymer micelles and modular liposomes
Block copolymer systems can provide high encapsulation efficacy with unique structural stability of the encapsulated macromolecules.[87–89] A polymeric backbone equipped with pH responsive charge switchability can provide enhanced gene delivery efficacy. Initially, a disulfide conjugatable diblock copolymer system, poly[(HPMA-co-PDSMA)-b-(PAA-co-DMAEMA-co-BMA)], with cationic dimethylaminoethyl methacrylate (DMAEMA) groups were developed for complexation and delivery of siRNA.[90] However, with an understanding of known toxicity issues from the positively charged DMAEMA groups, the polymer structure was modified by replacing some of the DMAEMA groups with PDS methacrylamide and hydrophilic 2-hydroxypropyl methacrylamide (HPMA) block.[91] Here, the PDS units were employed for disulfide mediated covalent conjugation with thiolated GAPDH and negative control siRNAs, which were subsequently released via DTT triggered thiol-disulfide exchange reaction in presence of sodium dodecyl sulfate surfactant.[53, 91–93] Also, protonation of DMAEMA at the micellar core and propylacrylate (PAA) moieties triggered the charge reversal under acidic condition. In combination with lipid penetrating butylmethacrylate (BMA) groups, this mechanism was found to help in the membrane destabilization activity, essentially required to boost the endosomal escape phenomena. The polymer system was found to deliver GAPDH siRNA, reduce the corresponding mRNA and protein to 11% and 35%, respectively after 48 h of incubation with negligible cellular toxicity.
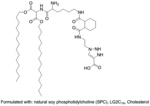
Ideally, similar balance in positive and negative charges can also be exploited in lipid-based modular assemblies. To this end, a liposome based siRNA delivery system was developed wherein the pKa of the assembly could be tuned via balancing the cationic amino acid-based lipid to anionic propiolic acid contents (Figure 10a).[94] After initial complexation with the cationic lipid and siRNA, the liposomes were modified with propiolic acid to exhibit ζ potentials of −19 mV and 10 mV at pH 7.4 and 6.5, respectively. The system was thoroughly investigated to show efficient tumor uptake, silencing of targeted survivin gene and safety profile in hepatocellular carcinoma bearing ICR mice model.
Figure 10.
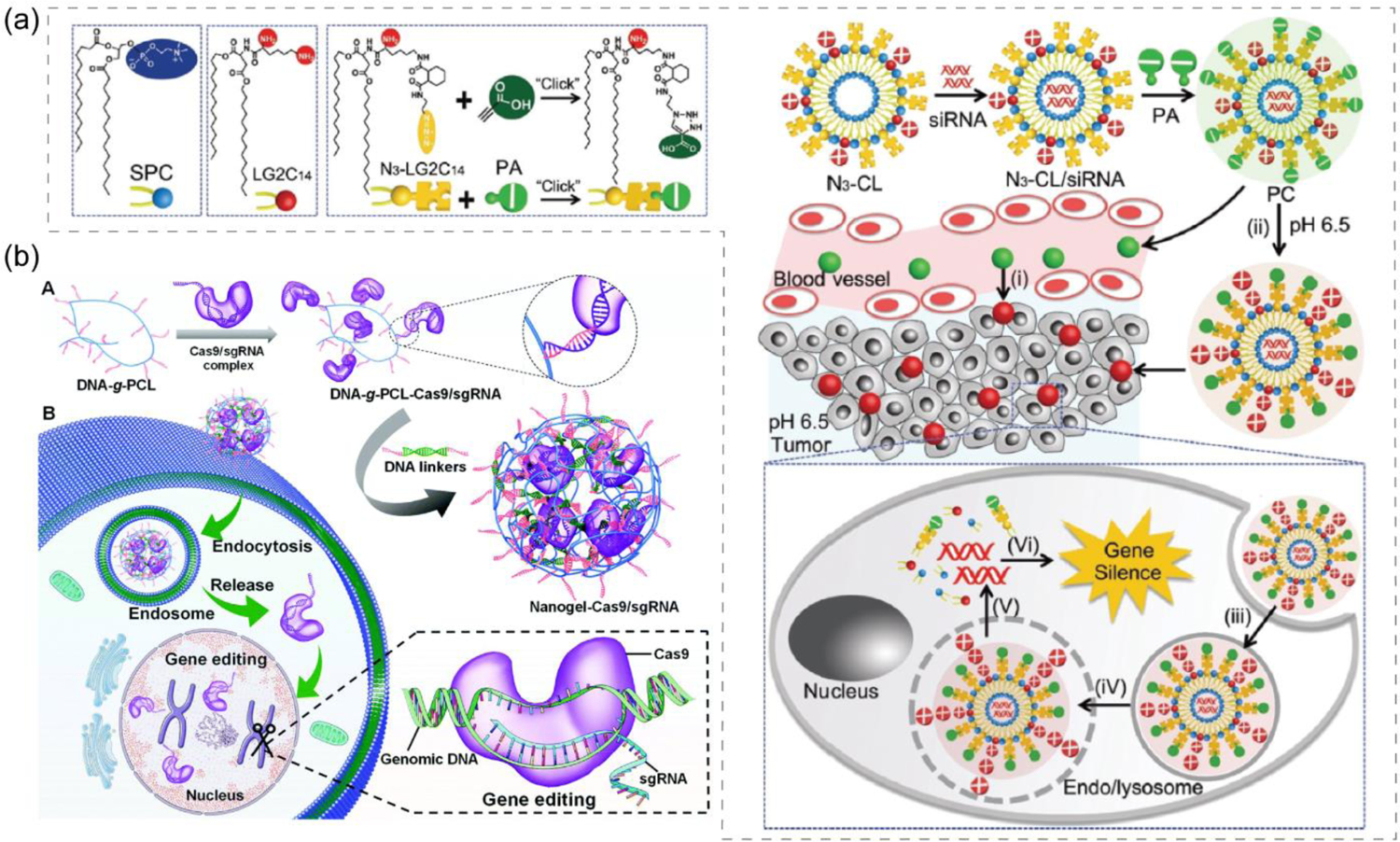
(a) Modular liposome assembled with pH sensitive charge converting complex for siRNA delivery, wherein anionic complex reverses to cationic under acidic tumor environment to enhance uptake and then nucleic acid is released to cytosol via a endosome membrane fusion mechanism (reproduced with permission from reference 94, Copyright 2018 Royal Society of Chemistry); (b) DNA-g-PCL nanogel formation to deliver CRISPR/Cas9 for intracellular gene editing (reproduced with permission from reference 109, Copyright 2019 Royal Society of Chemistry).
2.4.3. Anionic nucleotide lipid to complex nucleic acids: 3’- and 5’- phosphate modified versions
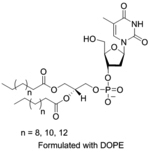
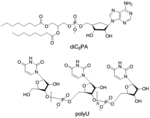
To alleviate the cytotoxicity of the cationic lipids, an interesting strategy was developed utilizing the charge-reversal lipid platform, discussed under ‘supramolecular assembly comprising lipids’ section. This strategy was further extended where instead of utilizing cationic charge, an anionic headgroup from a lipid (1,2-diacyl-sn-glycerol) was covalently joined to a nucleotide (3′-phosphate nucleotide lipid) which favored complexation with DNA in presence of a metal ion, Ca2+.[95] Synthesized thymidine-3′-monophosphate nucleotide incorporated anionic amphiphile showed complexation with DNA over 1 mM of Ca2+ concentration. Since the intracellular concentration of Ca2+ is a much lower (~μM) compared to the extracellular space, liberation of DNA is expected inside cytosol through the dissociation of the metal-bridged complexes.[96] Compared to non-nucleotide lipid controls, the designed anionic lipid showed low cytotoxicity and optimal transfection efficacy. While binding interactions between this anionic 3′-phosphate nucleotide lipid and DNA were governed by the presence of divalent metal ions, another promising approach for nucleic acid complexation could be based on highly specific and self-controlled molecular recognition phenomena. An elegant example of such oligonucleotide binding was reported with anionic micelles of 5′-phosphate modified dioctanoylphosphatidyladenosine (diC8PA) and polyuridylic acid (polyU) without the presence of metal ions.[97–98] Followed by this, another similar approach was reported for complexation with single (ss) and double stranded (ds) DNA with the anionic liposomes of POP-Ade and helper zwitterionic POPC (1-palmitoyl-2-oleoyl-phosphatidyladenosine or -choline) lipids in presence of Ca2+ salt.[99] Interestingly, the control anionic lipid without nucleic headgroup, POPG (1-palmitoyl-2-oleoyl-sn-phosphatidyl-glycerol), failed to show any interaction with ss-DNA even at higher concentrations of Ca2+, but managed to engage in moderate association with dsDNA giving rise to a different liquid-crystalline structural feature.
2.4.4. Charge reversal in metal polymer complexes
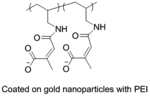
In the context of nucleic acid delivery, another widely researched approach includes Au nanoparticles coated with long chain charged linkers. Generally, nucleic acids were loaded into Au nanoparticles through thiol linkers or by electrostatic interaction with cationic gold nanoparticles.[100–103] In a similar approach as in ternary PIC micelles, PEI coated Au nanoparticles were covered with a charge-reversing cis-aconitic anhydride functionalized poly(allylamine) (PAH-Cit) and siRNA via layer-by-layer assembly process.[104–105] The charge reversal polymer coated nanoparticles were found to perform better in eGFP and lamin A/C gene suppression compared to the non-charge reversing control complex and commercial PEI. Due to the anionic nature, cytotoxicity of nanoparticles was found to be low when evaluated via MTT assay.[104] To further optimize the efficiency of the aforementioned system, a negatively charged core-shell nanoparticle was formed via LbL assembly starting with chitosan stabilized Au and sequentially coating PAH-Cit, PEI and siRNA, respectively.[106] The vehicle demonstrated high siRNA binding affinity and pH responsive disassembly at pH 5.5 releasing 79% of loaded siRNA compared to 23% release at pH 7.4. The developed nanoparticles demonstrated high MDR1 gene silencing efficacy with 80% reduction in mRNA level. Also, cytotoxicity evaluation in multiple cell lines resulted in comparable survival rate as with untreated cells.
2.5. Anionic DNA-polymer brush
Although cationic agents are effectively used to condense nucleic acid in a nanoparticle prior to delivery, a plausible alternative idea was to employ anionic nucleic acid-based systems to encapsulate nucleic acid cargoes. Spherical nucleic acid and nucleic acid nanostructures have provided inspiring examples to complex nucleic acids via covalent or molecular recognition-based interactions in comparison to traditional electrostatic interactions. Inspired by several nucleic acid-based architectures like, spherical nucleic acids and DNA nanostructures,[103, 107] a DNA-grafted polycaprolactone (DNA-g-PCL) based framework was synthesized to encapsulate siRNA which acted as a crosslinker when modified with single stranded overhangs on both sides complementary to the DNA in the framework.[108] Although there is no apparent charge conversion here as the system remain anionic even after releasing negatively charged siRNA, this could be classified as a delivery vehicle with anionic-to-anionic converting system. The developed DNA nanogel system was reported to successfully silence PLK1 gene expressions through siRNA-mediated RNAi mechanism under both in vitro and in vivo conditions. The strategy was successfully extended to deliver CRISPR/Cas9 system after molecular recognition-based complexation between DNA-g-PCL and Cas9/sgRNA where the sgRNA was incorporated with complementary tail to DNA (Figure 10b).[109] When delivered in eGFP expressing HeLa cells, the non-cationic vector was able to provide efficient gene editing via knocking down eGFP expressions by ~21%.
2.6. Ionizable lipids: pH dependent charge conversion from cationic-to-neutral-to-cationic
Permanently cationic lipids such as N-[1-(2,3-dioleyloxy)-propyl]-N,N,N-trimethylammonium chloride (DOTMA), have been widely utilized to complex negatively charged nucleic acid.[110] When formulated with helper lipid like DOPE to make liposomes, the system delivered efficient gene transfection in COS-7 cells using pSV2cat DNA. Nevertheless, the major drawbacks associated with permanently cationic lipids include cellular toxicity, aggregation with erythrocytes, triggering immune response via non-specific interaction with toll-like receptors, and rapid plasma clearance, which can be attributed to the higher positive surface potential of the nano-carriers.[111] Consequently, the focus has been shifted to the development of ionizable lipids comprising a hydrophobic tail, a linker group and a tertiary amine headgroup which can be protonated below a certain pH. Being positively charged at lower pH (<pKa), ionizable lipids are competent to complex with the negatively charged nucleic acids, while it switches to neutral or slightly positive at the physiological pH 7.4, thereby minimizing the cellular toxicity with efficient gene transfection ability. DODAP, the very first ionizable lipid (pKa ≈ 6.6–7), showed enhanced encapsulation of oligonucleotides (~ 70%) in acidic condition.[112] Recently, there have been many studies that explicate the structure-activity relationship to optimize these systems for efficient delivery of nucleic acid based therapeutics for clinical applications.[113] Among the first generation ionizable lipids, linoleyl hydrocarbon tail comprising 1,2-dilinoleyloxy-N,N-dimethyl-3-aminopropane (DLinDMA) lipid showed promising results in C57BL/6 mice model by delivering Factor VII siRNA.[114] The study revealed a low ED50 value (1 mg/Kg) associated with the ionizable lipid DLinDMA. Encouraged by these results, ionizable lipids with varying hydrophobic tail length and unsaturation, different linker length, and polyamine headgroups were systematically investigated. A subsequent lipid DLinDAP showed decreased potency in gene transfection which was due to higher possibility of hydrolysis of ester groups under in vivo conditions, and thus had compromised protection towards the encapsulated oligonucleotides. Later, improved versions of ionizable lipids 2,2-dilinoleyl-4-(2-dimethylaminoethyl)- [1,3]-dioxolane (DLin-KC2-DMA) and the most potent heptatriaconta-6,9,28,31-tetraen-19-yl 4-(dimethylamino)-butanoate (DLin-MC3-DMA) were developed which showed a significant decrease in the ED50 values of 0.1 mg/kg and 0.03 mg/Kg, respectively.[113, 115] More details about the ionizable lipids can be found in previous review articles.[116–119]
The chemical scaffolds along with their charge conversion strategies discussed in this review are summarized in Table 1.
Table 1.
Classification of nucleic acid delivery systems based on charge-conversion strategies
| Classifications (based on charge conversion) | System Name | Chemical Structure | Charge Alteration Mechanism | Encapsulated Nucleic Acid | References |
|---|---|---|---|---|---|
| ‘+’ to ‘0’ | Poly(pyridyldisulfide-ethylene glycol methacrylate) |
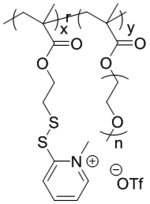
|
Crosslinking with disulfide | dsTuba1a | Biomacromolecules 2019, 20 (1), 435–442; Ind. Eng. Chem. Res. 2019, 58 (17), 6982–6991 |
| ‘+’ to ‘0’ | Poly(pyridyldisulfide-dodecyl methacrylate) coated with lipids, L-siP |
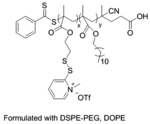
|
Crosslinking with disulfide | Cy3-siRNA, PLK1 siRNA, MDR1 siRNA | ACS Appl. Mater. Interfaces 2019, 11 (28), 24971–24983 |
| ‘+’ to ‘0’ | Poly(hydroxypropyl methacrylamide-dimethylaminoethyl-co-pyridyldithioethylamine methacrylamide)-b-polyethylene glycol) |
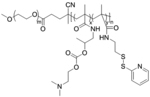
|
Through hydrolysis | pDNA (pCMV-luc or pCMV-eGFP) | J. Controlled Release 2013, 169 (3), 246–256; J. Controlled Release 2014, 195, 162–175 |
| ‘+’ to ‘0’ | pHDP-PEG-FA |
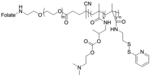
|
Through hydrolysis | anti-Luc siRNA, LV2 siRNA (scrambled siRNA) | Mol. Pharmaceutics 2015, 12 (1), 150–61 |
| ‘+’ to ‘0’ | Oligo(carbonate-b-a-amino ester) CART |
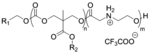
|
pH triggered self- immolation | eGFP mRNA, Fluc mRNA, Cy5-EGFP mRNA | Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (4), E448-E456; Biomacromolecules 2018, 19 (7), 2812–2824; Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (26), E5859-E5866 |
| ‘+’ to ‘0’ | Oligo(serine ester)-based charge-altering releasable transporters (Ser-CARTs) |
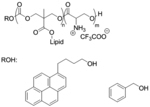
|
pH triggered self-immolation | eGFP mRNA, Fluc mRNA, Cy5-EGFP mRNA | J. Am. Chem. Soc. 2019, 141 (21), 8416–8421 |
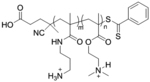
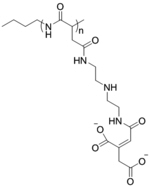
| ‘+’ to ‘±’ | Ester substituted linear poly(ethylenimine) |
|
Ester hydrolysis | pDNA (pEGFP-N1) | Macromolecules 2005, 38 (19), 7907–7914 |
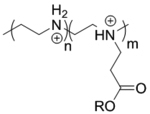
| ‘+’ to ‘±’ | Poly[3-aminopropylmethacrylamide-co-N,N(dimethylamino)ethylacrylate] |
|
Charge shifting hydrolysis | pDNA | ACS Omega 2020, 5 (16), 9114–9122; Macromolecules 2020, 53 (9), 3514–3523 |
| ‘+’ to ‘±’ | N-[3-((4-acetoxybenzyl)oxy)-3-oxopropyl]-N-methyl-quaternized PEI |
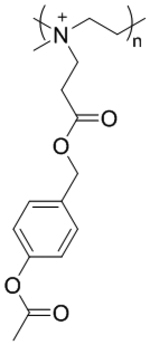
|
Esterase catalyzed hydrolysis | pDNA for TRAIL | Adv. Mater. 2016, 28 (48), 10613–10622 |
| ‘+’ to ‘±’ | Crosslinked polyethylene imine-phenylboronic acid-aginate (CrossPPA) |
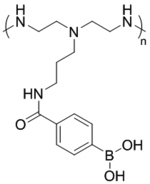
|
ATP triggered charge reversal | Bc12-siRNA | Theranostics 2018, 8 (17), 4604–4619 |
| ‘+’ to ‘±’ | PEI-FPBA-Dopa-Chol micelles (PFCDM) |
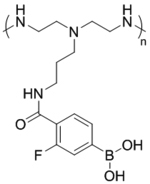
|
ATP triggered charge reversal | siScr (scrambled siRNA), FAM-siSCR, cy3-siScr, siEGFP, siBC12 | ACS Appl. Mater. Interfaces 2018, 10 (38), 32026–32037; Biomacromolecules 2018, 19 (9), 3776–3787 |
| ‘+’ to ‘-’ | Poly[(2-acryloyl)ethyl(p-boronic acid benzyl)diethylammonium bromide] (B-PDEAEA) |
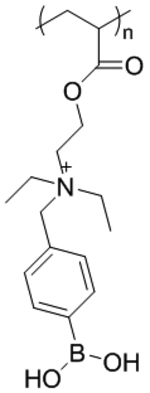
|
ROS triggered charge reversal | pDNA (EGFP, TRAIL) | Adv. Mater. 2016, 28 (9), 1743–1752; Biomacromolecules 2018, 19 (6), 2308–2319 |
| ‘+’ to ‘-’ | [2,3-Bis-(11-benzyloxycarbonyl-undecanoyloxy)-propyl]-trimethyl-ammonium; iodide |
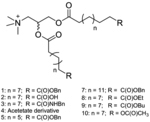
|
Esterase catalyzed hydrolysis | pDNA | J. Am. Chem. Soc. 2004, 126 (39), 12196–12197; Bioconjugate Chem. 2008, 19, 418–420; Bioconjugate Chem. 2011, 22 (4), 690–699 |
| ‘+’ to ‘-’ | Lysine–Glycine–Glycine (KGG)-Cx - NPE |
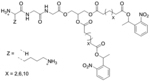
|
Light triggered degradation | pDNA, siRNA | Chem. Phys. Lipids 2016, 196, 52–60 |
| ‘-’ to ‘+’ | Poly(N-(N’-[(N”-cis-aconityl)- 2-aminoethyl]-2-aminoethyl)aspartamide); pAsp-(DET-Aco) |
|
pH dependent degradation | Luc-pDNA, YFP pDNA, cy5 pDNA | J. Am. Chem. Soc. 2007, 129 (17), 5362–5363 |
| ‘-’ to ‘0/+’ | Poly(ethylene glycol)-b-poly(L -lysine); PEG-b-PLys |
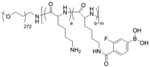
|
ATP responsive charge shifting | Cy5-siRNA, PLK1-siRNA | Angew. Chem. Int. Ed. 2012, 51 (43), 10751–10755; Macromol. Biosci. 2018, 18 (1), 1700357 |
| ‘-’ to ‘+’ | Poly[(HPMA-co-PDSMA)-b-(PAA-co-DMAEMA-co-BMA)] |
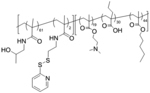
|
pH dependent charge reversal | GAPDH-siRNA, SCR-siRNA | Bioconjugate Chem. 2013, 24 (3), 398–407 |
| ‘-’ to ‘+’ | Proiolic acid modified N3-LG2C14 |
|
pH dependent charge shift | Survivin-siRNA (cpusiRNA), GAPDH-siRNA | Biomater. Sci. 2018, 6 (11), 3075–3084 |
| ‘-’ to ‘+’ | PAH-Cit |
|
pH dependent charge altering | Cy5 siRNA Lamin A/C siRNA |
AIChE J. 2018, 64 (3), 810–821; ACS Nano 2010, 4 (9), 5505–5511 |
| ‘-’ to ‘-/0’ | Thymidine-3′-(1,2-dipalmitoyl-sn-glycero-3-phosphate) diC16-3′-dT; 5’-phosphate modified dioctanoylphosphatidyladenosine (diC8PA) and polyuridylic acid (polyU) |
|
Ca2+ concentration dependent | pDNA (EGFP) | Bioconjugate Chem. 2009, 20 (9), 1765–1772; Adv. Mater. 2018, 30 (11), 1705078 |
|
Angew. Chem. Int. Ed. 2007, 46 (17), 3070–3073; J. Am. Chem. Soc. 2007, 129 (38), 11664–11665 |
||||
| ‘-’ to ‘-’ | DNA-g-PCL |
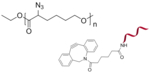
|
- | EGFP siRNA PLK1 siRNA |
Angew. Chem. Int. Ed. 2018, 57 (12), 3064–3068; Nanoscale 2019, 11 (37), 17211–17215 |
| ‘0’ to ‘+’ to ‘0’ | DODAP |
|
pH dependent charge altering | Antisense oligodeoxynucleotides (ODN) | Biochim. Biophys. Acta, Biomembr. 2001, 1510 (1–2), 152–166 |
| ‘0’ to ‘+’ to ‘0’ | DLin-DMA |
|
pH dependent charge altering | ApoB siRNA Factor VII siRNA |
Nat. Biotechnol. 2010, 28 (2), 172–176 |
| ‘0’ to ‘+’ to ‘0’ | DLin-MC3-DMA |
|
pH dependent charge altering | Nanomedicine 2013, 9 (2), 233–246; ACS Nano 2017, 11 (8), 7572–7586 |
3. Fate of charge-converting nucleic acid carriers in cellular uptake and endosomal escape
3.1. Effect of nanoparticle surface charge
One of the major bottlenecks for successful nucleic acid delivery is poor cellular uptake of nucleic acid carriers. Cellular uptake of nanocarriers is generally affected by four key parameters: charge, size, shape, and hydrophobicity.[39, 120] Understandably, surface charge would be the primary driving force in establishing an interaction between the carrier and the cell membrane.[121–122] In case of nonphagocytic cells, cationic carriers possess an advantage in cellular uptake, whereby enhanced interactions occur with the exposed negatively charged phosphate groups of hydrophobic lipid bilayer in cell membrane. In contrast, neutral or anionic nanocarriers do not own this specific privileged interaction, but could impart higher degrees of stability and residence time in circulation due to repulsive interactions with serum components, ultimately improving uptake efficiency. For phagocytic cells, it has been found that anionic nanoparticles were taken up to a higher degree because of the presence of SR-AI/II scavenger receptors.[123] When comparing cationic and decationized pHDP-PEG polyplexes, it was found that the decationized polyplex showed lower luciferase expression due to a decreased cellular uptake with respect to the cationic polyplex.[55] However, decationized polyplexes were still internalized by the cells with reduced non-specific uptake which was predominantly observed with the cationic version of the polyplex.[56] In contrast, a near neutral pDNA-PEI-mBSA polyplex, where ethylamine modified BSA was coated around a pDNA-PEI complex, was found to promote higher transfection (52%, ζ potential: ~2 mV) compared to the fully cationic pDNA-PEI polyplex (32%, ζ potential: ~9 mV) in 293T cells. Despite of high negative charge, several systems like spherical nucleic acids have been reported to possess high cellular internalization efficacy.[103] Likewise, PIC micelles, viz. DNA/pAsp(DET)/pAsp(DET-Aco) ternary polyplexes which were found to possess ζ potential of −40 mV at pH 7.4 were taken up by cells efficiently.[83] The transfection efficiency was also multi-fold superior compared to a commercial linear PEI based transfection agent, ExGen 500. Similarly, highly negatively-charged system like anionic DNA-g-PCL brushes also revealed high cellular uptake of siRNA or CRISPR/Cas-9 systems for gene silencing and editing, respectively.[108–109] These discussions on nanoparticle surface charge towards cellular uptake suggest that additional factors, apart from mere electrostatic interactions, are in play.
3.2. Direct uptake vs. receptor-mediated endocytosis
Cellular uptake of nanoparticles occurs via two pathways: (a) direct membrane penetration; or (b) endocytosis.[124] Direct cytosolic localization is highly desirable, yet a less understood process with the key driving force being the interactions between chemical functionalities on the surface of carriers and cell membrane.[125–126] By incorporating specific chemical moieties on nanoparticle surface, intracellular trafficking of materials could be promoted. Examples include direct conjugation or noncovalent complex formation of cell penetrating peptides (CPPs) to pass nucleic acid pharmaceuticals into the cytosol.[127] While the exact mechanism is unclear and still debated, few interesting hypotheses reported in literature are summarized next. First is the membrane-thinning model wherein positively charged CPP and negatively charged proteoglycans interact to generate a carpet-like structure followed by creation of permeable hole.[128] The second mechanism is about the pore formation where transportation happens via creation of toroidal or barrel-stave pores based on the mode of insertion of CPPs into the cell membrane.[128] The concentration of CPP also plays an important role in determining the cellular pathways. A higher concentration of CPP was found to be essential for direct translocation of cargo whereas, at low concentration, it moved through endocytic pathways followed by endosomal escape.[129] Recently, a synthetic mimic of poly-arginine, called as cell penetrating polydisulfide (CPD), had been introduced to significantly improve the transfection efficiency.[130–132] The polydisulfide backbone covalently interacts with cell surface thiols and enhances the cellular uptake. Upon internalization to cytosol, the polydisulfides get cleaved in presence of excess glutathione and the cargo is released. A βCD-CPD was shown to synergistically co-deliver miRNAs and campothecin (CPT) intracellularly in mammalian cell within 2 h without notable endosomal entrapment.[133]
However, a rather well-reported predominant uptake pathway for nanoparticles is via endocytosis which is broadly classified into two key pathways: phagocytosis and pinocytosis.[134] Phagocytosis is mostly observed in immune cells, such as macrophages, to engulf and digest cellular debris, foreign molecules, remove apoptotic cells and bacteria. Pinocytosis, on the other hand, is very common in most cells and is categorized into macropinocytosis, clathrin-mediated endocytosis, caveolae-mediated endocytosis and clathrin/caveolae independent endocytosis.[135] In micropinocytosis, the membrane wraps around and fuses with the particles to form a macropinosome to enter the cell. Whereas in clathrin-mediated endocytosis, the clathrin protein sheathes the nanoparticles to form “coated pit” and uptake occurs through the coated vesicles. Charge-reversing amphiphile/DNA lipoplexes were found to be uptaken mostly by micropinocytosis, as ~90% uptake was reduced upon the treatment of NaN3/2-deoxyglucose in CHO cells.[136] This is in contrast with the fact that cationic lipids are often observed to prefer clathrin-mediated pathways. For instance, PQDEA/pLUCI polyplexes followed a clathrin-dependent endocytic pathway.[67] When charge-converting nanoparticles were treated with different uptake inhibitors, chlorpromazine revealed significant inhibition in uptake suggesting a clathrin-dependent pathway. Similar observations were noted for B-PDEAEA polyplexes which also followed clathrin-mediated endocytosis pathway.[66] However, lipid coated crosslinked L-siP nanoassemblies were found to be predominantly uptaken by micropinocytosis in DU-145 cells, whereas in MDA-MB-231 and HeLa cell lines, both clathrin-dependent and micropinocytosis were operative.[53] Although caveolin-mediated endocytosis was observed for other nucleic acid carriers,[137] this specific pathway was rather rare for charge-converting nucleic acid carriers. Clathrin/caveolae independent endocytosis is not well understood and is most likely made up of several interdependent pathways.
Targeting through cell specific surface receptors is also possible and has served as a viable approach for nucleic acid delivery.[138–140] A folate ligand decorated decationized pHDP-PEG polyplex was developed to target folate overexpressed HeLa and OVCAR-3 cells.[56] Another report investigated PBA functionalized CrossPPA polyplex to target sialic acid found on many tumor cells (e.g., 4T1).[73] An integrin αvβ3, overexpressed during angiogenesis in endothelial cells, was utilized as target to deliver antiangiogenic gene in tumor environment via a polymerized lipid-based nanoparticle system.[141] Utilizing the preferred interactions between hyaluronic acid with the overexpressed CD44 receptor on cancer cells, cellular uptake was improved via receptor mediated uptake of HA-PEI/siRNA polyplexes.[76]
3.3. Factors influencing endosomal escape of nucleic acid delivery agents
Once internalized through the endocytosis pathway, drug carriers are entrapped inside the vesicle-like enclosure, endosome. Escape from the endosome might be the biggest barrier that many macromolecular delivery strategies encounter.[142–144] Although the mechanisms of escape are not fully understood and have been disputed in several articles, some popular theories of escape are pore formations, membrane fusion or destabilization, particle swelling, and proton sponge effect (umbrella theory).[142] Cationic carriers that contain amines like polyethyleneimine (PEI) were reported to cause endosomal buffering to induce proton sponge effect.[82, 145] The amines act as buffers and capture protons at low endosomal pH. To balance the charge, chloride ions are pumped into the endosome which causes osmotic imbalance and influx of water triggering the rupture of endosomes. Cationic carriers that do not contain amines are generally less efficient at escape in comparison to amine containing versions. PEI and HA-ATP conjugated polyplexes were found to escape endosome by enhanced buffering ability of phosphate group on ATP.[76] HA-PEI/FAM-siRNA polyplexes showed colocalized yellow fluorescence indicating endo/lysosomal entrapment, and after 6 h the separation of green and red channels revealed successful release of polyplexes from the trapped state.
Mere presence of positive charge may not influence proton sponge effect to favor endosomal escape. However, cationic carriers with charge-altering capability have dual advantages of having higher charge driven cellular uptake and endosomal escape feature due to the charge conversion. A self-immolative CART system (CART D13:A11) with a degradable backbone was found to escape endosome and showed eGFP expression after corresponding mRNA delivery, whereas the non-self-immolative variants (D13:G12 guanidinium-based and D13:Pip13 ammonium-based) failed to show efficient gene expression even though all of them had significant cellular uptake (Figure 11a).[60] While co-treated with concanamycin A, a preventor for endosomal acidification and CART system, the mRNA expression did not reduce significantly eliminating the endosomal buffering pathway for escape. Also, co-treatment with chloroquine, an endosomal acidification or buffering enhancer, did not have much influence suggesting a plausible osmotic rupture mechanism might be in place. The mixed-lipid CART variant also showed enhanced endosomolytic behavior in lymphocytes presumed to be due to the presence of two hydrophobic lipid tails.[64] In another example, PQDEA/pLUCI polyplexes induced osmotic swelling and resulted in lysosomal rupture in HeLa cells.[67] Although PQDEA had quaternary amines, those were presumably hydrolyzed in presence of esterases and formed tertiary amine containing PDEA intermediate with strong buffering capacity that enabled cytosolic release of DNA.
Figure 11.
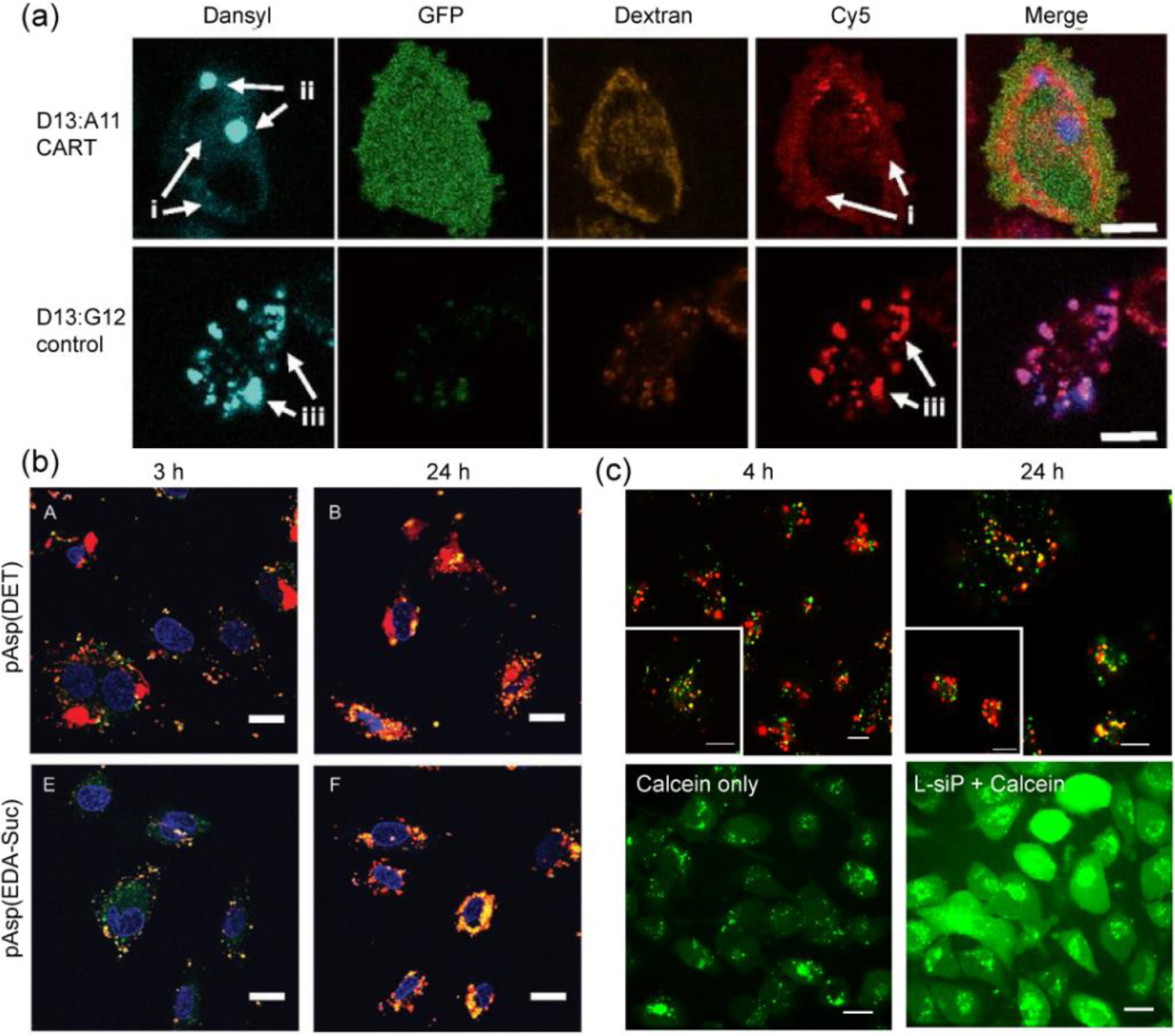
(a) Confocal microscopy of Cy5-mRNA treated HeLa cells stained with (TRITC)-Dextran4400 endosomal marker showing escape for D13:A11 CART system and colocalization for non-self-immolative control D13:G12 system, scale: 10 μm (reproduced with permission from reference 60, Copyright 2017 The National Academy of Sciences of the USA); (b) HUVEC cells transfected with Cy5-pDNA through pAsp(DET) and pAsp(EDA-Suc) polyplexes showing efficient endosomal escape after 24 h for only pAsp(DET) polyplexes, endosome/lysosomes were stained with Lysotracker green, scale: 20 μm (reproduced with permission from reference 83, Copyright 2008 Wiley-VCH Verlag GmbH & Co.); (c) top: Endosomal colocalization (after 4 h) and escape (after 24 h) of cy3-siRNA delivered via L-siP nanoassembly, endosome stained with Lysotracker blue (pseudo-colored as green), scale: 20 μm; bottom: Calcein assay showing release of calcein dye from endosomes to cytosol only in presence of L-siP nanoassembly, while calcein remain entrapped for control cells, scale: 10 μm (reproduced with permission from reference 53, Copyright 2019 American Chemical Society).
Another well explored mechanism of escape is via membrane destabilization, wherein nanoparticles are designed to expose complimentary functional groups to endosomal membrane for initiating disruption followed by release of nucleic acids into cytosol. Charge-converting pAsp(DET) polyplexes were reported to possess endosomolytic or membrane destabilizing capability releasing ~80% of encapsulated pDNA, whereas non-charge-converting polyplex pAsp(EDA-Suc) remained entrapped (Figure 11b).[83] The cytosolic release of Cy5 tagged pDNA was confirmed by confocal microscopy via colocalization studies with endosome-tracking dyes. Utilization of fusogenic lipid decorating the nanoparticle surface has often been found to enable endosomal escape operated via fusion with anionic membrane of endosome.[26] Based on this idea, L-siP15/1 nanoassembly was constructed with charge-converting polymer and siRNA encased in zwitterionic PEG and fusogenic DOPE lipids.[53] The nanoassemblies encapsulated with cy5-siRNA were found to be colocalized with lysotracker blue at 4 h in MDA-MB-231 cells, and separation of red and blue fluorescence channels were observed after 24 h (Figure 11c). Decrease in colocalization ratio from 0.65 at 4 h to 0.34 at 24 h indicated endosomal escape of siRNA, presumably governed by the fusogenicity of DOPE lipid. This was further confirmed by calcein assay where a diffused green fluorescence throughout the cytosol confirmed the endosomolytic behavior of L-siP particles (Figure 11c). Although anionic carriers do not possess specific mechanisms for efficient endosomal escape, this can be altered by the charge-reversal strategies to enhance evasion of endosome. Propiolic acid was utilized in a liposomal siRNA delivery system to impart a negative surface charge at neutral pH, which in acidic tumor environment reversed to positive for increased cell uptake.[94] Also, the initially entrapped siRNA was released in the cytosol after 6 h of incubation, as revealed from the colocalization studies with the endo-lysosome marker lysotracker. Although pHP-PEG polyplexes do not inherently possess efficient endosomal escape capability, those systems have showed promising gene silencing efficacy in presence of an endosome disrupting agent chloroquine,[54, 56] suggesting possible improvements via incorporating endosomolytic helper units, like, butyl methacrylate or peptide sequences.[57] In case of anionic nucleic acid based carriers such as DNA-g-PCL brushes, trafficking of siRNA was explored from early to late endosome and subsequent separation of siRNA was observed from the polymer-DNA delivery vector to release in cytosol within 24 h.[108] Although the process is similar to the spherical nucleic acid systems, further exploration is needed to reveal the exact mechanism of escape.
Table 2 summarizes the endocytosis and escape mechanisms for the charge converting systems discussed in this review.
Table 2.
Mechanism of cellular uptake and endosomal escape for charge-reversing nucleic acid delivery systems
| System Name | Encapsulated Nucleic Acid | Uptake Mechanism | Escape Mechanism | Release Stage | References |
|---|---|---|---|---|---|
| Poly(N-(N’-[(N”-cis-aconityl)-2-aminoethyl]-2-aminoethyl)aspartamide) [(pAsp(DET-Aco)]; Poly(N-[N’-(2-aminoethyl)-2-aminoethyl]aspartamide) [pAsp(DET)] | pDNA | Endocytosis | Endosome disruption (Membrane destabilization) | Endosome | Angew. Chem. Int. Ed. 2008, 407, 5163–5166 |
| Lipid-like amphiphile with ammonium headgroup and benzyl ester terminated lipophilic acyl chains | pDNA for β-gal, GFP encoding DNA | Macropinocytosis | - | - | J. Am. Chem. Soc. 2004, 126, 39, 12196–12197; Mol. Pharmaceutics 2011, 8, 758–766 |
| Poly[(2-acryloyl)ethyl(p-boronic acid benzyl)diethylammonium bromide] [B-PDEAEA] coated with DOPE and DC-Chol lipids | DNA | Clathrin-mediated endocytosis | Endosomal disruption by fusogenic DOPE (presumably) | Lysosome | Adv. Mater. 2016, 28, 1743–1752 |
| Polymer with quaternary amines carrying N-propionic 4-acetoxybenzyl ester | pLUCI pTRAIL |
Endocytosis | Endosomal disruption by fusogenic DOPE (presumably) | Lysosome | Adv. Mater. 2016, 28, 10613–10622 |
| Oligo(carbonate-b-α-amino ester) [CART] | EGFP mRNA |
Endocytosis | Osmotic rupture | Endosome | PNAS 114, no. 4 (2017): E448-E456. |
| Poly(N-[2-(acryloyloxy)ethyl]-N-[p-acetyloxyphenyl]-N,N-diethylammonium chloride) [PQDEA] | pLUCI pTRAIL |
Clathrin-mediated endocytosis | Osmotic swelling | Lysosome | Biomacromolecules 2018, 19, 2308–2319 |
| DNA-grafted polycaprolactone [DNA-g-PCL] | siRNA | Endocytosis | - | Early/Late Endosome | Angew. Chem. Int. Ed. 2018, 57, 3064–3068 |
| Hyaluronic acid- ethylenimine [HA-PEI] | siRNA | Receptor mediated endocytosis via HA-CD44 interaction | ATP dependent endosomolysis | Endo/Lysosome | Biomacromolecules 2018, 19, 3776–3787 |
| PEI-FPBA-Chol-Dopa micelle [PFCDM] | siRNA | Endocytosis | ATP dependent endosomolysis (presumably) | Endo/Lysosome | ACS Appl. Mater. Interfaces 2018, 10, 32026–32037 |
| Propiolic acid decorated cationic lipid LG2C14 | siRNA | Macropintocytosis and Clathrin-mediated pathways | Membrane fusion | Endo/Lysosome | Biomater. Sci., 2018, 6, 3075–3084 |
| Poly(pyridyldisulfide-dodecyl methacrylate) coated with DOPE-DSPE/PEG lipids [L-siP] | siRNA | Macropinocytosis and Clathrin-mediated endocytosis | Endosomal disruption by fusogenic DOPE | Endosome | ACS Appl. Mater. Interfaces 2019, 11, 24971–24983 |
| Poly[3-aminopropylmethacrylamide-co-N,N-(dimethylamino)ethylacrylate] [PAD] | DNA | Endocytosis | pH buffering and disruption of endosomal membrane | Endosome | ACS Omega 2020, 5, 9114–9122 |
| Polybutadiene-b-poly(methacrylic acid)-b-poly(2-(dimethylamino)ethyl methacrylate) [(BMAAD)] | pDNA | Endocytosis | Particle swelling followed by membrane disruption | Endosome | ACS nano, 2013, 7(11), pp.9621–9631 |
4. Outlook
4.1. Relevance in drug design and clinical importance
The goal next to designing and optimizing delivery vehicles with nucleic acids is to test through clinical trials before those can be cleared for usage in larger patient population. In this context, most of the charge-conversion strategies are still evolving and in the earlier stages of development. The most popular approach in clinical trials (see Table 3 consisting major nucleic acid clinical candidates) is to utilize ionizable lipid nanoparticles that would become cationic in acidic environment to increase cellular uptake and endosomal escape.[118, 146] In another scenario, nucleic acids are chemically modified in order to increase stability and are self-delivered.[23, 147] It was observed that cholesterol and lipophilic long chain fatty acid conjugated siRNAs were better in cellular uptake and gene silencing.[148] Moreover, cholesterol conjugated and HDL modified siRNAs showed 8–15 times higher apoB gene silencing compared to only cholesterol modified siRNA control. However, HDL modified siRNAs were rather delivered to various organs like kidney, liver, gut and other steroidogenic tissues through scavenger receptor SR-BI mediated uptake, whereas LDL modification only directed the siRNAs to liver. Similarly, another study reported an α-tocopherol conjugated siRNA for silencing apoB gene in the liver.[149] Examples of other ligands include oligo-arginine modified CD7 targeting siRNA for HIV-1 infection[150] and N-acetylgalactosamine (GalNAc) conjugated siRNAs that targets asialoglycoprotein receptor in liver.[151–152] Many of the ionizable LNP therapies were terminated during clinical trials for immunogenic issues (targeting ApoB for hypercholesterolemia-NCT00927459, Ebola-NCT02041715), showing room for further improvements. Here, we will majorly focus on the systems that are designed to contain delivery vehicles and are being considered in clinical trials.
Table 3.
A list of major nucleic acid candidates being evaluated in clinical trials
| Nucleic Acid | Name | Company | Nanoparticle System/Formulation | Disease | Clinical Trial Phase | ID Number |
|---|---|---|---|---|---|---|
| ASO | Pelacarsen (AKCEA-APO(a)-LRx) | Ionis, Akcea, and Novartis | Gen 2+ (LICA) | Hyperlipoproteinemia(a) and cardiovascular disease | Phase 2/3 | NCT03070782 |
| ASO | AKCEA-TTR-LRx | Ionis and Akcea | Gen 2+ (LICA) | Hereditary transthyretin-mediated amyloid polyneuropathy (ATTR) | Phase 3 | NCT04136184 |
| ASO | Vupanorsen | Ionis, Akcea, and Pfizer | Gen 2+ (LICA) (3 GalNAcs are attached on the 5’ end of modified ASO) | Cardiovascular disease | Phase 2 | NCT04459767 |
| ASO | AKCEA-APOCIII-LRx | Ionis and Akcea | Gen 2+ (LICA) | Cardiovascular disease/familial chylomicronemia syndrome | Phase 2 | NCT03385239 |
| ASO | IONIS-FB-LRx | Ionis and Roche | Gen 2+ (LICA) | IgA nephropathy | Phase 2 | NCT04014335 |
| ASO | IONIS-AGT-LRx | Ionis | Gen 2+ (LICA) | Hypertension | Phase 2 | NCT04083222 |
| ASO | IONIS-FXI-LRx | Ionis and Bayer | Gen 2+ (LICA) | Clotting disorders | Phase 2 | NCT03582462 |
| ASO | IONIS-GHR-LRx | Ionis | Gen 2+ (LICA) | Acromegaly | Phase 2 | NCT03548415 |
| ASO | IONIS-FB-LRx | Ionis and Roche | Gen 2+ (LICA) | Primary IgA nephropathy | Phase 2 | NCT04014335 |
| ASO | IONIS-FB-LRx | Ionis and Roche | Gen 2+ (LICA) | Primary IgA nephropathy | Phase 2 | NCT04014335 |
| ASO | IONIS-PKK-LRx | Ionis | Gen 2+ (LICA) | Hereditary angiodema | Phase 2 | NCT04030598 |
| ASO | IONIS-TMPRSS6-LRx | Ionis | Gen 2+ (LICA) | β-Thalassemia | Phase 2 | NCT04059406 |
| ASO | IONIS-GCGRRx | Ionis and Suzhou-Ribo | Gen 2+ (modified ASO) | Diabetes | Phase 2 | no results |
| ASO | Tofersen | Ionis and Biogen | Gen 2+ (modified ASO) | Amyotrophic lateral sclerosis (ALS) | Phase 3 | NCT02623699 |
| ASO | Tominersen | Ionis and Roche | Gen 2+ (modified ASO) | Huntington’s disease | Phase 3 | NCT03842969 |
| ASO | IONIS-C9Rx (BIIB078) | Ionis and Biogen | Gen 2+ (modified ASO) | Amyotrophic lateral sclerosis (ALS) | Phase 1/2 | NCT03626012 |
| ASO | IONIS-MAPTRx (BIIB080) | Ionis and Biogen | Gen 2+ (modified ASO) | Alzheimer’s disease & frontotemporal degeneration | Phase 2 | NCT03186989 |
| ASO | ION859 (BIIB094) | Ionis and Biogen | Gen 2+ (modified ASO) | Parkinson’s disease | Phase 1/2 | NCT03976349 |
| ASO | ION464 (BIIB101) | Ionis and Biogen | Gen 2+ (modified ASO) | Multiple system atrophy & Parkinson’s disease | Phase 1/2 | NCT04165486 |
| ASO | IONIS-HBVRx | Ionis and GSK | Gen 2+ (modified ASO) | Hepatitis B | Phase 2 | NCT02981602 |
| ASO | Tominersen (RO7234292) | Hoffmann-La Roche and Ionis | Gen 2+ (modified ASO) | Huntington’s disease | Phase 3 | NCT03761849 |
| ASO | Tominersen (RO7234292) | Hoffmann-La Roche and Ionis | Gen 2+ (modified ASO) | Huntington’s disease | Phase 3 | NCT03761849 |
| ASO | IONIS-DNM2–2.5Rx | Ionis and Dynacure | Gen 2.5 (modified ASO) | Centronuclear myopathy | Phase 2 | no results |
| ASO | IONIS-AR-2.5R | Ionis and Suzhou-Ribo | Gen 2.5 (modified ASO) | Prostate cancer | Phase 2 | NCT02144051 |
| ASO | Danvatirsen | Ionis and AstraZeneca | Gen 2.5 (modified ASO) | Cancer | Phase 2 | NCT02983578 |
| ASO | ION357/QR1123 | Ionis and ProQR Therapeutics | Gen 2.5 (modified ASO) | Autosomal dominant retinitis pigmentosa | Phase 1/2 | NCT04123626 |
| ASO (AON) | QR-313 | Wings Therapeutics | Hydrogel | Epidermolysis bullosa dystrophica | Phase 1/2 | NCT03605069 |
| ASO | BP1001 | Bio-Path Holdings Inc. | Liposome (DOPC based, neutrally charged) | Acute myeloid leukemia | Phase 2 | NCT02781883 |
| ASO | Sepofarsen (QR-110) | ProQR Therapeutics | - | Leber’s congenital amaurosis | Phase 2/3 | NCT03913143 |
| ASO | GTX-102 | GeneTX Biotherapeutics LLC and Ultragenyx Pharmaceutical Inc | - | Angelman syndrome | Phase 1/2 | NCT04259281 |
| ASO | QR-010 | ProQR Therapeutics | - | Cystic fibrosis | Phase 1/2 | NCT02532764 |
| ASO | WVE-120102 | Wave Life Sciences Ltd. | - | Huntington’s disease | Phase 1/2 | NCT03225846 |
| ASO | WVE-120101 | Wave Life Sciences Ltd. | - | Huntingtons disease | Phase 1/2 | NCT03225833 |
| ASO | Inotersen- IONIS-TTRRX/ISIS-420915 | Ionis, Brigham and Women’s Hospital | - | TTR Amyloidosis | Phase 2/3 | NCT03702829 |
| ASO | QR-421a | ProQR Therapeutics | - | Retinitis Pigmentosa, Usher syndrome type 2 | Phase 1/2 | NCT03780257 |
| ASO | STK-001 | Stoke Therapeutics | - | Dravet syndrome | Phase 1/2 | NCT04442295 |
| siRNA | ARO-AAT | Arrowhead Pharmaceuticals | TRiM (GalNAc-siRNA conjugates) | Alpha 1 liver disease | Phase 2/3 | NCT03945292 |
| siRNA | ARO-APOC3 | Arrowhead Pharmaceuticals | TRiM (GalNAc-siRNA conjugates) | Hypertrglyceridemia, Familial chylomicronemia | Phase 1/2 | NCT03783377 |
| siRNA | ARO-ANG3 | Arrowhead Pharmaceuticals | TRiM (GalNAc-siRNA conjugates) | Dyslipidemia | Phase 1/2 | NCT03747224 |
| siRNA | ARO-HSD | Arrowhead Pharmaceuticals | (TRiM)-ligand conjugate | Non-alcoholic steatohepatitis (NASH) | Phase 1/2 | NCT04202354 |
| siRNA | Tivanisiran (SYL1001) | Sylentis | Self-delivered (Hybridized single strand complementary RNA) | Dry eye disease | Phase 3 | NCT03108664 |
| siRNA | BMT101 | Hugel | Self-delivered | Hypertrophic scar | Phase 2 | NCT04012099 |
| siRNA | Inclisiran | Novartis Pharmaceuticals | GalNAc conjugate (Modified siRNA conjugated to triantennary N-acetylgalactosamine carbohydrates) | Homozygous familial hypercholesterolemia | Phase 3 | NCT03851705 |
| siRNA | Lumasiran | Alnylam Pharmaceuticals | GalNAc conjugate (ESC) | Primary hyperoxaluria type 1 | Phase 3 (commercial) | NCT03681184 |
| siRNA | DCR-PHXC | Dicerna Pharmaceuticals | GalNAc conjugate (GalXC) | Primary hyperoxaluria type 1 and 2 | Phase 2 | NCT03847909 |
| siRNA | Fitusiran | Genzyme (Sanofi) and Alnylam | GalNAc conjugate (N-acetylgalactosamine conjugation) | Hemophelia A, B; Rare bleeding diseases | Phase 3 | NCT03417245 |
| siRNA | Vutrisiran | Alnylam Pharmaceuticals | GalNAc conjugate (ESC) | Transthyretin amyloidosis (ATTR) | Phase 3 | NCT03759379 |
| siRNA | Cemdisiran | Alnylam Pharmaceuticals | GalNAc conjugate (N-acetylgalactosamine conjugation) | IgA nephropathy (IgAN), Berger disease, Glomerulonephritis, IgA | Phase 2 | NCT03841448 |
| siRNA | siG12D LODER | Silenseed Ltd | Polymer matrix (poly (lactic-co-glycolic) acid (PLGA)) | Locally advanced pancreatic cancer | Phase 2 | NCT01676259 |
| mRNA | mRNA-1273 | Moderna | Ionizable LNP (SM-102, PEG-2000-DMG, DSPC, Cholesterol) | SARS-COV-2 | Phase 3 | NCT04405076 |
| mRNA | mRNA-4157 | Moderna | Ionizable LNP | Melanoma | Phase 2 | NCT03897881 |
| mRNA | mRNA-1647 | Moderna | Ionizable LNP | Cytomegalovirus | Phase 2 | NCT04232280 |
| mRNA | mRNA 2416 | Moderna | Ionizable LNP | Relapsed/refractory solid tumor malignancies, lymphoma | Phase 1/2 | NCT03323398 |
| mRNA | mRNA-3704 | Moderna | Ionizable LNP | Methylmalonic acidemia | Phase 1/2 | NCT03810690 |
| mRNA | mRNA 2416 | Moderna | Ionizable LNP | Relapsed/Refractory solid tumor malignancies, Lymphoma | Phase 1/2 | NCT03323398 |
| mRNA | mRNA-3704 | Moderna | Ionizable LNP | Methylmalonic acidemia | Phase 1/2 | NCT03810690 |
| mRNA | BI 1361849 (CV9202) | CureVac, Boehringer Ingelheim, Ludwig Cancer Research | Combination of modified mRNA & protamine complexed mRNA | Metastatic non-small cell lung cancer | Phase1/2 | NCT03164772 |
| mRNA | W_pro1 | BioNTech RNA Pharmaceuticals | Lipoplex | Metastatic castration resistant prostate cancer | Phase1/2 | NCT04382898 |
| mRNA | PF-07302048 (BNT162) | Pfizer and BioNTech | LNP (ALC-0315, ALC-0159, DSPC, & cholesterol) | SARS-CoV-2 (COVID-19) | Phase 2/3 | NCT04368728 |
| DNA | AD26.COV2.S | Johnson & Johnson | Adenovirus serotype 26 vector | SARS-CoV-2 | Phase 3 | NCT04614948 |
| DNA | AZD1222(ChAdOx1 nCoV-19) | AstraZeneca, Oxford University | Adenovirus vector | SARS-CoV-2 | Phase 3 | NCT04516746 |
| DNA | Ad5-nCoV | CanSino Biologics | Adenovirus type-5 vector | SARS-CoV-2 | Phase 3 | NCT04540419 |
| DNA | PF-06838435 (SPK-9001) | Pfizer and Spark Therapeutics | Adeno-associated viral vector (AAV-Spark100) | Hemophilia B | Phase 3 | NCT03861273 |
| DNA | SPK-8011 | Spark Therapeutics | Adeno-associated viral vector (AAV-Spark200) | Hemophilia A | Phase 1/2 | NCT03003533 |
| DNA | SPK-3006 | Spark Therapeutics | Adeno-associated viral vector | Pompe disease | Phase 1/2 | NCT04093349 |
| DNA | AAV2/8-LSPhGAA | Asklepios Biopharmaceuticals | Adeno-associated viral vector | Pompe disease | Phase 1/2 | NCT03533673 |
LICA: Ligand Conjugated Antisense, DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine, TRiM: Targeted RNAi Molecule, ESC: Enhanced Stability Chemistry, SM-102: heptadecan-9-yl-8-((2-hydroxyethyl)(6-oxo-6-(undecyloxy)hexyl)amino)octanoate, PEG-2000-DMG: 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000, DSPC: 1,2 distearoyl-sn-glycero-3-phosphocholine, LNP: Lipid Nanoparticle, ALC-0315: ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate), ALC-0159: 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide
4.1.1. Ionizable lipids and polymeric nanoparticles for nucleic acids therapeutics in clinical trials
Popular clinical candidates for nucleic acid formulations comprise LNPs made out of ionizable or cationic lipids whose chemical designs vary depending on the type of nucleic acid and formulation requirements. Alnylam Pharmaceuticals and Arbutus Biopharma have established three generations of LNP-based systems with DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane), DLin-MC3-DMA ((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate) and L319 (di((Z)-non-2-en-1-yl) 9-((4-(dimethylamino) butanoyl) oxy) heptadecanedioate) as the key lipid components for safe and effective delivery.[153] The first successful RNAi drug on the market was Onpattro consisting of an siRNA that had 2’-O-methyl modifications and a 2’-deoxythymidine dinucleotide overhang on the 3’ end.[154] Onpattro’s delivery system is an ionizable lipid nanoparticle formulation that includes DLin-MC3-DMA with cholesterol, DSPC and PEG-C-DMG.[155] The designed lipid has a pKa ~6.4 so that it is cationic at acidic pH and apparently neutral at physiological pH to avoid charge-based toxicity. Onpattro downregulates the mRNA that synthesizes transthyretin protein to treat hereditary disease transthyretin-mediated amyloidosis (hATTR) and was a major milestone for RNA therapeutics.[156] Local Drug EluteR or LODER, introduced by Silenseed, is a biodegradable polymeric delivery vehicle consisting a copolymer of poly(lactic-co-glycolic) acid (PLGA) that can slowly release the RNAi drug locally.[157] LODER is used to produce siG12D-LODER system that is in phase 2 clinical trial to help target KRAS in pancreatic cancer. The platform is also being developed to aid in the delivery of other RNA therapeutics. While LNP-based formulations are not limited to siRNA delivery, recent promising results from the clinical trials on COVID-19 vaccines utilized LNP formulations comprising mRNAs. Importantly, the recent development of mRNA-1273 vaccine against novel COVID-19 is based on ionizable LNP formulations which contains SM-102 (likely to be heptadecan-9-yl-8-((2-hydroxyethyl)(6-oxo-6-(undecyloxy)hexyl)amino)octanoate), PEG2000-DMG (1,2-dimyristoyl-rac-glycero3-methoxypolyethylene glycol-2000) and DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine).[158] Pfizer and BioNTech have also used mRNA-LNP formulation for the COVID-19 vaccine utilizing lipid components ALC-0315, ALC-0159 and 1,2-distearoyl-sn-glycero-3-phosphocholine (DPSC).[159] These lipids contain tertiary amines which become positively charged under physiological pH to deliver better efficacy in comparison to the quaternary amines. Both LNP-based mRNA vaccines developed by Moderna and Pfizer/BioNTech target the perfusion stabilized spike protein for SARS-CoV-2.[160–162] Alongside, several other vaccine candidates for SARS-CoV-2 currently in clinical trials consist of ionizable lipids, specifically those based on the RNA platform.[163–164] Similarly, mRNA-1893 vaccine from Moderna for the Zika virus is also based on LNP formulation and is currently in Phase 1 clinical trial. The LNP is composed of an ionizable lipid, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol and PEG-lipid.[165] Another promising example includes BP1001 or Prexigebersen, an antisense drug encapsulated in a neutrally charged liposome, to target growth factor receptor bound protein 2 (Grb2) to potentially kill leukemia cells.[166]
4.1.2. Ligand conjugates for nucleic acid therapeutics in clinical trials
Covalent conjugation of ligands to the nucleic acids has resulted many promising clinical drug candidates. Arrowhead had developed a delivery system, named as dynamic polyconjugates (DPC), featuring a membrane-active polymer capable of evading acidic endosomes and releasing nucleic acid via a targeted delivery mechanism. The DPC was composed of a nucleic acid, a shielding agent (PEG), and a targeting ligand (N-acetylgalactosamine, NAG) – all of which were reversibly conjugated to a poly(vinyl ether) polymer consisting butyl and amino vinyl ethers.[167] While both shielding agent and hepatocyte targeting ligand were connected via pH-sensitive carboxylated dimethyl maleic acid (CDM) bonds, the siRNA was conjugated via reversible disulfide linkage to the polymer backbone. The pH-sensitive CDM moieties were cleaved under acidic endo-lysosomal pH leading to the generation of positive charge that helped to disrupt endosomal membrane via proton-sponge mechanism. Subsequently, the siRNA drug was delivered in cytosol via disruption of disulfide bond. Moving forward, Arrowhead had developed a better alternative, called TRiM (Targeted RNAi Molecule) technology[168] with enhanced pharmacokinetic performance and built-in targeting ligands, viz., GalNAc, αvβ6 ligands and RGD peptides to deliver nucleic acids in hepatocytes, lung epithelial cells and cancer cells, respectively.
Recently, GalNAc - a ligand for the asialoglycoprotein receptor expressed predominantly on the surface of hepatocytes have become one of the most promising methods for the delivery of nucleic acids.[151–152, 169] Typically, nucleic acids are covalently conjugated to the trivalent or tetravalent GalNAc moieties via an optimized linker. Alnylam’s second approved siRNA drug, Givlaari, contains GalNAc conjugate wherein the siRNA drug is attached to a tetravalent GalNAc to target hepatocytes. The drug reduces ALAS1 to treat acute intermittent porphyria.[170] Many other companies e.g., Dicerna, Arbutus and Ionis have also embraced the technology for numerous nucleic acid cargoes.[171–173] Notably, the RNAi technology GalXC™ by Dicerna utilized multivalent GalNAc functionalities attached to the dicer-substrate siRNA (DsiRNA) generating a tetraloop configuration.[174]
Ionis Pharmaceuticals has developed a nucleic acid delivery platform that consists of modified ASO and Ligand Conjugated Antisense or LICA technology. The LICA technology has a ligand that is able to target a cellular receptor with high specificity to deliver an antisense drug. The Ionis pipeline furnished Gen 2+, Gen 2+ LICA, Gen 2.5, and Gen 2.5 LICA consisting phosphorothioate, methoxyethyl (MOE) gapmer and 2’,4’-constrained ethyl (cEt) gapmer chemistries for ASO modifications.[175] The 2’-MOE (Gen 2) modifications increased the binding affinity of ASO, whereas the cEt modifications (Gen 2.5) favored hybridization with complementary RNA or pre-mRNA sequences.[175] Phosphorothioate modifications were also used to protect the ASO from nucleases and to improve cellular uptake efficacy.[176] This technology was utilized in Tominersen in a partnership with Roche to combat Huntington’s disease and is currently in phase 3 clinical trials.[176] Akcea Therapeutics had teamed up with Ionis Pharmaceuticals to use their LICA technology to deliver the antisense drug (AKCEA-TTR-LRx) for the treatment of TTR amyloidosis.[177] Empowered by similar technology, AKCEA-APOCIII-LRx drug was used for cardiovascular diseases to target APOC3 mRNA which inhibited apoC-III production reducing triglyceride levels (also see Table 3).[178] Latest technologies including Gen 2.5 LICA from Ionis consist of an ASO with phosphothiorate constrained cEt modifications and conjugated to GalNAc.[175, 179] Other ASO drugs with Gen 2.5 technology includes IONIS-DNM2–2.5Rx (for centronuclear myopathy) and IONIS-ENAC-2.5Rx (for cystic fibrosis) and ION839 (for non-alcoholic steatohepatitis, NASH).[180–182]
Delivery aids in the form of ionizable lipids, degradable polymer matrix or ligand conjugates are filling up the requirements of the rigorous clinical assessments. Though a significant number of clinical candidates are being evaluated, as reported in Table 3, the delivery vectors only satisfy the bare-minimum requirements for a successful drug candidate. In this prospect, charge-conversion strategies could serve to mitigate some of the stumbling blocks which are briefly discussed in the next section.
4.2. Pitfalls and scope of charge-conversion approaches
It has been well demonstrated that the removal of positive charge from the nanocarriers results in improved circulation time and biodistribution,[55] which are beneficial for utilization of these nanocarriers as a potential candidate for in vivo gene delivery applications. However, structural optimization of these carriers might be necessary in order to improve the efficiency of these systems. One of the reasons for lower transfection efficiency of non-cationic polyplexes might be due to compromised release profile of encapsulated nucleic acids under physiological conditions. Some of the promising strategies to address this might be by incorporating charge converting moieties in the nanostructures, as discussed in this review. The conversion of the surface charge from positive to negative can enhance forced release of nucleic acids in the intracellular compartments which in turn improves the gene transfection efficiency of the systems.[66–67] Another important reason of traditionally using polycations for nucleic acid delivery is due to their membrane disrupting properties, which can be directly correlated with the mechanism of transition through plasma and nuclear membranes.[183] Thus, in order to maintain the safety promises it is important that the nanocarriers lack membrane damage properties, which can help in minimizing cellular toxicity. However, this could seriously hamper the ability of these nanocarriers to escape the endosome in some cases, which can significantly lower the gene delivery efficiency of these systems.[82, 184] One elegant strategy to encounter this problem could be to use neutral lipids to encase the polymeric nanocarriers.[53] As specific lipids such as DOPE are known to have fusogenic properties,[185] it can significantly enhance the cellular uptake as well as endosomal escape of the nanocarriers improving desired gene expression or silencing efficacy.
As with any nucleic acid drug candidate for clinical consideration, the major concerns are still related to the transfection efficiency and biosafety of the systems.[186] Charge-reversing strategies adopted so far, logically the ones converting from cationic to neutral or anionic species, have demonstrated promising results by lowering the toxicity of the delivery vehicle and have shown efficient gene transfection in both in vitro and in vivo experiments. However, the biological safety issues at the sub-cellular levels are far from complete in understanding. Charge-alteration strategies which depend on specific environmental stimuli, such as pH, are associated with the risk of cargo leakage at off-target locations due to heterogeneous biological milieu. Similarly, anionic carrier materials converting to cationic species might have the possibility of reintroducing charge driven toxicity.
5. Conclusion
Although nucleic acid-based therapeutics are being developed over a couple of decades, this field has been rejuvenated after recent approvals of RNAi based medicines, potentially bolstering the strength of such therapeutic platform. Completion of the human genome project has tremendously helped in understanding the physical and functional perspectives of genetic information.[187–188] This, in turn, has helped to decipher the molecular mechanisms of complex diseases and led to discoveries of novel therapeutics. As confidence has grown, prediction of nucleic acid-based medication is becoming a reality. Though encouraging, the rate limiting step in the development of such drugs remain the same, delivery to the target site in active form. Addressing this, many ground-breaking research efforts have proposed unique carrier designs to transport nucleic acid cargoes, that majorly rely on the charge based molecular encapsulation strategies. This review specifically highlights the interesting recent developments of charge-switching approaches. As nucleic acids are negatively charged, a logical idea is to design complementary positively charged materials to effectively condense nucleic acids and produce well-defined stable complexes. However, existence of such cationic charge in the delivery vector poses a serious threat due to its interaction with the negatively charged serum and cellular components, increasing cytotoxicity. In contrast, positively charged materials also have beneficial effect in endosomal escape, one of the serious bottlenecks for any biologics-based drug development. Thus, the nucleic acid carrier designs are typically influenced by the fine balance between efficacy, stability and cytotoxicity.
However, recent developments of strategies that consider alteration of charges through built-in molecular mechanisms in the drug carriers can address this unmet need. The concept of charge alteration for nucleic acid delivery can be grossly divided into two categories, majorly focusing on the starting charge of the carrier. While charge conversion from cationic to neutral or anionic addresses the alarming concerns of cytotoxicity, the reverse approach of anionic to cationic conversion introduces the flexibility to create the cationic charge on demand for enhancing endosomal escape. This review summarizes several such interesting strategies with a particular focus on the underlying mechanisms of the charge conversion processes. In the field of nucleic acid drug delivery agents, charge reversal strategies are comparatively new and thus are still in the early developmental stages. Clinical translation of such methods would require considerable research investigations involving in vivo studies of toxicity, efficacy and immunogenicity with appropriate disease models. Acknowledging the optimistic early research results, focus is currently shifting from conventional cationic charge-based complexation approaches to alternate molecular designs accommodating demands of complex human diseases with specific target areas of action. In summary, charge-alteration based approaches have the potential to address the evolving needs of nucleic acid-based therapeutics establishing much smarter and fail-safe drug delivery platforms.
Acknowledgements
We are thankful for the support from the U.S. Army Research Office (W911NF-15-1-0568) and the National Institutes of Health (GM-136395, T32GM108556, T32GM008515).
References
- [1].Opalinska JB, Gewirtz AM, Nat. Rev. Drug Discovery 2002, 1, 503–514. [DOI] [PubMed] [Google Scholar]
- [2].Mitragotri S, Burke PA, Langer R, Nat. Rev. Drug Discovery 2014, 13, 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sridharan K, Gogtay NJ, Br. J. Clin. Pharmacol 2016, 82, 659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang F, Zuroske T, Watts JK, Nat. Rev. Drug Discovery 2020, 19, 441–442. [DOI] [PubMed] [Google Scholar]
- [5].Dammes N, Peer D, Trends Pharmacol. Sci 2020, 41, 755–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Saumet A, Mathelier A, Lecellier CH, Biomed Res. Int 2014, 2014, 642916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Parsel SM, Grandis JR, Thomas SM, Oncogene 2016, 35, 3217–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yamakawa K, Nakano-Narusawa Y, Hashimoto N, Yokohira M, Matsuda Y, Int. J. Mol. Sci 2019, 20, 4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Frederickson RM, Mol. Ther 2005, 12, 775–776. [DOI] [PubMed] [Google Scholar]
- [10].Tan X, Jia F, Wang P, Zhang K, J. Controlled Release 2020, 323, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Egli M, Manoharan M, Acc. Chem. Res 2019, 52, 1036–1047. [DOI] [PubMed] [Google Scholar]
- [12].Reineke TM, Raines RT, Rotello VM, Bioconjugate Chem. 2019, 30, 261–262. [DOI] [PubMed] [Google Scholar]
- [13].Merkel O, Amiji M, in Advances and Challenges in the Delivery of Nucleic Acid Therapeutics (Volume 1), Future Medicine, 2015, pp. 2–4. [Google Scholar]
- [14].Scholz C, Wagner E, J. Controlled Release 2012, 161, 554–565. [DOI] [PubMed] [Google Scholar]
- [15].Spagnou S, Miller AD, Keller M, Biochemistry 2004, 43, 13348–13356. [DOI] [PubMed] [Google Scholar]
- [16].Blakney AK, Yilmaz G, McKay PF, Becer CR, Shattock RJ, Biomacromolecules 2018, 19, 2870–2879. [DOI] [PubMed] [Google Scholar]
- [17].Lächelt U, Wagner E, Chem. Rev 2015, 115, 11043–11078. [DOI] [PubMed] [Google Scholar]
- [18].Jiang ZW, Thayumanavan S, Adv. Ther 2020, 3, 1900206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jiang L, Vader P, Schiffelers RM, Gene Ther. 2017, 24, 157–166. [DOI] [PubMed] [Google Scholar]
- [20].Van Bruggen C, Hexum JK, Tan Z, Dalal RJ, Reineke TM, Acc. Chem. Res 2019, 52, 1347–1358. [DOI] [PubMed] [Google Scholar]
- [21].Chen CM, Yang ZJ, Tang XJ, Med. Res. Rev 2018, 38, 829–869. [DOI] [PubMed] [Google Scholar]
- [22].Gallas A, Alexander C, Davies MC, Puri S, Allen S, Chem. Soc. Rev 2013, 42, 7983–7997. [DOI] [PubMed] [Google Scholar]
- [23].Khvorova A, Watts JK, Nat. Biotechnol 2017, 35, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bramsen JB, Laursen MB, Nielsen AF, Hansen TB, Bus C, Langkjaer N, Babu BR, Hojland T, Abramov M, Van Aerschot A, Odadzic D, Smicius R, Haas J, Andree C, Barman J, Wenska M, Srivastava P, Zhou C, Honcharenko D, Hess S, Muller E, Bobkov GV, Mikhailov SN, Fava E, Meyer TF, Chattopadhyaya J, Zerial M, Engels JW, Herdewijn P, Wengel J, Kjems J, Nucleic Acids Res. 2009, 37, 2867–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Crooke ST, Wang S, Vickers TA, Shen W, Liang XH, Nat. Biotechnol 2017, 35, 230–237. [DOI] [PubMed] [Google Scholar]
- [26].Nguyen J, Szoka FC, Acc. Chem. Res 2012, 45, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Verdera HC, Kuranda K, Mingozzi F, Mol. Ther 2020, 28, 723–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nayak S, Herzog RW, Gene Ther. 2010, 17, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Roy R, Jerry DJ, Thayumanavan S, Biomacromolecules 2009, 10, 2189–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Durymanov M, Reineke J, Front. Pharmacol 2018, 9, 00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bonner DK, Zhao XY, Buss H, Langer R, Hammond PT, J. Controlled Release 2013, 167, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sizovs A, Xue L, Tolstyka ZP, Ingle NP, Wu Y, Cortez M, Reineke TM, J. Am. Chem. Soc 2013, 135, 15417–15424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Majumder P, Bhunia S, Bhattacharyya J, Chaudhuri A, J. Controlled Release 2014, 180, 100–108. [DOI] [PubMed] [Google Scholar]
- [34].Majumder P, Bhunia S, Chaudhuri A, Chem. Commun 2018, 54, 1489–1492. [DOI] [PubMed] [Google Scholar]
- [35].Tan Z, Jiang Y, Zhang W, Karls L, Lodge TP, Reineke TM, J. Am. Chem. Soc 2019, 141, 15804–15817. [DOI] [PubMed] [Google Scholar]
- [36].Wu CN, Li JH, Wang WD, Hammond PT, ACS Nano 2018, 12, 6504–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ball RL, Hajj KA, Vizelman J, Bajaj P, Whitehead KA, Nano Lett. 2018, 18, 3814–3822. [DOI] [PubMed] [Google Scholar]
- [38].Hunter AC, Adv. Drug Delivery Rev 2006, 58, 1523–1531. [DOI] [PubMed] [Google Scholar]
- [39].Frohlich E, Int. J. Nanomed 2012, 7, 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zheng M, Pavan GM, Neeb M, Schaper AK, Danani A, Klebe G, Merkel OM, Kissel T, ACS Nano 2012, 6, 9447–9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kurrikoff K, Veiman KL, Kunnapuu K, Peets EM, Lehto T, Parnaste L, Arukuusk P, Langel U, Sci. Rep 2017, 7, 17056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Verhoef JJF, Anchordoquy TJ, Drug Delivery Transl. Res 2013, 3, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ikeda Y, Nagasaki Y, J. Appl. Polym. Sci 2014, 131, 40293. [Google Scholar]
- [44].Lu X, Zhang K, Nano Res. 2018, 11, 5519–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hatakeyama H, Akita H, Harashima H, Adv. Drug Delivery Rev 2011, 63, 152–160. [DOI] [PubMed] [Google Scholar]
- [46].Li Y, Kroger M, Liu WK, Biomaterials 2014, 35, 8467–8478. [DOI] [PubMed] [Google Scholar]
- [47].Adli M, Nat. Commun 2018, 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang H, La Russa M, Qi LS, Annu. Rev. Biochem 2016, 85, 227–264. [DOI] [PubMed] [Google Scholar]
- [49].Jiang ZW, Cui W, Prasad P, Touve MA, Gianneschi NC, Mager J, Thayumanavan S, Biomacromolecules 2019, 20, 435–442. [DOI] [PubMed] [Google Scholar]
- [50].Jiang ZW, Cui W, Mager J, Thayumanavan S, Ind. Eng. Chem. Res 2019, 58, 6982–6991. [Google Scholar]
- [51].Molineux IJ, Panja D, Nat. Rev. Microbiol 2013, 11, 194–204. [DOI] [PubMed] [Google Scholar]
- [52].Dimitrov DS, Nat. Rev. Microbiol 2004, 2, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dutta K, Bochicchio D, Ribbe AE, Alfandari D, Mager J, Pavan GM, Thayumanavan S, ACS Appl. Mater. Interfaces 2019, 11, 24971–24983. [DOI] [PubMed] [Google Scholar]
- [54].Novo L, van Gaal EV, Mastrobattista E, van Nostrum CF, Hennink WE, J. Controlled Release 2013, 169, 246–256. [DOI] [PubMed] [Google Scholar]
- [55].Novo L, Rizzo LY, Golombek SK, Dakwar GR, Lou B, Remaut K, Mastrobattista E, van Nostrum CF, Jahnen-Dechent W, Kiessling F, Braeckmans K, Lammers T, Hennink WE, J. Controlled Release 2014, 195, 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Novo L, Mastrobattista E, van Nostrum CF, Hennink WE, Bioconjugate Chem. 2014, 25, 802–812. [DOI] [PubMed] [Google Scholar]
- [57].Novo L, Takeda KM, Petteta T, Dakwar GR, van den Dikkenberg JB, Remaut K, Braeckmans K, van Nostrum CF, Mastrobattista E, Hennink WE, Mol. Pharmaceutics 2015, 12, 150–161. [DOI] [PubMed] [Google Scholar]
- [58].Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA, Himansu S, Schafer A, Ziwawo CT, DiPiazza AT, Dinnon KH, Elbashir SM, Shaw CA, Woods A, Fritch EJ, Martinez DR, Bock KW, Minai M, Nagata BM, Hutchinson GB, Wu K, Henry C, Bahl K, Garcia-Dominguez D, Ma L, Renzi I, Kong WP, Schmidt SD, Wang L, Zhang Y, Phung E, Chang LA, Loomis RJ, Altaras NE, Narayanan E, Metkar M, Presnyak V, Liu C, Louder MK, Shi W, Leung K, Yang ES, West A, Gully KL, Stevens LJ, Wang N, Wrapp D, Doria-Rose NA, Stewart-Jones G, Bennett H, Alvarado GS, Nason MC, Ruckwardt TJ, McLellan JS, Denison MR, Chappell JD, Moore IN, Morabito KM, Mascola JR, Baric RS, Carfi A, Graham BS, Nature 2020, 586, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hassett KJ, Benenato KE, Jacquinet E, Lee A, Woods A, Yuzhakov O, Himansu S, Deterling J, Geilich BM, Ketova T, Mihai C, Lynn A, McFadyen I, Moore MJ, Senn JJ, Stanton MG, Almarsson O, Ciaramella G, Brito LA, Mol. Ther.--Nucleic Acids 2019, 15, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].McKinlay CJ, Vargas JR, Blake TR, Hardy JW, Kanada M, Contag CH, Wender PA, Waymouth RM, Proc. Natl. Acad. Sci. U. S. A 2017, 114, E448–E456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Benner NL, Near KE, Bachmann MH, Contag CH, Waymouth RM, Wender PA, Biomacromolecules 2018, 19, 2812–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhi D, Zhang S, Wang B, Zhao Y, Yang B, Yu S, Bioconjugate Chem. 2010, 21, 563–577. [DOI] [PubMed] [Google Scholar]
- [63].Koynova R, Tenchov B, Wang L, MacDonald RC, Mol. Pharm 2009, 6, 951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].McKinlay CJ, Benner NL, Haabeth OA, Waymouth RM, Wender PA, Proc. Natl. Acad. Sci. U. S. A 2018, 115, E5859–E5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Benner NL, McClellan RL, Turlington CR, Haabeth OAW, Waymouth RM, Wender PA, J. Am. Chem. Soc 2019, 141, 8416–8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu X, Xiang J, Zhu D, Jiang L, Zhou Z, Tang J, Liu X, Huang Y, Shen Y, Adv. Mater 2016, 28, 1743–1752. [DOI] [PubMed] [Google Scholar]
- [67].Qiu N, Gao J, Liu Q, Wang J, Shen Y, Biomacromolecules 2018, 19, 2308–2319. [DOI] [PubMed] [Google Scholar]
- [68].Liu X, Yang JW, Miller AD, Nack EA, Lynn DM, Macromolecules 2005, 38, 7907–7914. [Google Scholar]
- [69].Rinkenauer AC, Schallon A, Günther U, Wagner M, Betthausen E, Schubert US, Schacher FH, ACS Nano 2013, 7, 9621–9631. [DOI] [PubMed] [Google Scholar]
- [70].Ros S, Freitag JS, Smith DM, Stover HDH, ACS Omega 2020, 5, 9114–9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ros S, Wang JX, Burke NAD, Stover HDH, Macromolecules 2020, 53, 3514–3523. [Google Scholar]
- [72].Qiu N, Liu X, Zhong Y, Zhou Z, Piao Y, Miao L, Zhang Q, Tang J, Huang L, Shen Y, Adv. Mater 2016, 28, 10613–10622. [DOI] [PubMed] [Google Scholar]
- [73].Zhou Z, Zhang Q, Zhang M, Li H, Chen G, Qian C, Oupicky D, Sun M, Theranostics 2018, 8, 4604–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].A. P. N. f. C.-C. P. i. R. to, G. Endosomal pHSpringsteen, Wang BH, Tetrahedron 2002, 58, 5291–5300. [Google Scholar]
- [75].Zhou Z, Li C, Zhang M, Zhang Q, Qian C, Oupicky D, Sun M, ACS Appl. Mater. Interfaces 2018, 10, 32026–32037. [DOI] [PubMed] [Google Scholar]
- [76].Zhou Z, Zhang M, Liu Y, Li C, Zhang Q, Oupicky D, Sun M, Biomacromolecules 2018, 19, 3776–3787. [DOI] [PubMed] [Google Scholar]
- [77].LaManna CM, Lusic H, Camplo M, McIntosh TJ, Barthelemy P, Grinstaff MW, Acc. Chem. Res 2012, 45, 1026–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Prata CA, Zhao Y, Barthelemy P, Li Y, Luo D, McIntosh TJ, Lee SJ, Grinstaff MW, J. Am. Chem. Soc 2004, 126, 12196–12197. [DOI] [PubMed] [Google Scholar]
- [79].Zhang XX, Prata CAH, McIntosh TJ, Barthelemy P, Grinstaff MW, Bioconjugate Chem. 2010, 21, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhang XX, Prata CAH, Berlin JA, McIntosh TJ, Barthelemy P, Grinstaff MW, Bioconjugate Chem. 2011, 22, 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hersey JS, LaManna CM, Lusic H, Grinstaff MW, Chem. Phys. Lipids 2016, 196, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bus T, Traeger A, Schubert US, J. Mater. Chem. B 2018, 6, 6904–6918. [DOI] [PubMed] [Google Scholar]
- [83].Lee Y, Miyata K, Oba M, Ishii T, Fukushima S, Han M, Koyama H, Nishiyama N, Kataoka K, Angew. Chem. Int. Ed 2008, 47, 5163–5166. [DOI] [PubMed] [Google Scholar]
- [84].Lee Y, Fukushima S, Bae Y, Hiki S, Ishii T, Kataoka K, J. Am. Chem. Soc 2007, 129, 5362–5363. [DOI] [PubMed] [Google Scholar]
- [85].Naito M, Ishii T, Matsumoto A, Miyata K, Miyahara Y, Kataoka K, Angew. Chem. Int. Ed 2012, 51, 10751–10755. [DOI] [PubMed] [Google Scholar]
- [86].Naito M, Yoshinaga N, Ishii T, Matsumoto A, Miyahara Y, Miyata K, Kataoka K, Macromol. Biosci 2018, 18, 1700357. [DOI] [PubMed] [Google Scholar]
- [87].Adams ML, Lavasanifar A, Kwon GS, J. Pharm. Sci 2003, 92, 1343–1355. [DOI] [PubMed] [Google Scholar]
- [88].Cabral H, Miyata K, Osada K, Kataoka K, Chem. Rev 2018, 118, 6844–6892. [DOI] [PubMed] [Google Scholar]
- [89].Jhaveri AM, Torchilin VP, Front. Pharmacol 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS, J. Controlled Release 2009, 133, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lundy BB, Convertine A, Miteva M, Stayton PS, Bioconjugate Chem. 2013, 24, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dutta K, Kanjilal P, Das R, Thayumanavan S, Angew. Chem. Int. Ed 2021, 60, 1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Dutta K, Hu D, Zhao B, Ribbe AE, Zhuang J, Thayumanavan S, J. Am. Chem. Soc 2017, 139, 5676–5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sun Q, Tang C, Su Z, Du J, Shang Y, Xue L, Zhang C, Biomater. Sci 2018, 6, 3075–3084. [DOI] [PubMed] [Google Scholar]
- [95].Khiati S, Pierre N, Andriamanarivo S, Grinstaff MW, Arazam N, Nallet F, Navailles L, Barthelemy P, Bioconjugate Chem. 2009, 20, 1765–1772. [DOI] [PubMed] [Google Scholar]
- [96].Baillet J, Desvergnes V, Hamoud A, Latxague L, Barthélémy P, Adv. Mater 2018, 30, 1705078. [DOI] [PubMed] [Google Scholar]
- [97].Banchelli M, Berti D, Baglioni P, Angew. Chem. Int. Ed 2007, 46, 3070–3073. [DOI] [PubMed] [Google Scholar]
- [98].Milani S, Bombelli FB, Berti D, Baglioni P, J. Am. Chem. Soc 2007, 129, 11664–11665. [DOI] [PubMed] [Google Scholar]
- [99].Montis C, Milani S, Berti D, Baglioni P, J. Colloid Interface Sci 2012, 373, 57–68. [DOI] [PubMed] [Google Scholar]
- [100].Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA, Science 2006, 312, 1027–1030. [DOI] [PubMed] [Google Scholar]
- [101].Ghosh P, Han G, De M, Kim CK, Rotello VM, Adv. Drug Delivery Rev 2008, 60, 1307–1315. [DOI] [PubMed] [Google Scholar]
- [102].Elbakry A, Zaky A, Liebl R, Rachel R, Goepferich A, Breunig M, Nano Lett. 2009, 9, 2059–2064. [DOI] [PubMed] [Google Scholar]
- [103].Cutler JI, Auyeung E, Mirkin CA, J. Am. Chem. Soc 2012, 134, 1376–1391. [DOI] [PubMed] [Google Scholar]
- [104].Guo S, Huang Y, Jiang Q, Sun Y, Deng L, Liang Z, Du Q, Xing J, Zhao Y, Wang PC, Dong A, Liang XJ, ACS Nano 2010, 4, 5505–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Xie X, Xu S, Pi P, Cheng J, Wen X, Liu X, Wang S, AIChE J. 2018, 64, 810–821. [Google Scholar]
- [106].Han L, Zhao J, Zhang X, Cao W, Hu X, Zou G, Duan X, Liang X-J, ACS Nano 2012, 6, 7340–7351. [DOI] [PubMed] [Google Scholar]
- [107].Hu Q, Li H, Wang L, Gu H, Fan C, Chem. Rev 2019, 119, 6459–6506. [DOI] [PubMed] [Google Scholar]
- [108].Ding F, Mou QB, Ma Y, Pan GF, Guo YY, Tong GS, Choi CHJ, Zhu XY, Zhang C, Angew. Chem. Int. Ed 2018, 57, 3064–3068. [DOI] [PubMed] [Google Scholar]
- [109].Ding F, Huang XG, Gao XH, Xie M, Pan GF, Li QF, Song J, Zhu XY, Zhang C, Nanoscale 2019, 11, 17211–17215. [DOI] [PubMed] [Google Scholar]
- [110].Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M, Proc. Natl. Acad. Sci. U. S. A 1987, 84, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Peer D, Adv. Drug Delivery Rev 2012, 64, 1738–1748. [DOI] [PubMed] [Google Scholar]
- [112].Semple SC, Klimuk SK, Harasym TO, Dos Santos N, Ansell SM, Wong KF, Maurer N, Stark H, Cullis PR, Hope MJ, Scherrer P, Biochim. Biophys. Acta, Biomembr 2001, 1510, 152–166. [DOI] [PubMed] [Google Scholar]
- [113].Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen JX, Du XY, Butler D, Eltepu L, Matsuda S, Narayanannair JK, Rajeev KG, Hafez IM, Akinc A, Maier MA, Tracy MA, Cullis PR, Madden TD, Manoharan M, Hope MJ, Angew. Chem. Int. Ed 2012, 51, 8529–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Semple SC, Akinc A, Chen JX, Sandhu AP, Mui BL, Cho CK, Sah DWY, Stebbing D, Crosley EJ, Yaworski E, Hafez IM, Dorkin JR, Qin J, Lam K, Rajeev KG, Wong KF, Jeffs LB, Nechev L, Eisenhardt ML, Jayaraman M, Kazem M, Maier MA, Srinivasulu M, Weinstein MJ, Chen QM, Alvarez R, Barros SA, De S, Klimuk SK, Borland T, Kosovrasti V, Cantley WL, Tam YK, Manoharan M, Ciufolini MA, Tracy MA, de Fougerolles A, MacLachlan I, Cullis PR, Madden TD, Hope MJ, Nat. Biotechnol 2010, 28, 172–176. [DOI] [PubMed] [Google Scholar]
- [115].Lin PJ, Tam YY, Hafez I, Sandhu A, Chen S, Ciufolini MA, Nabi IR, Cullis PR, Nanomedicine 2013, 9, 233–246. [DOI] [PubMed] [Google Scholar]
- [116].Rietwyk S, Peer D, ACS Nano 2017, 11, 7572–7586. [DOI] [PubMed] [Google Scholar]
- [117].Kanasty R, Dorkin JR, Vegas A, Anderson D, Nat. Mater 2013, 12, 967–977. [DOI] [PubMed] [Google Scholar]
- [118].Kulkarni JA, Cullis PR, van der Meel R, Nucleic Acid Ther. 2018, 28, 146–157. [DOI] [PubMed] [Google Scholar]
- [119].Cullis PR, Hope MJ, Mol. Ther 2017, 25, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zhao JC, Stenzel MH, Polym. Chem 2018, 9, 259–272. [Google Scholar]
- [121].Mislick KA, Baldeschwieler JD, Proc. Natl. Acad. Sci. U. S. A 1996, 93, 12349–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Vercauteren D, Rejman J, Martens TF, Demeester J, De Smedt SC, Braeckmans K, J. Controlled Release 2012, 161, 566–581. [DOI] [PubMed] [Google Scholar]
- [123].Chao Y, Karmali PP, Mukthavaram R, Kesari S, Kouznetsova VL, Tsigelny IF, Simberg D, ACS Nano 2013, 7, 4289–4298. [DOI] [PubMed] [Google Scholar]
- [124].Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, Brown D, Alkilany AM, Farokhzad OC, Mahmoudi M, Chem. Soc. Rev 2017, 46, 4218–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Madani F, Lindberg S, Langel U, Futaki S, Graslund A, J. Biophys 2011, 2011, 414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Jiang Y, Huo S, Mizuhara T, Das R, Lee YW, Hou S, Moyano DF, Duncan B, Liang XJ, Rotello VM, ACS Nano 2015, 9, 9986–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Nakase I, Akita H, Kogure K, Gräslund A, Langel Ü, Harashima H, Futaki S, Acc. Chem. Res 2012, 45, 1132–1139. [DOI] [PubMed] [Google Scholar]
- [128].Singh T, Murthy ASN, Yang HJ, Im J, Drug Delivery 2018, 25, 1996–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sahni A, Qian Z, Pei D, ACS Chem. Biol 2020, 15, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Abegg D, Gasparini G, Hoch DG, Shuster A, Bartolami E, Matile S, Adibekian A, J. Am. Chem. Soc 2017, 139, 231–238. [DOI] [PubMed] [Google Scholar]
- [131].Yu C, Qian L, Ge J, Fu J, Yuan P, Yao SC, Yao SQ, Angew. Chem. Int. Ed 2016, 55, 9272–9276. [DOI] [PubMed] [Google Scholar]
- [132].Gasparini G, Matile S, Chem. Commun 2015, 51, 17160–17162. [DOI] [PubMed] [Google Scholar]
- [133].Yang W, Yu C, Wu C, Yao SQ, Wu S, Polym. Chem 2017, 8, 4043–4051. [Google Scholar]
- [134].Kettiger H, Schipanski A, Wick P, Huwyler J, Int. J. Nanomed 2013, 8, 3255–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Conner SD, Schmid SL, Nature 2003, 422, 37–44. [DOI] [PubMed] [Google Scholar]
- [136].Zhang X-X, Allen PG, Grinstaff M, Mol. Pharmaceutics 2011, 8, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Zhou Z, Li H, Wang K, Guo Q, Li C, Jiang H, Hu Y, Oupicky D, Sun M, ACS Appl. Mater. Interfaces 2017, 9, 14576–14589. [DOI] [PubMed] [Google Scholar]
- [138].Vaughan HJ, Green JJ, Tzeng SY, Adv. Mater 2020, 32, 1901081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Majumder P, Bioengineering 2018, 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Xiao Y, Shi K, Qu Y, Chu B, Qian Z, Mol. Ther.--Methods Clin. Dev 2019, 12, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA, Science 2002, 296, 2404–2407. [DOI] [PubMed] [Google Scholar]
- [142].Smith SA, Selby LI, Johnston APR, Such GK, Bioconjugate Chem. 2019, 30, 263–272. [DOI] [PubMed] [Google Scholar]
- [143].Lonn P, Kacsinta AD, Cui XS, Hamil AS, Kaulich M, Gogoi K, Dowdy SF, Sci. Rep 2016, 6, 32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Brock DJ, Kondow-McConaghy HM, Hager EC, Pellois JP, Bioconjugate Chem. 2019, 30, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Peeler DJ, Sellers DL, Pun SH, Bioconjugate Chem. 2019, 30, 350–365. [DOI] [PubMed] [Google Scholar]
- [146].Yonezawa S, Koide H, Asai T, Adv. Drug Delivery Rev 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Ligtenberg MA, Pico de Coana Y, Shmushkovich T, Yoshimoto Y, Truxova I, Yang Y, Betancur-Boissel M, Eliseev AV, Wolfson AD, Kiessling R, Mol. Ther 2018, 26, 1482–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M, Nat. Biotechnol 2007, 25, 1149–1157. [DOI] [PubMed] [Google Scholar]
- [149].Nishina K, Unno T, Uno Y, Kubodera T, Kanouchi T, Mizusawa H, Yokota T, Mol. Ther 2008, 16, 734–740. [DOI] [PubMed] [Google Scholar]
- [150].Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, Laouar A, Yao J, Haridas V, Habiro K, Yang YG, Jeong JH, Lee KY, Kim YH, Kim SW, Peipp M, Fey GH, Manjunath N, Shultz LD, Lee SK, Shankar P, Cell 2008, 134, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Debacker AJ, Voutila J, Catley M, Blakey D, Habib N, Mol. Ther 2020, 28, 1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Springer AD, Dowdy SF, Nucleic Acid Ther. 2018, 28, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y, Liang XJ, Signal Transduction Targeted Ther. 2020, 5, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Nat. Biotechnol 2018, 36, 775–775. [DOI] [PubMed] [Google Scholar]
- [155].Garber K, Nat. Biotechnol 2018, 36, 777–778. [DOI] [PubMed] [Google Scholar]
- [156].Akinc A, Maier MA, Manoharan M, Fitzgerald K, Jayaraman M, Barros S, Ansell S, Du XY, Hope MJ, Madden TD, Mui BL, Semple SC, Tam YK, Ciufolini M, Witzigmann D, Kulkarni JA, van der Meel R, Cullis PR, Nat. Nanotechnol 2019, 14, 1084–1087. [DOI] [PubMed] [Google Scholar]
- [157].Ramot Y, Rotkopf S, Gabai RM, Zorde Khvalevsky E, Muravnik S, Marzoli GA, Domb AJ, Shemi A, Nyska A, Toxicol. Pathol 2016, 44, 856–865. [DOI] [PubMed] [Google Scholar]
- [158].Nat. Biotechnol 2021, 39, 1.33376248 [Google Scholar]
- [159].Lowe D, “RNA Vaccines And Their Lipids” can be found under https://blogs.sciencemag.org/pipeline/archives/2021/01/11/rna-vaccines-and-their-lipids, 2021.
- [160].“Safety and Immunogenicity Study of 2019-nCoV Vaccine (mRNA-1273) for Prophylaxis of SARS-CoV-2 Infection (COVID-19)” can be found under https://clinicaltrials.gov/ct2/show/NCT04283461, 2020.
- [161].Walsh EE, Frenck RW Jr., Falsey AR, Kitchin N, Absalon J, Gurtman A, Lockhart S, Neuzil K, Mulligan MJ, Bailey R, Swanson KA, Li P, Koury K, Kalina W, Cooper D, Fontes-Garfias C, Shi PY, Tureci O, Tompkins KR, Lyke KE, Raabe V, Dormitzer PR, Jansen KU, Sahin U, Gruber WC, Engl N. J. Med 2020, 383, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC, Engl N. J. Med 2020. [Google Scholar]
- [163].Krammer F, Nature 2020, 586, 516–527. [DOI] [PubMed] [Google Scholar]
- [164].Lundstrom K, Front. Genome Ed 2020, 2, 579297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, Ciaramella G, Diamond MS, Cell 2017, 168, 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].“Clinical Trial of BP1001 in Combination With With Venetoclax Plus Decitabine in AML” can be found under https://clinicaltrials.gov/ct2/show/NCT02781883, 2020.
- [167].Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, Roesch PL, Bertin SL, Reppen TW, Chu Q, Blokhin AV, Hagstrom JE, Wolff JA, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 12982–12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Wong SC, Cheng W, Hamilton H, Nicholas AL, Wakefield DH, Almeida A, Blokhin AV, Carlson J, Neal ZC, Subbotin V, Zhang G, Hegge J, Bertin S, Trubetskoy VS, Rozema DB, Lewis DL, Kanner SB, Mol. Cancer Ther 2018, 17, 140–149. [DOI] [PubMed] [Google Scholar]
- [169].Khorev O, Stokmaier D, Schwardt O, Cutting B, Ernst B, Bioorg. Med. Chem 2008, 16, 5216–5231. [DOI] [PubMed] [Google Scholar]
- [170].Yasuda M, Gan L, Chen B, Kadirvel S, Yu C, Phillips JD, New MI, Liebow A, Fitzgerald K, Querbes W, Desnick RJ, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 7777–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF, Riney S, Booten SL, Murray SF, Gaus H, Crosby J, Lima WF, Guo S, Monia BP, Swayze EE, Seth PP, Nucleic Acids Res. 2014, 42, 8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Lee ACH, Heyes J, Ye X, Holland R, Thi EP, Wood M, Judge A, Snead NM, Martin A, Sofia MJ, J. Hepatol 2018, 68, S18–S18. [Google Scholar]
- [173].Dicerna, “Corporate Overview” can be found under http://investors.dicerna.com/static-files/b0fe07db-a91e-4be5-a422-03d9fd6ab93e, 2021.
- [174].“Dicerna Receives Breakthrough Therapy Designation for DCR-PHXC for Treatment of Primary Hyperoxaluria Type 1 (PH1)” can be found under https://investors.dicerna.com/news-releases/news-release-details/dicerna-receives-breakthrough-therapy-designation-dcr-phxc, 2021.
- [175].Wu CH, Wang Y, Ma M, Mullick AE, Crooke RM, Graham MJ, Daugherty A, Lu HS, Biosci. Rep 2019, 39, BSR20180201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Kordasiewicz Holly B., Stanek Lisa M., Wancewicz Edward V., Mazur C, McAlonis Melissa M., Pytel Kimberly A., Artates Jonathan W., Weiss A, Cheng Seng H., Shihabuddin Lamya S., Hung G, Bennett CF, Cleveland Don W., Neuron 2012, 74, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Ackermann EJ, Guo S, Booten S, Alvarado L, Benson M, Hughes S, Monia BP, Amyloid 2012, 19, 43–44. [DOI] [PubMed] [Google Scholar]
- [178].Alexander VJ, Xia S, Hurh E, Hughes SG, O’Dea L, Geary RS, Witztum JL, Tsimikas S, Eur. Heart J 2019, 40, 2785–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].“Optimizing antisense” can be found under https://www.ionispharma.com/ionis-innovation/optimizing-antisense/, 2020.
- [180].Tasfaout H, Buono S, Guo S, Kretz C, Messaddeq N, Booten S, Greenlee S, Monia BP, Cowling BS, Laporte J, Nat. Commun 2017, 8, 15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Crosby JR, Zhao C, Jiang C, Bai D, Katz M, Greenlee S, Kawabe H, McCaleb M, Rotin D, Guo S, Monia BP, J. Cystic Fibrosis 2017, 16, 671–680. [DOI] [PubMed] [Google Scholar]
- [182].Lindén D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andréasson A-C, Sasidharan K, Madeyski-Bengtson K, Zurek M, Mancina RM, Lindblom A, Bjursell M, Böttcher G, Ståhlman M, Bohlooly-Y M, Haynes WG, Carlsson B, Graham M, Lee R, Murray S, Valenti L, Bhanot S, Åkerblad P, Romeo S, Mol. Metab 2019, 22, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Grandinetti G, Smith AE, Reineke TM, Mol. Pharm 2012, 9, 523–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Degors IMS, Wang CF, Rehman ZU, Zuhorn IS, Acc. Chem. Res 2019, 52, 1750–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Haque ME, McIntosh TJ, Lentz BR, Biochemistry 2001, 40, 4340–4348. [DOI] [PubMed] [Google Scholar]
- [186].Wirth T, Parker N, Yla-Herttuala S, Gene 2013, 525, 162–169. [DOI] [PubMed] [Google Scholar]
- [187].Hood L, Rowen L, Genome Med. 2013, 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Collins FS, McKusick VA, JAMA 2001, 285, 540–544. [DOI] [PubMed] [Google Scholar]