

Abstract
Nucleotide excision repair (NER) is a versatile DNA repair pathway essential for the removal of a broad spectrum of structurally diverse DNA lesions arising from a variety of sources, including UV irradiation and environmental toxins. Although the core factors and basic stages involved in NER have been identified, the mechanisms of the NER machinery are not well understood. This review summarizes our current understanding of the mechanisms and order of assembly in the core global genome (GG-NER) pathway.
Introduction
Nucleotide excision repair (NER) is an essential DNA repair pathway that is responsible for eliminating a wide range of lesions from genomic DNA. Defects in NER result in the genetic disorder xeroderma pigmentosum (XP), which is characterized by an extreme hypersensitivity to sunlight and a greater than 2000-fold increase in skin cancer [1, 2]. NER is a highly dynamic process that operates through the sequential assembly and action of more than 20 proteins at the DNA lesion. Biochemical and cell biological studies of XP patient cell lines deficient in the seven NER-specific XP proteins (XPA-XPG) have led to an understanding of the basic steps of NER and individual functions of each protein [3, 4]. There are two main NER pathways, which diverge by the mechanisms of initial damage recognition and their associated spectra of disease phenotypes: (i) transcription-coupled NER (TC-NER), in which lesions are detected by RNA polymerase II stalling on the transcribed strand [5] and (ii) global genome (GG)-NER, where lesions are detected throughout the genome. The specifics of TC-NER have been covered extensively elsewhere [6–8], so this minireview focuses on GG-NER. Defects in GG-NER that lead to XP are also associated with increased incidence of internal tumors and accelerated neurodegeneration and aging in some cases [9].
Human GG-NER has been reconstituted in vitro with six core factors: XPC-human Rad23 homolog B (HR23B), transcription factor IIH (TFIIH), XPA, replication protein A (RPA), XPG and XPF-excision repair cross complementation group 1 (ERCC1) [10–12]. Although the exact order of assembly of these factors at sites of damage is not yet fully resolved, the trajectory of the core GG-NER machinery can be summarized in four phases (Figure 1). It is generally understood that XPC-HR23B recognizes the presence of damage in the DNA due to preferential interaction with destabilized DNA [13–15]. For less helix-distorting lesions such as cyclopyrimidine dimers (CPDs), the damage recognition process additionally requires the DDB1-DDB2 E3 ubiquitin ligase complex [16, 17], but this complex is dispensable for the core NER reaction in vitro. Once XPC binds to the damaged duplex, it recruits TFIIH to the lesion through specific protein-protein interactions [18, 19]. The two helicase subunits of TFIIH, XPB and XPD, fulfill key roles in NER: XPB melts the DNA duplex [20, 21], thereby allowing XPD to load and track along the damaged strand in the 5’ - 3’ direction until it stalls at the lesion, which is best explained as arising from an inability to pass through the DNA binding channel of the helicase [22–27]. A characteristic feature of lesions repaired by NER is the presence of local flexibility or helical kinks in the DNA [28]. While XPC appears to sense increased DNA strand flexibility induced by a lesion and does not directly interact with the lesion, XPD directly interacts with and validates the presence of the lesion through its translocation on the DNA. Importantly, the two steps of damage recognition and validation are independent of the exact chemical structure of the lesion, providing a molecular basis for the very broad substrate specificity of NER. After creation of an unwound ‘bubble’ in the DNA and assembly of the XPA-RPA scaffold, the XPG and XPF-ERCC1 nucleases are recruited. XPF-ERCC1 incises the DNA 5’ to the lesion, replication proteins initiate gap-filling synthesis, and following 3’ incision by the XPG nuclease, filling of the gap is completed and the resulting nick sealed [29–32].
Figure 1. The trajectory of dynamic assembly of the core GG-NER machinery.
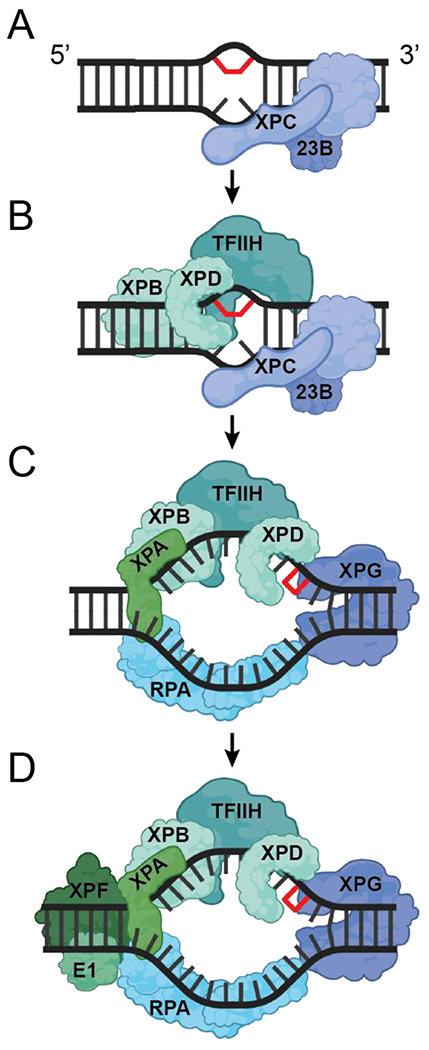
Four steps are identified: (A) damage recognition, (B) damage verification, (C) pre-incision, and (D) dual incision.
Phase 1: Recognition of DNA damage
XPC functions as the key initiator of damage recognition in GG-NER [33, 34], ultimately leading to the opening of duplex DNA around the lesion and recruitment of additional NER factors [35, 36]. XPC is found tightly associated with HR23B [37] and Centrin 2 [38]. Although XPC alone is sufficient in cell-free NER systems for the damage recognition step, both HR23B [39, 40] and Centrin 2 [41] significantly enhance the activity and stability of XPC.
XPC-HR23B binds tightly to duplex DNA that is destabilized by base modifications, adducts and other distorting lesions. The first insights into how XPC-RAD23B binds DNA were provided by a crystal structure of the yeast orthologue Rad4–Rad23 complex bound to duplex DNA containing a cyclobutane pyrimidine dimer (CPD) lesion [42]. Yeast Rad4 inserts a β-hairpin into the DNA major groove such that the damaged base pairs are flipped out of the helical stack and make no direct contacts with the protein. Instead, Rad4 interacts with the undamaged strand 3’ to the lesion. This suggests an indirect recognition mechanism in which XPC senses DNA destabilization rather than direct recognition of a specific lesion type, enabling NER to act on a wide range of bulky and non-bulky lesions. Following initial damage detection by XPC, additional factors verify the presence of the lesion prior to incision (vide infra).
While NER acts on UV-induced DNA cross-links (CPDs or 6-4 pyrimidine-pyrimidone photodimers (6-4PPs)), XPC is inefficient at detecting lesions like these that lead to only minor distortions of the DNA helix [43, 44]. In this case, the initial recognition of damage involves the UV DNA damage binding (UV-DDB) complex, a heterodimer of two subunits, DDB1 and XPE (DDB2). XPE interacts directly with damaged DNA [17, 45, 46] and in fact exhibits a higher affinity and specificity for CPDs than XPC [47]. In contrast to XPC, XPE inserts a β-hairpin into the minor groove of the duplex and directly contacts the damaged strand [48, 49]. UV-DDB is part of the larger CUL4-RBX1 E3 ubiquitin-ligase [50] that ubiquitinates several DNA binding proteins including XPC, histones and UV-DDB itself. Ubiquitination of XPE reduces its affinity for damage sites but has no effect on the affinity of XPC, thus facilitating the handoff of the lesion from UV-DDB to XPC and subsequent recruitment of TFIIH [17, 51]. UVDDB-CUL4 also ubiquitinates histones H3 and H4, which in turn improves accessibility of chromatin to XPC and other NER factors [52].
After initial lesion recognition, XPC recruits TFIIH to the lesion via interactions with the p62 and XPB subunits of TFIIH (Table 1) [18, 53]. A cryo-EM structure of an ‘initiation complex’ containing the yeast homologues of XPC-HR23B-Centrin 2 (Rad4-Rad23-Rad33), plus the seven-subunit TFIIH core and a dsDNA substrate containing a carcinogenic 2-acetylaminofluorene adduct, which confirms that TFIIH binds two distinct motifs in the yeast Rad4 N- and C-terminal regions (Figure 2A) [54]. The duplex DNA is locally unwound and positioned between Rad4 and the Ssl2 (yeast XPB) helicase subunit of TFIIH, with Rad4 on the 3′ and TFIIH on the 5′ side of the lesion. Interestingly, the dsDNA is not yet engaged by the XPD helicase in this structure, indicating that additional configurational changes in TFIIH must occur as the DNA unwinding and lesion verification proceeds.
Table 1.
Summary of interactions of the core NER factors.
| Protein | Interacting Residues or Subunits | Interacting Partner |
|---|---|---|
| XPC | 154-331, 492-940 | XPA |
| 494-741 | RAD23B | |
| 606-742 | DNA | |
| 816-940 | TFIIH (XPB and p62) | |
| 847-866 | Centrin 2 | |
| TFIIH | p62 1-108, XPB | XPC |
| XPD, p44, p62, XPB | DNA | |
| XPD, XPB and p52 | XPG | |
| p8, p52 | XPA | |
| XPA | 29-46 | RPA32C |
| 67-77 | ERCC1 | |
| 98-129 | RPA70A | |
| 98-239 | DNA | |
| 153-176 | RPA70B | |
| 161-170 | PCNA | |
| 185-226 | DDB2 | |
| 226-273 | TFIIH (p8 and p52) | |
| unknown | XPC-RAD23B | |
| RPA | RPA 14 : 70-121 | DNA |
| unknown | XPG | |
| RPA 32 : 40-174 | DNA | |
| RPA 32 : 237-270 | XPA | |
| RPA 70 : 181-442 | DNA | |
| RPA 70 : 236-382 | XPA, XPF | |
| XPG | 1-65 | TFIIH (p44 and p62) |
| unknown | RPA | |
| unknown | XPB | |
| unknown | XPD | |
| unknown | DNA | |
| XPF | 1-824 | RPA |
| 824-905 | ERCC1 | |
| unknown | DNA | |
| ERCC1 | 67-80 | XPA |
| 96-227 | DNA | |
| 224-245, 293-297 | XPF |
Figure 2. Cryo-EM structures of TFIIH complexe.
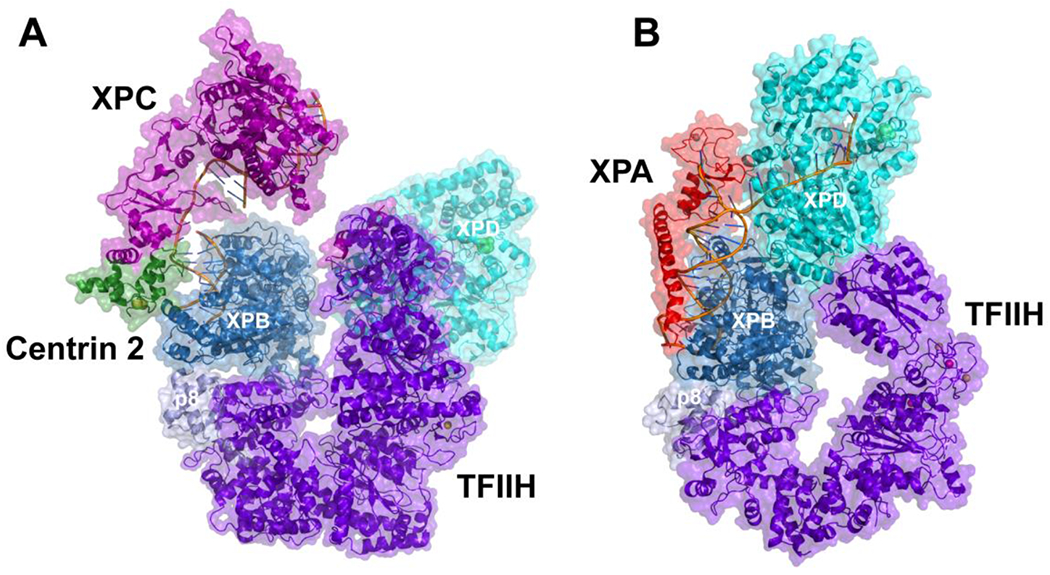
(A) Yeast TFIIH and XPC-HR23B-Cen2. (B) Human TFIIH and XPA.
Phase 2: DNA unwinding and verification of the site of damage
TFIIH is a central element of NER responsible for unwinding the duplex DNA around the site of damage and verifying the presence of the lesion. It contains ten subunits with a seven-subunit core complex and a three-subunit cyclin-dependent kinase (CDK) activating kinase (CAK) module. The core complex contains the XPB and XPD ATP-dependent helicases and five non-enzymatic subunits (p62, p52, p44, p34, p8). The CAK module is comprised of CDK7, cyclin H, and MAT1 subunits. NER requires only the core complex, using the two critical XPB and XPD helicases to extend the initial opening of the duplex by XPC and ultimately generate the 24-30 nucleotide asymmetric NER bubble around the lesion [55–58].
X-ray crystal structures of XPB [59, 60] and XPD [22–24, 27] have been reported. XPB contains two RecA-like domains, a DNA-damage recognition domain (DRD) domain, and an N-terminal extension domain (NTE) crucial for activity [59]. XPB is a dsDNA translocase that binds opposite to the damaged strand and unwinds the duplex in the 3′ to 5′ direction [59]. It is also involved in the recruitment of TFIIH by XPC [61]. XPD contains two RecA-like domains (RecA1 and RecA2) with conserved helicase motifs, a 4FeS cluster domain, and an additional Arch domain [22]. One of the most remarkable structural features of XPD is the presence of a DNA binding pore created by the Arch domain, the FeS domain and the RecA1 domain [62]. XPD, a ssDNA translocase, binds 5’ to the lesion on the damaged strand and moves along the DNA in the 5’-3’ direction until it stalls at the damaged site. Thus, XPD serves as the critical damage verification element [63, 64]. While both helicases are crucial for NER, their activity is also influenced by the presence of non-enzymatic TFIIH subunits [65]; XPB activity is upregulated by the p52 subunit [66] and XPD by interactions with the p44 subunit [67]. Recent studies have also shown that p8 is an essential factor for duplex opening, perhaps through its role in recruiting XPA [68, 69].
Structures of TFIIH determined by cryo-electron microscopy (EM) have revolutionized understanding of its mechanism of action [54, 70–73]. Among these, the structure of human TFIIH in the absence of substrate revealed the overall horseshoe-shaped architecture that is dominated by the side-by-side orientation of the helicase subunits, enforced by their extensive interfaces [71]. Additional contacts to other subunits include p52 and p8 with XPB, and p62, p44, and MAT1 with XPD. This network of inter-subunit interactions likely serves to modulate XPB and XPD activity [70, 71]. For example, the p62 subunit sterically interferes with and blocks the DNA binding pore of the XPD RecA1 domain [71, 74]. MAT1 is a non-globular protein that forms extensive contacts with the XPD Arch domain and tightly regulates its function [75, 76]. The long α-helix of MAT1 also separates XPB and XPD and reduces the inter-domain mobility in XPD. A high-resolution crystal structure of the Arch domain of XPD in complex with MAT1 shows that MAT1 renders XPD inactive by shielding residues essential for its helicase activity, affecting the DNA binding of XPD and interactions of XPD with XPG [76]. These observations suggest that the p62 and MAT1 subunits need to be displaced during NER to allow more conformational flexibility for XPD to bind and translocate along DNA for lesion verification.
Another breakthrough cryo-EM structure was that of TFIIH with XPA on a model NER bubble substrate, which provided many additional insights into the mechanism of action of TFIIH during NER (Figure 2B) [72]. This structure clarified the drastic shift in the arrangement of TFIIH subunits needed for DNA unwinding [71], including displacing the CAK module that ultimately allows XPD to engage ssDNA. In addition, this structure provided insight into how XPA enhances TFIIH specificity [64, 72, 77], as well as pointed to a potentially critical role of XPA as a wedge that pries apart base pairs as the helicases ‘pull’ the duplex DNA forward, as proposed by Tsutakawa et al.[78]. This structure also suggests that XPA enhances XPB activity by bridging the XPB ATPase lobes to the p52 and p8 subunits and facilitates DNA opening through contacts with XPD and ssDNA. Even though the cryo-EM complex of TFIIH was formed in the presence of XPA and XPG, XPG was not visible in the structure. However, its location near the XPD-MAT1 interaction site was inferred by chemical crosslinking, suggesting that XPG competes with MAT1 to interact with XPD [72]. These results in turn suggest that XPG can facilitate lesion scanning by blocking the kinase module interaction site on XPD, which would significantly enhance XPD’s helicase activity, consistent with the report that XPG greatly stimulates XPD [72].
These cryo-EM structures have informed models to explain how TFIIH unwinds DNA and transitions from the initial destabilization of the duplex to the fully opened NER bubble. In a structure of yeast TFIIH and XPC in complex with a lesion-containing duplex, XPC secures both the 3’ end of the DNA and TFIIH to prevent DNA rotation (Figure 2A) [54]. With the DNA held in place by XPC, XPB is then able to exert torque on the DNA duplex as it translocates in an ATP-dependent manner. The β-hairpin unit BHD3 of XPC positions itself between the two DNA strands leading to strand separation and facilitating unwinding. In this configuration, the dsDNA is far from XPD and the DNA binding pore of XPD is blocked by a plug element, hence XPD is unable to translocate. In contrast, in the structure of the complex with XPA, the plug is displaced so XPD can translocate on the ssDNA (Figure 2B) [72]. This transition requires a significant movement of the XPD and p44 subunits of ~80 Å in the 3’ direction to enable binding of ssDNA in the XPD DNA binding channel. The ssDNA threads along the DNA channel in close proximity to the FeS cluster and residues lining the pore interact with the sugar-phosphate backbone and aromatic residues proof-read DNA bases [72]. The ATPase motors in XPD drag the ssDNA through the channel to scan for lesions [79]. When XPD encounters a bulky lesion, both XPB and XPD are stalled and XPA was found to significantly enhance this lesion-induced stalling [62, 64].
Phase 3: Completing assembly of the pre-incision complex
Following DNA unwinding and lesion verification, the NER scaffolding is stabilized and the pre-incision complex (PIC) is formed [34, 35, 81]. As noted, current evidence suggests that XPA is recruited first by TFIIH to aid in generating the NER bubble [35, 64, 82, 83], although some studies suggest that XPA and RPA are recruited together [84, 85] and one early study suggested that RPA can be recruited to the damage site without the presence of XPA [86]. In all models, XPG is recruited through interactions with TFIIH and RPA, displacing XPC to complete formation of the PIC [81, 87, 88].
XPA and RPA play crucial roles to coordinate and ensure the accurate positioning of proteins in the PIC. Despite its small size (273 residues), XPA interacts with many NER factors, including TFIIH, RPA, XPC-HR23B, DDB2, and XPF-ERCC1 [89], as well as proliferating cell nuclear antigen (PCNA) required for gap-filling synthesis [13, 90–95]. XPA has a central globular DNA binding domain (DBD) spanning residues 98 to 239 [96] with a zinc binding motif and unordered, flexible N-terminal (1-98) and C-terminal (240-273) regions that mediate protein-protein interactions with other NER factors. XPA is positioned at the ss–dsDNA junction of the NER bubble [97, 98]. Most structural models place XPA at the junction 5′ to the lesion as it is known to interact with endonuclease XPF-ERCC1 [72, 90], although we have recently shown that binding to both the 5’ and 3’ junction is sterically feasible and proposed there may be switching between junctions over the course of the dynamic NER trajectory [99].
The XPA-RPA scaffolding stabilizes the PIC and is essential for NER [85, 99, 100]. RPA is a heterotrimer of RPA70, RPA32 and RPA14 subunits. It contains four DNA binding OB-fold domains (70A-70B-70C-32D) that can bind up to 30 nucleotides, which coincides with the size of the NER bubble [31, 101–103]. RPA binds to the undamaged strand of the NER bubble, protecting it from incoming nucleases [104, 105]. XPA and RPA make contact at two distinct sites: a stronger site that involves a motif in the XPA N-terminal domain and the RPA32C winged-helix protein recruitment domain [106], and a weaker site involving the tandem high affinity ssDNA binding domains RPA70AB and the XPA DBD [99].
XPG is understood to be recruited to the PIC through interactions with TFIIH and RPA (Table 1). XPG is a member of the FEN-1 (flap endonuclease) family of nucleases. The XPG active site consists of the highly conserved N-terminal (N)- and internal (I)-nuclease region separated by a large spacer region of 600 residues, the extended length of which is unique to XPG [107, 108]. This spacer region influences the substrate preference of XPG for ss-dsDNA junctions [109–112] and mediates essential interactions with the XPB, XPD, p62, and p44 subunits of TFIIH [111, 113, 114] and RPA [115–117]. The presence of XPG, but not its nuclease activity, was shown to stabilize the NER bubble and facilitate the first incision 5’ to the lesion by XPF-ERCC1 [118].
Phase 4: Dual incision
Removal of the damaged DNA depends on the concerted action of the two structure-specific endonucleases XPF-ERCC1 and XPG. These nucleases were first identified as part of the NER pathway but are now known to act in multiple DNA transactions [108, 119–121]. In NER, XPF-ERCC1 and XPG make incisions 5’ and 3’ to the lesion on the damaged strand, respectively [122, 123]. This dual incision and removal of approximately 24-30 nucleotides in the damaged strand is accompanied by gap filling synthesis, which completes the repair process. Because these nucleases introduce dangerous nicks in the DNA, their dual action is carefully coordinated by interactions by post-translational modifications and interactions with other NER factors, which regulate the order of assembly and catalytic activity during NER.
The order of assembly at the lesion and the incisions by XPF-ERCC1 and XPG were debated for many years, with conflicting studies conducted using multiple approaches including in vitro reconstitution and in vivo cell biological studies, different substrates, and various combinations of NER proteins [124–131]. In the currently accepted model based on the consensus of biochemical and biological studies, XPG is recruited before XPF-ERCC1, even though XPF-ERCC1 incises the damaged strand first; the XPF-ERCC1 5’ incision requires the presence of XPG but not its catalytic activity [129]. Because XPG catalytic activity is also not required for initiation of gap-filling repair synthesis starting from the exposed 3’-OH generated by XPF-ERCC1, it is generally understood that repair synthesis initiates prior to the 3’ incision by XPG [127]. Strengths of this model include high-fidelity to appropriately license and regulate the dual incision while minimizing potential DNA breaks due to exposure of the undamaged strand of ssDNA.
The XPF-ERCC1 complex is likely recruited through interactions between XPA and the central domain of ERCC1 (Table 1) [90, 132, 133] to form the incision complex. XPF is a member of the Mus81 nuclease family, with an archaeal SF2 helicase-like domain in the N-terminus, a central nuclease domain containing a conserved V/IERKX3D motif [134], and two C-terminal helix-turn-helix (HhH) domains (HhH2) [135]. Similarly, ERCC1 contains a central domain with high structural homology to the XPF nuclease domain, and two C-terminal HhH domains [135]. XPF functions as an obligate heterodimer with ERCC1 [136–140]. Dimerization is mediated by interaction between the HhH2 domains in each protein and is required to stabilize both proteins for efficient nuclease activity [136, 137, 141, 142]. The presence of RPA and direct interaction with XPA also enhance incision activity [115, 122, 143].
XPF-ERCC1 exhibits a preference for DNA junction substrates [115, 122, 124], with DNA binding mediated by the HhH2, XPF nuclease, and ERCC1 central domains [144]. In the current model for DNA binding, the HhH2 domains of XPF and ERCC1 each bind ssDNA near the 5’ junction with the ERCC1 central domain binding specifically to the 5’ arm of ssDNA [142, 145–147]. A recent cryo-EM structure of XPF-ERCC1 bound to a stem-loop DNA substrate suggests a model in which DNA binding relieves an auto-inhibited state where the XPF helical domain blocks the active site [148]. This auto-inhibited state would inhibit nuclease activity until XPF-ERCC1 is stabilized by the NER scaffold and the appropriate DNA substrate is bound. With XPG already in place, the incision complex would then be fully assembled and the XPF-ERCC1 nuclease activated.
The structural and biochemical characterization of the XPG catalytic domain [149] suggests that XPG has a similar structure and function to the FEN superfamily with a conserved active site consisting of carboxylates in a β-sheet core and basic residues near a pair of gating helices [150]. These active site gating helices play a key role in activating the XPG nuclease, by undergoing a disorder-to-order transition induced by DNA binding [151–153]. Interestingly, XPG can exist as a stable homodimer that could bind both ss-ds DNA junctions in the NER bubble, but with only one subunit positioned for its 3’ incision activity [149]. During NER, the ‘other’ subunit could be displaced by TFIIH [87]. An alternate model positions the second XPG subunit with XPF-ERCC1 on the same dsDNA arm, which could serve to regulate the timing of the 3’ incision after the 5’ incision by XPF-ERCC1.
Overview
The long-term goal of biochemical and structural NER research is to elucidate how this complex multi-protein machine recognizes and repairs DNA lesions in our genome. A substantial amount of data has been accumulated over many years, including exciting new 3D structures determined by single particle cryo-EM that provide unprecedented views of the molecular mechanisms of NER factors. While providing fertile ground for hypotheses about NER function and the search for potential NER-targeted chemotherapeutics, these studies fall short of the ultimate objective to truly understand the mechanism of action of the NER machinery. Nevertheless, the outlook for the future is bright. With the development of systems to produce NER factors and a seemingly endless variety of NER substrates, the prospect of building a series of complexes that mimic the NER trajectory is clearly at hand. Characterization of these complexes by a broad range of biochemical, structural and biophysical techniques is anticipated to bring us much closer to our long sought-after objective and provide invaluable information for future efforts directed to NER-targeted drug discovery and advancing the predictive power of NER-focused cancer precision medicine.
Perspective.
(i). The importance of the field
Fundamental knowledge of the mechanism of action of NER machinery is highly desirable as a means to design new therapeutic strategies to inhibit upregulation of NER activity in response to anti-cancer therapies that function by damaging DNA.
(ii). Summary of the current thinking
Despite many years of study and revolutionary new 3D structures that have advanced understanding of the assembly and activity of NER factors, the mechanism of action of the finely tuned NER multi-protein machinery remains an enigma.
(iii). Future directions
Recent progress in the production of the primary factors and substrates required to reconstitute NER in vitro presents tantalizing new possibilities for generating and structurally characterizing the complete trajectory of the multi-step nucleotide excision repair pathway.
Acknowledgements
We thank Orlando Scharer, John Tainer and Susan Tsutakawa for many informative and insightful discussions. NER research in the Chazin laboratory is supported by the US National Institutes of Health (R01 CA092584 and P01 CA092584).
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Lindahl T and Wood RD, Quality control by DNA repair. Science, 1999. 286(5446): p. 1897–1905. [DOI] [PubMed] [Google Scholar]
- 2.Cleaver J, Defective repair replication of DNA in xeroderma pigmentosum. nature, 1968. 218(5142): p. 652–656. [DOI] [PubMed] [Google Scholar]
- 3.Marteijn JA, et al. , Understanding nucleotide excision repair and its roles in cancer and ageing. Nature reviews Molecular cell biology, 2014. 15(7): p. 465–481. [DOI] [PubMed] [Google Scholar]
- 4.Schärer OD, Nucleotide excision repair in eukaryotes. Cold Spring Harbor perspectives in biology, 2013. 5(10): p. a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tornaletti S and Hanawalt PC, Effect of DNA lesions on transcription elongation. Biochimie, 1999. 81(1-2): p. 139–146. [DOI] [PubMed] [Google Scholar]
- 6.Vermeulen W and Fousteri M, Mammalian Transcription-Coupled Repair. Cold Spring Harb Perspect Biol, 2013. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanawalt PC and Spivak G, Transcription-coupled DNA repair: two decades of progress and surprises. Nature reviews Molecular cell biology, 2008. 9(12): p. 958–970. [DOI] [PubMed] [Google Scholar]
- 8.van den Heuvel D, et al. , Transcription-Coupled DNA Repair: From Mechanism to Human Disorder. Trends in Cell Biology, 2021. [DOI] [PubMed] [Google Scholar]
- 9.Marteijn JA, et al. , Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol, 2014. 15(7): p. 465–81. [DOI] [PubMed] [Google Scholar]
- 10.Aboussekhra A, et al. , Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell, 1995. 80(6): p. 859–868. [DOI] [PubMed] [Google Scholar]
- 11.Araújo SJ, et al. , Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes & development, 2000. 14(3): p. 349–359. [PMC free article] [PubMed] [Google Scholar]
- 12.Mu D, et al. , Reconstitution of Human DNA Repair Excision Nuclease in a Highly Defined System (*). Journal of Biological Chemistry, 1995. 270(6): p. 2415–2418. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, et al. , Kinetic gating mechanism of DNA damage recognition by Rad4/XPC. Nature communications, 2015. 6(1): p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shell SM, et al. , Xeroderma pigmentosum complementation group C protein (XPC) serves as a general sensor of damaged DNA. DNA repair, 2013. 12(11): p. 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugasawa K, et al. , A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes & development, 2001. 15(5): p. 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer ES, et al. , The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell, 2011. 147(5): p. 1024–1039. [DOI] [PubMed] [Google Scholar]
- 17.Sugasawa K, et al. , UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell, 2005. 121(3): p. 387–400. [DOI] [PubMed] [Google Scholar]
- 18.Okuda M, et al. , Structural insight into the mechanism of TFIIH recognition by the acidic string of the nucleotide excision repair factor XPC. Structure, 2015. 23(10): p. 1827–1837. [DOI] [PubMed] [Google Scholar]
- 19.Yokoi M, et al. , The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. Journal of Biological Chemistry, 2000. 275(13): p. 9870–9875. [DOI] [PubMed] [Google Scholar]
- 20.Fan L, et al. , Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Mol Cell, 2006. 22(1): p. 27–37. [DOI] [PubMed] [Google Scholar]
- 21.Oksenych V, et al. , Molecular insights into the recruitment of TFIIH to sites of DNA damage. The EMBO journal, 2009. 28(19): p. 2971–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan L, et al. , XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell, 2008. 133(5): p. 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuper J, et al. , Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. The EMBO journal, 2012. 31(2): p. 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, et al. , Structure of the DNA repair helicase XPD. Cell, 2008. 133(5): p. 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathieu N, et al. , DNA quality control by a lesion sensor pocket of the xeroderma pigmentosum group D helicase subunit of TFIIH. Current Biology, 2013. 23(3): p. 204–212. [DOI] [PubMed] [Google Scholar]
- 26.Pugh RA, Wu CG, and Spies M, Regulation of translocation polarity by helicase domain 1 in SF2B helicases. The EMBO journal, 2012. 31(2): p. 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolski SC, et al. , Crystal structure of the FeS cluster–containing nucleotide excision repair helicase XPD. PLoS biology, 2008. 6(6): p. e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isaacs RJ and Spielmann HP, A model for initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA repair, 2004. 3(5): p. 455–464. [DOI] [PubMed] [Google Scholar]
- 29.Tsodikov OV, et al. , Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. The EMBO journal, 2007. 26(22): p. 4768–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tripsianes K, et al. , The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure, 2005. 13(12): p. 1849–1858. [DOI] [PubMed] [Google Scholar]
- 31.De Laat WL, et al. , DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes & development, 1998. 12(16): p. 2598–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fanning E, Klimovich V, and Nager AR, A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic acids research, 2006. 34(15): p. 4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugasawa K, et al. , Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Molecular cell, 1998. 2(2): p. 223–232. [DOI] [PubMed] [Google Scholar]
- 34.Volker M, et al. , Sequential assembly of the nucleotide excision repair factors in vivo. Molecular cell, 2001. 8(1): p. 213–224. [DOI] [PubMed] [Google Scholar]
- 35.Riedl T, Hanaoka F, and Egly JM, The comings and goings of nucleotide excision repair factors on damaged DNA. The EMBO journal, 2003. 22(19): p. 5293–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tapias A, et al. , Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. Journal of Biological Chemistry, 2004. 279(18): p. 19074–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masutani C, et al. , Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. The EMBO journal, 1994. 13(8): p. 1831–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Araki M, et al. , Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. Journal of Biological Chemistry, 2001. 276(22): p. 18665–18672. [DOI] [PubMed] [Google Scholar]
- 39.Sugasawa K, et al. , HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Molecular and cellular biology, 1996. 16(9): p. 4852–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masutani C, et al. , Identification and characterization of XPC-binding domain of hHR23B. Molecular and cellular biology, 1997. 17(12): p. 6915–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishi R, et al. , Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Molecular and cellular biology, 2005. 25(13): p. 5664–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min J-H and Pavletich NP, Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature, 2007. 449(7162): p. 570–575. [DOI] [PubMed] [Google Scholar]
- 43.Nemzow L, et al. , XPC: Going where no DNA damage sensor has gone before. DNA repair, 2015. 36: p. 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emmert S, et al. , The xeroderma pigmentosum group C gene leads to selective repair of cyclobutane pyrimidine dimers rather than 6-4 photoproducts. Proceedings of the National Academy of Sciences, 2000. 97(5): p. 2151–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fitch ME, et al. , In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. Journal of Biological Chemistry, 2003. 278(47): p. 46906–46910. [DOI] [PubMed] [Google Scholar]
- 46.Moser J, et al. , The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA repair, 2005. 4(5): p. 571–582. [DOI] [PubMed] [Google Scholar]
- 47.Wittschieben BØ, Iwai S, and Wood RD, DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. Journal of Biological Chemistry, 2005. 280(48): p. 39982–39989. [DOI] [PubMed] [Google Scholar]
- 48.Matsumoto S, et al. , DNA damage detection in nucleosomes involves DNA register shifting. Nature, 2019. 571(7763): p. 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scrima A, et al. , Structural basis of UV DNA-damage recognition by the DDB1–DDB2 complex. Cell, 2008. 135(7): p. 1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Groisman R, et al. , The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell, 2003. 113(3): p. 357–367. [DOI] [PubMed] [Google Scholar]
- 51.Ribeiro-Silva C, et al. , Ubiquitin and TFIIH-stimulated DDB2 dissociation drives DNA damage handover in nucleotide excision repair. Nature communications, 2020. 11(1): p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, et al. , Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Molecular cell, 2006. 22(3): p. 383–394. [DOI] [PubMed] [Google Scholar]
- 53.Uchida A, et al. , The carboxy-terminal domain of the XPC protein plays a crucial role in nucleotide excision repair through interactions with transcription factor IIH. DNA repair, 2002. 1(6): p. 449–461. [DOI] [PubMed] [Google Scholar]
- 54.van Eeuwen T, et al. , Cryo-EM structure of TFIIH/Rad4–Rad23–Rad33 in damaged DNA opening in nucleotide excision repair. Nature communications, 2021. 12(1): p. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Compe E and Egly J-M, TFIIH: when transcription met DNA repair. Nature reviews Molecular cell biology, 2012. 13(6): p. 343–354. [DOI] [PubMed] [Google Scholar]
- 56.Egly J-M and Coin F, A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA repair, 2011. 10(7): p. 714–721. [DOI] [PubMed] [Google Scholar]
- 57.Fuss JO and Tainer JA, XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA repair, 2011. 10(7): p. 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coin F, et al. , Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Molecular cell, 2008. 31(1): p. 9–20. [DOI] [PubMed] [Google Scholar]
- 59.Fan L, et al. , Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Molecular cell, 2006. 22(1): p. 27–37. [DOI] [PubMed] [Google Scholar]
- 60.Hilario E, et al. , Structure of the C-terminal half of human XPB helicase and the impact of the disease-causing mutation XP11BE. Acta Crystallographica Section D: Biological Crystallography, 2013. 69(2): p. 237–246. [DOI] [PubMed] [Google Scholar]
- 61.Bernardes de Jesus BM, et al. , Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC. Molecular and cellular biology, 2008. 28(23): p. 7225–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Camenisch U, et al. , Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nature structural & molecular biology, 2006. 13(3): p. 278–284. [DOI] [PubMed] [Google Scholar]
- 63.Mathieu N, Kaczmarek N, and Naegeli H, Strand-and site-specific DNA lesion demarcation by the xeroderma pigmentosum group D helicase. Proceedings of the National Academy of Sciences, 2010. 107(41): p. 17545–17550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li C-L, et al. , Tripartite DNA lesion recognition and verification by XPC, TFIIH, and XPA in nucleotide excision repair. Molecular cell, 2015. 59(6): p. 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin YC, Choi WS, and Gralla JD, TFIIH XPB mutants suggest a unified bacterial-like mechanism for promoter opening but not escape. Nature structural & molecular biology, 2005. 12(7): p. 603–607. [DOI] [PubMed] [Google Scholar]
- 66.Coin F, Oksenych V, and Egly J-M, Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Molecular cell, 2007. 26(2): p. 245–256. [DOI] [PubMed] [Google Scholar]
- 67.Coin F, et al. , Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nature genetics, 1998. 20(2): p. 184–188. [DOI] [PubMed] [Google Scholar]
- 68.Giglia-Mari G, et al. , Dynamic interaction of TTDA with TFIIH is stabilized by nucleotide excision repair in living cells. PLoS biology, 2006. 4(6): p. e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Theil AF, et al. , Disruption of TTDA results in complete nucleotide excision repair deficiency and embryonic lethality. PLoS genetics, 2013. 9(4): p. e1003431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greber BJ, et al. , The cryo-electron microscopy structure of human transcription factor IIH. Nature, 2017. 549(7672): p. 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greber BJ, et al. , The complete structure of the human TFIIH core complex. Elife, 2019. 8: p. e44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kokic G, et al. , Structural basis of TFIIH activation for nucleotide excision repair. Nature communications, 2019. 10(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schilbach S, et al. , Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature, 2017. 551(7679): p. 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Constantinescu-Aruxandei D, et al. , Mechanism of DNA loading by the DNA repair helicase XPD. Nucleic acids research, 2016. 44(6): p. 2806–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abdulrahman W, et al. , ARCH domain of XPD, an anchoring platform for CAK that conditions TFIIH DNA repair and transcription activities. Proceedings of the National Academy of Sciences, 2013. 110(8): p. E633–E642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peissert S, et al. , In TFIIH the Arch domain of XPD is mechanistically essential for transcription and DNA repair. Nature communications, 2020. 11(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park C-H, et al. , The General Transcription-Repair Factor TFIIH Is Recruited to the Excision Repair Complex by the XPA Protein Independent of the TFIIE Transcription Factor (*). Journal of Biological Chemistry, 1995. 270(9): p. 4896–4902. [DOI] [PubMed] [Google Scholar]
- 78.Tsutakawa SE, et al. , Envisioning how the prototypic molecular machine TFIIH functions in transcription initiation and DNA repair. DNA repair, 2020. 96: p. 102972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.White MF, Structure, function and evolution of the XPD family of iron–sulfur–containing 5’→ 3’ DNA helicases. 2009, Portland Press Ltd. [DOI] [PubMed] [Google Scholar]
- 80.Sontz PA, et al. , DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proceedings of the National Academy of Sciences, 2012. 109(6): p. 1856–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wakasugi M and Sancar A, Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proceedings of the National Academy of Sciences, 1998. 95(12): p. 6669–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sugasawa K, et al. , Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Molecular cell, 2009. 36(4): p. 642–653. [DOI] [PubMed] [Google Scholar]
- 83.Evans E, et al. , Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. The EMBO journal, 1997. 16(3): p. 625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li L, et al. , An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Molecular and cellular biology, 1995. 15(10): p. 5396–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saijo M, Takedachi A, and Tanaka K, Nucleotide excision repair by mutant xeroderma pigmentosum group A (XPA) proteins with deficiency in interaction with RPA. Journal of Biological Chemistry, 2011. 286(7): p. 5476–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rademakers S, et al. , Xeroderma pigmentosum group A protein loads as a separate factor onto DNA lesions. Molecular and Cellular Biology, 2003. 23(16): p. 5755–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Araújo SJ, Nigg EA, and Wood RD, Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Molecular and cellular biology, 2001. 21(7): p. 2281–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dunand-Sauthier I, et al. , The spacer region of XPG mediates recruitment to nucleotide excision repair complexes and determines substrate specificity. Journal of Biological Chemistry, 2005. 280(8): p. 7030–7037. [DOI] [PubMed] [Google Scholar]
- 89.Yang Z, et al. , Specific and efficient binding of Xeroderma pigmentosum complementation group A to double-strand/single-strand DNA junctions with 3 ‘- and/or 5 ‘-ssDNA branches. Biochemistry, 2006. 45(51): p. 15921–15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li L, et al. , Specific association between the human DNA repair proteins XPA and ERCC1. Proceedings of the National Academy of Sciences, 1994. 91(11): p. 5012–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nocentini S, et al. , DNA damage recognition by XPA protein promotes efficient recruitment of transcription factor II H. Journal of Biological Chemistry, 1997. 272(37): p. 22991–22994. [DOI] [PubMed] [Google Scholar]
- 92.You J-S, Wang M, and Lee S-H, Biochemical analysis of the damage recognition process in nucleotide excision repair. Journal of Biological Chemistry, 2003. 278(9): p. 7476–7485. [DOI] [PubMed] [Google Scholar]
- 93.Bunick CG, et al. , Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry, 2006. 45(50): p. 14965–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wakasugi M, et al. , Physical and functional interaction between DDB and XPA in nucleotide excision repair. Nucleic acids research, 2009. 37(2): p. 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gilljam KM, et al. , Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PloS one, 2012. 7(11): p. e49199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sugitani N, et al. , Redefining the DNA-binding domain of human XPA. Journal of the American Chemical Society, 2014. 136(31): p. 10830–10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fadda E, Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Computational and structural biotechnology journal, 2016. 14: p. 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sugitani N, et al. , XPA: A key scaffold for human nucleotide excision repair. DNA repair, 2016. 44: p. 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Topolska-Woś AM, et al. , A key interaction with RPA orients XPA in NER complexes. Nucleic acids research, 2020. 48(4): p. 2173–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li L, et al. , An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol Cell Biol, 1995. 15(10): p. 5396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen R and Wold MS, Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays, 2014. 36(12): p. 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brosey CA, et al. , A new structural framework for integrating replication protein A into DNA processing machinery. Nucleic acids research, 2013. 41(4): p. 2313–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brosey CA, et al. , Functional dynamics in replication protein A DNA binding and protein recruitment domains. Structure, 2015. 23(6): p. 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Krasikova YS, et al. , Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucleic acids research, 2010. 38(22): p. 8083–8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Patrick SM and Turchi JJ, Replication protein A (RPA) binding to duplex cisplatin-damaged DNA is mediated through the generation of single-stranded DNA. Journal of Biological Chemistry, 1999. 274(21): p. 14972–14978. [DOI] [PubMed] [Google Scholar]
- 106.Mer G, et al. , Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell, 2000. 103(3): p. 449–456. [DOI] [PubMed] [Google Scholar]
- 107.Lieber MR, The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays, 1997. 19(3): p. 233–40. [DOI] [PubMed] [Google Scholar]
- 108.Scherly D, et al. , Complementation of the DNA repair defect in xeroderma pigmentosum group G cells by a human cDNA related to yeast RAD2. Nature, 1993. 363(6425): p. 182–5. [DOI] [PubMed] [Google Scholar]
- 109.Hohl M, et al. , Structural determinants for substrate binding and catalysis by the structure-specific endonuclease XPG. J Biol Chem, 2003. 278(21): p. 19500–8. [DOI] [PubMed] [Google Scholar]
- 110.Evans E, et al. , Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J, 1997. 16(3): p. 625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dunand-Sauthier I, et al. , The spacer region of XPG mediates recruitment to nucleotide excision repair complexes and determines substrate specificity. J Biol Chem, 2005. 280(8): p. 7030–7. [DOI] [PubMed] [Google Scholar]
- 112.Hohl M, et al. , Domain swapping between FEN-1 and XPG defines regions in XPG that mediate nucleotide excision repair activity and substrate specificity. Nucleic Acids Res, 2007. 35(9): p. 3053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Iyer N, et al. , Interactions involving the human RNA polymerase II transcription/nucleotide excision repair complex TFIIH, the nucleotide excision repair protein XPG, and Cockayne syndrome group B (CSB) protein. Biochemistry, 1996. 35(7): p. 2157–67. [DOI] [PubMed] [Google Scholar]
- 114.Thorel F, et al. , Definition of a short region of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol, 2004. 24(24): p. 10670–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Matsunaga T, et al. , Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nuclease. J Biol Chem, 1996. 271(19): p. 11047–50. [DOI] [PubMed] [Google Scholar]
- 116.He Z, et al. , RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature, 1995. 374(6522): p. 566–9. [DOI] [PubMed] [Google Scholar]
- 117.Bardwell L, et al. , Yeast RAD3 protein binds directly to both SSL2 and SSL1 proteins: implications for the structure and function of transcription/repair factor b. Proc Natl Acad Sci U S A, 1994. 91(9): p. 3926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Staresincic L, et al. , Coordination of dual incision and repair synthesis in human nucleotide excision repair. The EMBO journal, 2009. 28(8): p. 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Westerveld A, et al. , Molecular cloning of a human DNA repair gene. Nature, 1984. 310(5976): p. 425–9. [DOI] [PubMed] [Google Scholar]
- 120.Park CH, et al. , Purification and Characterization of the Xpf-Ercc1 Complex of Human DNA-Repair Excision Nuclease. Journal of Biological Chemistry, 1995. 270(39): p. 22657–22660. [DOI] [PubMed] [Google Scholar]
- 121.O’Donovan A, et al. , Isolation of active recombinant XPG protein, a human DNA repair endonuclease. J Biol Chem, 1994. 269(23): p. 15965–8. [PubMed] [Google Scholar]
- 122.Bessho T, et al. , Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J Biol Chem, 1997. 272(6): p. 3833–7. [DOI] [PubMed] [Google Scholar]
- 123.O’Donovan A, et al. , XPG endonuclease makes the 3′ incision in human DNA nucleotide excision repair. Nature, 1994. 371(6496): p. 432–5. [DOI] [PubMed] [Google Scholar]
- 124.Mu D, Hsu DS, and Sancar A, Reaction mechanism of human DNA repair excision nuclease. J Biol Chem, 1996. 271(14): p. 8285–94. [DOI] [PubMed] [Google Scholar]
- 125.Mu D, et al. , Characterization of reaction intermediates of human excision repair nuclease. J Biol Chem, 1997. 272(46): p. 28971–9. [DOI] [PubMed] [Google Scholar]
- 126.Riedl T, Hanaoka F, and Egly JM, The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J, 2003. 22(19): p. 5293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Staresincic L, et al. , Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J, 2009. 28(8): p. 1111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Volker M, et al. , Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell, 2001. 8(1): p. 213–24. [DOI] [PubMed] [Google Scholar]
- 129.Wakasugi M, Reardon JT, and Sancar A, The non-catalytic function of XPG protein during dual incision in human nucleotide excision repair. J Biol Chem, 1997. 272(25): p. 16030–4. [DOI] [PubMed] [Google Scholar]
- 130.Wakasugi M and Sancar A, Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc Natl Acad Sci U S A, 1998. 95(12): p. 6669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ziani S, et al. , Sequential and ordered assembly of a large DNA repair complex on undamaged chromatin. J Cell Biol, 2014. 206(5): p. 589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Orelli B, et al. , The XPA-binding domain of ERCC1 is required for nucleotide excision repair but not other DNA repair pathways. J Biol Chem, 2010. 285(6): p. 3705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tsodikov OV, et al. , Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J, 2007. 26(22): p. 4768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Enzlin JH and Scharer OD, The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J, 2002. 21(8): p. 2045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fagbemi AF, Orelli B, and Scharer OD, Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair (Amst), 2011. 10(7): p. 722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gaillard PHL and Wood RD, Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair. Nucleic Acids Res, 2001. 29(4): p. 872–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sijbers AM, et al. , Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res, 1996. 24(17): p. 3370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Biggerstaff M, Szymkowski DE, and Wood RD, Co-correction of the ERCC1, ERCC4 and xeroderma pigmentosum group F DNA repair defects in vitro. EMBO J, 1993. 12(9): p. 3685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sijbers AM, et al. , Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell, 1996. 86(5): p. 811–22. [DOI] [PubMed] [Google Scholar]
- 140.van Vuuren AJ, et al. , Evidence for a repair enzyme complex involving ERCC1 and complementing activities of ERCC4, ERCC11 and xeroderma pigmentosum group F. EMBO J, 1993. 12(9): p. 3693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.de Laat WL, et al. , Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res, 1998. 26(18): p. 4146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tripsianes K, et al. , The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure, 2005. 13(12): p. 1849–58. [DOI] [PubMed] [Google Scholar]
- 143.de Laat WL, et al. , DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev, 1998. 12(16): p. 2598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Su Y, et al. , Multiple DNA binding domains mediate the function of the ERCC1-XPF protein in nucleotide excision repair. J Biol Chem, 2012. 287(26): p. 21846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tsodikov OV, et al. , Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proc Natl Acad Sci U S A, 2005. 102(32): p. 11236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nishino T, et al. , Structural and functional analyses of an archaeal XPF/Rad1/Mus81 nuclease: asymmetric DNA binding and cleavage mechanisms. Structure, 2005. 13(8): p. 1183–92. [DOI] [PubMed] [Google Scholar]
- 147.Newman M, et al. , Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J, 2005. 24(5): p. 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jones M, et al. , Cryo-EM structures of the XPF-ERCC1 endonuclease reveal how DNA-junction engagement disrupts an auto-inhibited conformation. Nature communications, 2020. 11(1): p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tsutakawa SE, et al. , Human XPG nuclease structure, assembly, and activities with insights for neurodegeneration and cancer from pathogenic mutations. Proceedings of the National Academy of Sciences, 2020. 117(25): p. 14127–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.González-Corrochano R, et al. , The crystal structure of human XPG, the xeroderma pigmentosum group G endonuclease, provides insight into nucleotide excision DNA repair. Nucleic acids research, 2020. 48(17): p. 9943–9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tsutakawa SE, et al. , Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell, 2011. 145(2): p. 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Orans J, et al. , Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell, 2011. 145(2): p. 212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hohl M, et al. , Structural determinants for substrate binding and catalysis by the structure-specific endonuclease XPG. Journal of Biological Chemistry, 2003. 278(21): p. 19500–19508. [DOI] [PubMed] [Google Scholar]