

Abstract
The β-galactosidase from the Antarctic gram-negative bacterium Pseudoalteromonas haloplanktis TAE 79 was purified to homogeneity. The nucleotide sequence and the NH2-terminal amino acid sequence of the purified enzyme indicate that the β-galactosidase subunit is composed of 1,038 amino acids with a calculated Mr of 118,068. This β-galactosidase shares structural properties with Escherichia coli β-galactosidase (comparable subunit mass, 51% amino sequence identity, conservation of amino acid residues involved in catalysis, similar optimal pH value, and requirement for divalent metal ions) but is characterized by a higher catalytic efficiency on synthetic and natural substrates and by a shift of apparent optimum activity toward low temperatures and lower thermal stability. The enzyme also differs by a higher pI (7.8) and by specific thermodynamic activation parameters. P. haloplanktis β-galactosidase was expressed in E. coli, and the recombinant enzyme displays properties identical to those of the wild-type enzyme. Heat-induced unfolding monitored by intrinsic fluorescence spectroscopy showed lower melting point values for both P. haloplanktis wild-type and recombinant β-galactosidase compared to the mesophilic enzyme. Assays of lactose hydrolysis in milk demonstrate that P. haloplanktis β-galactosidase can outperform the current commercial β-galactosidase from Kluyveromyces marxianus var. lactis, suggesting that the cold-adapted β-galactosidase could be used to hydrolyze lactose in dairy products processed in refrigerated plants.
Enzymes from psychrophilic organisms are in general quite efficient in compensating for the reduction of reaction rates induced by low temperatures through improvement of the turnover number (kcat) or of the physiological efficiency (kcat/Km). It is thought that optimization of the catalytic parameters originates from a higher flexibility of crucial parts of the molecular edifice, providing an enhanced ability to undergo conformational changes at low energy cost during catalysis. Cold-adapted enzymes are also characterized by a thermal instability which is regarded as a consequence of their conformational flexibility (6). The gain in reaction rate which usually covers the temperature range from 0 to 30°C is due to a decrease in the activation energy, induced by a decrease in the activation enthalpy, itself partially compensated by an unfavorable modification of the activation entropy compared to mesophilic enzymes (13). The adaptation of the molecular structure mainly consists in a decrease of the number of strength of intramolecular interactions and in some cases in a better accessibility of the catalytic cavity (7).
In the context of the study of protein adaptation to low temperatures, an Antarctic bacterial strain producing a β-galactosidase was collected in an environment displaying an average temperature of −1°C. β-d-Galactosidase (β-d-galactoside galactohydrolase; EC 3.2.1.23) catalyzes the hydrolysis of β-1,4-d galactosidic linkages. This enzyme is widely distributed in nature, being found in numerous microorganisms and in plant and animal tissues (30). Escherichia coli β-galactosidase is one of the most thoroughly studied enzyme because the Lac operon has played a central role in the elucidation of the genetic control of gene regulation in E. coli (2, 17). This enzyme is composed of four identical protomers with a molecular mass of 116,248 Da (9), each containing one catalytic site (4); each active site is made up of elements from two different subunits (11). Elucidation of its three-dimensional structure (11) provides an excellent foundation for examining and comparing the structures of other β-galactosidases.
The enzyme catalyzing the hydrolysis of lactose into its constituent monosaccharides, glucose and galactose, has attracted the attention of researchers and the dairy industry because of nutritional (lactose intolerance), technological (crystallization), and environmental (pollution) problems associated with this major milk carbohydrate (29). Cold β-galactosidases capable of hydrolyzing lactose in milk or whey at low temperatures have been studied to some extent (28), but information remains sparse. For treatment of milk, pH and temperature are the most important conditions for sustained enzyme activity. An ideal β-galactosidase should be active at pH 6.7 to 6.8 and at 4 to 8°C during shipping and storage. In this respect, we report here the characterization of the cold-active β-galactosidase from the Antarctic bacteria Pseudoalteromonas haloplanktis.
MATERIALS AND METHODS
Bacterial strain and culture conditions.
The Antarctic bacterium P. haloplanktis TAE 79 was isolated from seawater on necrosed algae at the J. S. Dumont d'Urville Antarctic station (60°40'S; 40°01'E). Screening for β-galactosidase activity was carried out on L-agar plates containing per liter, 10 g of Bacto Tryptone, 5 g of yeast extract, 25 g of sea salts, and 17 g of agar (Difco, Detroit, Mich.), with 0.2% lactose and 32 mg of X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) with or without 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Growth properties were studied in modified L broth (10 g of tryptone, 5 g of yeast extract, and 30 g of sea salts in 1 liter at pH 7.5) containing 2% lactose. E. coli β-galactosidase was from Sigma (G2513).
Enzyme assay.
Assay of β-galactosidase activity was carried out using 3 mM ONPG (o-nitrophenyl-β-galactopyranoside) as a chromogenic substrate in 100 mM sodium phosphate buffer–1 mM MgCl2–100 mM 2-mercaptoethanol, pH 7.5. Activities were recorded in a thermostated Uvikon 860 Spectrophotometer (Kontron, Zurich, Switzerland) at 25°C and calculated on the basis of an extinction coefficient for o-nitrophenol of 3.5 mM−1 cm−1 at 410 nm (16). Assays for activity on lactose were carried out in the same buffer, but the reaction was stopped by boiling the sample for 3 min, and the galactose dehydrogenase assay was used to measure the amount of galactose released by the enzyme (24). The specific activity is defined as the number of micromoles of galactose released per minute per milligram of protein. Km values were recorded using substrate concentrations ranging from 0.1 to 20 Km.
Hydrolysis of lactose in milk.
Kluyveromyces marxianus var. lactis β-galactosidase was from Gist-Brocades (MA Delft, The Netherlands). Five micrograms of β-galactosidase from K. marxianus or from P. haloplanktis was added to 500 μl of diluted skimmed milk, and the mixture was incubated at the desired temperature. The reaction was stopped by boiling the sample for 3 min, and 0.5% sulfosalicylic acid was then added for protein precipitation. The sample was neutralized with NaOH and filtered. Determination of lactose and d-galactose was carried out using the lactose/d-galactose UV method of Boehringer Mannheim (Mannheim, Germany).
β-Galactosidase purification.
The Antarctic strain was cultivated at 4°C for 5 days in 2 liters of modified L broth containing 2% lactose. After 44 h, the culture was induced by 1 mM IPTG and reincubated for a further 68 h. The cells were harvested by centrifugation at 12,000 × g for 60 min at 4°C and resuspended in 200 ml of buffer A (50 mM 3-morpholinepropanesulfonic acid [MOPS], 1 mM MgCl2, 1 mM MnCl2, 10 mM 2-mercaptoethanol [pH 7.5]). The cell extract was prepared by cell disintegration using an LH-SGI Inceltech (Wokingham Berkshire, England) disruptor, then phenylmethylsulfonyl fluoride (1 mM, final concentration) was added to the crude extract, and debris was removed by centrifugation at 15,000 × g for 30 min. The supernatant was then treated for 2 h with protamine sulfate at a final concentration of 1 g/liter to remove nucleic acids. After centrifugation for 30 min at 27,000 × g, the supernatant was dialyzed twice against 2 liters of buffer A and then loaded on a DEAE-Sepharose column (2.5 by 35 cm) equilibrated in the same buffer and eluted with an NaCl linear gradient (500 to 500 ml, 0 to 1 M NaCl). Fractions containing β-galactosidase activity were pooled and concentrated to 20 ml by ultrafiltration on a 100-kDa molecular mass limit PTHK membrane (Millipore, Bedford, Mass.), followed by two cycles of dilution with 50 ml of buffer A and concentration. The sample was then loaded to an affinity matrix (26) of agarose (3.5 by 5 cm) derivatized with p-aminobenzyl-1-thio-β-d-galactopyranoside (Sigma A0414). After a wash with 200 ml of 1 M KCl in buffer A, elution was carried out using 100 mM lactose–1 M KCl in buffer A. The active fractions were pooled and applied on a Sephadex G-25 column (1.8 by 20 cm) equilibrated with buffer A.
Analytical procedures.
Protein concentrations were determined by the method of Bradford (3), using bovine serum albumin (Pierce, Rockford, Ill.) as a standard. For the purified enzymes, the extinction coefficients at 280 nm used were 241,590 M−1 cm−1 for β-galactosidase from E. coli and 195,000 M−1 cm−1 for β-galactosidase from P. haloplanktis. The NH2-terminal amino acid sequence of the P. haloplanktis β-galactosidase was determined using a pulsed liquid-phase protein sequencer (Procise 492; Applied Biosystems Foster City, Calif.). Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and isoelectric focusing were run essentially as described by the supplier of the electrophoresis equipment (Hoefer Scientific Instruments, San Francisco, Calif.). The activation energy (Ea) was determined from the slope (−Ea/R) of the Arrhenius plot, and the thermodynamic activation parameters of the reaction (in kilojoules per mole) were calculated according to the following equations (13):
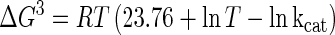 |
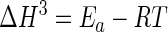 |
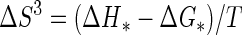 |
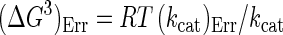 |
Subscript “Err” denotes standard deviation. Results are based on three activity determinations at 10 different temperatures.
The Ca2+ binding constant was determined in 120 mM MOPS–90 mM KCl–2 mM EDTA–5 mM MgCl2 (pH 7.0) containing 5 μg of β-galactosidase from either E. coli or P. haloplanktis TAE 79. Several samples were prepared in which the free Ca2+ concentration was set from pCa2+ 8.3 to 3.5 upon addition of CaCl2 (40 mM) and according to the computer program (23), making use of the Smith and Martel stability constants (25). After an incubation time of 10 min, ONPG (30 mM) was added and β-galactosidase activity was recorded. Hill equation was used to fit the data points as described elsewhere (10). The Mg2+ binding constant was measured under identical conditions in 120 mM MOPS–90 mM KCl–2 mM EDTA (pH 7.0) but in the absence of added Ca2+ (free Ca2+ below pCa 10). Free Mg2+ (pMg2+ from 7 to 2) was set using a 500 mM MgCl2 stock solution.
DNA purification and cloning.
DNA from P. haloplanktis was isolated by a modification of the method of Brahamsha and Greenberg (3a). Lysozyme concentration was increased to 1 mg/ml, and the cells were treated for 30 min at 37°C. The extract was then incubated for 1 h at 55°C in 0.5% SDS containing proteinase K (1 μg/ml, final concentration). The resulting lysate was then extracted three times with an equal volume of phenol-chloroform (50% [vol/vol]) followed by chloroform extraction. DNA was precipitated with ethanol and suspended in TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8]).
The genomic DNA was digested with Sau3AI, HindIII, PstI, or SphI, and the resulting fragments were inserted into the corresponding sites of pSP73 (Promega). The ligated DNA was transformed in E. coli DH5α, and clones were selected on L-agar plates containing 100 μg of ampicillin/ml, 0.01% X-Gal, and 100 μM IPTG. After 2 days at 25°C, β-galactosidase-positive colonies appeared blue. The DNA fragment containing the β-galactosidase gene (9 kb) was subcloned into the polylinker of pSP73 by digestion with EcoRI and plasmid self-ligation. For DNA sequencing, the subclone EcoRI was ligated in pK19 (21). DNA sequencing was done by the chromosome walking technique with 5′ fluorescein-labeled primers. The products of the sequencing reaction were analyzed on an ALF DNA sequencer (Pharmacia). Synthetic oligonucleotides used as primers were from Eurogentec S.A. (Liege, Belgium).
Construction of the β-galactosidase expression vector.
The lacZ gene was amplified by PCR using Vent DNA polymerase (New England Biolabs, Beverley, Mass.) with primer 5′-GCAACAGGAATACATATGACCTCTTTACAGCAC-3′, which contains an engineered NdeI site (underlined), and reverse primer 5′-GTAAACAGGTTAAGTTGTAATCCCCCCAG-3′, which contains the stop codon (underlined). The PCR product was cloned into the pPCR-Script Amp SK(+) cloning vector with PCR-Script Amp cloning kit (Stratagene, La Jolla, Calif.), transformed in E. coli RR1 cells, and sequenced. This construction was then digested separately with NdeI plus SalI and SalI plus XhoI, and the two fragments corresponding to the β-galactosidase gene were then ligated into the NdeI and XhoI sites of the expression vector pET22b (Novagen, Madison, Wis.). The resulting recombinant plasmid was transformed in E. coli NovaBlue(DE3) competent cells.
Production and purification of the recombinant β-galactosidase.
The recombinant β-galactosidase was produced using the expression T7 system (27). E. coli NovaBlue competent cells (Novagen) carrying the expression vector pET22b-β-galactosidase were grown at 18°C in L broth containing 100 mg of ampicillin liter. When A595 reached 0.6, expression of the enzyme was induced by IPTG to a final concentration of 1 mM, and the cells were further cultivated at 18°C for 20 h. The cells were then harvested by centrifugation and resuspended in buffer A. The recombinant enzyme was purified by the procedure described for the wild-type enzyme.
Thermal unfolding.
Heat-induced unfolding of the wild-type and recombinant β-galactosidase in buffer A was analyzed by fluorescence spectroscopy. The change in fluorescence emission at 330 nm was recorded after excitation at 280 nm using an SLW-Aminco spectrophotometer (Aminco, Rochester, N.Y.) at a scan rate of 1°C/min. The pre- and posttransition baseline slopes were used to normalize the raw data as described elsewhere (20).
Nucleotide sequence accession number.
The EMBL accession number for the sequence reported in this article is AJ131635.
RESULTS
Selection of P. haloplanktis and culture conditions.
About 300 bacterial isolates collected in Antarctica were screened for X-Gal hydrolysis on plates at 4°C. P. haloplanktis TAE 79 displayed the highest intracellular β-galactosidase activity and was selected for further analysis. Culture medium containing 20 g of lactose and 30 g of sea salts in 1 liter of L broth appeared to be the optimal medium for both growth and β-galactosidase production. Addition of sea salts to the culture medium enhanced the growth of P. haloplanktis TAE 79 by a factor of 10, whereas the addition of 1 mM IPTG to the culture medium after 44 h increases β-galactosidase activity by a factor of 2 to 3. P. haloplanktis TAE 79 grows well between 0 and 25°C but not at 30°C. Temperatures higher than 4°C induced faster growth; however, β-galactosidase activity at the stationary phase decreased concomitantly with the increase in culture temperature (8, 6, and 4 U/ml at 4, 12, and 25°C, respectively). Thus, both the highest cell density and maximal β-galactosidase activity were obtained at 4°C. Accordingly, P. haloplanktis TAE 79 can be defined as a psychrophilic microorganism (18).
Purification and characterization of P. haloplanktis β-galactosidase.
The different purification steps and recovery are shown in Table 1. Affinity chromatography on p-aminobenzyl-1-thio-β-d-galactopyranoside agarose was required to remove remaining contaminants and denatured β-galactosidase. Following this procedure, the enzyme is 99% pure as determined by SDS-PAGE and has an estimated apparent molecular mass of 118 kDa. Ultrafiltration trials showed that the β-galactosidase is retained on an ultrafiltration membrane with a cutoff of 300 kDa. The isoelectric point of P. haloplanktis β-galactosidase was determined as 7.8 by isoelectric focusing. This value is higher than the acidic pI of 4.6 for E. coli β-galactosidase (30). Both enzymes have a broad pH stability, ranging from 6.5 to 10 in Michaelis's barbital sodium acetate buffer and Sorensen's glycine II buffer. In the Good's series {50 mM MOPS, morpholineethanesulfonic acid (MES), Tris, or (2-[N-cyclohexylamino]ethanesulfonic acid)}, the enzyme stability is slightly higher in MOPS buffer at pH 7.5 and in MES buffer at pH 7.
TABLE 1.
Purification of the intracellular β-galactosidase from a 2-liter culture of P. haloplanktis
| Purification step | Vol (ml) | Protein (mg) | Total activity (U)a | Sp act (U/mg) | Recovery (%) | Purification (fold) |
|---|---|---|---|---|---|---|
| Cell extract | 200 | 1,480 | 3,477 | 2.3 | 100 | 1 |
| DEAE-agarose | 80 | 178 | 1,550 | 8.7 | 45 | 3.7 |
| Affinity chromatography | 6 | 9 | 1,254 | 138.2 | 36 | 58.8 |
One unit of β-galactosidase activity is defined as the amount of enzyme required to release 1 μmol of o-nitrophenol per min at pH 7.3 and at 20°C.
The pH optimum for the P. haloplanktis β-galactosidase activity was found to be pH 8.5, which is similar to that of the E. coli enzyme (pH 8.0). Both P. haloplanktis and E. coli β-galactosidase require divalent cations for optimal activity. Addition of 5 mM EDTA into the reaction mixture results in 90% loss of the initial activity. Excess Mg2+, Mn2+, Li2+, and Ca2+ restored the activity of both enzymes. By contrast, the presence of 1 mM Zn2+, Cu2+, and Ni2+ in the reaction medium strongly inhibited P. haloplanktis β-galactosidase. The binding constants for Mg2+ and Ca2+ have been determined by activation kinetics (Fig. 1). Calcium titration yielded a slightly lower affinity for the P. haloplanktis enzyme (Ka = 1.2 106 M−1) than for E. coli β-galactosidase (Ka = 6.2 106 M−1), whereas affinity for Mg2+ was the same for both enzymes (Ka = 2.5 105 M−1). Optimal activity of P. haloplanktis and E. coli β-galactosidases was obtained with 40 to 100 mM 2-mercaptoethanol in the reaction medium. At these concentrations, the reducing agent stimulated twofold the activity of both enzymes.
FIG. 1.

Magnesium-dependent activity curves for P. haloplanktis (●) and E. coli (○) β-galactosidases. Activity was recorded using the synthetic substrate ONPG. pMg2+ = −log [Mg2+].
Thermodependence, stability, and kinetic parameters of P. haloplanktis β-galactosidase.
The effect of temperature on β-galactosidase activity was determined by assaying the enzyme at various temperatures from 5 to 60°C using ONPG as a substrate. The psychrophilic β-galactosidase shows a shift of the apparent optimal temperature of activity by about 10°C toward low temperatures compared to the E. coli enzyme (Fig. 2A). At 20°C, the kcat of the P. haloplanktis enzyme is twice as high as that of the E. coli enzyme. These curves have been used to construct Arrhenius plots and to calculate the activation energy parameters of the reaction (Table 2). The lower free energy of activation (ΔG*) of the psychrophilic β-galactosidase correlates well with its higher specific activity. However, the contributions of the enthalpy term (ΔH*) and of the entropy term (TΔS*) to ΔG* differ in psychrophilic and mesophilic enzymes. As shown in Fig. 2B, the apparent Km for ONPG sharply increases at temperatures higher than 15°C in the case of P. haloplanktis β-galactosidase. As a result, the physiological efficiency or specificity constant (kcat/Km) is markedly affected at these temperatures, whereas this ratio is about three times higher for the cold-active enzyme at 10°C. The kinetic parameters of both enzymes were also determined at 25°C with lactose as a substrate (Table 3). Hydrolysis of the natural substrate by the cold-active β-galactosidase was much more efficient regarding both reaction rate and apparent affinity. As a result, the kcat/Km ratio of the cold-adapted enzyme was 90 times higher than that of the E. coli β-galactosidase.
FIG. 2.

Thermodependence of the activity of P. haloplanktis (○) and E. coli (●) β-galactosidases. Shown are turnover number (A) and physiological efficiency (B) as a function of temperature, using ONPG as a substrate.
TABLE 2.
Kinetic and thermodynamic activation parameters of β-galactosidases from P. haloplanktis and E. coli at 20°C, using ONPG as a substrate
| Species | Mean ± SD
|
||||
|---|---|---|---|---|---|
| kcat (s−1) | Ea (kJ mol−1) | ΔG* (kJ mol−1) | ΔH* (kJ mol−1) | TΔS* (kJ mol−1 K−1) | |
| P. haloplanktis | 203 ± 11 | 20.8 ± 1.4 | 58.7 ± 0.1 | 18.3 ± 1 | −40.4 ± 0.9 |
| E. coli | 99.5 ± 2 | 26 ± 0.9 | 60.5 ± 0.05 | 23.6 ± 1 | −36.9 ± 0.9 |
TABLE 3.
Kinetic parameters for P. haloplanktis and E. coli β-galactosidases determined at 25°C, using the natural substrate lactose
| Species | kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) |
|---|---|---|---|
| P. haloplanktis | 33 | 2.4 | 13.7 |
| E. coli | 2 | 13 | 0.15 |
Comparison of lactose removal in milk was carried out using identical concentrations of P. haloplanktis β-galactosidase and of the current commercial Kluyveromyces lactis β-galactosidase from yeast. After 30 min of incubation at 25°C, 26% of milk lactose was hydrolyzed by P. haloplanktis β-galactosidase and 16% was hydrolyzed by the commercial enzyme. After 50 min of incubation at 4°C, 33% of milk lactose was hydrolyzed by the psychrophilic enzyme and only 12% was hydrolyzed by the yeast β-galactosidase.
Cloning and nucleotide sequence of the P. haloplanktis β-galactosidase gene.
The plasmid, pSP73, used for cloning lacks the lacZα fragment which could complement the deleted E. coli DH5α β-galactosidase. From colonies screened at 25°C, we obtained three β-galactosidase-positive colonies, all carrying a PstI-cleaved genomic DNA fragment of nearly 9 kb. Based on blue color development on plate, an EcoRI-PstI fragment was found to be the smallest fragment encoding β-galactosidase activity. Within this 5,088-bp fragment, we found a singlelarge open reading frame, starting with an ATG codon at nucleotide 1531 and ending with a TAG at nucleotide 4649. The first NH2-terminal amino acids of the native protein determined by Edman degradation are recognized following the ATG codon (Fig. 3). Therefore, the protein contains 1,038 amino acids with a calculated Mr of 118,068. Upstream from the ATG codon, a partial open reading frame showing homology with the lactose operon transcription activator from Staphylococcus xylosus was found on the complementary strand (1).
FIG. 3.

Alignment of amino acid sequences of bacterial β-galactosidases from E. coli, P. haloplanktis (TAE79), Arthrobacter strain B7 (Artsp) and Thermotoga maritima (Thema). Arrows indicate residues of the catalytic site (Glu 461, Glu 537, Met 502 and Tyr 503).
The deduced amino acid sequence of the P. haloplanktis β-galactosidase showed 51% identity with E. coli LacZ. The alignment showed that the proposed active-site residues in E. coli LacZ; i.e., Glu 461, Glu 537, Met 502, and Tyr 503 (14) are conserved in the P. haloplanktis sequence. Alignment with other β-galactosidases showed significant homology surrounding Glu 461 and Glu 537, forming the consensus sequences WSLGNE and ILCEYAHAMGN, respectively (Fig. 3). β-Galactosidase protein sequence analysis allowed identification of structural features encountered in psychrophilic enzymes. The cold-active β-galactosidase is characterized by an arginine content (3.8% versus 6.5%) and a Arg/Arg + Lys ratio (0.5% versus 0.77%) lower than those calculated for E. coli β-galactosidase. The proline content is also lower for the psychrophilic enzyme (4.4% versus 6.2%), whereas its glycine content is higher within the 15 amino acids surrounding the catalytic residue Glu 461. Alignment with E. coli LacZ showed three insertions in the P. haloplanktis β-galactosidase. These insertions of four, five, and nine additional residues occur after residues Glu 76, Gln 632, and Asn 736 respectively.
Heterologous expression in E. coli and thermal unfolding.
The coding sequence of the P. haloplanktis β-galactosidase was cloned at the NdeI site of plasmid pET22b and expressed in E. coli. The N-terminal amino acid sequence determined by Edman degradation shows that the first amino acid of the recombinant enzyme is the expected threonine. Measured at 25°C, the specific activity of the recombinant enzyme was similar to that of the wild-type β-galactosidase. Thermal inactivation of the recombinant β-galactosidase, compared with the wild-type and mesophilic enzymes, was analyzed by recording the residual activity after various incubation times at 45°C (Fig. 4). The half-lives of activity of the wild-type and the recombinant enzymes are similar; both enzymes exhibit a highly reduced thermostability compared to the mesophilic counterpart from E. coli. Heat-induced unfolding of the wild-type, the recombinant, and the E. coli β-galactosidases was monitored by fluorescence spectroscopy. Wild-type and recombinant enzymes have comparable melting points (48 and 49.8°C, respectively), which are lower than that of the E. coli enzyme (56.5°C). The three β-galactosidases display similar cooperative transition (Fig. 5). However, both the activity and the stability of the recombinant enzyme are slightly lower than those of the wild-type β-galactosidase. The occurrence of marginal misfolding, resulting from the expression at 18°C for instance, cannot be ruled out.
FIG. 4.

Thermal stability of activities of β-galactosidases from E. coli (●) and P. haloplanktis (○) and of the recombinant enzyme (▴) at 45°C. Enzymes were incubated for the indicated periods of time, and residual activities were determined using ONPG as a substrate.
FIG. 5.

Thermal unfolding of β-galactosidases from P. haloplanktis (●) and E. coli (▴) and of the recombinant enzyme (○). The fraction of the protein in the unfolded state (fu) was calculated as follows: fu = (yF − y)/(yF − yu), where yF and yu are the fluorescence intensities of the native and fully unfolded states, respectively, and y is the fluorescence intensity at a given temperature.
DISCUSSION
In the context of the study of the adaptation of psychrophilic enzymes to low temperatures, we have characterized a β-galactosidase from an Antarctic bacterial strain. P. haloplanktis, a gram-negative bacterium, displays the characteristics of a microorganism adapted to cold (18). Indeed, it does not grow at temperatures higher than 25°C and shows optimal cell development and maximal β-galactosidase production at 4°C, a temperature close to that of the natural environment.
The intracellular β-galactosidase produced by the Antarctic strain shares structural properties with the mesophilic β-galactosidase from E. coli. The subunit mass is comparable to that of the E. coli enzyme. Under native conditions, the enzyme is a multimer, since it is concentrated by an ultrafiltration membrane of 300-kDa cutoff, and is probably a tetramer, as shown for E. coli β-galactosidase (11). P. haloplanktis β-galactosidase is a metalloenzyme with a strict requirement for divalent metal ions as also shown for the E. coli β-galactosidase (30). Indeed, the three-dimensional structure of E. coli β-galactosidase displays two bound Mg2+ per monomer (11). Identical binding constants determined for both enzymes indicate that Mg2+ ion is also essential for psychrophilic β-galactosidase activity. The two β-galactosidases exhibit similar optimal pH values for stability and activity, as well as identical 2-mercaptoethanol dependence and cysteine content.
Sequence alignment of the P. haloplanktis enzyme with other LacZ β-galactosidases showed the conservation of the amino acid residues involved in catalysis. In the three-dimensional structure of β-galactosidase from E. coli, the acid/base catalyst Glu 461, the nucleophile Glu 537, and the accessory catalysts Met 502, Tyr 503, and Arg 388 are found close to each other and form the active site pocket (11). All of these residues are also conserved in the P. haloplanktis sequence.
However, the cold β-galactosidase displays a lower apparent optimum temperature of activity (Fig. 2A), a weaker thermal stability of activity (Fig. 4), and reduced conformational stability (Fig. 5) than the E. coli enzyme. Moreover, over the temperature range of 0 to 40°C, the turnover (kcat) of P. haloplanktis β-galactosidase toward ONPG is higher than that of the E. coli enzyme. This difference in favor of the psychrophilic enzyme is dramatically increased when the natural substrate lactose is used (15-fold at 25°C). The thermodynamic parameters (Table 2) are consistent with the fact that the activated state of the enzyme-substrate complex is reached through a minimum of enthalpy change, therefore rendering the reaction less temperature dependent compared to E. coli β-galactosidase. The higher activation entropy change, as shown by P. haloplanktis β-galactosidase, has been tentatively related to the improved active site plasticity of cold-active enzymes (8, 13). The physiological efficiency or specificity constant kcat/Km is generally a better indication of catalytic evolution than kcat alone, especially in the case of intracellular enzymes that catalyze their reaction at substrate concentrations close to the Km (7). With lactose as substrate, P. haloplanktis β-galactosidase optimizes kcat/Km by decreasing Km and increasing kcat. With ONPG as the substrate, the Km values at low temperatures are comparable for both P. haloplanktis and E. coli enzymes. Interestingly, the Km for the natural substrate is drastically optimized compared to that for ONPG. This confirms that small synthetic substrates may have quite distinct binding modes compared to natural ones (7).
The alignment of the amino acid sequence of P. haloplanktis β-galactosidase with that of E. coli β-galactosidase shows three insertions of four, five, and nine residues. These insertions could contribute to increase the flexibility of the solvent-exposed molecular surface, as also suggested in the case of subtilisin S41 (5), or to reduce interactions between monomers (22). Nevertheless, the involvement of insertions or deletions in cold adaptation is strongly specific to each enzyme type and cannot be generalized (6). The difference in pI between the psychrophilic and mesophilic β-galactosidases reveals a distinct pattern of ionizable side chains. This has been related to altered interactions with the solvent in cold-adapted enzymes (7). The lower Ca2+ binding constant determined for the psychrophilic β-galactosidase could contribute to the reduced thermal stability compared to the mesophilic enzyme (7). Indeed, weak Ca2+ coordination is involved in the less compact conformation of psychrophilic metalloenzymes (7). The multivalent character of arginine, forming up to five weak interactions with surrounding residues, accounts for its low occurrence in many psychrophilic enzymes and in enzymes of low stability in general (15). As a matter of fact, the Arg content of P. haloplanktis β-galactosidase is low. For instance, Arg 282 can stabilize the active site of E. coli β-galactosidase. This residue is substituted by a lysine in P. haloplanktis β-galactosidase. Substitutions of lysine with arginine were shown to improve the thermal stability of structurally unrelated proteins (19). The psychrophilic enzyme also shows a lower proline content. The cyclic structure of proline severely restricts the rotations about its N-Cα bond and greatly reduces the number of possible local conformations of the polypeptide backbone (14). Finally, it has been suggested that the stacking of Gly around the catalytic residues, as demonstrated by P. haloplanktis β-galactosidase, improves the active site flexibility (12). A detailed analysis of these possible determinants of heat lability and high activity awaits the availability of a three-dimensional structure.
Trials in milk demonstrated that the P. haloplanktis β-galactosidase is superior to the current commercial enzyme from K. marxianus var. lactis with respect to hydrolyzing lactose in milk, especially at low temperatures. This property confers a promising potential to the psychrophilic enzyme for lactose removal in milk and dairy products at low temperatures. In addition, we have shown that the heat-labile β-galactosidase can be expressed in a mesophilic host grown at moderate temperatures while keeping the wild-type properties. This prerequisite for large-scale production reinforces the biotechnological potential of P. haloplanktis β-galactosidase.
ACKNOWLEDGMENTS
We thank N. Gerardin-Otthiers and R. Marchand for expert technical assistance and Tony Collins for carefully reading the manuscript.
This work was supported by the EU, through network contracts CT94051 and CT97-0131, concerted action Bio 4-CT95-0017, and Biotech program Bio 4-CT96-0051, by the Ministère de l'Education, de la Recherche et de la Formation, concerted action ARC 93/98-170, and by the Region Walonne, conventions 1828 and 9613492. Support of the FNRS is also acknowledged (contract 2.4523.97 to C. Gerday). We also thank the Institut Français de Recherche et Technologie Polaire for generously accommodating year after year our research fellows at the Antarctic Station J. S. Dumont d'Urville in Terre Adelie.
REFERENCES
- 1.Bassias J, Brückner R. Regulation of lactose utilization genes in Staphylococcus xylosus. J Bacteriol. 1998;180:2273–2279. doi: 10.1128/jb.180.9.2273-2279.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckwith J R, Zipser D. The lactose operon. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1970. [Google Scholar]
- 3.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 3a.Brahamsha B, Greenberg E P. Complementation of a trpE deletion in Escherichia coli by Spirochaeta aurantia DNA encoding anthranilate synthetase component I activity. J Bacteriol. 1987;169:3764–3769. doi: 10.1128/jb.169.8.3764-3769.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohn M. Contributions of studies on the β-galactosidase of Escherichia coli to our understanding of enzyme synthesis. Bacteriol Rev. 1957;21:140–168. doi: 10.1128/br.21.3.140-168.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davail S, Feller G, Narinx E, Gerday C. Cold adaptation of proteins. Purification, characterization, and sequence of the heat-labile subtilisin from the antarctic psychrophile Bacillus TA41. J Biol Chem. 1994;269:17448–17453. [PubMed] [Google Scholar]
- 6.Feller G, Arpigny J L, Narinx E, Gerday C. Molecular adaptations of enzymes from psychrophilic organisms. Comp Biochem Physiol. 1997;118A:495–499. [Google Scholar]
- 7.Feller G, Gerday C. Psychrophilic enzymes: molecular basis of cold adaptation. Cell Mol Life Sci. 1997;53:830–841. doi: 10.1007/s000180050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fields P A, Somero G N. Hot spots in cold-adaptation: localized increases in conformational flexibility in lactate dehydrogenase A4 orthologs of Antarctic notothenioid fishes. Proc Natl Acad Sci USA. 1998;95:11476–11481. doi: 10.1073/pnas.95.19.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fowler A V, Zabin I. The amino acid sequence of beta-galactosidase of Escherichia coli. Proc Natl Acad Sci USA. 1977;74:1507–1510. doi: 10.1073/pnas.74.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grabarek Z, Leavis P C, Gergely J. Calcium binding to the low affinity sites in troponin C induces conformational changes in the high affinity domain. J Biol Chem. 1986;261:608–613. [PubMed] [Google Scholar]
- 11.Jacobson R H, Zhang X J, DuBose R F, Matthews B W. Three-dimensional structure of beta-galactosidase from E. coli. Nature. 1994;369:761–766. doi: 10.1038/369761a0. [DOI] [PubMed] [Google Scholar]
- 12.Karplus P A, Shultz G E. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72:212–213. [Google Scholar]
- 13.Lonhienne T, Gerday C, Feller G. Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim Biophys Acta. 2000;1543:1–10. doi: 10.1016/s0167-4838(00)00210-7. [DOI] [PubMed] [Google Scholar]
- 14.Matthews B W, Nicholson H, Becktel W J. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc Natl Acad Sci USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menendez-Arias L, Argos P. Engineering protein thermal stability. Sequence statistics point to residue substitutions in alpha-helices. J Mol Biol. 1989;206:397–406. doi: 10.1016/0022-2836(89)90488-9. [DOI] [PubMed] [Google Scholar]
- 16.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. pp. 352–355. [Google Scholar]
- 17.Miller J H, Reznikoff W S. The operon. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1978. [Google Scholar]
- 18.Morita R Y. Psychrophilic bacteria. Bacteriol Rev. 1975;39:144–167. doi: 10.1128/br.39.2.144-167.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mrabet N T, van den Broeck A, van den Brande I, Stanssens P, Laroche Y, Lambeir A M, Matthijssens G, Jenkins J, Chiadmi M, van Tilbeurgh H, et al. Arginine residues as stabilizing elements in proteins. Biochemistry. 1992;31:2239–2253. doi: 10.1021/bi00123a005. [DOI] [PubMed] [Google Scholar]
- 20.Pace C N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 21.Pridmore R D. New and versatile cloning vectors with kanamycin-resistance marker. Gene. 1987;56:309–312. doi: 10.1016/0378-1119(87)90149-1. [DOI] [PubMed] [Google Scholar]
- 22.Rentier-Delrue F, Moyens S, Lion M, Martial J A. Sequence of the triosephosphate isomerase-encoding gene isolated from the thermophile Bacillus stearothermophilus. Gene. 1993;134:137–138. doi: 10.1016/0378-1119(93)90188-9. [DOI] [PubMed] [Google Scholar]
- 23.Robertson S P, Potter J D, Rouslin W. The Ca2+ and Mg2+ dependence of Ca2+ uptake and respiratory function of porcine heart mitochondria. J Biol Chem. 1982;257:1743–1748. [PubMed] [Google Scholar]
- 24.Schachter H. Enzymic microassays for d-mannose, d-glucose, d-galactose, l-fucose, and d-glucosamine. Methods Enzymol. 1975;41:3–10. doi: 10.1016/s0076-6879(75)41003-5. [DOI] [PubMed] [Google Scholar]
- 25.Smith R M, Martel A E. Critical stability constants. Vol. 1. 1974. pp. 204–272. , and vol. 2, p. 276–285. Plenum Press, London, England. [Google Scholar]
- 26.Steers E, Jr, Cuatrecasas P, Pollard H B. The purification of beta-galactosidase from Escherichia coli by affinity chromatography. J Biol Chem. 1971;246:196–200. [PubMed] [Google Scholar]
- 27.Studier F W, Moffatt B A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 28.Trimbur D E, Gutshall K R, Prema P, Brenchley J E. Characterization of a psychrotrophic Arthrobacter gene and its cold-active β-galactosidase. Appl Environ Microbiol. 1994;60:4544–4552. doi: 10.1128/aem.60.12.4544-4552.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Triveni P S. Beta-galactosidase technology: a solution to the lactose problem. Crit Rev Food Technol. 1975;5:325–354. [Google Scholar]
- 30.Wallenfels K, Weil R. Beta-galactosidase. In: Boyer P D, editor. The enzymes. Vol. 7. New York, N.Y: Academic Press; 1972. pp. 617–663. [Google Scholar]