

Abstract
Norepinephrine exerts powerful influences on the metabolic, neuroprotective and immunoregulatory functions of astrocytes. Until recently, all effects of norepinephrine were believed to be mediated by receptors localized exclusively to the plasma membrane. However, recent studies in cardiomyocytes have identified adrenergic receptors localized to intracellular membranes, including Golgi and inner nuclear membranes, and have shown that norepinephrine can access these receptors via transporter-mediated uptake. We recently identified a high-capacity norepinephrine transporter, organic cation transporter 3 (OCT3), densely localized to outer nuclear membranes in astrocytes, suggesting that adrenergic signaling may also occur at the inner nuclear membrane in these cells. Here, we used immunofluorescence and western blot to show that β1-adrenergic receptors are localized to astrocyte inner nuclear membranes; that key adrenergic signaling partners are present in astrocyte nuclei; and that OCT3 and other catecholamine transporters are localized to astrocyte plasma and nuclear membranes. To test the functionality of nuclear membrane β1-adrenergic receptors, we monitored real-time protein kinase A (PKA) activity in astrocyte nuclei using a fluorescent biosensor. Treatment of astrocytes with norepinephrine induced rapid increases in PKA activity in the nuclear compartment. Pretreatment of astrocytes with inhibitors of catecholamine uptake blocked rapid norepinephrine-induced increases in nuclear PKA activity. These studies, the first to document functional adrenergic receptors at the nuclear membrane in any central nervous system cell, reveal a novel mechanism by which norepinephrine may directly influence nuclear processes. This mechanism may contribute to previously described neuroprotective, metabolic and immunoregulatory actions of norepinephrine.
Keywords: Norepinephrine, adrenergic receptor, G protein-coupled receptors, monoamine transporter, protein kinase A, nucleus, subcellular localization
Graphical Abstract

1. INTRODUCTION
Norepinephrine is a key mediator of integrated central nervous system responses to stressful and arousing stimuli. In addition to its well-characterized influences on neuronal function and synaptic plasticity, norepinephrine has powerful effects on core astrocyte functions, including neuroprotective (Junker et al., 2002), immunomodulatory (Laureys et al., 2014), and metabolic support functions (Coggan et al., 2018). These effects, mediated by G-protein-coupled α- and β-adrenergic receptors, include both rapid changes in enzyme activity and delayed changes in gene expression. For example, norepinephrine induces rapid activation of cytosolic glycogen phosphorylase, leading to increases in glycogen breakdown and lactate release (Sorg and Magistretti, 1991; Hertz et al., 2010; Coggan et al., 2018) and gradual increases in the expression of genes that allow adaptive re-synthesis of glycogen (Sorg and Magistretti, 1991). These integrated cellular responses to norepinephrine require the propagation of adrenergic receptor signaling events to cytosolic and nuclear targets. Dysregulation of noradrenergic signaling and of astrocyte function have been implicated in both neuropsychiatric and neurodegenerative disorders, including Alzheimer’s disease and multiple sclerosis (Keyser et al., 2010; Santello et al., 2019). Determining the mechanisms by which norepinephrine-induced signals reach both cytosolic and nuclear locations is critical for a complete understanding of noradrenergic regulation of cellular function under normal and pathological conditions.
As G-protein-coupled receptors, the adrenergic receptors influence cellular function through signal transduction pathways that alter the phosphorylation of proteins throughout the cell. The specific effect of an extracellular ligand on a given target cell depends not only on the number and class of receptors expressed by the cell, but also on the localization of those receptors in relation to intracellular effectors. Recent studies in cardiomyocytes have demonstrated that, in addition to the plasma membrane, β1-adrenergic receptors (β1-ARs) may also be localized to, and activated at, intracellular membranes including Golgi apparatus and inner nuclear membranes, and that receptors in distinct cellular locations mediate distinct cellular responses to catecholamines (Boivin et al., 2006; Vaniotis et al., 2011; Wu et al., 2014; Irannejad et al., 2017; Dahl et al., 2018; Wang et al., 2021). These studies have also shown that access of norepinephrine, a cationic and therefore membrane-impermeable ligand, to intracellular β1-ARs requires carrier-mediated uptake (Wright et al., 2008; Irannejad et al., 2017; Wang et al., 2021). We recently demonstrated that organic cation transporter 3 (OCT3), a high-capacity transporter for norepinephrine and other monoamines, is densely expressed in the outer nuclear membranes of astrocytes in the rat and mouse brain (Gasser et al., 2017), suggesting that nuclear adrenergic signaling may also occur in the central nervous system. If present, nuclear adrenergic receptors would represent a powerful mechanism underlying previously described effects of norepinephrine on astrocyte gene expression and physiology.
We hypothesized that, as in cardiomyocytes, β1-ARs are localized to the inner nuclear membrane in mouse astrocytes, and that activation of these receptors by norepinephrine leads to activation of intranuclear Gs-mediated signaling. We further hypothesized that norepinephrine-induced activation of inner nuclear membrane β1-ARs requires transporter-mediated uptake. To test these hypotheses, we used immunofluorescence and immunoblot methods to examine the subcellular localization of β1-AR and its signaling partners in murine astrocytes. We identified a population of β1-ARs localized to astrocyte inner nuclear membranes and obtained evidence that key components of Gs-mediated signaling are localized to the nuclear compartment. We also identified multiple catecholamine transporters in addition to OCT3, localized to plasma and nuclear membranes in astrocytes, that may allow norepinephrine to cross these membranes and activate nuclear β1-ARs. To test the functionality of these receptors and the dependence of their activation on transporter activity, we monitored the effects of norepinephrine on the activity of protein kinase A (PKA) in astrocyte nuclei in real time using a recombinant fluorescent biosensor. Our findings provide evidence of a previously undescribed population of functional β1-ARs localized to the inner nuclear membrane in astrocytes. These receptors represent a powerful mechanism by which norepinephrine may more directly influence nuclear processes including the regulation of gene expression, and may contribute to the metabolic, neuroprotective, and immunomodulatory actions of norepinephrine in astrocytes.
2. MATERIALS AND METHODS
2.1. Animals
Wild-type C57BL/6 mice were bred in-house or obtained from Envigo (Madison, WI, USA) and housed in a temperature-controlled environment on a 12:12 light:dark cycle with ad libitum access to food and water. Animal care and all experimental procedures were approved by the Marquette University Institutional Animal Care and Use Committee (IACUC #AR-210 and 3869) and adhered to all Animal Care Guidelines of the United States National Institutes of Health (NIH). This study was not pre-registered, and no blinding or randomization was performed.
2.2. Primary astrocyte cell culture
Primary astrocyte cultures were derived from cerebral cortices of postnatal day 1 to 2 C57BL/6 mouse pups obtained either from an in-house colony or from pregnant female mice purchased from Envigo. Pups of both sexes were included in all astrocyte cultures. Cultures were prepared as described in Uliasz et al (Uliasz et al., 2011). Briefly, pups were anesthetized with isoflurane (Piramal Critical Care Cat# 66794-017). After loss of paw withdrawal, pups were decapitated, and heads were briefly immersed in ethanol. Brains were removed and placed into Dissection Medium (Hank’s Balanced Salt Solution without calcium, magnesium, phenol red, or sodium bicarbonate (Gibco Cat# 14185052) containing (in mM): glucose, 27.8; sucrose, 20.5; HEPES, 1; pH 7.4). Meninges were removed and cerebral cortices were dissected, minced, and dissociated in (0.025% Trypsin (Corning Cat# 25-054-CI) in Dissection Medium at 37°C for 30 minutes. Cells were pelleted by brief centrifugation, resuspended by gentle trituration in Astrocyte Plating Medium (Minimum Essential Medium Eagle with Earle’s salts without L-glutamine and phenol red (MEM, Corning Cat# 17-305CV), containing (in mM): glucose, 15.8; sodium bicarbonate, 3.2; 10% fetal bovine serum (R&D Systems Cat# S11550), 10% iron-supplemented calf serum (R&D Systems Cat# S11950), penicillin (50 μg/mL)/streptomycin (50 I.U./mL) (Corning Cat# 300-002-CI), 2 mM L-glutamine (Gibco Cat# 25030-081), and 0.01 μg/mL epidermal growth factor (Gibco Cat# 53003-018)), and plated. After 10 days in culture, confluent cells were treated with 8 μM cytosine arabinoside (ara-C, Sigma-Aldrich Cat# C1768), an anti-mitotic agent, to limit microglial contamination. After 5 days, medium was replaced with Growth Medium (MEM containing 15.8 mM glucose, 3.2 mM sodium bicarbonate, 10% FBS, penicillin (50 μg/mL)/streptomycin (50 I.U./mL), 2 mM L-glutamine) and changed weekly. Astrocytes were harvested for subcellular fractionation or plated for immunofluorescence or excitation ratio imaging 3-5 weeks after initial culture. One day before all experiments, astrocytes were treated with 75 mM L-leucine methyl ester (Sigma-Aldrich Cat# L1002) to further deplete microglia.
2.3. Antibodies and plasmids
Antibodies used in western blot and immunofluorescence studies are described in Table 1. The PMAT antibody was the generous gift of Dr. Joanne Wang. The pcDNA3 Flag β1-adrenergic-receptor plasmid was a gift from Dr. Robert Lefkowitz (Addgene plasmid # 14698; http://n2t.net/addgene:14698 ; RRID:Addgene_14698). The ExRai-AKAR-NLS plasmid was a gift from Dr. Jin Zhang.
Table 1.
Primary antibodies used in western blot and immunofluorescence experiments.
| Antigen | RRID | Source | Cat # | Host | Concentration/Dilution |
|---|---|---|---|---|---|
| ATP1A1 | AB_2060976 | Thermo Fisher Scientific | MA3-928 | Ms | W: 0.374 μg/mL (1:5000) |
| β1-AR (C-terminal) | AB_2289444 | Thermo Fisher Scientific/Abcam | PA1-049/ab3442 | Rb | W, IF: 1:1000 |
| β1-AR (ECL2) | AB_2340886 | Alomone Labs | AAR-023 | Rb | W: 1.7 μg/mL (1:500) IF: 2.125 μg/mL (1:400, IF) |
| Dopamine transporter (DAT) | AB_1586991 | Millipore-Sigma | AB2231 | Rb | W: 1 μg/mL (1:1000) |
| FLAG | AB_262044 | Millipore-Sigma | F1804 | Ms | IF: 2 μg/mL (1:500) |
| GAPDH | AB_10615768 | Millipore-Sigma | AB2302 | Ckn | W: 0.067 μg/mL (1:15000) |
| GFAP | AB_11212597 | Millipore-Sigma | MAB360 | Ms | IF: 1:1600 |
| GFAP | AB_10001722 | Novus Biologicals | NB300-141 | Rb | IF: 1:2000 |
| Gsα | AB_2868457 | Abcam | ab235956 | Rb | IF: 15 μg/mL (1:200) |
| Gsα | AB_631538 | Santa Cruz Biotechnology | sc-823 | Rb | W: 0.4 μg/mL (1:500) |
| Lamin A/C | AB_10545756 | Cell Signaling Technology | 4777S | Ms | W: 3.5 ng/mL (1:2000) IF: 17.5 ng/mL (1:400) |
| Lamin A/C | AB_2572338 | Boster Biological Technology | M00438-2 | Ckn | IF: 1:1000 |
| NET | AB_2571639 | MAb Technologies | NET 05-2 | Ms | W: 1:1000 |
| PKA catalytic subunit α | AB_398293 | BD Transduction Laboratories | 610980 | Ms | W: 0.25 μg/mL (1:1000) |
| PKA Regulatory subunit I | AB_397566 | BD Transduction Laboratories | 610165 | Ms | W: 0.25 μg/mL (1:1000) IF: 0.5 μg/mL (1:500) |
| PKA Regulatory subunit IIα | AB_2819176 | GeneTex | GTX35228 | Rb | W, IF: 3.29 μg/mL (1:1000) |
| PMAT “P469” | none | Gift from Joanne Wang | N/A | Rb | IF: 1:1000 |
| OCT2 | AB_1621334 | Alpha Diagnostic International | OCT21-A | Rb | W: 0.5 μg/mL (1:2000) IF: 10 μg/mL (1:100) |
| OCT3 | AB_1622571 | Alpha Diagnostic International | OCT31-A | Rb | W, IF: 1 μg/mL (1:500) |
W: Western blot; IF: Immunofluorescence.
2.4. General immunofluorescence and microscopy
Initial studies examining the potential nuclear localization of β1-ARs and their signaling partners used immunofluorescence in astrocytes treated with Triton X-100, a detergent which permeabilizes all cellular membranes, allowing antibodies to have access to proteins in all cellular compartments. Cultured astrocytes were plated on glass-bottomed chamber slides (MatTek, Ashland, MA, USA Cat# CCS4; or Millicell EZ Slide, Millipore-Sigma, Burlington, MA, USA Cat# PEZGS0416). 5-7 days after plating, astrocytes were fixed (PBS + 2% paraformaldehyde, 10 min, 4°C), rinsed in 0.05 M PBS, and permeabilized by incubation in blocking buffer (0.05 M PBS, 0.3 M glycine, 5% donkey serum) containing Triton X-100 (0.1%; Sigma-Aldrich Cat# X100). Astrocytes were then rinsed and incubated (overnight, 4°C) in 0.05 M PBS containing the primary antibody of interest (see Table 1 for antibody and dilution information). After thorough rinsing, astrocytes were incubated (2 h, room temperature) in PBS containing 0.1% Triton X-100 (PBST) containing the appropriate fluorophore-conjugated secondary antibody (see Table 2 for antibody and dilution information). In experiments in which a second antigen was labeled, cells were thoroughly rinsed and incubated in blocking buffer with 0.1% Triton X-100 (20 min, room temperature) prior to incubation with primary and secondary antibodies as above. After final rinses, cells were dried and cover-slipped using fluorescent mounting medium (DAPI Fluoromount-G, SouthernBiotech, Birmingham, AL, USA Cat# 0100-20; or EverBrite, Biotium, Fremont, CA, USA, Cat# 23001 or 23002). In some studies, nuclei were counterstained using DAPI. Photomicrographs were captured using a Nikon 80i microscope fitted with an ORCA-Flash 4.0 LT digital camera (Hamamatsu Photonics, Japan) linked to a computer running NIS Elements software (Nikon Instruments, Melville, NY).
Table 2.
Secondary antibodies used in western blot and immunofluorescence experiments.
| Antibody | RRID | Source | Cat# | Concentration/Dilution |
|---|---|---|---|---|
| Donkey Anti-Mouse Alexa Fluor 488 | AB_2341099 | Jackson ImmunoResearch Laboratories | 715-545-151 | IF: 0.35 μg/mL (1:2000) |
| Donkey Anti-Rabbit Alexa Fluor 594 | AB_2340621 | Jackson ImmunoResearch Laboratories | 711-585-152 | IF: 0.375 μg/mL (1:2000) |
| Donkey Anti-Chicken Alexa Fluor 488 | AB_2340375 | Jackson ImmunoResearch Laboratories | 703-545-155 | IF: 0.375 μg/mL (1:2000) |
| Donkey Anti-Mouse Alexa Fluor 594 | AB_2340855 | Jackson ImmunoResearch Laboratories | 715-585-151 | IF: 0.375 μg/mL (1:2000) |
| Goat anti-Rabbit Alexa Fluor Plus 800 | AB_2633284 | Invitrogen | A32735 | W: 0.133 μg/mL (1:15000) |
| Goat anti-Mouse Alexa Fluor Plus 680 | AB_2633278 | Invitrogen | A32729 | W: 0.133 μg/mL (1:15000) |
| Donkey anti-Mouse IRDye 680LT | AB_10715072 | LI-COR Biosciences | 926-68022 | W: 0.067 μg/mL (1:15000) |
W: Western blot; IF: Immunofluorescence.
2.5. Immunofluorescence with differential detergent permeabilization
Immunofluorescence signal observed over the nucleus in Triton-permeabilized cells could originate from antibodies bound to proteins localized to a number of sites. For transmembrane proteins like the β1-AR or membrane-associated proteins like G protein subunits, the signal could originate from target protein in one or more of the following locations: a) plasma membrane overlying the nucleus; b) inner or outer nuclear membrane; or c) perinuclear endoplasmic reticulum or Golgi. To better identify the origin of immunofluorescence signals over astrocyte nuclei, we compared the immunostaining patterns observed in Triton-permeabilized astrocytes with those observed in astrocytes permeabilized with digitonin, a reagent that preferentially permeabilizes cholesterol-rich membranes, leaving nuclear membranes essentially intact ((Jamur and Oliver, 2009)). Cultured astrocytes plated on glass-bottomed chamber slides were fixed as described above and permeabilized by incubation in blocking buffer containing either 0.1% Triton X-100 or 10 μg/mL digitonin (0.001%, Sigma-Aldrich Cat# D141). Astrocytes were then rinsed and incubated (overnight, 4°C) in 0.05 M PBS containing the primary antibody of interest (see Table 1 for antibody and dilution information). After thorough rinsing, astrocytes were incubated (2 h, room temperature) in PBS containing 0.1% Triton X-100 (PBST) containing the appropriate fluorophore-conjugated secondary antibody (see Table 2 for antibody and dilution information). Triton X-100 was included in this incubation to allow secondary antibody to reach all cellular compartments. To identify nuclei, cells were then thoroughly rinsed and incubated in blocking buffer with 0.1% Triton X-100 (20 min, room temperature) prior to incubation with an antibody against type A/C nuclear lamins, followed by rinsing and incubation with secondary antibody as above. Photomicrographs of cells in the two detergent conditions were captured with identical exposure times.
Each detergent comparison experiment was repeated at least three times (biological replicates) using cells cultured from distinct mouse litters. For quantification, nuclear fluorescence intensity data were collected from photomicrographs captured using a Nikon A1R+ laser scanning confocal microscope. Fluorescence intensities were collected from separate images from two channels (laser frequencies): one for the protein of interest (the target), and one for the nuclear marker. For each biological replicate and permeabilization condition, photomicrographs were captured from 3 separate regions of the slide. Images were analyzed using NIS Elements AR software (Nikon Instruments) as follows. Nuclei were identified using only the nuclear marker channel and set as regions of interest (ROIs) using a binary threshold. Any eligible nuclei missed by thresholding were manually selected. Nuclei of dividing cells and nuclei touching image borders were manually excluded. Fluorescence intensity data in both the target and the nuclear marker channels were exported to an Excel spreadsheet. To control for differences in nuclear size, total fluorescence intensity (area under the curve) for each ROI in the target channel was divided by total fluorescence intensity in the nuclear marker channel. This value was termed “nuclear immunofluorescence”. The effects of detergent condition on normalized nuclear immunofluorescence were examined using the following methods:
Nuclear immunofluorescence values for each target from all ROIs across all biological replicates were pooled for each detergent condition. Since each biological replicate produced a large number of observations that varied across replications and were not normally distributed, a bootstrap analysis was conducted in R version 4.1.3 and RStudio (2022.02.0, Build 443 ©2009-2022) for each target protein to determine the effect of detergent on nuclear immunofluorescence. Individual observations within each detergent condition were resampled with replacement 10,000 times and pooled to generate means with constant sample size (n=150 per target protein; n=50 per biological replicate). For each iteration, a mean difference (Mean Triton - Mean Digitonin) was calculated and a frequency distribution was empirically derived to generate confidence intervals and percentile boundaries. The reported p values are drawn directly from this frequency distribution and represent the percent of the mean differences that are less than the 2.5th or greater than the 97.5th percentile, or equal to 0 (i.e., with α=0.05 and a two-tailed test, the observed probability that the true mean difference is equal to zero under the null hypothesis).
The size of the effect of detergent on nuclear immunofluorescence was estimated by calculating Cohen’s d using the equation , where M1 and M2 are the means of the two detergent groups, and SD1 and SD2 are the standard deviations of the two groups. Effect size (d) was reported to avoid over-reliance on the P value as the sole determinant of significance (biological or statistical), especially given the power advantage of large numbers of observations to detect small effect sizes.
To examine the reproducibility of the detergent effect across biological replicates, we calculated mean nuclear immunofluorescence for the two detergent conditions in each biological replicate. This resulted in n=3 replicates per detergent group per target. For each target, the mean nuclear immunofluorescence values for the two detergent conditions in each replicate were plotted as paired observations to clearly show the direction of the detergent effect.
To confirm that the two detergents differentially permeabilized astrocyte plasma and nuclear membranes, we probed Triton- and digitonin-permeabilized astrocytes with antibodies against type A/C nuclear lamins, proteins which are localized to the inner surface of the inner nuclear membrane (Gruenbaum et al., 2005). In these studies, nuclear ROIs were identified by staining with DAPI and normalized nuclear immunofluorescence was calculated by dividing lamin fluorescence intensity by DAPI fluorescence intensity.
2.6. Perfusions and immunofluorescence in mouse brain tissue
Adult female C57BL/6 mice (n = 4, Envigo) were deeply anesthetized by intraperitoneal injection of sodium pentobarbital (100 mg/kg). Pentobarbital was chosen for perfusions because of its long duration of action. After loss of righting reflex and paw withdrawal, mice were transcardially perfused with ice-cold 0.05 M phosphate-buffered saline followed by 4% paraformaldehyde in 0.1 M sodium phosphate buffer (PB, pH 7.4). Following perfusion, brains were removed and post-fixed in the 4% paraformaldehyde solution for 12 hours at 4°C and rinsed twice in 0.1 M PB for 12 hours. Brains were then incubated in 30% sucrose in 0.1 M PB for approximately 72 hours followed by rapid freezing in dry-ice-chilled liquid isopentane and storage at −80 °C until sectioning. Forebrain sections (25 μm) were cut across the coronal plane using a cryostat (Leica Biosystems, Buffalo Grove, IL, USA), and stored in cryoprotectant (30% ethylene glycol/20% glycerol (w/w) in 0.05 M PB, pH 7.4) at −20 °C until immunostaining.
After rinsing in PBS, sections were incubated (20 min, room temperature) in blocking buffer (0.05 M PBS, 0.3% Triton X-100, 0.3 M glycine, 5% donkey serum), and then rinsed prior to incubation (overnight, room temperature) with C-terminal-directed anti-β1-AR antibody in 0.1% PBST with 5% donkey serum. Sections were rinsed the next day and incubated with Alexa Fluor 594-conjugated donkey anti-rabbit IgG (1:2000; Jackson ImmunoResearch RRID:AB_2340621) in PBS containing 0.1% Triton-X100 for 2 h. Sections were then rinsed, incubated in blocking buffer, and incubated with anti-GFAP antibody in 0.1% PBST with 5% donkey serum as described above. The following day sections were rinsed, incubated 2 h with Alexa Fluor 488-conjugated donkey anti-mouse IgG (1:2000; Jackson ImmunoResearch RRID:AB_2341099) in 0.1% PBST, rinsed, and mounted onto SuperFrost microscope slides. After drying, sections were cover-slipped with fluorescent mounting medium with DAPI as above. Photomicrographs were acquired using a Nikon 80i microscope fitted with an ORCA-Flash 4.0 LT digital camera (Hammamatsu Photonics, Japan) linked to a computer running NIS Elements-BR software (Nikon Instruments).
2.7. Subcellular fractionation, gel electrophoresis and Western blot
Cytosolic, nuclear, and plasma membrane proteins were purified from cultured astrocytes using a commercially available kit (Qproteome Cell Compartment Kit; Qiagen, Germantown, MD, USA, Cat# 37502) according to the manufacturer’s instructions. Protein concentration in each fraction was determined (Pierce BCA Protein Assay, Thermo Fisher Scientific Cat# 23225). Subcellular fractions were prepared for electrophoresis by addition of Bolt LDS sample buffer and Bolt sample reducing agent (Invitrogen Cat#s B0007 and B0009) followed by heating at 37°C for 30 min. Proteins (approximately 6 μg/sample) were electrophoresed on 4-12% Bis-Tris polyacrylamide gels (Invitrogen Cat# NW04122) in 1X Bolt MOPS SDS Running Buffer with 0.1% Bolt antioxidant (Invitrogen Cat#s B0001 and BT0005) at 200V for 55 minutes. They were then electroblotted (25V, 0.13A. 17W 1.5 hours) onto Immobilon-FL polyvinylidene difluoride membranes (Millipore-Sigma Cat# IPFL07810) in 1X Bolt Transfer Buffer (Invitrogen Cat# BT00061) with 0.1% Bolt antioxidant using a wet transfer apparatus (Thermo Fisher Scientific). Membranes were dried overnight at room temperature. The following day, membranes were briefly re-wet in 100% methanol, followed by rinsing and blocking (Odyssey TBS Blocking Buffer, LI-COR Biosciences, Lincoln, NE, Cat# 927-5000), and incubated overnight at 4°C in blocking buffer containing 0.1% Tween-20 (Thermo Fisher Scientific Cat# BP3337) and primary antibodies of interest (see Table 1). Blots were then rinsed and incubated (2 h, room temperature) with species-specific secondary antibodies conjugated to either Alexa Fluor Plus 680 or Alexa Fluor Plus 800 (see Table 2 for antibody and dilution information) in blocking buffer containing 0.1% Tween-20 and 0.01% sodium n-dodecyl sulfate (Millipore-Sigma Cat# 428018). After rinsing, digital images of the fluorescent bands were captured using an Odyssey Fc Imaging System (LI-COR Biosciences). Images were captured at each wavelength and saved as separate files. Data for individual antigens are presented separately as black-and-white images. Each immunoblot was repeated at least three times with proteins from independent astrocyte cultures.
2.8. Excitation ratio imaging
Cultured astrocytes were transfected (Lipofectamine 3000, Invitrogen Cat# L3000-008) with a plasmid driving the expression of ExRai-AKAR-NLS, a single-fluorophore biosensor that combines a PKA substrate sequence with the FHA1 domain of AKAR (A-kinase activity reporter), a circularly permuted GFP, and a nuclear localization sequence (NLS). The fluorophore has two discrete excitation peaks: one centered around 400 nm and another around 509 nm, with a shoulder at 480 nm. Phosphorylation of the PKA substrate sequence results in increased efficacy of 480 nm-induced excitation and decreased efficacy of 400 nm-induced excitation. Thus, an increase in kinase activity is observed as an increase in the 488-/402-nm excitation ratio (Mehta et al., 2018), which was the primary endpoint of the experiment. Excitation ratio imaging experiments were conducted 24 hours after transfection. Thirty minutes before imaging, cells were rinsed with serum-free medium and incubated in medium containing one of two treatments: either A) a cocktail of the following catecholamine transport inhibitors: corticosterone (500 μM complexed with hydroxypropyl β-cyclodextrin (HBC) (Sigma-Aldrich Cat# C-174) to inhibit OCT2 (IC50 = 500 nM (Gründemann et al., 1998a)), OCT3 (IC50 = 120 nM (Gründemann et al., 1998b)) and PMAT (IC50 = 400 μM (Engel et al., 2004)); atomoxetine (10 μM; Thermo Fisher Scientific Cat# A23571G) to inhibit NET; and GBR 12909 (10 μM; Tocris Bioscience Cat# 0421) to inhibit DAT or B) vehicle (2.4 mg/mL HBC, MP Biomedicals Cat# 153540). Dishes containing live astrocytes were then transferred to the temperature-controlled stage of a Nikon A1R confocal microscope to monitor excitation ratio responses to norepinephrine. Images at two excitation wavelengths (402 and 488 nm) were collected every 20 seconds for 3 minutes prior to, and for 30 minutes after, bath application of norepinephrine (Millipore Sigma Cat# A9512) to a final concentration 50 nM. This concentration was chosen based on in vivo studies demonstrating that peak extracellular norepinephrine concentrations reach approximately 300 nM after robust electrical stimulation of the medial forebrain bundle (Park et al., 2011). This experiment was conducted on matched pairs (Groups: Vehicle and Transport Inhibitors) of dishes from 5 separate primary astrocyte culture preparations.
Nuclear fluorescence intensities for each excitation wavelength were normalized based on the average intensity during the 3-minute baseline period for each nucleus. A ratio was then calculated by dividing the normalized intensity at 488 nm by the normalized intensity at 402 nm. Thus, increases in PKA are exhibited as increases in the ratio. Before examining group differences, any nuclei with excessive ratio drift during the baseline period were eliminated. The slope of the ratio during the baseline period was calculated for each nucleus, and the standard deviation of all slopes was calculated. Nuclei with baseline slopes exceeding two standard deviations of the mean were eliminated from subsequent analyses (one nucleus from the Transport Inhibitors group and two nuclei from the Vehicle group). For the remaining nuclei (n = 17 in the Transporter Inhibitors group and n=19 in the Vehicle group), average excitation ratios were calculated for the 3-minute baseline period and for each of three 10-min post-norepinephrine periods.
2.9. Statistical analysis of ratiometric imaging
The average excitation ratios for the two groups (Vehicle vs. Transport Inhibitors) were analyzed with a mixed ANOVA across the 4 periods (Baseline vs. 0-10 vs. 10-20 vs. 20-30 minutes) using StatSoft Statistica (RRID: SCR_014213). Prior to the ANOVA, normality of the data was determined by generating a plot of the actual vs the predicted residuals (QQ plot) using GraphPad Prism (RRID:SCR_002798). As the resulting plot showed only minor deviations from linearity, we proceeded with mixed ANOVA. Because we anticipated that the largest effect of the inhibitors would occur during the early post-norepinephrine periods, we conducted two planned interaction contrasts comparing the baseline periods with the first post-norepinephrine period (Baseline vs. 0-10 minutes); and comparing the first two post-norepinephrine periods (0-10 vs 10-20 minutes). Further pairwise comparisons were made post hoc using Tukey’s HSD. Graphs were prepared using GraphPad Prism.
Data availability
All data supporting the findings of this study are available from the corresponding author upon request.
3. RESULTS
3.1. OCT3 is localized to astrocyte nuclei.
We previously demonstrated that OCT3 is localized to the outer nuclear membrane in astrocytes and neurons in the brain (Gasser et al., 2017). To confirm that OCT3 is localized to the nuclear envelope in cultured astrocytes as it is in situ, we used an antibody directed against a peptide in the large intracellular loop of mouse and rat OCT3 (Alpha Diagnostic International Cat# OCT31-A, RRID:AB_1622571; (Gorboulev et al., 2005)) in immunofluorescence. The specificity of this antibody has been confirmed previously in immunofluorescence applications in brain tissue from wild-type and OCT3-knockout mice and in cells exogenously expressing OCT3 (Lips et al., 2005; Vialou et al., 2004). In our studies, dense punctate OCT3-like immunoreactivity was observed over the nucleus in nearly all astrocytes in culture (Fig. 1A–C). OCT3-like immunoreactivity was also observed, at lower density, over astrocyte cell bodies.
Fig. 1. OCT3 is localized to astrocyte nuclei.

Fluorescence photomicrographs depicting organic cation transporter 3 (OCT3) immunoreactivity (magenta in A, B; white in C); GFAP immunoreactivity (green in A only); and lamin A/C immunoreactivity (green in B only) in primary cultured mouse astrocytes. Box in (B) indicates area shown at higher magnification in (C). OCT3-like immunoreactivity is localized over apparent nuclei in all GFAP+ astrocytes (A). At higher magnification (B, C), OCT3-like immunoreactivity is observed over cell bodies (arrowheads in B, C) and nuclei (arrows in B, C). Images are representative of immunostaining patterns observed in n = 5 cell cultures from separate litters. Scale bar, 80 μm (A), 25 μm (B), 6.25 μm (C).
3.2. β1-adrenergic receptor is localized to both plasma and inner nuclear membranes of astrocytes
To visualize the subcellular localization of β1-AR in astrocytes, we used two different commercially available antisera, each directed against a distinct epitope of the receptor, in immunofluorescence studies. One antibody (Thermo Fisher Scientific Cat# PA1-049, RRID:AB_2289444 (also sold as Abcam cat #ab3442, RRID:AB_10890808) is directed against an amino acid sequence in the C-terminal tail of mouse β1-AR which is an intracellular epitope in receptors localized to the plasma membrane. The specificity of this antibody has been confirmed previously in immunofluorescence in cells treated with β1-AR siRNA (Irannejad et al., 2017). The second antibody (ECL2; Alomone Labs Cat# AAR-023, RRID:AB_2340886) is directed against an amino acid sequence in the second extracellular loop of the plasma membrane-localized receptor. The specificity of this antibody has been confirmed previously in western blot using conditional Adrb1 knockdown tissue (Evans et al., 2020). Astrocyte nuclei were identified either by counterstaining with the DNA stain DAPI, or by immunolabeling with an antibody against type A/C nuclear lamins.
The two β1-AR antibodies produced similar patterns of immunofluorescence in Triton-permeabilized GFAP+ astrocytes (Fig. 2A, B). Punctate β1-AR-like immunoreactivity (β1-like ir) was diffusely distributed over astrocyte somata and densely localized over nuclei. Putative nuclear β1-like ir was reliably observed in nearly all cultured astrocytes. This pattern of staining was observed in 11 repeated assays with the C-terminal antibody and 7 repeated assays with the ECL2 antibody (each replicate used astrocyte cultures from distinct mouse litters). β1-AR-like-ir was not observed over nuclei or somata when immunofluorescence was conducted using primary antibodies which had been pre-adsorbed with an excess of the respective antigenic peptide (extracellular loop 2 (AAR-023 Control Peptide, Alomone) or C-terminus (Custom PA1-049 Control Peptide, Thermo Fisher Scientific, Project# MDB100511.1, Ref# A2381-1)) (Fig. 2C–F).
Fig. 2. β1-adrenergic receptor localization in astrocytes.
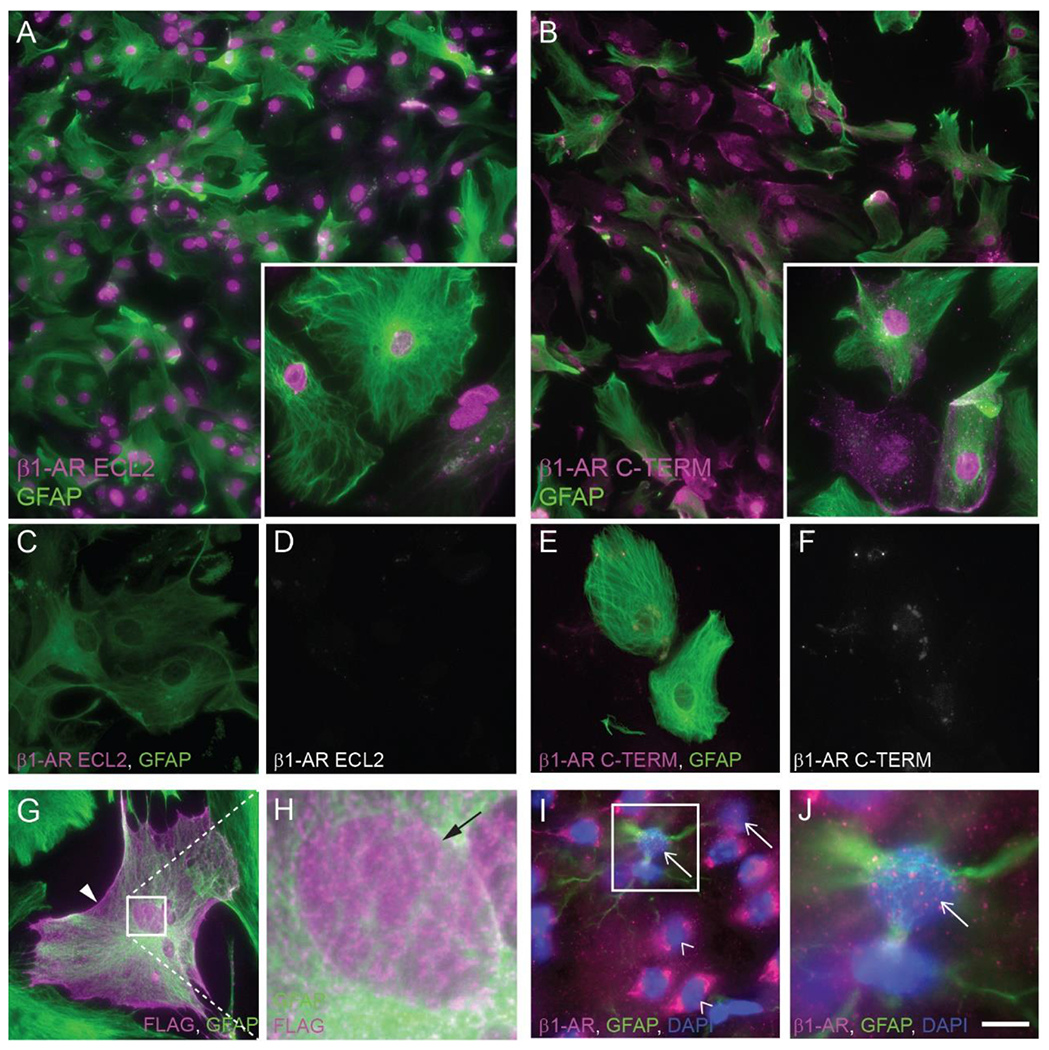
Fluorescence photomicrographs depicting β1-AR immunoreactivity in mouse primary astrocytes. β1-AR (magenta) was detected with an antibody against extracellular loop 2 (A, C, D) or the C-terminal tail (B, E, F) of the receptor. GFAP immunoreactivity is shown in green. Antibodies used in C, D, E, and F were pre-adsorbed with the immunizing peptide prior to application to cells. (G, H) Immunofluorescence of primary mouse astrocytes transfected with a FLAG-tagged human β1-AR and incubated with antibodies against FLAG peptide (magenta) and GFAP (green). Box in G indicates area shown at higher magnification in H. Arrowhead in G indicates cell body-localized FLAG (putative β1-AR) immunoreactivity. Arrow in H indicates nuclear FLAG (putative β1-AR) immunoreactivity. (I, J) Fluorescence photomicrographs depicting β1-AR (magenta) and GFAP (green) immunoreactivity in mouse cortical tissue. DAPI labeling of DNA is depicted in blue. Arrows indicate β1-AR-immunoreactive nuclei. Empty arrowheads indicate β1-AR-immunonegative nuclei. Insets in A, B are higher magnification images from the same experiment. Images are representative of immunostaining patterns observed in n ≥ 3 cell cultures from separate litters (except for C-D and E-F, for which n = 2 or 1, respectively, separate litters) or n=4 female mouse brains (I, J). Scale bar, 62.5 μm (A, B), 25 μm (C-G, insets in A, B); 3.8 μm (H); 20 μm (I); 6.8 μm (J).
The observation of nuclear β1-like immunoreactivity using two different antibodies, each directed against a distinct epitope of the receptor, strongly suggests that the observed immunoreactivity represents actual β1-AR. However, we wished to confirm the observation using a technique that does not rely on commercially available anti-β1-AR antibodies. To this end, cultured astrocytes were transfected (Lipofectamine 3000; Invitrogen) for 5 hours at 37°C with a plasmid directing the expression of a recombinant FLAG-tagged human β1-AR (Tang et al., 1999), followed by rinsing and incubation for 72 hours. To visualize the localization of the FLAG-tagged β1-AR, fixed, Triton-permeabilized astrocytes subjected to immunofluorescence using an anti-FLAG primary antibody (Table 1) carried out as described above. Punctate anti-FLAG immunoreactivity was observed over nuclei and cell bodies of transfected astrocytes (Fig. 2G, H).
To determine whether nuclear β1-like ir is present in mouse cortical astrocytes in situ, we incubated fixed, frozen brain sections of mouse forebrain with the C-terminal-directed β1-AR antibody. β1-AR-like immunoreactivity was observed over both nuclei and somata in mouse cortical tissue (Fig. 2I, J). Identification of astrocyte nuclei is difficult in sectioned brain tissue using GFAP-immunoreactivity, as cortical astrocytes in situ express low levels of GFAP (Verkhratsky and Nedergaard, 2018) and because astrocyte processes extend three-dimensionally throughout the tissue, making it difficult to determine definitively whether a given nucleus belonged to a GFAP+ cell. However, β1-AR-like immunostaining was repeatedly observed over nuclei that were obviously surrounded by GFAP-immunofluorescence.
The immunofluorescence signal observed over nuclei could represent receptors localized to inner (INM) or outer (ONM) nuclear membrane, plasma membrane, or peri-nuclear Golgi or endoplasmic reticulum. To begin to determine the extent to which nuclear immunofluorescence originates from receptors localized to the inner or outer nuclear membranes, we conducted immunostaining using each of the two β1-AR antibodies on primary astrocytes permeabilized with either Triton X-100 or digitonin. Triton X-100, which solubilizes all cellular membranes, allows free access of antibodies to all cellular compartments. Digitonin at the concentration used here preferentially permeabilizes cholesterol-rich membranes like the plasma membrane, leaving inner and outer nuclear membranes mainly intact (Adam et al., 1990). Thus, in digitonin-permeabilized cells, antibodies could not enter the perinuclear space (between outer and inner nuclear membranes) or the nuclear compartment and could not label any antigens in those spaces. To confirm that these two detergents differentially permeabilized astrocyte membranes, we conducted immunostaining using an antibody to type A/C nuclear lamins, proteins localized to the inner surface of the INM (Gruenbaum et al., 2005), in Triton- and digitonin-permeabilized astrocytes. Dense, uniform lamin A/C immunostaining was observed in nuclei of all Triton-permeabilized astrocytes but was almost completely lost in digitonin-permeabilized astrocytes (Fig. 3A). Bootstrapping analysis on the mean difference in nuclear immunofluorescence between the two detergent groups indicated that nuclear lamin A/C-like immunofluorescence was significantly lower in digitonin- than in Triton-permeabilized astrocytes (Fig. 3B; p < 0.0001). The effect of detergent was large (Cohen’s d = 3.04), and the direction of the detergent effect was the same in every biological replicate (Fig. 3B).
Fig. 3. β1-adrenergic receptors are localized to the astrocyte inner nuclear membrane.
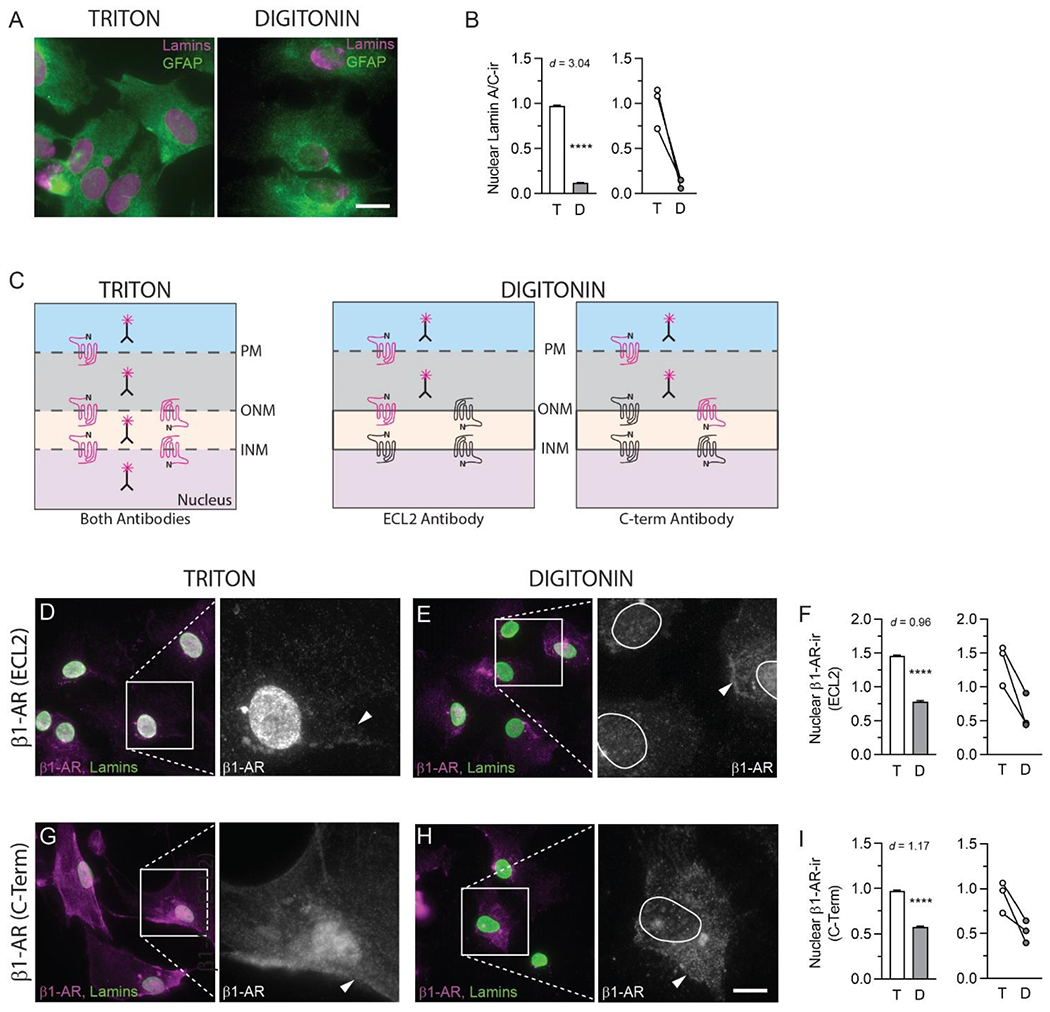
(A) Fluorescence photomicrographs of formaldehyde-fixed mouse primary astrocytes permeabilized with Triton X-100 or digitonin and incubated with antibodies against GFAP (green) and lamin A/C (magenta). Scale bar, 25 μm. (B) Quantification of nuclear lamin A/C immunofluorescence in Triton- and digitonin-permeabilized astrocytes. The bar graph on the left depicts mean ± s.d. nuclear lamin A/C-like immunofluorescence from all ROIs across all biological replicates for each detergent condition. The line graph on the right depicts mean nuclear lamin A/C-like immunofluorescence for each detergent in each of the three biological replicates. Lines connect mean values for the two detergent conditions in the same biological replicate. (C) Diagram depicting the predicted staining patterns resulting from each of two β1-AR antibodies, one targeting extracellular loop 2 (ECL2) and the other targeting the C-terminal tail, in astrocytes permeabilized with either Triton X-100 or digitonin. Nuclear membrane β1-ARs are depicted in both possible receptor orientations. Red receptors indicate predicted positive immunofluorescence. (D, E, G, H) Fluorescence photomicrographs depicting β1-AR (magenta or white) and lamin A/C (green) immunoreactivity in mouse primary astrocytes. Astrocytes were initially permeabilized with either Triton X-100 (D, G) or digitonin (E, H) and incubated with antibodies directed against the extracellular loop 2 (D, E) or C-terminal domain (G, H) of β1-AR. After β1-AR antibody incubation, all cells were re-permeabilized with Triton X-100 and incubated with an antibody against Lamin A/C to label nuclei. Arrowheads indicate β1-AR-immunoreactive perikarya. Nuclei are outlined in white for the insets in E and H. Images are representative of immunostaining patterns observed in n ≥ 3 cell cultures from separate litters. For the pairs of images in D, E, G and H, scale bars represent 25 μm for the left-hand image and 9.25 μm for the right-hand image. (F, I) Quantification of nuclear β1-AR-like immunofluorescence in Triton- and digitonin-permeabilized astrocytes (ECL2 antibody in F, C-Term antibody in I). Bar graphs on the left depict mean ± s.d. nuclear β1-AR-like immunofluorescence from all ROIs across all biological replicates for each detergent condition. Line graphs on the right depict mean nuclear β1-AR-like immunofluorescence for each detergent in each of the three biological replicates. Lines connect mean values for the two detergent conditions in the same biological replicate. Cohen’s d values are shown to indicate the size of the effect. **** P< 0.0001.
The diagram in Figure 3C depicts the staining patterns predicted to result from immunofluorescence experiments using each of the two β1-AR antibodies in Triton- and digitonin-permeabilized astrocytes based upon the potential membrane localizations and orientations of the receptors. Briefly, in digitonin-permeabilized cells, β1-ARs localized to INM would not be labeled by either antibody, while β1-AR localized to ONM, or to peri-nuclear Golgi or ER would be labeled by one of the two antibodies depending on the membrane orientation of the receptor.
For both β1-AR antibodies, the dense punctate nuclear immunofluorescence observed in Triton-permeabilized astrocytes was markedly reduced in digitonin-permeabilized astrocytes (Fig. 3D, E, G, H). Bootstrapping this mean difference revealed that nuclear β1-AR-like immunofluorescence intensities obtained with both antibodies were significantly lower in digitonin- than in Triton-permeabilized astrocytes (ECL2 antibody: P<0.0001, Cohen’s d = 0.96; C-TERM antibody: P<0.0001, Cohen’s d = 1.17). The direction of the detergent effect was the same in every biological replicate (Fig. 3F, I). While both β1-AR antibodies yielded immunostaining at both plasma and nuclear membranes, the patterns of staining observed using the two antibodies were slightly different. The ECL2 antibody produced very strong nuclear immunofluorescence, but only weak cell body staining (Fig 2A, 3D, 3E). In contrast, the C-terminal antibody produced similar immunofluorescence intensities over cell bodies and nuclei (Fig 2B, 3G, 3H).
To further confirm nuclear localization of β1-AR, we purified plasma membrane and nuclear proteins from primary astrocyte cultures (see Methods) and conducted western blots. We tested the effectiveness of the fractionation protocol by conducting western blots on subcellular fractions using antibodies to type A/C nuclear lamins (nuclear proteins) and the ATP1A1 subunit of the sodium-potassium ATPase (plasma membrane protein). Immunoreactivity for type A/C nuclear lamins appeared in whole cell lysates and nuclear fractions, but not in plasma membrane fractions, while immunoreactivity for the ATP1A1 subunit of the sodium-potassium ATPase appeared in whole cell lysates and plasma membrane fractions, but not in nuclear fractions (Fig. 4A). We assessed the specificity of the two β1-AR antibodies by conducting western blots on commercially available lysates of wild-type HeLa cells and HeLa cells in which the gene for β1-AR had been knocked out by CRISPR/Cas9-mediated modification (Abcam, Cat# ab257108). Several bands appeared in both wild-type and β1-AR knockout extracts, but a single β1-AR-immunoreactive band of approximately 50 kDa molecular weight (Predicted MW 51 kDa UniProt ID: P08588 (human)) was observed in wild-type, but not β1-AR knockout lysates using both antibodies (Fig. 4B). In western blots of astrocyte subcellular fractions, a β1-AR-immunoreactive band of approximately 50 kDa molecular weight was observed in whole cell lysates and nuclear fractions using both the C-terminal and ECL2-directed antibodies (Fig. 4B, Predicted MW 50.5 kDa, UniProt ID: P34971 (mouse)).
Fig. 4. β1-adrenergic receptor immunoreactivity in astrocyte subcellular fractions.
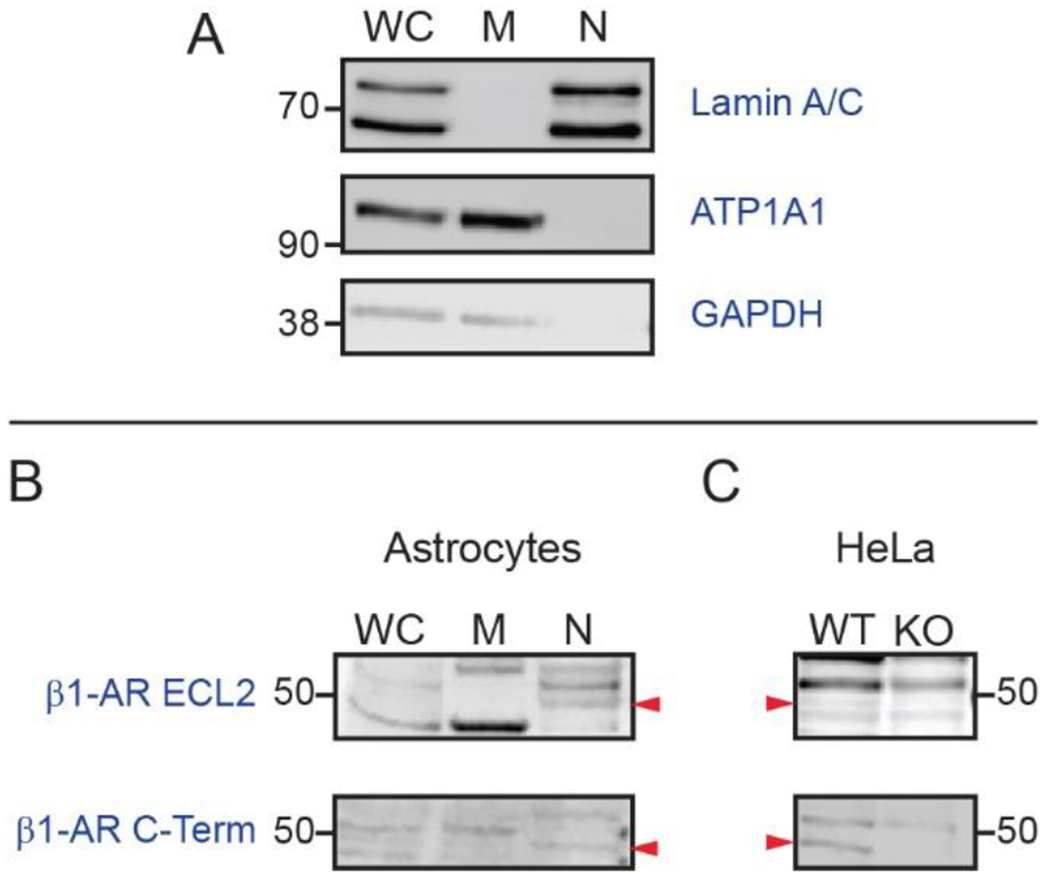
(A) Validation of subcellular fractionation protocol. Immunoblotting of proteins from whole cell lysate, membrane, and nuclear fractions using antibodies against lamin A/C, sodium/potassium ATPase subunit alpha-1 (ATP1A1), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Images are representative of immunostaining patterns observed in n = 4 cell cultures from separate litters. (B) β1-AR immunoblotting of whole cell lysate, membrane, and nuclear proteins from primary cultured astrocytes using antibodies directed against either ECL2 (top) or the C-terminal tail (bottom) of β1-AR. Arrowheads indicate the position of immunoreactive bands of the predicted molecular weight. Images are representative of patterns observed in n = 3 blots of proteins from astrocytes cultured from independent mouse litters. (C) Validation of β1-AR antibodies. β1-AR immunoblotting of whole cell lysates from wild-type (WT) and β1-AR knockout (KO) HeLa cells. Position of 50-kDa MW marker is indicated. Arrowheads indicate position of β1-AR-immunoreactive bands which are present in WT, but not in KO lysates.
3.3. Gsα and Gs-coupled signaling components are localized to the nucleus in cultured astrocytes.
To begin testing the hypothesis that nuclear membrane-localized β1-ARs are functionally coupled to intranuclear G-protein-mediated signaling pathways, we used immunofluorescence with differential detergent permeabilization to examine the potential nuclear localization of key components of canonical G-protein-coupled receptor signaling, specifically Gαs and PKA regulatory subunit isoforms I and II (PKA-RI, PKA-RIIα) (see Table 1 for details). Nuclei of cultured astrocytes were identified by immunolabeling with an antibody against nuclear lamins A and C. Decisions of which isoforms of PKA subunits to examine were based on astrocyte gene expression data from the brain RNASeq database (Zhang et al., 2014).
We observed dense Gsα-like immunofluorescence over nuclei, and more diffuse, punctate immunofluorescence over somata of Triton-permeabilized astrocytes (Fig. 5A). Nuclear Gsα-like immunofluorescence was almost completely absent in digitonin-permeabilized astrocytes, while immunofluorescence over the soma remained (Fig. 5B). Bootstrapping analysis indicated that mean nuclear Gsα-like immunofluorescence was significantly lower in digitonin- than in Triton-permeabilized astrocytes (Fig. 5C, P < 0.0001). The effect of digitonin was large (Cohen’s d = 2.37), and the direction of detergent effect was the same in every biological replicate (Fig. 5C). In western blots of proteins from astrocyte subcellular fractions (Fig. 5J), Gsα-ir bands of approximately 45 and 50 kDa (Predicted MW 45.6 kDa, UniProt P63094) were observed in both nuclear and plasma membrane fractions. The molecular weights of these bands are consistent with those of distinct Gαs isoforms that have been previously reported, with 45 kDa corresponding to the canonical isoform (Robishaw et al., 1986). A prominent intermediate sized band was observed in membrane, but not nuclear, fractions.
Fig. 5. Nuclear localization of β-adrenergic signaling components in mouse astrocytes.
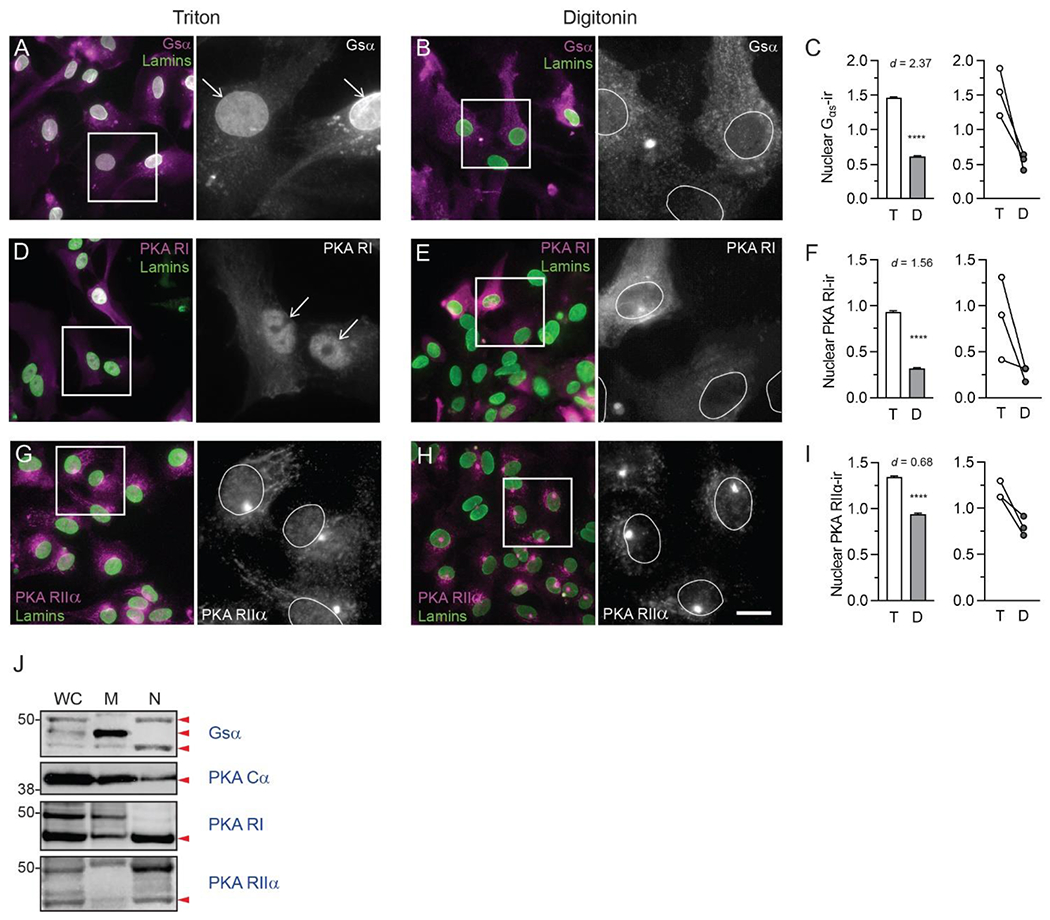
Fluorescence photomicrographs of fixed mouse primary astrocytes permeabilized with either Triton X-100 or digitonin, followed by incubation with antibodies directed against adrenergic receptor downstream signaling components (magenta in dual color images, white in single color). All astrocytes were then permeabilized again with Triton X-100 and incubated with an antibody against lamins A and C (green) to label nuclei. Boxes indicate areas shown at higher magnification to the right. Positions of nuclei are indicated with arrows or white ovals. Images are representative of immunostaining patterns observed in n ≥ 3 cell cultures from separate litters for each of the following proteins: (A, B) Alpha subunit of the stimulatory G protein (Gsα); (D, E) PKA regulatory subunit I (PKA RI); and (G,H) PKA regulatory subunit IIα (PKA RIIα). In each pair of images, scale = 25 μm in the dual-color image, 9.25 μm in grayscale image. (C, F, I) Quantification of nuclear immunofluorescence for each target protein in Triton- and digitonin-permeabilized astrocytes. Bar graphs on the left depict mean ± s.d. nuclear immunofluorescence for each target protein from all ROIs across all biological replicates for each detergent condition. Line graphs on the right depict target protein mean nuclear immunofluorescence in each of the three biological replicates. Lines connect mean values for the two detergent conditions from the same biological replicate. Cohen’s d values are shown to indicate the size of the effect. **** P< 0.0001. (J) Immunoblotting of whole cell lysate, membrane, and nuclear proteins from primary mouse astrocytes using antibodies against Gsα, PKA catalytic subunit (PKA Cα), PKA RI, and PKA RIIα. Red arrowheads indicate positions of predicted molecular weights for the protein of interest. All images are representative of immunostaining patterns observed in n = 3 cell cultures from separate litters.
PKA-RI-like immunoreactivity was observed over cell bodies and nuclei of Triton-permeabilized astrocytes, with dense staining observed over nuclei (Fig. 5D). In digitonin-permeabilized astrocytes, cell body PKA-RI immunostaining was similar to that observed in Triton-permeabilized astrocytes, but nuclear immunostaining appeared to decrease (Fig. 5E). Bootstrapping this mean difference indicated that nuclear PKA-RI-like immunofluorescence was significantly lower in digitonin- than in Triton-permeabilized astrocytes (Fig. 5F, P < 0.0001). The effect of detergent was large (Cohen’s d = 1.56), and the direction of the detergent effect was the same in all biological replicates (Fig. 5F). In western blots, a PKA-RI-ir band of approximately 45 kDa (predicted PKA-RI molecular weight = 43 kDa UniProt ID: Q9DBC7) was observed in both membrane and nuclear fractions, while a 50-kDa band was observed only in membrane fractions (Fig. 5J).
Punctate PKA-RIIα-like immunoreactivity was distributed evenly over cell bodies and nuclei of Triton-permeabilized astrocytes, and at higher density in endomembrane (ER or Golgi)-like profiles (Fig. 5G). Cell body and endomembrane PKA-RIIα-like immunoreactivity was also observed in digitonin-permeabilized astrocytes, but nuclear immunoreactivity was markedly decreased (Fig. 5H). Bootstrapping analysis of the mean difference between the two detergents indicated that nuclear PKA-RIIα immunofluorescence was significantly lower in digitonin-permeabilized than in Triton-permeabilized astrocytes (Fig. 5I, P < 0.0001). The effect of detergent was moderate (Cohen’s d = 0.68), and the direction of the effect was the same in all biological replicates (Fig. 5I). In western blots, PKA-RIIα-ir bands of approximately 45 kDa (predicted PKA-RII-α molecular weight = 45 kDa, UniProt ID: P12367) were observed in whole cell lysate, membrane, and nuclear fractions (Fig. 5J). We observed a PKA catalytic subunit (alpha isoform, PKA Cα)-immunoreactive band of approximately 40 kDa molecular weight (predicted molecular weight = 40 kDa, UniProt ID: P05132) in western blots of astrocyte nuclear and membrane fractions (Fig. 5J).
3.4. Catecholamine transporters are localized to astrocyte nuclei and plasma membranes.
Access of norepinephrine, which is cationic and membrane-impermeable at physiological pH, to β1-ARs localized to inner nuclear membranes would require carrier-mediated transport across both plasma and outer nuclear membranes. While our studies indicate that OCT3 could potentially mediate transport across both membranes, it is possible that additional catecholamine transporters are involved. To begin to characterize more completely the mechanisms by which norepinephrine may access nuclear β1-AR, we examined the localization of two additional uptake2 transporters: organic cation transporter 2 (OCT2) and the plasma membrane monoamine transporter (PMAT), and of the uptake1 catecholamine transporters NET (norepinephrine transporter) and DAT (dopamine transporter), using immunofluorescence (exclusively on Triton-permeabilized astrocytes) and western blot.
OCT2-like-immunoreactivity was observed over nuclei and somata of Triton-permeabilized astrocytes (Fig. 6A,B). OCT2-ir bands of approximately 60 kDa molecular weight (predicted molecular weight = 61.8 kDa, UniProt ID: O70577) were observed in membrane and nuclear fractions (Fig. 6E). PMAT-like immunoreactivity was also observed over both nuclei and somata of Triton-permeabilized astrocytes (Fig. 6C, D). In western blots examining distribution of uptake1 transporters, a NET-like-ir band of approximately 45-kDa molecular weight was observed in both membrane and nuclear fractions and a 60-kDa band was observed only in nuclear fractions (Fig. 6E). Non-glycosylated hNET migrates in western blots at approximately 46-50 kDa (Melikian et al., 1996), with glycosylated forms appearing at 54- and 70 kDa (Melikian et al., 1996; Matthies et al., 2009). DAT-ir bands of 50 and 75 kDa were observed in both nuclear and membrane fractions (Fig. 6E). Fully glycosylated DAT appears in western blots as a broad band centered around 75-85 kDa molecular weight, while non-glycosylated DAT appears at 40-50 kDa (Li et al., 2004).
Fig. 6. Localization of catecholamine transporters in mouse astrocytes.
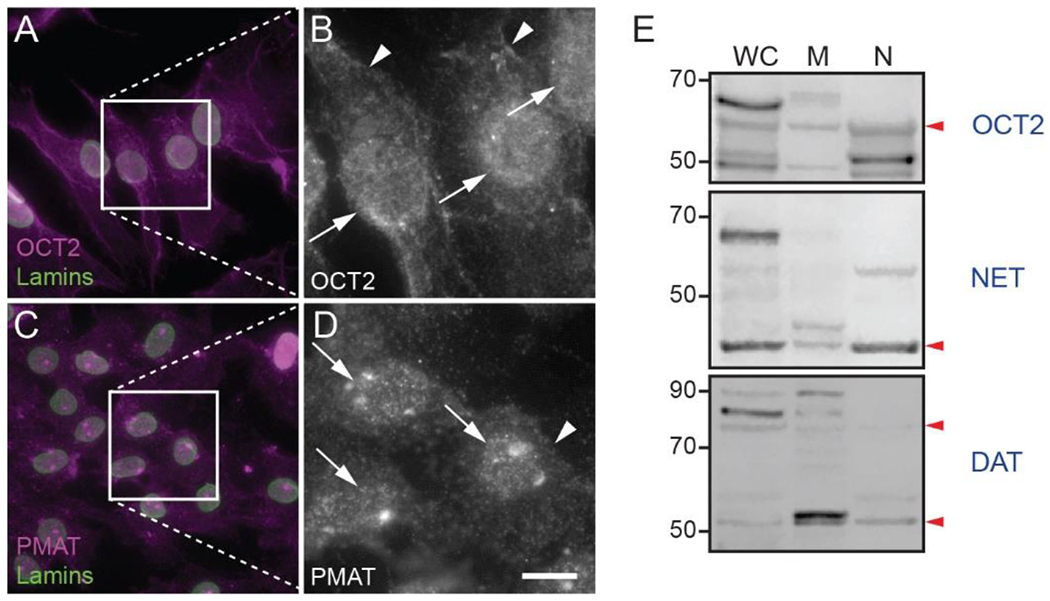
(A-D) Fluorescence photomicrographs of mouse primary astrocytes permeabilized with Triton X-100 and incubated with antibodies against lamin A/C (green) and catecholamine transporters (magenta and white). Images are representative of immunostaining patterns observed in n ≥ 3 cell cultures from separate litters for each of the following transporters: (A, B) organic cation transporter 2 (OCT2); (C, D) plasma membrane monoamine transporter (PMAT). Arrowheads indicate immunoreactivity in perikarya. Arrows indicate putative nuclear immunoreactivity. Scale bar in (D), 25 μm in all dual-color images, 9.25 μm in all grayscale images. (E) Immunoblotting of whole cell lysate, membrane, and nuclear proteins from primary mouse astrocytes using antibodies against OCT2, NET and DAT. Red arrowheads indicate approximate positions of predicted molecular weight bands.
3.5. Norepinephrine induces rapid and prolonged increases in nuclear PKA activity.
The localization of β1-ARs to inner nuclear membranes and the evidence that G-proteins and their signaling partners occur in astrocyte nuclei suggest that nuclear receptors are capable of activating downstream cAMP signaling. The presence of OCT3 and other catecholamine transporters in both plasma and nuclear membranes is consistent with the hypothesis that access to and activation of nuclear membrane β1-ARs is gated by transporter activity. To determine whether nuclear β1-ARs are functionally coupled to signaling machinery and to test the hypothesis that activation of nuclear receptors is transporter-gated, we transfected astrocytes with plasmids driving the expression of a nuclear localized PKA activity sensor, ExRai-AKAR-NLS, to allow monitoring of nuclear PKA activity in real time. This sensor consists of a PKA substrate sequence and a phosphoamino acid-binding domain fused with the circularly permuted GFP from GCaMP3. This recombinant protein displays two excitation peaks (~400 and ~509 nm with a shoulder at ~480 nm), both of which emit at ~515 nm. Phosphorylation of the PKA substrate sequence induces a conformational change in the sensor that results in a decrease in 400 nm-induced emission and an increase in 488 nm-induced emission, resulting in a large increase in the “excitation ratio” (Mehta et al., 2018). We examined the effects of bath-applied norepinephrine (50 nM) on nuclear excitation ratios in astrocytes in the presence or absence of catecholamine transport inhibitors. Transfected astrocytes were pretreated with either vehicle or a cocktail of catecholamine transport inhibitors including atomoxetine (NET inhibitor), GBR 12909 (DAT inhibitor), and corticosterone at a concentration shown to inhibit OCT3, OCT2, and PMAT (Gründemann et al., 1998b; Engel et al., 2004; Duan and Wang, 2010). After the preincubation period, excitation ratiometric data were collected every 20 seconds for a 3-minute baseline period, after which norepinephrine was bath applied to all astrocytes and ratiometric data were collected every 20 seconds for an additional 30 minutes. Increases in nuclear PKA were detected as increases in the excitation ratio. Norepinephrine-induced increases in nuclear excitation ratio were observed in both vehicle- and transport inhibitor-pretreated cells, but the kinetics of the initial responses in the two groups were different. In vehicle-pretreated cells, norepinephrine treatment produced rapid and robust increases in nuclear excitation ratio over the initial 10-minute period that continued to increase for the entire 30-minute duration of norepinephrine treatment (representative traces and ratio images in Fig. 7A, B; mean responses in Fig. 7C, D). In transport inhibitor-treated cells, norepinephrine-induced increases in nuclear excitation ratio did not appear until the second 10-minute post-treatment period, after which they continued to increase for the remainder of norepinephrine treatment. A 2 (Group: Vehicle vs Inhibitors) x 4 (Period: Baseline vs 0-10 vs 10-20 vs 20-30 minutes) ANOVA revealed higher average ratios in the Vehicle group relative to the Inhibitors group, F(1, 34) = 8.18, p = 0.007, and an increase in the ratio across time periods, F(1, 102) = 64.93, p < 0.0001, as well as an interaction between group and time period, F(1, 102) = 3.31, p = 0.023 (Fig. 7E). Planned interaction contrasts showed that excitation ratios increased more in the Vehicle group from baseline to 0-10 minutes relative to the Inhibitors group, F(1, 34) = 38.47, p < 0.0001, consistent with the hypothesis that the rapid nuclear PKA response is dependent on catecholamine transport. However, across the subsequent time periods (0-10 minutes vs. 10-20 minutes), the rate of change in nuclear PKA was similar between the two groups (F(1, 34) = 0.89, p = 0.35). A Tukey’s HSD analysis supported this conclusion, showing that excitation ratios increased across every post-NE time period in the vehicle-pretreated group (p < 0.025), but only exhibited a significant elevation above baseline after 10-minutes of norepinephrine exposure in the transport inhibitor-pretreated group (p < 0.004). Taken together, the results suggest that rapid nuclear PKA responses (within 10 minutes) in astrocytes are dependent on carrier-mediated transport of norepinephrine, and that more gradual increases in nuclear PKA signaling (after 10 minutes) are not.
Fig. 7. Rapid norepinephrine-induced increases in nuclear PKA activity require catecholamine transporter function.
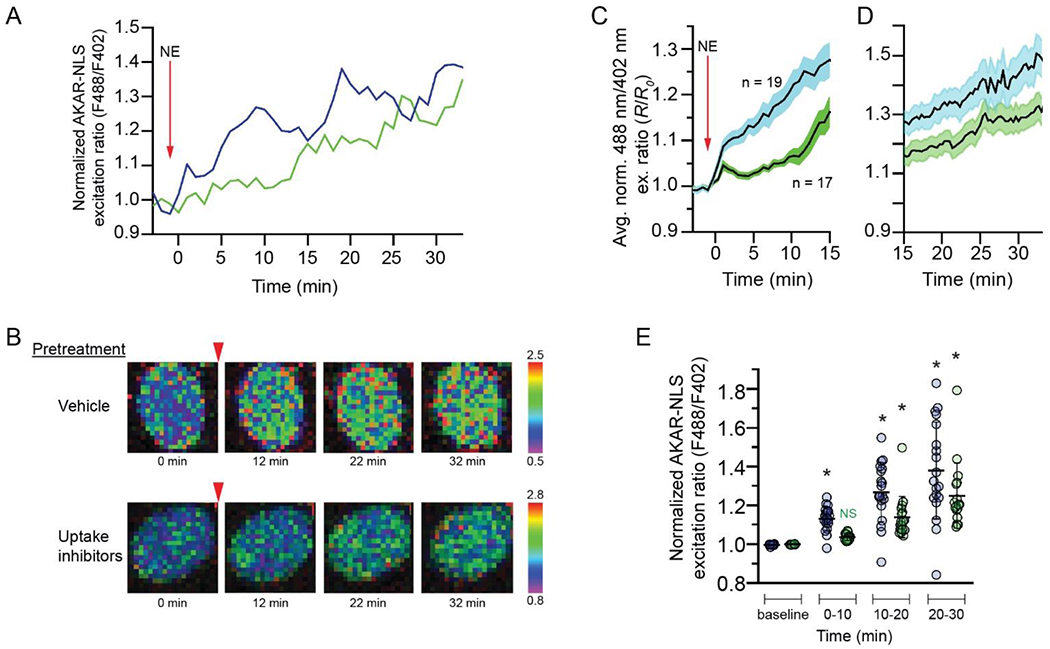
(A) Representative time courses of nuclear PKA responses to 50 nM norepinephrine in ExRai-AKAR-NLS-expressing astrocytes pretreated with vehicle (blue) or catecholamine transport inhibitors (green). Curves are plotted as 488/402 nm excitation ratios normalized with respect to time 0. (B) Pseudocolor images of the ExRai-AKAR-NLS ratio responses of the astrocytes from A. Warmer colors indicate higher excitation ratios as shown in the scales to the right of the images. Red arrowheads indicate the time of norepinephrine application. Pretreatment condition is indicated to the left of the images. (C, D) Average time courses (t = −3 to 15 min in C; 15-35 min in D) of nuclear PKA responses to 50 nM norepinephrine in ExRai-AKAR-NLS-expressing astrocytes pretreated with vehicle (blue, n = 19 cells from 5 separate astrocyte cultures) or transport inhibitors (green, n = 17 cells from 5 separate astrocyte cultures). Curves are plotted as 488/402 nm excitation ratios normalized with respect to time 0. Solid lines represent the mean; shaded areas represent s.e.m. (E) Normalized nuclear PKA responses to 50 nM norepinephrine of vehicle (blue)- or transport inhibitor (green)-pretreated ExRai-AKAR-NLS-expressing astrocytes (same cells shown in (C)). Ratiometric responses obtained during the indicated time periods were pooled and averaged for each cell. Thick, horizontal lines represent the mean and error bars represent SD. * indicates significantly different from the baseline excitation ratio for that group (p < 0.05). NS – not significant compared to baseline excitation ratio for that group.
4. DISCUSSION
These studies provide evidence that the inner nuclear membrane is an adrenergic signaling platform in astrocytes. They reveal a population of functional, G-protein-coupled β1-adrenergic receptors localized to the inner nuclear membrane and indicate that norepinephrine accesses these receptors through the actions of catecholamine transporters localized to plasma and outer nuclear membranes. Treatment of astrocytes with norepinephrine leads to rapid increases in nuclear PKA activity that are blocked by catecholamine uptake inhibitors, suggesting that norepinephrine-induced increases in nuclear PKA activity are mediated by nuclear receptors. Nuclear β1-adrenergic receptors represent a powerful mechanism by which norepinephrine may directly influence nuclear processes, leading to profound effects on gene expression that may contribute to neuroprotective, immunomodulatory, and metabolic regulatory actions of norepinephrine in astrocytes.
Growing numbers of studies have demonstrated that G-protein-coupled receptors can initiate signaling cascades from intracellular locations including the nuclear membrane (Campden et al., 2015; Jong et al., 2018). Our studies provide evidence that β1-ARs are localized to both plasma and nuclear membranes in astrocytes. The fact that nuclear immunofluorescence was observed using two separate antibodies, directed against distinct intra- and extracellular-facing epitopes of the receptor, and that exogenously expressed FLAG-tagged β1-AR was also observed at the nucleus strongly indicate that the immunofluorescence observed does represent actual β1-ARs. This staining pattern, with both nuclear and plasma membrane β1-like ir, is similar to that observed in previous studies of H9C2 cells using the C-terminal antibody (Irannejad et al, 2017). Importantly in those studies, nuclear and plasma membrane β1-like immunoreactivity was eliminated in cells treated with β1-AR siRNA.
It is expected that nuclear β1-ARs, like their counterparts at the plasma membrane, are transmembrane proteins and thus could be localized to inner, outer, or both nuclear membranes. The punctate pattern of immunofluorescence we observed for β1-ARs as well as OCTs and other transporters in astrocyte nuclei is different from the dense, uniform immunofluorescence pattern of the intranuclear protein Lamin A/C, but is similar to immunofluorescence patterns observed with other transmembrane proteins, including Lamin B receptor, a transmembrane protein localized to the inner nuclear membrane (Filesi et al., 2005). We did observe differences in the relative intensity of nuclear β1-like immunofluorescence resulting from the ECL2 and the C-terminus antibodies (Fig 2 A, B; Fig. 3D, G). This phenomenon may be due to differences in the avidity of antibody/antigen binding between the two antibodies, or to biological differences including differences in accessibility of the respective epitopes. Further studies are required to address these differences. In addition to differences in nuclear labeling intensity, we also observed apparently weaker cell body immunofluorescence with the ECL2 antibody. We believe that this phenomenon results a technical issue stemming from the fact that optimal exposure times required to capture nuclear fluorescence were shorter with the ECL2 than with the C-terminal antibody. Because the nuclear signal resulting from the ECL2 antibody was so strong, it was difficult to capture cell body immunostaining without overexposing nuclei.
The results of our differential detergent permeabilization studies, in which the nuclear immunofluorescence observed in Triton-permeabilized astrocytes with antibodies directed against both intra- and extracellular epitopes of the receptor was lost when astrocytes were permeabilized with digitonin, indicate that β1-ARs are localized primarily to inner, rather than outer, nuclear membranes of cultured astrocytes. If nuclear labeling represented receptors localized to the outer nuclear membrane or to peri-nuclear ER or Golgi, nuclear fluorescence resulting from at least one of the two antibodies would be unchanged by digitonin permeabilization. Although previous studies suggested potential nuclear localization of β1-ARs in some neurons (Milner et al., 2000; Cox et al., 2008), the present findings are the first to document specific localization of adrenergic receptors to the inner nuclear membrane in any CNS cell type. They suggest that, in addition to actions at the plasma membrane, norepinephrine may initiate Gs-mediated signaling cascades in the nuclear compartment of astrocytes. They are similar to findings in cardiomyocytes, in which α- and β- adrenergic receptors are localized to the nuclear envelope, where they activate Gq- and Gs-coupled signaling pathways (Vaniotis et al., 2011; Wu et al., 2014).
The results of our immunolocalization studies combined with the demonstration of rapid norepinephrine-induced increases in nuclear PKA activity, suggest that β1-ARs localized to astrocyte nuclear membranes are functionally coupled to Gs and downstream signaling components. Both immunofluorescence and western blot indicate that a significant amount of Gαs in astrocytes is localized to the nuclear compartment. This pattern of immunofluorescence is consistent with studies of Gαs in H9C2 cells (He et al., 2014; Irannejad et al., 2017) and mouse embryonic fibroblasts (He et al., 2014). If it functions similarly in the nuclear compartment as it does at the cell surface, nuclear Gαs would be expected to associate with the inner surface of the INM. The pattern of Gαs immunoreactivity we observed in astrocyte nuclei, distributed uniformly across the nuclear space, was similar to that observed for type A/C nuclear lamins, proteins that are closely associated with the inner surface of the INM. Thus, this pattern is consistent with association of Gαs with the INM in astrocytes. The western blot banding patterns we observed suggest that distinct Gαs isoforms may localize to nuclear and plasma membrane sites.
Consistent with previous studies that demonstrated the existence of a cAMP-activable pool of PKA holoenzyme resident in nuclei of HEK-293 cells (Sample et al., 2012; Clister et al., 2019), our studies suggest that a subset of PKA holoenzyme resides in the nuclei of unstimulated astrocytes. Both PKA RI and RIIα were localized to astrocyte nuclei, with RI more strongly localized to nuclei and RIIα distributed more evenly between plasma membrane and nuclei. PKA RIIα also appeared in putative endomembrane profiles, and was densely localized to a perinuclear structure, likely the centrosome. Our findings are consistent with previous studies demonstrating that PKA RIIα localizes to centrosomes and Golgi apparatus (Keryer et al., 1999), and to the nucleus (Clister et al., 2019). Together, the results of our immunolocalization studies suggest that, while β1-ARs are localized to both cell surface and nuclear membranes, receptors at the two sites may initiate distinct signaling processes. The distribution and density of immunostaining for key components of the canonical Gs signaling machinery, including Gαs and PKA subunits, differed between nuclei and cell bodies, consistent with the emerging understanding that PKA can be organized into signaling microdomains organized by A-Kinase-Anchoring Proteins (AKAPs) with distinct cellular localizations (Torres-Quesada et al., 2017). Thus, nuclear and plasma membrane-localized receptors may initiate distinct signaling processes which mediate different aspects of the overall cellular response to norepinephrine. Additional studies will be required to fully characterize signaling processes activated by the two populations of receptors.
The present studies provide evidence that multiple catecholamine transporters may gate access of norepinephrine to nuclear receptors. They confirm our previous observation that OCT3 is localized to plasma and nuclear membranes (Gasser et al., 2017), and they suggest potential roles for OCT2 and PMAT, as well as the uptake1 transporters DAT and NET, in gating activation of nuclear receptors. They are consistent with studies demonstrating that inhibition or genetic knockout of OCT3 decreases the activation of nuclear alpha-adrenergic receptors (Wright et al., 2008) and Golgi-localized β1-ARs (Irannejad et al., 2017; Wang et al., 2021). While the present studies are the first to suggest nuclear membrane localization of OCT2, PMAT, and NET, they are not the first to suggest that DAT may be localized to the nucleus. Immunogold electron microscopic examination of neurons in the substantia nigra revealed DAT localized to both inner and outer nuclear membranes (Hersch et al., 1997; Block et al., 2015). The data from the present studies do not allow us to determine the specific nuclear membrane (inner or outer) localization of any transporters. Neither do they provide information about the orientation of any of the transporters in the membrane. Additional studies, including transport assays in isolated nuclei, will be required to fully characterize the localization and function of nuclear membrane catecholamine transporters. The role of a given transporter in either allowing or limiting the activation of nuclear β1-ARs would depend on its localization to the inner or outer nuclear membrane and on its orientation in those membranes. Localization of monoamine transporters to the nuclear envelope is also interesting in light of recent studies documenting the phenomenon of histone monoaminylation, in which dopamine and serotonin, both substrates of the uptake2 transporters, are enzymatically conjugated to histones and modulate gene expression (Farrelly et al., 2019; Chan and Maze, 2020).
Our functional studies provide strong evidence that nuclear membrane β1-ARs are functionally coupled to downstream signaling machinery. We observed two classes of nuclear PKA responses to norepinephrine based on their sensitivity to pretreatment with inhibitors of catecholamine transport: a transporter-dependent, early response, and a transporter-independent, delayed response. The transporter-dependent response was rapid, detectable within the first minutes of norepinephrine treatment, and was absent in cells pre-treated with uptake inhibitors. The transporter-independent response was more gradual, only becoming significant during the second 10-minute time block after norepinephrine application and was unaffected by uptake inhibition. The rapid onset of early, transporter-dependent responses suggests that they are mediated by PKA that is resident to the nucleus and activated in response to nuclear membrane β1-AR-initiated Gs signaling, and not by PKA activated at the plasma membrane and diffusing to the nucleus. This is consistent with studies examining the kinetics of nuclear PKA activation in response to cAMP generated either at the plasma membrane or in the nucleus (Sample et al., 2012). In those studies, when cAMP was generated at the plasma membrane, nuclear PKA activity increased gradually, with a half time (t1/2) of approximately 20 minutes and was mediated by catalytic subunits which had been activated at the plasma membrane and diffused into the nucleus. When cAMP was generated within the nucleus, nuclear PKA activity increased rapidly, with t1/2 of 3-4 minutes, and appeared to be mediated by nuclear-resident PKA. In our studies, the rapid onset of the transporter-dependent increase in PKA activity suggests that it reflects activation of nuclear-resident PKA in response to locally generated cAMP. The delayed onset of the transporter-independent PKA response in the present studies suggests that it reflects the activity of cytosolic PKA activated in response to plasma-membrane-localized β-AR receptor activation. It is important to note that in our studies norepinephrine was not washed off after it was applied, meaning that cells were exposed to the agonist for the entire duration of the 30-minute monitoring period. This likely explains the long duration of norepinephrine-induced increases in nuclear PKA activity. It is likely that nuclear PKA responses to a brief pulse of norepinephrine would have been more transient, and that the delayed (transporter-independent) responses may not have been as robust. Further studies will be required to examine the responses of astrocytes to briefer pulses of norepinephrine that may better reflect physiological conditions.
The ability of inhibitors of catecholamine uptake to block norepinephrine-induced increases in nuclear PKA activity strongly suggests that nuclear PKA responses are mediated exclusively by receptors resident in the inner nuclear membrane. However, other interpretations are possible. One possibility is that inhibition of norepinephrine uptake in our experiments facilitated the activation of cell surface α2-adrenergic receptors, which are expressed in murine astrocytes (Zhang et al., 2014). Previous studies have demonstrated that activation of α2-adrenergic receptors can inhibit β-AR-induced activation of adenylyl cyclase (Northam et al., 1989). This could explain the delayed increase in nuclear PKA activity we observed in cells treated with uptake inhibitors. Further studies are needed to explore this possibility.
Norepinephrine exerts powerful and pervasive actions in the central nervous system. In astrocytes, these effects include neuroprotective, immunomodulatory, and metabolic regulatory actions, many of which involve regulation of nuclear processes. Activation of astrocyte β-adrenergic receptors induces phosphorylation of RNA Polymerase II (Lee and Jungmann, 1981), histones (Harrison et al., 1980), and CREB (Koppel et al., 2018), and regulates the expression of neurotrophic factors (Juric et al., 2008; Koppel et al., 2018), metabolic enzymes (Pellegri et al., 1996; Allaman et al., 2000) and immunoregulatory proteins (Feinstein et al., 2002; Gavrilyuk et al., 2002; Madrigal et al., 2009). Our findings suggest a novel and powerful mechanism by which norepinephrine may initiate these and other actions in astrocytes. They add to the growing body of evidence that G-protein-coupled receptors can be localized to, and activated at, endomembrane sites (Jong et al., 2018). β1-ARs at nuclear and plasma membranes may mediate distinct aspects of integrated cellular responses to norepinephrine and contribute uniquely to the actions of norepinephrine in astrocytes.
MAIN POINTS:
β1-adrenergic receptors and their signaling partners are localized to astrocyte nuclei. Norepinephrine-induced activation of nuclear β1 receptors leads to rapid increases in nuclear PKA activity and requires transmembrane catecholamine transport.
ACKNOWLEDGEMENTS
We are indebted to Dr. Jin Zhang for the ExRai-AKAR-NLS expression plasmid. We thank Colleen Lavin and Dr. Suresh Kumar for excellent technical assistance. This study was supported by grant DA032895 to PG from the National Institutes of Health, and by support from the Charles E. Kubly Mental Health Research Center and the Just Live Foundation.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- Adam SA, Marr RS, Gerace L. 1990. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J Cell Biology 111:807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, Pellerin L, Magistretti PJ. 2000. Protein targeting to glycogen mRNA expression is stimulated by noradrenaline in mouse cortical astrocytes. Glia 30:382–391. [PubMed] [Google Scholar]
- Block ER, Nuttle J, Balcita-Pedicino JJ, Caltagarone J, Watkins SC, Sesack SR, Sorkin A. 2015. Brain Region-Specific Trafficking of the Dopamine Transporter. J Neurosci 35:12845–12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin B, Lavoie C, Vaniotis G, Baragli A, Villeneuve L-R, Ethier N, Trieu P, Allen BG, Hébert TE. 2006. Functional β-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res 71:69–78. [DOI] [PubMed] [Google Scholar]
- Campden R, Audet N, Hébert TE. 2015. Nuclear G protein signaling: new tricks for old dogs. Journal of cardiovascular pharmacology 65:110–22. [DOI] [PubMed] [Google Scholar]
- Chan JC, Maze I. 2020. Nothing Is Yet Set in (Hi)stone: Novel Post-Translational Modifications Regulating Chromatin Function. Trends Biochem Sci 45:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clister T, Greenwald EC, Baillie GS, Zhang J. 2019. AKAP95 Organizes a Nuclear Microdomain to Control Local cAMP for Regulating Nuclear PKA. Cell Chem Biol 26:885–891.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggan JS, Keller D, Calì C, Lehväslaiho H, Markram H, Schürmann F, Magistretti PJ. 2018. Norepinephrine stimulates glycogenolysis in astrocytes to fuel neurons with lactate. Plos Comput Biol 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DJ, Racca C, Lebeau FEN. 2008. β-adrenergic receptors are differentially expressed in distinct interneuron subtypes in the rat hippocampus. J Comp Neurol 509:551–565. [DOI] [PubMed] [Google Scholar]
- Dahl EF, Wu SC, Healy CL, Harsch BA, Shearer GC, O’Connell TD. 2018. Subcellular compartmentalization of proximal Gαq-receptor signaling produces unique hypertrophic phenotypes in adult cardiac myocytes. J Biol Chem 293:8734–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan H, Wang J. 2010. Selective Transport of Monoamine Neurotransmitters by Human Plasma Membrane Monoamine Transporter and Organic Cation Transporter 3. Journal of Pharmacology and Experimental Therapeutics 335:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Zhou M, Wang J. 2004. Identification and Characterization of a Novel Monoamine Transporter in the Human Brain. J Biol Chem 279:50042–50049. [DOI] [PubMed] [Google Scholar]
- Evans AK, Ardestani PM, Yi B, Park HH, Lam RK, Shamloo M. 2020. Beta-adrenergic receptor antagonism is proinflammatory and exacerbates neuroinflammation in a mouse model of Alzheimer’s Disease. Neurobiol Dis 146:105089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrelly LA, Thompson RE, Zhao S, Lepack AE, Lyu Y, Bhanu NV, Zhang B, Loh Y-HE, Ramakrishnan A, Vadodaria KC, Heard KJ, Erikson G, Nakadai T, Bastle RM, Lukasak BJ, Zebroski H, Alenina N, Bader M, Berton O, Roeder RG, Molina H, Gage FH, Shen L, Garcia BA, Li H, Muir TW, Maze I. 2019. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567:535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein DL, Heneka MT, Gavrilyuk V, Russo C, Weinberg G, Galea E. 2002. Noradrenergic regulation of inflammatory gene expression in brain. Neurochemistry International 41:357–365. [DOI] [PubMed] [Google Scholar]
- Filesi I, Gullotta F, Lattanzi G, D’Apice MR, Capanni C, Nardone AM, Columbaro M, Scarano G, Mattioli E, Sabatelli P, Maraldi NM, Biocca S, Novelli G. 2005. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics 23:150–158. [DOI] [PubMed] [Google Scholar]
- Gasser PJ, Hurley MM, Chan J, Pickel VM. 2017. Organic cation transporter 3 (OCT3) is localized to intracellular and surface membranes in select glial and neuronal cells within the basolateral amygdaloid complex of both rats and mice. Brain Struct Funct 222:1913–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilyuk V, Russo CD, Heneka MT, Pelligrino D, Weinberg G, Feinstein DL. 2002. Norepinephrine Increases IκBα Expression in Astrocytes. J Biol Chem 277:29662–29668. [DOI] [PubMed] [Google Scholar]
- Gorboulev V, Shatskaya N, Volk C, Koepsell H. 2005. Subtype-Specific Affinity for Corticosterone of Rat Organic Cation Transporters rOCT1 and rOCT2 Depends on Three Amino Acids within the Substrate Binding Region. Molecular Pharmacology 67:1612–1619. [DOI] [PubMed] [Google Scholar]
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. 2005. The nuclear lamina comes of age. Nat Rev Mol Cell Bio 6:21–31. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Köster S, Kiefer N, Breidert T, Engelhardt M, Spitzenberger F, Obermüller N, Schömig E. 1998a. Transport of Monoamine Transmitters by the Organic Cation Transporter Type 2, OCT2. J Biol Chem 273:30915–30920. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Schechinger B, Rappold GA, Schömig E. 1998b. Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nature neuroscience 1:349–51. [DOI] [PubMed] [Google Scholar]
- Harrison JJ, Suter P, Suter S, Jungmann RA. 1980. Isoproterenol-induced selective phosphorylative modification in vivo of rat C6 glioma cell histones. Biochem Bioph Res Co 96:1253–1260. [DOI] [PubMed] [Google Scholar]
- He X, Zhang L, Chen Y, Remke M, Shih D, Lu F, Wang H, Deng Y, Yu Y, Xia Y, Wu X, Ramaswamy V, Hu T, Wang F, Zhou W, Burns DK, Kim SH, Kool M, Pfister SM, Weinstein LS, Pomeroy SL, Gilbertson RJ, Rubin JB, Hou Y, Wechsler-Reya R, Taylor MD, Lu QR. 2014. The G protein α subunit Gαs is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat Med 20:1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Yi H, Heilman CJ, Edwards RH, Levey AI. 1997. Subcellular localization and molecular topology of the dopamine transporter in the striatum and substantia nigra. J Comp Neurology 388:211–227. [PubMed] [Google Scholar]
- Hertz L, Lovatt D, Goldman SA, Nedergaard M. 2010. Adrenoceptors in brain: Cellular gene expression and effects on astrocytic metabolism and [Ca2+]i. Neurochem Int 57:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M, Zastrow M von. 2017. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol 13:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamur MC, Oliver C. 2009. Immunocytochemical Methods and Protocols. Methods Mol Biology 588:63–66. [DOI] [PubMed] [Google Scholar]
- Jong YI, Harmon SK, O’Malley KL. 2018. GPCR signalling from within the cell. Brit J Pharmacol 175:4026–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker V, Becker A, Hühne R, Zembatov M, Ravati A, Culmsee C, Krieglstein J. 2002. Stimulation of β-adrenoceptors activates astrocytes and provides neuroprotection. Eur J Pharmacol 446:25–36. [DOI] [PubMed] [Google Scholar]
- Juric DM, Loncar D, Carman-Krzan M. 2008. Noradrenergic stimulation of BDNF synthesis in astrocytes: mediation via alpha1- and beta1/beta2-adrenergic receptors. Neurochemistry international 52:297–306. [DOI] [PubMed] [Google Scholar]
- Keryer G, Skålhegg BS, Landmark BF, Hansson V, Jahnsen T, Taskén K. 1999. Differential Localization of Protein Kinase A Type II Isozymes in the Golgi–Centrosomal Area. Exp Cell Res 249:131–146. [DOI] [PubMed] [Google Scholar]
- Keyser JD, Laureys G, Demol F, Wilczak N, Mostert J, Clinckers R. 2010. Astrocytes as potential targets to suppress inflammatory demyelinating lesions in multiple sclerosis. Neurochem Int 57:446–450. [DOI] [PubMed] [Google Scholar]
- Koppel I, Jaanson K, Klasche A, Tuvikene J, Tiirik T, Pärn A, Timmusk T. 2018. Dopamine cross-reacts with adrenoreceptors in cortical astrocytes to induce BDNF expression, CREB signaling and morphological transformation. Glia 66:206–216. [DOI] [PubMed] [Google Scholar]
- Laureys G, Gerlo S, Spooren A, Demol F, Keyser JD, Aerts JL. 2014. β2-adrenergic agonists modulate TNF-α induced astrocytic inflammatory gene expression and brain inflammatory cell populations. J Neuroinflamm 11:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Jungmann RA. 1981. Isoproterenol-induced selective phosphorylation invivo of the 214,000 dalton subunit of rat C6 glioma cell RNA polymerase II. Biochem Bioph Res Co 102:538–544. [DOI] [PubMed] [Google Scholar]
- Li L-B, Chen N, Ramamoorthy S, Chi L, Cui X-N, Wang LC, Reith MEA. 2004. The Role of N-Glycosylation in Function and Surface Trafficking of the Human Dopamine Transporter. J Biol Chem 279:21012–21020. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Leza JC, Polak P, Kalinin S, Feinstein DL. 2009. Astrocyte-derived MCP-1 mediates neuroprotective effects of noradrenaline. The Journal of neuroscience : the official journal of the Society for Neuroscience 29:263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies HJ, Han Q, Shields A, Wright J, Moore JL, Winder DG, Galli A, Blakely RD. 2009. Subcellular localization of the antidepressant-sensitive norepinephrine transporter. Bmc Neurosci 10:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Zhang Y, Roth RH, Zhang J, Mo A, Tenner B, Huganir RL, Zhang J. 2018. Single-fluorophore biosensors for sensitive and multiplexed detection of signalling activities. Nat Cell Biol 20:1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikian HE, Ramamoorthy S, Tate CG, Blakely RD. 1996. Inability to N-glycosylate the human norepinephrine transporter reduces protein stability, surface trafficking, and transport activity but not ligand recognition. Mol Pharmacol 50:266–76. [PubMed] [Google Scholar]
- Milner TA, Shah P, Pierce JP. 2000. beta-adrenergic receptors primarily are located on the dendrites of granule cells and interneurons but also are found on astrocytes and a few presynaptic profiles in the rat dentate gyrus. Synapse (New York, NY) 36:178–93. [DOI] [PubMed] [Google Scholar]
- Northam WJ, Bedoy CA, Mobley PL. 1989. Pharmacological identification of the α-adrenergic receptor type which inhibits the β-adrenergic activated adenylate cyclase system in cultured astrocytes. Glia 2:129–133. [DOI] [PubMed] [Google Scholar]
- Park J, Takmakov P, Wightman RM. 2011. In vivo comparison of norepinephrine and dopamine release in rat brain by simultaneous measurements with fast-scan cyclic voltammetry. J Neurochem 119:932–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegri G, Rossier C, Magistretti PJ, Martin J-L. 1996. Cloning, localization and induction of mouse brain glycogen synthase. Mol Brain Res 38:191–199. [DOI] [PubMed] [Google Scholar]
- Robishaw JD, Smigel MD, Gilman AG. 1986. Molecular basis for two forms of the G protein that stimulates adenylate cyclase. J Biological Chem 261:9587–90. [PubMed] [Google Scholar]
- Sample V, DiPilato LM, Yang JH, Ni Q, Saucerman JJ, Zhang J. 2012. Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nat Chem Biol 8:375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Toni N, Volterra A. 2019. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci 22:154–166. [DOI] [PubMed] [Google Scholar]
- Sorg O, Magistretti PJ. 1991. Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res 563:227–233. [DOI] [PubMed] [Google Scholar]
- Tang Y, Hu LA, Miller WE, Ringstad N, Hall RA, Pitcher JA, DeCamilli P, Lefkowitz RJ. 1999. Identification of the endophilins (SH3p4/p8/p13) as novel binding partners for the β1-adrenergic receptor. Proc National Acad Sci 96:12559–12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Quesada O, Mayrhofer JE, Stefan E. 2017. The many faces of compartmentalized PKA signalosomes. Cell Signal 37:1–11. [DOI] [PubMed] [Google Scholar]
- Uliasz TF, Hamby ME, Jackman NA, Hewett JA, Hewett SJ. 2011. Astrocytes, Methods and Protocols. Methods Mol Biology 814:61–79. [DOI] [PubMed] [Google Scholar]
- Vaniotis G, Duca D, Trieu P, Rohlicek CV, Hébert TE, Allen BG. 2011. Nuclear β-adrenergic receptors modulate gene expression in adult rat heart. Cellular Signalling 23:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M. 2018. Physiology of Astroglia. Physiol Rev 98:239–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shi Q, Li M, Zhao M, Reddy GR, Teoh J, Xu B, Zhu C, Ireton KE, Srinivasan S, Chen S-L, Gasser PJ, Bossuyt J, Hell JW, Bers DM, Xiang YK. 2021. Intracellular β1-Adrenergic Receptors and Organic Cation Transporter 3 Mediate Phospholamban Phosphorylation to Enhance Cardiac Contractility. Circ Res 128:246–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CD, Chen Q, Baye NL, Huang Y, Healy CL, Kasinathan S, O’Connell TD. 2008. Nuclear α1-Adrenergic Receptors Signal Activated ERK Localization to Caveolae in Adult Cardiac Myocytes. Circ Res 103:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Dahl EF, Wright CD, Cypher AL, Healy CL, O’Connell TD. 2014. Nuclear localization of a1A-adrenergic receptors is required for signaling in cardiac myocytes: an “inside-out” a1-AR signaling pathway. Journal of the American Heart Association 3:e000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. 2014. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J Neurosci 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data supporting the findings of this study are available from the corresponding author upon request.