

Abstract
The clinical course of chronic lymphocytic leukemia (CLL) is highly variable. Immunoglobulin heavy chain variable region (IgHV) mutation status is among the most important prognostic factors, with unmutated IgHV associated with inferior outcomes. CLL presumably arises from mature B cells. However, we hypothesized that IgHV unmutated CLL could arise early in B cell differentiation. We prospectively studied 29 patients with mutated and 88 with unmutated IgHV CLL for the presence of CD34+CD19+ cells harboring CLL chromosomal abnormalities. CD34+CD19+ cells were never detected in mutated CLL. In contrast, a small but distinct population of CD34+CD19+ cells harboring the CLL chromosomal abnormality was present in 86/88 patients with unmutated IgHV across all cytogenetic subtypes. Moreover, the CD34+CD19+ cells generated a 3.8 ± 0.7 fold CLL cell expansion over 3–4 weeks in cultures containing IL-3 and IL-2. Unmutated IgHV CLL appears to arise in CD34+ B cells, which perhaps contributes to its poorer prognosis.
Keywords: Chronic lymphocytic leukemia (CLL), stem cells, CD34, CD19
Introduction
CLL is caused by the monoclonal expansion of mature-appearing B cells that progressively accumulate in the blood, lymph nodes, and bone marrow [1,2]. CLL cells not only express the normal B cell markers CD19 and CD20, but also CD5 and CD23. Cytogenetics and immunoglobulin heavy chain variable region (IgHV) mutation status are two of the most important biological prognostic factors for risk assessment and choice of therapy [2]. Interphase fluorescence in situ hybridization (FISH) performed on CLL cells identifies cytogenetic abnormalities in more than 80% of all cases [3]. Patients with deletions of 17p or 11q generally display the worst prognoses, and del 13q the best.
It remains unclear why the 40–50% of CLL patients with unmutated IgHV exhibit significantly more aggressive disease leading to poorer outcomes [4,5]. In normal B cell development, IgHV mutation status represents B cells at different stages of B-cell differentiation: mutated IgHV is characteristic of post-germinal center B cells while unmutated IgHV characterize B cells that have not yet entered lymph node germinal centers [4,5]. Nevertheless, the cellular origins of CLL remain somewhat a matter of debate [6,7]. Most data suggest that cytogenetics and IgHV mutational status are independent and complementary risk factors [8-10]. Accordingly, data from our group [11,12] and others [13,14] suggest that IgHV mutational status influences the prognosis within cytogenetic groups; thus, mutated del 17p and del 11q CLL patients have better prognoses than the more common unmutated patients with these deletions, and unmutated del 13q CLL patients do worse than the typical mutated del 13q patients.
Although classic CLL cytogenetic mutations have never been identified in CD34+ cells, CLL patients have been shown to harbor mutations that could predispose to cancer in nonmalignant cells exhibiting an immature (CD34+CD19−) hematopoietic phenotype [15-18]. The presence of such mutations suggest that CLL patients may have a predisposition toward malignant transformation anywhere within the hierarchy of lympho-hematopoietic development. As exemplified by acute and chronic myeloid leukemias, cancer-initiating events often occur in immature CD34+ progenitors. These so-called leukemia stem cells continue to at least partially differentiate into mature progeny that form the preponderance of, as well as characterize, the leukemia [19]. Thus, we hypothesized that unmutated IgHV CLL could arise in rare immature (CD34+CD19+) B cell progenitors that give rise to the disease-defining mature CD5+ B lymphocytes.
Materials and methods
Human samples
Blood was obtained from 88 patients with unmutated IgHV and 29 with mutated CLL seen at the John Hopkins Sidney Kimmel Comprehensive Cancer Center. Patients granted informed consent as approved by the Johns Hopkins Medical Institutes Institutional Review Board. All patients had active CLL, although treatment status varied from untreated to having received multiple lines of therapy. CD34+ cells were initially enriched by density centrifugation (density <1.078; Ficoll-Paque; Pharmacia, Piscataway, NJ, USA) using the CD34 progenitor isolation kit and the VarioMACS Separator (Miltenyi Biotec, Auburn, CA, USA).
Flow cytometric analyses
The following monoclonal antibodies were used: mouse anti–human CD34-fluourescein isothyocyanate, CD19-allophycocyanin, CD5–brilliant violet 421, and CD38 APC-cyanine 7 (all antibodies from BD PharMingen, San Diego, CA). For some experiments, CD34-brilliant violet 421 was used, and the results were the same. After the addition of 2 μg/mL propidium iodide (BD PharMingen) to discriminate dead cells, cells were analyzed and/or sorted with a FACSAria (BD Biosciences, San Jose, CA) or a MoFlo Legacy (Beckman Coulter, Indianapolis, IN). CD34+CD19+, CD34−CD19+, and CD34+CD19− fractions were sorted onto slides for FISH analysis and into tubes for purity checks. Following sorting, the fractions were found to be ≥96% pure.
Cultures
When available, 2500 – 40,000 additional CD34+CD19+ cells were sorted into tubes for cell culture. The culture media consisted of RPMI (Gibco, Gaithersburg, MD) + 20% FBS (Sigma-Aldrich, St, Louis, MO), 2% L-glutamine (Sigma-Aldrich), 1% pen-strep (Sigma-Aldrich) with 10ug/ml IL-2 and IL-3 (both from GeminiBio, Riverside, DA) added every two days. Cells were cultured for 3-4 weeks and counted at the end of the experiment. Live cells were analyzed using the same makers as before and dropped onto FISH slides for analysis of appropriate cytogenetic markers.
FISH and IgHV mutation analyses
As part of their routine clinical care, all patients had IgHV analyses at a single CLIA-certified reference laboratory (Quest), and all but 3 had FISH at the Johns Hopkins CLIA-certified Clinical Cytogenetics Laboratory. An IgHV nonhomologous sequence of <2% was defined as unmutated and an IgHV nonhomologous sequence of ≥2% was defined as mutated. FISH on the isolated and cultured cells was performed and analyzed by the Johns Hopkins Kimmel Cancer Center Cytogenetics Core using probes specific for the patient’s known cytogenetic abnormality according to the manufacturer’s guidelines (Abbott Molecular): 11 cen (D11Z1)/11q22.3 (ATM), 12 cen (D12Z3)/12q15 (MDM2), 13q14 (D13S319)/13q34(LAMP1), and 17 cen (TP53)/17p13.1 (D17Z1).
Xenograft studies
Isolated CD34+CD19+ cells demonstrating the CLL-specific chromosomal abnormality by FISH were injected intravenously into 10-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ male mice (The Jackson Laboratory, stock no. 013062) 48 h after intraperitoneal injection of Clophosome-A clodronate liposomes (100 μl/mouse) as previously described [20]. Peripheral blood was collected from the mice at days about 24, 55, and 78 days after the injection.
Results
Patient characteristics
We studied 117 patients with CLL: 88 with unmutated IgVH and 29 with mutated. The unmutated patients had the following cytogenetic abnormalities on FISH: del 11q (including 14 with additional cytogenetic abnormalities, often del 13q) in 24, trisomy 12 in 23, del 17p (including 11 with additional abnormalities, often del 13q) in 22, del 13q as sole abnormality in 15, 1 without abnormalities, and 3 unknown. The treatment status was available in 66 of these 88 patients: 33 were treatment naive, 19 were on active treatment, and 14 had relapsed after therapy. We also studied 29 CLL patients with mutated IgVH. Del 13q was the sole abnormality in 13, six patients had del 17p (5 as a sole abnormality), four had trisomy 12, two del 11q, and four no detectable cytogenetic abnormalities. All the mutated patients were treatment naïve.
Most unmutated, but not mutated, IgHV CLL patients harbored a distinct CD34+CD19+ population
CD34+ cells represent only about 0.2% of peripheral blood cells in normal [21] and 0–0.2% in patients with active CLL, depending on degree of lymphocytosis [22]. Thus, we first enriched for circulating CD34+ cells using an immuno-magnetic column (Miltenyi), which increased the CD34+ cell frequency about 30–100 fold (Figures 1 and 2). None of the 29 patients with mutated CLL harbored a circulating CD34+CD19+ population (Figure 1). The small population of CD34+CD19− cells apparently were normal hematopoietic progenitors, since they never harbored CLL-specific cytogenetic abnormalities.
Figure 1.
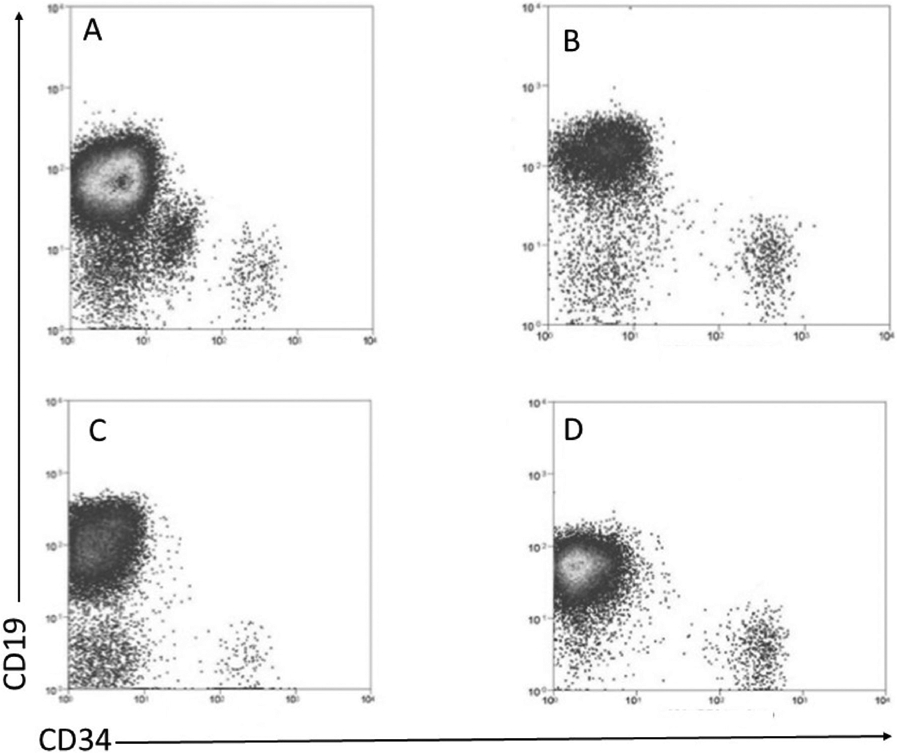
Flow cytometric analysis of peripheral blood cells from 4 representative CLL patients with mutated IgHV for CD19 and CD34, after CD34 selection: (A) del 13q, (B) del 11q, (C) del 17p, and (D) trisomy 12. The vertical and horizontal lines represent to CD19 and CD34 gates, respectively.
Figure 2.
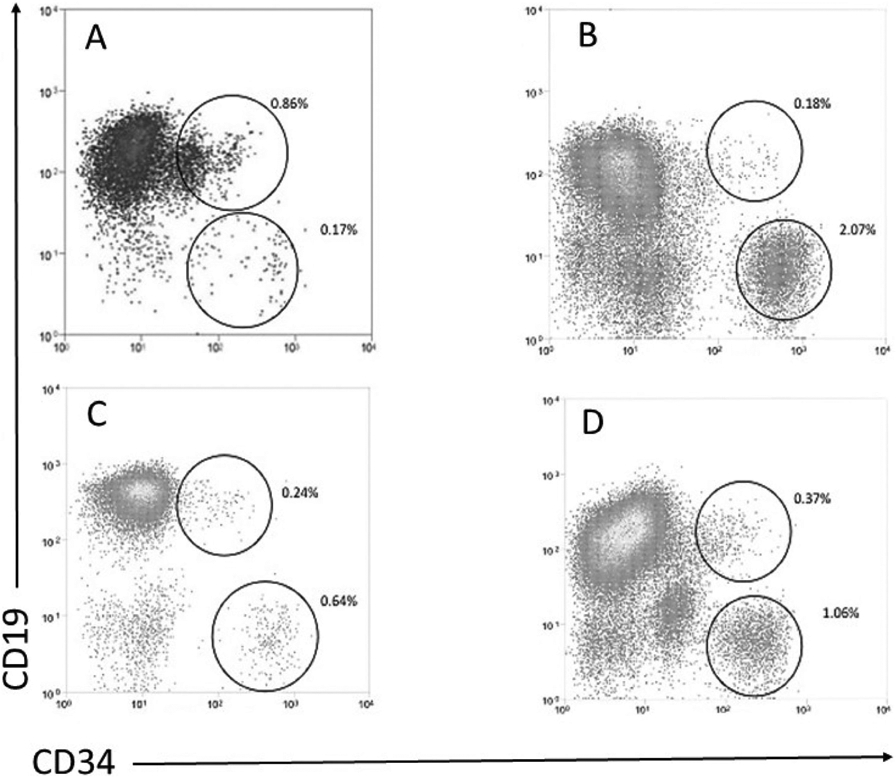
Flow cytometric analysis peripheral blood cells from 4 representative CLL patients with unmutated IgHV, after CD34 selection: (A) del 13q, (B) del 11q, (C) del 17p, and (D) trisomy 12. The circles represent the CD34+CD19+ and CD34+CD19− populations and their % after the CD34 selection. The vertical and horizontal lines represent to CD19 and CD34 gates, respectively.
In contrast, there was a small but easily detectable population of circulating CD34+CD19+ cells (Figure 2) that also expressed CD5 (Figure 3), in 86/88 unmutated patients; the population was not found in two patients with trisomy 12, one untreated and one undergoing treatment. Although only accounting for 0.28 ± 0.07% of the circulating CD19+ B cells in the unmutated CLL patients, the CD34+CD19+ fraction represented 52 ± 8% of the circulating CD34 cells compared to 10% in normal [21]. When enough CD34+CD19+ cells from unmutated CLL patients could be isolated for FISH analysis, the CLL-specific chromosomal abnormality was detected in 99 ± 1% of the cells, including those with del 13q. As in the mutated CLL patients, the isolated CD34+CD19− populations did not harbor CLL-specific abnormalities.
Figure 3.
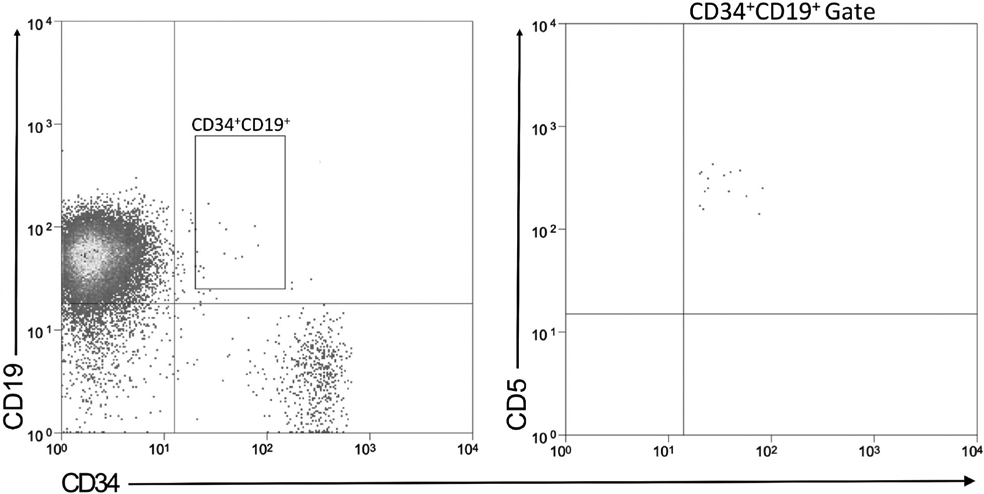
CD5 expression on CD34+CD19+ CLL cells. Peripheral blood cells from a representative patient with unmutated CLL were analyzed for (A) CD34 and CD19 expression, and (B) the CD34+CD19+ cells for CD5 expression.
CD34+CD19+ CLL cells extensively expand in vitro
CLL initiating (i.e. stem) cells should have the capacity to proliferate and generate CD19+ CLL cells. To date, both in vitro and in vivo models appear to primarily support just survival, or at best limited expansion, of CLL cells [23-27], but these studies assessed bulk CLL populations not specifically CD34+CD19+ cells. CLL cells express a variety of cytokine receptors, including IL-2 and IL-3, although the function of these receptors has been unclear [28]. Moreover, normal mouse [29,30] and human [31] B cell progenitors survive and differentiate in vitro in response to IL-3, especially in combination with IL-2, although cellular expansion is limited. Thus, we studied the effects of IL-3 plus IL-2 specifically on the proliferation of the isolated CD34−CD19+ CLL cells in vitro. B cell populations were isolated from six IgVH unmutated and four mutated CLL patients and cultured in the two cytokines. In all 10 cases, purity of the isolated fractions were ≥96%. As expected [24-28], the CD34−CD19+ populations from both the unmutated and mutated patients, as well as the CD34+CD19+ cells from the mutated CLL patients failed to expand and died over 7-10 days in culture. However, the CD34+CD19+ from the 6 unmutated CLL patients not only remained viable, but showed a 5.6 (range 4.3 – 7.3) fold increase in cell numbers compared to input cells over 3-4 weeks in culture (Figure 4). Flow cytometry demonstrated the majority of expanded cells to be CD34−CD19+, with 66.7 ± 8% of the cells harboring the CLL-specific chromosomal abnormality by FISH (Figure 4). This translated into a 3.8 (range 2.1 – 6.3) fold increase in CD19+ CLL cells (Figure 4). Similar to other reports [15], the isolated CD34+CD19+ CLL cells from 3 patients (2000 cells per mice from one patient and 13,000 per mice for the other two based on cell recovery) failed to proliferate in immunodeficient mice.
Figure 4.

CD34+CD19+ CLL cells from unmutated CLL patients expand and generate CLL cells in culture. Sorted CD34+CD19+ cells from 6 patients with unmutated CLL and cytogenetic abnormalities detectable by FISH were placed in liquid culture with 10ug/ml IL-2 and IL-3. The purity of the isolated CD34+CD19+ CLL cells was ≥96% for all 6 patients. The total cell expansion is shown in first column, and total FISH + cell expansion based on the % FISH + cells after culture (66.7 ± 8% of the cells harbored the CLL-specific chromosomal abnormality) is shown in the second column.
Discussion
Normal CD34+CD19+ cells, also commonly known as early hematogones, are so infrequent in the blood of normal adults that their presence is used to validate the quality of bone marrow aspirates [32]. Similarly, we were unable to detect a distinct population of CD34+CD19+ cells in patients with mutated CLL In contrast, we found a distinct population of circulating CD34+CD19+ cells harboring CLL-specific cytogenetic abnormalities in 86/88 IgHV unmutated patients. Although the CD34+CD19+ subset represented only a small fraction of the total CLL cells, this population was about 5-fold larger than the circulating CD34+CD19+ compartment in normals. Importantly, these CD34+CD19+ CLL cells were able to generate a nearly 4-fold expansion of characteristic (CD34−) CD19+ CLL cells harboring the CLL-specific chromosomal abnormalities, after 3 weeks in culture with IL-3 and IL-2. In contrast, bulk CLL cells at best minimally expand in either in vitro or in vivo models [23-27], and accordingly, the CD34−CD19+ cells from both the unmutated and mutated CLL patients failed to expand.
IgHV mutational status and cytogenetics are arguably the two most important biomarkers in CLL [8-10], and in combination these two biomarkers alone have been shown to be strongly prognostic [10]. Importantly, they are independent risk factors in most prognostic models [8-10]. Thus, it is perhaps not surprising that the difference between mutated and unmutated CLL regarding the presence of clonal CD34+CD19+ cells was independent of cytogenetics; i.e. patients with del 17p mutated CLL did not harbor these cells, while those with del 13q unmutated CLL did.
A hierarchical structure similar to the normal tissue of origin is maintained in many malignancies, such that most of the tumor cells primarily represent the differentiated progeny of rare primitive progenitors or cancer stem/initiating cells [19]. Leukemias represent some of the best-studied examples of this cancer stem cell concept [19]. Our results for the first time suggest that unmutated CLL arises from primitive CD34+ B lineage cells, following the pattern of most leukemias. Previously, genetic mutations that could predispose to cancer have been identified in nonmalignant CD34+ cells from CLL patients [15-18]. In addition, immunodeficient mice transplanted with normal, non-transformed CD34+CD38− hematopoietic progenitors from CLL patients generated oligoclonal B cell populations that were unrelated to the patients’ CLL [15]. Thus, CD34+ cells from CLL patients may predispose to lympho-hematopoietic cancer development, similar to inherited cancer predispositions predisposing to cancers in multiple tissues. However to our knowledge, CD34+ cells have never been shown to be the actual cell of origin for CLL. The frequency of the CD34+CD19+ cells within the CLL clone is similar to that reported for cancer stem cells in other malignancies [19].
We and others [15] found CD34+CD19+ cells from CLL patients were unable engraft mice [15]. Although growth in immunodeficient mice has been used as the gold standard for leukemia stem cells, it is now recognized that many leukemia stem cells are unable to grow in current immunodeficient mouse models [19,33]. The mouse engraftment assay may reflect more accurately the proliferative potential of the leukemia cells and/or their interactions with the mouse microenvironment, than it does their role in disease maintenance and relapse [33].
Unmutated CLL’s more primitive cell of origin may in part explain its poorer prognosis relative to mutated disease, including the rare, but well-documented clonal transformation of CLL into CD34+ B lineage ALL [34]. Similar to most B cell non-Hodgkin lymphomas, CLL with mutated IgHV appears to arise in cells with a phenotype similar to normal circulating B cells [35]. Small lymphocytic lymphoma is the term generally reserved for CLL that lacks a leukemic phase. However, since other B cell lymphomas (e.g. diffuse large B cell, mantle cell, and marginal zone) maintain the lymphoma mantra even when found circulating in the blood, perhaps small lymphocytic lymphoma may be a more appropriate name for mutated CLL regardless of its tissue distribution.
Funding
This work was supported by NIH [Grants P01 CA225618 and P30 CA006973].
Footnotes
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Burger JA, O’Brien S. Evolution of CLL treatment - from chemoimmunotherapy to targeted and individualized therapy. Nat Rev Clin Oncol. 2018;15(8):510–527. [DOI] [PubMed] [Google Scholar]
- [2].Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760. [DOI] [PubMed] [Google Scholar]
- [3].Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. [DOI] [PubMed] [Google Scholar]
- [4].Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847. [PubMed] [Google Scholar]
- [5].Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated IgVH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854. [PubMed] [Google Scholar]
- [6].Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117(6):1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seifert M, Sellmann L, Bloehdorn J, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209(12):2183–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Parikh SA, Strati P, Tsang M, et al. Should IGHV status and FISH testing be performed in all CLL patients at diagnosis? A systematic review and meta-analysis. Blood. 2016;127(14):1752–1760. [DOI] [PubMed] [Google Scholar]
- [9].The International CLL-IPI Working Group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol. 2016;17:779–790. [DOI] [PubMed] [Google Scholar]
- [10].Delgado J, Doubek M, Baumann T, et al. Chronic lymphocytic leukemia: a prognostic model comprising only two biomarkers (IGHV mutational status and FISH cytogenetics) separates patients with different outcome and simplifies the CLL-IPI. Am J Hematol. 2017;92(4):375–380. [DOI] [PubMed] [Google Scholar]
- [11].Gladstone DE, Swinnen L, Kasamon Y, et al. Importance of immunoglobulin heavy chain variable region mutational status in del(13q) chronic lymphocytic leukemia. Leuk Lymphoma. 2011;52(10):1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gladstone DE, Blackford A, Cho E, et al. The importance of IGHV mutational status in del(11q) and del(17p) chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2012;12(2):132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tam CS, Shanafelt TD, Wierda WG, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo clinic experience. Blood. 2009;114(5):957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Van Dyke DL, Werner L, Rassenti LZ, et al. The dohner fluorescence in situ hybridization prognostic classification of chronic lymphocytic leukaemia (CLL): the CLL research consortium experience. Br J Haematol. 2016;173(1):105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kikushige Y, Ishikawa F, Toshihiro Miyamoto T, et al. Self-Renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246–259. [DOI] [PubMed] [Google Scholar]
- [16].Damm F, Mylonas E, Cosson A, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014;4(9):1088–1101. [DOI] [PubMed] [Google Scholar]
- [17].Di Ianni M, Baldoni S, Del Papa B, et al. NOTCH1 is aberrantly activated in chronic lymphocytic leukemia hematopoietic stem cells. Front Oncol. 2018;8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Quijada-Álamo M, Hernández-Sánchez M, Robledo C, et al. Next-generation sequencing and FISH studies reveal the appearance of gene mutations and chromosomal abnormalities in hematopoietic progenitors in chronic lymphocytic leukemia. J Hematol Oncol. 2017;10(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sharrow AC, Ghiaur G, Jones RJ. Cancer stem cell principles. In: Bast RC, Markman M, Hawk E, Tsimberidou A-M, Kurzrock R, Anderson KC, editors. Targeted therapy in translational cancer research. John Wiley & Sons, Inc.; 2015. pp 39–46. [Google Scholar]
- [20].Zeisberger SM, Odermatt B, Marty C, et al A. H et al. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95(3):272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bender JG, Unverzagt KL, Walker DE, et al. Identification and comparison of CD34-positive cells and their subpopulations from normal peripheral blood and bone marrow using multicolor flow cytometry. Blood. 1991;77(12):2591–2596. [PubMed] [Google Scholar]
- [22].Berger M, Rapatel C, Boiret N, et al. Peripheral progenitor cells (CFU-GM, BFU-E, CD34) are increased in untreated chronic lymphocyte leukemia patients: stage B and C display a particularly high CD34+ cell count. Leuk Lymphoma. 1999;35(5–6):587–591. [DOI] [PubMed] [Google Scholar]
- [23].Herman SEM, Wiestner A. Preclinical modeling of novel therapeutics in chronic lymphocytic leukemia: the tools of the trade. Semin Oncol. 2016;43(2):222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Panayiotidis P, Jones D, Ganeshaguru K, et al. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br J Haematol. 1996;92(1):97–103. [DOI] [PubMed] [Google Scholar]
- [25].Kurtova AV, Balakrishnan K, Chen R, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mongini PK, Gupta R, Boyle E, et al. TLR-9 and IL-15 synergy promotes the in vitro clonal expansion of chronic lymphocytic leukemia B cells. JI. 2015;195(3):901–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dürig J, Ebeling P, Grabellus F, et al. A novel nonobese diabetic/severe combined immunodeficient xenograft model for chronic lymphocytic leukemia reflects important clinical characteristics of the disease. Cancer Res. 2007;67(18):8653–8661. [DOI] [PubMed] [Google Scholar]
- [28].Trentin L, Zambello R, Agostini C, et al. Expression and regulation of tumor necrosis factor, interleukin-2, and hematopoietic growth factor receptors in B-cell chronic lymphocytic leukemia. Blood. 1994;84(12):4249–4256. [PubMed] [Google Scholar]
- [29].Palacios R, Henson G, Steinmetz M, et al. Interleukin-3 supports growth of mouse pre-B-cell clones in vitro. Nature. 1984;309(5964):126–131. [DOI] [PubMed] [Google Scholar]
- [30].Palacios R, Steinmetz M. IL-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell. 1985;41(3):727–734. [DOI] [PubMed] [Google Scholar]
- [31].Tadmori W, Feingersh D, Clark SC, et al. Human recombinant IL-3 stimulates B cell differentiation. J Immunol. 1989;142(6):1950–1955. [PubMed] [Google Scholar]
- [32].Sevilla DW, Colovai AI, Emmons FN, et al. Hematogones: a review and update. Leuk Lymphoma. 2010;51(1):10–19. [DOI] [PubMed] [Google Scholar]
- [33].Karantanos T, Jones RJ. Acute myeloid leukemia stem cell heterogeneity and its clinical relevance. Vol. 1139. In: Birbrair A, editor. Advances in Experimental Medicine and Biology. Springer New York LLC; 2019. p. 153–169. [DOI] [PubMed] [Google Scholar]
- [34].Yun S, Zhang L, Patel MR, et al. Transformation of chronic lymphocytic leukemia into B-cell acute lymphoblastic leukemia. Blood. 2018;131(11):1258–1261. [DOI] [PubMed] [Google Scholar]
- [35].Johnsen HE, Bergkvist KS, Schmitz A, Myeloma Stem Cell Network (MSCNET), et al. Myeloma stem cell network (MSCNET). cell of origin associated classification of B-cell malignancies by gene signatures of the normal B-cell hierarchy. Leuk Lymphoma. 2014;55(6):1251–1260. [DOI] [PubMed] [Google Scholar]