

Abstract
Numerous epithelial–mesenchymal transition (EMT) characteristics have now been demonstrated to participate in tumor development. Indeed, EMT is involved in invasion, acquisition of stem cell properties, and therapy‐associated resistance of cancer cells. Together, these mechanisms offer advantages in adapting to changes in the tumor microenvironment. However, recent findings have shown that EMT‐associated transcription factors (EMT‐TFs) may also be involved in DNA repair. A better understanding of the coordination between the DNA repair pathways and the role played by some EMT‐TFs in the DNA damage response (DDR) should pave the way for new treatments targeting tumor‐specific molecular vulnerabilities, which result in selective destruction of cancer cells. Here we review recent advances, providing novel insights into the role of EMT in the DDR and repair pathways, with a particular focus on the influence of EMT on cellular sensitivity to damage, as well as the implications of these relationships for improving the efficacy of cancer treatments.
Keywords: DNA damage response, DNA repair, epithelial–mesenchymal transition, synthetic lethality, ZEB1
Novel insights into the role of epithelial to mesenchymal transition in the DNA damage response and repair pathways, as well as the implications of these relationships for improving the efficacy of cancer treatments.
1. INTRODUCTION
Epithelial to mesenchymal transition (EMT) is a physiologically reversible process essential for key embryonic steps. In adults, this conversion is activated during physiological processes, such as wound healing, and may be involved in pathological aspects of fibrosis and cancer. EMT is a major driver of cellular plasticity allowing cells to remodel, reshape, and acquire enhanced motility and stemness properties without genetic modification. This transition is orchestrated by multiple signaling pathways, transcription factors, and chromatin‐remodelers. Key EMT‐associated transcription factors (EMT‐TFs) are divided into three main families: the zinc‐finger E‐box binding protein, including ZEB1 and ZEB2, the SNAI family, encompassing SNAIL and SLUG, and the TWIST family, containing TWIST1. Nevertheless, the switch from one transcriptional state to another is sustained by chromatin remodeling, also known as chromatin plasticity. In such events, the ZEB1 promoter status may be crucial as it maintains a bivalent chromatin configuration that enables the cell to respond readily to extracellular signals. 1 A major aspect of chromatin plasticity is its impact on DNA damage signaling and DNA repair. In this review, we will concentrate on double‐strand break (DSB) repair as a major cause of cancer‐related genomic instability.
The proper coordination of DNA synthesis with other aspects of chromatin structure regulation are important for efficient DNA replication and repair. Defects in this coordination can trigger replication stress and further chromosome rearrangements. To maintain genome integrity, cancer cells depend heavily on the modulation of both the chromatin environment and the DNA damage response (DDR) (please refer to Section 2). Then, to ensure DSB repair, cells rely on four mechanisms, two dominant, mainly faithful repair mechanisms, and two error‐prone processes (please refer to Section 3.1). Modulation of the choice of DSB pathway is presented depending on chromatin topology, cell type, cell cycle commitment, transcription (please refer to Section 3.2), and EMT (please refer to Section 3.3). This strategy is hijacked during resistance to cancer treatment, as it was shown that DNA damage signaling and repair pathways contributed significantly to intrinsic or acquired drug resistance, 2 providing key target mechanisms in cancer. Indeed, some tumors harbor defects in one of the DSB repair processes, thereby rendering them dependent on backup pathways to repair broken DNA. This vulnerability can be targeted by synthetic lethality approaches (please refer to Section 4). Here, we discuss recent findings on the coordination of multiple DNA repair pathways and synthetic lethality approaches with a focus on EMT.
2. EPITHELIAL TO MESENCHYMAL TRANSITION AND REGULATION OF DDR/DNA DAMAGE SIGNALING
The DDR is a complex signaling pathway that senses DNA damage and mobilizes the subsequent cascade of DNA repair pathways. 3 The apical sensor kinase for DSBs is Ataxia‐telangiectasia‐mutated (ATM). ATM acts as a very first sensor by interacting with the DSB‐binding complex MRN (MRE11, RAD50, and NBS1). ATM also promotes the recruitment of the poly(ADP‐ribose)polymerase 1 (PARP1) to produce poly(ADP‐ribose) (PAR) polymers and extend DNA damage signaling. 4 ATM instantly autophosphorylates and rapidly triggers a phosphorylation cascade, targeting downstream effectors such as the histone H2A variant H2AX that, upon phosphorylation at Serine 139, forms the γH2AX mark of damaged chromatin that acts as a platform to recruit DNA repair proteins. 5 It has been shown that heterochromatic DSBs require ATM kinase signaling to be repaired and this ATM dependency is correlated with increased chromatin complexity rather than the damage itself. 6
Recent investigations have highlighted the implication of some EMT‐TFs in DDR regulation through their physical interaction with large chromatin protein remodeling complexes, advocating for a broader range of functions for EMT‐TFs compared with those specifically restricted to orchestrating the expression of epithelial/mesenchymal genes. Zhang et al. 7 identified ZEB1 as a target of ATM in response to DNA damage. The phosphorylated ZEB1 was shown to directly interact with the USP7 deubiquitylating enzyme, surprisingly triggering the stabilization of CHK1 to promote homologous recombination (HR)‐dependent DNA repair. Indeed, knocking‐down ZEB1 decreased CHK1 protein abundance, but had no effect on CHK2. This pioneering work established a link between EMT and DDR, and unveiled an association between ZEB1 and radioresistance. Additionally, ZEB1 forms a complex with p300/pCAF to activate the ATM promoter, therefore participating in a positive feedback loop with ATM and increasing the DNA repair capacity in response to radio or chemotherapy. 8 Radiation was found to stabilize the ZEB1 protein, but had no effect on SNAIL, SLUG, or TWIST proteins. 7 , 9 An interplay between miRNAs, ZEB1 and DNA damage signaling pathways was identified, in which phosphorylated ZEB1 following irradiation could repress the transcription of its own negative regulator, miR‐205, but not miR‐200c. 10
Aside from the strong implication of ZEB1, other EMT‐TFs are also involved in the DDR. The E3 ubiquitin ligase RNF8 ubiquitinates TWIST1, which promotes its nuclear translocation and then modulates DDR pathways, leading to increased expression of HR genes. 11 In turn, it has been shown that RNF8 is a key player in DDR by modulating ATM activation and the DNA damage response. 12 In addition, impairing PARP1 induced EMT, in particular by triggering ZEB1 expression. 13
3. EPITHELIAL TO MESENCHYMAL TRANSITION AND DSB REPAIR PATHWAY CHOICE
3.1. Double‐strand break repair pathways
Double‐strand breaks are mainly repaired by canonical nonhomologous end joining (c‐NHEJ) and HR, but single‐strand annealing (SSA) and the more recently characterized Alternative‐End Joining pathways (Alt‐EJ) 14 also contribute (Figure 1).
FIGURE 1.
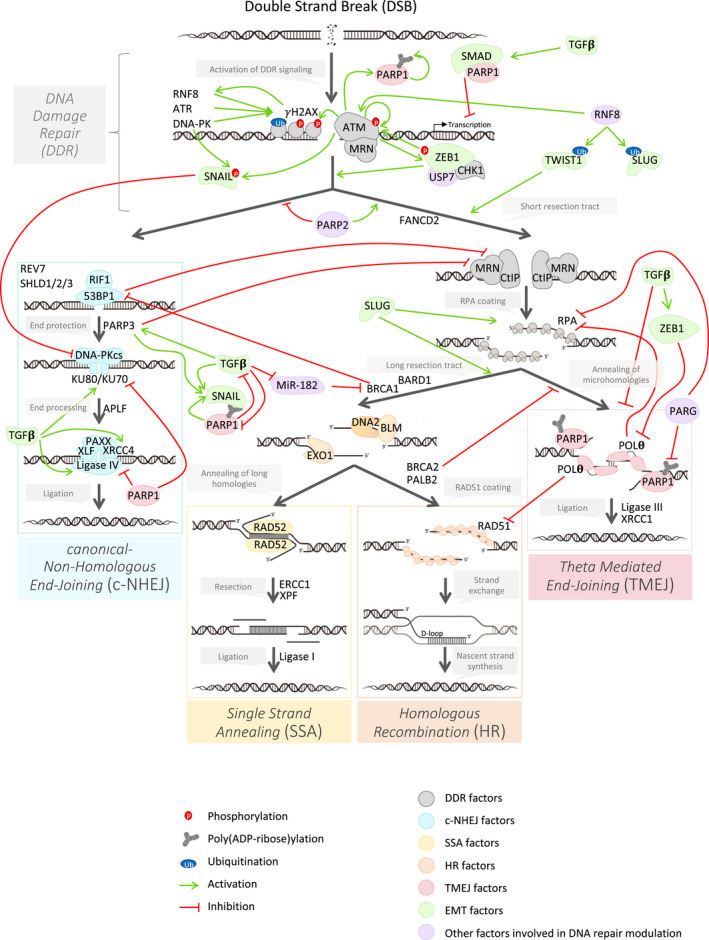
Influence of epithelial to mesenchymal transition (EMT) on the modulation of the DNA repair pathway choice. This schematic representation describes the strong complexity of the modulation of the DNA damage response (DDR) signaling (in gray) as well as the four major DNA double‐strand break repair pathways: c‐NHEJ (in blue), SSA (in yellow), HR (in orange) and TMEJ (in red). This regulation occurs from the DDR signaling, to orientate the repair toward one of these four pathways. Other factors external to the DNA repair pathways (in purple) influence this choice, notably by post‐translational modification (phosphorylation, PARylation, ubiquitination). As shown in this figure, EMT factors (in green) are key modulators of pathways choice
The c‐NHEJ is a rapid and high‐capacity pathway that joins two DNA ends with no complementary base pairing. HR, SSA, or Alt‐EJ enable DSB repair after DNA resection initiated by the MRN complex. 15 In recent years, the notion of Alt‐EJ has emerged. The most extensively described idea is called microhomology‐mediated end joining (MMEJ) or theta‐mediated end joining (TMEJ), an error‐prone repair pathway in which DNA polymerase theta uses microhomologies to perform end joining repair. Several proteins have been described in human models to execute TMEJ, such as DNA polymerase θ (coded by POLQ gene), 16 PARP1, XRCC1, and DNA ligase IIIα (LIG3). 17 , 18 , 19 POLQ depletion increases micronuclei, 20 , 21 suggesting the reliance on TMEJ for repair of certain types of lesions. Nevertheless, the protective effect of POLθ on genomic integrity is associated with an enhanced genomic instability 22 in cancer cells, highlighted recently in the work by Prodhomme and colleagues, 23 even if TMEJ is associated with a protective role in noncancer models. 21
3.2. Modulating the choice of DNA repair pathway
It has long been accepted that the nature of the DSB is the first determinant to influence the choice of the DNA repair pathway. 24 The choice of repair pathway also depends on the environment of the break, chromatin status and nuclear position (heterochromatin, euchromatin, centrosome, telomere, etc.) 25 and the phase of the cell cycle. 26 , 27 , 28
In human cells, a blunt DNA‐end DSB is preferentially repaired by c‐NHEJ. However, DSBs arising from a fork collapse or DNA‐end are not directly manageable by ligation, and initially require a resection step making DNA ends available for HR, SSA, or TMEJ. 29 While the HR mechanism is active in S/G2, TMEJ appears to be used more in the S phase, although theoretically TMEJ is used during the whole cell cycle, as for c‐NHEJ. 27 , 28 Key factors of DNA DSB repair also regulate the choice of DNA repair pathway. The switch from c‐NHEJ to HR and vice versa has been largely studied. For example, 53BP1 promotes c‐NHEJ by blocking CtIP‐dependent DNA resection. 30 Inversely, BRCA1 promotes HR by 53BP1 dephosphorylation and RIF1 release. 31
The continual identification of novel players completes this initial binary model. Indeed, BRCA2, main actor of HR, can stabilize replication protein A (RPA) proteins to inhibit TMEJ activity. 29 , 32 Conversely, POLθ, a key factor in TMEJ, also modulates HR activity. 33 Indeed, POLθ interacts directly with RAD51, limits RAD51–ssDNA nucleofilament assembly, and therefore suppresses the HR pathway in favor of TMEJ. 34 Moreover, the helicase domain of POLθ has the ability to remove the loading of RPA and stimulates annealing of ssDNA, an essential step to switch from the HR to the TMEJ pathway. 33 , 35 In addition, FANCD2, required for fork protection and fork restart in BRCA1/2‐deficient tumors, promotes POLθ recruitment to lesions and promotes TMEJ repair. 36 Finally, in the G1 phase of the cell cycle, loss of 53BP1 or RIF1 enhances the recruitment of BRCA1, CtIP/MRE11‐dependent end resection and RPA, but not of RAD51, therefore promoting TMEJ activity but not that of HR. 37 Taken together, these findings highlight a close link between HR and TMEJ pathways and the loss of HR activity to encourage the cell to rely on the TMEJ pathway.
Finally, the synthesis of PAR polymers, catalyzed by PARPs, is a powerful regulator of several DSB repair pathways and, of note, PARP1 is the major producer of cellular PAR. 38 PARP1, PARP2, and PARP3 activities increase in response to DSBs. 39 , 40 , 41 , 42 PARP1 is a player in the TMEJ pathway 17 and seems to be a platform regulating the balance between HR and TMEJ. For example, PARP1 seems to promote mutagenic TMEJ repair by inhibiting c‐NHEJ repair mediated by the inhibitory action of PARP1‐Ku70 and PARP1‐Lig4 on the BRCT domain of PARP1. 43 , 44 PARP2 plays an important role in the regulation of DSB repair pathway choice. 39 PARP2 limits 53BP1 accumulation onto DNA lesions, facilitating the CtIP‐dependent DNA‐end resection and thereby limiting c‐NHEJ activity. 39 Finally, PARP3 limits DNA‐end resection and promotes c‐NHEJ activity by its direct interaction with PARylated Ku70/Ku80 and induces an imbalance between the specific pathways of BRCA1 and 53BP1. 38 , 41 , 45 Last, PAR glycohydrolase (PARG), a strong regulator of PARP activity due to its ability to degrade PAR from PARylated proteins, limits c‐NHEJ pathway DNAPK‐cs dependent activity. 46 Moreover, the loss of PARG, in particular PARG‐2 in Caenorhabditis elegans, was reported to increase TMEJ activity. 47
3.3. Epithelial to mesenchymal transition and DNA repair
Over the last few years, many studies have shown that EMT, particularly EMT‐TFs, are involved in the choice of DNA DSB repair pathways (Figure 1). EMT is associated with chemoresistance and radioresistance, among others, through the acquisition of stem cell properties. As previously mentioned, ZEB1 promotes DDR, but also the DNA repair choice itself. This function of ZEB1, initiated by phosphorylation and stabilization of ZEB1 by ATM, leads to increased HR pathway activity. 7 Along the same lines, transforming growth factor‐β (TGF‐β) signaling, known to induce EMT and to activate EMT‐TF expression, 48 such as that of ZEB, SNAI and TWIST families, 49 , 50 supports HR activity. 51 Indeed, TGF‐β signaling inhibits miR‐182, which represses both BRCA1, necessary for HR, and FOXO3, required for ATM kinase activity. Consistently, compromised TGF‐β signaling impairs HR proficiency. 52 Finally, more recently, the EMT‐TF ZEB1 was revealed as a direct regulator of DSB repair. Indeed, ZEB1 is a negative regulator of TMEJ, which may explain why some tumors with stem cell properties display low genomic instability. 53 , 54 POLQ expression, mainly associated with genomic instability and TMEJ activity, is lower in ZEB1‐expressing claudin‐low tumors characterized by a subnormal genomic landscape. ZEB1 represses the POLQ promoter, which consequently limits TMEJ activity 23 and therefore most probably favors HR and/or c‐NHEJ. These results were confirmed by recent work from the Barcellos‐Hoff team. Indeed, in this article, the authors show that not only that TGF‐β is able to limit the activity of TMEJ by inhibiting POLQ expression, but also by inhibiting PARP1 and LIG1. 55 TGF‐β also enhances c‐NHEJ activity, as demonstrated by Kim and colleagues, 56 who showed that TGF‐β treatment increased the levels of LIG4, XRCC4 and KU70/KU80. In conclusion, ZEB1 and possibly other EMT‐TFs, seem to inhibit error‐prone repair and encourage faithful DNA repair.
Additionally, repair factors have been shown to both induce or prevent EMT. First, the PARP family plays an important role in the modulation of EMT. Indeed, PARP1 deficiency results in the acquisition of an EMT phenotype during tumorigenesis. 57 Conversely, impairing PARP1 upregulates TGF‐β and SMAD pathways, which induce EMT and increase the levels of ZEB1 and SNAIL. In 2011, Rodriguez et al. revealed a new regulatory mechanism of SNAIL by PARP1, involving its post‐translational stabilization by PARylation. 58 This functional interaction highlights the importance of PARP1 activity in the control of SNAIL activation with major consequences on malignant transformation through EMT. Moreover, Kumar et al. recently showed that PARP1 facilitates EMT in non–small‐cell lung cancer through the induction of ZEB1 and the SMAD pathway. 59 PARP3, another actor of the PARP family, also promotes EMT by TGF‐β induction, cell motility, and chemoresistance in mammary epithelial cells. 60 Finally, the kinase DNA‐PKcs, a key actor in c‐NHEJ, is responsible for SNAIL stabilization by phosphorylation at Ser100. Consequently, the kinase activity of DNA‐PKcs is inhibited by phospho‐SNAIL, resulting in the inhibition of c‐NHEJ. 61 Downstream of ATM activation, SLUG is essential for the activation of RPA32 (subunit of RPA with RPA70 and RPA14), resulting in efficient HR‐mediated DSB repair. 62 In conclusion, a strong interconnection exists between EMT and DNA DSB repair.
4. EPITHELIAL TO MESENCHYMAL TRANSITION AND SYNTHETIC LETHALITY APPROACHES BASED ON DSB REPAIR PATHWAYS TARGETING
As described above, when a specific DDR pathway is inactivated, cancer cells become dependent on other DDR pathways to overcome the deleterious effects of DNA damage. This is a rationale for using a synthetic lethality approach in the context of a genetic deficiency in a DDR pathway and a drug that targets the fallback repair pathway. Theoretically, synthetic lethality‐based drugs should exhibit a high therapeutic index and have been proposed as promising anticancer treatments. The most successful examples are PARP inhibitors (PARPi; Figure 2). PARP inhibition causes synthetic lethality with deficiency in tumor suppressor BRCA1 or BRCA2 genes. 63 , 64 PARPi repress the repair of single‐strand breaks (SSB), which results in DSBs when the cell enters S phase of the cell cycle. Because DSBs in S/G2 phases are repaired by HR, cells with intact HR survive upon PARP inhibition. In contrast, BRCA1/2‐deficient cancer cells cannot repair DSBs by HR and progress to cell death (Figure 2). Following approval of olaparib, several PARPi, such as niraparib, rucaparib, and talazoparib, have also been approved by the Food and Drug Administration in the USA. The United States indications for the first‐in‐class PARPi, olaparib, include breast, ovarian, pancreatic and prostate cancers with HR deficiency (referred to as HRD) and platinum‐sensitive ovarian cancer. As companion diagnostics, Myriad HRD assay and Foundation Medicine loss of heterozygosity (LOH) assay predict HRD. PARPi also have the so‐named PARP trapping activities, which stabilize the SSB‐PARP complex and cause catastrophic DNA damage. Among PARPi, talazoparib exhibits the most potent PARP trapping activity, whereas that of veliparib, still under clinical tests, is at the lowest level.
FIGURE 2.
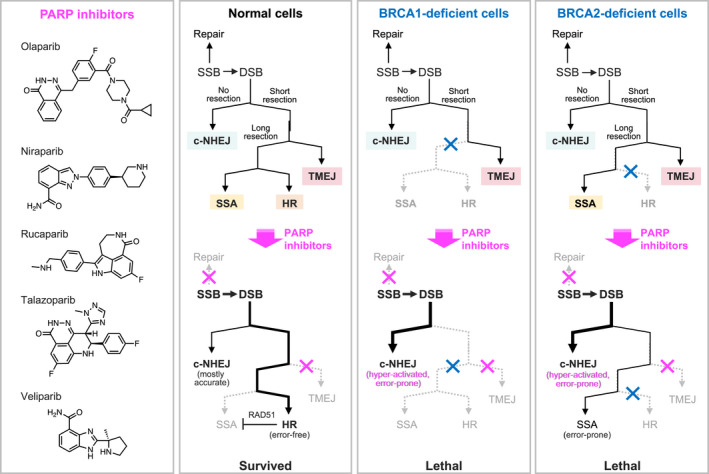
Poly(ADP‐ribose)polymerase (PARP) inhibitor‐driven synthetic lethality in the contexts of DNA damage repair deficiencies. PARP inhibitors repress single‐strand break (SSB) repair and base excision repair, as well as TMEJ. While normal cells can repair the resulting double‐strand breaks (DSBs) via the error‐free HR pathway, HR‐deficient cells (e.g., BRCA1/2‐deficient cells) cannot escape from these deleterious effects. In BRCA2‐deficient cells, high dependency on SSA is lethal because this repair system is error prone and causes cell catastrophe. The PARP trapping activity, which is the most marked for talazoparib and the weakest for veliparib, also leads to DNA damage and yields therapeutic efficacies
PARPi resistance emerges in patients within a year of treatment. Mechanisms for resistance to PARPi include restoration of HR by frame‐revertant mutations in BRCA1/2 65 or inactivation of c‐NHEJ factors (53BP1 and shieldin components, such as REV7), 66 , 67 PARP1 loss‐of‐function mutations 68 enhanced the drug efflux by P‐glycoprotein/ABCB1 overexpression 69 and replication fork stabilization by SLFN11 downregulation. 70 The role of EMT in the occurrence of drug resistance has been widely described. 71 PARPi treatments are no exception, and EMT appears as a new PARPi resistance mechanism. 72 It has been recently shown that the combination of niraparib, cisplatin, and downregulation of TWIST1 re‐sensitized ovarian cells to niraparib. 73
Because PARPi induce replicative stress, PARPi‐resistant cancer cells often acquire a high dependency on the ATR/CHK1/WEE1 pathway. This suggests that this pathway may be a promising target to overcome PARPi resistance.
Currently, an ATR inhibitor, VE‐821, and a CHK1 inhibitor, prexasertib, have shown these types of results in ovarian cancer cells. 74 , 75 VE‐821 disrupts both RAD51 loading to DSBs and fork protection in PARPi‐resistant BRCA1‐deficient cancer cells. ATR inhibitors are also effective in ATM‐mutated gastric cancer and chronic lymphocytic leukemia, as ATR and ATM pathways bear a synthetic lethal relationship. 76 , 77 Furthermore, the combination with gemcitabine and an ATR inhibitor, berzosertib, extended the progression‐free survival of platinum‐resistant high‐grade serous ovarian cancer patients compared with gemcitabine treatment alone in a phase 2 clinical trial. 78 Mechanistically, gemcitabine causes replicative stress, whereas inhibition of the ATR/CHK1/WEE1 pathway prevents the replicative stress response, leading to cytotoxicity.
Recently, some studies have investigated the role of EMT in response to ATR inhibitors, including VE‐821. Rapidly after treatment with VE‐821, treated pancreatic cancer cell lines have increased migratory ability, decreased protein level of E‐cadherin, and increased protein levels of vimentin and ZEB1, suggesting an increase in EMT upon loss of ATR. 79 , 80 , 81 The combined loss of ATR and ZEB1 then significantly reduces cell survival. 80 This latest study also shows that ZEB1 inhibition promotes CHK1 phosphorylation, suggesting that the dual inhibition of ZEB1 and CHK1 may be an essential treatment strategy in the treatment of ZEB1‐expressing tumors. Another study has already shown, previously, the close link between CHK1 and ZEB1. In 2014, Peijing Zhang et al. 7 conversely showed that the loss of ZEB1 in mouse embryonic fibroblasts (MEFs) led to a decrease in protein levels of CHK1, but that the simultaneous loss of ZEB1 and CHK1 led to sensitization of cells to radiotherapy.
HR deficiency provides additional opportunities for synthetic lethality‐based therapies. As described above, the DSB repair mechanisms involving end resection include HR, SSA, and Alt‐EJ or TMEJ. Accordingly, HRD cancer cells often gain a hyperdependence on SSA and TMEJ pathways. As TMEJ requires POLθ, POLθ inhibition induces synthetic lethality in HRD epithelial ovarian cancers. 34 From a diagnostic perspective, POLθ upregulation is one of the hallmarks of HRD and TMEJ dependency, which could be used as a predictive biomarker for response to PARP and POLθ inhibitors. EMT can lead to inefficiency of POLθ inhibitors as we have shown that ZEB1 expression leads to TMEJ repression. 23 RAD52 binding to ssDNA is required for the SSA repair mechanism. A small‐compound, AICAR, which disrupts the RAD52/ssDNA interaction and SSA, preferentially inhibits the growth of BRCA1/2‐deficient cancer cells. 82 Therefore, protein–protein interactions (PPIs) and protein–DNA interactions (PDIs) may also constitute promising therapeutic targets in DSB repair pathways. However, unlike the design of enzyme inhibitors, the development of PPI/PDI modifiers remains challenging due to the difficulty in blocking specific macromolecular interactions using small compounds. Recent technologies, such as proteolysis‐targeting chimera (PROTAC), may offer the opportunity to target these nonenzymatic molecules.
Accumulating evidence indicates that DSB repair pathways are affected by oncogenic alterations of epigenetic regulators. For example, gain‐of‐function mutations of isocitrate dehydrogenase‐1 and ‐2 (IDH1/2) foster enzymes to produce an oncometabolite, 2‐hydroxyglutarate (2‐HG), instead of α‐ketoglutarate (α‐KG). This metabolic alteration represses the α‐KG‐dependent histone lysine demethylases KDM4A and KDM4B, which are required for DSB repair. Therefore, IDH1/2 mutations cause HRD, providing a therapeutic opportunity for PARPi‐mediated synthetic lethality. 83 IDH1/2 mutants also induce EMT with high levels of ZEB1 and knockdown of IDH1 mutant form is sufficient to reverse many characteristics of EMT. 84 , 85 Targeted inhibitors of IDH1 are currently under trials for glioma cancer. 86
Overexpression of CARM1, an arginine methyltransferase, in ovarian cancer induces the epigenetic silencing of the shieldin component MAD2L2/REV7 in a histone lysine methyltransferase EZH2‐dependent manner. In this cellular context, EZH2 inhibition derepresses MAD2L2/REV7 expression and switches the DSB repair pathway from HR to c‐NHEJ. EZH2 and PARPi, GSK126, and olaparib, and therefore respectively exhibit a synergetic antitumor effect in orthotopic and patient‐derived xenograft models. 87
However, EZH2 knockdown by GSK126 induced EMT‐like changes in ovarian cancer cells. The EMT‐TF ZEB2 was upregulated in cells treated with this EZH2 inhibitor. Furthermore, TGF‐β enhanced the expression of ZEB2 in EZH2 siRNA‐ or GSK126‐treated cells. 88
These observations indicate that transcriptional reprograming EMT may affect cell vulnerability to DDR‐directed drugs and be worth monitoring to predict and even enhance the therapeutic impact of DDR‐directed drugs.
5. CONCLUSION
To conclude, in addition to the direct competition for access to a given DNA damage locus, many complex mechanisms influence the use of one repair pathway over another, such as the nature of the DNA lesion (single‐strand or DSB, intercrosslink, mismatch, G‐quadruplex, etc.), the location of the lesion (heterochromatin, euchromatin, centrosome, telomere, etc.), the cell cycle, the environment (hypoxia, the immune system, etc.). 24 More recently, EMT has also been described as a potent regulator of DDR signaling and DSB repair pathways, as presented in this review.
Epithelial to mesenchymal transition‐TFs, such as ZEB1, may influence DNA repair pathway choice directly, by regulating POLQ expression for instance, but also indirectly in response to environmental stress during tumorigenesis. For example, even if the role of hypoxia as a modulator of DNA repair pathways via ZEB1, or via EMT, has yet to be demonstrated, hypoxia‐inducible factor 1 alpha (HIF‐1α), the main hypoxia‐induced factor, is known to induce ZEB1 and EMT. 89 We propose that part of the role of hypoxia in modulating DNA repair choice may be regulated by ZEB1.
Overall, we proposed that the role of EMT is to limit the establishment of high genomic instability 23 , 53 by the non‐use of a mutagenic repair pathway. Therefore, some DNA damage cannot be dealt with and a loss of genomic integrity is observed, as evidenced by the high level of micronuclei in EMT‐TF‐expressing cells. 23 These conclusions can be extended to other EMT‐TFs and EMT or DNA damage other than DSBs. ZEB2 increases the nucleotide excision repair (NER) pathway activity. In colorectal cancer, ZEB2 enhances resistance of cancer cells to oxaliplatin‐induced DNA damage, by increasing NER capacity by upregulation of the ERCC1 gene. 90 Again, in this case, ZEB2‐mediated EMT is instrumental in increasing genomic stability and cell survival.
Through all these roles, EMT, and more precisely ZEB1, induces a strong resistance to conventional treatments in cancer patients. Indeed, ZEB1 has frequently been associated with the mechanisms of resistance to radiotherapy or chemotherapy. 7 EMT inhibitors, such as ZEB1 inhibitors, could therefore be promising treatment options for blocking tumor initiation, reducing the occurrence of metastases, but also for modulating DNA repair and therefore sensitizing tumors to radiotherapy or chemotherapy.
CONFLICT OF INTEREST
HS received a research grant from the Nippon Foundation and is Associate Editor and Secretary for Editor‐in‐Chief of Cancer Science. The other authors have no competing interest to disclose.
ACKNOWLEDGMENTS
We thank B. Manship and A. Senaratne for useful comments on the manuscript.
Moyret‐Lalle C, Prodhomme MK, Burlet D, et al. Role of EMT in the DNA damage response, double‐strand break repair pathway choice and its implications in cancer treatment. Cancer Sci. 2022;113:2214–2223. doi: 10.1111/cas.15389
Caroline Moyret‐Lalle and Mélanie K Prodhomme contributed equally.
Funding information
This work was supported by the Ligue contre le Cancer (EL2016.LNCC/AIP and EL2019.LNCC/AIP), ANR‐17‐CONV‐0002, and ANR‐10‐LABX‐0061. This work was also supported by JSPS KAKENHI (20K21555 and 20H04789), AMED P‐CREATE (21cm0106184h0001) and funding from Nippon Foundation to HS
Contributor Information
Hiroyuki Seimiya, Email: hseimiya@jfcr.or.jp.
Agnès Tissier, Email: agnes.tissier@inserm.fr.
REFERENCES
- 1. Chaffer CL, Marjanovic ND, Lee T, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154(1):61‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmed SU, Carruthers R, Gilmour L, Yildirim S, Watts C, Chalmers AJ. Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem‐like cells. Cancer Res. 2015;75(20):4416‐4428. [DOI] [PubMed] [Google Scholar]
- 3. Jackson SP, Bartek J. The DNA‐damage response in human biology and disease. Nature. 2009;461(7267):1071‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ko HL, Ren EC. Functional aspects of PARP1 in DNA repair and transcription. Biomolecules. 2012;2(4):524‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA‐PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64(7):2390‐2396. [DOI] [PubMed] [Google Scholar]
- 6. Berger ND, Stanley FKT, Moore S, Goodarzi AA. ATM‐dependent pathways of chromatin remodelling and oxidative DNA damage responses. Philos Trans R Soc B Biol Sci. 2017;372:20160283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang P, Wei Y, Wang L, et al. ATM‐mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16:864‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X, Zhang Z, Zhang Q, et al. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. 2018;9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin Y, Bai X, Zhou W, He Y, Wu Y, Wang X. Radiation exposure triggers the progression of triple negative breast cancer via stabilizing ZEB1. Biomed Pharmacother. 2018;107:1624‐1630. [DOI] [PubMed] [Google Scholar]
- 10. Zhang P, Wang L, Rodriguez‐Aguayo C, et al. miR‐205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat Commun. 2014;5:5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee H‐J, Li C‐F, Ruan D, et al. The DNA damage transducer RNF8 facilitates cancer chemoresistance and progression through twist activation. Mol Cell. 2016;63(6):1021‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu J, Chen Y, Lu L‐Y, et al. Chfr and RNF8 synergistically regulate ATM activation. Nat Struct Mol Biol. 2011;18:761‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pu H, Horbinski C, Hensley PJ, Matuszak EA, Atkinson T, Kyprianou N. PARP‐1 regulates epithelial‐mesenchymal transition (EMT) in prostate tumorigenesis. Carcinogenesis. 2014;35(11):2592‐2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wyatt DW, Feng W, Conlin MP, et al. Essential roles for polymerase θ‐mediated end joining in the repair of chromosome breaks. Mol Cell. 2016;63:662‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Truong LN, Li Y, Shi LZ, et al. Microhomology‐mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double‐strand breaks in mammalian cells. Proc Natl Acad Sci USA. 2013;110(19):7720‐7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Newman JA, Cooper CDO, Aitkenhead H, Gileadi O. Structure of the helicase domain of DNA polymerase theta reveals a possible role in the microhomology‐mediated end‐joining pathway. Structure. 2015;23(12):2319‐2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Audebert M, Salles B, Calsou P. Involvement of Poly(ADP‐ribose) polymerase‐1 and XRCC1/DNA Ligase III in an alternative route for DNA double‐strand breaks rejoining. J Biol Chem. 2004;279(53):55117‐55126. [DOI] [PubMed] [Google Scholar]
- 18. Muvarak N, Kelley S, Robert C, et al. c‐MYC generates repair errors via increased transcription of alternative‐NHEJ factors, LIG3 and PARP1, in tyrosine kinase‐activated leukemias. Mol Cancer Res. 2015;13(4):699‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. Homology and enzymatic requirements of microhomology‐dependent alternative end joining. Cell Death Dis. 2015;6(3):e1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shima N, Munroe RJ, Schimenti JC. The mouse genomic instability mutation chaos1 Is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol. 2004;24:10381‐10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yousefzadeh MJ, Wyatt DW, Takata K, et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014;10:e1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Schendel R, van Heteren J, Welten R, Tijsterman M. Genomic scars generated by polymerase theta reveal the versatile mechanism of alternative end‐joining. PLoS Genet. 2016;12(10):e1006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prodhomme MK, Pommier RM, Franchet C, et al. EMT transcription factor ZEB1 represses the mutagenic POLθ‐mediated end‐joining pathway in breast cancers. Can Res. 2021;81:1595‐1606. [DOI] [PubMed] [Google Scholar]
- 24. Scully R, Panday A, Elango R, Willis NA. DNA double‐strand break repair‐pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20(11):698‐714. doi: 10.1038/s41580-019-0152-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schep R, Brinkman EK, Leemans C, et al. Impact of chromatin context on Cas9‐induced DNA double‐strand break repair pathway balance. Mol Cell. 2021;81:2216‐2230. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frit P, Barboule N, Yuan Y, Gomez D, Calsou P. Alternative end‐joining pathway(s): Bricolage at DNA breaks. DNA Repair. 2014;17:81‐97. [DOI] [PubMed] [Google Scholar]
- 27. Iliakis G, Mladenov E, Mladenova V. Necessities in the processing of DNA double strand breaks and their effects on genomic instability and cancer. Cancers. 2019;11:1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Her J, Bunting SF. How cells ensure correct repair of DNA double‐strand breaks. J Biol Chem. 2018;293:10502‐10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahrabi S, Sarkar S, Pfister SX, et al. A role for human homologous recombination factors in suppressing microhomology‐mediated end joining. Nucleic Acids Res. 2016;44(12):5743‐5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bunting SF, Callén E, Wong N, et al. 53BP1 inhibits homologous recombination in brca1‐deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Isono M, Niimi A, Oike T, et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017;18(2):520‐532. [DOI] [PubMed] [Google Scholar]
- 32. Han J, Ruan C, Huen MSY, et al. BRCA2 antagonizes classical and alternative nonhomologous end‐joining to prevent gross genomic instability. Nat Commun. 2017;8(1):1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mateos‐Gomez PA, Kent T, Deng SK, et al. The helicase domain of Polθ counteracts RPA to promote alt‐NHEJ. Nat Struct Mol Biol. 2017;24:1116‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ceccaldi R, Liu JC, Amunugama R, et al. Homologous‐recombination‐deficient tumours are dependent on Polθ‐mediated repair. Nature. 2015;518:258‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jenkins T, Northall SJ, Ptchelkine D, et al. The HelQ human DNA repair helicase utilizes a PWI‐like domain for DNA loading through interaction with RPA, triggering DNA unwinding by the HelQ helicase core. NAR Cancer. 2021;3(1):zcaa043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kais Z, Rondinelli B, Holmes A, et al. FANCD2 maintains fork stability in BRCA1/2‐deficient tumors and promotes alternative end‐joining DNA repair. Cell Rep. 2016;15(11):2488‐2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bakr A, Köcher S, Volquardsen J, et al. Impaired 53BP1/RIF1 DSB mediated end‐protection stimulates CtIP‐dependent end resection and switches the repair to PARP1‐dependent end joining in G1. Oncotarget. 2016;7(36):57679‐57693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beck C, Robert I, Reina‐San‐Martin B, Schreiber V, Dantzer F. Poly(ADP‐ribose) polymerases in double‐strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329:18‐25. [DOI] [PubMed] [Google Scholar]
- 39. Fouquin A, Guirouilh‐Barbat J, Lopez B, Hall J, Amor‐Guéret M, Pennaneach V. PARP2 controls double‐strand break repair pathway choice by limiting 53BP1 accumulation at DNA damage sites and promoting end‐resection. Nucleic Acids Res. 2017;45(21):12325‐12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amé J‐C, Rolli V, Schreiber V, et al. PARP‐2, a novel mammalian DNA damage‐dependent Poly(ADP‐ribose) polymerase. J Biol Chem. 1999;274(25):17860‐17868. [DOI] [PubMed] [Google Scholar]
- 41. Rulten SL, Fisher AEO, Robert I, et al. PARP‐3 and APLF function together to accelerate nonhomologous end‐joining. Mol Cell. 2011;41(1):33‐45. [DOI] [PubMed] [Google Scholar]
- 42. Boehler C, Gauthier LR, Mortusewicz O, et al. Poly(ADP‐ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc Natl Acad Sci USA. 2011;108(7):2783‐2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hochegger H, , Dejsuphong D, Fukushima T, et al. Parp‐1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006;25(6):1305‐1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paddock MN, Bauman At, Higdon R, Kolker E, Takeda S, Scharenberg AM. Competition between PARP‐1 and Ku70 control the decision between high‐fidelity and mutagenic DNA repair. DNA Repair. 2011;10(3):338‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gravells P, Neale J, Grant E, et al. Radiosensitization with an inhibitor of poly(ADP‐ribose) glycohydrolase: a comparison with the PARP1/2/3 inhibitor olaparib. DNA Repair. 2018;61:25‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bae W, Park JH, Lee M, Park HW, Koo H. Hypersensitivity to DNA double‐strand breaks associated with PARG deficiency is suppressed by exo‐1 and polq‐1 mutations in Caenorhabditis elegans . FEBS J. 2020;287:1101‐1115. [DOI] [PubMed] [Google Scholar]
- 48. Zhang J, Tian XJ, Zhang H, et al. TGF‐beta‐induced epithelial‐to‐mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal. 2014;7:ra91. [DOI] [PubMed] [Google Scholar]
- 49. Lindley LE, Briegel KJ. Molecular characterization of TGFβ‐induced epithelial‐mesenchymal transition in normal finite lifespan human mammary epithelial cells. Biochem Biophys Res Comm. 2010;399(4):659‐664. [DOI] [PubMed] [Google Scholar]
- 50. Taube JH, Herschkowitz JI, Komurov K, et al. Core epithelial‐to‐mesenchymal transition interactome gene‐expression signature is associated with claudin‐low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010;107(35):15449‐15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Q, Ma L, Jones T, et al. Subjugation of TGFb signaling by human papilloma virus in head and neck squamous cell carcinoma shifts DNA repair from homologous recombination to alternative end joining. Clin Cancer Res. 2018;24:6001‐6014. [DOI] [PubMed] [Google Scholar]
- 52. Martinez‐Ruiz H, Illa‐Bochaca I, Omene C, et al. A TGF ‐miR‐182‐BRCA1 axis controls the mammary differentiation hierarchy. Sci Signal. 2016;9:ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morel A‐P, Ginestier C, Pommier RM, et al. A stemness‐related ZEB1‐MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat Med. 2017;23:568‐578. [DOI] [PubMed] [Google Scholar]
- 54. Pommier RM, Sanlaville A, Tonon L, et al. Comprehensive characterization of claudin‐low breast tumors reflects the impact of the cell‐of‐origin on cancer evolution. Nat Commun. 2020;11:3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu Q, Palomero L, Moore J, et al. Loss of TGFB signaling increases alternative end‐joining DNA repair that sensitizes to genotoxic therapies across cancer types. Sci Transl Med. 2021;13:eabc4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim MR, Lee J, An YS, et al. TGFβ1 protects cells from γ‐IR by enhancing the activity of the NHEJ repair pathway. Mol Cancer Res. 2015;13(2):319‐329. [DOI] [PubMed] [Google Scholar]
- 57. Lönn P, van der Heide LP, Dahl M, et al. PARP‐1 attenuates smad‐mediated transcription. Mol Cell. 2010;40(4):521‐532. [DOI] [PubMed] [Google Scholar]
- 58. Rodríguez MI, González‐Flores A, Dantzer F, Collard J, de Herreros AG, Oliver FJ. Poly(ADP‐ribose)‐dependent regulation of Snail1 protein stability. Oncogene. 2011;30(42):4365‐4372. [DOI] [PubMed] [Google Scholar]
- 59. Kumar M, Jaiswal RK, Prasad R, et al. PARP‐1 induces EMT in non‐small cell lung carcinoma cells via modulating the transcription factors Smad4, p65 and ZEB1. Life Sci. 2021;269:118994. [DOI] [PubMed] [Google Scholar]
- 60. Karicheva O, Rodriguez‐Vargas JM, Wadier N, et al. PARP3 controls TGFβ and ROS driven epithelial‐to‐mesenchymal transition and stemness by stimulating a TG2‐Snail‐E‐cadherin axis. Oncotarget. 2016;7(39):64109‐64123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pyun B‐J, Seo HR, Lee H‐J, et al. Mutual regulation between DNA‐PKcs and snail1 leads to increased genomic instability and aggressive tumor characteristics. Cell Death Dis. 2013;4(2):e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gross KM, Zhou W, Breindel JL, et al. Loss of slug compromises DNA damage repair and accelerates stem cell aging in mammary epithelium. Cell Rep. 2019;28:394‐407. e6. [DOI] [PubMed] [Google Scholar]
- 63. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434(7035):913‐917. [DOI] [PubMed] [Google Scholar]
- 64. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 65. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2‐mutated cancers. Nature. 2008;451(7182):1116‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jaspers JE, Kersbergen A, Boon U et al. Loss of 53BP1 Causes PARP inhibitor resistance in Brca1 ‐mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68‐81. doi: 10.1158/2159-8290.CD-12-0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double‐strand break resection and affects PARP inhibition. Nature. 2015;521:541‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pettitt SJ, Krastev DB, Brandsma I, et al. Genome‐wide and high‐density CRISPR‐Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9(1):1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Henneman L, van Miltenburg MH, Michalak EM, et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1‐deficient metaplastic breast cancer. Proc Natl Acad Sci USA. 2015;112(27):8409‐8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lok BH, Gardner EE, Schneeberger VE, et al. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res. 2017;23:523‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moyret‐Lalle C, Pommier R, Bouard C, Nouri E, Richard G, Puisieux A. Cancer cell plasticity and metastatic dissemination. Med Sci (Paris). 2016;32:725‐731. [DOI] [PubMed] [Google Scholar]
- 72. Ordonez LD, Hay T, McEwen R, et al. Rapid activation of epithelial‐mesenchymal transition drives PARP inhibitor resistance in Brca2 ‐mutant mammary tumours. Oncotarget. 2019;10:2586‐2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bahar E, Kim J‐Y, Kim D‐C, Kim H‐S, Yoon H. Combination of niraparib, cisplatin and twist knockdown in cisplatin‐resistant ovarian cancer cells potentially enhances synthetic lethality through ER‐stress mediated mitochondrial apoptosis pathway. Int J Mol Sci. 2021;22:3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yazinski SA, Comaills V, Buisson R, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor‐resistant BRCA‐deficient cancer cells. Genes Dev. 2017;31(3):318‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Parmar K, Kochupurakkal BS, Lazaro J‐B, et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high‐grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin Cancer Res. 2019;25(20):6127‐6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kwok M, Davies N, Agathanggelou A, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53‐ or ATM‐defective chronic lymphocytic leukemia cells. Blood. 2016;127(5):582‐595. [DOI] [PubMed] [Google Scholar]
- 77. Min A, Im S‐A, Jang H, et al. AZD6738, a novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Mol Cancer Ther. 2017;16(4):566‐577. [DOI] [PubMed] [Google Scholar]
- 78. Konstantinopoulos PA, Cheng SC, Wahner Hendrickson AE, et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum‐resistant high‐grade serous ovarian cancer: a multicentre, open‐label, randomised, phase 2 trial. Lancet Oncol. 2020;21(7):957‐968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Song N, Bai M, Che X, et al. PD‐L1 upregulation accompanied with epithelial–mesenchymal transition attenuates sensitivity to ATR inhibition in p53 mutant pancreatic cancer cells. Med Oncol. 2020;37:47. [DOI] [PubMed] [Google Scholar]
- 80. Song N, Jing W, Li C, et al. ZEB1 inhibition sensitizes cells to the ATR inhibitor VE‐821 by abrogating epithelial–mesenchymal transition and enhancing DNA damage. Cell Cycle. 2018;17:595‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang W‐Y, Cao YX, Zhou X, et al. HMGA2 gene silencing reduces epithelial‐mesenchymal transition and lymph node metastasis in cervical cancer through inhibiting the ATR/Chk1 signaling pathway. Am J Transl Res. 2018;10:3036‐3052. [PMC free article] [PubMed] [Google Scholar]
- 82. Sullivan K, Cramer‐Morales K, McElroy DL, et al. Identification of a Small Molecule Inhibitor of RAD52 by Structure‐Based Selection. PLoS One. 2016;11(1):e0147230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sulkowski PL, Corso CD, Robinson ND, et al. 2‐Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9:eaal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nesvick CL, Zhang C, Edwards NA, et al. ZEB1 expression is increased in IDH1‐mutant lower‐grade gliomas. J Neurooncol. 2016;130:111‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Grassian AR, Lin F, Barrett R, et al. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/MicroRNA (miR)‐200‐dependent epithelial‐mesenchymal transition (EMT). J Biol Chem. 2012;287:42180‐42194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21:373‐380. [PubMed] [Google Scholar]
- 87. Karakashev S, Fukumoto T, Zhao B, et al. EZH2 inhibition sensitizes CARM1‐high, homologous recombination proficient ovarian cancers to PARP inhibition. Cancer Cell. 2020;37(2):157‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cardenas H, Zhao J, Vieth E, Nephew KP, Matei D. EZH2 inhibition promotes epithelial‐to‐mesenchymal transition in ovarian cancer cells. Oncotarget. 2016;7:84453‐84467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhu J, Huang Z, Zhang M, et al. HIF‐1α promotes ZEB1 expression and EMT in a human bladder cancer lung metastasis animal model. Oncol Lett. 2018;15:3482‐3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sreekumar R, Al‐Saihati H, Emaduddin M, et al. The ZEB2‐dependent EMT transcriptional programme drives therapy resistance by activating nucleotide excision repair genes ERCC1 and ERCC4 in colorectal cancer. Mol Oncol. 2021;15:2065‐2083. [DOI] [PMC free article] [PubMed] [Google Scholar]