

Abstract
Irinotecan is a topoisomerase inhibitor, widely used in treatment of malignancies including pancreatic ductal adenocarcinoma (PDAC) as part of the FOLFIRINOX regimen prescribed as a first‐line treatment in several countries. However, irinotecan has not been successfully introduced as a second‐line treatment for pancreatic cancer and few randomized clinical studies have evaluated its added value. Efficacy of liposomal irinotecan (nal‐IRI) combined with 5‐fluorouracil and leucovorin (5‐FU/LV) was reported in the phase III NAPOLI‐1 trial in metastatic PDAC following failure of gemcitabine‐based therapy. Several features of nal‐IRI pharmacokinetics (PK) could result in better outcomes versus nonliposomal irinotecan. Irinotecan is a prodrug that is converted to active SN‐38 by carboxylesterase enzymes and inactivated by cytochrome P450 3A4/3A5. SN‐38 is inactivated by UGT1A1 enzymes. Individual variations in their expression and activity could influence enhanced localized irinotecan activity and toxicity. Liposomal irinotecan exploits the enhanced permeability and retention effect in cancer, accumulating in tumor tissues. Liposomal irinotecan also has a longer half‐life and higher area under the concentration‐time curve (0–∞) than nonliposomal irinotecan, as the liposomal formulation protects cargo from premature metabolism in the plasma. This results in irinotecan activation in tumor tissue, leading to enhanced cytotoxicity. Importantly, despite the longer exposure, overall toxicity for nal‐IRI is no worse than nonliposomal irinotecan. Liposomal irinotecan exemplifies how liposomal encapsulation of a chemotherapeutic agent can alter its PK properties, improving clinical outcomes for patients. Liposomal irinotecan is currently under investigation in other malignancies including biliary tract cancer (amongst other gastrointestinal cancers), brain tumors, and small‐cell lung cancer.
Keywords: carcinoma, pancreatic ductal, chemotherapy, drug delivery system, irinotecan, liposome
Liposomal encapsulation of chemotherapeutic agents can modify their pharmacokinetic properties to improve clinical outcomes for patients. Liposomal irinotecan (nal‐IRI) exemplifies the benefits of this technology, leading to enhanced antitumor activity without increases in toxicity versus nonliposomal irinotecan. While approved for use in metastatic pancreatic ductal adenocarcinoma that has progressed on gemcitabine treatment, nal‐IRI is also under investigation in various other malignancies, and could result in better outcomes for patients with these tumors.

Abbreviations
- 5‐FU
5‐fluorouracil
- ABC
adenosine‐triphosphate binding cassette
- AUC
area under the concentration–time curve
- C avg
average concentration
- CES
carboxylesterase
- C max
maximum concentration
- CYP3A4/5
cytochrome P450 3A4/3A5
- EPR
enhanced permeability and retention
- FOLFIRI
floinic acid, fluorouracil, irinotecan
- FOLFIRINOX
folinic acid, fluorouracil, irinotecan, oxaliplatin
- LV
leucovorin
- mCRC
metastatic colorectal cancer
- mPDAC
advanced/metastatic pancreatic ductal adenocarcinoma
- nal‐IRI
liposomal irinotecan
- ORR
objective response rate
- OS
overall survival
- PDAC
pancreatic ductal adenocarcinoma
- PFS
progression‐free survival
- PK
pharmacokinetics
- SN‐38
7‐ethyl‐10‐hydroxycamptothecin
- SN‐38G
10‐O‐glucuronyl‐SN‐38
- SNP
single nucleotide polymorphism
- t1/2
half‐life
- TAM
tumor‐associated macrophage
- thr
threshold
- tIRI
total irinotecan
- TOP1
topoisomerase I
- tSN‐38
total SN‐38
- UGT1A
uridine diphosphateglucuronosyl transferase 1A family
- uSN‐38
unencapsulated SN‐38
- WT
wild type
1. INTRODUCTION
Liposomal carriers have been used to deliver anticancer drugs directly to tumors. Liposome deposition in tumors is thought to be facilitated by tumor blood vessel immaturity and leakiness, as well as by impaired lymphatic drainage at the tumor site (EPR). Reducing systemic drug exposure relative to tumor exposure in this manner might also improve safety. 1 , 2 Examples include liposomal doxorubicin and nal‐IRI (Onivyde® Servier (outside the USA and Taiwan; Les Laborotoires Servier SAS, 50 Rue Carnot, Suresnes 92284, France); Ipsen (within the USA; 106 Allen Road, Basking Ridge, NJ 07920, USA); PharmaEngine (Taiwan; 11F, 10 Minsheng East Road, Sec. 3, Taipei 104, Taiwan)). 3 , 4
Pancreatic ductal adenocarcinoma is a tumor with limited treatment options. Most patients present with metastatic disease at diagnosis and are ineligible for surgery. Advanced/metastatic pancreatic cancer is characterized by rapid clinical deterioration, thus chemotherapeutic treatment following progression on front‐line therapy could be limited to palliative chemotherapy. 5 Second‐line treatment options remain limited and depend on the first‐line treatment used. Liposomal irinotecan is an IV liposomal formulation that encapsulates the TOP1 inhibitor irinotecan in a lipid bilayer vesicle. 6 , 7 , 8 Treatment with nal‐IRI+5‐FU/LV was associated with significantly improved outcomes for patients with mPDAC versus 5‑FU/LV, including OS, median PFS, and ORR in the NAPOLI‐1 study. 4 Recent data have also shown improved PFS and OS outcomes for patients with biliary tract cancer receiving nal‐IRI+5‐FU/LV as second‐line therapy. 9
We aim to provide an overview of the available PK data on nal‐IRI, highlighting differences from nonliposomal irinotecan.
2. IRINOTECAN AS AN ANTICANCER AGENT
In the US, irinotecan is indicated in first‐line therapy for mCRC, with or without 5‐FU/ LV, and in patients with recurrent or progressive disease following initial fluorouracil‐based treatment. 10 Irinotecan has shown activity in other cancer types. 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 Clinical trial data for use of nonliposomal irinotecan as second‐line therapy for PDAC after disease progression with gemcitabine‐based treatment have shown varying degrees of activity. 19 , 20 , 21 , 22
2.1. METABOLISM OF IRINOTECAN
2.1.1. Irinotecan activation and inactivation
Nonliposomal irinotecan is a water‐soluble prodrug that inhibits the TOP1 cation, mainly through its active metabolite SN‐38, by stabilizing the TOP1/DNA complex, leading to DNA strand breaks, cell replication inhibition, and eventual cell death. 23 , 24 SN‐38 shows up to 1000‐fold increased TOP1 inhibitory activity versus irinotecan (Figure 1). 24 Irinotecan and its metabolites are excreted through a hepatobiliary pathway into the feces and urine by ABC transporters. 25 , 26 The inactive SN‐38 metabolite, SN‐38G, can be reactivated to SN‐38 by β‐glucuronidases in the human colorectum. Increased levels of tumor β‐glucuronidases could contribute to tumor SN‐38 exposure in vivo. 27
FIGURE 1.
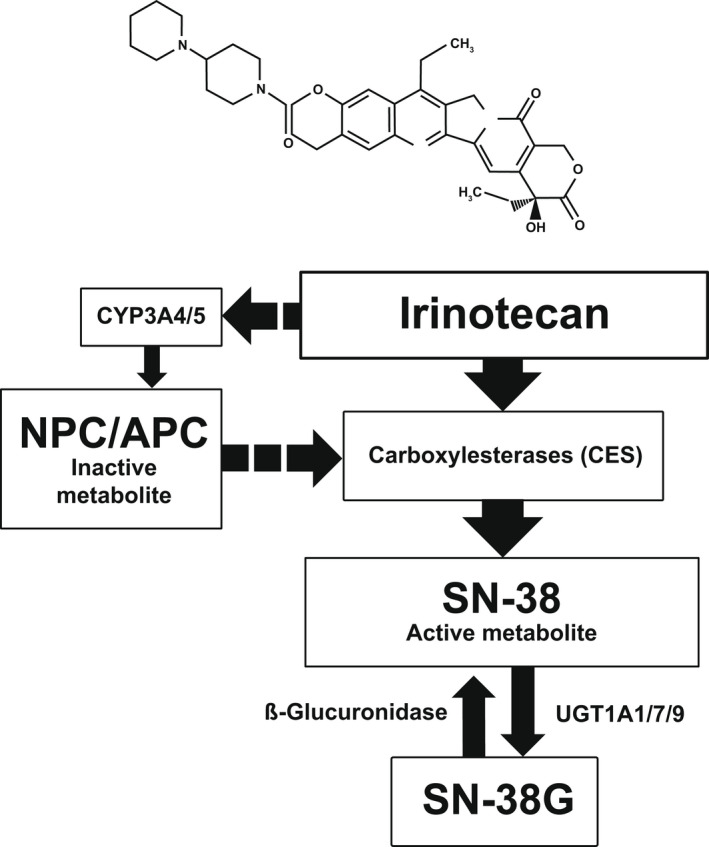
Overview of the metabolic pathway of irinotecan. In humans, the metabolic activation of irinotecan is primarily carried out through hydrolysis by carboxylesterase type 1 and 2 enzymes (CES1 and CES2, respectively), found in the liver, with activation also taking place in plasma, the small intestine, and tumor tissue. 65 , 66 , 69 , 70 CES2 is the major contributor of 7‐ethyl‐10‐hydroxycamptothecin (SN‐38) production from irinotecan and shows higher activity than CES1 in vitro. 71 In humans, irinotecan is metabolized and inactivated by cytochrome P450 3A4/3A5 (CYP3A4/5), which convert irinotecan to inactive 7‐ethyl‐10‐[4‐N‐(5‐aminopentanoicacid)‐1‐piperidino] carbonyloxycamptothecin (APC), 7‐ethyl‐10‐(4‐amino‐1‐piperidino)carbonyloxycamptothecin (NPC), and metabolites M2 and M4. 72 APC and NPC may be converted to SN‐38 by CES. 73 Alternatively, SN‐38 is inactivated by glucuronidation to a β‐glucuronic acid conjugate (10‐O‐glucuronyl‐SN‐38 [SN‐38G]) by hepatic uridine diphosphateglucuronosyl transferase 1A (UGT1A) enzymes, in particular, UGT1A1, UGT1A7, and UGT1A9. 32 , 74 SN‐38G can be re‐activated to SN‐38 by β‐glucuronidases present in the human colorectum, and increased levels of β‐glucuronidases in tumors may contribute to tumor SN‐38 exposure in vivo 27
2.1.2. Individual variations in enzymes involved in irinotecan metabolism
Irinotecan response varies among patients, possibly due to variation in expression of enzymes involved in its elimination. Liver CYP3A4 levels vary depending on environmental, rather than genetic, factors. 28 CYP3A4 status (defined as 6β‐hydroxycortisol / cortisol ratio) was found to be a predictor of diarrhea in patients with mCRC receiving a combination of irinotecan and 5‐FU. 29
UGT1A enzymatic activity or expression levels show interpatient variability based on genetic predisposition. Examples include individuals with Gilbert's syndrome who typically carry UGT1A1*28 promoter variants, or UGT1A1*6, UGT1A7*3, or UGT1A9*1b variants. 30 These factors are typically associated with reduced SN‐38 glucuronidation rates with nonliposomal irinotecan, with increased risk for gastrointestinal and bone marrow toxicities. 31 , 32 , 33 , 34 , 35 UGT1A1*28 is predictive of neutropenia in patients with mCRC receiving nonliposomal irinotecan and 5‐FU, and is associated with elevated plasma bilirubin. 29
Nonliposomal irinotecan‐induced delayed‐type diarrhea has been correlated with the presence of at least one UGT1A1*28 allele. 36 UGT1A1*28 7/7 homozygosity is a risk factor for hematological toxicity in patients receiving irinotecan, depending on administration schedule. 37
UGT1A1*28 homozygosity also correlates with SN‐38 concentrations in a nonliposomal irinotecan dose‐dependent manner. 31 , 38 UGT1A1*6 polymorphisms are observed in East Asians, but not in Caucasians. Glucuronidation activity of UGT1A1*6 is decreased to a similar extent as UGT1A1*28, and therefore, UGT1A1*6 is as important as UGT1A1*28 in East Asians. There are therefore gene–dose effects of UGT1A1*6 or *28 on glucuronidation activity, SN‐38 exposure, and neutropenia, as the two polymorphisms are mutually exclusive. 35 Although the effect of UGT1A1*28 on nonliposomal irinotecan toxicity was not suggested at lower doses in Caucasian patients, 37 Asian patients harboring UGT1A1*6 or *28 experienced severe irinotecan toxicity at lower doses. 35 , 39 It is recommended to reduce irinotecan dose for UGT1A1*6 or *28 homozygotes and those harboring both UGT1A1*6 and *28 30 , 40 ; the FDA recommended dose reduction is only for patients homozygous for UGT1A1*28.
Asian studies have focused on UGT1A1 polymorphism‐associated toxicity. A study of 48 patients from China, including 8 with unresectable PDAC and 12 with unresectable biliary tract cancer receiving FOLFIRI or irinotecan monotherapy, used direct sequencing to identify UGT1A1*28/*6 polymorphisms. 41 Patients homozygous or heterozygous for UGT1A1*6 polymorphisms were more likely to develop grade III/IV neutropenia versus patients with a WT genotype. Patients heterozygous or homozygous for UGT1A1*6 and UGT1A1*28 polymorphisms were more likely to experience grade III/IV neutropenia versus patients with a double WT genotype.
The BioBank Japan project analyzed 651 patient records (102 cases and 549 controls of various malignancies), and found that UGT1A1*6 homozygosity was predictive of adverse irinotecan reactions. 42 A meta‐analysis of 1652 patient records from nine studies (eight from Asia) of patients with colorectal cancer associated UGT1A1*6 polymorphism with late‐onset diarrhea and severe neutropenia. 43 ABCB1 (P‐glycoprotein) gene polymorphisms affect renal irinotecan clearance, with the ABCB1*8 genotype being independently associated with the irinotecan PK profile to a lesser extent than UGT1A1*28. 44 , 45 One CES2 promoter region SNP that appears to result in decreased enzyme activity, and therefore decreased irinotecan activation, has been identified. 46 Several SNPs identified in Japanese patients (1A>T; Met[1]Leu; 100 C>T, 424G>A, IVS8‐2A>G) were found to reduce activity of CES2, either by reduced mRNA transcription or loss of enzyme activity. 47 , 48
3. LIPOSOMAL IRINOTECAN
nal‐IRI consists of pegylated liposomal particles (111 nm diameter) encapsulating an irinotecan sucrosofate salt payload. The drug to phospholipid ratio is 473 mg irinotecan‐HCl/mmol phospholipid, and the phospholipid composition of the liposome is distearoylphosphatidylcholine, cholesterol, and pegylated 1,2‐distearoyl‐sn‐glycero‐3‐phosphorylethanolamine in a molar ratio of 3:2:0.015. 7 Liposomal encapsulation keeps irinotecan in circulation for longer before metabolic conversion to its active metabolite SN‐38, leading to an improved pharmacokinetic profile. Approximately 95% of the irinotecan payload is retained within liposomes 24 h after nal‐IRI administration, allowing for high drug load and increased plasma t 1/2 versus nonliposomal irinotecan (Table 1). 7 , 49 Analysis of patient‐derived PDAC xenografts in immunocompromised mice has shown that nal‐IRI has a higher therapeutic index than nonliposomal irinotecan (20 vs. 5) and prolongs time to reach tumor volume of 600 mm3 (90.5 vs. 60.6 days). 50
TABLE 1.
Effects of the liposomal encapsulation of irinotecan in preclinical models 7
| Advantage of nal‐IRI encapsulation | Nonliposomal irinotecan | nal‐IRI |
|---|---|---|
| Prolonged exposure in plasma | Irinotecan and SN‐38 cleared from circulation within 8 h | Irinotecan and SN‐38 remained in circulation within >50 h |
| Prolonged exposure in tumor xenograft models | >90% irinotecan cleared from tumors in 24 h; SN‐38 exposure in tumors <48 h | Irinotecan persisted in tumors at >10,000 nmol/L for 168 h; prolonged SN‐38 exposure above activity threshold for up to 168 h |
| Dose needed to achieve similar SN‐38 exposure in plasma and tumors in tumor xenograft models | 50 mg/kg | 10 mg/kg |
| Enhanced tumor growth inhibition in animal models | ~40% | ~110% |
Abbreviations: nal‐IRI, liposomal irinotecan; SN‐38, 7‐ethyl‐10‐hydroxycamptothecin.
Preclinical experiments using human histiocytic lymphoma cell lines (U937) indicated that TAMs, which express CES, can convert irinotecan to SN‐38. 7 Liposomal irinotecan appears to preferentially accumulate in tumor tissue through EPR, resulting in gradual accumulation in the tumor stroma. 7 This results in continual SN‐38 release through local TAM CES activity, as macrophages can take up nal‐IRI liposomes, releasing their cargo and allowing access to CES enzymes. Tumor cells also express CES, and this CES might also be able to act on any irinotecan that has been locally released. 7 Variability in PDAC CES expression could thus influence the response to irinotecan‐based treatments. 51 Conversely, nonliposomal irinotecan can be transported in and out of tissues with a short plasma t 1/2, reducing SN‐38 duration in tumors. Additionally, neutral plasma pH results in lactone ring opening, decreasing topoisomerase inhibition. 47 , 49 In preclinical human colon (HT‐29) and breast (BT474) cancer xenograft models, nal‐IRI showed superior efficacy versus nonliposomal irinotecan. 49 In preclinical, ex vivo, time‐course assays, nal‐IRI conversion to SN‐38 by nude mouse‐derived macrophages required at least 24 h and was complete after 72 h. 49
nal‐IRI showed significant and lasting tumor growth inhibition in a preclinical HT‐29 mouse xenograft model versus nonliposomal irinotecan. 7 Moreover, nal‐IRI treatment resulted in longer intratumor SN‐38 exposure and increased circulatory time in patient plasma compared with nonliposomal irinotecan, resulting in increased time above the tumor growth inhibition threshold. Computational PK modelling predicted that similar exposure of HT‐29 mouse xenograft tumors to SN‐38 from nal‐IRI could be achieved at one‐fifth of the dose versus nonliposomal irinotecan, with similar AUC, but longer tumor exposure above threshold and a higher efficacy (Table 2). 6 , 7 , 52 Similar effects have been seen in mouse xenograft models of breast cancer brain metastases. 53
TABLE 2.
Overview of pharmacokinetic parameters (total irinotecan) for nonliposomal irinotecan and liposomal irinotecan (nal‐IRI) in patients with locally advanced of metastatic gastric or gastroesophageal junction adenocarcinoma
| Nonliposomal irinotecan | nal‐IRI | |
|---|---|---|
| AUC0–∞ (plasma), h ng/ml 6 | 24,155 | 1,651,508 |
| C max (plasma), ng/ml 6 | 4,265 | 60,842 |
| t 1/2 (plasma), h 6 | 7.7 | 21.2 |
AUC0–∞ (area under the concentration–time curve between t = 0 and t = infinity) and C max (maximum concentration) were normalized to dose level in the source study. t1/2 (half‐life).
3.1. Clinical PK properties of nal‐IRI
Phase I PK data from patients with advanced solid tumors receiving nal‐IRI (alone or with 5‐FU/LV) showed a lower C max, prolonged t 1/2, and higher SN‐38 AUC (all in plasma) versus patients receiving nonliposomal irinotecan. Additionally, slow release of irinotecan from liposomes over time was suggested. 52 , 54
In a phase II study in patients with gastric cancer, the SN‐38 t 1/2 and AUC were increased with nal‐IRI versus nonliposomal irinotecan, while a lower C max was maintained. 6 Further analysis indicated that nal‐IRI had a tIRI (sum of irinotecan in liposomes and free irinotecan) C max 13.4‐times higher, a t 1/2 2.0‐times longer, and an AUC from time 0–∞ 46.2‐times greater than nonliposomal irinotecan. 55
In a clinical trial that evaluated nal‐IRI‐mediated tumor delivery in biopsies collected 72 h following administration (70 mg/m2), tumor tIRI was 0.5‐times higher than that observed in plasma. Tumor tSN‐38 was 6‐times higher than in plasma, and the tumor tSN‐38 : tIRI (a measure of the extent of conversion) was 8‐times higher than in plasma. 8
3.2. Liposomal irinotecan exposure−efficacy association
In the NAPOLI‐1 nal‐IRI+5‐FU/LV arm, longer OS and PFS were associated with higher Cavg of tIRI, tSN‐38, and uSN‐38, as well as with longer time when SN‐38 is above the threshold concentration of 0.03 ng/ml (tuSN38>thr), with the strongest association noted for tuSN38>thr. 55 In a population PK modelling analysis of nal‐IRI using plasma samples from patients with various tumors (including colorectal, gastric, and pancreatic cancer) from six studies (n = 353), higher Cavg and longer tuSN38>thr was associated with longer OS and PFS in patients with PDAC receiving nal‐IRI+5‐FU/LV. This was also associated with an increased ORR; however, Cmax was not associated with OS in these patients. 55
3.3. Liposomal irinotecan exposure−safety association
In a phase II study in patients with mPDAC receiving nal‐IRI, pharmacogenetic analysis of patient samples (n = 28) for genetic polymorphisms in UGT1A1 and UGT1A9 did not find any correlation with toxicities, although the patient numbers are likely too small to identify any relationship. 56 In the NAPOLI‐1 trial, of seven patients positive for the UGT1A1*28 polymorphism, five began treatment at a reduced starting dose and received the full planned dose of nal‐IRI in subsequent treatment cycles. 4
A recent population PK analysis found that UGT1A1*28 was not a significant predictor of SN‐38 levels following a nal‐IRI dose, with the authors proposing that liposomal encapsulation lowered irinotecan release rate, avoiding increased plasma SN‐38. 55 Additionally, a higher probability for neutropenia incidence and severity with higher uSN‐38 Cmax was observed. The association with neutropenia was stronger for uSN‐38 Cmax than for tSN‐38 Cmax. 55 It was also stronger for uSN‐38 Cmax than Cavg. A higher incidence and severity of diarrhea was associated with higher tIRI Cmax. This effect was observed in Asian subpopulations but mostly in Caucasians. It is important to note that UGT1A1*6 polymorphism was not assessed, despite 42% patients in the study being of East Asian origin.
In NAPOLI‐1, this association was only observed in the nal‐IRI monotherapy arm, presumably due to the higher nal‐IRI dose used (100 mg/m2 every 3 weeks vs. 70 mg/m2 nal‐IRI every 2 weeks in the nal‐IRI+5‐FU/LV combination arm), resulting in higher tIRI Cmax for patients receiving nal‐IRI monotherapy. 4 , 55 Differences in observed neutropenia and diarrhea rates among Caucasian and Asian patients in NAPOLI‐1 can be attributed to racial differences in the tIRI and uSN‐38 Cmax, and potentially UGT1A1*6 genotypes that were not detected due to the study design. 55 Japanese patients experienced more FOLFIRINOX (i.e., including nonliposomal irinotecan) related toxicities than Caucasian patients despite exclusion of patients homozygous for UGT1A1*28/*6 polymorphisms (or heterozygous for both). 57
A phase II study of nal‐IRI+5‐FU/LV treatment in Japanese patients with mPDAC found no unexpected increases in rates of diarrhea or neutropenia versus the NAPOLI‐1 trial. 58 Importantly, only three patients in the nal‐IRI+5‐FU/LV arm had relevant UGT1A1 mutations. The nal‐IRI dose was the same in both this study and NAPOLI‐1 (80 mg/m2, equivalent to 70 mg/m2 irinotecan free base), however the nal‐IRI dose was reduced for patients homozygous for UGT1A1*28 and UGT1A1*6 polymorphisms.
Dose modification did not significantly influence survival outcomes in a post‐hoc analysis of patients from NAPOLI‐1 who underwent protocol‐specified dose modification; for example, OS with dose modification was 8.4 months versus 6.7 months without (hazard ratio 0.89; 95% confidence interval, 0.59–1.35), 59 suggesting nal‐IRI dose modification is a feasible strategy to maintain clinical benefit. In Japan, dosing of nal‐IRI is reduced according to UGT1A1 genotype 58 ; this aligns with the NAPOLI‐1 study where patients homozygous for the UGT1A1*28 allele were initially treated with a 20 mg/m2 dose reduction of nal‐IRI before building up to a full dose in the absence of any toxic effects.
3.4. Potential for future clinical development of nal‐IRI
The properties of nal‐IRI have made it attractive for targeting various cancers. A retrospective study of 14 patients with metastatic biliary tract cancer showed that second‐line treatment with nal‐IRI+5‐FU/LV resulted in half of the patients achieving disease control, suggesting efficacy in this population. 60 The phase II NIFE trial is currently underway, comparing use of nal‐IRI+5‐FU/LV with gemcitabine + cisplatin for treatment of locally advanced or metastatic biliary tract adenocarcinoma 61 ; the NALIRICC trial is also ongoing, comparing nal‐IRI+5‐FU/LV with 5‐FU/LV in biliary tract cancer. 60 Data from the NIFTY trial have recently been presented, with improved OS and PFS outcomes for patients with biliary tract cancer receiving nal‐IRI+5‐FU/LV as a second‐line treatment. 9 Data from preclinical models of small‐cell lung cancer has shown that nal‐IRI has antitumor activity at clinically relevant dose levels, with partial or complete responses observed in tumors derived from several cell line models, and improved survival outcomes in mouse models (vs. irinotecan and topotecan). 62 Liposomal irinotecan also had activity in the second‐line setting following topotecan failure. 62
Preclinical data from animal models of breast cancer brain metastases showed that liposomes preferentially accumulate in metastatic lesions after crossing the blood–brain barrier. 53 Moreover, nal‐IRI treatment results in increased accumulation of both irinotecan and SN‐38 in brain metastases. A phase I study of nal‐IRI in 29 patients with metastatic breast cancer showed that patients with central nervous system disease (n = 10) receiving nal‐IRI had an ORR of 30%, indicating that nal‐IRI could be a viable treatment option in this population. 63
A phase I dose escalation study was carried out in patients with high‐grade glioma, with WT UGT1A1 (n = 16) versus UGT1A1*28 heterozygotes (n = 18); this found a maximum tolerated nal‐IRI dose of 120 mg/m2 for WT, and 150 mg/m2 for heterozygous patients, with nal‐IRI safety and toxicity signals similar to previous observations. 64 Further investigation will support understanding of potential nal‐IRI activity in this context.
Expression profiling using a variety of human tumor tissue and liver samples showed that CES2 expression correlated with irinotecan conversion to SN‐38. 65 High CES2 expression levels in tumor tissue correlated with increased OS in patients with resectable and borderline resectable PDAC receiving neoadjuvant therapy with FOLFIRINOX (combination of irinotecan, 5‐FU, oxaliplatin, and LV). 51 High levels of CES activity in the small intestine suggest that irinotecan‐associated delayed diarrhea is at least partly caused by local conversion of irinotecan to SN‐38. 66 A better understanding of CES2 levels could support individualized dosing of nal‐IRI.
Cationic liposomes can stimulate dendritic cell activation in vitro, potentially by promoting expression of costimulatory molecules. 67 This might create opportunities to combine liposomal formulations such as nal‐IRI with immune checkpoint inhibitors. 68 However, immune recognition of the liposomal formulation could result in clearance of the drug and therefore reduced delivery of the irinotecan payload.
4. CONCLUSION
The advent of liposomal agents such as nal‐IRI has led to improvements in drug formulations with altered PK profiles compared with their parent compounds, which translate into different efficacy and safety profiles in clinical practice. Compared with nonliposomal irinotecan, nal‐IRI generally shows increased exposure and prolonged retention (with a preference for accumulation in tumor cells), and has been shown to have increased plasma half‐life among other desirable PK properties in multiple preclinical, in silico, and clinical studies. Ongoing refinement of delivery modes at the nano scale holds the promise that compounds with undesirable toxicity could become targetable at the tumor environment, reducing the incidence of negative off‐target effects and improving efficacy.
CONFLICTS OF INTEREST
GM has received honoraria from Servier for advisory board meetings and conferences. FI holds a patent for UGT1A1 genotyping and is an Abbvie employee and stockholder. HM is an editorial board member of Cancer Science.
AUTHOR CONTRIBUTIONS
GM, FI, and HM contributed to the literature searches, drafting the manuscript, and critically reviewing the content. GM, FI, and HM approved the final version of the manuscript for submission.
ACKNOWLEDGMENTS
Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Ashfield MedComms GmbH, Mannheim, Germany, an Ashfield Health company, and was funded by Servier.
Milano G, Innocenti F, Minami H. Liposomal irinotecan (Onivyde): Exemplifying the benefits of nanotherapeutic drugs. Cancer Sci. 2022;113:2224–2231. doi: 10.1111/cas.15377
Funding information
Medical writing support was funded by Servier
REFERENCES
- 1. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751‐760. [DOI] [PubMed] [Google Scholar]
- 2. Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363‐1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barenholz Y. Doxil® — the first FDA‐approved nano‐drug: lessons learned. J Control Release. 2012;160:117‐134. [DOI] [PubMed] [Google Scholar]
- 4. Wang‐Gillam A, Li CP, Bodoky G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine‐based therapy (NAPOLI‐1): a global, randomised, open‐label, phase 3 trial. Lancet. 2016;387:545‐557. [DOI] [PubMed] [Google Scholar]
- 5. Ducreux M, Cuhna AS, Caramella C, et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2015;26(Suppl 5):v56‐68. [DOI] [PubMed] [Google Scholar]
- 6. Roy AC, Park SR, Cunningham D, et al. A randomized phase II study of PEP02 (MM‐398), irinotecan or docetaxel as a second‐line therapy in patients with locally advanced or metastatic gastric or gastro‐oesophageal junction adenocarcinoma. Ann Oncol. 2013;24:1567‐1573. [DOI] [PubMed] [Google Scholar]
- 7. Kalra AV, Kim J, Klinz SG, et al. Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 2014;74:7003‐7013. [DOI] [PubMed] [Google Scholar]
- 8. Ramanathan RK, Korn RL, Raghunand N, et al. Correlation between ferumoxytol uptake in tumor lesions by MRI and response to nanoliposomal irinotecan in patients with advanced solid tumors: a pilot study. Clin Cancer Res. 2017;23:3638‐3648. [DOI] [PubMed] [Google Scholar]
- 9. Yoo C, Kim K‐P, Kim I, et al. Liposomal irinotecan (nal‐IRI) in combination with fluorouracil (5‐FU) and leucovorin (LV) for patients with metastatic biliary tract cancer (BTC) after progression on gemcitabine plus cisplatin (GemCis): multicenter comparative randomized phase 2b study (NIFTY). J Clin Oncol. 2021;39:Abstract 4006. [Google Scholar]
- 10. Pfizer . Camptosar® (Pfizer), US Prescribing Information.
- 11. Verschraegen CF. Irinotecan for the treatment of cervical cancer. Oncology (Williston Park). 2002;16:32‐34. [PubMed] [Google Scholar]
- 12. Langer CJ. The global role of irinotecan in the treatment of lung cancer: 2003 update. Oncology (Williston Park). 2003;17:30‐40. [PubMed] [Google Scholar]
- 13. Perez EA, Hillman DW, Mailliard JA, et al. Randomized phase II study of two irinotecan schedules for patients with metastatic breast cancer refractory to an anthracycline, a taxane, or both. J Clin Oncol. 2004;22:2849‐2855. [DOI] [PubMed] [Google Scholar]
- 14. Bouché O, Raoul JL, Bonnetain F, et al. Randomized multicenter phase II trial of a biweekly regimen of fluorouracil and leucovorin (LV5FU2), LV5FU2 plus cisplatin, or LV5FU2 plus irinotecan in patients with previously untreated metastatic gastric cancer: a Federation Francophone de Cancerologie Digestive Group Study–FFCD 9803. J Clin Oncol. 2004;22:4319‐4328. [DOI] [PubMed] [Google Scholar]
- 15. Tsuda H, Takatsuki K, Ohno R, et al. Treatment of adult T‐cell leukaemia‐lymphoma with irinotecan hydrochloride (CPT‐11). CPT‐11 Study Group on Hematological Malignancy. Br J Cancer. 1994;70:771‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohno R, Okada K, Masaoka T, et al. An early phase II study of CPT‐11: a new derivative of camptothecin, for the treatment of leukemia and lymphoma. J Clin Oncol. 1990;8:1907‐1912. [DOI] [PubMed] [Google Scholar]
- 17. Wagner LM. Fifteen years of irinotecan therapy for pediatric sarcoma: where to next? Clin Sarcoma Res. 2015;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palmerini E, Jones RL, Setola E, et al. Irinotecan and temozolomide in recurrent Ewing sarcoma: an analysis in 51 adult and pediatric patients. Acta Oncol. 2018;57:958‐964. [DOI] [PubMed] [Google Scholar]
- 19. Ulrich‐Pur H, Raderer M, Verena Kornek G, et al. Irinotecan plus raltitrexed vs raltitrexed alone in patients with gemcitabine‐pretreated advanced pancreatic adenocarcinoma. Br J Cancer. 2003;88:1180‐1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoo C, Hwang JY, Kim JE, et al. A randomised phase II study of modified FOLFIRI.3 vs modified FOLFOX as second‐line therapy in patients with gemcitabine‐refractory advanced pancreatic cancer. Br J Cancer. 2009;101:1658‐1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yi SY, Park YS, Kim HS, et al. Irinotecan monotherapy as second‐line treatment in advanced pancreatic cancer. Cancer Chemother Pharmacol. 2009;63:1141‐1145. [DOI] [PubMed] [Google Scholar]
- 22. Zaniboni A, Aitini E, Barni S, et al. FOLFIRI as second‐line chemotherapy for advanced pancreatic cancer: a GISCAD multicenter phase II study. Cancer Chemother Pharmacol. 2012;69:1641‐1645. [DOI] [PubMed] [Google Scholar]
- 23. Kaneda N, Nagata H, Furuta T, Yokokura T. Metabolism and pharmacokinetics of the camptothecin analogue CPT‐11 in the mouse. Cancer Res. 1990;50:1715‐1720. [PubMed] [Google Scholar]
- 24. Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN‐38, a metabolite of the camptothecin derivative CPT‐11, in the antitumor effect of CPT‐11. Cancer Res. 1991;51:4187‐4191. [PubMed] [Google Scholar]
- 25. Slatter JG, Schaaf LJ, Sams JP, et al. Pharmacokinetics, metabolism, and excretion of irinotecan (CPT‐11) following I.V. infusion of [(14)C]CPT‐11 in cancer patients. Drug Metab Dispos. 2000;28:423‐433. [PubMed] [Google Scholar]
- 26. de Jong FA, Kitzen JJ, de Bruijn P, Verweij J, Loos WJ. Hepatic transport, metabolism and biliary excretion of irinotecan in a cancer patient with an external bile drain. Cancer Biol Ther. 2006;5:1105‐1110. [DOI] [PubMed] [Google Scholar]
- 27. Tobin P, Clarke S, Seale JP, et al. The in vitro metabolism of irinotecan (CPT‐11) by carboxylesterase and beta‐glucuronidase in human colorectal tumours. Br J Clin Pharmacol. 2006;62:122‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A‐mediated metabolism. Adv Drug Deliv Rev. 2002;54:1271‐1294. [DOI] [PubMed] [Google Scholar]
- 29. Rouits E, Charasson V, Pétain A, et al. Pharmacokinetic and pharmacogenetic determinants of the activity and toxicity of irinotecan in metastatic colorectal cancer patients. Br J Cancer. 2008;99:1239‐1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fujiwara Y, Minami H. An overview of the recent progress in irinotecan pharmacogenetics. Pharmacogenomics. 2010;11:391‐406. [DOI] [PubMed] [Google Scholar]
- 31. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43‐47. [DOI] [PubMed] [Google Scholar]
- 32. Iyer L, King CD, Whitington PF, et al. Genetic predisposition to the metabolism of irinotecan (CPT‐11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN‐38) in human liver microsomes. J Clin Invest. 1998;101:847‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rivory LP, Haaz MC, Canal P, Lokiec F, Armand JP, Robert J. Pharmacokinetic interrelationships of irinotecan (CPT‐11) and its three major plasma metabolites in patients enrolled in phase I/II trials. Clin Cancer Res. 1997;3:1261‐1266. [PubMed] [Google Scholar]
- 34. Zhang X, Yin JF, Zhang J, Kong SJ, Zhang HY, Chen XM. UGT1A1*6 polymorphisms are correlated with irinotecan‐induced neutropenia: a systematic review and meta‐analysis. Cancer Chemother Pharmacol. 2017;80:135‐149. [DOI] [PubMed] [Google Scholar]
- 35. Minami H, Sai K, Saeki M, et al. Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese: roles of UGT1A1*6 and *28. Pharmacogenet Genomics. 2007;17:497‐504. [DOI] [PubMed] [Google Scholar]
- 36. de Jong FA, Kehrer DF, Mathijssen RH, et al. Prophylaxis of irinotecan‐induced diarrhea with neomycin and potential role for UGT1A1*28 genotype screening: a double‐blind, randomized, placebo‐controlled study. Oncologist. 2006;11:944‐954. [DOI] [PubMed] [Google Scholar]
- 37. Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. UGT1A1*28 genotype and irinotecan‐induced neutropenia: dose matters. J Natl Cancer Inst. 2007;99:1290‐1295. [DOI] [PubMed] [Google Scholar]
- 38. Stewart CF, Panetta JC, O'Shaughnessy MA, et al. UGT1A1 promoter genotype correlates with SN‐38 pharmacokinetics, but not severe toxicity in patients receiving low‐dose irinotecan. J Clin Oncol. 2007;25:2594‐2600. [DOI] [PubMed] [Google Scholar]
- 39. Han JY, Lim HS, Shin ES, et al. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non‐small‐cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol. 2006;24:2237‐2244. [DOI] [PubMed] [Google Scholar]
- 40. Pharmaceutical and Medical Devices Agency . PMDA Review Report of Pembrolizumab; 2013.
- 41. Yang C, Liu Y, Xi WQ, et al. Relationship between UGT1A1*6/*28 polymorphisms and severe toxicities in Chinese patients with pancreatic or biliary tract cancer treated with irinotecan‐containing regimens. Drug Des Devel Ther. 2015;9:3677‐3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hikino K, Ozeki T, Koido M, et al. Comparison of effects of UGT1A1*6 and UGT1A1*28 on irinotecan‐induced adverse reactions in the Japanese population: analysis of the Biobank Japan Project. J Hum Genet. 2019;64:1195‐1202. [DOI] [PubMed] [Google Scholar]
- 43. Zhu X, Ma R, Ma X, Yang G. Association of UGT1A1*6 polymorphism with irinotecan‐based chemotherapy reaction in colorectal cancer patients: a systematic review and a meta‐analysis. Biosci Rep. 2020;40:BSR20200576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sai K, Kaniwa N, Itoda M, et al. Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype‐dependent renal clearance of irinotecan. Pharmacogenetics. 2003;13:741‐757. [DOI] [PubMed] [Google Scholar]
- 45. Mathijssen RH, de Jong FA, van Schaik RH, et al. Prediction of irinotecan pharmacokinetics by use of cytochrome P450 3A4 phenotyping probes. J Natl Cancer Inst. 2004;96:1585‐1592. [DOI] [PubMed] [Google Scholar]
- 46. Charasson V, Bellott R, Meynard D, Longy M, Gorry P, Robert J. Pharmacogenetics of human carboxylesterase 2, an enzyme involved in the activation of irinotecan into SN‐38. Clin Pharmacol Ther. 2004;76:528‐535. [DOI] [PubMed] [Google Scholar]
- 47. Kubo T, Kim SR, Sai K, et al. Functional characterization of three naturally occurring single nucleotide polymorphisms in the CES2 gene encoding carboxylesterase 2 (HCE‐2). Drug Metab Dispos. 2005;33:1482‐1487. [DOI] [PubMed] [Google Scholar]
- 48. Kim SR, Sai K, Tanaka‐Kagawa T, et al. Haplotypes and a novel defective allele of CES2 found in a Japanese population. Drug Metab Dispos. 2007;35:1865‐1872. [DOI] [PubMed] [Google Scholar]
- 49. Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66:3271‐3277. [DOI] [PubMed] [Google Scholar]
- 50. Barbier S, Lezmi S, Beaufils B, et al. Differentiation of liposomal irinotecan from dose‐dense non‐liposomal irinotecan in patient‐derived pancreatic cancer xenograft tumor models. J Clin Oncol. 2020;38:e16724. [Google Scholar]
- 51. Capello M, Lee M, Wang H, et al. Carboxylesterase 2 as a determinant of response to irinotecan and neoadjuvant FOLFIRINOX therapy in pancreatic ductal adenocarcinoma. J Natl Cancer Inst. 2015;107:djv132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chiang NJ, Chao TY, Hsieh RK, et al. A phase I dose‐escalation study of PEP02 (irinotecan liposome injection) in combination with 5‐fluorouracil and leucovorin in advanced solid tumors. BMC Cancer. 2016;16:907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mohammad AS, Griffith JI, Adkins CE, et al. Liposomal irinotecan accumulates in metastatic lesions, crosses the blood‐tumor barrier (BTB), and prolongs survival in an experimental model of brain metastases of triple negative breast cancer. Pharm Res. 2018;35:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang TC, Shiah HS, Yang CH, et al. Phase I study of nanoliposomal irinotecan (PEP02) in advanced solid tumor patients. Cancer Chemother Pharmacol. 2015;75:579‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Adiwijaya BS, Kim J, Lang I, et al. Population pharmacokinetics of liposomal irinotecan in patients with cancer. Clin Pharmacol Ther. 2017;102:997‐1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ko AH, Tempero MA, Shan YS, et al. A multinational phase 2 study of nanoliposomal irinotecan sucrosofate (PEP02, MM‐398) for patients with gemcitabine‐refractory metastatic pancreatic cancer. Br J Cancer. 2013;109:920‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Okusaka T, Ikeda M, Fukutomi A, et al. Phase II study of FOLFIRINOX for chemotherapy‐naïve Japanese patients with metastatic pancreatic cancer. Cancer Sci. 2014;105:1321‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ueno M, Nakamori S, Sugimori K, et al. nal‐IRI+5‐FU/LV versus 5‐FU/LV in post‐gemcitabine metastatic pancreatic cancer: randomized phase 2 trial in Japanese patients. Cancer Med. 2020;9:9396‐9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen LT, Macarulla T, Blanc JF, et al. Early dose reduction/delay and the efficacy of liposomal irinotecan with fluorouracil and leucovorin in metastatic pancreatic ductal adenocarcinoma (mPDAC): a post hoc analysis of NAPOLI‐1. Pancreatology. 2021;21:192‐199. [DOI] [PubMed] [Google Scholar]
- 60. Taghizadeh H, Unseld M, Schmiderer A, et al. First evidence for the antitumor activity of nanoliposomal irinotecan with 5‐fluorouracil and folinic acid in metastatic biliary tract cancer. Cancer Chemother Pharmacol. 2020;86:109‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perkhofer L, Berger AW, Beutel AK, et al. Nal‐IRI with 5‐fluorouracil (5‐FU) and leucovorin or gemcitabine plus cisplatin in advanced biliary tract cancer – the NIFE trial (AIO‐YMO HEP‐0315) an open label, non‐comparative, randomized, multicenter phase II study. BMC Cancer. 2019;19:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leonard SC, Lee H, Gaddy DF, et al. Extended topoisomerase 1 inhibition through liposomal irinotecan results in improved efficacy over topotecan and irinotecan in models of small‐cell lung cancer. Anticancer Drugs. 2017;28:1086‐1096. [DOI] [PubMed] [Google Scholar]
- 63. Sachdev JC, Munster P, Northfelt DW, et al. Phase I study of liposomal irinotecan in patients with metastatic breast cancer: findings from the expansion phase. Breast Cancer Res Treat. 2021;185:759‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Clarke JL, Molinaro AM, Cabrera JR, et al. A phase 1 trial of intravenous liposomal irinotecan in patients with recurrent high‐grade glioma. Cancer Chemother Pharmacol. 2017;79:603‐610. [DOI] [PubMed] [Google Scholar]
- 65. Xu G, Zhang W, Ma MK, McLeod HL. Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin Cancer Res. 2002;8:2605‐2611. [PubMed] [Google Scholar]
- 66. Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of irinotecan by a human intestinal carboxylesterase. Cancer Res. 2000;60:4725‐4728. [PubMed] [Google Scholar]
- 67. Vangasseri DP, Cui Z, Chen W, Hokey DA, Falo LD Jr, Huang L. Immunostimulation of dendritic cells by cationic liposomes. Mol Membr Biol. 2006;23:385‐395. [DOI] [PubMed] [Google Scholar]
- 68. Inglut CT, Sorrin AJ, Kuruppu T, et al. Immunological and toxicological considerations for the design of liposomes. Nanomaterials (Basel). 2020;10:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Guichard S, Terret C, Hennebelle I, et al. CPT‐11 converting carboxylesterase and topoisomerase activities in tumour and normal colon and liver tissues. Br J Cancer. 1999;80:364‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shingyoji M, Takiguchi Y, Watanabe‐Uruma R, et al. In vitro conversion of irinotecan to SN‐38 in human plasma. Cancer Sci. 2004;95:537‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Senter PD, Beam KS, Mixan B, Wahl AF. Identification and activities of human carboxylesterases for the activation of CPT‐11, a clinically approved anticancer drug. Bioconjug Chem. 2001;12:1074‐1080. [DOI] [PubMed] [Google Scholar]
- 72. Santos A, Zanetta S, Cresteil T, et al. Metabolism of irinotecan (CPT‐11) by CYP3A4 and CYP3A5 in humans. Clin Cancer Res. 2000;6:2012‐2020. [PubMed] [Google Scholar]
- 73. Sanghani SP, Quinney SK, Fredenburg TB, Davis WI, Murry DJ, Bosron WF. Hydrolysis of irinotecan and its oxidative metabolites, 7‐ethyl‐10‐[4‐n‐(5‐aminopentanoic acid)‐1‐piperidino] carbonyloxycamptothecin and 7‐ethyl‐10‐[4‐(1‐piperidino)‐1‐amino]‐carbonyloxycamptothecin, by human carboxylesterases ces1a1, CES2, and a newly expressed carboxylesterase isoenzyme, CES3. Drug Metab Dispos. 2004;32:505‐511. [DOI] [PubMed] [Google Scholar]
- 74. de Jong FA, de Jonge MJ, Verweij J, Mathijssen RH. Role of pharmacogenetics in irinotecan therapy. Cancer Lett. 2006;234:90‐106. [DOI] [PubMed] [Google Scholar]