

Abstract
Immortal cancer cell lines (CCLs) are the most widely used system for investigating cancer biology and for the preclinical development of oncology therapies. Pharmacogenomic and genome‐wide editing screenings have facilitated the discovery of clinically relevant gene–drug interactions and novel therapeutic targets via large panels of extensively characterised CCLs. However, tailoring pharmacological strategies in a precision medicine context requires bridging the existing gaps between tumours and in vitro models. Indeed, intrinsic limitations of CCLs such as misidentification, the absence of tumour microenvironment and genetic drift have highlighted the need to identify the most faithful CCLs for each primary tumour while addressing their heterogeneity, with the development of new models where necessary. Here, we discuss the most significant limitations of CCLs in representing patient features, and we review computational methods aiming at systematically evaluating the suitability of CCLs as tumour proxies and identifying the best patient representative in vitro models. Additionally, we provide an overview of the applications of these methods to more complex models and discuss future machine‐learning‐based directions that could resolve some of the arising discrepancies.
Keywords: cancer cell lines, computational biology, drug discovery, personalised medicine, pharmacogenomics
Subject Categories: Cancer, Computational Biology, Pharmacology & Drug Discovery
Cancer cell lines (CCLs) are widely used for analysing cancer and identifying therapies. This Review discusses the limitations of CCLs in representing tumours and considers computational methods that can systematically evaluate the suitability of CCLs as tumour proxies.
Cancer cell lines: a mainstay for cancer biology, drug discovery and large‐scale multi‐omic data generation
Since the first cultured cell line was established in 1951 from Henrietta Lacks' cervical cancer cells (Scherer et al, 1953), the use of immortalised cell lines as cancer in vitro models has become a pivotal tool for studying primary tumours. Cancer cell lines (CCLs) are widely used for therapy discovery, as they are easily amenable to experimental manipulation, and suitable for high‐throughput screens, supporting the generation of large‐scale perturbation data sets (McDonald et al, 2017; Meyers et al, 2017; Tsherniak et al, 2017; Behan et al, 2019), as well as comprehensive multi‐omic characterizations (Gillet et al, 2013; Ghandi et al, 2019; Fig 1).
Figure 1. Major public cell line‐based data sets with corresponding omics and reference publications.
The horizontal bars indicate the data type/omic type availability. Created with BioRender.com.
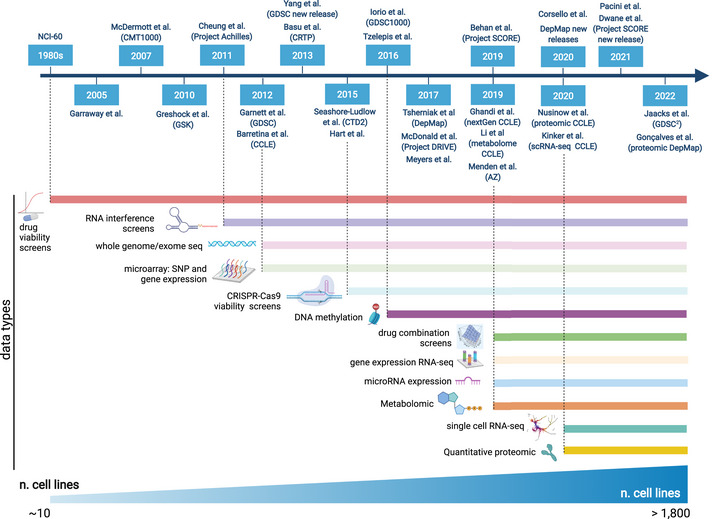
Currently, the use of large‐scale cell‐line‐based multi‐omic data sets is having a major impact on drug discovery and repositioning, facilitating the identification of genetic linkages between candidate drug targets and disease biology, thus increasing the likelihood of investigative compounds to progress through the different phases of clinical development (Wilding & Bodmer, 2014; Nelson et al, 2015; Corsello et al, 2020; Francies et al, 2020). Starting from the pioneer NCI‐60 panel, created in the 1980s (Shoemaker, 2006) and aimed at identifying compounds with tumour‐type‐specific growth‐inhibitory effects across 60 CCLs, next‐generation high‐throughput techniques have given rise to large‐scale pharmacogenomic screens (Sharma et al, 2010), in the attempt to dissect the relationship between cell viability reduction upon compounds' treatment and genetic features.
Besides extensively characterisations of the NCI‐60 panel (Garraway et al, 2005), tremendous effort has been and is still being made to assemble increasingly large CCL drug response data sets. These embody quite comprehensive tumour molecular heterogeneity together with viability reduction measurements of thousands of in vitro models in response to treatment with hundreds of compounds. Examples include the Cancer Cell Line Encyclopedia (CCLE; Barretina et al, 2012), the Genomics of Drug Sensitivity in Cancer (GDSC; Garnett et al, 2012; Yang et al, 2013), the Centre for Molecular Therapeutics 1000 (CMT1000; McDermott et al, 2007), Cancer Target Discovery and Development (CTD (Basu et al, 2013) and CTD2 (Seashore‐Ludlow et al, 2015)) and a study from (Greshock et al, 2010). In addition, comprehensive drug response data sets have been recently expanded to incorporate non‐oncology drugs (Corsello et al, 2020), and combinatorial treatments (Menden et al, 2019; Jaaks et al, 2022).
In parallel, CCL characterizations have expanded in the direction of multi‐omic data assembly to reveal regulatory mechanisms associated with cancer vulnerabilities arising from cancer driver genomic (as well as, epigenetic and transcriptomic) alterations. For instance, CCLE moved beyond the initial genomic and transcriptomic space and characterised RNA splicing, DNA methylation, microRNA expression, global histone modifications, proteomic and metabolomic quantitative profiles in more than 1,000 CCLs from multiple lineages and ethnicities (Ghandi et al, 2019; Li et al, 2019; Nusinow et al, 2020; preprint: Goncalves et al, 2022). Among those, a subset of 198 CCLs in 22 cancer types have been recently profiled by single‐cell RNA‐seq to study intra‐tumour heterogeneity (Kinker et al, 2020).
Various computational approaches have been used to jointly analyse these drug response data sets and the comprehensive multi‐omic characterisations of the CCLs, revealing molecular features that are informative and predictive of drug response, most often based on stratifying CCLs on the presence/absence of individual molecular features. This has allowed recovering established and identifying novel genomic (Basu et al, 2013; Seashore‐Ludlow et al, 2015; Iorio et al, 2016; Jaaks et al, 2022) as well as transcriptional (Garcia‐Alonso et al, 2018; Jaaks et al, 2022) markers of drug sensitivity, leading to new testable hypotheses and clinical trials. For instance, Ewing's Sarcoma lines were found to be hypersensitive to PARP inhibitors (Gill et al, 2015), leading to the proposed use of these inhibitors in combination with chemo/radiotherapy (Vormoor & Curtin, 2014). Canakinumab and spartalizumab are undergoing phase 1 clinical trial as a combinatorial treatment for clear cell renal carcinoma patients (NCT04028245) and entinostat (histone deacetylase inhibitor) is undergoing phase 2 in the treatment of neuroendocrine tumours (NCT03211988). Finally, CHEK1 inhibitors were found to act synergistically with chemotherapy (irinotecan) in microsatellite‐stable and KRAS‐TP53 double‐mutant colon cancer cells, both in vitro and in vivo (Jaaks et al, 2022).
Simultaneously, large‐scale RNA interference (RNAi) (Cheung et al, 2011; McDonald et al, 2017; Tsherniak et al, 2017) and genome‐wide CRISPR‐Cas9 knockout screens (Hart et al, 2015; Tzelepis et al, 2016; Meyers et al, 2017) performed on large panels of CCLs enabled the systematic identification of cancer genetic dependencies (i.e. genes necessary for cancer cell proliferation and survival, also called fitness genes) (Grimm, 2004). With higher efficiency and precision compared with RNAi (Evers et al, 2016), CRISPR‐based studies have elucidated the landscape of cancer vulnerabilities and unveiled novel and therapeutically exploitable synthetic‐lethalities (Chan et al, 2019), allowing the development of advanced bioinformatics methods for the identification and the prioritisation of new candidate therapeutic targets on a genome scale (Behan et al, 2019).
Increasing the level of complexity, more recent in vitro screens are focussing on digenic dependencies, uncovering compensatory relationships between pairs of genes and are starting to identify interactions that are synthetic lethal for cancer cell survival, and most often involve specific paralogous genes (Ito et al, 2021; Thompson et al, 2021). In addition, CCL‐based post‐perturbational transcriptomic data sets such as the Connectivity Map (Lamb, 2007; Bush et al, 2017; Ye et al, 2018) and related Web resources (Stathias et al, 2020) have been pivotal for computational drug discovery and repositioning (Pushpakom et al, 2018), and are now being increasingly assembled also at single‐cell resolution (McFarland et al, 2020; Srivatsan et al, 2020).
This ecosystem of CCL data sets is publicly accessible, actively curated and allows generating new hypotheses about the biology of cancer, its dependencies and response to therapy (Table 1). For instance, Cellosaurus (Bairoch, 2018) provides curated CCL metadata resources across multiple species. COSMIC (Tate et al, 2019) includes the Cell Lines Project dataset (Iorio et al, 2016), which collects exome sequencing data and molecular profiling of more than 1,000 CCLs. cBioPortal (Gao et al, 2013) allows users to interactively explore multidimensional cancer genomic and clinical data sets, including data visualisation and analytical options across genes, samples and data types, gathering both CCL and patient tumour information. The GDSC (Yang et al, 2013) and GDSC2 (Jaaks et al, 2022) databases are large public resources of drug sensitivity data derived from treating more than a thousand CCLs with hundreds of individual and pairs of compounds, respectively. These resources are also paired with Web portals equipped with interactive data exploration tools, aiming at facilitating the discovery of statistical associations between molecular features and differential treatment response to single or combinatorial therapies. The Cancer Dependency Map has continued to generate and refine data from the characterisation of increasingly larger CCL collections, now accounting for more than 1,800 in vitro models, and making the corresponding omics and CRISPR‐screening data available pre‐publication. Similarly, the Cell Model Passports portal (van der Meer et al, 2018) includes highly curated multi‐omic and clinical data sets derived from the characterisation of more than 1,900 CCLs and organoids. The Project Score (Dwane et al, 2021) database allows the exploration of systematic genome‐scale CRISPR‐Cas9 dropout screen results in a variety of CCLs. Finally, the Online Gene Essentiality Database (Gurumayum et al, 2021) contains gene fitness data for 91 species, encompassing more than 500 CCLs.
Table 1.
Portals providing access to large CCL‐based data sets and related in vitro models' curated annotations.
| Portal name | URL | Available info |
|---|---|---|
| Cellosaurus | https://web.expasy.org/cellosaurus/ | CCL names with synonyms, sex and age of the donor, and molecular charachteristics (MSI, doubling time etc). |
| Engineering procedure (gene KO or insertion), resistance to drug, known contaminations. | ||
| COSMIC | https://cancer.sanger.ac.uk/cosmic | Catalogue of cancer somatic mutations: variant type, gene fusions, CN variants, drug resistant mutations, GE and HypMet effects. |
| https://cancer.sanger.ac.uk/cell_lines |
CCLs' exome sequencing and other molecular profiles. |
|
| cBioPortal | https://www.cbioportal.org/ | Interactive exploration of genetic, epigenetic, gene expression, proteomic events and clinical data. Connection to disrupted pathways. |
| GDSC | https://www.cancerrxgene.org/ | CCLs' drug sensitivity and molecular markers of drug response. |
| GDSC2 | https://gdsc‐combinations.depmap.sanger.ac.uk/ | CCLs' drug combination sensitivity and related molecular markers. |
| DepMap | https://depmap.org/portal/ | Portal collecting multi‐omic data from the characterisation of 100s of CCLs (maintained at the Broad and other institutes). |
| CCLs' molecular, drug sensitivity, gene essentiality (from CRISPR‐Cas9 and RNAi screens) profiles. | ||
| CellModelPassport | https://cellmodelpassports.sanger.ac.uk/ | Portal with multi‐omic data from the characterisation of 100s of CCLs (maintained at the Wellcome Sanger institute). |
| CCLs' molecular, drug sensitivity, gene essentiality (from CRISPR‐Cas9 screens) profiles. | ||
| ProjectScore | https://score.depmap.sanger.ac.uk/ | Systematic genome‐scale CRISPR‐Cas9 drop‐out screens with exploration tools. |
| Online Gene Essentiality Database | https://v3.ogee.info/ | CCLs' gene essentiality profiles (from CRISPR‐Cas9 and RNAi screens). |
Despite initial concerns about inter‐study reproducibility (Haibe‐Kains et al, 2013), this plethora of resources has been proven consistent across institutes and publications, from a pharmacogenomic point of view (Cancer Cell Line Encyclopedia Consortium & Genomics of Drug Sensitivity in Cancer Consortium, 2015; Geeleher et al, 2016; Haverty et al, 2016), as well as when considering drug response profiling (Mpindi et al, 2016) and CRISPR‐Cas9 screens (Dempster et al, 2019). This agreement across studies has allowed their integration (Pacini et al, 2021), paving the way to large unified resources and inter‐study/institute Cancer Dependency Maps (Boehm et al, 2021). Compared with more recent cancer models such as patient‐derived xenografts (PDx) and patient‐derived organoids (PDO), the scalability and cost efficiency of CCLs is reflected by the larger volume and diversity of available data (Feng et al, 2021). Hence, it is likely that for the foreseeable future, CCLs will remain the main source of information for genomics‐guided and data‐driven preclinical development of cancer therapies (Francies et al, 2020), and for the discovery and validation of cancer genetics dependencies (Lin & Sheltzer, 2020). Nonetheless, CCLs have intrinsic and unsurmountable limitations, including the fact that they are cultured in 2D flat dishes, growing in cell culture media and lacking matching tumour microenvironment (TME) components. This poses questions about how reliably CCLs mimic patient tumours and the extent to which this represents an obstacle for the translation of CCL derived findings from‐bench‐to‐bedside.
If CCL characterizations, pharmacogenomic and genetic perturbation screenings are effective in the context of forward translation, which implies actualizing research discoveries into practice, reverse translation, that is the elucidation of the mechanistic basis of clinical observations, is a complementary and equally important need for successful drug development (Honkala et al, 2021). Hence, reverse translational practices such as the identification of clinically predictive features and their observational validation in real tumours is meaningless if it is not preceded by a correct selection of properly representative CCLs for each considered patient cohort.
In a precision medicine context, patients' genomic heterogeneity has been linked to differences in treatment response, and the efficacy of 75 FDA‐approved anti‐cancer drugs associated with 47 biomarkers across 25 cancer types (Feng et al, 2021). Indeed, efforts to genomically characterise tumour patients (International Cancer Genome Consortium et al, 2010; Cancer Genome Atlas Research Network et al, 2013) have also led to a comprehensive collection of data sets spanning across multiple omics, in some cases paired with clinical observations (Gao et al, 2013). This has allowed retrospectively validating to a certain extent some of the associations between molecular features and drug responses observed in CCLs. However, new CCL‐derived pharmacogenomic associations were not always confirmed in clinical trials. For example, the upregulation of IGFR1 found associated with tamoxifen resistance in breast cancer CCLs, exhibited the opposite behaviour in patients (Drury et al, 2011).
Leveraging the plethora of existing data, it is now possible to develop methods able to map CCLs to tumours, to identify CCLs that most closely resemble relevant patient characteristics to (1) achieve a better understanding of cancer mechanisms and (2) maximise the likelihood that virtual drug prescriptions discovered from CCL‐based studies are effective and beneficial for a specific patient segment.
Here, we first review the factors that might compromise how well CCLs represent primary tumours. We then discuss computational studies that investigate CCL resemblance to patient tumours, ranging from cancer‐specific investigations focussed on individual (or few) data modalities to more recent multi‐omic and pan‐cancer approaches. Finally, we offer an outlook on the use of machine learning methods in this context.
Factors that might compromise how well cell lines represent tumour characteristics
The relevance of the findings originating from CCL‐based studies and their translation in clinical applications have been long questioned, long before large‐scale screenings became widespread (Hughes et al, 2007; Gillet et al, 2013). This was due to several factors that potentially compromise the faithfulness of CCLs in representing the cancer patients they are intended to model (Fig 2).
Figure 2. Factors hampering the faithfulness of CCLs as tumour models.
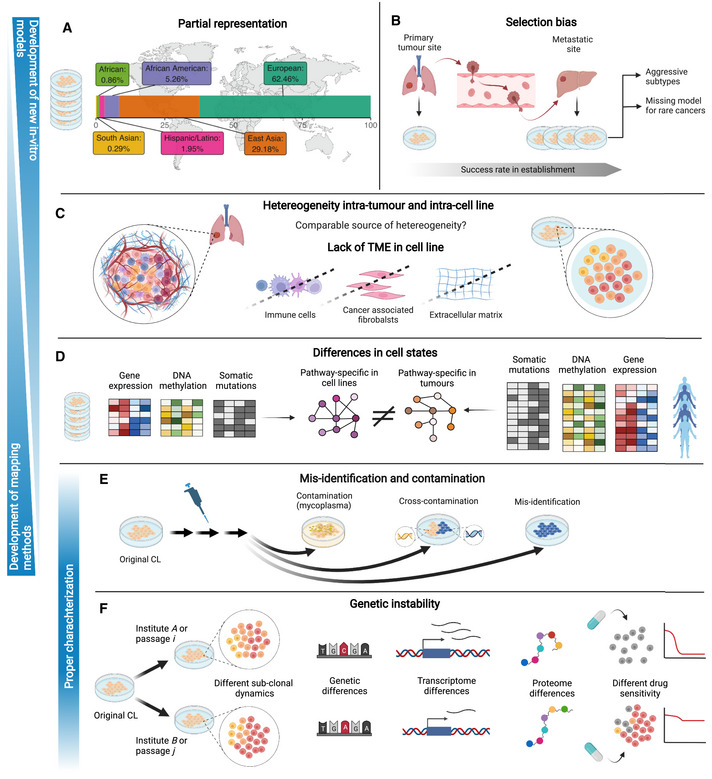
Panels A to E show issues that can be addressed by establishing new in vitro models (top to bottom) or by developing cell line‐tumour mapping methods (bottom to top). (A) Cell line biobanks are mostly derived from European and east Asian ancestries (data from Dutil et al, 2019). (B) Ease in establishing cell lines from more aggressive subtypes. (C) Intra‐tumour and intra‐cell lines dynamics, possibly reduced heterogeneity in cell lines that additionally do not include tumour microenvironment. (D) Differences in cell states among cell lines and tumour biobanks in terms of genetic, transcriptional, epigenomic and proteomic features that lead to differentially regulated pathways. (E) Contamination and mis‐identification due to lab conditions. Cells in blue represent a different donor. (F) Genetic instability in the same cell line due to different culture conditions or passaging can lead to divergences in genetic features, transcriptional and proteomic states and consequentially drug response. Created with BioRender.com.
Misidentification and contamination
The first issue arises from possible contaminations and misclassifications due to culturing the cells in the laboratory (Fig 2E). Cross‐contamination, for example cells from a foreign culture introduced accidentally in a CCL, is a well‐documented problem. Capes‐Davis et al (2010) performed a literature screening and reported that 360 CCLs from 68 references were cross‐contaminated, mostly intra‐species (90%), with the most common contaminant being HeLa cells (29%). Cross‐contamination cases are usually spotted by short tandem repeat (STR) profiling, generally performed for CCL authentication purposes, thus also suitable for the identification of mislabelled CCLs. Via STR profiling performed on 113 independent sources in China, 95 CCLs of the 380 tested were detected as cross‐contaminated, with 93.22% of the cases involving HeLa cells contamination (Ye et al, 2015). Another well‐known source of contamination is mycoplasma, a small parasitic bacterium which might be passed on by other contaminated cell cultures or laboratory personnel. RNA sequencing showed that 11% of 9,395 samples from hundreds of laboratories were indeed contaminated with mycoplasma (Olarerin‐George & Hogenesch, 2015).
Misidentification and misclassification errors arise when the gender, species, tissue or cell type, disease or CCL names are wrongly annotated and do not match the actual source. It was estimated that more than 30,000 scientific publications were affected by CCLs not being of the declared type (Horbach & Halffman, 2017). To tackle this significant problem, Cellosaurus (Bairoch, 2018) offers a CCL authentication system by STR profiling in the CLASTR (Cellosaurus STR similarity search) tool (Robin et al, 2020), which allows contrasting one or more STR profiles against that of 6,474 human CCLs. Despite being the international reference system for in vitro model authentication, STR profiling is still susceptible to heterogeneity within the same CCL that can occur due to differences in laboratory and culture conditions or genetic drifting, especially in the presence of microsatellite instability (Much et al, 2014).
Genetic drift, in vitro selection pressures and genetic instability
Genetic heterogeneity in CCLs of the same origin has been observed by several groups (Ben‐David et al, 2018; Liu et al, 2019b; Quevedo et al, 2020), aggravating differences in models that originated from the same donor (Fig 2F). For instance, Ben‐David et al (2018) reported on many CCLs exhibiting complex clonal dynamics and evolutionary pressures specific to in vitro culturing conditions. Ben‐David et al (2018) reported that this in turn impacted functional properties such as morphology, proliferative capacity, gene expression and drug response. A recent study from Quevedo et al (2020) explored genetic stability across the 3 largest pharmacogenomic studies that leveraged both RNA‐seq and SNP array data, finding discrepancies both intra‐ and inter‐institutions, and hinting that pharmacological delineation could have been derived at different passages and/or stocks, thus not properly defining CCLs' drug responses and being linked to different and variable transcriptional programmes. Indeed, within‐CCL genetic variability greatly impacts gene expression, even at the level of cancer‐related genes (Fasterius & Al‐Khalili Szigyarto, 2018). Genomic and transcriptomic variations can translate also into variations at the proteomic level, as it was shown while investigating 14 HeLa cells strains (Liu et al, 2019b). On the contrary, epigenetic changes driven by environmental factors (e.g. culture conditions) are also plausible, given the evidence of DNA methylation instability in human pluripotent stem cells (Weissbein et al, 2017). However, no study has analysed this aspect so far. In addition, the mutational variability in a CCL donor can lead to the continuous emergence of new subclones in that CCL (Ben‐David et al, 2018), which can lead to the emergence of drug resistance (Hata et al, 2016).
In general, given the intrinsic differences between human physiological environment and cell culture conditions, it sems rather unlikely that tumours and CCLs are subjected to the same evolutionary selective pressures. This increases the molecular divergence between cancer models and the tumours they were originated from (reviewed in Ben‐David et al, 2019).
The similarity of CCLs to their tumour of origin can be considered a non‐critical issue if CCLs are used just as a means for investigating intrinsic oncogenic mechanisms. However, diverse evolutionary mechanisms could contribute to significantly distancing cancer model populations from patient cohorts, not properly mimicking the focal aspects of oncogenic addiction in cancer patients and their triggered genetic dependencies. This can lead to misleading findings that would not be recapitulated in real tumours. On the contrary, unstable molecular features in an in vitro cohort also limit the faithfulness of its molecular characterisation, making collected data inaccurate across multiple strains, with possible false findings arising especially when using CCLs for harvesting biomarkers of drug response.
Selection bias
Existing biobanks and panels of in vitro cancer models are biased towards the preferential representation of certain cancer types and subtypes (Fig 2B). It has been reported that CCLs are more commonly derived from metastatic tumours due to their predisposition to grow successfully in vitro (Masters, 2000). The genetic changes accumulated by aggressive cancers are one possible explanation for their increased chances of growing indefinitely in vitro. Consequently, aggressive cancer subtypes tend to be overrepresented across CCL collections of a specific tissue. For example, breast CCLs are mostly derived from metastases rather than primary lesions (Burdall et al, 2003). In addition, CCL cohorts do not sufficiently represent all patient tumour subtypes (van Staveren et al, 2009; Klijn et al, 2015). This is a prominent problem for rare cancers which collectively make up 25% of cancer diagnoses each year (Greenlee et al, 2010) and for most of which no representative CCLs are available to date (Sharifnia et al, 2017).
Missing tumour microenvironment factors
CCLs are cultured in flat plastic dishes, fed with synthetic media enriched with bovine serum and they completely lack the tumour microenvironment (TME) that surrounds patients' cancer cells in vivo (Fig 2C). The TME includes non‐malignant cell types such as immune cells and fibroblasts, extracellular matrix and signalling proteins (Binnewies et al, 2018). The crosstalk between tumour cells and the surrounding TME enhances both tumorigenesis and tumour progression, and also plays a role in preventing therapy efficacy and increasing multidrug resistance (Klemm & Joyce, 2015; Baghban et al, 2020). Furthermore, recent studies have shown that cell culture media impacts genetic dependencies observed in CCLs (Cheteh et al, 2017; Li et al, 2019; Rossiter et al, 2021). Nevertheless, despite the lack of immune‐like cells or cancer fibroblasts, it was found that specific metabolites in human plasma‐like medium also influence the set of essential genes in CCLs detected in CRISPR‐based screens (Rossiter et al, 2021). Co‐culturing CCLs with cancer‐associated fibroblasts (CAF) or even CAF‐conditioned medium reduced response to chemotherapeutic treatments and conversely increased cell survival in prostate CCLs (Cheteh et al, 2017). This offers the possibility to reproduce in vitro some of the interactions occurring between cancer cells and the TME. More complete TME representations have been implemented via co‐culturing technologies in complex in vitro models. For instance, three‐dimensional patient‐derived organoids (PDO) have been co‐cultured with endogenous native infiltrating immune cell populations and non‐immune stromal elements, allowing in vitro immune oncology investigations (Neal et al, 2018). In addition, single‐cell analyses in PDOs from pancreatic cancers showed that TME signals drive malignant cell states and influence drug responses (Raghavan et al, 2021). Interestingly, Raghavan et al (2021) also demonstrated that ex vivo soluble micro‐environment can be manipulated to alter transcriptional states, demonstrating again that at least some TME components can be modelled in vitro.
Heterogeneity in tumours and cell lines
Individual patient tumours are typically highly heterogeneous in terms of their genetic, epigenetic, transcriptional, cell state and other phenotypic features (Marusyk et al, 2012; Jamal‐Hanjani et al, 2015). These different levels of intra‐tumour heterogeneity can arise from genetic instability followed by subclonal evolution, as well as epigenetic plasticity, diverse microenvironmental factors, and heterotypic interactions with immune and stromal cells (Hinohara & Polyak, 2019; Vitale et al, 2021). Recent work suggests that distinct genetic and molecular subtypes can often co‐exist within the same tumour (Patel et al, 2014; Roerink et al, 2018; preprint: Gavish et al, 2021; Raghavan et al, 2021). Such intra‐tumour heterogeneity plays a role in governing cancer progression and metastasis, as well as therapeutic response and resistance (Roider et al, 2020; Hong et al, 2019; Kim et al, 2018).
CCLs are typically believed to lack much of the representative heterogeneity of tumour cell populations due to the aforementioned in vitro culture conditions, lack of TME complexity and strong selective pressures induced by in vitro culturing that are thought to limit subclonal diversity. However, recent studies have casted doubt on the notion that CCLs models are made of homogenous, stable and clonal cell populations (Fig 2C).
Genetic heterogeneity in CCL models has been observed by different groups. For example, Ben‐David et al (2018) found that even single‐cell clones rapidly produce heterogeneous populations due to genetic instability. Minussi et al (2021) used single‐cell DNA‐Seq to characterise subclonal diversity in triple negative breast cancer and found that CCLs showed similar levels of subclonal diversity as tumours, and that this re‐emerged rapidly after single‐cell cloning. Similar subclonal dynamics have been observed to drive drug resistance in CCLs (Bhang et al, 2015). Single‐cell studies suggest that apart from genetic heterogeneity, CCL populations may additionally exhibit transcriptional heterogeneity, but not to the same extent as tumours. For example, recent pan‐cancer efforts aiming at characterising recurrent patterns of transcriptional heterogeneity in CCL models and tumours found that many of the transcriptional programmes driving intra‐tumour heterogeneity in patients were also observed in CCLs (Kinker et al, 2020; preprint: Gavish et al, 2021). Tumours exhibit significant heterogeneity also at the epigenetic level (Brocks et al, 2014). However, CCLs are largely underexplored at the epigenome level, and it remains to be determined how much of the transcriptional diversity observed in CCLs is rooted in their epigenetic heterogeneity.
Despite their complex and dynamic nature, CCLs unavoidably lack much of the tumour spatial organisation, cellular architecture and microenvironmental factors, and further understanding these similarities and differences in CCLs and tumours remains a key challenge. Homogenous in vitro models might be desirable for experimental studies of defined cancer types and states, as they allow pinpointing specific intrinsic molecular features. On the contrary, populations lacking representative sources of heterogeneity would fail to capture key aspects of patient tumours biology, dynamics and treatment response.
Differences in genomic and cell state
Comparisons of tumours and CCLs, at bulk or single‐cell level, have indicated discrepancies with respect to somatic mutations and copy number alteration (CNA) frequencies, transcriptional and epigenetic states, (Fig 2D). Bulk gene expression profiling has identified pathway‐specific differences (Sandberg & Ernberg, 2005; Ertel et al, 2006). Pathways upregulated in CCLs are generally involved in metabolic processes, including cell nucleotide metabolism and oxidative phosphorylation, whereas downregulated ones typically involve cell adhesion and communication. Based on SAGE (Serial Analysis of Gene Expression) technology (Stein et al, 2004), 62 genes selectively overexpressed in tumours were found to be enriched for immune response and complement pathways, reflecting the presence of stromal and immune components, as well as extracellular matrix proteins. On the contrary, protein synthesis pathways were found dominantly enriched among the 61 genes overexpressed in CCLs. In addition, 5′C‐phosphate‐G‐3′ (CpG) islands were found more hypermethylated in CCLs, with more than 57% of model‐specific hypermethylated loci not being found in primary tumours (Smiraglia et al, 2001).
When considering transcriptional components involved in multidrug resistance (MDR), CCLs were observed to exhibit upregulation of genes that would facilitate survival (Gillet et al, 2011). This implies that CCLs are selected during their establishment via the expression of genes that are connected to MDR most likely as a response to environmental adversity. In addition, CCLs have been reported to be more sensitive to cytotoxic drugs compared with solid tumours, possibly due to their faster proliferation rate and their lack of a TME, which has been found to reduce responsiveness to chemotherapeutics (Marin et al, 2008; Straussman et al, 2012).
At the genome level, genetic aberrations characterising primary tumour are generally preserved in tissue‐matching CCLs. However, CCLs also tend to acquire novel locus‐specific alterations, several of which are rarely or never observed in primary tumours (Greshock et al, 2007; Tsuji et al, 2010), and show a generally higher frequency of mutations (Domcke et al, 2013; Jiang et al, 2016).
Considering the intrinsic differences between CCLs and primary tumours, Iorio et al (2016) focussed on multi‐omic cancer functional events (CFEs), that is molecular features derived by processing more than 11,000 primary tumour samples across 29 tissues with state‐of‐the‐art software aiming at identifying cancer driver alterations (Gonzalez‐Perez et al, 2013; van Dyk et al, 2013). The CFEs encompassed somatic mutations in cancer genes (CGs), recurrently aberrant copy number segments (RACs) involving at least a gene and affected in at least 2.5% of subjects, and hypermethylated informative CpG sites (iCpGs) in gene promoters with consistent hypo‐/hyper‐methylation. The status of the identified CFEs was then observed in more than 1,000 CCLs. Interestingly, this revealed that all pan‐cancer RACs identified in patients occurred in at least one CCL, followed by 89% of iCpGs and 64% of CGs. However, the correlation between CFEs occurrences in CCLs and patient tumours was high on average but highly variable across cancer types.
Partial ancestry representation
Because available biobanks do not properly cover all ethnicities, CCLs are not representative of diverse ancestry (Fig 2A). This issue was clearly shown by Dutil et al (2019) using an interactive tool called ECLA (Estimated Cell Line Ancestry) that visualised ancestry of CCLs inferred from genome‐wide SNP array in the context of the 1,000 Genome project reference populations. Among more than 1,000 CCLs in CCLE and the COSMIC panels, European and East Asian account for 91.64% of the CCL ancestry. Moreover, 64 CLLs involve a discordant annotation between the genetically inferred ancestry and the self‐reported one.
This is a quite significant challenge considering that genetic variants associated with cancer risk could have a different effect across populations. For instance, variants detected in one population from genome‐wide association studies are not always recapitulated in a different ancestry or even display a different direction of association (Wang et al, 2018). One example is the rs2046210 variant at 6q25.1 in breast cancer, which is detected in asian and european women but not African‐American (Cai et al, 2011). A complete ancestry representation in in vitro models is essential for understanding how ethnicity differences impact cancer biology and to gain a comprehensive view of the underlying mechanisms.
Computational methods for comparing cell lines and primary tumours
Some of the limitations hampering a correct modelling of primary tumours by CCLs can be circumvented computationally, by in silico preprocessing the related data. Particularly, appropriately mapping representative CCLs onto specific tumour segments can be achieved in a genomically guided fashion most preferably considering reprofiling CCLs immediately before possible screens for genetic dependency/drug sensitivity. This way it is possible (1) to elucidate at least the biological mechanisms that are retained in CCLs and (2) to facilitate translating CCL‐derived findings (for example from genetic dependencies and drug responses) into potential treatments for the mapped patient cohorts. A correct CCL‐to‐tumour matching overcomes CCL “misidentification” issues, reduces the effects of different culture conditions and allows focussing on features that are relevant to primary tumours while putting less emphasis on CCL‐specific ones.
Interconnected objectives: integration, scoring and selection
Here, we review 22 studies that have leveraged multi‐omic data to compare the molecular characterisation of tumours and that of commercially available CCLs (Table 2 and Fig 3). To this end, we have classified these approaches based on three different but not mutually exclusive pursued objectives (Table 2 and Fig 4):
integration of cell lines and tumour data.
scoring , that is estimating a score to rank the quality of cell lines as tumour models.
assignment and selection of cell lines as representative models for defined tumour type/subtype, with a consequent identification of gaps, that is tumours lacking representative cell lines.
Table 2.
Methods/studies that map cell lines to tumours.
| Reference | Data Input | Multi‐omic integration | Application | Unsupervised/Supervised | Clustering/subtype | Method | ||
|---|---|---|---|---|---|---|---|---|
| CCL—Tumour integration | Scoring CCL | Selecting CCL | ||||||
| Warren et al (2021) (Celligner) | GE | – | Pan‐cancer | Unsupervised | Subtype | Contrastive PCA + Mutual Nearest Neighbour | Pearson corr. on aligned space | – |
| Assignment by k‐NN | ||||||||
| Peng et al (2021) (CancerCellNet) | GE | – | Pan‐cancer | Supervised | Subtype | – | Classification score | Multi‐class Random Forest |
| “Correct” class: classification score > thr in actual type | ||||||||
|
Sinha et al (2021) (TumorComparer) |
GE, CNA, Mut | Late | Pan‐cancer (independent) | Unsupervised | Subtype | – | Aggregated ranking of weighted Pearson's corr./Jaccard Index | – |
|
Zhang & Kschischo (2021) (MFmap) |
GE, CNA, Mut | Intermediate | Pan‐cancer (independent) | Supervised (subtype) | Subtype | ComBat (GE) | Cosine coefficient (latent space) | Neural network classifier on latent space |
| Concatenated VAE | ||||||||
| Fang et al (2021) | PE | – | Thyroid Carcinoma | Unsupervised | Subtype | – | Pearson's corr. | – |
|
Najgebauer et al (2020) (CELLector) |
CNA, Mut, HypMet | Early | Pan‐cancer (independent) | Unsupervised | Clustering | – | Signature length times fraction of samples in group | Eclat clustering |
| Map by decision tree | ||||||||
|
Salvadores et al (2020) (HyperTracker) |
GE, HypMet | Late | Pan‐cancer | Supervised | Subtype | ComBat | – | Binomial ridge regression |
| “Golden set” from matching data modalities | ||||||||
| Batchu et al (2020) | GE | – | Alveolar Rhabdomyosarcoma | Unsupervised | – | – | Spearman's corr. | – |
|
Yu et al (2019) (CompHealth) |
GE | – | Pan‐cancer (independent) | Unsupervised | Subtype | ComBat | Spearman's corr. | TCGA‐110‐CL panel: 5 highest score per type |
| Supervised (subtype) | ||||||||
| Liu, et al (2019a) | GE, CNA, Mut | – | Metastatic Breast Cancer | Unsupervised | Subtype | – | Spearman's corr. (GE and CNA) | – |
|
Ronen et al (2019) (Maui) |
GE, CNA, Mut | Intermediate | Colorectal cancer | Unsupervised | Clustering | Multimodal stacked VAE | Euclidean distance (latent space) | K‐means clustering (latent space) |
| at least 1 of 5 NN being tumour | ||||||||
| Zhao et al (2017) | GE, CNA, Mut | Late | Pan‐cancer (independent) | Unsupervised | Subtype | Distance weighted discrimination | Kendall Rank corr. (GE and CNA) | Similarity in at least 3 out of 4 modalities |
| Gene Ontology enrichment score | ||||||||
| Mutation presence | ||||||||
| Luebker et al (2017) | CNA, Mut | – | Melanoma | Unsupervised | – | – | Fraction of genome altered | – |
| Pearson's corr. (CN) | ||||||||
| Vincent & Postovit (2017) | GE | – | Melanoma | Unsupervised | Subtype | – | Pearson's corr. | – |
| Sinha et al (2017) | GE, CNA, Mut | – | Renal Cancer | Unsupervised | Clustering/Subtype | ComBat | – | Hierarchical clustering (Spearman corr., CN) |
| Supervised (subtype) | PAMR classifier (Spearman corr., GE) | |||||||
| Jiang et al (2016) | GE, CNA, Mut, PE | Late | Breast Cancer | Unsupervised | Clustering/Subtype | – | Sum Pearson corr. | Hierarchical clustering (PE, GE) |
| Sun & Liu (2015) | GE, CNA | Late | Breast Cancer | Unsupervised | Subtype | – | Aggregated ranking of Spearman's corr. | – |
| Vincent et al (2015) | GE | – | Breast Cancer | Unsupervised | Subtype | ‐ | Pearson's corr. (group specific) | ‐ |
| Chen et al (2015) | GE | – | Hepatocellar Carcinoma | Unsupervised | – | – | Spearman corr. | – |
| Sadanandam et al (2013) | GE | – | Colorectal cancer | Unsupervised | Clustering | Distance weighted discrimination | – | SAM and PAM for feature extraction |
| Consensus‐based NMF | ||||||||
| Domcke et al (2013) | GE, CNA, Mut | Late | Ovarian Cancer | Unsupervised | Subtype | ‐ | sum: CNA Pearson corr. and Mut presence/absence | GE for validation: hierarchical clustering |
| Virtanen et al (2002) | GE | – | Lung Cancer | Unsupervised | Clustering | Lowess normalisation | – | Hierarchical clustering |
| Comparison with known label | ||||||||
CCL, cancer cell line; CNA, copy number alterations; GE, gene expression; HypMetm DNA methylation; Mut, somatic mutations; PE, protein expression.
Figure 3. Number of studies classified based on the characteristic displayed on the x‐axis.
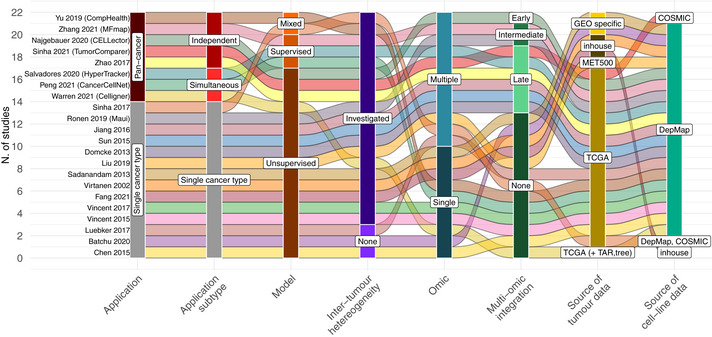
Each spline (alluvium) corresponds to a study in Table 2. “TAR” and “tree” abbreviations refer to TARGET and treehouse data set, respectively.
Figure 4. Aims of the major computational approaches proposed so far.
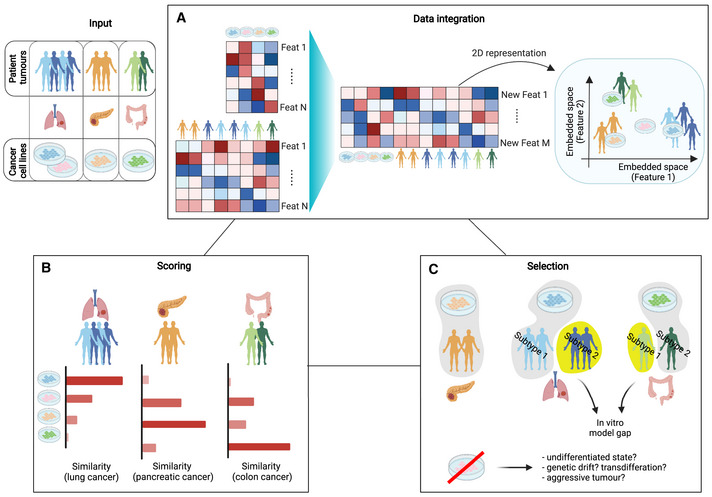
(A) Integration of cell lines and tumour in a common, comparable and visualisable feature space. (B) Scoring of cancer cell lines (CCLs) in terms of suitability in modelling a certain tumour population. (C) Selection of CCLs as proper model for tumour type/subtypes. Pursuing this objective can also highlight tumour populations lacking representative in vitro models and CCLs that diverge extensively from all the considered tumour populations. Created with BioRender.com.
While most of the studies (14 of 22) focus on a single cancer type, more recent ones are applied to a pan‐cancer context (8 of 22). Among those, only 3 describe methods analysing all cancer types with available data jointly, while the remaining ones are separately applied to individual cancer types (Pan‐cancer independent). Inter‐tumour heterogeneity within the same cancer type is accounted for in 19 of 22 studies either using prior known subtype labels (for example, consensus molecular subtypes) or determined in a data‐driven way via cluster analysis. In general, when comparing CCLs and tumours, the preferred approach is unsupervised (17 of 22), estimating global similarities between samples on a domain of informative features only. Instead, 3 of 22 methods adopt a supervised approach, building predictive models which are trained on primary tumours' data to capture a phenotype of interest (tumour type/subtype), then used to classify CCLs. Finally, 2 studies include both strategies, unsupervised for general CL‐tumour comparison and supervised for tumour subtype assignment.
Large‐scale publicly available databases, such the CCLE and those collected under the DepMap portals, are typically used as the source of CCL data (19 studies), plus an earlier application (Virtanen et al, 2002) used data related to CCLs that were subsequently collected in DepMap. Seventeen studies use TCGA as a source of primary tumours' data. Moreover, despite the developed methods not being strictly subordinate to the data type included (gene expression, somatic mutations, copy number alterations, DNA methylation and/or protein expression), we observed heterogenous choices of used omic(s). More importantly, only 9 of 22 studies address multi‐omic sources with disparate strategies for their integration. Here, we only focus on studies comparing tumours to CCLs, but we nevertheless note that 2 of the listed publications (Liu et al, 2019b; Peng et al, 2021) extend their methods to more complex models such as tumoroids and PDx, highlighting different representative performances and quality across model complexities.
Only 9 studies apply a data integration method that goes beyond a straightforward juxtaposition of the two data sources (CCLs and primary tumours) based on a common and/or most variable set of features and performing a data harmonisation in an integrated comparable space (Fig 4A and Table 2). This is usually implemented when handling gene expression data, via multi‐platform microarray integration methods (for example, distance weighted discrimination and lowess normalisation), or borrowing techniques for experimental batch correction, such as ComBat (Johnson et al, 2007) or mutual nearest neighbour (MNN) (Haghverdi et al, 2018). The benefits of data integration are clearly shown in (Salvadores et al, 2020; Warren et al, 2021), which display a 2D projection of combined CCLs' and primary tumours' data in an uncorrected and corrected version (i.e. contrastive PCA followed by MNN or quantile normalisation plus ComBat), with only the second case showing the two datasets properly mixed, while maintaining tissue type separations. A different approach is applied in (Ronen et al, 2019; Zhang & Kschischo, 2021), based on a variational autoencoder (VAE) that identifies, in an unsupervised manner, non‐linear latent factors from the initial feature space common to both CCLs and tumours.
The scoring objective is instead one of the most pursued across the examined methods (Fig 4B and Table 2). This goal is usually achieved through the use of a correlation (Spearman's or Pearson's) or similar metric (Kendall or Jaccard index, Euclidean distance or cosine coefficient) (Domcke et al, 2013; Chen et al, 2015; Sun & Liu, 2015; Vincent et al, 2015; Jiang et al, 2016; Luebker et al, 2017; Sinha et al, 2017, 2021; Vincent & Postovit, 2017; Liu et al, 2019a; Ronen et al, 2019; Batchu et al, 2020; Fang et al, 2021; Zhang & Kschischo, 2021), sometimes applied to a new “corrected” feature space (Warren et al, 2021). This similarity score is usually computed first sample‐wise, then for each CCL averaged across tumours from a given tumour type/subtype (usually matching that in the CCL annotation). The approaches that work this way build on unsupervised strategies that focus on a global similarity metric. Supervised methods, on the contrary, first build a classification model that can learn discriminative features between tumour types/subtypes and then examine and classify CCLs based on the status of these features. Although more appropriate for the selection tasks, classification “scores” can also be used as a quality estimation and hence to rank CCLs based on their representative quality (Peng et al, 2021). Regardless of whether a certain classification is correct, studies including a scoring objective can indicate the most fitting CCL for every tumour‐type/subtype under investigation.
With respect to the selection objective (Fig 4C and Table 2), not all methods proposed so far clearly pinpoint a set of CCLs as representative models for the tumour type under investigation. Although selection could be a consequence of scoring via the application of a filtering threshold on the estimated scores (Yu et al, 2019), this is usually not the case from correlation‐based studies. Instead, supervised methods more naturally assign a CCL to a tumour of interest and a representative set of CCLs is then obtained as those retaining their tissue identity following the classification (Salvadores et al, 2020; Peng et al, 2021).
One of unsupervised methods, CELLector (Najgebauer et al, 2020), initially clusters primary tumours in an unsupervised way based on the most co‐occurring combinations of genomic features (signatures) and subsequently assigns CCLs to a patient segment conditional to the presence of the underlying signature, without relying on a similarity score. Although a score is built as a product of the percentage of the tumour samples covered by a patient segment and the granularity of the underlying signature (in terms of number of accounted features), this is more representative of the patient subgroup “quality” and informativeness rather than the CCL ability to represent that patient cohort.
Generally, pursuing the selection objective helps also identifying tumour types and subtypes lacking adequate model representation, providing guidelines for new in vitro model development. Moreover, it boosts pharmacogenomic associations whose significance is diluted when accounting for low‐quality models (Najgebauer et al, 2020; Salvadores et al, 2020; Peng et al, 2021; Zhang & Kschischo, 2021). For instance, (Salvadores et al, 2020) identified drug sensitivity markers across cancer types using only “golden sets” of CCLs strongly resembling their cancer type of origin based on transcriptomic and epigenomic profiles. They found a higher number of pharmacogenomic associations across tumour types compared to using data from all available CCLs, including a previously unreported association between CDKN2A losses and camptothecin sensitivity in glioblastoma. On the contrary, removing non‐representative/low‐quality CCLs from pharmacogenomic associations studies filters out likely not relevant statistical interactions between drug responses and genomic features. For instance, in colorectal CCLs, BRAF mutations are no longer associated with dabrafenib responsiveness, consistent with what was observed in tumour patients. Najgebauer et al, 2020 used a different approach, where CCLs were grouped according to the genomic signatures underlying the patient segments they were mapped onto, and assessed for differential drug response across the resulting stratification. This yielded 88 unique drugs whose differential response is more significantly associated with a signature of genomic alterations than it is to individual genomic events. As an example of precision medicine application, this approach also shows that refining the subpopulation of KRAS mutated lung adenocarcinoma CCLs based on 2 complementary signatures (TP53 mutant and ARL17A promoter hypermethylation and the absence of those plus the absence of GSTT1 promoter hypermethylation, respectively) increases the differential drug response significance for the MEK1/2 inhibitor selumetinib and the BRAF inhibitor dabrafanib. Finally, scoring/selection objectives can identify cell state discrepancies between tumours and CCLs, as well as spot mis‐identified CCL clearly exhibiting features of a different tumour type, allowing for their reclassification in a pan‐cancer context.
Pan‐cancer approaches identify discrepancies in cell lines states
Among the 22 studies considered here, only 3 develop a pan‐cancer approach that simultaneously considers all CCLs and tumour types with available data (Salvadores et al, 2020; Peng et al, 2021; Warren et al, 2021), all focussing on transcriptional states and data. HyperTracker (Salvadores et al, 2020) and CancerCellNet (Peng et al, 2021) adopt a supervised approach to build a cancer type classifier using primary tumour data for training based on a one‐versus‐rest binomial ridge regression and a multiclass random forest, respectively. Cancer type labels are then predicted by the trained classifier for the CCLs and compared with tissue/cancer‐type labels they were originally annotated with. This allows to partition the analysed CCLs into (1) correctly classified (“golden/silver” set or “correct” class), (2) high confidently predicted to be derived from a cancer type different from the annotated one (“suspected” set or “other” class) and (3) ambiguously assigned to more than one cancer type, with low certainty or no concordance among multiple data types (“undetermined” set or “none/mixed” class). On the contrary, Celligner (Warren et al, 2021) uses an unsupervised approach, creating a “corrected” gene expression space through the simultaneous integration of CCLs and primary tumours across cancer types, hence allowing to detect distinct subpopulations and cross‐cancer‐types affinities. For each CCL, a comparison between its 25‐nearest tumour samples labels in the new space and the original annotation enables identifying differences in CCLs states. Only portions of analysed CCLs retain the original label state as correctly predicted across the considered studies (60% in (Salvadores et al, 2020; Warren et al, 2021)) and with variable results across cancer types. For instance, in (Peng et al, 2021), a CancerCellNet application shows that in only 6 of 20 cancer types more than 50% of CCLs assignment match their original label. Cancer types with poorly aligned CCLs are those that originated from pancreas, thyroid, oesophagus and central nervous system (CNS) tissues, consistently across studies.
Historically, glioblastoma (GBM) cell models exhibit a distinct transcriptional state than primary tumours and tend to lose their ability to differentiate when grown in serum‐containing media (Lee et al, 2006; Ledur et al, 2017). Indeed, CancerCellNet assigns the majority of GBM CCLs to the sarcoma type, similarly to Celligner that places 82% of CNS CCLs as part of an undifferentiated/mesenchymal group. In addition, when training the classifier on single‐cell RNA‐seq data, CancerCellNet (Peng et al, 2021) classifies 25 of 31 GBM lines as GBM neoplastic cells with 10 lines ambiguously being assigned also to the cancer‐tumour fibroblast class, possibly due to a derivation from a mesenchymal subtypes. Of note, low‐grade glioma (LGG) is consistently underrepresented across studies in CCLs collections even when considering co‐occurence of genomic cancer‐functional events, with 95% of tumour patients lacking a representative in vitro model (Najgebauer et al, 2020). Moreover, 91% of thyroid CCLs are also part of the transcriptionally based undifferentiated group (Warren et al, 2021), consistent with previous findings (Pilli et al, 2009).
Although an undifferentiated state is not a specific characteristic of pancreatic and oesophageal CCLs, the majority of CCLs annotated as being derived from these tissues are not predicted as adequate tumour models and further reasons related to this, for example propensity to metaplastic events, should be investigated (Wang & Souza, 2011). In general, a pan‐cancer approach, especially based on transcriptional levels, allows the identification of a common undifferentiated state, possibly representative of known tumour subtype (e.g. dedifferentiated melanoma), due to artefacts from 2D culture or indicative of a stem‐like state or an aggressive tumour cell state which is not detectable from bulk tumour data.
Another advantage of pan‐cancer approaches is that they can properly reclassify in vitro models when multiple sources are considered. For instance, in (Salvadores et al, 2020), HyperTracker identified a set of 43 CCLs with transcriptomic and epigenomic profiles significantly different from those of their originally annotated cancer types. A closer inspection re‐classified 22 of these CCLs, based on similarities spanning multiple data modalities, to a different cancer type. The authors provide two possible explanations for CCLs with discordant predicted/annotated tissue labels: misidentifications at the time of isolation or transdifferentiation. As label reassignment is supported in this study by multiple independently generated data omics, the authors conclude that the 22 lines were most likely misidentified. This is because transdifferentiation, that is CCL divergence during cell culture towards another cancer type, would be inclined to strongly affect the transcriptome/epigenome while having a reduced impact on the genetic component.
TumorComparer (Sinha et al, 2021) considers a weighted correlation metric to compare tumours and CCLs, across individual omics separately. This metric weights more cancer relevant features and results into similarity scores that are subsequently aggregated across omics layers. In this way, the authors identify 69 outlier CCLs (not sufficiently similar to any other tumour type) that need to be further investigated to determine whether they are from an undifferentiated state, or they have been mislabelled or possibly subjected to other molecular divergences. Finally, non‐pan‐cancer studies could also identify CCLs with discordant predicted/annotated labels which can be repurposed for studies investigating a cancer tissue different from that they were thought to model originally. For instance, Chen et al, 2015 detected 7 non‐liver CCLs whose molecular profile significantly correlates with that of hepatocellular carcinomas, and 2 or them (the pancreatic CCL TCC‐PAN2 and the stomach CCL FU97) even exhibit a higher level of similarity than actual liver CCLs. These two CCLs could be therefore used in hepatocellular carcinoma studies with a focus on transcriptomic data. In conclusion, pan‐cancer approaches proposed so far leverage the genomic differences between the cancer types and can identify CCLs with discordant states from their supposed tumour of origin.
Addressing inter‐tumour heterogeneity
Recent technological advances such as single‐cell sequencing are starting to shed light on intra‐tumour heterogeneity, that is genomic and physiological variations within a tumour gained by cell evolution under selective pressures and microenvironmentally driven epigenetic modulation (Jamal‐Hanjani et al, 2015; Hinohara & Polyak, 2019). Nevertheless, the prevailing understanding of patient tumour heterogeneity is still restricted to differences across patient genomic profiles, thus leading to disease subclassification. This inter‐tumour heterogeneity has been linked to differences in treatment response and used for the therapeutic management of different cancer types (Heiser et al, 2012; Ceccarelli et al, 2016; Liu et al, 2018). Consequently, an important task is to identify in vitro models most resembling a certain tumour molecular subtype to draw correct conclusions when examining drug efficacy and genetic dependencies, which might be specific to individual patient subcohorts. As an intrinsic characteristic of tumour cohorts, inter‐tumour heterogeneity is investigated in almost all studies that we have considered in this Review (Table 2 and Fig 3), either via unsupervised clustering of patient data (6 of 22) or leveraging their a priori defined molecular subclassification (15 of 22), sometimes at the CCL level (Domcke et al, 2013; Vincent et al, 2015; Fang et al, 2021). The strategies adopted to account for tumour heterogeneity are numerous and disparate. For instance, Celligner (Warren et al, 2021) examines the intra‐cluster variability in a corrected gene expression space that integrates CCLs and tumours, finding this reflective of known tumour subclassifications for breast, kidney, leukaemia and skin cancer. Conversely, supervised methods (Sinha et al, 2017; Yu et al, 2019; Salvadores et al, 2020; Peng et al, 2021; Zhang & Kschischo, 2021) use prior known subtype labels of patient tumours to build a classifier and then predict the subtype of CCLs, similarly to the strategy adopted in the pan‐cancer approaches described in the previous section. Unsupervised strategies based on a correlation metric ranked CCLs based on their average similarity to tumour subtypes (Sinha et al, 2021; Fang et al, 2021; Liu et al, 2019b; Vincent & Postovit, 2017). Molecular subtypes are still being established and refined to better capture disease progression, and prior known subtypes could be assigned through a human inspection, which is error prone. For that reason, it is still relevant to discover and integrate classification systems built on other genomic features. Indeed, two studies perform unsupervised patient clustering instead of relying on known partitions, Maui (Ronen et al, 2019) focussing on colorectal cancer and CELLector (Najgebauer et al, 2020) considering 16 cancer types independently. Maui (Ronen et al, 2019) applies a multimodal stacked variational autoencoder (VAE) to integrate CCLs and primary tumours in a latent space on which cluster analysis is then performed. Following this, the authors report that multi‐omic‐derived clustering is more powerful than transcriptionally derived consensus molecular subtyping (CMS (Guinney et al, 2015), widely used by the community) as it reveals distinct CNAs, mutation and methylation profiles not detected only based on the gene expression classification. In addition, Maui assigns CCLs to the closest group in a latent space that hence resembles these genomic changes. CELLector (Najgebauer et al, 2020) instead leverages clinical relevant genomic and epigenomic features (Iorio et al, 2016) and unveils inter‐tumour heterogeneity by partitioning patients samples based on the most frequently occurring sets of molecular signatures. CCLs are then assigned to patient subcohorts based on the collective presence or absence of these features.
The investigation of inter‐tumour heterogeneity also allows to detect patient subgroups lacking representative models, especially for strategies that include a selection objective. For example, Najgebauer et al, 2020 estimate that across cancer types and 14 TCGA cohorts (4,153 samples), 11.7% of patients belong to segments with no representative in vitro models in the CELLector search spaces built using CNAs and mutations in high confidence genes. This percentage varies across cancer types. In particular, LGG and prostate adenocarcinoma (PRAD) are the most underrepresented cancer types with 95% and 62% of patients without matching CCL models. Even for widely studied cancers such as LUAD, the large cohort of CCLs fail to represent 3% of patients characterised by mutation in KRAS and ATM and the absence of mutations in TP53 and STK1 genes. For annotated subtypes, Sinha et al, 2017 report that no kidney‐derived CCLs cluster with chromophobe renal cell carcinoma (RCC) (a more indolent and less prevalent subtype than other RCCs) based on CNAs. Accordingly, based on transcriptional data, the cluster of chromophobe RCC tumour samples does not incorporate any CCLs in the Celligner‐corrected space (Warren et al, 2021). Celligner also reveals that there is a underrepresentation of transitory melanoma subtype due to the fact that CCL derive from metastatic tumours rather than primary tumours. In a subtype classification implemented across 15 cancer types by HyperTracker (Salvadores et al, 2020), half of the tumour subtypes are not represented by any CCLs in kidney, bladder and brain cancer. In particular, 78% of GBM and LGG CCLs are assigned to a mesenchymal‐like type in agreement with other studies (Peng et al, 2021; Warren et al, 2021). To understand whether CancerCellNet classification was not successful when considering multiple cancer types (i.e. assigned to a “Mixed” or “None” prediction) due to the presence of a strong diverging subtype for a certain cancer type, Peng et al, 2021 performed a subtype classification for 11 cancer types, accounting also from subtypes defined from histology or molecular profiles. Interestingly, 25 CCLs (13% of the analysed cohort) without a successful classification in the general framework are in this case reliably classified as a specific subtype that hence exhibit features not shared across cancers from the same tissue. The CancerCellNet subtype classification also highlights the absence of representative CCLs for basal and secretory LUSC, terminal respiratory unit LUAD and indicates only one representative CCL for endometrioid carcinoma. These results are indicative of a selection bias towards deriving CCLs from aggressive tumour types. In a breast cancer analysis (Sun & Liu, 2015), a subset of CCLs shows low similarity to any of the breast tumour subgroups, most likely because they are derived from metastasis. Sinha et al, 2017 reported that kidney tumours clustering with kidney‐derived CCLs are representative of a more aggressive state, namely clear cell RCC. Vincent et al, 2015 show that breast CCLs of the more invasive basal subtype are transcriptionally more similar to their respective tumours than luminal CCLs.
Using a supervised approach based on nearest template prediction, Yu et al, 2019 built a gene expression‐based predictive model trained on primary tumour data and then inferred cancer subtype status in CCLs from nine cancer types. All subtypes had a predicted representative CCL; however, the proportions of representative CCLs across subtypes significantly differs in breast invasive carcinoma (BRCA), LUAD and skin cutaneous melanoma (SKCM). In particular, the predominant predicted classes for the CCLs are basal for BRCA, proximal inflammatory/proliferative for LUAD, keratin/mitf_low for SKCM, all corresponding to poor prognosis groups with medium‐to‐low survival rates. Finally, the subtype classification from HyperTracker presented in (Salvadores et al, 2020) finds a single predominant subtype predicted in CCL panels for liver, skin and thyroid cancers.
In conclusion, the selection bias in establishing in vitro models from more invasive cancer subtypes appears clearly from the inter‐tumour heterogeneity investigations of the reviewed methods, also highlighting many cases of cancer subtypes lacking representative in vitro models.
Unveiling biases in current cell line usage
In 9 of the studies discussed in this Review, the authors determine the number of times individual CCLs are mentioned across manuscripts published in peer‐reviewed journals (Sinha et al, 2021; Liu et al, 2019b; Yu et al, 2019; Zhao et al, 2017; Sinha et al, 2017; Jiang et al, 2016; Vincent et al, 2015; Chen et al, 2015; Domcke et al, 2013). This reveals that the most frequently used CCLs are not usually those more genomically similar to tumours. The reasons for this usage bias could lie on the ease in obtaining specific CCLs, their growing efficiency or a mere literature miscommunication.
Domcke et al, 2013 were the first to highlight this controversy for ovarian cancer. Focussing on high‐grade serous ovarian cancer (HGSOC), the most prevalent subtype, they found that the most used CCLs, SK‐OV‐3 and A2780 accounting for 60% of the total HGSOC CCL citations in the analysed literature (3,464 studies), greatly diverge from patient tumours when comparing CNAs profiles and the absence/presence of subtype specific mutations (e.g. TP53 mutations). The authors also highlight 12 CCLs more genomically similar to primary tumours which are generally less considered, with at the time only 1% of PubMed citations, and whose selection should be prioritised when establishing a new in vitro study. These results are also confirmed in a later study (Zhao et al, 2017) that additionally includes gene expression and functional similarity across gene ontology terms, and ranked SK‐OV‐3 and A2780 poorly in representing ovarian primary tumour. Screening eight cancer types, Zhao et al (2017) find further inconsistencies in intestine adenocarcinoma, with HT‐29 being the most cited CCL (∼ 18,000 PubMed citations) but ranking only at the 30th position based on molecular faithfulness to patient tumours. Similarly, the most used breast CCLs MCF‐7 and MDA‐MB‐231 (collectively accounting for > 53,000 PubMed citations) are also not high‐quality models of primary tumours, in contrast to the less cited T47D, SK‐BR‐3, MDA‐MB‐468 and BT483 CCLs. In line with this, Jiang et al, 2016 showed that the highly cited breast CCLs MCF‐7 and MDA‐MB‐231 only rank 17th and 21st as most similar to tumours based on a comparative correlation sum that combines gene expression, mutational profiles, CNAs and protein expression and instead assign to the less cited BT483 and T47D lines the highest similarity to tumour scores. MCF‐7 and MDA‐MB‐231 were also reported to be poorly representative of metastatic breast cancer by (Liu et al, 2019a), who for example reported MCF‐7 as reliably classifiable as of luminalB subtype. On the contrary, MDA‐MB‐231, which is used as a triple‐negative metastatic breast cancer model across many studies, could not be assigned to any subtype and ranked poorly in terms of correlation to basal‐like metastatic tumour samples. These findings were also independently confirmed using gene expression data (Vincent et al, 2015). This study who directly considered known CCL partitions in luminal/basal types and compared them with a similar classification of primary tumours, ranking MCF‐7 and T47D as 5th and 6th best models for luminal subtype and MDA‐MB‐231 in 17th position for basal subtype. Furthermore, Yu et al, 2019 highlight that the most used CCL for pancreatic adenocarcinoma (PAAD), MIA PaCa‐2 (∼ 1,000 PubMed citations), is the least transcriptionally similar to primary tumours, across a panel of 41 pancreatic CCLs, likely due to neuroendocrine differentiation. Discrepancies also arise for highly cited CCLs subtype annotations compared with their genomic features. For instance, the IGROV1 CCL is often quoted as HGSOC, but it is found more fitting as a model for endometrioid or ovarian clear cell carcinoma due to co‐occurrence of PIK3CA and PTEN mutations and expression‐based clustering in (Domcke et al, 2013).
In a study on renal cell carcinoma (RCC) subtypes based on CNAs profiles, Sinha et al (2017) find that ACHN is the third most cited CCL with a generic RCC annotation. However, it specifically clusters with the less prevalent papillary subtype, covering only 15% of RCC tumours. Overall, application of TumorComparer (Sinha et al, 2021) across 24 cancer types finds 69 CCLs detected as outliers based on an aggregated correlation from gene expression, CNAs and somatic mutations to their tumour of origin, of which 31 exceedingly 1,000 PubMed citations. Although, in this study, CCLs could be categorised as outliers even close to less frequent subtypes, these results are still indicative of biases in in vitro model selection. Of note, this phenomenon is not always occurring. For example, HepG2, a widely used hepatocellar carcinoma CCL, is reported as the highest quality model based on transcriptional correlation with patient tumours in (Chen et al, 2015).
It is important to stress that the performed citation searches are agnostic with respect to the usage of the cited models in the considered studies. Hence, false positives could be present due to publications using a CCL as a generic validation tool rather than for investigating cancer type/subtype‐specific mechanisms. As an example, (Gonçalves et al, 2021) use the HT‐29 CCL just as a tool model for testing the performances of a new CRISPR‐Cas9 library of guide RNAs. Nevertheless, these studies show that there is a clear bias from a literature search in specific CCLs usage and this highlights the importance of assessing the suitability of a CCL as a proper model of the tumour under investigation at the early stages of an experiment, without being drawn towards the easiest to retrieve or to grow in vitro models.
Challenges from tumour impurity
In constrast to in vitro models, tumours are surrounded by a TME composed of stromal, immune cells and extracellular matrix. Accounting for these factors while comparing tumours and CCLs is particularly challenging when using data derived from bulk experiments. Indeed, bulk experiments for profiling copy number alterations, gene expression and DNA methylation do not differentiate among malignant and non‐malignant cell types, rather giving a mixed view of all cells in the tumour sample. To understand the extent of malignant cell fraction in bulk data, computational methods estimating tumour purity have been developed (Carter et al, 2012; Yoshihara et al, 2013; Aran et al, 2015).
Although all the methods discussed in this Review analyse data from bulk experiments, tumour purity is investigated in 9 of 22 studies, mostly when considering gene expression (Vincent et al, 2015; Luebker et al, 2017; Vincent & Postovit, 2017; Liu et al, 2019b; Yu et al, 2019; Batchu et al, 2020; Salvadores et al, 2020; Peng et al, 2021; Warren et al, 2021). A frequent strategy is to exclude genes whose expression pattern across samples is found highly correlated with sample purity scores (or their surrogate), using an a priori decided filtering threshold (Vincent et al, 2015; Vincent & Postovit, 2017; Yu et al, 2019; Batchu et al, 2020). Yu et al (2019) show that this method alleviates a similarity bias: indeed, the elevated presence of stromal and immune cells decreases the similarity between tumours and CCLs. Nevertheless, the relationship between tumour sample purity and CCL‐tumour correlation became not significant if signatures of high‐impurity genes are removed from the comparison, and the expression patterns of the remaining ones are additionally corrected for purity scores. On the contrary, the contribution of the immune infiltrate component cannot be entirely removed, as it has been shown from differential analysis that the protein–protein interaction network of upregulated genes in primary tumour is still enriched for genes in the immune response pathway.
The same approach of removing genes whose expression pattern is highly correlated with impurity scores was applied in a study focussing on alveolar rhabdomyosarcoma (Batchu et al, 2020); however, this fails to alleviate differences between CCLs and primary tumour, as indicated by a principal component (PC) space inspection. Principal component analysis detects as major source of variability differences in TME in (Vincent et al, 2015; Vincent & Postovit, 2017). In particular, PCs computed on the juxtaposed CCL‐tumour gene expression data sets reveal a clear separation between the two data sources, with PC2 being correlated with lymphocyte density in melanoma (Vincent & Postovit, 2017) and PC1 with stromal scores in breast cancer (Vincent et al, 2015).
Furthermore, implementing filtering strategies aiming at limiting TME differences is not always performed via gene removal. In a melanoma study (Luebker et al, 2017), gene expression is used to estimate tumour cell fraction on a patient sample but then tumour samples with high tumour impurity are just removed from the analysis.
Without a prior knowledge of tumour purity but with a more sophisticated data integration step, Celligner (Warren et al, 2021) combines gene expression data for in vitro models and primary tumours in a multiple‐step procedure. First, a contrastive principal component analysis (cPCA) is applied to detect variability enriched in one data source with respect to the other and vice versa, and the first four cPCAs are removed. Then, mutual nearest neighbour (MNN) correction is applied using the CCL data as a reference. This ad hoc procedure highlights that tumour‐specific signatures associated with the first cPC are enriched in immune pathways and that the second cPC is highly correlated with tumour purity estimates. Despite accounting for the first four cPCs, TME effects still persist and are later captured by the MNN step.
Of note, two pan‐cancer‐supervised methods (Salvadores et al, 2020; Peng et al, 2021) do not focus on CCL‐tumour differences driven by normal cell contamination, but instead investigate whether tumour impurity interferes with model prediction accuracies. In particular, HyperTracker (Salvadores et al, 2020) integrates CCL and tumours via quantile normalisation plus a ComBat application (Leek et al, 2012), and CancerCellNet (Peng et al, 2021) converts gene expression matrices in binary gene‐pair formats, assigning 1 if the first gene in the pair has higher expression than the second gene within a sample. In both cases, the results show that purity does not affect the model estimates: HyperTracker AUPRC values are very similar when training on low or high purity TCGA samples, and CancerCellNet mean scores have only a marginal correlation with mean sample purity (correlation = 0.14).
Finally, single‐cell technologies provide a unique opportunity to clear up tumour infiltrating cells, allowing the comparison between CCLs and pure populations of malignant cells from a patient tumour (Vincent & Postovit, 2017; Peng et al, 2021). For instance, Vincent & Postovit (2017) show an improved correlation among CCLs and malignant cells from primary tumours in melanoma compared to accounting for all cell types (0.83 and 0.67 respectively).
In conclusion, while TME effects cannot be entirely removed from bulk experiments, a proper integration strategy can alleviate the immune and stromal related differences leading to more reliable cell‐lines versus tumours comparisons.
Feature selection strategies
The quality of CCLs also depends on features and biological states considered as relevant when they are compared with primary tumours. Most of the studies reviewed here focus more on comprehensive comparisons, aiming at assessing CCLs resemblance to tumours across the largest possible number of available features. In this respect, Celligner (Warren et al, 2021) uses the top 1,000 genes with the highest inter‐cluster variance within each data type. After initially addressing tumour‐CCL variability via cPCA, this tool analyses the remaining highest sources of variation that could discriminate against cancer types. In contrast, other pan‐cancer methods (Yu et al, 2019; Salvadores et al, 2020; Peng et al, 2021) focus on most variable features across cancer types when analysing tumour data. For example, CancerCellNet (Peng et al, 2021) selects genes coming up as highly differentially expressed when contrasting a cancer type versus all other samples. Similarly, HyperTracker (Salvadores et al, 2020) and CompHealth (Yu et al, 2019) select the 5,000 genes with the most variable expression pattern across all tumour samples. This type of filtering prioritises features that are discriminatory across cancer types from the perspective of tumour samples only, and subsequently leverages the expression of these genes observed in CCLs, to predict CCLs' cancer types or to compute a similarity‐to‐tumours score.
A distinctively different approach is TumorComparer (Sinha et al, 2021), which associates a weight to each multi‐omic feature while computing correlation scores between CCLs and primary tumours. An initial feature selection in this method is based on gene expression, CNAs and somatic mutations and outputs the 2,000 most variable features across all tumour types. Subsequently, TumorComparer assigns a weight to each feature in a 0–1 range based on their frequency of observation across primary tumours. Despite being a very useful framework due to the possibility of customising the feature weights (based on novel observational tumour data), assigning a bigger relevance to recurrent features in tumours might reward CCLs that are similar to very common cancer subtypes, possibly missing CCL that are good models of less recurrent ones. Still emphasising relevant features observed only in tumours, CELLector (Najgebauer et al, 2020) focuses on cancer functional events (CFEs) comprising recurrent mutated cancer genes, focal amplifications or deletions, and methylated gene promoters identified in patient tumours (Iorio et al, 2016). In cancer‐specific studies (Vincent et al, 2015; Jiang et al, 2016; Vincent & Postovit, 2017; Ronen et al, 2019; Batchu et al, 2020), features are instead filtered based on their variability observed jointly in the considered CCL and tumour data sets, weighting more features that are discriminative between in vitro models and patient tumours. Finally, in studies that include somatic mutations, the retained features are known (non‐synonymous) functional mutations present in both CCLs and tumours (Sinha et al, 2021; Najgebauer et al, 2020; Ronen et al, 2019; Zhao et al, 2017; Jiang et al, 2016) or a subset of the most relevant ones for a certain cancer type (Domcke et al, 2013).
In conclusion, as the aim of all the studies is to investigate the resemblance of CCLs to tumours, all the considered methods built on a wide range of features rather than reduced selections of oncogenic genes, but adopt disparate strategies to define a feature as informative.
Multi‐omic integration and discordant cell line selection
Among the reviewed studies, 5 data types are considered for matching CCLs to primary tumours: gene expression (GE), somatic mutations (Mut), copy number alterations (CNAs), DNA methylation (HypMet) and protein expression (PE) (Table 2). In particular, we observe a tendency to include gene expression, either to investigate CCL‐tumour resemblance or as an additional means of validation, with 19 of 22 studies using gene expression from microarrays and bulk or single‐cell RNA‐seq. Only 12 studies consider more than one data type, among which 9 apply a multi‐omic integration method to combine multiple data modalities.
Specifically, CELLector (Najgebauer et al, 2020) combines multi‐omic cancer functional events (CFEs) encoded as binary matrices for CNA, Mut and HypMet. This allows accounting for most of the analysed patients when assembling the CELLector signatures (recurrent combination of CFEs) and hence led to large percentages of patient samples that are represented by at least one CCLs. That said, late integration is the most frequently adopted strategy, with CCLs/tumour mappings performed separately across individual omics and then combined at a later stage. For instance, studies based on an unsupervised method aggregate the rankings of CCL‐tumours similarities obtained from each omic type (Sun & Liu, 2015; Zhao et al, 2017; Sinha et al, 2021) then sum the omic‐specific correlations (Jiang et al, 2016), or just incorporate correlation scores together with selected mutation occurrences (Domcke et al, 2013). Supervised methods, such as HyperTracker (Salvadores et al, 2020) on the other hand, build a predictive model for each omics data type then combine the classification results. This strategy prevents capturing interactions between omics and a proper representation of underlying biological mechanisms. A more refined strategy, intermediate integration, is developed in Maui (Ronen et al, 2019) and MFmap (Zhang & Kschischo, 2021). Briefly, Maui considers GE, CNA, and Mut omics and builds a multimodal stacked variational autoencoder (VAE) to represent tumour samples and CCLs in a low dimension latent space, with the latent factors regarded as higher‐order genomic features. Similarly, MFmap transforms GE, CNA and Mut data into a low dimensional latent space via VAE that is then applied on the concatenated omics. This intermediate strategy enables a joint integration of all available data and projects them onto a common shared space, although less interpretable than the original molecular features. For example, the application of Maui to colorectal cancer (CRC) (Ronen et al, 2019) allowed the identification of a more refined subgrouping compared with the widely adopted CMS classification (Guinney et al, 2015), which associates with more pronounced differences in biological pathway activities and survival outcomes.
The use of multi‐omics also unveils discrepancies arising across omic‐specific CCL/tumour matching cases, emphasising different biological mechanisms controlling genetic, transcriptional or epigenetic changes. For instance, Jiang et al (2016) compute Pearson's correlation scores between CCLs and patient tumours in breast cancer across four different data modalities and find very different ranges, with GE exhibiting the highest values, followed by CNA, Mut and PE. In accordance with these results, TumorComparer (Sinha et al, 2021) finds GE as the data modality with the widest range of CCL/tumour similarity across 24 cancer types, followed by CNAs and Muts. In addition, Sinha et al (2021) showed that only 18 of 594 CCLs can be consistently assigned to a cancer type with a normalised rank > 0.9 (meaning that the CCL is more similar to the tumours type of origin than 90% of the considered panel) for all the 3 data omics, while several CCLs had a rank > 0.9 only for a single omic data modality. This highlights for example that CCLs with high gene expression similarity might retain tissue‐specific expression but lack characteristic genomic features (mutations or CNAs). Likewise confirmed by Zhao et al (2017), CCL similarity rankings resulting from different data omics are discordant. In CompHealth (Yu et al, 2019), transcriptional correlation scores for ovarian CCLs are compared with the results by Domcke et al (2013) that are instead based on CNAs together with informative Muts presence. This highlights a significant consistency (Spearman's corr. = 0.59, P‐value = 5.84e‐05). However, the two studies (Domcke et al, 2013; Yu et al, 2019) disagree on the CCL that are most transcriptionally and genomically similar to tumours, with CAOV4 ranking 1st when considering transcriptomic data and only 9th when considering genomics.
By implementing a supervised approach, HyperTracker (Salvadores et al, 2020) compares GE and HypMet classification results. One hundred and thirty‐one of 614 CCLs (silver set) are discordantly assigned to a cancer type but with the outcome resulting from analysing only one omic concordant to the annotated label. In the same study, 67 CCLs (undetermined set) are discordantly classified across all omics. Although partially discordant, the authors show that CCLs in the silver set could still be informative: joining CCLs in the silver and golden sets (composed of CCLs with correctly classified across all omics) and considering the result in the context of drug response datasets significantly changes drug response selectivity, in cancer types such as PAAD, and increased the number of significant pharmacogenomic associations. Finally, despite Celligner (Warren et al, 2021) is based on GE only, the authors compared their k‐NN assignments built on corrected GE with the k‐NN resulting from computing Jaccard similarity scores on CFEs (Muts, CNAs, HypMet) and found a similar classification accuracy (60% and 61% respectively). Nevertheless, CCL rankings change substantially, indicating that the different omics are representative of different processes and states.
In summary, the discrepancies arising from data underlying different states highlights the necessity of a proper multi‐omic integration to comprehensively capture tumour mechanisms.
Extension to more complex in vitro and in vivo models
Complex models such as tumour organoids, patient‐derived xenografts (PDx) and genetically engineered mice (GEMM) have been compared with patient tumours in some of the studies we discuss in this Review. In particular, CancerCellNet (Peng et al, 2021) has also been applied to a collection of organoids, GEMM and PDx other than CCLs, to predict their cancer type and subtype in a supervised manner. Collectively, GEMM and organoids achieve the highest median correct classification scores in 4 of 5 tested cancer types, with organoids exhibiting the best classification rate, hence supposedly being the most appropriate tumour models. Indeed, compared with CCLs, GEMM are influenced by their native immune system and organoids benefit from cell–cell interactions arising from their 3D nature. Conversely, classification scores for PDxs demonstrated a bigger variability, with only few models yielding better scores than any of the organoids or GEMM. In the context of inter‐tumour heterogeneity, GEMM are the only models able to reflect a mixture of subtypes rather than modelling a single one, possibly due to a plasticity that is also influenced by the host environment. Although providing many insights, a proper comparison with different models derived from the same donor would be necessary to identify the most appropriate ones in representing patient tumours. Liu et al (2019a) also considered patient‐derived organoids for breast cancer and compared them to metastatic tumours, highlighting a better transcriptome resemblance compared with CCLs across breast cancer subtypes.
Beside highlighting the expected better performances of complex models in resembling tumours with respect to CCLs, these studies also demonstrate the adaptability of the CCLs versus tumours comparative analyses. While continuing to characterise increasingly larger collections of CCLs, the interest of the community is in parallel moving towards the development of large‐scale novel cancer models matched with original patient data. For instance, the recently established Human Cancer Models Initiative (HCMI) aims at creating a large collection of patient‐derived next‐generation cancer models that will include organoids and conditionally reprogrammed cells from diverse tumour subtypes and populations, with a particular attention to rare cancers that are at the moment widely unrepresented in in vitro models. Most importantly, collecting matched patients' samples and normal tissues will provide a unique opportunity to better understand the molecular changes introduced selectively in the in vitro model derivation phase.
Along the same lines, the PDXNet consortium has established more than 1,400 PDx samples with matched patient tumours, collecting sequencing data across multiple cancer types, and highlighting the suitability of this biobank for preclinical drug testing (Woo et al, 2021).
Looking at rare tumour models, a recent paediatric high‐grade glioma PDx collection comprising 21 models that included DNA methylation, mutation, and gene expression profiles was assembled from matched patient tumours (He et al, 2021). Analogous efforts are made to properly represent the interactive effect between in vitro models and TME, although so far at a smaller scale. For example, co‐culturing strategies for 3D patient‐derived organoids have been developed to include stromal and immune components (Neal et al, 2018; Tsai et al, 2018) and cell culture media have been modified to resemble human plasma (Cantor et al, 2017; Rossiter et al, 2021) or to include cancer‐associated fibroblasts (Cheteh et al, 2017).
Future directions
Advanced multi‐omic integration
Up to now, computational methods have generally focused on using particular feature types or employing simple strategies to combine features across modalities such as late or early integration.
However, different omics modalities encode for distinct but complementary information in cancer biology, with the genome regarded as the first affected layer when the tumour originates that constantly undergoes selective pressure, and the epigenome and transcriptome as more malleable and consequential states that are disrupted both by oncogene mutations and the environment. Because of the intricate interplay among the different biological components, and considering the large available collection of wide molecular characterizations, now more than ever we need integration methods able to learn a unified representation of cancer features. With a proper multi‐omic integration, the mapping of CCLs to tumours would focus on resembling as much as possible the underlying tumour‐specific mechanisms rather than the approximation of a single state.
Such considerations necessitate approaches that address the omic‐specific technical challenges (e.g. batch effects), while flexibly and efficiently integrating information across data modalities and potentially capturing variable information content across diverse data sets/cancer types. In this direction, Maui (Ronen et al, 2019) and MFmap (Zhang & Kschischo, 2021) are the only methods among the reviewed studies that adopt a joint representation of the multiple data modalities via a VAE approach. In general, similar challenges have emerged in the context of single‐cell methodologies and advanced methods have been proposed, focussing on the integration of multimodal simultaneously measured data (e.g. CITE‐seq or SHARE‐seq) such as MOFA+, totalVI and WNN (Argelaguet et al, 2020; Gayoso et al, 2021; Hao et al, 2021). For example, MOFA+ and totalVI, built on variational inference, are highly efficient in a large‐scale context and can incorporate multiple data omics measured across different batches while accounting for noise and technical biases of each omic modality. In the context of bridging CCLs and tumours, these methods could be adapted regarding different batches as tumour and in vitro model division.
Finally, none of the studies reviewed here employ a hierarchical integration strategy combining the different omic components from a regulatory point of view, despite the clear evidence of genetic drift leading to changes in gene expression and consequently drug screenings (Ben‐David et al, 2018). This sort of approach could reveal complex mechanisms that would otherwise remain undetected, building a regulatory network of a tumour and then mapping single CCLs in it leveraging conserved and shared mechanisms. Indeed, from a holistic point of view, Webber et al (2018) highlighted that inferred gene regulatory networks for tumours compared with those built on CCLs include preserved modules that are highly predictive of therapeutic response.
Transfer learning
With the goal of developing personalised treatments, translating in vitro measured key phenotypes to patient tumours rely on our ability to understand these relationships. In this context, transfer learning is a powerful tool to leverage drug sensitivity data in CCLs training machine learning models and transfer those predictions to patient tumours. Particularly prominent in the framework of image classification at pathologist level accuracy (Esteva et al, 2017; Coudray et al, 2018) and molecular subtyping from gene expression (Sevakula et al, 2019), transfer learning relies on the assumption that a predictive feature learned in a certain domain can be applied and adapted to a different but analogous one, even of limited sample size. Although promising, the main challenges are rooted in the inconsistencies between protocols and techniques used in different studies possibly leading to batch effects hard to be generalised, for example the way drug response is assessed. Specifically, transfer learning methods in model‐tumour context for drug sensitivity should consider fundamental differences between their genomic profiles, ultimately aiming at understanding which results can be robustly transferred, and how to optimally adjust model predictions.
In this context, pioneer works aim at learning a shared structure that can be leveraged for drug response prediction on patient tumour. For example, Geelher et al (Geeleher et al, 2014) used batch correction (ComBat) on gene and miRNA expression data between in vitro model and patient tumour to build a predictive model of drug sensitivity on CCLs and further validated the predicted drug response in primary tumours with respect to known clinical trial results. More recent methodologies such as PRECISE (Mourragui et al, 2019) and TRANSACT (Mourragui et al, 2021) first learn a shared feature subspace (linear and non‐linear, respectively) and then use it to build a predictive model for drug response. Sharifi‐Noghabi et al (2020) proposed an adversarial inductive transfer learning method that focuses on discrepancies in both gene expression (input) and drug response (output), adapting both aspects in the two different domains.
Across model domains, Ma et al (2021) developed a transfer learning method (few‐shot learning) based on a neural network trained to identify relevant input features (mutations and gene expression) for cell‐based screenings by optimising their transferability to patient‐derived tumour cells and patient‐derived xenografts, in a small sample size setting for the target domain. They also evaluated the ability of their transfer learning model to predict CRISPR‐Cas9 screening outcomes of CCLs considering as target domain another tissue with reduced sample size. A similar approach, from one large tissue to an underrepresented one, showed promising results in terms of drug synergy prediction (Kim et al, 2021). Finally, Villemin et al (2021) used a transfer learning strategy to identify biomarkers for basal A and B subtypes in breast CCLs in a supervised way, while iteratively adapting this model prediction to the tumour and integrating those that are classified with confidence.
Leveraging single‐cell data sets
The reviewed studies do not investigate intra‐tumour heterogeneity (with few exceptions (Vincent & Postovit, 2017; Peng et al, 2021)) neither do they address population heterogeneity in in vitro models. Nevertheless, large‐scale single‐cell genomics investigation could potentially help with resolving relationships between in vitro models and tumour cell populations more precisely and with greater resolution. First, comparing in vitro models and tumours at the single‐cell level could mitigate the limitations arising from tumour purity by comparing CCLs solely with malignant cells. Second, single‐cell studies could enable a more fine‐grained mapping of the relationship of the subclonal populations and cell states in heterogeneous tumour populations to representative in vitro models, effectively delineating each tumour as a mixture of in vitro models. For example, Gambardella et al (2022) created a single‐cell atlas of 32 breast CCLs and showed that the single‐cell transcriptional profile from a single patient could be mapped into the in vitro model atlas to assign a CCL model to each patient's cells. Strikingly, the tumours were found to be highly heterogeneous as none was mapped into a single CCL and they were overall represented by a mixture of models.
Pan‐cancer tumour single‐cell atlas studies could also reveal the predominant sources of intra‐tumour heterogeneity and their relationship to different in vitro models. Indeed, pan‐cancer single‐cell characterisation of CCLs (Kinker et al, 2020) has revealed that many of the recurrent drivers of transcriptional heterogeneity in tumours (preprint: Gavish et al, 2021) are also observed in CCLs, suggesting that individual CCLs might be evaluated as models for different components of intra‐tumour heterogeneity.
More generally, the development of large single‐cell atlases for both in vitro models and tumour patients offer a unique opportunity to create an integrated reference, enabling direct comparisons of tumours and models with single‐cell resolution.
Increasing interpretability
Despite the advantages in obtaining a similarity score representative for each cell type, the majority of the methods developed so far do not investigate the underlying factors that are driving these similarities (with the exception of CELLector (Najgebauer et al, 2020)). To adequately interpret the results arising from a matching procedure, we need interpretable methods, with the goal of understanding the shared mechanisms between tumours and a CCL and possibly their connection to treatment responses or genetic dependencies. Network and pathway approaches could be leveraged to enhance interpretability. These approaches use information from Reactome, Gene Ontology and protein–protein interaction databases to guide a more biologically relevant comparison of the underlying cell states.
Filling the gap in ancestry representation
As previously noted, currently available in vitro models are not representative of the human population and are mostly skewed towards European and Asian cohorts (Dutil et al, 2019). New in vitro models should be generated keeping in mind panel heterogeneity (e.g. HCMI), as ethnicity is reflected in differences in mutation frequency and transcriptional signatures that may cover specific mechanisms that are not present in the current panel. On the contrary, before a reasonable size can be reached to reliably build models for each ancestry and because of the heterogeneity of the concept itself, it would be important to also address this aspect in new computational approaches, for example via linear mixed models, a concept established in genome‐wide association studies (Loh et al, 2015).
Concluding remarks
The methods developed so far guided the evaluation of CCLs as proper tumour models, identified problematic cell lines showing putative misclassification as well as undifferentiation, directed towards CCLs representative of tumour subpopulation (known or estimated) and pointed out gaps in in vitro models. These studies provide a systematic framework to assess tumour patient populations lacking in vitro models and allow guiding gap‐filling efforts, consequently generating new hypotheses for under‐represented groups.
In all the reviewed studies, the assessment of the in vitro CCL as a proper model is built upon its closeness to the selected tumour population based on genomic features. Although resembling the tumour as close as possible can reveal both shared mechanisms and aid the translational medicine process, a feature‐specific approach could be preferred for targeted purposes. For example, in vitro models of microsatellite instability might be useful to identify dependencies, regardless of their similarity to primary tumours. In addition, the field still lacks methods capable of properly handling and integrating multi‐omic datasets. Matching CCLs to primary tumour from a more “global” perspective can only be addressed via methods that take into consideration the landscape of available multi‐omic data and the discrepancies observed across individual omic comparisons.
Looking forward, characterisation at the single‐cell level could circumvent some of the intrinsic limitations of CCLs repurposing them as more close models of specific tumour cellular states and intra‐tumour heterogeneity components. Despite all the limitations, we envision that the large data collection available for CCLs, readily available to the entire community via public repositories, will still be widely used in the future to model and understand cancer mechanisms and to aid early anti‐cancer drug discovery. On the contrary, large panels of newly developed models will help recapitulating and validating mechanisms that are impossible to observe in the actual available 2D models, among which genomic features characteristic solely of the model establishment, three‐dimensional assembly, the interplay with TME, drug assimilation and half‐life in blood streams.
Developing computational methods to align and compare existing or newly generated cancer models and tumour patients will continue to play a pivotal role for the effective use of these models in investigating the biology of cancer, as well as for contributing to the realisation of the personalised medicine paradigm.
Author contributions
Lucia Trastulla: Conceptualization; visualization; writing – original draft; writing – review and editing. Javad Noorbakhsh: Writing – original draft. Francisca Vazquez: Writing – original draft; writing – review and editing. James McFarland: Writing – original draft; writing – review and editing. Francesco Iorio: Conceptualization; supervision; funding acquisition; visualization; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
F.I. receives funding from Open Targets, a public‐private initiative involving academia and industry, and performs consultancy for the joint AstraZeneca‐CRUK functional genomics centre and for Mosaic Therapeutics. J.M. receives funding from Dependency Map Consortium. F.V. receives funding from Dependency Map Consortium and Novo ventures. J.N. is a shareholder of Kojin Therapeutics.
Acknowledgements
This work was supported by the Wellcome Trust grant number 206194/Z/17/Z, by the Open Targets Consortium (grant number OTAR2‐055) and the Dependency Map Consortium.
Mol Syst Biol. (2022) 18: e11017
References
- Aran D, Sirota M, Butte AJ (2015) Systematic pan‐cancer analysis of tumour purity. Nat Commun 6: 8971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argelaguet R, Arnol D, Bredikhin D, Deloro Y, Velten B, Marioni JC, Stegle O (2020) MOFA+: a statistical framework for comprehensive integration of multi‐modal single‐cell data. Genome Biol 21: 111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghban R, Roshangar L, Jahanban‐Esfahlan R, Seidi K, Ebrahimi‐Kalan A, Jaymand M, Kolahian S, Javaheri T, Zare P (2020) Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal 18: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A (2018) The Cellosaurus, a cell‐line knowledge resource. J Biomol Tech 29: 25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D et al (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S et al (2013) An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154: 1151–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchu S, Kellish AS, Hakim AA (2020) Assessing alveolar rhabdomyosarcoma cell lines as tumor models by comparison of mRNA expression profiles. Gene 760: 145025 [DOI] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M et al (2019) Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568: 511–516 [DOI] [PubMed] [Google Scholar]
- Ben‐David U, Beroukhim R, Golub TR (2019) Genomic evolution of cancer models: perils and opportunities. Nat Rev Cancer 19: 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R et al (2018) Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560: 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, Singh AP, Kao I, Rakiec D, Shaw P et al (2015) Studying clonal dynamics in response to cancer therapy using high‐complexity barcoding. Nat Med 21: 440–448 [DOI] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand‐Rosenberg S, Hedrick CC et al (2018) Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24: 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm JS, Garnett MJ, Adams DJ, Francies HE, Golub TR, Hahn WC, Iorio F, McFarland JM, Parts L, Vazquez F (2021) Cancer research needs a better map. Nature 589: 514–516 [DOI] [PubMed] [Google Scholar]
- Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, Oakes C, Zucknick M, Lipka DB, Weischenfeldt J et al (2014) Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep 8: 798–806 [DOI] [PubMed] [Google Scholar]
- Burdall SE, Hanby AM, Lansdown MRJ, Speirs V (2003) Breast cancer cell lines: friend or foe? Breast Cancer Res 5: 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush EC, Ray F, Alvarez MJ, Realubit R, Li H, Karan C, Califano A, Sims PA (2017) PLATE‐seq for genome‐wide regulatory network analysis of high‐throughput screens. Nat Commun 8: 105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Wen W, Qu S, Li G, Egan KM, Chen K, Deming SL, Shen H, Shen C‐Y, Gammon MD et al (2011) Replication and functional genomic analyses of the breast cancer susceptibility locus at 6q25.1 generalize its importance in women of Chinese, Japanese, and European ancestry. Cancer Res 71: 1344–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Cell Line Encyclopedia Consortium , Genomics of Drug Sensitivity in Cancer Consortium (2015) Pharmacogenomic agreement between two cancer cell line data sets. Nature 528: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM (2013) The cancer genome atlas Pan‐cancer analysis project. Nat Genet 45: 1113–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Abu‐Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr, Lewis CA, Sabatini DM (2017) Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169: 258–272.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capes‐Davis A, Theodosopoulos G, Atkin I, Drexler HG, Kohara A, RAF ML, Masters JR, Nakamura Y, Reid YA, Reddel RR et al (2010) Check your cultures! A list of cross‐contaminated or misidentified cell lines. Int J Cancer 127: 1–8 [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA et al (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30: 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164: 550–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Shibue T, JM MF, Gaeta B, Ghandi M, Dumont N, Gonzalez A, JS MP, Li T, Zhang Y et al (2019) WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568: 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Sirota M, Fan‐Minogue H, Hadley D, Butte AJ (2015) Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med Genomics 8: S5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheteh EH, Augsten M, Rundqvist H, Bianchi J, Sarne V, Egevad L, Bykov VJ, Östman A, Wiman KG (2017) Human cancer‐associated fibroblasts enhance glutathione levels and antagonize drug‐induced prostate cancer cell death. Cell Death Dis 8: e2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC et al (2011) Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage‐specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A 108: 12372–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, Humeidi R, Peck D, Wu X, Tang AA et al (2020) Discovering the anticancer potential of non‐oncology drugs by systematic viability profiling. Nat Cancer 1: 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyö D, Moreira AL, Razavian N, Tsirigos A (2018) Classification and mutation prediction from non‐small cell lung cancer histopathology images using deep learning. Nat Med 24: 1559–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill‐Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H et al (2019) Agreement between two large pan‐cancer CRISPR‐Cas9 gene dependency data sets. Nat Commun 10: 5817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Sinha R, Levine DA, Sander C, Schultz N (2013) Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 4: 2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury SC, Detre S, Leary A, Salter J, Reis‐Filho J, Barbashina V, Marchio C, Lopez‐Knowles E, Ghazoui Z, Habben K et al (2011) Changes in breast cancer biomarkers in the IGF1R/PI3K pathway in recurrent breast cancer after tamoxifen treatment. Endocr Relat Cancer 18: 565–577 [DOI] [PubMed] [Google Scholar]
- Dutil J, Chen Z, Monteiro AN, Teer JK, Eschrich SA (2019) An interactive resource to probe genetic diversity and estimated ancestry in cancer cell lines. Cancer Res 79: 1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwane L, Behan FM, Gonçalves E, Lightfoot H, Yang W, van der Meer D, Shepherd R, Pignatelli M, Iorio F, Garnett MJ (2021) Project score database: a resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res 49: D1365–D1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyk E, Reinders MJT, Wessels LFA (2013) A scale‐space method for detecting recurrent DNA copy number changes with analytical false discovery rate control. Nucleic Acids Res 41: e100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel A, Verghese A, Byers SW, Ochs M, Tozeren A (2006) Pathway‐specific differences between tumor cell lines and normal and tumor tissue cells. Mol Cancer 5: 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteva A, Kuprel B, Novoa RA, Ko J, Swetter SM, Blau HM, Thrun S (2017) Dermatologist‐level classification of skin cancer with deep neural networks. Nature 542: 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers B, Jastrzebski K, Heijmans JPM, Grernrum W, Beijersbergen RL, Bernards R (2016) CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol 34: 631–633 [DOI] [PubMed] [Google Scholar]
- Fang L, Liu Y‐J, Zhang Y‐W, Pan Z‐F, Zhong L‐K, Jiang L‐H, Wang J‐F, Zheng X‐W, Chen L‐Y, Huang P et al (2021) Comparison of proteomics profiles between xenografts derived from cell lines and primary tumors of thyroid carcinoma. J Cancer 12: 1978–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasterius E, Al‐Khalili Szigyarto C (2018) Analysis of public RNA‐sequencing data reveals biological consequences of genetic heterogeneity in cell line populations. Sci Rep 8: 11226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng F, Shen B, Mou X, Li Y, Li H (2021) Large‐scale pharmacogenomic studies and drug response prediction for personalized cancer medicine. J Genet Genomics 48: 540–551 [DOI] [PubMed] [Google Scholar]
- Francies HE, McDermott U, Garnett MJ (2020) Genomics‐guided pre‐clinical development of cancer therapies. Nat Cancer 1: 482–492 [DOI] [PubMed] [Google Scholar]
- Gambardella G, Viscido G, Tumaini B, Isacchi A, Bosotti R, di Bernardo D (2022) A single‐cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat Commun 13: 1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alonso L, Iorio F, Matchan A, Fonseca N, Jaaks P, Peat G, Pignatelli M, Falcone F, Benes CH, Dunham I et al (2018) Transcription factor activities enhance markers of drug sensitivity in cancer. Cancer Res 78: 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J et al (2012) Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483: 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J et al (2005) Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436: 117–122 [DOI] [PubMed] [Google Scholar]
- Gavish A, Tyler M, Simkin D, Kovarsky D, Nicolas Gonzalez Castro L, Halder D, Chanoch‐Myers R, Laffy J, Mints M, Greenwald AR et al (2021) The transcriptional hallmarks of intra‐tumor heterogeneity across a thousand tumors. bioRxiv 10.1101/2021.12.19.473368 [PREPRINT] [DOI] [Google Scholar]
- Gayoso A, Steier Z, Lopez R, Regier J, Nazor KL, Streets A, Yosef N (2021) Joint probabilistic modeling of single‐cell multi‐omic data with totalVI. Nat Methods 18: 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Cox NJ, Huang RS (2014) Clinical drug response can be predicted using baseline gene expression levels and in vitrodrug sensitivity in cell lines. Genome Biol 15: R47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Gamazon ER, Seoighe C, Cox NJ, Huang RS (2016) Consistency in large pharmacogenomic studies. Nature 540: E1–E2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané‐Valbuena J, Kryukov GV, Lo CC, ER MD 3rd, Barretina J, Gelfand ET, Bielski CM, Li H et al (2019) Next‐generation characterization of the Cancer Cell Line Encyclopedia. Nature 569: 503–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SJ, Travers J, Pshenichnaya I, Kogera FA, Barthorpe S, Mironenko T, Richardson L, Benes CH, Stratton MR, McDermott U et al (2015) Combinations of PARP inhibitors with temozolomide drive PARP1 trapping and apoptosis in Ewing's sarcoma. PLoS One 10: e0140988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet J‐P, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, Patel C, Orina JN, Eliseeva TA, Singal V et al (2011) Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti‐cancer drug resistance. Proc Natl Acad Sci U S A 108: 18708–18713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet J‐P, Varma S, Gottesman MM (2013) The clinical relevance of cancer cell lines. J Natl Cancer Inst 105: 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves E, Poulos RC, Cai Z, Barthorpe S, Manda S, Lucas N, Beck A, Bucio‐Noble D, Dausmann M, Hall C et al (2022) Pan‐cancer proteomic map of 949 human cell lines reveals principles of cancer vulnerabilities. bioRxiv 10.1101/2022.02.26.482008 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves E, Thomas M, Behan FM, Picco G, Pacini C, Allen F, Vinceti A, Sharma M, Jackson DA, Price S et al (2021) Minimal genome‐wide human CRISPR‐Cas9 library. Genome Biol 22: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Perez A, Perez‐Llamas C, Deu‐Pons J, Tamborero D, Schroeder MP, Jene‐Sanz A, Santos A, Lopez‐Bigas N (2013) IntOGen‐mutations identifies cancer drivers across tumor types. Nat Methods 10: 1081–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee RT, Goodman MT, Lynch CF, Platz CE, Havener LA, Howe HL (2010) The occurrence of rare cancers in U.S. adults, 1995–2004. Public Health Rep 125: 28–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, McNeil E, Moy C, Wegrzyn R, Auger K et al (2010) Molecular target class is predictive of in vitro response profile. Cancer Res 70: 3677–3686 [DOI] [PubMed] [Google Scholar]
- Greshock J, Nathanson K, Martin A‐M, Zhang L, Coukos G, Weber BL, Zaks TZ (2007) Cancer cell lines as genetic models of their parent histology: analyses based on array comparative genomic hybridization. Cancer Res 67: 3594–3600 [DOI] [PubMed] [Google Scholar]
- Grimm S (2004) The art and design of genetic screens: mammalian culture cells. Nat Rev Genet 5: 179–189 [DOI] [PubMed] [Google Scholar]
- Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P et al (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 21: 1350–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurumayum S, Jiang P, Hao X, Campos TL, Young ND, Korhonen PK, Gasser RB, Bork P, Zhao X‐M, He L‐J et al (2021) OGEE v3: Online GEne Essentiality database with increased coverage of organisms and human cell lines. Nucleic Acids Res 49: D998–D1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, Marioni JC (2018) Batch effects in single‐cell RNA‐sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36: 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe‐Kains B, El‐Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJWL, Quackenbush J (2013) Inconsistency in large pharmacogenomic studies. Nature 504: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen‐Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M et al (2021) Integrated analysis of multimodal single‐cell data. Cell 184: 3573–3587.e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet‐Turcotte A, Sun S et al (2015) High‐resolution CRISPR screens reveal fitness genes and genotype‐specific cancer liabilities. Cell 163: 1515–1526 [DOI] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez‐Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HC, Krishnamurthy Radhakrishna V et al (2016) Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22: 262–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, Neve RM, Martin S, Settleman J, Yauch RL et al (2016) Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 533: 333–337 [DOI] [PubMed] [Google Scholar]
- He C, Xu K, Zhu X, Dunphy PS, Gudenas B, Lin W, Twarog N, Hover LD, Kwon C‐H, Kasper LH et al (2021) Patient‐derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high‐grade glioma. Nat Commun 12: 4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiser LM, Sadanandam A, Kuo W‐L, Benz SC, Goldstein TC, Ng S, Gibb WJ, Wang NJ, Ziyad S, Tong F et al (2012) Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A 109: 2724–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinohara K, Polyak K (2019) Intratumoral heterogeneity: more than just mutations. Trends Cell Biol 29: 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SP, Chan TE, Lombardo Y, Corleone G, Rotmensz N, Bravaccini S, Rocca A, Pruneri G, KR ME, Coombes RC et al (2019) Single‐cell transcriptomics reveals multi‐step adaptations to endocrine therapy. Nat Commun 10: 3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkala A, Malhotra SV, Kummar S, Junttila MR (2021) Harnessing the predictive power of preclinical models for oncology drug development. Nat Rev Drug Discov 21: 99–114 [DOI] [PubMed] [Google Scholar]
- Horbach SPJM, Halffman W (2017) The ghosts of HeLa: how cell line misidentification contaminates the scientific literature. PLoS One 12: e0186281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes P, Marshall D, Reid Y, Parkes H, Gelber C (2007) The costs of using unauthenticated, over‐passaged cell lines: how much more data do we need? Biotechniques 43: 575, 577–8, 581–2 passim [DOI] [PubMed] [Google Scholar]
- International Cancer Genome Consortium , Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabé RR, Bhan MK, Calvo F, Eerola I et al (2010) International network of cancer genome projects. Nature 464: 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H et al (2016) A landscape of pharmacogenomic interactions in cancer. Cell 166: 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Young MJ, Li R, Jain S, Wernitznig A, Krill‐Burger JM, Lemke CT, Monducci D, Rodriguez DJ, Chang L et al (2021) Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway‐driven cancers. Nat Genet 53: 1664–1672 [DOI] [PubMed] [Google Scholar]
- Jaaks P, Coker EA, Vis DJ, Edwards O, Carpenter EF, Leto SM, Dwane L, Sassi F, Lightfoot H, Barthorpe S et al (2022) Effective drug combinations in breast, colon and pancreatic cancer cells. Nature 603: 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal‐Hanjani M, Quezada SA, Larkin J, Swanton C (2015) Translational implications of tumor heterogeneity. Clin Cancer Res 21: 1258–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Zhang S, Yazdanparast A, Li M, Pawar AV, Liu Y, Inavolu SM, Cheng L (2016) Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genomics 17: 525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8: 118–127 [DOI] [PubMed] [Google Scholar]
- Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, Crosetto N, Foukakis T, Navin NE (2018) Chemoresistance evolution in triple‐negative breast cancer delineated by single‐cell sequencing. Cell 173: 879–893.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Zheng S, Tang J, Jim Zheng W, Li Z, Jiang X (2021) Anticancer drug synergy prediction in understudied tissues using transfer learning. J Am Med Inform Assoc 28: 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, JM MF, Warren A, Rodman C, Roth JA, Bender SA et al (2020) Pan‐cancer single‐cell RNA‐seq identifies recurring programs of cellular heterogeneity. Nat Genet 52: 1208–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm F, Joyce JA (2015) Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol 25: 198–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J et al (2015) A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 33: 306–312 [DOI] [PubMed] [Google Scholar]
- Lamb J (2007) The connectivity map: a new tool for biomedical research. Nat Rev Cancer 7: 54–60 [DOI] [PubMed] [Google Scholar]
- Ledur PF, Onzi GR, Zong H, Lenz G (2017) Culture conditions defining glioblastoma cells behavior: WHAT is the impact for novel discoveries? Oncotarget 8: 69185–69197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W et al (2006) Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum‐cultured cell lines. Cancer Cell 9: 391–403 [DOI] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD (2012) The sva package for removing batch effects and other unwanted variation in high‐throughput experiments. Bioinformatics 28: 882–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, Souza A, Pierce K, Keskula P, Hernandez D et al (2019) The landscape of cancer cell line metabolism. Nat Med 25: 850–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Sheltzer JM (2020) Discovering and validating cancer genetic dependencies: approaches and pitfalls. Nat Rev Genet 21: 671–682 [DOI] [PubMed] [Google Scholar]
- Liu Y, Mi Y, Mueller T, Kreibich S, Williams EG, Van Drogen A, Borel C, Frank M, Germain P‐L, Bludau I et al (2019) Multi‐omic measurements of heterogeneity in HeLa cells across laboratories. Nat Biotechnol 37: 314–322 [DOI] [PubMed] [Google Scholar]
- Liu K, Newbury PA, Glicksberg BS, Zeng WZD, Paithankar S, Andrechek ER, Chen B (2019) Evaluating cell lines as models for metastatic breast cancer through integrative analysis of genomic data. Nat Commun 10: 2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez‐Vega F, Seoane JA, Farshidfar F, Bowlby R, Islam M et al (2018) Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 33: 721–735.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P‐R, Tucker G, Bulik‐Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B et al (2015) Efficient Bayesian mixed‐model analysis increases association power in large cohorts. Nat Genet 47: 284–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebker SA, Zhang W, Koepsell SA (2017) Comparing the genomes of cutaneous melanoma tumors to commercially available cell lines. Oncotarget 8: 114877–114893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Fong SH, Luo Y, Bakkenist CJ, Shen JP, Mourragui S, LFA W, Hafner M, Sharan R, Peng J et al (2021) Few‐shot learning creates predictive models of drug response that translate from high‐throughput screens to individual patients. Nat Cancer 2: 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin JJG, Castaño B, Martinez‐Becerra P, Rosales R, Monte MJ (2008) Chemotherapy in the treatment of primary liver tumours. Cancer Ther 6: 711–728 [Google Scholar]
- Marusyk A, Almendro V, Polyak K (2012) Intra‐tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12: 323–334 [DOI] [PubMed] [Google Scholar]
- Masters JR (2000) Human cancer cell lines: fact and fantasy. Nat Rev Mol Cell Biol 1: 233–236 [DOI] [PubMed] [Google Scholar]
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D et al (2007) Identification of genotype‐correlated sensitivity to selective kinase inhibitors by using high‐throughput tumor cell line profiling. Proc Natl Acad Sci U S A 104: 19936–19941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K et al (2017) Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large‐scale, deep RNAi screening. Cell 170: 577–592.e10 [DOI] [PubMed] [Google Scholar]
- McFarland JM, Paolella BR, Warren A, Geiger‐Schuller K, Shibue T, Rothberg M, Kuksenko O, Colgan WN, Jones A, Chambers E et al (2020) Multiplexed single‐cell transcriptional response profiling to define cancer vulnerabilities and therapeutic mechanism of action. Nat Commun 11: 4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer D, Barthorpe S, Yang W, Lightfoot H, Hall C, Gilbert J, Francies HE, Garnett MJ (2018) Cell model passports—a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res 47: D923–D929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menden MP, Wang D, Mason MJ, Szalai B, Bulusu KC, Guan Y, Yu T, Kang J, Jeon M, Wolfinger R et al (2019) Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat Commun 10: 2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S et al (2017) Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat Genet 49: 1779–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minussi DC, Nicholson MD, Ye H, Davis A, Wang K, Baker T, Tarabichi M, Sei E, Du H, Rabbani M et al (2021) Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature 592: 302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourragui S, Loog M, van de Wiel MA, Reinders MJT, Wessels LFA (2019) PRECISE: a domain adaptation approach to transfer predictors of drug response from pre‐clinical models to tumors. Bioinformatics 35: i510–i519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourragui SMC, Loog M, Vis DJ, Moore K, Manjon AG, van de Wiel MA, Reinders MJT, Wessels LFA (2021) Predicting patient response with models trained on cell lines and patient‐derived xenografts by nonlinear transfer learning. Proc Natl Acad Sci U S A 118: e2106682118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpindi JP, Yadav B, Östling P, Gautam P, Malani D, Murumägi A, Hirasawa A, Kangaspeska S, Wennerberg K, Kallioniemi O et al (2016) Consistency in drug response profiling. Nature 540: E5–E6 [DOI] [PubMed] [Google Scholar]
- Much M, Buza N, Hui P (2014) Tissue identity testing of cancer by short tandem repeat polymorphism: pitfalls of interpretation in the presence of microsatellite instability. Hum Pathol 45: 549–555 [DOI] [PubMed] [Google Scholar]
- Najgebauer H, Yang M, Francies HE, Pacini C, Stronach EA, Garnett MJ, Saez‐Rodriguez J, Iorio F (2020) CELLector: GENOMICS‐guided selection of cancer in vitro models. Cell Syst 10: 424–432.e6 [DOI] [PubMed] [Google Scholar]
- Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou S‐H, Salahudeen AA, Smith AR et al (2018) Organoid modeling of the tumor immune microenvironment. Cell 175: 1972–1988.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J et al (2015) The support of human genetic evidence for approved drug indications. Nat Genet 47: 856–860 [DOI] [PubMed] [Google Scholar]
- Nusinow DP, Szpyt J, Ghandi M, Rose CM, ER MD 3rd, Kalocsay M, Jané‐Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M et al (2020) Quantitative proteomics of the cancer cell line encyclopedia. Cell 180: 387–402.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olarerin‐George AO, Hogenesch JB (2015) Assessing the prevalence of mycoplasma contamination in cell culture via a survey of NCBI's RNA‐seq archive. Nucleic Acids Res 43: 2535–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacini C, Dempster JM, Boyle I, Gonçalves E, Najgebauer H, Karakoc E, van der Meer D, Barthorpe A, Lightfoot H, Jaaks P et al (2021) Integrated cross‐study datasets of genetic dependencies in cancer. Nat Commun 12: 1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL et al (2014) Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Gleyzer R, Tai W‐H, Kumar P, Bian Q, Isaacs B, da Rocha EL, Cai S, DiNapoli K, Huang FW et al (2021) Evaluating the transcriptional fidelity of cancer models. Genome Med 13: 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli T, Prasad KV, Jayarama S, Pacini F, Prabhakar BS (2009) Potential utility and limitations of thyroid cancer cell lines as models for studying thyroid cancer. Thyroid 19: 1333–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C et al (2018) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 18: 41–58 [DOI] [PubMed] [Google Scholar]
- Quevedo R, Smirnov P, Tkachuk D, Ho C, El‐Hachem N, Safikhani Z, Pugh TJ, Haibe‐Kains B (2020) Assessment of genetic drift in large pharmacogenomic studies. Cell Syst 11: 393–401.e2 [DOI] [PubMed] [Google Scholar]
- Raghavan S, Winter PS, Navia AW, Williams HL, DenAdel A, Lowder KE, Galvez‐Reyes J, Kalekar RL, Mulugeta N, Kapner KS et al (2021) Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184: 6119–6137.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin T, Capes‐Davis A, Bairoch A (2020) CLASTR: The Cellosaurus STR similarity search tool ‐ a precious help for cell line authentication. Int J Cancer 146: 1299–1306 [DOI] [PubMed] [Google Scholar]
- Roerink SF, Sasaki N, Lee‐Six H, Young MD, Alexandrov LB, Behjati S, Mitchell TJ, Grossmann S, Lightfoot H, Egan DA et al (2018) Intra‐tumour diversification in colorectal cancer at the single‐cell level. Nature 556: 457–462 [DOI] [PubMed] [Google Scholar]
- Roider T, Seufert J, Uvarovskii A, Frauhammer F, Bordas M, Abedpour N, Stolarczyk M, Mallm J‐P, Herbst SA, Bruch P‐M et al (2020) Dissecting intratumour heterogeneity of nodal B‐cell lymphomas at the transcriptional, genetic and drug‐response levels. Nat Cell Biol 22: 896–906 [DOI] [PubMed] [Google Scholar]
- Ronen J, Hayat S, Akalin A (2019) Evaluation of colorectal cancer subtypes and cell lines using deep learning. Life Sci Alliance 2: e201900517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiter NJ, Huggler KS, Adelmann CH, Keys HR, Soens RW, Sabatini DM, Cantor JR (2021) CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell Metab 33: 1248–1263.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, LCG O, Lannon WA, Grotzinger C, Del Rio M et al (2013) A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med 19: 619–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadores M, Fuster‐Tormo F, Supek F (2020) Matching cell lines with cancer type and subtype of origin via mutational, epigenomic, and transcriptomic patterns. Sci Adv 6: eaba1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Ernberg I (2005) The molecular portrait of in vitro growth by meta‐analysis of gene‐expression profiles. Genome Biol 6: R65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer WF, Syverton JT, Gey GO (1953) Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med 97: 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seashore‐Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J et al (2015) Harnessing connectivity in a large‐scale small‐molecule sensitivity dataset. Cancer Discov 5: 1210–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevakula RK, Singh V, Verma NK, Kumar C, Cui Y (2019) Transfer learning for molecular cancer classification using deep neural networks. IEEE/ACM Trans Comput Biol Bioinform 16: 2089–2100 [DOI] [PubMed] [Google Scholar]
- Sharifi‐Noghabi H, Peng S, Zolotareva O, Collins CC, Ester M (2020) AITL: Adversarial inductive transfer learning with input and output space adaptation for pharmacogenomics. Bioinformatics 36: i380–i388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifnia T, Hong AL, Painter CA, Boehm JS (2017) Emerging opportunities for target discovery in rare cancers. Cell Chem Biol 24: 1075–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Haber DA, Settleman J (2010) Cell line‐based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer 10: 241–253 [DOI] [PubMed] [Google Scholar]
- Shoemaker RH (2006) The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 6: 813–823 [DOI] [PubMed] [Google Scholar]
- Sinha R, Luna A, Schultz N, Sander C (2021) A pan‐cancer survey of cell line tumor similarity by feature‐weighted molecular profiles. Cell Reports Methods 1: 100039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Winer AG, Chevinsky M, Jakubowski C, Chen Y‐B, Dong Y, Tickoo SK, Reuter VE, Russo P, Coleman JA et al (2017) Analysis of renal cancer cell lines from two major resources enables genomics‐guided cell line selection. Nat Commun 8: 15165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiraglia DJ, Rush LJ, Frühwald MC, Dai Z, Held WA, Costello JF, Lang JC, Eng C, Li B, Wright FA et al (2001) Excessive CpG Island hypermethylation in cancer cell lines versus primary human malignancies. Hum Mol Genet 10: 1413–1419 [DOI] [PubMed] [Google Scholar]
- Srivatsan SR, JL MF‐F, Ramani V, Saunders L, Cao J, Packer J, Pliner HA, Jackson DL, Daza RM, Christiansen L et al (2020) Massively multiplex chemical transcriptomics at single‐cell resolution. Science 367: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathias V, Turner J, Koleti A, Vidovic D, Cooper D, Fazel‐Najafabadi M, Pilarczyk M, Terryn R, Chung C, Umeano A et al (2020) LINCS Data Portal 2.0: next generation access point for perturbation‐response signatures. Nucleic Acids Res 48: D431–D439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Staveren WCG, Weiss Solís DY, Hébrant A, Detours V, Dumont JE, Maenhaut C (2009) Human cancer cell lines: experimental models for cancer cells in situ? For cancer stem cells? Biochim Biophys Acta 1795: 92–103 [DOI] [PubMed] [Google Scholar]
- Stein WD, Litman T, Fojo T, Bates SE (2004) A serial analysis of gene expression (SAGE) database analysis of chemosensitivity: comparing solid tumors with cell lines and comparing solid tumors from different tissue origins. Cancer Res 64: 2805–2816 [DOI] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily‐Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT et al (2012) Tumour micro‐environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487: 500–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Liu Q (2015) Deciphering the correlation between breast tumor samples and cell lines by integrating copy number changes and gene expression profiles. Biomed Res Int 2015: 901303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E et al (2019) COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47: D941–D947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson NA, Ranzani M, van der Weyden L, Iyer V, Offord V, Droop A, Behan F, Gonçalves E, Speak A, Iorio F et al (2021) Combinatorial CRISPR screen identifies fitness effects of gene paralogues. Nat Commun 12: 1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, Dwinell MB, Hunt B, Evans DB, Gershan J et al (2018) Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 18: 335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill‐Burger JM et al (2017) Defining a Cancer Dependency Map. Cell 170: 564–576.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji K, Kawauchi S, Saito S, Furuya T, Ikemoto K, Nakao M, Yamamoto S, Oka M, Hirano T, Sasaki K (2010) Breast cancer cell lines carry cell line‐specific genomic alterations that are distinct from aberrations in breast cancer tissues: comparison of the CGH profiles between cancer cell lines and primary cancer tissues. BMC Cancer 10: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis K, Koike‐Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M et al (2016) A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep 17: 1193–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemin J‐P, Lorenzi C, Cabrillac M‐S, Oldfield A, Ritchie W, Luco RF (2021) A cell‐to‐patient machine learning transfer approach uncovers novel basal‐like breast cancer prognostic markers amongst alternative splice variants. BMC Biol 19: 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent KM, Findlay SD, Postovit LM (2015) Assessing breast cancer cell lines as tumour models by comparison of mRNA expression profiles. Breast Cancer Res 17: 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent KM, Postovit L‐M (2017) Investigating the utility of human melanoma cell lines as tumour models. Oncotarget 8: 10498–10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen C, Ishikawa Y, Honjoh D, Kimura M, Shimane M, Miyoshi T, Nomura H, Jones MH (2002) Integrated classification of lung tumors and cell lines by expression profiling. Proc Natl Acad Sci U S A 99: 12357–12362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, Shema E, Loi S, Galluzzi L (2021) Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 27: 212–224 [DOI] [PubMed] [Google Scholar]
- Vormoor B, Curtin NJ (2014) Poly(ADP‐ribose) polymerase inhibitors in Ewing sarcoma. Curr Opin Oncol 26: 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Qian F, Zheng Y, Ogundiran T, Ojengbede O, Zheng W, Blot W, Nathanson KL, Hennis A, Nemesure B et al (2018) Genetic variants demonstrating flip‐flop phenomenon and breast cancer risk prediction among women of African ancestry. Breast Cancer Res Treat 168: 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DH, Souza RF (2011) Biology of Barrett's esophagus and esophageal adenocarcinoma. Gastrointest Endosc Clin N Am 21: 25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren A, Chen Y, Jones A, Shibue T, Hahn WC, Boehm JS, Vazquez F, Tsherniak A, McFarland JM (2021) Global computational alignment of tumor and cell line transcriptional profiles. Nat Commun 12: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber JT, Kaushik S, Bandyopadhyay S (2018) Integration of tumor genomic data with cell lines using multi‐dimensional network modules improves cancer pharmacogenomics. Cell Syst 7: 526–536.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbein U, Plotnik O, Vershkov D, Benvenisty N (2017) Culture‐induced recurrent epigenetic aberrations in human pluripotent stem cells. PLoS Genet 13: e1006979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding JL, Bodmer WF (2014) Cancer cell lines for drug discovery and development. Cancer Res 74: 2377–2384 [DOI] [PubMed] [Google Scholar]
- Woo XY, Giordano J, Srivastava A, Zhao Z‐M, Lloyd MW, de Bruijn R, Suh Y‐S, Patidar R, Chen L, Scherer S et al (2021) Conservation of copy number profiles during engraftment and passaging of patient‐derived cancer xenografts. Nat Genet 53: 86–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR et al (2013) Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41: D955–D961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen C, Qin J, Liu J, Zheng C (2015) Genetic profiling reveals an alarming rate of cross‐contamination among human cell lines used in China. FASEB J 29: 4268–4272 [DOI] [PubMed] [Google Scholar]
- Ye C, Ho DJ, Neri M, Yang C, Kulkarni T, Randhawa R, Henault M, Mostacci N, Farmer P, Renner S et al (2018) DRUG‐seq for miniaturized high‐throughput transcriptome profiling in drug discovery. Nat Commun 9: 4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres‐Garcia W, Treviño V, Shen H, Laird PW, Levine DA et al (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4: 2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chen B, Aran D, Charalel J, Yau C, Wolf DM, van't Veer LJ, Butte AJ, Goldstein T, Sirota M (2019) Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat Commun 10: 3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kschischo M (2021) MFmap: a semi‐supervised generative model matching cell lines to tumours and cancer subtypes. PLoS One 16: e0261183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Liu Y, Wei Y, Yan Z, Zhang Q, Wu C, Chang Z, Xu Y (2017) Optimization of cell lines as tumour models by integrating multi‐omics data. Brief Bioinform 18: 515–529 [DOI] [PubMed] [Google Scholar]