

Abstract
The interpretation of ion mobility coupled to mass spectrometry (IM-MS) data to predict unknown structures is challenging and depends on accurate theoretical estimates of the molecular ion collision cross section (CCS) against a buffer gas in a low or atmospheric pressure drift chamber. The sensitivity and reliability of computational prediction of CCS values depend on accurately modeling the molecular state over accessible conformations. In this work, we developed an efficient CCS computational workflow using a machine learning model in conjunction with standard DFT methods and CCS calculations. Furthermore, we have performed Traveling Wave IM-MS (TWIMS) experiments to validate the extant experimental values and assess uncertainties in experimentally measured CCS values. The developed workflow yielded accurate structural predictions and provides unique insights into the likely preferred conformation analyzed using IM-MS experiments. The complete workflow makes the computation of CCS values tractable for a large number of conformationally flexible metabolites with complex molecular structures.
Graphical Abstract
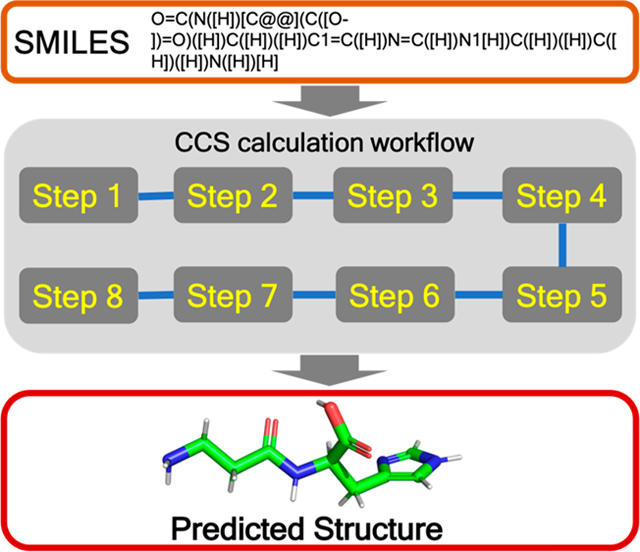
INTRODUCTION
The metabolome is the total collection of biologically active small molecules with molecular weights lower than about ~1.5 kDa.1,2 This includes endogenous molecules that are biosynthesized by metabolic networks in "primary metabolism", specialized "secondary metabolite" signaling, or defense molecules, molecules derived from diet or environmental exposures (the exposome), and molecules derived from the biosynthetic interactions with associated microbes (the microbiome). Metabolites, both known and unknown, have structures that span a wide chemical structure space.3,4 The chemical space of metabolites of 100 atoms or less is on the order of ~1060 unique molecules, which makes it a grand challenge to explore and characterize metabolomes using experimental and theoretical tools.5–7 Metabolomics utilizes sophisticated experimental and computational technologies for the identification of biologically relevant metabolites.8,9 Identification of metabolites is important in order to understand metabolic diseases, develop precision medicine, and elucidate the pathways altered in complex metabolic networks.10–12 The experimental annotation of metabolites is performed by matching chemical features against databases or, if available, with appropriate chemical standards.13,14 Currently, this last approach is nontrivial as the vast majority of metabolites do not have such standards. For example, the HMDB database contains a small percentage of the total number of metabolites across multiple organisms, and most of the of molecules in the HMDB do not have authentic chemical standards.8,15 Further, ~1% of available compounds in the U.S. Environmental Protection Agency (EPA) Distributed Structure-Searchable Toxicity (DSSTox) Database, PubChem, ChemSpider, and the American Chemical Society’s CAS databases, which in aggregate contain millions of chemicals,16,17 have known chemical structures. Even the most powerful analytical technologies such as nuclear magnetic resonance spectroscopy (NMR)18–21 and mass spectrometry (MS)22–24 can have difficulties in confidently identifying unknown compounds for a number of technical reasons. Therefore, it is tremendously challenging to build a complete experimental map of any given metabolome, considering the large gaps in our knowledge of metabolite structures.
Ion mobility coupled to mass spectrometry (IM-MS) has emerged as an effective tool to study the structures of unknown compounds while providing high selectivity and fast multidimensional separations.25–27 Ion mobility spectrometry (IMS) separates gas-phase ions based on differences in their rotationally averaged collisional cross-section (CCS) values. The advantage of calculating the molecular surface area or CCS as compared to other properties such as MS/MS spectra or chromatographic retention time is that CCS can be measured to a relative standard deviation (RSD) ranging from 0.25 to 6.0% depending on the instrument employed, thereby providing a unique physiochemical property for structure elucidation of target metabolites.28,29 Isomeric metabolites that commonly exist in biological samples can, in many cases, be accurately distinguished by CCS values. More importantly, CCS values are reproducible and relatively insensitive toward instrumental resources and laboratory environments. McLean and co-workers30,31 and Baker and co-workers32 have used standard chemicals to construct experimental CCS databases with >1000 CCS values.
Data curation efforts of the MS community have contributed positively to the sharing of mass spectra, an approach that could be mimicked for IM data. One example is the MassBank database, with a wide user base and contributors from many different countries.33 The MassBank of North America and the European MassBank34 have further accelerated the sharing of mass spectral data for annotated metabolites. These data servers include autocuration of spectra and chemical structure information (InChI keys). On the other hand, the GNPS35 spectral database utilizes a crowd-sourcing approach to annotate unknown compounds. However, these resources are still reported in different formats, and at present, there are no standardized procedures for data collection, collation, standardization, and sharing.30,32
Over the past decade, IM-MS36 has emerged as an advanced technique for the analysis of metabolites.37–40 IM-MS experiments enable measurement of both mass and structural features within milliseconds. Four main types of IM-MS technologies are used widely: traveling wave (TWIMS),41,42 drift tube (DTIM),38,43 trapped IM (TIMS),44 and differential mobility MS (DMS).45 IM-MS uses different types of ionization techniques which include matrix assisted laser desorption ionization (MALDI), electrospray ionization (ESI), and atmospheric pressure chemical ionization (APCI). Liquid-phase separations such as capillary electrophoresis, gas chromatography, and supercritical fluid chromatography can also be integrated into IM-MS systems. Although different experimental methods are used to measure CCS, the typical experimental uncertainty and differences between platforms amount to ~3%, which is the accepted threshold for comparing predicted and experimental CCS values.
In silico technologies have shown promise for the comprehensive identification of compounds and as a way by which to expand database content through the introduction of accurate theoretical information.46–49 High throughput and comprehensive identification of metabolites can afford a deeper understanding of the role that small molecules play in a given biological system. Importantly, in silico approaches are "standard free" and, in principle, can provide high quality predictions for a range of properties. Many advances in in silico approaches for structure determination have been reported, including NMR chemical shifts50,51 and spin coupling, chromatographic retention times,52,53 ion mobility CCS,49,54 and tandem mass spectra.55,56 Renslow et al. have developed a quantum chemistry-based pipeline (ISiCLE) for CCS calculations that produces CCS predictions in good agreement with experimental values.49 To identify the protonated and deprotonated state of a molecule, ISiCLE considers the lowest energy conformation of the molecule. More recently, Grimme et al. have shown that the lowest energy protonated state does not necessarily give the best agreement with experimental CCS values.57 Other high energy states were found to also be important in order to obtain accurate CCS predictions. This finding suggests that in order to obtain the most accurate CCS predictions all of the protonated and deprotonated states should be correctly modeled. Furthermore, ISiCLE uses simulated annealing molecular dynamics (MD) methods to generate a number of temporally correlated structures, which are then subjected to QM optimization, making this a computationally expensive approach.58 MD trajectories use molecular mechanics for the Hamiltonian. Moreover, because of errors in small-molecule force fields there is no guarantee that the global minimum conformation will be identified.59 Investing computational resources on the DFT refinement of MD-generated initial geometries is not necessarily optimal. Hopkins et al. performed CCS calculation using Mobcal-MPI with 25 different computational methods, showing an accuracy of CCS predictions of ~3.0% relative to experimental values.60 However, this approach does not extensively map the conformational space of a given molecule, which is known to be important for CCS prediction.
Two basic approaches currently exist for predicting IM CCS values. Appropriately constructed machine learning approaches greatly accelerate CCS calculation and have modest CPU needs. However, training set dependencies limit the application of this approach and the quality of results. On the other hand, standard quantum mechanics (QM) methods are very reliable but computationally intensive. In this work, we have developed a CCS calculation workflow that combines the best features of both machine learning and standard QM methods. We have compared our calculated CCS values computed using our in silico workflow with the available database results to confirm their accuracy. Twenty metabolites and their possible molecular states (protonated/deprotonated/neutral) were explored in this benchmark. Furthermore, we performed our own set of TWIMS experiments to measure CCS values for the 20 metabolites considered in this study. The CCS values calculated using our workflow were in good agreement with available experimental values in the literature (±3%), with the values determined herein (±3%), and with the average or consensus values between the two experimental values (±3%). This high-throughput workflow was automated via a series of Python scripts and has the potential to compute CCS values for large numbers of metabolites to enhance structural analysis in metabolomics.
METHODS
The developed workflow consists of several distinct steps (see Figure 1). Initially, we determine the molecular protonation state of the corresponding gas-phase ions (typically [M + H]+ or [M − H]−) and then generate the conformations for these using the RDKit toolkit.61–63 All generated conformations undergo geometry optimization using a QM-based ML model called ASE_ANI64–66 followed by an unsupervised clustering step using in-house unsupervised clustering code (viz. AutoGraph)67 to obtain structurally distinct conformations. Standard DFT geometry optimization and atomic charge calculation are then performed on a representative conformation from each cluster at the B3LYP/6–31+G(d,p) and B3LYP/6–311++G(d,p) level of theory, respectively, using the Gaussian 16 software package.68–71 The input file for CCS calculation is prepared by extracting the geometry and atomic charges from the DFT computations. Finally, the roomtemperature N2-based trajectory method is used to calculate the average collisional cross-sectional areas using the HPCCS code developed by Zanotto et al.72,73 and predict the structure of the target metabolites by comparing the computed results using a Boltzmann-weighted average over multiple conformers with experimental CCS values. We describe details of each step of the workflow in the Supporting Information. The TWIMS CCS experimental values are also reported in the Supporting Information, together with instrumental parameters (Tables S1 and S2). The workflow is integrated in our freely available Web server, www.pomics.org.
Figure 1.
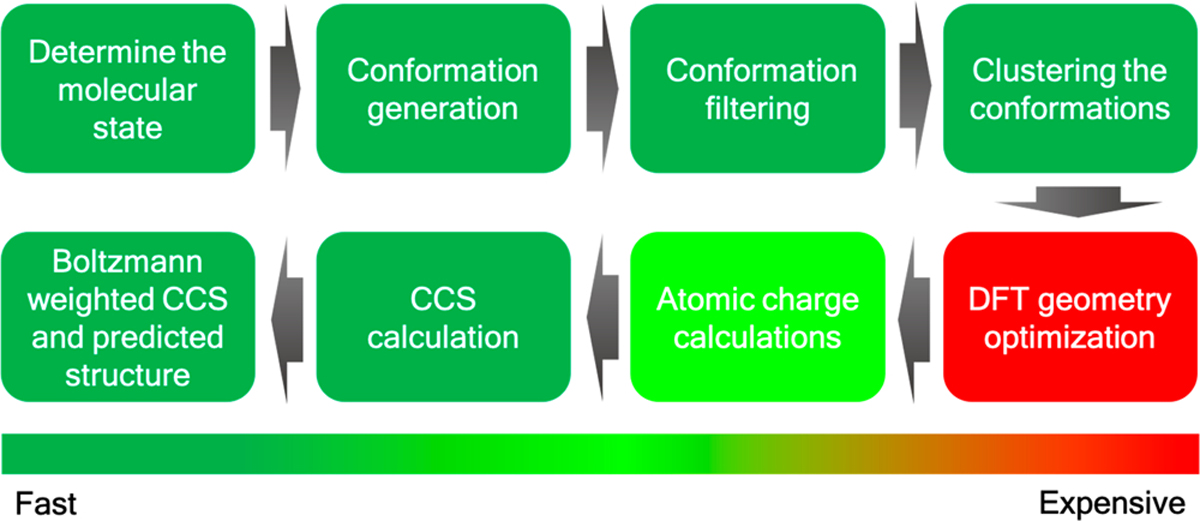
In silico collisional cross section calculation workflow.
RESULTS AND DISCUSSION
The goal of this study was to develop an in silico workflow to accurately calculate the IM CCS value of an unknown metabolite in order to assign its 3D structure and filter out false positives more easily. This workflow involves an eightstep process starting from protonation-state determination to Boltzmann-weighted structure assignment, as illustrated in Figure 1. To validate the proposed workflow, we computed the CCS values for 20 metabolites (see Figure S1) with atom counts ranging from 10 to 40 and compared the computed results to literature CCS values and to CCS values measured in this work. Comparisons against literature values and the TWIMS experimental data are summarized in Table S3.
The number of molecular states for each metabolite are given in Table 1. As an example, for carnosine we have generated five states including one neutral, three monoprotonated ions, and one doubly protonated ion. As our aim was structure elucidation, we performed a rigorous study covering a range of likely molecular states. Apart from the pKa determination, chemical intuition can also play an important role in predicting likely protonation states of a particular metabolite. Each of the molecular states was then converted into a 1D SMILES string and subjected to conformation generation using the RDKit conformation generation tool. We only requested 1000 conformers, but this number could be increased or decreased depending on the application or on the desired coverage of the conformational space. The total number of conformations generated for each metabolite are depicted in Table 1, and it can be seen that for conformationally rigid species few conformers are generated, while for more flexible molecules many more conformations are produced. The AutoGraph clustering method greatly reduces the conformational complexity by lumping related conformations into distinct clusters. The five predicted molecular states for carnosine (viz. models 1–5) generated 990, 994, 994, 994, and 996 conformers, respectively, which after ANI-1ccx minimization and clustering represent 16, 13, 15, 12, and 14 unique conformations, respectively. A representative cluster of the carnosine molecule is depicted in Figure 2. The results for the remaining 19 metabolites are summarized in Table 1. Optimization of large number of generated conformation using a ML model followed by structural similarity based clustering helps to identify relevant conformations in a large conformational space and greatly reduces the number of QM geometry optimization yielding a computationally efficient workflow without compromising accuracy.
Table 1.
Compound Name, Molecular State, Molecular Charge, Number of Atoms, Number of Conformations, Number of Clusters, Boltzmann-Weighted CCS Values, and the Percent Error between the In Silico Predicted CCS Value As Compared to the Corresponding Experimental Consensus CCS Valuea
| no. | compd name | molecular state | molecular charge | no. of atoms | conformation no. | cluster no. | Boltzmann-weighted CCS | error (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | carnosine | model 1 | 0 | 30 | 990 | 16 | 137.15 | 9.40 |
| model 2 | 1 | 31 | 994 | 13 | 166.54 | 9.91 | ||
| model 3 | 1 | 31 | 994 | 15 | 163.57 | 8.27 | ||
| model 4 | 1 | 31 | 994 | 12 | 150.21 | 0.11 | ||
| model 5 | 2 | 32 | 996 | 14 | 217.5 | 31.02 | ||
| 2 | L-anserine | model 1 | 0 | 33 | 998 | 8 | 141.8 | 8.49 |
| model 2 | 1 | 34 | 996 | 9 | 159.74 | 3.70 | ||
| model 3 | 1 | 34 | 996 | 10 | 163.96 | 6.18 | ||
| 3 | abscisic acid | model 1 | 0 | 39 | 1000 | 13 | 154.38 | 5.44 |
| model 2 | 1 | 40 | 1000 | 9 | 162.86 | 0.05 | ||
| model 3 | 1 | 40 | 1000 | 10 | 166.43 | 2.19 | ||
| 4 | O-succinyl-l-homoserine | model 1 | 0 | 28 | 1000 | 21 | 134.4 | 7.80 |
| model 2 | 1 | 29 | 1000 | 17 | 145.45 | 0.39 | ||
| 5 | L-tyrosine | model 1 | 0 | 24 | 961 | 9 | 118.68 | 19.97 |
| model 2 | −1 | 23 | 902 | 10 | 148.53 | 4.14 | ||
| 6 | L-citrulline | model 1 | 0 | 25 | 1000 | 14 | 122.85 | 10.18 |
| model 2 | −1 | 24 | 1000 | 17 | 141.86 | 4.59 | ||
| model 3 | −1 | 26 | 999 | 17 | 141.96 | 4.65 | ||
| 7 | quinolinic acid | model 1 | 0 | 17 | 87 | 7 | 105.66 | 27.81 |
| model 2 | 1 | 18 | 66 | 5 | 142.14 | 5.00 | ||
| 8 | nicotinic acid | model 1 | 0 | 14 | 21 | 3 | 90.78 | 40.42 |
| model 2 | 1 | 15 | 18 | 3 | 132.17 | 3.55 | ||
| 9 | guanidinoacetic acid | model 1 | 0 | 15 | 989 | 10 | 90.15 | 40.92 |
| model 2 | 1 | 16 | 792 | 6 | 131.49 | 3.38 | ||
| model 3 | 1 | 16 | 997 | 6 | 131.1 | 3.10 | ||
| model 4 | 1 | 16 | 998 | 10 | 130.21 | 2.43 | ||
| 10 | citramalic acid | model 1 | 0 | 18 | 998 | 11 | 98.98 | 22.53 |
| model 2 | −1 | 17 | 999 | 10 | 122.52 | 1.01 | ||
| model 3 | −1 | 17 | 995 | 9 | 119.62 | 1.39 | ||
| model 4 | −1 | 17 | 997 | 9 | 121.58 | 0.24 | ||
| 11 | N-methyl-l-gIutam.ate | model 1 | 0 | 22 | 1000 | 14 | 105.34 | 25.17 |
| model 2 | 1 | 23 | 999 | 17 | 129.3 | 1.98 | ||
| 12 | serotonin | model 1 | 0 | 25 | 987 | 7 | 131.36 | 0.38 |
| model 2 | 1 | 26 | 914 | 8 | 165.04 | 20.11 | ||
| model 3 | 1 | 26 | 901 | 4 | 161.6 | 18.41 | ||
| 13 | L-mimosine | model 1 | 0 | 24 | 935 | 8 | 127.93 | 12.00 |
| model 2 | 1 | 25 | 919 | 10 | 150.9 | 5.05 | ||
| model 3 | 1 | 25 | 958 | 9 | 145.36 | 1.43 | ||
| 14 | L-tryptophan | model 1 | 0 | 27 | 934 | 9 | 136.84 | 9.62 |
| model 2 | 1 | 28 | 919 | 11 | 161.55 | 7.14 | ||
| model 3 | 1 | 28 | 924 | 10 | 159.69 | 6.06 | ||
| 15 | L-ornithine | model 1 | 0 | 21 | 999 | 14 | 110.09 | 16.81 |
| model 2 | 1 | 22 | 1000 | 12 | 127.39 | 0.95 | ||
| model 3 | 1 | 22 | 997 | 11 | 126.63 | 1.55 | ||
| 16 | N,N-dimethylglycine | model 1 | 0 | 16 | 999 | 7 | 100.1 | 25.42 |
| model 2 | 1 | 17 | 1000 | 6 | 118.19 | 6.23 | ||
| 17 | kynurenine | model 1 | 0 | 27 | 993 | 10 | 137.15 | 7.64 |
| model 2 | 1 | 28 | 978 | 8 | 146.94 | 0.47 | ||
| 18 | L-asparagine | model 1 | 0 | 17 | 997 | 10 | 111.86 | 15.03 |
| model 2 | 1 | 18 | 991 | 7 | 124.49 | 3.36 | ||
| model 3 | 1 | 18 | 999 | 8 | 128.93 | 0.20 | ||
| 19 | L-2-aminoadipic acid | model 1 | 0 | 22 | 999 | 16 | 112.99 | 16.44 |
| model 2 | 1 | 23 | 1000 | 14 | 129.56 | 1.55 | ||
| 20 | glutamine | model 1 | 0 | 20 | 1000 | 12 | 115.5 | 13.14 |
| model 2 | 1 | 21 | 1000 | 9 | 129.92 | 0.58 |
Nitrogen gas was used as the drift gas.
Figure 2.
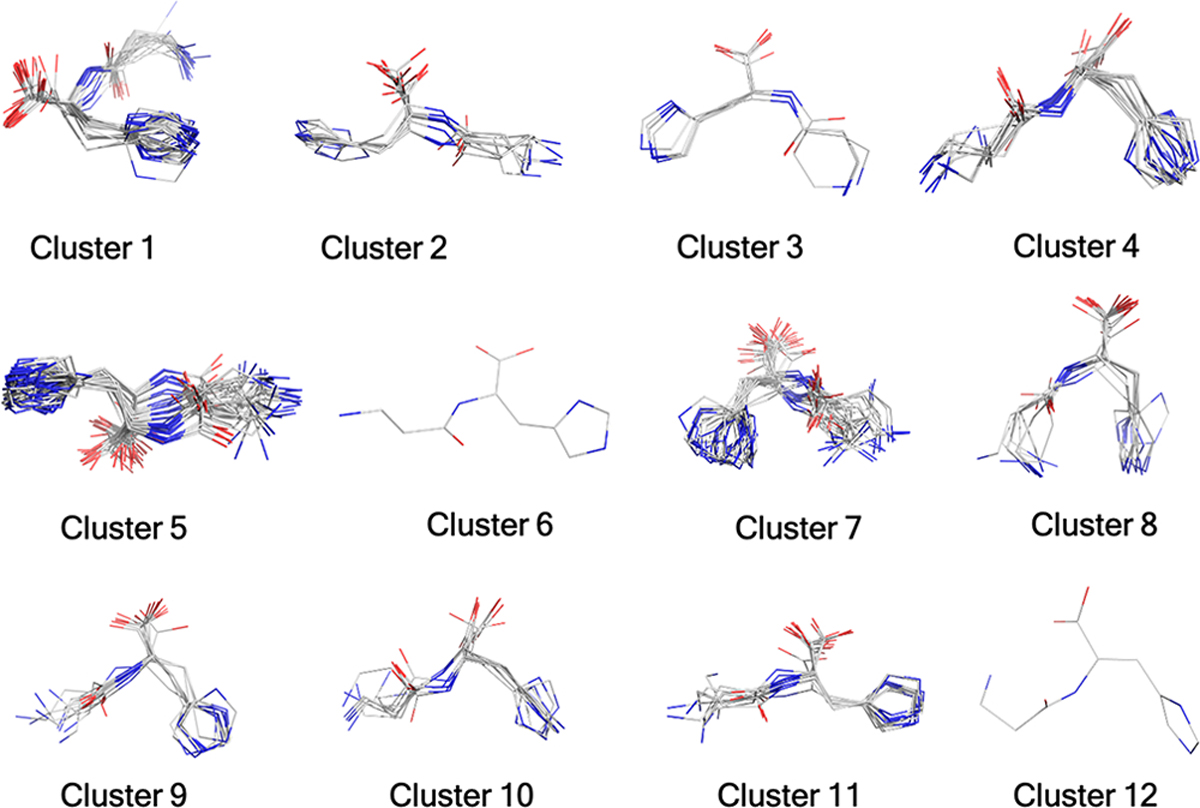
Structure clusters for carnosine (Model 4) as an example. A total of 994 ANI optimized conformers yielded 12 clusters.
To assign the structure correctly and to understand the dependency of the CCS values on the molecular state, we compared the calculated CCS values with literature value, and the CCS values measured in this work and report the quality of the results in the form of the percent error. The results are tabulated in Table 2 for the 20 molecules in each of their molecular states (neutral and charged). For carnosine, the consensus CCS value for the [M + H]+ ionic species is 150.1 Å2. Therefore, it is expected from the in silico modeling that one of the three singly protonated species should best match experiment as this is the species being detected experimentally. For carnosine model 4, where the terminal amine group is protonated (Figure 3), a computed CCS value of 150.21 Å2 was calculated using our workflow. The CCS error was 0.11% relative to the consensus experimental value indicating that the prediction in this case is well within the experimental error estimated as ±3% in Table 2.29 The other two singly protonated species (models 2 and 3, 166.54 and 163.57 Å2, respectively) have 9.9% and 8.2% errors relative to experiment. We were further interested in calculating the CCS values of the neutral species (model 1) and the doubly protonated species (model 5) to gain more insight into the effect structure plays in the predicted CCS values, despite these not being experimentally detectable. When compared to the CCS value of monoprotonated carnosine the neutral and doubly protonated models exhibited percent errors of 9.4% and 31.0%, respectively. From this analysis we concluded that the [M + H]+ ion corresponds to model 4. For the remaining systems we only investigated the singly charged protonated or deprotonated species (be it positive or negative) and the neutral species. In the absence of experimental electrospray data indicating multiple charges, we did not explore other higher charge states for the remaining 19 metabolites.
Table 2.
Experimental Uncertainties of CCS Values for the Studied Metabolites (Å2)a
| no. | compound name | experimental CCS (Literature) | experimental CCS (this work) | error (%)b | consensus |
|---|---|---|---|---|---|
| 1 | camosine | 152.2 | 147.9 | 2.89 | 150.1 |
| 2 | L-anserine | 156.0 | 151.7 | 2.81 | 153.9 |
| 3 | abscisic acid | 160.6 | 165.0 | 2.69 | 162.8 |
| 4 | O-succinyl-l-homoserine | 147.5 | 142.3 | 3.63 | 144.9 |
| 5 | L-tyrosine | 145.8 | 139.0 | 4.86 | 142.4 |
| 6 | L-citrulline | 139.5 | 131.2 | 6.33 | 135.4 |
| 7 | quinolinic acid | 139.0 | 131.1 | 6.01 | 135.1 |
| 8 | nicotinic acid | 128.4 | 126.6 | 1.38 | 127.5 |
| 9 | guanidinoacetic acid | 126.9 | 127.2 | 0.25 | 127.1 |
| 10 | citramalic acid | 124.9 | 117.7 | 6.09 | 121.3 |
| 11 | N-methyl-l-glutam.ate | 133.7 | 130.0 | 2.85 | 131.9 |
| 12 | serotonin | 151.9 | 144.2 | 5.34 | 148.1 |
| 13 | L-mimosine | 143.5 | 143.1 | 0.26 | 143.3 |
| 14 | L-tryptophan | 143.5 | 145.8 | 1.60 | 144.7 |
| 15 | L-ornithine | 129.8 | 127.4 | 1.88 | 128.6 |
| 16 | N,N-dimethylglycine | 123.9 | 127.2 | 2.59 | 125.6 |
| 17 | kynurenine | 151.1 | 144.2 | 4.76 | 147.7 |
| 18 | L-asparagine | 131.5 | 125.8 | 4.56 | 128.7 |
| 19 | L-2-aminoadipic Acid | 131.5 | 131.6 | 0.05 | 131.6 |
| 20 | glutamine | 133.5 | 127.9 | 4.34 | 130.7 |
Nitrogen gas was used as the drift gas for CCS measurement. The average of two experimental data, i.e., consensus CCS values, is also reported.
Average % of error is 3.3 ± 2
Figure 3.
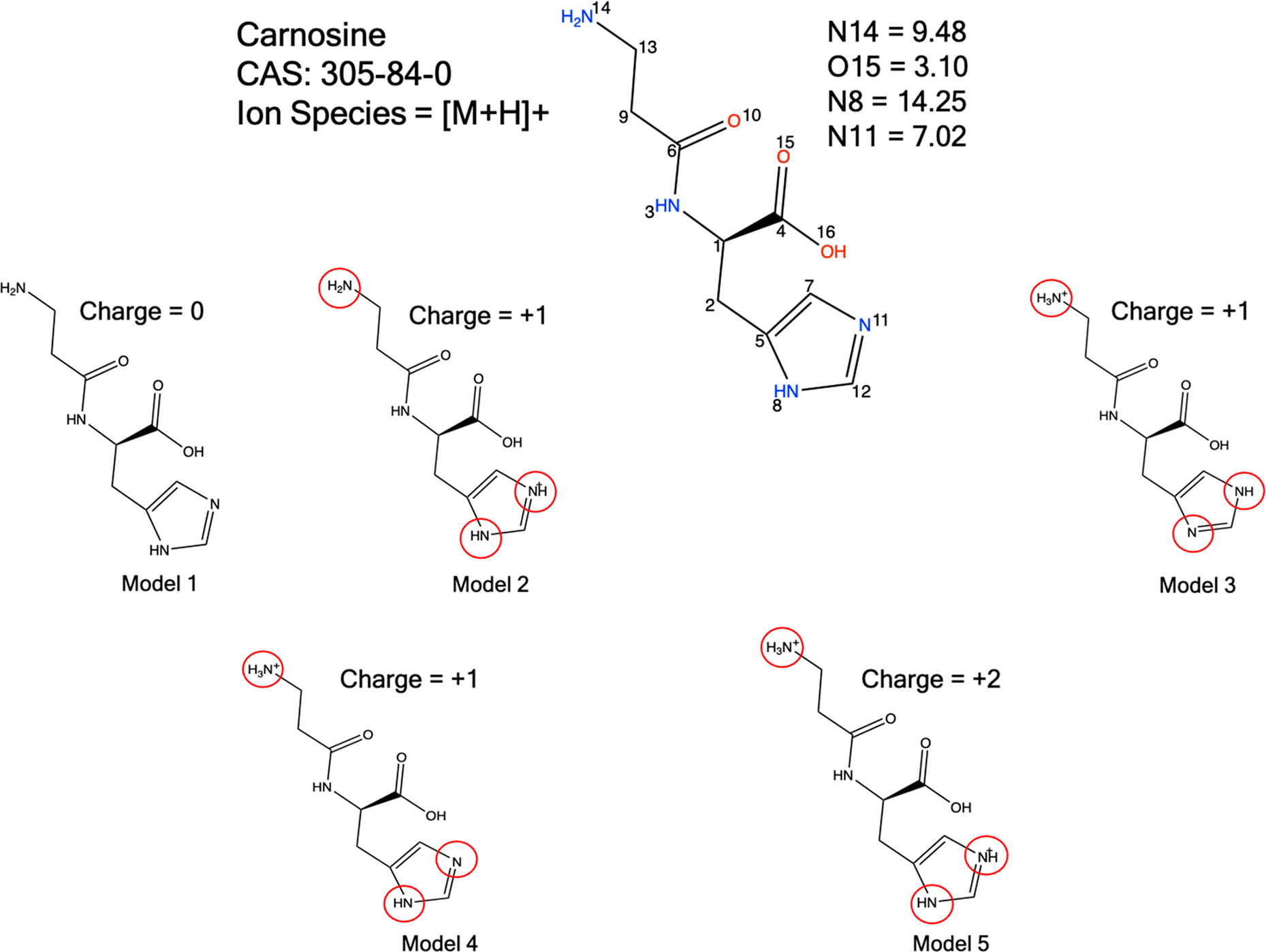
Protonation state is modeled by pKa prediction and chemical intuition. Only cationic species were generated to match the electrospray ionization mode. The pKa is calculated using the EPIK module implemented in the Schrödinger software suite.
TWIMS-MS experiments exhibit excellent between-lab and between-run reproducibility but require calibration to a set of compounds of known CCS values in order to calculate CCS experimental values.74 These known values are most typically obtained using drift tube ion mobility (DTIM) and the Mason–Schamp equation,25 though DTIM measured CCS values can vary largely between laboratories and instruments. For example, Hines et al. measured the CCS of the polyalanine hexamer to be 190.8 Å2, while Picache et al. measured 194.0 Å2, a 1.6% difference.75,76 Much of the error calculated for our experimental TWIMS CCS values may be attributed to di?erences between the DTIM system used to measure CCS values for our polyalanine calibration75 and the DTIM system used to generate the database used for our comparison with select metabolites.32 For this reason, it is essential to calibrate TWIMS measurements using CCS values from a single DTIM database if the goal is to match unknown compounds to CCS measurements against that same database.
To further validate the results obtained from this workflow, we performed the CCS computation for additional metabolites with different molecular states. Depending on the electrospray ionization mode used for IM-MS experiments with each analyte, either the [M + H]+ or [M − H]− molecular states were generated. The neutral state was always considered for each metabolite essentially as an internal reference in that the neutral molecule should not match experiment well at all. A total of 52 states were generated for the 19 metabolites (excluding the five for carnosine), and we executed the workflow to obtain the computed CCS values. The final step of this workflow is to Boltzmann average the CCS values of the multiple conformers.
The Boltzmann weighting step takes the total energies and uses them to weight each individual conformer to determine how much it contributes to the observed CCS value. To illustrate this, we use model 4 of carnosine as an exemplar (see Table 3). A similar analysis for the other molecular states of carnosine and the 19 other metabolites is reported in the Supporting Information (see Tables S4–S57). It is observed from Table 3 that the most stable conformation of model 4 is conformation number 3, which contributes 99% to the Boltzmann-weighted CCS value. Conformation number 11 is 2.55 kcal mol−1 higher in energy than conformation 3, and this high energy conformer contributes only 1% to the Boltzmann-weighted CCS value of 150.21 Å2. The remaining high energy conformers negligibly contribute to the computed CCS value. We observe that, in many cases, molecules can exist in multiple conformations having an effect in CCS calculations. Therefore, in our experience, it is always better to consider Boltzmann average properties rather than the lowest energy state. For example, model 3 of L-tryptophan (see Table S43) has two conformations (4 and 9) with relative populations of 54% and 45% in the gas phase, respectively. Conformations 4 and 9 have CCS values of 166.44 and 156.03 Å2, and the experimentally reported value is 154.22 Å2. The Boltzmann average CCS value is 159.69 Å2. Therefore, the Boltzmann average value gives a significantly better prediction of the CCS value relative to the global minimum. In our previous work on NMR chemical shift calculation50 for metabolites, we observed that high-energy conformations had a subtle impact on the computed NMR chemical shifts, and in order to obtain high-resolution predictions, the entire ensemble was essential. Beyond giving accurate CCS values, the ensembles themselves give molecularlevel insights into the conformational space of molecules, which in many cases can be quite informative in their own right.
Table 3.
Conformation Number, Relative Energy, Mole Fraction, and the CCS Value for Carnosine (Model 4, Protonated Species)
| conformation no. | relative energy (kcal/mol) | mol fraction | CCS value (Å2) |
|---|---|---|---|
| 1 | 25.25 | 0.00 | 169.05 |
| 2 | 28.35 | 0.00 | 172.56 |
| 3 | 0.00 | 0.99 | 150.21 |
| 4 | 28.76 | 0.00 | 170.15 |
| 5 | 25.05 | 0.00 | 170.87 |
| 6 | 22.69 | 0.00 | 169.19 |
| 7 | 8.86 | 0.00 | 160.17 |
| 8 | 14.49 | 0.00 | 160.97 |
| 9 | 12.31 | 0.00 | 160.86 |
| 10 | 22.97 | 0.00 | 167.64 |
| 11 | 2.55 | 0.01 | 150.15 |
| 12 | 13.56 | 0.00 | 163.02 |
To further understand the conformational energy surface, we have further investigated the AutoGraph clustering of the ANI-1ccx optimized conformations. Figure 4 shows the clustering of carnosine (model 4) based on weighted degree and energy values. This figure confirms that both conformations 3 and 11 are in the low energy basin. Cluster regions with the QM energies are shown in Figure S3, which further confirms that conformations 3 and 11 are low energy conformations and, hence, the most probable structures of carnosine. From this analysis we predict that conformation 3 is the most likely structure of the carnosine as it traverses the IM device along with a small fraction of conformation 11. This suggests that our protocol not only can return a CCS value but can provide molecular level insights into the metabolite under study.
Figure 4.
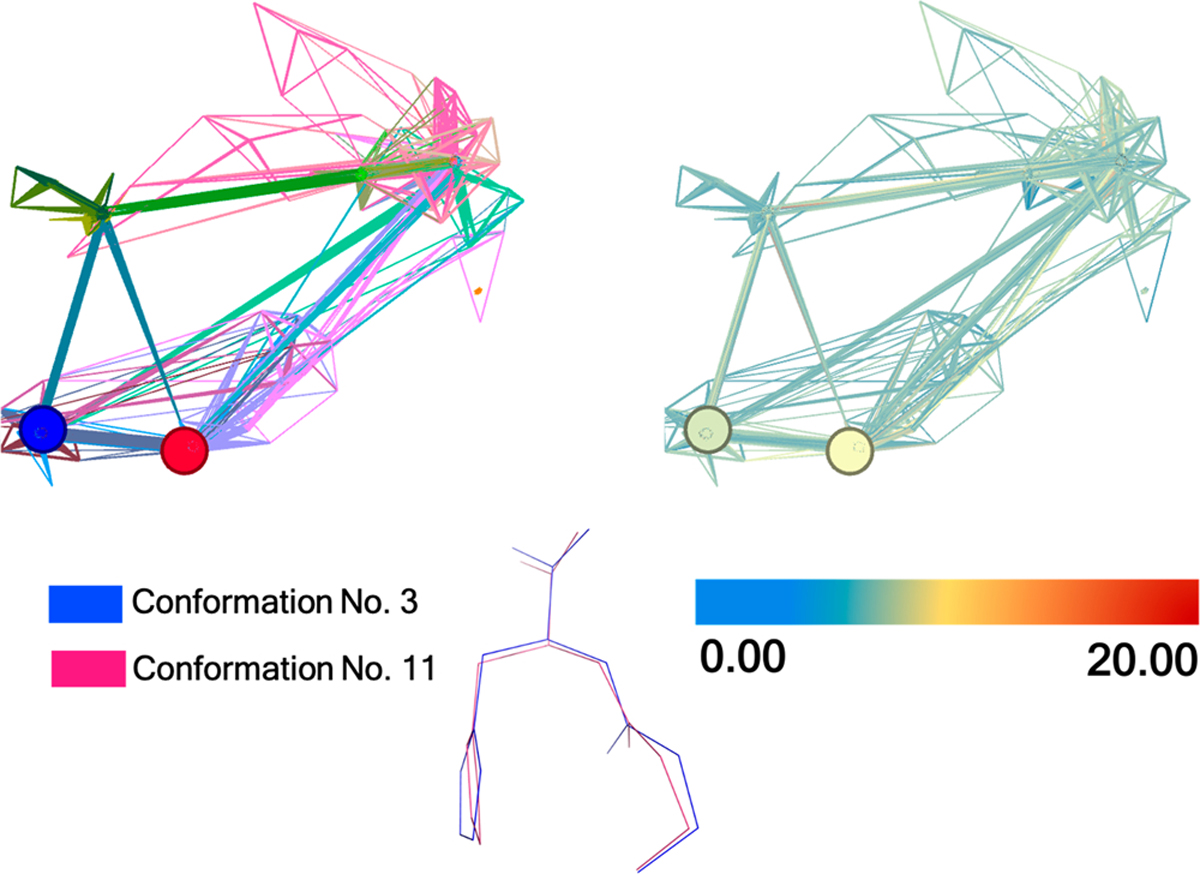
Unsupervised clustering of carnosine (model 4) by AutoGraph. Twelve different colors represent the 12 cluster regions (Left). Low to high energies (ANI-1ccx potential) are represented by blue to red, respectively, and the relative energy value is in kcal mol−1 (right). Figure generated with the Gephi software.
Using the best matching CCS value with experiment for all 20 metabolites yields an average error of 2.2% (within experimental uncertainties), suggesting that our computed CCS values are highly reliable and will lead to highly accurate CCS and structure predictions for unknown metabolites.
CONCLUSIONS
This article introduces a robust in silico workflow to accurately predict the CCS values of small molecules. Moreover, we also evaluated the experimental CCS values and compared them with available literature values to establish that the experimental variation between laboratories is ~±3%, which along with other analyses28,29 establishes the reproducibility of experimental values. The computational workflow is an eightstep process, and the pipeline utilizes the best aspects of force fields, machine learning QM and QM methods to achieve highly accurate and reliable results. Accurate CCS prediction primarily depends on the molecular state (protonated/deprotonated) of a metabolite. Hence, thorough characterization of relevant molecular states yields CCS predictions within 3% of experimental values. The unsupervised clustering method included in this workflow reduces the possibility of human bias and error in cluster selection. The QM-ML model and clustering technique makes this protocol more computationally efficient. All of the steps can be processed in an automated way using series of Python-based scripts, making this protocol even more useful in building an in silico library of predicted CCS values and can be used to assign the structure of an unknown metabolite. Our newly developed workflow maintains an excellent balance between accuracy and computational cost, and we anticipate that this advanced protocol will be a useful tool for structure prediction for the metabolomics community and other communities studying small molecular species. It should be mentioned here that structure elucidation of any unknown compound, including metabolites, is challenging and only one data point is very unlikely to identify an unknown. CCS prediction is only one part of a larger workflow using multiple techniques that include retention time matching, MS/MS databases, isotopic cluster assignments, NMR, and even MSn. Hence, accurate structure elucidation involves determining a range of properties both experimentally and computationally. CCS calculations, however, can reduce the molecular space and can be used as a filter to remove false positives. When coupled with other techniques, it can lead to the more reliable annotation of unknown metabolites.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the high-performance computing center (HPCC) at Michigan State University for providing computational resources. A.S.E., K.M.M., and F.M.F. acknowledge support from NIH 1U2CES030167-01. F.M.F. also acknowledges support by 1R01CA218664-01.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.1c00315.
Details of the CCS workflow steps, benchmark study of the computational cost of our workflow, TWIMS experimental data and instrument parameters, Boltzmann-weighted CCS values, data uncertainty analysis, clustering by AutoGraph (PDF)
Contributor Information
Susanta Das, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Kiyoto Aramis Tanemura, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Laleh Dinpazhoh, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Mithony Keng, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Christina Schumm, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Lydia Leahy, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Carter K Asef, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
Markace Rainey, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
Arthur S. Edison, Departments of Genetics and Biochemistry, Institute of Bioinformatics and Complex Carbohydrate Center, University of Georgia, Athens, Georgia 30602, United States.
Facundo M. Fernández, School of Chemistry and Biochemistry and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
Kenneth M. Merz, Jr., Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
REFERENCES
- (1).Saorin A; Di Gregorio E; Miolo G; Steffan A; Corona G Emerging Role of Metabolomics in Ovarian Cancer Diagnosis. Metabolites 2020, 10 (10), 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wishart DS; Feunang YD; Marcu A; Guo AC; Liang K; Vazquez-Fresno R; Sajed T; Johnson D; Li C; Karu N; Sayeeda Z; Lo E; Assempour N; Berjanskii M; Singhal S; Arndt D; Liang Y; Badran H; Grant J; Serra-Cayuela A; Liu Y; Mandal R; Neveu V; Pon A; Knox C; Wilson M; Manach C; Scalbert A HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018, 46 (D1), D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jones OAH Illuminating the dark metabolome to advance the molecular characterisation of biological systems. Metabolomics 2018, 14 (8), 101. [DOI] [PubMed] [Google Scholar]
- (4).Athersuch T Metabolome analyses in exposome studies: Profiling methods for a vast chemical space. Arch. Biochem. Biophys. 2016, 589, 177–86. [DOI] [PubMed] [Google Scholar]
- (5).Dobson CM Chemical space and biology. Nature 2004, 432 (7019), 824–8. [DOI] [PubMed] [Google Scholar]
- (6).Fiehn O Metabolomics - The link between genotypes and phenotypes. Plant Molecular Biology 2002, 48 (1–2), 155–171. [PubMed] [Google Scholar]
- (7).Sumner LW; Mendes P; Dixon RA Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry 2003, 62 (6), 817–36. [DOI] [PubMed] [Google Scholar]
- (8).Markley JL; Bruschweiler R; Edison AS; Eghbalnia HR; Powers R; Raftery D; Wishart DS The future of NMR-based metabolomics. Curr. Opin Biotechnol 2017, 43, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bingol K; Bruschweiler R Knowns and unknowns in metabolomics identified by multidimensional NMR and hybrid MS/NMR methods. Curr. Opin Biotechnol 2017, 43, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Berendsen RL; Pieterse CM; Bakker PA The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17 (8), 478–86. [DOI] [PubMed] [Google Scholar]
- (11).Griffin JL; Bollard ME Metabonomics: its potential as a tool in toxicology for safety assessment and data integration. Curr. Drug Metab 2004, 5 (5), 389–98. [DOI] [PubMed] [Google Scholar]
- (12).Nicholson JK; Connelly J; Lindon JC; Holmes E Metabonomics: a platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov 2002, 1 (2), 153–61. [DOI] [PubMed] [Google Scholar]
- (13).Nicholson JK; Wilson ID Opinion: understanding ‘global’ systems biology: metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov 2003, 2 (8), 668–76. [DOI] [PubMed] [Google Scholar]
- (14).Daviss B Growing pains for metabolomics: the newest ‘omic science is producing results—and more data than researchers know what to do with. Scientist 2005, 19 (8), 25. [Google Scholar]
- (15).Tulp M; Bohlin L Functional versus chemical diversity: is biodiversity important for drug discovery? Trends Pharmacol. Sci. 2002, 23 (5), 225–231. [DOI] [PubMed] [Google Scholar]
- (16).Schymanski EL; Singer HP; Slobodnik J; Ipolyi IM; Oswald P; Krauss M; Schulze T; Haglund P; Letzel T; Grosse S; Thomaidis NS; Bletsou A; Zwiener C; Ibanez M; Portoles T; de Boer R; Reid MJ; Onghena M; Kunkel U; Schulz W; Guillon A; Noyon N; Leroy G; Bados P; Bogialli S; Stipanicev D; Rostkowski P; Hollender J Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Anal Bioanal Chem. 2015, 407 (21), 6237–55. [DOI] [PubMed] [Google Scholar]
- (17).Richard AM; Gold LS; Nicklaus MC Chemical structure indexing of toxicity data on the internet: moving toward a flat world. Curr. Opin Drug Discov. Devel. 2006, 9 (3), 314–25. [PubMed] [Google Scholar]
- (18).Nicholson JK; Foxall PJ; Spraul M; Farrant RD; Lindon JC 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal. Chem. 1995, 67 (5), 793–811. [DOI] [PubMed] [Google Scholar]
- (19).Nicholson JK; Lindon JC; Holmes E ‘Metabonomics’ understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29 (11), 1181–9. [DOI] [PubMed] [Google Scholar]
- (20).Beckonert O; Keun HC; Ebbels TM; Bundy J; Holmes E; Lindon JC; Nicholson JK Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc 2007, 2 (11), 2692–703. [DOI] [PubMed] [Google Scholar]
- (21).Yu Z; Li P; Merz KM Using Ligand-Induced Protein Chemical Shift Perturbations To Determine Protein-Ligand Structures. Biochemistry 2017, 56 (18), 2349–2362. [DOI] [PubMed] [Google Scholar]
- (22).Dettmer K; Aronov PA; Hammock BD Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007, 26 (1), 51–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78 (3), 779–87. [DOI] [PubMed] [Google Scholar]
- (24).Soga T; Ohashi Y; Ueno Y; Naraoka H; Tomita M; Nishioka T Quantitative Metabolome Analysis Using Capillary Electrophoresis Mass Spectrometry. J. Proteome Res. 2003, 2 (5), 488–494. [DOI] [PubMed] [Google Scholar]
- (25).Dodds JN; Baker ES Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom. 2019, 30 (11), 2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mairinger T; Causon TJ; Hann S The potential of ion mobility-mass spectrometry for non-targeted metabolomics. Curr. Opin Chem. Biol. 2018, 42, 9–15. [DOI] [PubMed] [Google Scholar]
- (27).Paglia G; Astarita G Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nat. Protoc. 2017, 12 (4), 797–813. [DOI] [PubMed] [Google Scholar]
- (28).Bijlsma L; Bade R; Celma A; Mullin L; Cleland G; Stead S; Hernandez F; Sancho JV Prediction of Collision Cross-Section Values for Small Molecules: Application to Pesticide Residue Analysis. Anal. Chem. 2017, 89 (12), 6583–6589. [DOI] [PubMed] [Google Scholar]
- (29).Gabelica V; Shvartsburg AA; Afonso C; Barran P; Benesch JLP; Bleiholder C; Bowers MT; Bilbao A; Bush MF; Campbell JL; Campuzano IDG; Causon T; Clowers BH; Creaser CS; De Pauw E; Far J; Fernandez-Lima F; Fjeldsted JC; Giles K; Groessl M; Hogan CJ Jr.; Hann S; Kim HI; Kurulugama RT; May JC; McLean JA; Pagel K; Richardson K; Ridgeway ME; Rosu F; Sobott F; Thalassinos K; Valentine SJ; Wyttenbach T Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38 (3), 291–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Picache JA; Rose BS; Balinski A; Leaptrot KL; Sherrod SD; May JC; McLean JA Collision cross section compendium to annotate and predict multi-omic compound identities. Chem. Sci. 2019, 10 (4), 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nichols CM; Dodds JN; Rose BS; Picache JA; Morris CB; Codreanu SG; May JC; Sherrod SD; McLean JA Untargeted Molecular Discovery in Primary Metabolism: Collision Cross Section as a Molecular Descriptor in Ion Mobility-Mass Spectrometry. Anal. Chem. 2018, 90 (24), 14484–14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zheng X; Aly NA; Zhou Y; Dupuis KT; Bilbao A; Paurus VL; Orton DJ; Wilson R; Payne SH; Smith RD; Baker ES A structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry. Chem. Sci. 2017, 8 (11), 7724–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Horai H; Arita M; Kanaya S; Nihei Y; Ikeda T; Suwa K; Ojima Y; Tanaka K; Tanaka S; Aoshima K; Oda Y; Kakazu Y; Kusano M; Tohge T; Matsuda F; Sawada Y; Hirai MY; Nakanishi H; Ikeda K; Akimoto N; Maoka T; Takahashi H; Ara T; Sakurai N; Suzuki H; Shibata D; Neumann S; Iida T; Tanaka K; Funatsu K; Matsuura F; Soga T; Taguchi R; Saito K; Nishioka T MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45 (7), 703–14. [DOI] [PubMed] [Google Scholar]
- (34).Stravs MA; Schymanski EL; Singer HP; Hollender J Automatic recalibration and processing of tandem mass spectra using formula annotation. J. Mass Spectrom. 2013, 48 (1), 89–99. [DOI] [PubMed] [Google Scholar]
- (35).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; Porto C; Bouslimani A; Melnik AV; Meehan MJ; Liu WT; Crusemann M; Boudreau PD; Esquenazi E; Sandoval-Calderon M; Kersten RD; Pace LA; Quinn RA; Duncan KR; Hsu CC; Floros DJ; Gavilan RG; Kleigrewe K; Northen T; Dutton RJ; Parrot D; Carlson EE; Aigle B; Michelsen CF; Jelsbak L; Sohlenkamp C; Pevzner P; Edlund A; McLean J; Piel J; Murphy BT; Gerwick L; Liaw CC; Yang YL; Humpf HU; Maansson M; Keyzers RA; Sims AC; Johnson AR; Sidebottom AM; Sedio BE; Klitgaard A; Larson CB; P CAB; Torres-Mendoza D; Gonzalez DJ; Silva DB; Marques LM; Demarque DP; Pociute E; O’Neill EC; Briand E; Helfrich EJN; Granatosky EA; Glukhov E; Ryffel F; Houson H; Mohimani H; Kharbush JJ; Zeng Y; Vorholt JA; Kurita KL; Charusanti P; McPhail KL; Nielsen KF; Vuong L; Elfeki M; Traxler MF; Engene N; Koyama N; Vining OB; Baric R; Silva RR; Mascuch SJ; Tomasi S; Jenkins S; Macherla V; Hoffman T; Agarwal V; Williams PG; Dai J; Neupane R; Gurr J; Rodriguez AMC; Lamsa A; Zhang C; Dorrestein K; Duggan BM; Almaliti J; Allard PM; Phapale P; Nothias LF; Alexandrov T; Litaudon M; Wolfender JL; Kyle JE; Metz TO; Peryea T; Nguyen DT; VanLeer D; Shinn P; Jadhav A; Muller R; Waters KM; Shi W; Liu X; Zhang L; Knight R; Jensen PR; Palsson BO; Pogliano K; Linington RG; Gutierrez M; Lopes NP; Gerwick WH; Moore BS; Dorrestein PC; Bandeira N Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34 (8), 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Eiceman GA; Karpas Z Ion Mobility Spectrometry. CRC Press., 2005. [Google Scholar]
- (37).Kanu AB; Dwivedi P; Tam M; Matz L; Hill HH Jr. Ion mobility-mass spectrometry. J. Mass Spectrom. 2008, 43 (1), 1–22. [DOI] [PubMed] [Google Scholar]
- (38).May JC; McLean JA Ion mobility-mass spectrometry: timedispersive instrumentation. Anal. Chem. 2015, 87 (3), 1422–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Basit A; Pontis S; Piomelli D; Armirotti A Ion mobility mass spectrometry enhances low-abundance species detection in untargeted lipidomics. Metabolomics 2016, 12 (3), 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).May JC; Gant-Branum RL; McLean JA Targeting the untargeted in molecular phenomics with structurally-selective ion mobility-mass spectrometry. Curr. Opin. Biotechnol. 2016, 39, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Giles K; Williams JP; Campuzano I Enhancements in travelling wave ion mobility resolution. Rapid Commun. Mass Spectrom. 2011, 25 (11), 1559–66. [DOI] [PubMed] [Google Scholar]
- (42).Paglia G; Astarita G Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nat. Protoc 2017, 12 (4), 797–813. [DOI] [PubMed] [Google Scholar]
- (43).Kaplan K; Graf S; Tanner C; Gonin M; Fuhrer K; Knochenmuss R; Dwivedi P; Hill HH Jr. Resistive glass IMTOFMS. Anal. Chem. 2010, 82 (22), 9336–43. [DOI] [PubMed] [Google Scholar]
- (44).Michelmann K; Silveira JA; Ridgeway ME; Park MA Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass. Spectrom. 2015, 26 (1), 14–24. [DOI] [PubMed] [Google Scholar]
- (45).Basanta M; Jarvis RM; Xu Y; Blackburn G; Tal-Singer R; Woodcock A; Singh D; Goodacre R; Thomas CL; Fowler SJ Non-invasive metabolomic analysis of breath using differential mobility spectrometry in patients with chronic obstructive pulmonary disease and healthy smokers. Analyst 2010, 135 (2), 315–20. [DOI] [PubMed] [Google Scholar]
- (46).Kaufmann A; Butcher P; Maden K; Walker S; Widmer M Practical application of in silico fragmentation based residue screening with ion mobility high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2017, 31 (13), 1147–1157. [DOI] [PubMed] [Google Scholar]
- (47).Zhou Z; Luo M; Chen X; Yin Y; Xiong X; Wang R; Zhu ZJ Ion mobility collision cross-section atlas for known and unknown metabolite annotation in untargeted metabolomics. Nat. Commun. 2020, 11 (1), 4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Colby SM; Nunez JR; Hodas NO; Corley CD; Renslow RR Deep Learning to Generate in Silico Chemical Property Libraries and Candidate Molecules for Small Molecule Identification in Complex Samples. Anal. Chem. 2020, 92 (2), 1720–1729. [DOI] [PubMed] [Google Scholar]
- (49).Colby SM; Thomas DG; Nunez JR; Baxter DJ; Glaesemann KR; Brown JM; Pirrung MA; Govind N; Teeguarden JG; Metz TO; Renslow RS ISiCLE: A Quantum Chemistry Pipeline for Establishing in Silico Collision Cross Section Libraries. Anal. Chem. 2019, 91 (7), 4346–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Das S; Edison AS; Merz KM Jr. Metabolite Structure Assignment Using In Silico NMR Techniques. Anal. Chem. 2020, 92 (15), 10412–10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yesiltepe Y; Nunez JR; Colby SM; Thomas DG; Borkum MI; Reardon PN; Washton NM; Metz TO; Teeguarden JG; Govind N; Renslow RS An automated framework for NMR chemical shift calculations of small organic molecules. J. Cheminform 2018, 10 (1), 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Vinaixa M; Schymanski EL; Neumann S; Navarro M; Salek RM; Yanes O Mass spectral databases for LC/MS- and GC/MS-based metabolomics: State of the field and future prospects. Trac-Trends in Analytical Chemistry 2016, 78, 23–35. [Google Scholar]
- (53).Randazzo GM; Tonoli D; Strajhar P; Xenarios I; Odermatt A; Boccard J; Rudaz S Enhanced metabolite annotation via dynamic retention time prediction: Steroidogenesis alterations as a case study. J. Chromatogr B Analyt Technol. Biomed Life Sci. 2017, 1071, 11–18. [DOI] [PubMed] [Google Scholar]
- (54).Plante PL; Francovic-Fontaine E; May JC; McLean JA; Baker ES; Laviolette F; Marchand M; Corbeil J Predicting Ion Mobility Collision Cross-Sections Using a Deep Neural Network: DeepCCS. Anal. Chem. 2019, 91 (8), 5191–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wolf S; Schmidt S; Muller-Hannemann M; Neumann S In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics 2010, 11 (1), 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Bocker S Searching molecular structure databases using tandem MS data: are we there yet? Curr. Opin Chem. Biol. 2017, 36, 1–6. [DOI] [PubMed] [Google Scholar]
- (57).Koopman J; Grimme S From QCEIMS to QCxMS: A Tool to Routinely Calculate CID Mass Spectra Using Molecular Dynamics. J. Am. Soc. Mass Spectrom. 2021, 32 (7), 1735–1751. [DOI] [PubMed] [Google Scholar]
- (58).Hawkins PCD Conformation Generation: The State of the Art. J. Chem. Inf Model 2017, 57 (8), 1747–1756. [DOI] [PubMed] [Google Scholar]
- (59).Kanal IY; Keith JA; Hutchison GR A sobering assessment of small-molecule force field methods for low energy conformer predictions. Int. J. Quantum Chem. 2018, 118 (5), e25512. [Google Scholar]
- (60).Ieritano C; Hopkins WS Assessing collision cross section calculations using MobCal-MPI with a variety of commonly used computational methods. Materials Today Communications 2021, 27, 102226. [Google Scholar]
- (61).Ebejer JP; Morris GM; Deane CM Freely available conformer generation methods: how good are they? J. Chem. Inf Model 2012, 52 (5), 1146–58. [DOI] [PubMed] [Google Scholar]
- (62).Riniker S; Landrum GA Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf Model 2015, 55 (12), 2562–74. [DOI] [PubMed] [Google Scholar]
- (63).Landrum G, RDKit: Open-source cheminformatics. 2006, https://www.rdkit.org/.
- (64).Smith JS; Nebgen BT; Zubatyuk R; Lubbers N; Devereux C; Barros K; Tretiak S; Isayev O; Roitberg AE Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10 (1), 2903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Smith JS; Isayev O; Roitberg AE ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chemical Science 2017, 8 (4), 3192–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Rufa DA; Bruce Macdonald HE; Fass J; Wieder M; Grinaway PB; Roitberg AE; Isayev O; Chodera JD, Towards chemical accuracy for alchemical free energy calculations with hybrid physics-based machine learning/molecular mechanics potentials. bioRxiv 2020, 2020.07.29.227959. [Google Scholar]
- (67).Tanemura KA; Das S; Merz KM Jr. AutoGraph: Autonomous Graph-Based Clustering of Small-Molecule Conformations. J. Chem. Inf Model 2021, 61 (4), 1647–1656. [DOI] [PubMed] [Google Scholar]
- (68).Becke AD Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. [Google Scholar]
- (69).Zhao Y; Truhlar DG Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41 (2), 157–167. [DOI] [PubMed] [Google Scholar]
- (70).Ditchfield R; Hehre WJ; Pople JA Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54 (2), 724–728. [Google Scholar]
- (71).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ, Gaussiañ16 {R}evision {C}.01. 2016, Gaussian 16 Revision C.01. [Google Scholar]
- (72).Zanotto L; Heerdt G; Souza PCT; Araujo G; Skaf MS High performance collision cross section calculation-HPCCS. J. Comput. Chem. 2018, 39 (21), 1675–1681. [DOI] [PubMed] [Google Scholar]
- (73).Heerdt G; Zanotto L; Souza PCT; Araujo G; Skaf MS, Collision Cross Section Calculations Using HPCCS. In Ion Mobility-Mass Spectrometry: Methods and Protocols, Paglia G; Astarita G, Eds. Springer US: New York, NY, 2020; pp 297–310. [DOI] [PubMed] [Google Scholar]
- (74).Nye LC; Williams JP; Munjoma NC; Letertre MPM; Coen M; Bouwmeester R; Martens L; Swann JR; Nicholson JK; Plumb RS; McCullagh M; Gethings LA; Lai S; Langridge JI; Vissers JPC; Wilson ID A comparison of collision cross section values obtained via travelling wave ion mobility-mass spectrometry and ultra high performance liquid chromatography-ion mobility-mass spectrometry: Application to the characterisation of metabolites in rat urine. J. Chromatogr. A 2019, 1602, 386–396. [DOI] [PubMed] [Google Scholar]
- (75).Hines KM; Ross DH; Davidson KL; Bush MF; Xu L Large-Scale Structural Characterization of Drug and Drug-Like Compounds by High-Throughput Ion Mobility-Mass Spectrometry. Anal. Chem. 2017, 89 (17), 9023–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Forsythe JG; Petrov AS; Walker CA; Allen SJ; Pellissier JS; Bush MF; Hud NV; Fernandez FM Collision cross section calibrants for negative ion mode traveling wave ion mobility-mass spectrometry. Analyst 2015, 140 (20), 6853–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.