

Abstract
Background
Hereditary angioedema (HAE) is a rare disease characterized by recurrent attacks of severe swellings of the skin and submucosa. More than 900 variants of the SERPING1 gene associated with HAE have been identified. However, only approximately 50 variants have been identified in the Chinese population. This study aimed to update the mutational spectrum in Chinese HAE patients and provide evidence for the accurate diagnosis of HAE.
Methods
A total of 97 unrelated HAE patients were enrolled in the study. Sanger sequencing and multiple ligation-dependent probe amplification analysis were used to identify the variants in the SERPING1 gene. The variants were reviewed in a number of databases, including the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/) and the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/variants/SERPING1). The American College of Medical Genetics and Genomics-Association for Molecular Pathology (ACMG-AMP) criteria was used to determine the pathogenicity of the variants.
Results
Of the 97 patients, 76 different variants were identified in 90 of them and no disease-causing variants were identified in the remaining 7 patients. Among the 76 variants, 35 variants were novel and submitted to ClinVar. Missense and in-frame variants were the most common variants (36.8%), followed by frameshift (28.9%), nonsense (14.5%), splice site (13.2%) variants, and gross deletions/duplications (6.6%).
Conclusions
Our findings broaden the mutational spectrum of SERPING1 and provide evidence for accurate diagnosis and predictive genetic counseling.
Supplementary Information
The online version contains supplementary material available at 10.1186/s41065-022-00242-z.
Keywords: C1 inhibitor, Hereditary angioedema, Mutational analysis, Phenotype, SERPING1
Background
Hereditary angioedema (HAE; OMIM #106100) is a rare, life-threatening disease characterized by unpredictable skin and submucosal swelling [1]. HAE can be caused by C1 inhibitor (C1-INH) deficiency (Type 1 HAE, HAE-1), C1-INH dysfunction (Type 2 HAE, HAE-2), or other mechanisms (HAE with normal C1-INH, HAE-nC1-INH). The genetic defect in HAE-1/2 are mutations in the SERPING1 gene [1]. HAE-nC1-INH was first described in 2000, [2] and the pathogenic genes were gradually identified as F12, [3] angiopoietin-1 (ANGPT1), [4] plasminogen (PLG), [5] kininogen (KNG1), [6] myoferlin (MYOF), [7] and heparan sulfate-glucosamine 3-O-sulfotransferase 6 (HS3ST6) [8].
The first case of HAE in China was diagnosed by our research team in 1980, and related studies have gradually been conducted in China since then [9]. However, to date, the SERPING1 gene is the only pathogenic gene known to be related to HAE in the Chinese population. The SERPING1 gene encodes a highly glycosylated plasma protein, C1-INH, which irreversibly encapsulates the proteases of the kallikrein-kinin-system such as plasma kallikrein, factor XIIa, and factor XIIf in its molecule [1]. This process, termed suicide inactivation, incapacitates multiple proteases involved in the kallikrein-kinin system, complement, fibrinolysis, and coagulation pathways [1]. In HAE-1/2, the kallikrein-kinin system is overactivated and produces large amounts of bradykinin. Bradykinin mediates the active transfer of fluid into localized tissues by binding to the bradykinin B2 receptor, causing angioedema [10]. HAE-1 is caused by diverse variants of the SERPING1 gene [11]. Missense, nonsense, frameshift, and splicing defect mutations on the SERPING1 gene cause misfolded or truncated protein products [12]. Patients with HAE-1 are characterized by low antigenic and functional C1-INH levels [12]. However, HAE-2 occurs mainly due to missense mutations in exon 8 of the SERPING1 gene. Such mutations affects the reactive loop and reduces the inhibitory effect of C1-INH on target proteins [1]. Patients with HAE-2 are characterized by normal or elevated antigenic but low functional C1-INH levels [13].
To date, 611 variants of the SERPING1 gene were reported in the Human Gene Mutation Database (HGMD® Professional 2021.4, http://www.hgmd.cf.ac.uk/), and 962 variants were reported in the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/variants/SERPING1). Most of the variants recorded in both databases overlapped. However, in China, the HAE genotype has not been adequately studied. Our previous study reported the clinical characteristics and mutational spectrum in 48 Chinese HAE patients [14]. In these 48 patients, 25 novel mutations and 3 novel single nucleotide polymorphisms (SNPs) were identified [14]. However, no large deletions and insertions have been reported in Chinese HAE patients. Therefore, the present study aimed to provide additional information on the genotype of HAE among Chinese patients to promote the accurate diagnosis of HAE.
Materials and methods
Study subjects
From 2013 to 2022, 97 unrelated patients diagnosed with HAE at the Department of Allergy, Peking Union Medical College Hospital, were enrolled in the study. The diagnosis of HAE was made accroding to the US HAEA medical advisory board 2020 guidelines for the management of hereditary angioedema [15] and the international WAO/EAACI guideline for the management of hereditary angioedema [13]. In brief, HAE was diagnosed according to the typical clinical manifestation of recurrent swelling and abnormal laboratory test results. Patients with reduced C1-INH concentration and function as well as reduced C4 levels were diagnosed as having HAE-1. Patients with normal or elevated C1-INH concentration, reduced C1-INH function and reduced C4 levels were diagnosed as having HAE-2. Sixty-six unrelated healthy individuals were included as controls. All the subjects signed an informed consent form under a research protocol approved by the Research and Ethics Board of the Peking Union Medical College Hospital (Approval number: HS-2402).
Complement measurements
Serum C1-INH antigen levels were measured using the BNTM II System (Dade Behring Marburg GmbH, Marburg, Germany). Levels of functional C1-INH were determined by using a Chromogenic Kit (Immuno Chrom, Vienna, Austria), whereas serum levels of complement 4 (C4) were determined via immunonephelometry using C4 Reagent, 29,100 Test Cartridge (Beckman Coulter, CA, USA).
Polymerase chain reaction (PCR) amplification and sequencing analysis
Genomic DNA was isolated from EDTA-containing whole blood using the QIAamp DNA Blood Mini Kit (Qiagen GmbH, Hilden, Germany). The concentrations of DNA were detected at 260 nm and 280 nm using a laboratory spectrophotometer (Thermo Fisher Scientific, USA). Polymerase chain reaction (PCR) was used to amplify the promoter, noncoding exon 1, seven coding exons, and intron–exon boundaries of the SERPING1 gene. Primers and PCR conditions are shown in supplemental Table S1. PCR products were sequenced using ABI 3730xl DNA Sequencer (Applied Biosystems, Foster, VA, USA) after purification. Sequence alignment comparison was performed with the reference sequence (NCBI Reference Sequence: NM_000062.2). Multiplex ligation-dependent probe amplification (MLPA) analysis was also applied. The SALSA® MLPA® Probemix P243-B1 SERPING1-F12 kit (MRC-Holland, Amsterdam, The Netherlands) was used to detect deletions or duplications of one or more exons of SERPING1. MLPA data were analyzed using the COFFALYSER® software (MRC-Holland). SERPING1 variants were numbered according to the recommended CDS numbering system provided by the Human Genome Variation Society (HGVS, http://www.hgvs.org/rec.html). For amino acid positions, historical numbering based on the 478-residue mature protein was used. These variants were reviewed in public databases, including the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/) and the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/variants/SERPING1), to determine whether they have been previously reported as pathogenic.
In silico analysis
The VarCards web server (http://159.226.67.237/sun/varcards/) was used to interpret novel missense variations. It is a comprehensive clinical and genetic database for coding variants in the human genome. This web server predicts the effect of missense mutations on proteins through 23 in silico predictive algorithms, including SIFT, Polyphen-2, and MutationTaster [16]. For intron variants, we used ESE finder 3.0 (http://krainer01.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home) to predict the effect on alternative splicing.
Results
The average age of the 97 enrolled HAE patients was 36.0 years. Of the 97 patients, 96.9%, 69.1%, and 66.0% reported skin, gastrointestinal, and laryngeal swellings, respectively. More than 90% of patients were receiving danazol or tranexamic acid for long-term prophylaxis at the time of the study. Specifically, because the first-line long-term prophylaxes recommended by the international WAO/EAACI guidelines were not approved in China during the study period, these patients were being treated with danazol except for 2 patients younger than 16 years of age who were being treated with tranexamic acid. In addition, 8.2% of the patients had other complications, 2 patients had allergic rhinitis, 2 patients had nephritis, 1 patient had hypertension, 1 patient had hyperlipidemia, 1 patient had hepatitis B, and 1 patient had Sjögren's syndrome (Table 1).
Table 1.
Demographic characteristics and clinical manifestations of 97 unrelated Chinese HAE patients
| Clinical characteristics | Mean ± SD or n (%) |
|---|---|
| Female | 56 (57.7) |
| Age (years) | 36.0 ± 12.7 |
| HAE-1 | 91 (93.8) |
| HAE-2 | 6 (6.2) |
| Positive family history | 64 (66.0) |
| Onset age (years) | 18.4 ± 9.4 |
| Skin edema | 94 (96.9) |
| Gastrointestinal edema | 67 (69.1) |
| Laryngeal edema | 64 (66.0) |
| Need for long-term prophylaxis | 88 (90.7) |
| Have other complications | 8 (8.2) |
We identified 76 different variants in 90 unrelated HAE patients, 35 of which were novel (Table 2). None of the novel variants were detected in the 66 unrelated controls. Novel variants were submitted to ClinVar with ClinVar accession numbers from SCV001977563 to SCV001977593, SCV002043721, and SCV002522446. The pathogenicity classification was based on the ACMG-AMP variant classification criteria [17]. In addition, seven patients were not identified as carrying disease-causing variants. Their demographic characteristics and clinical presentation can be seen in Table S2. Therefore, the sensitivity of the sequencing method applied in this study was 92.8% in Chinese HAE patients.
Table 2.
SERPING1 mutations identified in Chinese HAE patients
| Region | DNA change | Consequences | Predictive algorithm | Laboratory test | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SIFTa | Polyphen-2b | MutationTasterc | Clinical classificationd | No. of patients | C1-INH protein (g/L)e | C4 protein (g/L)f | References | |||
| Exon 2 | c.1A > G | p.(Met1Val) | 0.022 | 0.941 | DC(1) | pxathogenic | 1 | 0.04 | 0.015 | [18] |
| Exon 2 | c.44del | p.(Leu15Argfs*64) | likely pathogenic | 1 | 0.04 | 0.01 | [9] | |||
| Exon 2 | c.49G > A | p.(Gly17Arg) | 0.003 | 0.011 | DC(1) | likely pathogenic | 2 | 0.05 | 0.06 | [19] |
| 0.16 | 0.043 | |||||||||
| Exon 3 | c.74del | p.(Asn25Metfs*54) | pathogenic | 1 | 0.09 | 0.093 | This study | |||
| Exon 3 | c.100C > A | p.(Pro34Thr) | 0.136 | 0.975 | P(1) | VUS | 1 | 0.13 | 0.053 | This study |
| Exon 3 | c.120_121del | Increased exon 3 skipping | pathogenic | 1 | 0.04 | 0.015 | [20] | |||
| Exon 3 | c.172_181del | p.(Pro58Argfs*18) | DC(1) | pathogenic | 1 | 0.03 | 0.006 | This study | ||
| Exon 3 | c.197dup | p.(Thr67Aspfs*15) | pathogenic | 1 | 0.05 | 0.218 | This study | |||
| Exon 3 | c.229A > T | p.(Lys77*) | DCA(1) | pathogenic | 1 | 0.04 | 0.024 | This study | ||
| Exon 3 | c.232del | p.(Ile78*) | pathogenic | 1 | 0.03 | 0.005 | This study | |||
| Exon 3 | c.322C > T | p.(Gln108*) | DCA(1) | pathogenic | 1 | 0.04 | 0.033 | [21] | ||
| Exon 3 | c.377del | p.(Pro126Leufs*22) | pathogenic | 1 | 0.11 | 0.06 | This study | |||
| Exon 3 | c.403_404del | p.(His136Phefs*120) | pathogenic | 1 | 0.09 | 0.094 | [11] | |||
| Exon 3 | c.508 T > C | p.(Ser170Pro) | 0 | 1 | DC(1) | pathogenic | 1 | 0.04 | 0.011 | [22] |
| Exon 3 | c.509C > T | p.(Ser170Phe) | 0 | 1 | DC(1) | likely pathogenic | 1 | 0.06 | 0.041 | [23] |
| Exon 3 | c.538C > T | p.(Gln180*) | DCA(1) | likely pathogenic | 1 | 0.06 | 0.04 | This study | ||
| Exon 3 | c.550G > T | Exon 3 skipping | 0 | 1 | DC(1) | pathogenic | 2 | 0.05 | 0.002 | [20] |
| 0.08 | 0.076 | |||||||||
| Exon 3 | c.550G > A | Exon 3 skipping | 0 | 1 | DC(1) | pathogenic | 1 | 0.04 | 0.028 | [24] |
| Intron 3 | c.550 + 1G > T | Splicing defect | pathogenic | 1 | 0.05 | 0.016 | This study | |||
| Intron 3 | c.550 + 1G > A | Splicing defect | pathogenic | 1 | 0.20 | 0.046 | [25] | |||
| Exon 4 | c.623dup | p.(Ala209Glyfs*48) | pathogenic | 1 | 0.15 | 0.200 | This study | |||
| Exon 4 | c.635dup | p.(Phe213Leufs*44) | pathogenic | 1 | 0.07 | 0.061 | This study | |||
| Exon 4 | c.666_667del | p.(Gln223Aspfs*33) | pathogenic | 1 | 0.06 | 0.111 | [12] | |||
| Exon 4 | c.669_670del | p.(Gln223Hisfs*33) | pathogenic | 1 | 0.05 | 0.003 | [26] | |||
| Exon 4 | c.673_675del | p.(Phe225del) | pathogenic | 1 | 0.08 | 0.054 | This study | |||
| Intron 4 | c.685 + 1G > T | Splicing defect | pathogenic | 1 | 0.04 | 0.018 | [12] | |||
| Intron 4 | c.686-1G > A | Splicing defect | pathogenic | 2 | 0.04 | 0.030 | [27] | |||
| 0.07 | 0.052 | |||||||||
| Exon 5 | c.708 T > G | p.(Phe236Leu) | 0.007 | 0.999 | DC(0.907) | likely pathogenic | 1 | 0.12 | 0.329 | This study |
| Exon 5 | c.733_736dup | p.(Ser246Lysfs*12) | pathogenic | 1 | 0.05 | 0.046 | This study | |||
| Exon 5 | c.744_745del | p.(Arg249Serfs*7) | pathogenic | 2 | 0.03 | 0.032 | [28] | |||
| 0.03 | 0.028 | |||||||||
| Exon 5 | c.779dup | p.(Leu261Alafs*44) | pathogenic | 1 | 0.07 | 0.059 | This study | |||
| Exon 5 | c.785dup | p.(Asn263Glnfs*42) | pathogenic | 1 | 0.07 | 0.054 | This study | |||
| Exon 5 | c.816_818del | p.(Asn272del) | pathogenic | 1 | 0.07 | 0.025 | [29] | |||
| Exon 6 | c.941_942insTC | p.(Phe315Profs*7) | pathogenic | 1 | 0.06 | 0.058 | This study | |||
| Exon 6 | c.951dup | p.(Ser318Leufs*10) | pathogenic | 1 | 0.04 | 0.014 | This study | |||
| Exon 6 | c.983_984delinsC | p.(Lys328Thrfs*13) | pathogenic | 1 | 0.04 | 0.030 | This study | |||
| Exon 6 | c.1019del | p.(Leu340*) | pathogenic | 1 | 0.03 | 0.002 | This study | |||
| Intron 6 | c.1030-2A > G | Splicing defect | pathogenic | 1 | 0.04 | 0.037 | [30] | |||
| Exon 7 | c.1051del | p.(His351Thrfs*3) | pathogenic | 1 | 0.06 | 0.020 | This study | |||
| Exon 7 | c.1094dup | p.(His365Glnfs*4) | pathogenic | 2 | 0.05 | 0.019 | This study | |||
| 0.05 | 0.073 | |||||||||
| Exon 7 | c.1100 T > G | p.(Leu367Arg) | 0.001 | 1 | DC(1) | likely pathogenic | 1 | 0.07 | 0.082 | This study |
| Exon 7 | c.1121 T > C | p.(Leu374Pro) | 0.006 | 1 | DC(1) | VUS | 1 | 0.07 | 0.054 | ClinVar VCV000426682.2 |
| Exon 7 | c.1157_1158del | p.(Leu386Argfs*38) | pathogenic | 1 | 0.06 | 0.024 | [30] | |||
| Exon 7 | c.1186del | p.(Leu396*) | pathogenic | 1 | 0.08 | 0.063 | This study | |||
| Exon 7 | c.1192C > G | p.(Leu398Val) | 0.141 | 0.902 | P(1) | likely pathogenic | 1 | 0.09 | 0.101 | This study |
| Exon 7 | c.1193 T > G | p.(Leu398Arg) | 0.002 | 1 | P(0.915) | likely pathogenic | 1 | 0.05 | 0.016 | This study |
| Exon 7 | c.1223A > G | p.(Asp408Gly) | 0.006 | 0.999 | DC(0.987) | pathogenic | 1 | 0.07 | 0.022 | [31] |
| Intron 7 | c.1249 + 2 T > C | Splicing defect | pathogenic | 1 | 0.04 | 0.034 | This study | |||
| Intron 7 | c.1250-2A > G | Splicing defect | pathogenic | 1 | 0.25 | 0.026 | [21] | |||
| Exon 8 | c.1269 T > A | p.(Tyr423*) | DC(1) | pathogenic | 1 | 0.03 | 0.01 | This study | ||
| Exon 8 | c.1289 T > C | p.(Leu430Pro) | 0.001 | 1 | DC(1) | pathogenic | 1 | 0.08 | 0.071 | [12] |
| Exon 8 | c.1289 T > G | p.(Leu430Arg) | 0.001 | 0.999 | DC(0.999) | likely pathogenic | 1 | 0.05 | 0.019 | This study |
| Exon 8 | c.1312del | p.(Val438Phefs*12) | pathogenic | 1 | 0.05 | 0.060 | [11] | |||
| Exon 8 | c.1340 T > C | p.(Leu447Pro) | 0.001 | 1 | DC(1) | pathogenic | 1 | 0.12 | 0.142 | [26] |
| Exon 8 | c.1342G > T | p.(Glu448*) | DC(1) | pathogenic | 1 | 0.05 | 0.049 | [14] | ||
| Exon 8 | c.1351G > A | p.(Glu451Lys) | 0.042 | 1 | DC(1) | pathogenic | 1 | 0.05 | 0.060 | [32] |
| Exon 8 | c.1356_1357del | p.(Val454Glyfs*18) | pathogenic | 1 | 0.05 | 0.018 | [33] | |||
| Exon 8 | c.1373C > T | p.(Ala458Val) | 0.004 | 1 | P(0.960) | pathogenic | 1 | 0.13 | 0.053 | [34] |
| Exon 8 | c.1379C > G | p.(Ser460Cys) | 0.005 | 1 | P(1) | likely pathogenic | 1 | 0.14 | 0.044 | This study |
| Exon 8 | c.1396C > T | p.(Arg466Cys) | 0.003 | 0.969 | DC(1) | pathogenic | 1 | 0.56 | 0.007 | [35] |
| Exon 8 | c.1396C > A | p.(Arg466Cys) | 0.013 | 0.255 | DC(1) | pathogenic | 1 | 0.64 | 0.027 | [36] |
| Exon 8 | c.1397G > T | p.(Arg466Leu) | 0.007 | 0.067 | DC(1) | pathogenic | 1 | 0.35 | 0.016 | [37] |
| Exon 8 | c.1397G > A | p.(Arg466His) | 0.005 | 0.666 | DCA(1) | pathogenic | 3 | 0.37 | 0.021 | [35] |
| 0.36 | 0.043 | |||||||||
| 0.37 | 0.015 | |||||||||
| Exon 8 | c.1420C > T | p.(Gln474*) | DC(1) | pathogenic | 1 | 0.04 | 0.008 | [38] | ||
| Exon 8 | c.1422G > C | p.(Gln474His) | 0.2 | 0.996 | P(0.725) | likely pathogenic | 1 | 0.05 | 0.009 | This study |
| Exon 8 | c.1423C > T | p.(Gln475*) | DC(1) | pathogenic | 1 | 0.06 | 0.109 | [38] | ||
| Exon 8 | c.1424A > C | p.(Gln475Pro) | 0.006 | 0.998 | DC(0.961) | likely pathogenic | 1 | 0.09 | 0.101 | This study |
| Exon 8 | c.1425G > T | p.(Gln475His) | 0.074 | 0.174 | DC(0.865) | likely pathogenic | 1 | 0.05 | 0.003 | This study |
| Exon 8 | c.1480C > T | p.(Arg494*) | DC(1) | pathogenic | 7 | 0.05 | 0.050 | [24] | ||
| 0.06 | 0.056 | |||||||||
| 0.05 | 0.048 | |||||||||
| 0.07 | 0.084 | |||||||||
| 0.08 | 0.066 | |||||||||
| 0.05 | 0.101 | |||||||||
| 0.05 | 0.054 | |||||||||
| Exon 8 | c.1481G > T | p.(Arg494Leu) | 0.004 | 0.999 | DC(0.994) | pathogenic | 2 | 0.05 | 0.128 | [12] |
| 0.05 | 0.011 | |||||||||
| Exon 8 | c.1492C > T | p.(Pro498Ser) | 0 | 1 | DC(1) | pathogenic | 1 | 0.05 | 0.031 | [24] |
| Exon 1–2 | Deletion of exon 1–2 | pathogenic | 1 | 0.04 | 0.031 | [26] | ||||
| Exon 3–4 | Duplication of exon 3–4 | pathogenic | 1 | 0.05 | 0.100 | This study | ||||
| Exon 4 | Deletion of exon 4 | pathogenic | 3 | 0.04 | 0.049 | [21] | ||||
| 0.07 | 0.045 | |||||||||
| 0.04 | 0.018 | |||||||||
| Exon 1–4 | Deletion of exon 1–4 | pathogenic | 1 | 0.04 | 0.058 | [39] | ||||
| Exon 2–4 | Deletion of exon 2–4 | pathogenic | 1 | 0.06 | 0.041 | This study | ||||
a SIFT® (Sorting Intolerant From Tolerant) is a program that predicts whether an amino acid substitution affects protein function. SIFT scores range from 0.0 (harmful) to 1.0 (tolerable). This score can be interpreted as follows: variants with scores in the range of 0.0 to 0.05 are considered to be harmful
b PolyPhen-2® (Polymorphism Phenotyping v2) is a tool which predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations. The score can be interpreted as follows: 0.0 to 0.15 – Variants with scores in this range are predicted to be benign; 0.15 to 1.0 – Variants with scores in this range are possibly damaging; 0.85 to 1.0 – Variants with scores in this range are more confidently predicted to be damaging
c Mutation Taster® is a free web-based application to evaluate DNA sequence variants for their disease-causing potential. In order to predict the potential pathogenicity of an alteration, each variant is distributed between Disease Causing (DC), according to NCBI ClinVar, and Polymorphism (P), according to the 1000 Genomes Project, with corresponding probability in brackets
d Clinical classification is based on the ACMG-AMP criteria for variant classification supporting pathogenicity
eThe normal range of C1-INH protein is 0.21–0.39 g/L
fThe normal range of C4 protein is 0.100–0.400 g/L
Missense and in-frame mutations
We identified 26 missense variants in 28 unrelated patients, 10 of which were novel (Table 2). A total of 16 previously described missense variants, including c.1A > G;p.(Met1Val), c.49G > A;p.(Gly17Arg), c.508 T > C;p.(Ser170Pro), and etc., were identified in the present study. In HAE-2, four previously reported variants were detected. These four variants include c.1396C > T;p.(Arg466Cys), c.1396C > A;p.(Arg466Cys), c.1397G > T; p.(Arg466Leu), and c.1397G > A; p.(Arg466His). All of these variants are located in exon 8 and affect Arg466. Arg466 has been identified as a critical residue of the active center loop, and variants of codon 466 are the most frequent cause of HAE-2. HAE-2 patients carrying the Arg466 variants had normal or elevated antigenic but low functional C1-INH levels, and low C4 levels (Table S3).
In addition, 10 novel variants c.100C > A;p.(Pro34Thr), c.708 T > G;p.(Phe236Leu), c.1100 T > G;p.(Leu367Arg), c.1192C > G;p.(Leu398Val), c.1193 T > G;p.(Leu398Arg), c.1289 T > G;p.(Leu430Arg), c.1379C > G;p.(Ser460Cys), c.1422G > C;p.(Gln474His), c.1424A > C;p.(Gln475Pro), and c.1425G > T;p.(Gln475His) were identified. Mutations c.1192C > G;p.(Leu398Val) and c.1424A > C;p.(Gln475Pro) were detected in the same family, and all three symptomatic members of this lineage carried both mutations (Figure S1). However, there was a significant clinical heterogeneity in these three patients. One patient developed the disease at age 14, had previous skin, gastrointestinal and laryngeal edema; whereas the other two patients first developed HAE at ages 25 and 23, respectively, both presented only with gastrointestinal edema. Therefore, the genetic variant was not sufficient to explain the phenotype of the patients. The cause of the biological plausibility of the difference in clinical phenotype in the 3 individuals with the same 2 variants remains to be investigated. In addition, a novel in-frame deletion c.673_675del;p.(Phe225del) was identified in one patient.
Nonsense and frameshift mutations
A total of 11 nonsense and 22 frameshift mutations were identified in 41 unrelated patients. Among these mutations, 6 nonsense and 14 frameshift mutations were newly identified (see Table 2). For c.744_745del;p.(Arg249Serfs*7) and c.1094dup;p.(His365Glnfs*4), each of them was uniquely identified in a total of 2 patients. The previously reported c.1480C > T;p.(Arg494*) was identified in 7 patients. Other nonsense and frameshift mutations were found in different patients.
Splicing mutations
We detected 10 splicing mutations in 12 patients, 2 of which had not been reported previously. Mutation c.686-1G > A presented in 2 unrelated patients. This mutation may affect alternative splicing of SERPING1 through regulation of serine/arginine splicing factor (SRSF) 1 [40]. Mutation c.1030-2A > G may affect SRSF2-mediated splicing. In particular, the splicing effects of c.550G > T, c.550G > A, and c.120_121del have been demonstrated to increase exon 3 skipping and the three mutations are therefore not considered as real missense or deletion variants [20].
Gross deletions and duplications
MLPA analysis revealed gross deletion of exon 4 in 3 patients. Exon 4 of SERPING1 is a hotspot for large fragment deletions because Alu has a high repeat density in introns 3 and 4 [41]. The Alu sequences are movable repetitive elements which may represent hotspots for nonhomologous recombination leading to various hereditary diseases [11]. In addition, large deletion of exon 1 and 2, exon 2 – 4, and exon 1 – 4 was identified in 3 patients, respectively. Duplication of exon 3 and 4 was identified in 1 patient.
Polymorphism
Six previously described rs28362945 (c.51 + 101G > A), rs1005510 (c.52-130C > T), rs11546660 (c.167 T > C), rs11229063 (c.685 + 88G > A), rs2511988 (c.1030-20A > G), and rs4926 (c.1438G > A) SNPs were identified in this study.
Family study
In this study, 35 novel variants were identified. To support the pathogenicity of the novel variants, family studies were conducted. Unfortunately, only 5 patients carrying the novel variants provided blood samples from their family members. The c.172_181del, c.229A > T, c.232del, c.1051del, c.1192C > G, and c.1424A > C variants co-segregated with the disease in these five families. Of the remaining novel variants, 16 were nonsense variants or frameshift variants. These 16 novel variants create premature stop codons, which result in the termination of mRNA translation and the synthesis of truncated protein products [42]. These premature stop codons are all located upstream of the reactive loop (RCL) of C1-INH [42]. As a result, the synthesized protein will lack the RCL and could not identify the target proteases, resulting in a lack of inhibitory function of C1-INH [42]. In addition, there are also 2 novel large defects and 2 novel splicing mutations. Accroding to the ACMG-AMP criteria, nonsense, frameshift, canonical ± 1 or 2 splice sites, initiation codon, and large defects can provide very strong evidence of pathogenicity [17]. The 2 large defects mutations, the 2 splicing mutations, and the 16 nonsense or frameshift variants can be classified as pathogenic accroding to the ACMG-AMP criteria. Therefore, family studies have not been undertaken for the total 20 novel variants already classified as pathogenic. For the remaining 9 novel missense and in-frame mutations, we did not perform family studies because patient carrying the 9 novel missense and in-frame mutations refused to provide parental blood samples or were unable to provide blood samples due to the death of both parents.
Discussion
In this study, 76 different variants were identified in 90 unrelated Chinese patients with HAE, 46.1% of which were novel. This study expands the mutational spectrum of HAE and provides evidence for accurate diagnosis and predictive genetic counseling. The sensitivity of mutational analyses applied in this study is 92.8%, which is comparable with other studies reporting a sensitivity of ≥ 82% [21, 43, 44]. We failed to find disease-causing mutations in seven patients. One possible reason is that the disease-causing mutations are located outside the sequenced region. Mutations in deep intronic region of HAE have been reported earlier. Sofia Vatsiou et al. reported the novel mutation c.-22-155G > T in intron 1 of the SERPING1 gene, which was curated as pathogenic according to the American College of Medical Genetics and Genomics 2015 guidelines [45]. In the same year, Pavla Hujová et al. reported c.1029 + 384A > G, a novel deep intronic mutation in intron 6, is responsible for HAE. This mutation results in de novo donor splice site creation and subsequent pseudoexon inclusion [46].
The mutations that cause HAE are diverse in the Chinese population. Missense and in-frame mutations are the most common types of mutations, accounting for 36.8% of all mutations, followed by frameshift mutations (28.9%), nonsense mutations (14.5%), splice site mutations (13.2%), and gross deletions and duplications (6.6%) (Fig. 1). Figure 2 shows the disease-causing variants that have been identified in Chinese HAE patients [9, 14, 42]. Of these variants, 22 were found in exon 8, accounting for nearly one-third of all variants (Figure S2). Exon 8 appears to be a mutational hotspot in Chinese HAE patients as well as HAE patients from other countries [47]. This may be due to the fact that exon 8 contains the critical hinge region and the reaction center of the C1 inhibitor molecule [48]. Frameshift mutations and nonsense mutations account for nearly half of all mutations. These 2 mutation types result in a premature stop codon, or a nonsense codon in the transcribed mRNA [49]. When ribosomes encounter a premature stop codon, a nonsense-mediated mRNA decay is triggered, which results in a truncated, incomplete, and usually nonfunctional protein product [49]. Our previous study also showed that frameshift mutations and nonsense mutations led to a reduced expression of SERPING1 mRNA in peripheral blood [42].
Fig. 1.
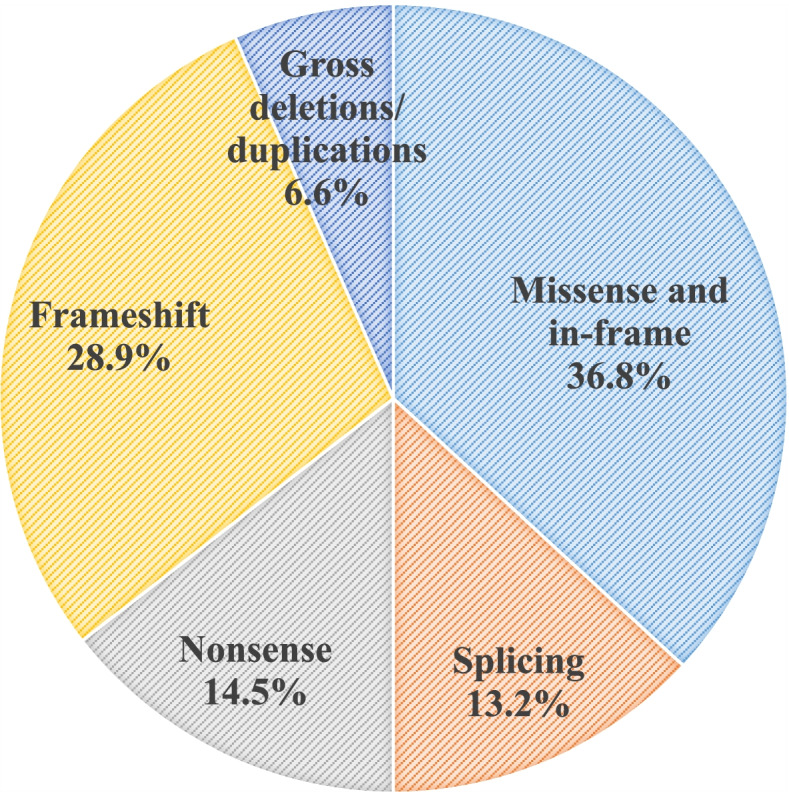
Distribution of the SERPING1 mutation types in Chinese HAE patients
Fig. 2.

Genetic variants identified in Chinese HAE patients. Exons are numbered 1–8 and are drawn to scale in blue, with coding sequence in dark blue. Variants in blue font are reported in this study; variants in black font are reported in other studies; variants in dark red font are identified in HAE-2
In the Chinese HAE cohort, patients with HAE-1 and HAE-2, respectively, accounted for 93.8% and 6.2% of all HAE patients. The increased proportion of HAE-2 compared with our previously reported 1.27% may be a more comprehensive response to the characteristics of Chinese HAE patients after expanding the sample size [9, 50]. The updated proportion of HAE-2 in China is comparable to that found in Denmark, Spain, and Germany [43, 44, 51]. Besides, the percentage of gastrointestinal edema in Chinese HAE patients was 69.1%, which was higher compared with our previous report of 34.2% [9, 50]. The percentage reported in the current study is higher than that in Japan (45.0%) [52] and Taiwan, China (18.2%), [53] but lower than those in Western countries (German: 97.0% and Danish: 96.1%) [51, 54]. Moreover, the higher percentage of gastrointestinal edema reported in this study indicates that HAE is gradually being recognized in China, and more patients with gastrointestinal edema are being diagnosed. In addition to gastrointestinal edema, the percentage of laryngeal edema in Chinese HAE patients in the current study (66.0%) was also slightly higher compared with that of our previous report (58.9%) [50].
Several limitations should be noticed. First, this is a single center analysis although the patients are from all over China. Second, we did not find HAE patients with normal C1-INH levels. This does not mean that the SERPING1 gene is the only pathogenic gene in Chinese HAE patients. Given the vast population of China, it is possible that there are HAE patients with normal C1-INH levels that have not been identified because of the absence of commercially available biomarkers. Third, we failed to find disease-causing mutations in seven HAE patients. The pathogenic mutations in these patients may be located outside of the sequenced region. The intronic region of SERPING1 will be sequenced in a subsequent study. Fourth, although we reported 35 novel mutations, functional validation was not performed, which can be explored in future studies.
Conclusions
HAE is a rare disease with great heterogeneity in both genotype and clinical phenotype. This study updated the clinical phenotypic characteristics and mutational spectrum of Chinese HAE patients. 41 previously reported variants and 35 novel variants were identified in 90 unrelated patients. This study provides more comprehensive information on the genetic characteristics and clinical presentation of Chinese HAE patients.
Supplementary Information
Additional file 1: Table S1. Primers and PCR conditions. Table S2. Demographic characteristics and clinical manifestations of 7 patients without genetic variants. Table S3. The antigenic and functional C1-INH levels and C4 levels corresponding to the genetic variants of HAE-2. Figure S1. Mutations c.1192C>G;p.(Leu398Val) and c.1424A>C;p.(Gln475Pro) were detected in all three symptomatic members of this lineage. Figure S2. Proportional distribution of variants on the SERPING1 gene.
Acknowledgements
We appreciate all the cooperation from the HAE patients and healthy controls included in this study.
Authors' contributions
Xue Wang made substantial contributions to acquisition, analysis, and interpretation of data, and drafted the manuscript; Shubin Lei, Yingyang Xu, and Shuang Liu also made considerable contributions to the analysis and interpretation of data; Yu-Xiang Zhi made substantial contributions to conception and design, revised the manuscript critically for important intellectual content, and gave final approval of the version to be published. The author(s) read and approved the final manuscript.
Funding
This project was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1–003).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
All the subjects signed an informed consent under a research protocol approved by the research and ethics board of the Peking Union Medical College Hospital (Approval number: HS-2402).
Consent for publication
All the subjects signed informed consent regarding publishing their data.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Busse PJ, Christiansen SC. Hereditary Angioedema. N Engl J Med. 2020;382(12):1136–1148. doi: 10.1056/NEJMra1808012. [DOI] [PubMed] [Google Scholar]
- 2.Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356(9225):213–217. doi: 10.1016/S0140-6736(00)02483-1. [DOI] [PubMed] [Google Scholar]
- 3.Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343(4):1286–1289. doi: 10.1016/j.bbrc.2006.03.092. [DOI] [PubMed] [Google Scholar]
- 4.Bafunno V, Firinu D, D'Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141(3):1009–1017. doi: 10.1016/j.jaci.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 5.Bork K, Wulff K, Steinmuller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2018;73(2):442–450. doi: 10.1111/all.13270. [DOI] [PubMed] [Google Scholar]
- 6.Bork K, Wulff K, Rossmann H, Steinmuller-Magin L, Braenne I, Witzke G, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy. 2019;74(12):2479–2481. doi: 10.1111/all.13869. [DOI] [PubMed] [Google Scholar]
- 7.Ariano A, D'Apolito M, Bova M, Bellanti F, Loffredo S, D'Andrea G, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy. 2020;75(11):2989–2992. doi: 10.1111/all.14454. [DOI] [PubMed] [Google Scholar]
- 8.Bork K, Wulff K, Mohl BS, Steinmuller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. 2021;148:1041–1048. doi: 10.1016/j.jaci.2021.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Liu S, Xu Y, Liu Y, Zhi Y. Hereditary angioedema: a Chinese perspective. Eur J Dermatol. 2019;29(1):14–20. doi: 10.1684/ejd.2018.3487. [DOI] [PubMed] [Google Scholar]
- 10.Cugno M, Nussberger J, Cicardi M, Agostoni A. Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol. 2003;3(3):311–317. doi: 10.1016/S1567-5769(02)00162-5. [DOI] [PubMed] [Google Scholar]
- 11.Ponard D, Gaboriaud C, Charignon D, Ghannam A, Wagenaar-Bos IGA, Roem D, et al. SERPING1 mutation update: Mutation spectrum and C1 Inhibitor phenotypes. Hum Mutat. 2020;41(1):38–57. doi: 10.1002/humu.23917. [DOI] [PubMed] [Google Scholar]
- 12.Pappalardo E, Caccia S, Suffritti C, Tordai A, Zingale LC, Cicardi M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: functional and structural correlates. Mol Immunol. 2008;45(13):3536–3544. doi: 10.1016/j.molimm.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygoren-Pursun E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy. 2022;00:1–30.
- 14.Xu YY, Zhi YX, Yin J, Wang LL, Wen LP, Gu JQ, et al. Mutational spectrum and geno-phenotype correlation in Chinese families with hereditary angioedema. Allergy. 2012;67(11):1430–1436. doi: 10.1111/all.12024. [DOI] [PubMed] [Google Scholar]
- 15.Busse PJ, Christiansen SC, Riedl MA, Banerji A, Bernstein JA, Castaldo AJ, et al. US HAEA Medical Advisory Board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract. 2021;9(1):132–50. doi: 10.1016/j.jaip.2020.08.046. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Shi L, Zhang K, Zhang Y, Hu S, Zhao T, et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018;46(D1):D1039–D1048. doi: 10.1093/nar/gkx1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Germenis AE, Margaglione M, Pesquero JB, Farkas H, Cichon S, Csuka D, et al. International Consensus on the Use of Genetics in the Management of Hereditary Angioedema. J Allergy Clin Immunol Pract. 2020;8(3):901–911. doi: 10.1016/j.jaip.2019.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Kesim B, Uyguner ZO, Gelincik A, Mete Gokmen N, Sin AZ, Karakaya G, et al. The Turkish Hereditary Angioedema Pilot Study (TURHAPS): the first Turkish series of hereditary angioedema. Int Arch Allergy Immunol. 2011;156(4):443–450. doi: 10.1159/000323915. [DOI] [PubMed] [Google Scholar]
- 19.Xu YY, Zhi YX. A Compound Mutation (c.953C<G and c.49G<A) Aggravates Functional Impairments of C1-INH in Hep G2 cells. Allergy Asthma Immunol Res. 2018;10(3):285–6. doi: 10.4168/aair.2018.10.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grodecka L, Hujova P, Kramarek M, Krsjakova T, Kovacova T, Vondraskova K, et al. Systematic analysis of splicing defects in selected primary immunodeficiencies-related genes. Clin Immunol. 2017;180:33–44. doi: 10.1016/j.clim.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Gosswein T, Kocot A, Emmert G, Kreuz W, Martinez-Saguer I, Aygoren-Pursun E, et al. Mutational spectrum of the C1INH (SERPING1) gene in patients with hereditary angioedema. Cytogenet Genome Res. 2008;121(3–4):181–188. doi: 10.1159/000138883. [DOI] [PubMed] [Google Scholar]
- 22.Pappalardo E, Cicardi M, Duponchel C, Carugati A, Choquet S, Agostoni A, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol. 2000;106(6):1147–1154. doi: 10.1067/mai.2000.110471. [DOI] [PubMed] [Google Scholar]
- 23.Faiyaz-Ul-Haque M, Al-Gazlan S, Abalkhail HA, Al-Abdulatif A, Toulimat M, Peltekova I, et al. Novel and recurrent mutations in the C1NH gene of Arab patients affected with hereditary angioedema. Int Arch Allergy Immunol. 2010;151(2):149–154. doi: 10.1159/000236005. [DOI] [PubMed] [Google Scholar]
- 24.Verpy E, Biasotto M, Brai M, Misiano G, Meo T, Tosi M. Exhaustive mutation scanning by fluorescence-assisted mismatch analysis discloses new genotype-phenotype correlations in angiodema. Am J Hum Genet. 1996;59(2):308–319. [PMC free article] [PubMed] [Google Scholar]
- 25.Kalmar L, Bors A, Farkas H, Vas S, Fandl B, Varga L, et al. Mutation screening of the C1 inhibitor gene among Hungarian patients with hereditary angioedema. Hum Mutat. 2003;22(6):498. doi: 10.1002/humu.9202. [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Lera A, Garrido S, Roche O, Lopez-Trascasa M. SERPING1 mutations in 59 families with hereditary angioedema. Mol Immunol. 2011;49(1–2):18–27. doi: 10.1016/j.molimm.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 27.Veronez CL, Mendes AR, Leite CS, Gomes CP, Grumach AS, Pesquero JB, et al. The panorama of primary Angioedema in the Brazilian population. J Allergy Clin Immunol Pract. 2021;9(6):2293–304. doi: 10.1016/j.jaip.2020.11.039. [DOI] [PubMed] [Google Scholar]
- 28.Guryanova I, Suffritti C, Parolin D, Zanichelli A, Ishchanka N, Polyakova E, et al. Hereditary angioedema due to C1 inhibitor deficiency in Belarus: epidemiology, access to diagnosis and seven novel mutations in SERPING1 gene. Clin Mol Allergy. 2021;19(1):3. doi: 10.1186/s12948-021-00141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bissler JJ, Cicardi M, Donaldson VH, Gatenby PA, Rosen FS, Sheffer AL, et al. A cluster of mutations within a short triplet repeat in the C1 inhibitor gene. Proc Natl Acad Sci U S A. 1994;91(20):9622–9625. doi: 10.1073/pnas.91.20.9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashimura C, Kiyohara C, Fukushi JI, Hirose T, Ohsawa I, Tahira T, et al. Clinical and genetic features of hereditary angioedema with and without C1-inhibitor (C1-INH) deficiency in Japan. Allergy. 2021;76(11):3529–3534. doi: 10.1111/all.15034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinho A, Mendes J, Simoes O, Nunes R, Gomes J, Dias Castro E, et al. Mutations analysis of C1 inhibitor coding sequence gene among Portuguese patients with hereditary angioedema. Mol Immunol. 2013;53(4):431–434. doi: 10.1016/j.molimm.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Rijavec M, Korosec P, Silar M, Zidarn M, Miljkovic J, Kosnik M. Hereditary angioedema nationwide study in Slovenia reveals four novel mutations in SERPING1 gene. PLoS ONE. 2013;8(2):e56712. doi: 10.1371/journal.pone.0056712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nabilou S, Pak F, Alizadeh Z, Fazlollahi MR, Houshmand M, Ayazi M, et al. Genetic Study of Hereditary Angioedema Type I and Type II (First Report from Iranian Patients: Describing Three New Mutations). Immunol Invest. 2020;51(1):170-81. [DOI] [PubMed]
- 34.Siddique Z, McPhaden AR, Whaley K. Type II hereditary angio-oedema associated with two mutations in one allele of the C1-inhibitor gene around the reactive-site coding region. Hum Hered. 1992;42(5):298–301. doi: 10.1159/000154086. [DOI] [PubMed] [Google Scholar]
- 35.Skriver K, Radziejewska E, Silbermann JA, Donaldson VH, Bock SC. CpG mutations in the reactive site of human C1 inhibitor. J Biol Chem. 1989;264(6):3066–3071. doi: 10.1016/S0021-9258(18)94031-7. [DOI] [PubMed] [Google Scholar]
- 36.Obtulowicz K, Ksi AT, Bogdali A, Dyga W, Czarnobilska E, Juchacz A. Genetic variants of SERPING1 gene in Polish patients with hereditary angioedema due to C1 inhibitor deficiency. Cent Eur J Immunol. 2020;45(3):301–309. doi: 10.5114/ceji.2020.101252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frangi D, Aulak KS, Cicardi M, Harrison RA, Davis AE., 3rd A dysfunctional C1 inhibitor protein with a new reactive center mutation (Arg-444–>Leu) FEBS Lett. 1992;301(1):34–36. doi: 10.1016/0014-5793(92)80204-T. [DOI] [PubMed] [Google Scholar]
- 38.Speletas M, Szilagyi A, Psarros F, Moldovan D, Magerl M, Kompoti M, et al. Hereditary angioedema: molecular and clinical differences among European populations. J Allergy Clin Immunol. 2015;135(2):570–573. doi: 10.1016/j.jaci.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Duponchel C, Di Rocco C, Cicardi M, Tosi M. Rapid detection by fluorescent multiplex PCR of exon deletions and duplications in the C1 inhibitor gene of hereditary angioedema patients. Hum Mutat. 2001;17(1):61–70. doi: 10.1002/1098-1004(2001)17:1<61::AID-HUMU7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 40.Paz S, Ritchie A, Mauer C, Caputi M. The RNA binding protein SRSF1 is a master switch of gene expression and regulation in the immune system. Cytokine Growth Factor Rev. 2021;57:19–26. doi: 10.1016/j.cytogfr.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoppa-Lyonnet D, Carter PE, Meo T, Tosi M. Clusters of intragenic Alu repeats predispose the human C1 inhibitor locus to deleterious rearrangements. Proc Natl Acad Sci U S A. 1990;87(4):1551–1555. doi: 10.1073/pnas.87.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu YY, Gu JQ, Zhi YX. Hereditary angioedema caused by a premature stop codon mutation in the SERPING1 gene. Clin Transl Allergy. 2020;10(1):53. doi: 10.1186/s13601-020-00360-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bygum A, Fagerberg CR, Ponard D, Monnier N, Lunardi J, Drouet C. Mutational spectrum and phenotypes in Danish families with hereditary angioedema because of C1 inhibitor deficiency. Allergy. 2011;66(1):76–84. doi: 10.1111/j.1398-9995.2010.02456.x. [DOI] [PubMed] [Google Scholar]
- 44.Roche O, Blanch A, Duponchel C, Fontan G, Tosi M, Lopez-Trascasa M. Hereditary angioedema: the mutation spectrum of SERPING1/C1NH in a large Spanish cohort. Hum Mutat. 2005;26(2):135–144. doi: 10.1002/humu.20197. [DOI] [PubMed] [Google Scholar]
- 45.Vatsiou S, Zamanakou M, Loules G, Psarros F, Parsopoulou F, Csuka D, et al. A novel deep intronic SERPING1 variant as a cause of hereditary angioedema due to C1-inhibitor deficiency. Allergol Int. 2020;69(3):443–449. doi: 10.1016/j.alit.2019.12.009. [DOI] [PubMed] [Google Scholar]
- 46.Hujova P, Soucek P, Grodecka L, Grombirikova H, Ravcukova B, Kuklinek P, et al. Deep Intronic Mutation in SERPING1 Caused Hereditary Angioedema Through Pseudoexon Activation. J Clin Immunol. 2020;40(3):435–446. doi: 10.1007/s10875-020-00753-2. [DOI] [PubMed] [Google Scholar]
- 47.Steiner UC, Keller M, Schmid P, Cichon S, Wuillemin WA. Mutational spectrum of the SERPING1 gene in Swiss patients with hereditary angioedema. Clin Exp Immunol. 2017;188(3):430–436. doi: 10.1111/cei.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanch A, Roche O, Lopez-Granados E, Fontan G, Lopez-Trascasa M. Detection of C1 inhibitor (SERPING1/C1NH) mutations in exon 8 in patients with hereditary angioedema: evidence for 10 novel mutations. Hum Mutat. 2002;20(5):405–406. doi: 10.1002/humu.9073. [DOI] [PubMed] [Google Scholar]
- 49.Amrani N, Sachs MS, Jacobson A. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol. 2006;7(6):415–425. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]
- 50.Xu YY, Jiang Y, Zhi YX, Yin J, Wang LL, Wen LP, et al. Clinical features of hereditary angioedema in Chinese patients: new findings and differences from other populations. Eur J Dermatol. 2013;23(4):500–504. doi: 10.1684/ejd.2013.2105. [DOI] [PubMed] [Google Scholar]
- 51.Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267–274. doi: 10.1016/j.amjmed.2005.09.064. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto T, Horiuchi T, Miyahara H, Yoshizawa S, Maehara J, Shono E, et al. Hereditary angioedema in Japan: genetic analysis of 13 unrelated cases. Am J Med Sci. 2012;343(3):210–214. doi: 10.1097/MAJ.0b013e31822bdb65. [DOI] [PubMed] [Google Scholar]
- 53.Lei WT, Shyur SD, Huang LH, Kao YH, Lo CY. Type I hereditary angioedema in Taiwan – clinical, biological features and genetic study. Asian Pac J Allergy Immunol. 2011;29(4):327–331. [PubMed] [Google Scholar]
- 54.Bygum A. Hereditary angio-oedema in Denmark: a nationwide survey. Br J Dermatol. 2009;161(5):1153–1158. doi: 10.1111/j.1365-2133.2009.09366.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Primers and PCR conditions. Table S2. Demographic characteristics and clinical manifestations of 7 patients without genetic variants. Table S3. The antigenic and functional C1-INH levels and C4 levels corresponding to the genetic variants of HAE-2. Figure S1. Mutations c.1192C>G;p.(Leu398Val) and c.1424A>C;p.(Gln475Pro) were detected in all three symptomatic members of this lineage. Figure S2. Proportional distribution of variants on the SERPING1 gene.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.