

Abstract
p53, encoded by the tumor suppressor gene TP53, is one of the most important tumor suppressor factors in vivo and can be negatively regulated by MDM2 through p53–MDM2 negative feedback loop. Abnormal p53 can be observed in almost all tumors, mainly including p53 mutation and functional inactivation. Blocking MDM2 to restore p53 function is a hotspot in the development of anticancer candidates. Till now, nine MDM2 inhibitors with different structural types have entered clinical trials. However, no MDM2 inhibitor has been approved for clinical application. This review focused on the discovery, structural modification, preclinical and clinical research of the above compounds from the perspective of medicinal chemistry. Based on this, the possible defects in MDM2 inhibitors in clinical development were analyzed to suggest that the multitarget strategy or targeted degradation strategy based on MDM2 has the potential to reduce the dose-dependent hematological toxicity of MDM2 inhibitors and improve their anti-tumor activity, providing certain guidance for the development of agents targeting the p53–MDM2 interaction.
Keywords: p53, MDM2, Inhibitor, Multi-target, Degrader
Introduction
Protein–protein interactions (PPIs) play an important role in almost all biological activities (such as DNA synthesis and cell signal transduction), which can promote or inhibit the occurrence, development and metastasis of tumors. Therefore, intervention of PPI is a potential strategy in the field of tumor therapy [1]. However, since PPI is an interaction between two proteins, the binding surfaces of PPI are usually different from those of common small-molecule targets. The characteristics of large binding surfaces and dispersed activity sites make the development of small-molecule inhibitors extremely difficult for PPIs, which have long been known as ‘undruggable targets’ [2]. In recent years, studies have found that most PPIs have ‘hot spots’ [3]. According to the strategy of structure-based design or virtual screening in their hot spots, small-molecule inhibitors targeting PPIs, such as VHL (von Hippel–Lindau)-HIF1α (hypoxia inducible factor-1α) and Keap1 (Kelch-like ECH-associated protein 1)-Nrf2 (nuclear factor E2-related factor 2) [4, 5], have been successfully developed and have ideal clinical application prospects.
The p53–MDM2 (mouse double minute 2) protein interaction is an important target in the development of anti-tumor drugs. As one of the most important tumor suppressors, the p53 protein is inactivated or mutated in more than 50% of cancer cells. It plays an important role in the regulation of tumor cell cycle, apoptosis and DNA repair directly or induces the expression of downstream targets. The activation of the p53-dependent pathway caused by internal or external cell stress signals affects the occurrence, development and metastasis of cancer cells and prevents the proliferation of damaged cells with carcinogenic potential. In addition, as a transcription factor, a variety of genes can be activated by p53 to promote these tumor-related specific processes. Several proteins are closely related to the regulation of the function of p53, such as MDM2, MDMX, TPSO (translocator protein), Bcl-2 (B-cell lymphoma-2), and NFAT1 (nuclear factor of activated T-cells 1). (Fig. 1) [6]. Studies have revealed that after inactivation of the TP53 gene (encoding p53) in mice, cells lacking functional p53 cannot respond appropriately to external stimuli, resulting in a high probability of tumors [7]. Therefore, the targets related to the regulation of p53 protein expression or activation are of great research value. The MDM2 protein is widely studied because it can directly regulate the expression of p53 protein through negative feedback.
Fig. 1.

Proteins related to the regulation of p53 function
The MDM2 gene locates on the long arm 13 ~ 14 of chromosome 12 (12q13 ~ 14), with a full length of 2372 kb, including 12 exons, encoding a protein containing 498 amino acids (Fig. 2). The MDM2 protein has four functional regions: region I includes approximately 100 amino acid residues at the N-terminus, and in addition to binding to the p53 protein, it can also directly bind to gene promoters to activate gene transcription; region II is a highly acidic region that can bind to ribosomal 15 protein and 5 s rRNA; region III contains a zinc finger structure, which has the activity of transcription factors and promotes cells from G1 phase to S phase; and region IV contains a RING finger structure that can mediate the interaction with p53 and bind to DNA or RNA to participate in cell cycle regulation and promote cell proliferation [8, 9]. The ATM-dependent phosphorylation of MDM2 allows the overexpressed MDM2 to bind p53 mRNA and promote p53 translation [10]. The negative feedback regulation of MDM2 protein has many mechanisms. First, the MDM2 protein can bind to the p53 protein through PPI to prevent its expression under stress conditions and then inactivate or weaken its transcriptional function. Second, the MDM2 protein has a unique RING domain, which can promote the transfer of p53 protein from the nucleus to the cytoplasm and reduce its accumulation in the nucleus, leading to the disappearance of p53 protein transcription function. Finally, although p53 is mainly polyubiquitinated by UBE4B (ubiquitination factor E4B, an E4 ligase) for proteasomal degradation, MDM2 can cooperate with MDMX (mouse double minute X) to kinetically enhance p53 polyubiquitination, resulting in p53 stability and the occurrence of tumors [11–13]. Therefore, regulation of the p53–MDM2 protein interaction can effectively regulate the expression and function of the p53 protein.
Fig. 2.

Structure of MDM2 gene and protein
The cocrystal structure of the N-terminal transcriptional activation region of MDM2 and p53 protein showed (PDB: 1YCR, Fig. 2) that the binding surface area of p53 and MDM2 protein was small, and there was an obvious binding cavity. Further verification by alanine mutation scanning has shown that the residues of three amino acids of p53, Phe19, Trp23 and Leu26 were deeply inserted into the binding pocket of p53 and MDM2, which were, respectively, so that they were closely bound by physical action [14]. According to these three clear binding sites, a variety of structural types of MDM2 inhibitors have been reported, among which nine have entered clinical trials (RG7112, RG7388/RO6839921, MK-8242, AMG232, SAR405838, DS-3032b, HDM201, NVP-CGM097 and APG-115) (Fig. 3), with a variety of scaffold types [15–18]. For six of these, the details of their discovery and the structural modification of the lead compounds have been reported. (RO6839921 is the inactive prodrug of RG7388.) The discovery process of HDM201 has been generally reported, while MK-8242 and DS-3032b have no specific literature to report the source of this molecule. Therefore, this review mainly focuses on the seven compounds with detailed research and development processes from the perspective of medicinal chemistry and summarizes the discovery of the lead compounds, structural optimization, and preclinical and clinical results.
Fig. 3.

Nine MDM2 inhibitors in clinical trials
RG7112
RG7112 was modified by Roche based on the structure of Nutlins and was the first to be introduced into clinical trials (Fig. 4) [19]. Nutlins have an imidazoline scaffold, which was reported to have been obtained through high-throughput screening in 2004 [20]. Nutlin-3a had the strongest activity, and its binding activity with MDM2 reached 90 nM. At the same time, the crystal structure of the complex of the first small molecule with MDM2 protein (PDB ID: 1RV1) was analyzed, and the binding mode of MDM2 with small-molecule inhibitors was revealed for the first time, which laid the foundation for the subsequent discovery of MDM2 inhibitors. Superimposition of Nutlin-2 on the crystal structure of the p53–MDM2 has shown that the main active sites of Nutlin-2 binding to MDM2 are consistent with the results of alanine mutation scanning experiments, which have shown that p53 primarily occupies the binding cavity in MDM2 through residues Phe19, Trp23 and Leu26, indicating that small-molecular compounds (such as Nutlin) can simulate the α-helix of p53 protein in solution or in vivo to occupy the key binding cavity of p53 and MDM2 protein, thereby inhibiting the activity of MDM2 protein and releasing p53 protein. Nutlin-3a can compete for binding sites in the N-terminus of MDM2 protein and p53 (Phe19, Trp23, Leu26) to block the binding between MDM2 and p53 and reduce the degradation of p53, which then results in induction of apoptosis of tumor cells, reversal of the immunosuppressive microenvironment and triggering of immunogenic cell death [21].
Fig. 4.

Discovery of RG7112
Nutlin-3a can activate wild-type p53 protein in tumor and normal cells at the same time and selectively inhibit tumor cells. Its antiproliferative activity against a variety of wild-type p53 inactivated tumor cells is 1–2 μM (including HCT116, RKO and SJSA-1), but its activity against mutant p53 tumor cells (MDA-MB-435 and SW480) is not obvious [22]. It has strong selectivity, which is similar to the results of the polypeptide inhibitors of MDM2 protein, which is wild-type p53 dependent. The in vivo efficacy showed that Nutlin-3a can activate p53 protein in a dose-dependent manner and induce the expression of p53-related genes (p21 and MDM2). At 200 mg/kg, twice a day, 20 days after oral administration of Nutlin-3a, the tumor growth inhibition rate reached 90%, with no obvious side effects [20].
Further structural optimization was carried out to maintain the most important structural characteristics of its binding to the MDM2 protein and investigate the effect of other side chain groups on its activity. The crystal structure of the complex of Nutlin-3a and MDM2 protein showed that its p-chlorophenyl was perfectly embedded in the active pockets of Leu26 and Trp23 and that isopropyloxy occupied the cavity of Phe19, demonstrating retention of the three key groups. To prevent the imidazoline ring from being oxidized to imidazoline ring, a methyl was added at its 4 and 5 positions. In vitro metabolism studies showed that the methoxy group of Nutlin-3a was unstable and was therefore substituted with tert-butyl. At the same time, to reduce the molecular weight of the target compound, the ethoxy was replaced with isopropyloxy, based on the crystal structure of Nutlin-2 and MDM2; after the above groups were replaced, the effects of exposure to solvent area were investigated, and the effects of different polar groups on their binding to MDM2 and the pharmacokinetic parameters were determined. Finally, through the activity and pharmacokinetic evaluations, it was determined that RG7112 had a stronger binding ability to MDM2 than Nutlin-3a with an enhanced cell activity and better pharmacokinetic parameters, making it more suitable for clinical trials. The crystal complex structure of RG7112 and Nutlin-3a with MDM2 protein is shown in Fig. 5. The binding mode in the three key cavities is nearly the same, which verifies the stability of the MDM2 protein active pocket.
Fig. 5.

A Comparison of binding patterns between Nutlin-2/p53 and MDM2 protein (PDB ID: 1RV1/1YCR); B comparison of protein binding patterns between Nutlin-3a/RG7112 and MDM2 (PDB ID: 4J3E/4IPF)
RG7112 has been demonstrated to have growth inhibition and killing effects on SJSA-1 osteosarcoma cells with a high expression of MDM2 protein in vitro. Dose-dependent cell cycle arrest was induced in HCT116 and SJSA1 cells at the G1 and G2/M phases [23]. In vivo experiments showed that RG7112 (25 ~ 200 mg/kg, oral administration) activated the p53 pathway in vivo and induced apoptosis of tumor cells. RG7112 (100 mg/kg−1, once a day, 5 days/week, for 3 weeks) reduced the tumor growth rate and improve the survival rate in the GBM model [24]. At present, RG7112 has been tested in a variety of clinical trials, mainly including chronic myeloid leukemia (CML), acute myeloid leukemia (AML), solid tumors and hematological tumors. Although the levels of p53 and downstream p21 in patients with liposarcoma treated with this drug were significantly increased, one case of PR and 14 cases of SD were found in 17 assessable patients. However, at least one adverse reaction occurred in all patients, including 12 serious adverse reactions, including neutropenia and thrombocytopenia in eight patients [25]. Another phase I clinical trial for leukemia patients also proved that RG7112 treatment can improve the expression level of p53 and downstream genes. In 30 patients, five cases of CR or PR and nine cases of SD were observed [26]. However, the follow-up test of RG7112 was not carried out due to the poor tolerability and relatively severe hematologic and gastrointestinal toxicities at the required high doses.
RG7388
Roche’s researchers observed that the two benzene rings bound to the active cavities of Trp23 and Leu26 in the structure of the reported MDM2 inhibitors were all cis-conformations, while MDM2 inhibitors with trans-conformations were not reported. Therefore, based on MI-219 [27, 28] and RG7112, these researchers designed and synthesized Compound 1 with trans-conformations of the two key benzene rings Fig. 6), in which cyano was crucial to maintain its conformation and activity. After structural optimization, the second MDM2 inhibitor (RG7388) to enter clinical trials was obtained [29].
Fig. 6.

Discovery of RG7388
The cocrystal structure of Compound 1 and MDM2 protein showed (PDB ID: 4JRG) (Fig. 7) that Compound 1 occupied the three key active cavities of MDM2 protein, and its binding activity with MDM2 protein was 196 nM (IC50, HTRF). Compound 1 showed moderate inhibitory activity against wild-type p53 tumor cells, but no obvious inhibitory activity against p53 mutant tumor cells. After a systematic structure–activity relationship study, Compound 2 showed higher selectivity and activity than Compound 1. However, due to the high clearance rates and low oral bioavailability of Compounds 1 and 2, the structure of the groups exposed to the solvent region was optimized to improve its PK parameters. Finally, the second MDM2 inhibitor, RG7388, was obtained, which had higher binding ability, cell activity, microsome stability and PK parameters. The binding activity of RG7388 to MDM2 reached 6 nM, and the antiproliferative activity against various tumor cells with high expression of wild-type p53 was approximately 300 nM. The selectivity over p53 mutant tumor cells was greater than 100 times. The oral bioavailability in mice reached 80%, half-life was 1.6 h, and the metabolic stability was better than those of Compounds 1 and 2.
Fig. 7.
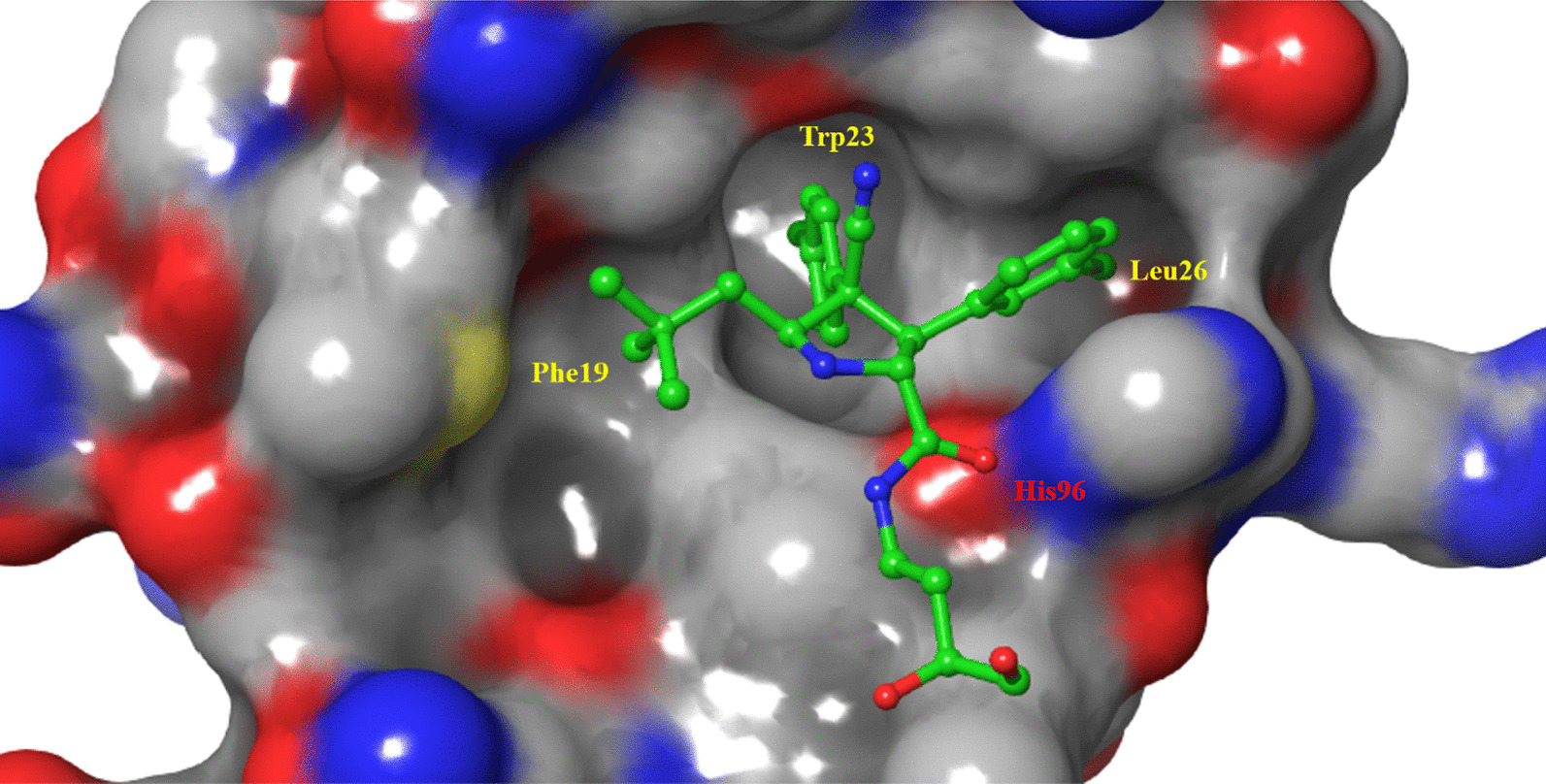
The binding mode of Compound 1 with MDM2 (PDB ID: 4JRG)
RG7388 can effectively activate the p53 pathway, lead to wild-type p53 expression, activate cell cycle arrest or apoptosis, and inhibit tumor proliferation in nude mouse neuroblastoma xenograft experiments [30]. In the SJSA xenograft model of mice, RG7388 (25 mg/kg, oral administration) led to tumor growth inhibition and regression, and induced apoptosis and antiproliferative effects [31]. According to the China Pharmaceutical Pipeline Monitor database (CPM), RG7388 has been assessed in 15 clinical trials, of which the world 's highest state of research and development is phase III clinical trials (NCT02545283) in combination with cytarabine in the treatment of relapsed or refractory AML. The preliminary results showed that the complete remission rate reached 25%, and the median remission time was approximately 6.4 months, which was related to the level of MDM2 protein before treatment [32]. However, the study was discontinued due to unexpected results.
RO6839921 is an inactive PEGylated prodrug of RG7388, which was designed to improve the exposure variability and pharmacokinetic characteristics of RG7388. Intravenous injection of RO6839921 at a safe dose showed good anti-tumor activity in osteosarcoma and AML xenograft models [33]. Although phase I studies demonstrated improved pharmacokinetic parameters of RG7388 for advanced solid tumors and AML, its safety was comparable to that of RG7388 and did not show sufficient advantages [34, 35].
SAR405838 (MI-77301) and APG-115
SAR405838 and APG-115 belong to spirooxindoles, which were developed by Wang Shaomeng's research group based on the protein binding of p53 and MDM2. The indole ring is the key to the binding between p53 and MDM2. The amino group in the indole ring can form hydrogen bonds with MDM2, and it was also found that the oxindole can perfectly simulate the binding mode of the Trp23 residue of p53 with MDM2 (Fig. 8). Taking into account the role of natural products in drug discovery, the team searched for the substructures of oxindole rings and found that many natural products have an oxindole ring (spirotryprostatin A and alstonisine). However, molecular docking showed that these natural products could not bind well to MDM2 protein, which keenly detected the possibility of spirooxindole rings as MDM2 inhibitors. As a rigid scaffold, spiropyrrolidine can be used as a carrier to simulate the three key amino acid residues of Phe19, Trp23 and Leu26. MI-5 was preliminarily obtained based on the structural design, and its three key residues that could simulate the binding of p53 to MDM2 were determined by molecular docking, but its binding ability was low (Ki = 8.46 μM). After replacing isobutyl for a 2,2-dimethylpropyl with stronger hydrophobic effect by structural optimization, compound MI-17, with a 98-fold activity enhancement, was obtained, which had strong selectivity and tumor inhibitory activity [27].
Fig. 8.

Discovery of MI77301
By comparing MI-17 with the most active peptide inhibitor at that time [36], it was found that the activity of MI-17 was nearly 100 times lower than that of the peptide inhibitor, indicating that there was still room for further optimization of MI-17. By analyzing the crystal complex structure of p53–MDM2, it was found that the Leu22 residue played a key role in the binding of p53 and MDM2 and that the Leu22 was located at a position not too deep into the binding pocket of MDM2. Therefore, based on MI-17, it can simulate the binding of Leu22 to MDM2 at the same time and increase polar groups to improve the physical and chemical properties of the target compounds. The binding activity of MI-63 with MDM2 was 3 nM, which was 2000 times higher than that of the p53 peptide (residues 13–29). MI-63 had high stereospecificity and selectively blocked the interaction of p53–MDM2 without affecting the interaction of Bcl-2-Bid. Its inhibitory effect on tumor was wild-type p53-dependent, and its toxicity to p53 knockout cells and normal cells was small. In addition, MI-63 can induce apoptosis of tumor cells in patients with chronic lymphocytic leukemia [37]. However, its PK constant is not suitable for in vivo studies. In order to improve the shortcomings of MI-63, the structure was optimized and MI-219 was obtained with the binding activity with MDM2 of 5 nM. The wild-type p53 protein was selectively activated in a variety of tumor cells, and the activity of mutant and knockout p53 tumor cells was not obvious. It had good oral bioavailability in both rats and mice and can activate p53 protein in mice. It can completely inhibit the growth of tumor cells in vivo, and however, it cannot cause tumor regression [28, 38].
Since the cell activity of MI-147 was higher than that of MI-219, structural optimization based on MI-147 was carried out to further improve its PK parameters and anti-tumor activity in vivo. Finally, MI-888 was obtained, which can completely and continuously eliminate the tumor. The trans conformation of the two benzene rings of MI-888 is the main reason for the enhanced binding ability of MI-888 to the MDM2 protein. Removing the fluorine substituent on the oxindole ring in MI-219 and retaining the fluorine substituent on the benzene ring can improve its PK parameters. At the same time, its polar tail extending into the solvent region can further improve its PK parameters in vivo and its binding ability to MDM2 protein (Ki = 0.44 nM). Its inhibitory activity against tumor cells was at the nM level in SJSA-1 and RS4;11 cells, and the selectivity for wild-type p53 over mutant or knockout p53 tumor cells was high, without significant toxic side effects [39].
MI-77301 (SAR405838) was obtained by simply replacing the groups exposed to the solvent region of MI-888, and its binding activity with MDM2 reached 0.88 nM, with a high selectivity and high specificity over other proteins [40]. The cocrystal structure of SAR405838 and MDM2 protein showed (Fig. 9) that, in addition to simulating the three key amino acid residues of p53–MDM2, SAR405838 is also capable of other additional interactions and induces the refolding of the N-terminal region of unstructured MDM2 to achieve its high-affinity binding to MDM2. Wild-type p53 can be effectively activated in vitro and in xenograft tumor tissues of leukemia or solid tumor, leading to p53-dependent cell cycle arrest and apoptosis. In well-tolerated dose regimens, SAR405838 can lead to durable tumor regression by complete tumor growth inhibition in SJSA-1, RS4;11, LNCaP and HCT-116 xenograft models.
Fig. 9.
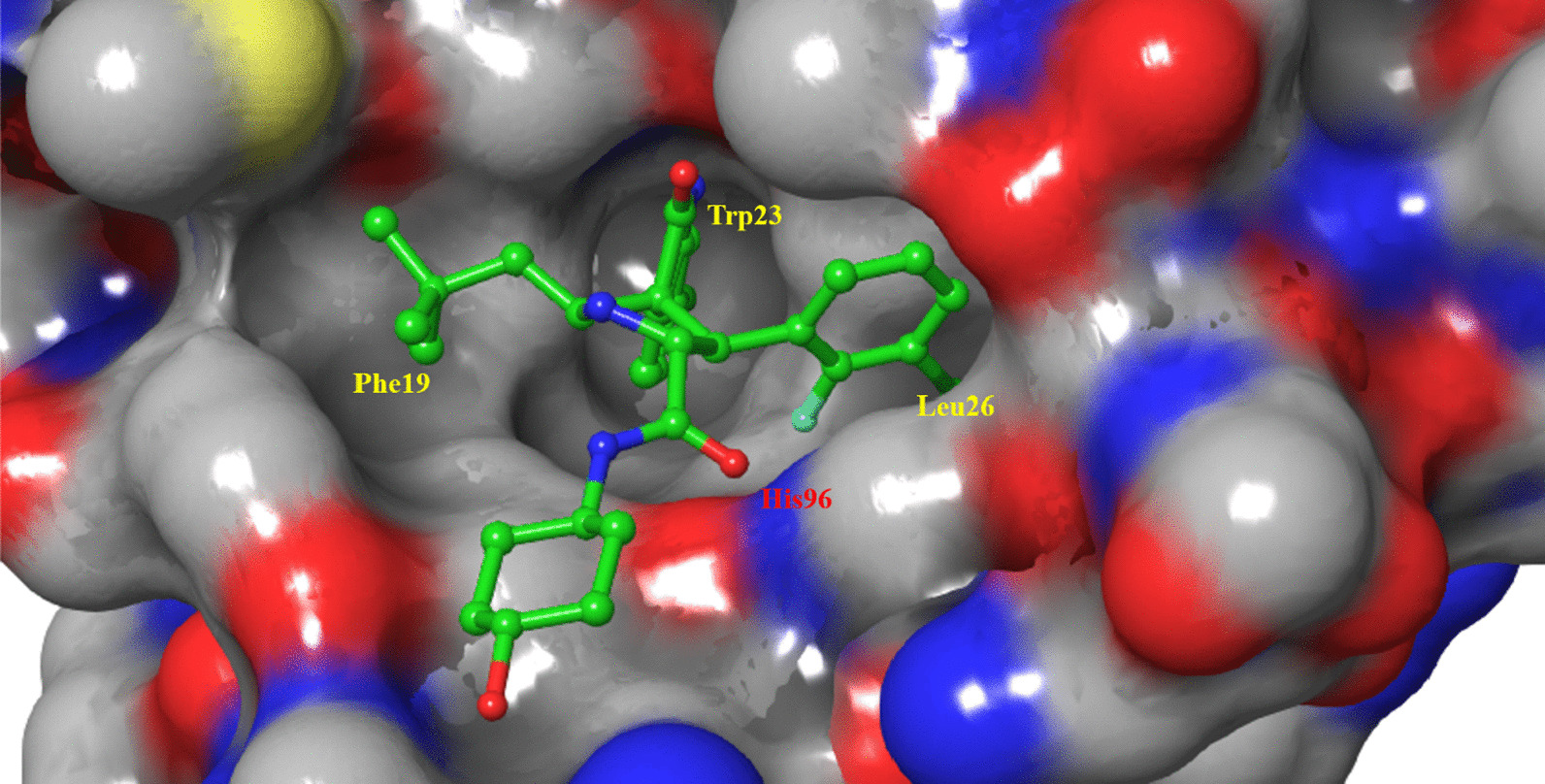
Binding pattern of MI77301 to MDM2 protein (PDB ID: 5TRF)
In the SJSA-1 model, SAR405838 was shown to effectively inhibit the growth of tumors with a high selectivity over p53 mutant or deleted cancer cell lines [40]. SAR405838 effectively inhibited cell growth and induced dose-dependent apoptosis in ABTR1 and ABTR2 sublines [41]. With a well-tolerated dose scheme, SAR405838 achieved persistent tumor regression or completely inhibited tumor growth in the xenograft models of SJSA1, RS411, LNCaP and HCT-116 mice [40]. Phase I clinical trials for patients with advanced solid tumors demonstrated that SAR405838 had good safety and pharmacokinetics, but was not able to be administered at the planned weekly maximum tolerance dose [42]. Further exploration of its potential in combination with the MEK1/2 (MAP kinase kinase 1/2) inhibitor pimasertib in the treatment of locally advanced or metastatic solid tumors did not find significant drug interactions [43]. However, its anti-tumor activity suggests inhibition of the MAPK (mitogen-activated protein kinase) pathway, while restoring p53 activity has potential significance in malignant tumors with wild-type TP53 and MAPK mutations.
The team further determined that the above spiroindole compounds could be transformed into four diastereoisomers in protonic solvents [44]. The possible mechanism was that the pyrrolidine ring could be opened by the anti-Mannich reaction, after which the above four diastereoisomers were formed by the Mannich reaction cyclization. Among them, IV was the most stable and most active diastereoisomer. However, the activity instability of the above MDM2 inhibitors was caused by the differential isomerization. Based on the above reasons, after the introduction of two identical substituents at C2, there were two kinds of diastereoisomers. Studies have shown that when the C2 was the introduced substituent, the compound was more likely to transform to the trans conformation (stable conformation). Finally, the structure was optimized to obtain MI-1061, and the binding activity of MI-1061 to MDM2 was 0.16 nM (Ki). It can effectively activate p53 protein and induce apoptosis in SJSA-1 xenograft tumor in mice, as well as tumor regression. As a result, a defect with high chemical stability in the solvent and breaking through the first generation of spirooxindole MDM2 inhibitors was obtained [45].
Compound 3 was used as the lead compound for a systematic structure–activity relationship study to obtain APG-115 (Fig. 10), which showed a strong binding ability with MDM2 protein (Ki < 1 nM) [46]. It could activate p53 protein in a variety of tumor cells and inhibit cell proliferation at nM concentrations, with an enhancement in its selectivity over p53 knockout tumor cells. APG-115 was very stable in solution and had optimal oral pharmacokinetic parameters. Single-dose oral administration of APG-115 could effectively activate the p53 protein in SJSA-1 xenograft tumor in mice and cause complete and permanent regression of the tumor, as well as in RS4;11 AML model.
Fig. 10.

Discovery of APG-115
APG-115 has obvious activity and drug administration advantages and overcomes the pharmaceutical defects of a slow isomerization reaction and poor solubility in a neutral environment in the early clinical compound SAR405838, with an activity greater than 10 times that of SAR405838. APG-115 affects progression by inducing G0/G1 arrest in AGS and MKN45 cells with wild-type p53, activating p53 to enhance the radiosensitivity of AGS and MKN45 cells, and inducing concentration-dependent G2/M arrest and S phase reduction in p53 wild-type cell lines (TPC-1 and KTC-1). In vivo experiments showed that APG-115 combined with radiotherapy could enhance the radioanti-tumor effect against gastric adenocarcinoma [47]. Phase I trials showed that APG-115 had good tolerance, controllable adverse reactions and good anti-tumor activity [48] and demonstrated the potential to mediate drug resistance in immunotherapy [49, 50]. A phase I trial in a Chinese population showed that APG-115 had good anti-tumor activity in the treatment of patients with MDM2 amplification and TP53-WT liposarcoma [51].
AMG232
Researchers of Amgen designed and synthesized a series of six-membered ring scaffolds based on the structure of the existing MDM2 inhibitors (Fig. 11). After preliminary evaluation, it was determined that the morpholinone scaffold could well simulate the three key amino acid residues (IC50 = 5.4 μM) of p53 binding to MDM2, and the introduction of a benzyl group at the C2 position enhanced the activity of Compound 5. However, its cocrystal structure with MDM2 showed that it did not occupy the Leu26 cavity. Simulated spirooxindole compounds, the introduction of 3-chlorobenzene can better occupy the Leu26 cavity, and through the π–π bond with His96 binding, while the activity had a certain enhancement; further introduction of an acetic acid group at C2 indicated that it enhanced the binding ability of the compound to MDM2 through electrostatic interaction with the His96 residue [52].
Fig. 11.

Discovery of AMG232
Further studies showed that the stereo conformation of the compound was also crucial for its binding to MDM2. The morpholinone scaffold was replaced by a piperidone scaffold, and the conformation was fixed to obtain Compound 9. The cocrystallization of Compound 9 with MDM2 protein showed that cyclopropyl penetrated into the Phe19 cavity in the downwards direction. Therefore, the introduction of a Boc group at the position adjacent to cyclopropyl to fix the conformation of cyclopropyl significantly improved its IC50, and a methyl group at the 3 position was also introduced to stabilize the conformation. The structure–activity relationship of the lead compound was studied systematically. The substitution of the ester site for hydroxymethyl improved the activity to 1.1 nM, resulted in a low clearance rate (CL = 0.03 L/H/Kg) with higher target selectivity than Compound 11, and reduced the inhibition rates of CYP450, CYP3A4 and hPXR. Substituting its hydroxyl group with cyclopropylsulfonamide allowed binding of the compound to the Gly58 amino acid residue near the cavity of Phe19, further improving its affinity for MDM2 protein and its cellular activity. However, Compounds 13 and 14 had high clearance rates in rats and human hepatocytes. Then, the sulfonamide group was replaced by a more stable sulfone group, and its cocrystal structure with MDM2 protein was shown (Fig. 12). As expected, the compound could occupy the three key active cavities, occupy the Gly58 cavity and interact electrostatically effect with His96. Its metabolic stability was further improved, its intrinsic clearance rate was reduced, and its affinity for MDM2 protein and cell activity was also improved [53].
Fig. 12.

Binding pattern of Compound 12 with MDM2 protein (PDB ID: 4OAS)
On this basis, simple structural optimization was carried out to obtain AMG232 [54], which had better PK parameters in rhesus monkeys, low inhibitory activities against CYP, CYP3A4 and PXR, low internal clearance rates in human hepatocytes and rats, and 2500 times higher selectivity for tumor cells with high expression of wild-type p53 than that for p53 knockout tumor cells. In the SJSA-1 osteosarcoma model, its anti-tumor activity was obvious, and it could significantly promote tumor regression without obvious toxicity or side effects [54, 55]. In addition, AMG232 can be combined with other cytotoxic drugs to improve its anti-tumor activity without obvious side effects [56]. Based on these findings, researchers have carried out more in-depth research, but no other compounds have entered clinical trials [57–60].
AMG232, MEK inhibitors and DNA damage-induced chemotherapy have synergistic tumor-killing effects, and combination therapy can significantly increase anti-AML effects in vivo [61]. Compared with RG7112, the selectivity of AMG232 for p53 wild-type cells was higher than that for p53 mutant cells, and glioblastoma stem cells were highly sensitive to AMG232 [62]. In addition, the combined application of AMG232 and anti-PD-1 antibodies can enhance the killing effect of T cells on tumor cells and had the potential to overcome the drug resistance of immunotherapy [63]. In a phase I trial, AMG232 showed good safety and pharmacokinetic parameters in patients with p53 WT advanced solid tumor or multiple myeloma and could control the progression of the disease [64]. A phase I clinical trial on metastatic cutaneous melanoma demonstrated that AMG232 combined with trametinib or dabrafenib had a tolerated dose, safety and pharmacokinetic parameters, and early anti-tumor activity [65]. A Phase I trial on AML patients showed that although AMG232 had serious gastrointestinal adverse reactions at high doses, it had objective pharmacokinetic parameters, targeting and clinical activity [66].
NVP-CGM097
Dihydroisoquinolinone was selected as the scaffold of MDM2 inhibitors by virtual screening (Fig. 13). After preliminary structural optimization, Compound 17 was obtained, and its binding activity with MDM2 protein was 8 nM (TR-FRET). The anti-tumor activity in SJSA-1 cells was at the μM level. The cocrystal structure of Compound 17 with MDM2 protein showed that it had binding modes that were different from the existing MDM2 inhibitors. Butoxy and chlorobenzene occupied Leu26 and Trp23 cavities, respectively. Structure–activity relationship studies showed that these two parts had small structural modification potential. In the Phe19 cavity, groups can be introduced to enhance its binding to the cavity, and its 4-pyridine ring is surrounded by Tyr-67, Met-62 and Gln-72 amino acids of MDM2, which can significantly enhance its binding to MDM2 [67].
Fig. 13.

Discovery of NVP-CGM097
Through molecular docking, further studies on the binding of the Phe19 cavity showed that the substitution of the pyridine ring in the compound for naphthylamine or naphthyl compounds effectively enhanced its binding ability to the surrounding amino acids. For example, when the pyridine ring was substituted with piperidine and other rings, its affinity to MDM2 protein and cell activity were significantly enhanced, and at the same time, it showed good ADME parameters in vitro. Compound 18 was selected for PK experiments in rats. Compound 18 had typical PK parameters of tertiary amine compounds, with a wide distribution in vivo and long half-life. However, its in vivo clearance rate was low. The low oral bioavailability indicated that the absorption of the agent was not completely related to its in vitro solubility/permeability balance. It was also observed that its terminal dimethylamino group had a great influence on the activity. Based on this, the effects of different cyclic derivatives on their activities were further investigated. When this group was derivatized with imidazolidinone or piperazinone, the activity of the target compound was the strongest with good PK parameters and reduced in vivo distribution and half-life, and retained an intermediate in vivo clearance rate. Compared with compound 18, it had better absorption efficiency and oral bioavailability. Finally, NVP-CGM097 was obtained by ammonia methylation of piperazinone to enhance its permeability, which showed good cell activity, metabolic stability and PK parameters [68]. When comparing the binding ability of NVP-CGM097 and Nutlin-3a to MDM2 protein in different species, Nutlin-3a had little difference in affinity to MDM2 protein in different species, while NVP-CGM097 had great difference. The binding ability of NVP-CGM097 to human MDM2 was 16 times higher than that to dog MDM2, while the difference between Nutlin-3a and other species was larger. It is speculated that it may be due to the difference in the Leu-54 and Leu-57 amino acids. Therefore, it is important to study the stability of Nutlin-3a in monkey liver microsomes and the PK parameters in monkeys. Therefore, NVP-CGM097 was determined to be the most suitable clinical candidate.
The cocrystal structure of NVP-CGM097 and MDM2 protein is shown in Fig. 14, which showed that the dihydroisoquinolinone scaffold is located in the middle of the MDM2 active pocket as a scaffold to connect the three key groups to occupy the three key active cavities of Leu26, Trp23 and Phe19. The Leu-26 pocket was filled with isopropyl ether and methyl ether. The ether oxygen group could form water-mediated H-bond interaction with the hydroxyl of Tyr-100 and the carbonyl of Gln-24. The carbonyl group in the scaffold was also involved in the formation of H-bonds with the carbonyl group of Phe-55, which is helpful for the overall affinity of the molecule. More importantly, it induced the conformational constraints of its adjacent N-aryl side chain, so that it had a correct torsion angle to ideally enter the Phe19 active cavity ideally. N-Methyl piperazine can bind to the entrance of the Phe19 cavity in the water-rich region, perfectly located between the protein wall, and significantly enhance its affinity with the MDM2 protein [68].
Fig. 14.
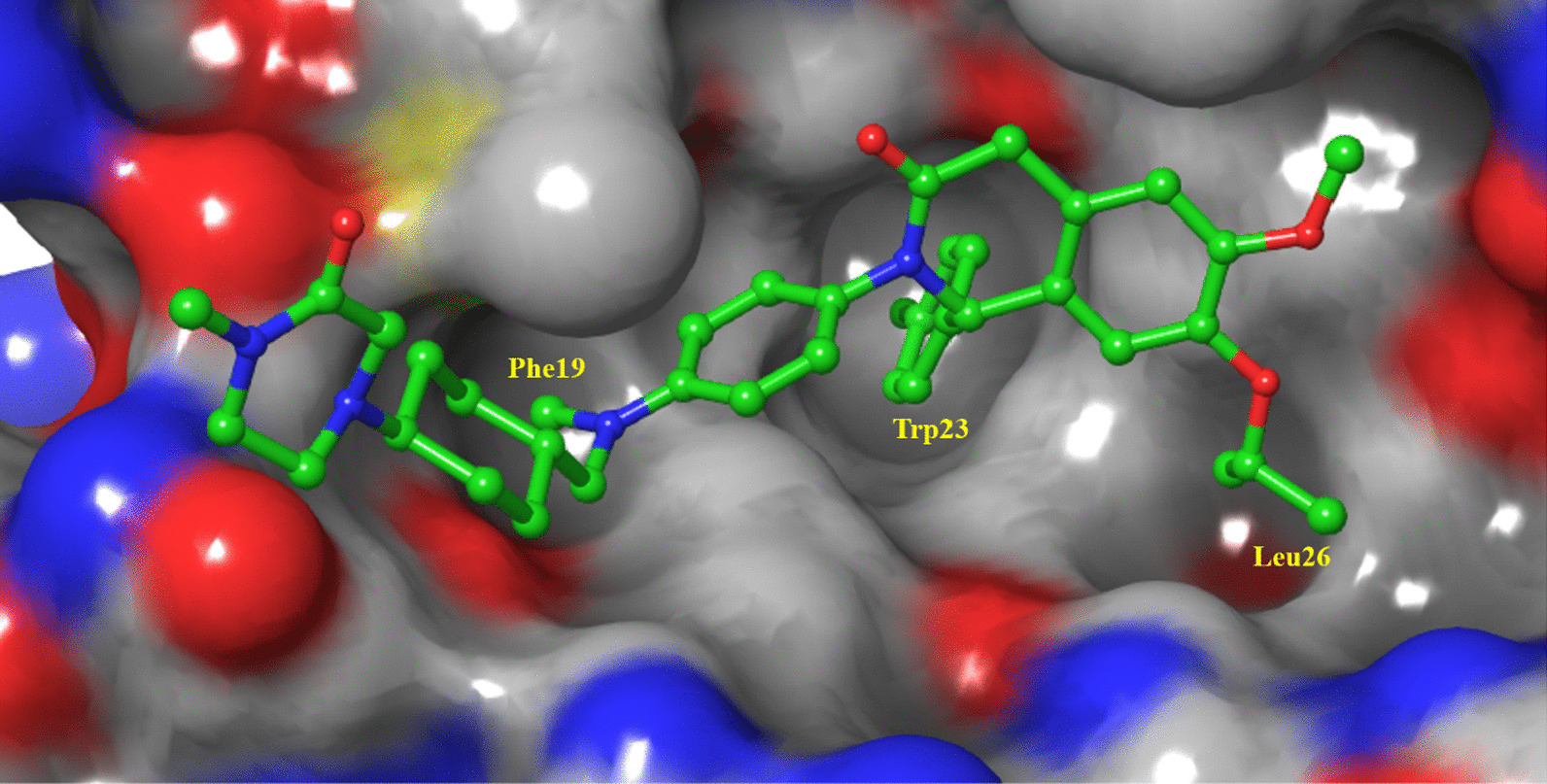
Binding pattern of NVP-CGM097 to MDM2 protein (PDB ID: 4ZYF)
NVP-CGM097 showed strong tumor inhibitory activity with derivative inhibitors in patients with B-cell acute lymphoblastic leukemia [69]. It had the effect of synergistic anti-BRAF (v-raf murine sarcoma viral oncogene homolog B1) mutation melanoma cell proliferation with RAF kinase inhibitor [70], and synergistic anti-ALK (anaplastic lymphoma kinase) mutation neuroblastoma with ALK inhibitor and prolonged survival [71]. The p53 mutant BON1 and NCI-H727 cells were resistant to NVP-CGM097, but when it was combined with 5-fluorouracil, it played a potential role in treating neuroendocrine tumors by increasing the expression of p53 and p21 [72]. NVP-CGM097 can also reverse the multidrug resistance of tumor caused by ABCB1 (ATP binding cassette subfamily B member 1) by blocking the drug efflux mediated by ABCB1 and improving the therapeutic effect of chemotherapy [73]. A phase I clinical trial demonstrated that NVP-CGM097 can control solid tumor progression by activating the p53 pathway and has acceptable pharmacokinetic parameters and safety [74].
HDM201
HDM201 was also designed by Novartis. The design inspiration was mainly derived from the conformational controversy of MDM2 protein binding to dihydroisoquinolinone compounds. Although the specific structural modification process of HDM201 has not been fully published, its research ideas have been reported in the literature. Therefore, the research ideas and the current research process are summarized to provide inspiration for the design of other types of MDM2 inhibitors.
When NVP-CGM097 binds to the MDM2 protein, the scaffold of dihydroisoquinolinone is not completely planar, and therefore, the combination of NVP-CGM097 and the MDM2 protein results in a conformational energy loss, which can be overcome by van der Waals forces or hydrogen bonding interactions with binding sites. However, it is reasonable to believe that the planar scaffold is beneficial for the binding of the compound to MDM2. Therefore, it was assumed that the five-membered lactam ring replaces the six-membered lactam ring of the dihydroisoquinolinone nucleus (Fig. 15). When it was fused with the aromatic ring, it was completely planar and highlighted the substituent, and this idea was verified by molecular simulation. Subsequently, Compound 19 was obtained, and its binding capacity with MDM2 was 1.5 μM. Through simple structural optimization, two methyl groups were introduced into the benzene ring to increase the steric hindrance, so that the two benzene rings were rotated to a suitable angle. After structural analysis, there were still modifiable sites for introduction of phenyl at the N2 position, which led to the discovery of Compound 20 with further improved binding ability to 0.13 nM and cell activity to 90 nM [75].
Fig. 15.

Discovery of HDM201
Subsequently, the effects of a pyrazole ring, pyrrole ring and imidazole ring in the scaffold on the activity of the compound were compared. It was found that the imidazole ring reduced the lipophilicity of the compound, and therefore, an intermediate scaffold was prepared with pyrrolidone and imidazole rings, which resulted in Compound 21. Metasite analysis showed that the methoxy group embedded in the Phe19 cavity was easy to oxidize and metabolize, which led to the rapid metabolic clearance of the compound. Therefore, it was proposed to increase the appropriate substituent to reduce its electronic density. Finally, Compound 22 was obtained, which was effective in the SJSA-1 tumor model without affecting animal weight, compared with NVP-CGM097, which had more potential as the clinical candidate [76]. Subsequent structural optimization has not been reported, but this structure is very similar to HDM201.
HDM201 could selectively induce cell cycle arrest and apoptosis of p53 wild-type tumors and had dose-dependent PKPD parameters to inhibit SJSA-1 in vivo growth [77–79]. In contrast to NVP-CGM097, HDM201 intermittent pulse high-dose treatment can induce PUMA (p53 upregulated modulator of apoptosis) expression and apoptosis in the preclinical model to achieve in vivo anti-tumor activity [80]. The PKPD model of HDM201 also demonstrated that the anti-tumor activity of HDM201 was not scheduled dependent, but was related to cumulative dose [81], suggesting the feasibility of clinical intermittent administration to reduce the common hematological toxicity of MDM2 inhibitors. HDM201 combined with FLT3 (Fms-like tyrosine kinase 3) inhibitor can specifically induce apoptosis and death of FLT3-ITD positive TP53 wild-type AML cells [82], indicating that HDM201 has the potential to be combined with other targeted agents. Phase I clinical results showed that HDM201 had good pharmacokinetic parameters and safety, and there was no significant difference between its safety and tumor type and treatment plan, and it was prone to thrombocytopenia [83]. Clinical research on single-dose administration is ongoing. HDM201 showed promising antileukemia activity in patients with wild-type p53 [84]. The phase Ib trial with AML patients showed that AML patients had a good tolerance and therapeutic response to HDM201 combined with Bcl-2 inhibitors [85]. The safety and clinical efficacy of HDM201 against liposarcoma in combination with LEE011 (a CDK4/6 inhibitor) were also confirmed [86]. In AML patients with relapse after allogeneic stem cell transplantation, HDM201 also had good tolerance and clinical activity [87].
Discussion
A study on the protein–protein interaction of p53–MDM2 showed that the expression of p53 protein could be regulated by MDM2. MDM2 can directly bind to the p53 protein, so that p53 protein would not be released and activated in time when stimulated, so that it could directly inactivate or weaken its transcriptional function. The unique RING domain of the MDM2 protein promotes the formation of dimers, which induces the transfer of the p53 protein from the nucleus to the cytoplasm, which abolishes its transcriptional function. At the same time, studies have shown that MDM2 can cooperate with E4 ligase to degrade p53. At present, nine small-molecule MDM2 inhibitors belonging to different structural types have entered clinical trials (Table 1). However, the clinical research data (such as for RG7112, RG7388 and AMG232) have shown that the progression of MDM2 inhibitors in clinical trials is hindered by various problems. For example, RG7112 was an outstanding candidate in preclinical research and demonstrated strong anti-tumor proliferation activity against various tumor cells. However, its efficacy in clinical research was not obvious, almost all tumor patients had no obvious tumor response, and nearly half of the patients had multiple serious adverse reactions (such as neutropenia and thrombocytopenia) [26]. A clinical study of RG7388 also showed similar phenomenon, with no obvious p53 activation and serious dose-dependent side effects [88]. In addition to the above problems, the mutation of ccfDNA (circulating cell-free DNA) was also found in the clinical study of AMG232. After continuous administration of medication, the mutation frequency gradually increased, and a variety of p53 mutations also occurred, resulting in severe drug resistance [65]. The main defects can be summarized as the following: (1) the clinical efficacy of MDM2 inhibitors is inconsistent with the results of in vitro studies, indicating that the underlying mechanism and a novel strategy need to be explored; (2) the hematological toxicity with activation of p53 in the bone marrow, which maybe solved by optimizing the dosing regimen (have been carried out by several clinical studies); (3) targeting MDM2 only has an inhibitory effect on p53 WT tumors, and however, p53 mutation can be found in more than 50% of cancers, and MDM2 inhibitors have been shown to induce p53 mutations in experimental systems.
Table 1.
Lists of small-molecule MDM2 inhibitors in clinical trials
| Drug | Disease | Combination | Phase | Status | Trial No |
|---|---|---|---|---|---|
| RG7112 (RO5045337) | Advanced solid tumors | I | Completed | NCT00559533 | |
| Hematologic neoplasm | I | Completed | NCT00623870 | ||
| Solid tumors | I | Completed | NCT01164033 | ||
| Sarcoma | Doxorubicin | Ib | Completed | NCT01605526 | |
| AML | Cytarabine | Ib | Completed | NCT01635296 | |
| Sarcoma | I | Completed | NCT01143740 | ||
| CML, neoplasms, AML | I | Completed | NCT01677780 | ||
| RG7388 (Idasanutlin) | Advanced malignancies, except leukemia | I | Completed | NCT01462175 | |
| Solid tumors | I | Completed | NCT03362723 | ||
| AML |
Idarubicin Daunorubicin Cytarabine |
I/Ib | Completed | NCT01773408 | |
| Relapsed and refractory AML | Cytarabine | III | Terminated | NCT02545283 | |
| Non-Hodgkin’s lymphoma |
Obinutuzumab Rituximab |
I/Ib | Terminated | NCT02624986 | |
| Relapsed and refractory AML | venetoclax | Ib | Completed | NCT02670044 | |
| Relapsed and refractory follicular lymphoma, relapsed and refractory diffuse large B-cell lymphoma |
Obinutuzumab Venetoclax Rituximab |
Ib/II | Terminated | NCT03135262 | |
| AML |
Cytarabine Daunorubicin |
Ib/II | Completed | NCT03850535 | |
| Breast cancer | Atezolizumab | I/II | Terminated | NCT03566485 | |
| Solid tumors | I | Completed | NCT02828930 | ||
| Polycythemia vera, essential thrombocythemia | Pegasys | I | Completed | NCT02407080 | |
| Neoplasms | Posaconazole | I | Completed | NCT01901172 | |
| AML, acute lymphocytic leukemia, neuroblastoma, solid tumors |
Cyclophosphamide Topotecan Fludarabine Cytarabine |
I/II | Recruiting | NCT04029688 | |
| Relapsed multiple myeloma |
Ixazomib Dexamethasone venetoclax |
I/II | Active, not recruiting | NCT02633059 | |
| Solid tumors |
Entrectinib Alectinib Atezolizumab Ipatasertib Trastuzumab emtansine Inavolisib Belvarafenib Pralsetinib |
II | Recruiting | NCT04589845 | |
| Colorectal cancer |
Regorafenib Atezolizumab Imprime PGG Bevacizumab Isatuximab Selicrelumab AB928 Genetic: LOAd703 |
I/II | Recruiting | NCT03555149 | |
| Glioblastoma |
APG101 Alectinib Atezolizumab Vismodegib Temsirolimus Palbociclib |
I/II | Recruiting | NCT03158389 | |
| AMG232 (KRT-232) | Advanced solid tumors, multiple myeloma | I | Completed | NCT01723020 | |
| AML | Trametinib | I | Completed | NCT02016729 | |
| Metastatic melanoma |
Trametinib Dabrafenib |
Ib/IIa | Completed | NCT02110355 | |
| AML, relapsed and refractory AML | Decitabine | I | Suspended | NCT03041688 | |
| Soft tissue sarcoma | Radiation therapy | Ib | Recruiting | NCT03217266 | |
| Polycythemia vera | Ruxolitinib | II | Active, not recruiting | NCT03669965 | |
| Relapsed multiple myeloma |
Carfilzomib Dexamethasone Lenalidomide |
I | Recruiting | NCT03031730 | |
| Brain cancer | Radiation therapy | I | Suspended | NCT03107780 | |
| AML |
Cytarabine Idarubicin HCI |
Ib | Recruiting | NCT04190550 | |
| APG-115 (AA-115) | Advanced solid tumors, lymphomas | I | Completed | NCT02935907 | |
| Metastatic melanomas, advanced solid tumors | Pembrolizumab | Ib/II | Recruiting | NCT03611868 | |
| Salivary gland carcinoma | Carboplatin | I/II | Recruiting | NCT03781986 | |
| AML, acute lymphocytic leukemia, neuroblastoma |
Azacitidine Cytarabine |
Ib | Recruiting | NCT04275518 | |
| AML | 5-Azacitidine | Ib/II | Recruiting | NCT04358393 | |
| Liposarcoma, advanced solid tumors | Toripalimab | Ib/II | Recruiting | NCT04785196 | |
| T-prolymphocytic leukemia | APG-2575 | IIa | Recruiting | NCT04496349 | |
| CGM-097 | Advanced solid tumors with TP53wt | I | Completed | NCT01760525 | |
| HDM201 | Liposarcoma | Ribociclib | Ib/II | Completed | NCT02343172 |
| Uveal melanoma | LXS196 | I | Completed | NCT02601378 | |
| Advanced solid and hematological TP53wt tumors | Ancillary treatment | I | Completed | NCT02143635 | |
| AML | I/II | Withdrawn | NCT03760445 | ||
| Advanced/metastatic colorectal cancer | Trametinib | I | Recruiting | NCT03714958 | |
| Myelofibrosis | Ruxolitinib | I/II | Recruiting | NCT04097821 | |
| Colorectal cancer, nonsmall cell lung carcinoma, triple negative breast cancer, renal cell carcinoma | Spartalizumab | I | Completed | NCT02890069 | |
| Malignant solid tumors | Ribociclib | II | Recruiting | NCT04116541 | |
| AML | Midostaurin | I | Recruiting | NCT04496999 | |
| AML, myelodysplastic syndromes | MBG453 | Ib | Recruiting | NCT03940352 | |
| DS-3032b (Milademetan) | Advanced solid tumors, lymphomas | I | Completed | NCT01877382 | |
| Relapsed and refractory AML | I | Completed | NCT03671564 | ||
| AML | Quizartinib | I | Terminated | NCT03552029 | |
| AML, myelodysplastic syndromes | 5-Azacitidine | I | Terminated | NCT023199369 | |
| AML, relapsed and refractory AML |
Cytarabine Venetoclax |
I/II | Completed | NCT03634228 | |
| Myeloma | I | Terminated | NCT02579824 | ||
| SAR405838 | Neoplasm malignant | Pimasertib | I | Completed | NCT01985191 |
| Neoplasm malignant | I | Completed | NCT016636479 | ||
| MK-8242 | AML | Cytarabine | I | Terminated | NCT01451437 |
| Solid tumors | I | Terminated | NCT01463696 |
When the cellular DNA is damaged, stimulated by abnormal growth signals, and affected by chemical drugs or ultraviolet rays, the p53 gene is transcribed and translated into protein, and various kinases are activated to phosphorylate the p53 protein. The accumulated phosphorylated p53 protein in the nucleus promotes cell cycle arrest for self-repair. While the MDM2 is overexpressed, the function of p53 in protecting the cell will be disrupted. Therefore, the clinical efficacy of MDM2 inhibitors may mainly rely on the reversible cell cycle arrest rather than direct induction of cell death through p53-dependent signaling pathway [89–91]. Taking Nutlin-3a as an example, the sensitivity of MDM2 inhibitors against cancer cells was influenced by more criteria other than p53 mutation [92]. The wild-type p53-PUMA pathway was found to have the potential to drive the metabolic switch of cancer cells, and wild-type p53 is required for the maintenance of cancer cell growth and glycolysis in several cancers [93], indicating that activation of p53 in some cases may have the opposite effect, which was consistent with results from other studies [94–96]. MDM2 inhibitors may promote the emergence of p53 mutations and lead to genomic instability through a p53-independent mechanism, resulting in acquired resistance against MDM2 inhibitors [97–99]. In addition, although missense mutations in MDM2 are rare, multiple isoforms of MDM2 protein generated by alternative promoters and alternative proteins have been observed, and the change in the sequence may disrupt the N-terminal p53-binding domain to reduce the efficacy of MDM2 inhibitors and promote tumor progression [100–103]. All of the above factors may be the reasons for the unsatisfactory clinical effects and development of resistance to MDM2 inhibitors. Although the biological classifier based on the genome-wide association has the potential to discriminate response to MDM2-inhibitor therapy [104], novel treatment strategies based on MDM2 need to be explored.
Studies have shown that although MDMX has no obvious regulatory effect on the expression of p53 protein, it can bind to MDM2 protein to form dimers, improve the activity of its E3 ubiquitin ligase and form the above RING domain, thereby further inactivating the p53 protein, which showed considerable efficacy with MDM2. Inhibition of MDM2 alone cannot completely release and activate p53 protein [25, 105], which can induce the overexpression of MDMX, thereby reducing the effect of MDM2 inhibitors and promoting drug resistance [106]. Meanwhile, in some tumors, such as liver cancer and breast cancer, the inactivation of p53 protein is mainly related to the overexpression of MDMX, and therefore, MDM2 inhibitors have shown little effect on such tumors [107]. Studies have shown that the application of MDM2/MDMX dual-target inhibitors can activate higher levels of p53 protein than single-target inhibitors with a better anticancer efficacy, indicating that the simultaneous inhibition of MDM2 and MDMX proteins can effectively regulate the expression of p53 protein. It seems that the development of MDM2/MDMX dual-target inhibitors may solve the problems existing in the clinical research of the above MDM2 inhibitors and has great practical significance for the treatment of related tumors [97, 108, 109].
However, at present, the development of MDM2/MDMX dual-target inhibitors is slow, although the interaction between p53 and MDM2/MDMX occurs through the same key amino acid residues (Phe19, Trp23 and Leu26), the binding cavity of MDMX is more flexible than that of MDM2, which makes the existing MDM2 inhibitors with rigid scaffolds unable to conform to the conformational flexibility in the active cavity of the MDMX protein. However, the MDMX inhibitor SJ-172550 was reported to be extremely unstable [110]. It easily deteriorates in solution, and the basis of its specific role is not clear. Only indole-acetylureas and pseudopeptides have been reported to have dual protein inhibitory effects onMDM2 and MDMX [111–113], with few structural types, unclear mechanisms of action, and no further studies.
In addition to small-molecule inhibitors, another promising class of MDM2/MDMX inhibitors is cell-penetrating stapled α-helical peptides. The most promising is ATSP-7041 and its analog ALRN-6924 [114, 115]. ALRN-6924 significantly improved survival in a xenograft model of AML [116]. Clinically, ALRN-6924 is being evaluated as a monotherapy and in combination with cytarabine in patients with hematological malignancies (NCT02909972) and has entered a Phase I/II clinical trial in patients with advanced solid tumors or with preserved wild-type p53 lymphoma (NCT02264613). ALRN-6924 was well tolerated, and the most common adverse side effect was gastrointestinal [115].
The dual-target inhibitor based on MDM2 is a promising way to solve the defects of MDM2 inhibitors. The MDM2/XIAP (X-linked inhibitor of apoptosis protein) dual-target inhibitor MX69 can simultaneously inhibit XIAP and activate p53 to exert anti-tumor activity. Importantly, it showed little effect on normal human hematopoietic function in vitro and was well tolerated in animal models [117], indicating that it had the potential to avoid adverse reactions in hematopoietic system of MDM2 inhibitors. He et al. discovered an oral dual inhibitor of MDM2 and HDAC (histone deacetylase), which had excellent anti-tumor effect in xenograft models [118]. Targeting MDM2/TPSO (translocator protein) [119], MDM2/PKC (protein kinase C) [120], MDM2/NF-kB (nuclear factor-kappa B) [121], MDM2/Bcl-2 (B-cell lymphoma-2) [122] and MDM2/NFAT1 (nuclear factor of activated T-cells 1) [123] had good anti-tumor activity and had the potential to reduce the adverse reactions of MDM2 inhibitors. Targeted MDM2 degradation also had the potential to reduce the dose-dependent hematological toxicity of MDM2 inhibitors [124–128]. In addition, MDM2 inhibitors have the potential to be incorporated into cyclotherapy, since low-dose MDM2 inhibitors could arrest the cell cycle and prevent S-Phase and M-Phase drug damage to p53 proficient normal cells. However, no multitarget inhibitors or degradation agents or cyclotherapies related to MDM2 have entered the clinical trials, suggesting that the related structures need to be further optimized to improve their physicochemical properties and promote their clinical development.
Conclusion
In summary, the design and development of anti-tumor drugs with new mechanisms targeting MDM2–p53 are one of the hotspots in the field of global tumor drug research and development. However, the specificity of MDM2–p53 protein interaction brings considerable difficulties to the development of small-molecule inhibitors, resulting in only a handful candidates under clinical trials, and no marketed drugs targeting MDM2. The multi-target strategy or targeted degradation strategy based on MDM2 has the potential to improve the clinical defects of MDM2 inhibitors, but more evidence is still needed to confirm its clinical application value.
Acknowledgments
Not applicable.
Abbreviations
- ABCB1
ATP binding cassette subfamily B member 1
- ALK
Anaplastic lymphoma kinase
- AML
Acute myeloid leukemia
- Bcl-2
B-cell lymphoma-2
- BRAF
V-raf murine sarcoma viral oncogene homolog B1
- CML
Chronic myeloid leukemia
- CPM
China Pharmaceutical Pipeline Monitor database
- FLT3
Fms-like tyrosine kinase 3
- HDAC
Histone deacetylase
- HIF1α
Hypoxia inducible factor-1α
- Keap1
Kelch-like ECH-associated protein 1
- MAPK
Mitogen-activated protein kinase
- MDM2
Mouse double minute 2
- MDMX
Mouse double minute X
- MEK
MAP kinase kinase
- NFAT1
Nuclear factor of activated T-cells 1
- NF-kB
Nuclear factor-kappa B
- Nrf2
Nuclear factor E2-related factor 2
- PKC
Protein kinase C
- PPI
Protein–protein interaction
- PUMA
P53 upregulated modulator of apoptosis
- TPSO
Translocator protein
- UBE4B
Ubiquitination factor E4B
- VHL
Von Hippel–Lindau
- XIAP
X-linked inhibitor of apoptosis protein
Author contributions
JY conceived the study; GH, ZHH, JYY, ZQ, DZQ performed literature searching and summary; YK and ZHH wrote the manuscript; and JY, YK, TL, and ZZH edited and revised the manuscript. All authors read and approved the final manuscript.
Funding
The work is supported by the National Natural Science Foundation of China (No. 8210131157), Wuxi Municipal Health Commission (Nos. Q202101, Q202167, M202167 and ZH202110), Wuxi Taihu Talent Project (Nos. WXTTP2020008 and WXTTP2021), Wuxi Medical Development Discipline Project (No. FZXK2021012), Jiangsu Research Hospital Association for Precision Medication (JY202105).
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Haohao Zhu, Hui Gao, Yingying Ji have contributed equally to this work
Contributor Information
Ying Jiang, Email: jiangying911010@163.com.
Kun Yao, Email: mhc2133@163.com.
Zhenhe Zhou, Email: zhouzh@njmu.edu.cn.
References
- 1.Arkin M. Protein–protein interactions and cancer: small molecules going in for the kill. Curr Opin Chem Biol. 2005;9(3):317–324. doi: 10.1016/j.cbpa.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the 'undruggable' cancer targets. Nat Rev Cancer. 2017;17(8):502–508. doi: 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo W, Wisniewski JA, Ji H. Hot spot-based design of small-molecule inhibitors for protein-protein interactions. Bioorg Med Chem Lett. 2014;24(11):2546–2554. doi: 10.1016/j.bmcl.2014.03.095. [DOI] [PubMed] [Google Scholar]
- 4.Buckley DL, Van Molle I, Gareiss PC, et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J Am Chem Soc. 2012;134(10):4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhuang C, Narayanapillai S, Zhang W, Sham YY, Xing C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J Med Chem. 2014;57(3):1121–1126. doi: 10.1021/jm4017174. [DOI] [PubMed] [Google Scholar]
- 6.Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. 2016;85:375–404. doi: 10.1146/annurev-biochem-060815-014710. [DOI] [PubMed] [Google Scholar]
- 7.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 8.Oh S, Lee MK, Chi SW. Single-molecule analysis of interaction between p53TAD and MDM2 using aerolysin nanopores. Chem Sci. 2021;12(16):5883–5891. doi: 10.1039/D1SC00386K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein AM, Biderman L, Tong D, et al. MDM2, MDMX, and p73 regulate cell-cycle progression in the absence of wild-type p53. Proc Natl Acad Sci USA. 2021;118(44):e2102420118. doi: 10.1073/pnas.2102420118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chibaya L, Karim B, Zhang H, Jones SN. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc Natl Acad Sci USA. 2021;118(4):e2003193118. doi: 10.1073/pnas.2003193118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hock AK, Vousden KH. The role of ubiquitin modification in the regulation of p53. Biochim Biophys Acta. 2014;1843(1):137–149. doi: 10.1016/j.bbamcr.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 12.Bang S, Kaur S, Kurokawa M. Regulation of the p53 family proteins by the ubiquitin proteasomal pathway. Int J Mol Sci. 2019;21(1):261. doi: 10.3390/ijms21010261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.do Patrocinio AB, Rodrigues V, Guidi Magalhães L. P53: Stability from the Ubiquitin-Proteasome System and Specific 26S Proteasome Inhibitors. ACS Omega. 2022;7(5):3836–3843. [DOI] [PMC free article] [PubMed]
- 14.Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 15.Wang S, Zhao Y, Aguilar A, Bernard D, Yang CY. Targeting the MDM2-p53 protein–protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb Perspect Med. 2017;7(5):a026245. doi: 10.1101/cshperspect.a026245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang Y, Liao G, Yu B. Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: advances and perspectives. Acta Pharm Sin B. 2020;10(7):1253–1278. doi: 10.1016/j.apsb.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kocik J, Machula M, Wisniewska A, Surmiak E, Holak TA, Skalniak L. Helping the released guardian: drug combinations for supporting the anticancer activity of HDM2 (MDM2) antagonists. Cancers (Basel) 2019;11(7):1014. doi: 10.3390/cancers11071014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58(3):1038–1052. doi: 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vu B, Wovkulich P, Pizzolato G, et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett. 2013;4(5):466–469. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 21.Guo G, Yu M, Xiao W, Celis E, Cui Y. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res. 2017;77(9):2292–2305. doi: 10.1158/0008-5472.CAN-16-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crane EK, Kwan SY, Izaguirre DI, et al. Nutlin-3a: a potential therapeutic opportunity for TP53 wild-type ovarian carcinomas. PLoS ONE. 2015;10(8):e0135101. doi: 10.1371/journal.pone.0135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tovar C, Graves B, Packman K, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73(8):2587–2597. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- 24.Verreault M, Schmitt C, Goldwirt L, et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin Cancer Res. 2016;22(5):1185–1196. doi: 10.1158/1078-0432.CCR-15-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray-Coquard I, Blay JY, Italiano A, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13(11):1133–1140. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 26.Andreeff M, Kelly KR, Yee K, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22(4):868–876. doi: 10.1158/1078-0432.CCR-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding K, Lu Y, Nikolovska-Coleska Z, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127(29):10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 28.Yu S, Qin D, Shangary S, et al. Potent and orally active small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2009;52(24):7970–7973. doi: 10.1021/jm901400z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding Q, Zhang Z, Liu JJ, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56(14):5979–5983. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- 30.Lakoma A, Barbieri E, Agarwal S, et al. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discov. 2015;1:15026. doi: 10.1038/cddiscovery.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higgins B, Glenn K, Walz A, et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin Cancer Res. 2014;20(14):3742–3752. doi: 10.1158/1078-0432.CCR-14-0460. [DOI] [PubMed] [Google Scholar]
- 32.Khurana A, Shafer DA. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388) Onco Targets Ther. 2019;12:2903–2910. doi: 10.2147/OTT.S172315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higgins B, Tovar C, Glen K, et al. Preclinical activity of MDM2 antagonist RO6839921, a pegylated prodrug for intravenous administration (abstract A156). Mol Cancer Ther. 2014;14(12 Supplement 2): A156.
- 34.Abdul Razak AR, Miller WH, Jr, Uy GL, et al. A phase 1 study of the MDM2 antagonist RO6839921, a pegylated prodrug of idasanutlin, in patients with advanced solid tumors. Investig New Drugs. 2020;38(4):1156–1165. doi: 10.1007/s10637-019-00869-2. [DOI] [PubMed] [Google Scholar]
- 35.Uy GL, Assouline S, Young AM, et al. Phase 1 study of the MDM2 antagonist RO6839921 in patients with acute myeloid leukemia. Investig New Drugs. 2020;38(5):1430–1441. doi: 10.1007/s10637-020-00907-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.García-Echeverría C, Chène P, Blommers MJ, Furet P. Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53. J Med Chem. 2000;43(17):3205–3208. doi: 10.1021/jm990966p. [DOI] [PubMed] [Google Scholar]
- 37.Saddler C, Ouillette P, Kujawski L, et al. Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood. 2008;111(3):1584–1593. doi: 10.1182/blood-2007-09-112698. [DOI] [PubMed] [Google Scholar]
- 38.Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105(10):3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y, Yu S, Sun W, et al. A potent small-molecule inhibitor of the MDM2-p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J Med Chem. 2013;56(13):5553–5561. doi: 10.1021/jm4005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Sun W, Zhao Y, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74(20):5855–5865. doi: 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffman-Luca CG, Ziazadeh D, McEachern D, et al. Elucidation of acquired resistance to Bcl-2 and MDM2 inhibitors in acute leukemia in vitro and in vivo. Clin Cancer Res. 2015;21(11):2558–2568. doi: 10.1158/1078-0432.CCR-14-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Jonge M, de Weger VA, Dickson MA, et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer. 2017;76:144–151. doi: 10.1016/j.ejca.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 43.de Weger VA, de Jonge M, Langenberg MHG, et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br J Cancer. 2019;120(3):286–293. doi: 10.1038/s41416-018-0355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Liu L, Sun W, et al. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J Am Chem Soc. 2013;135(19):7223–7234. doi: 10.1021/ja3125417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aguilar A, Sun W, Liu L, et al. Design of chemically stable, potent, and efficacious MDM2 inhibitors that exploit the retro-mannich ring-opening-cyclization reaction mechanism in spiro-oxindoles. J Med Chem. 2014;57(24):10486–10498. doi: 10.1021/jm501541j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aguilar A, Lu J, Liu L, et al. Discovery of 4-((3'R,4'S,5'R)-6″-Chloro-4'-(3-chloro-2-fluorophenyl)-1'-ethyl-2″-oxodispiro[cyclohexane-1,2'-pyrrolidine-3',3″-indoline]-5'-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J Med Chem. 2017;60(7):2819–2839. [DOI] [PMC free article] [PubMed]
- 47.Nakamaru K, Seki T, Tazaki K, et al. Abstract B5: Preclinical characterization of a novel orally-available MDM2 inhibitor DS-3032b: anti-tumor profile and predictive biomarkers for sensitivity. Cancer Res. 2015;14:B5. [Google Scholar]
- 48.Rasco DW, Lakhani NJ, Li Y, et al. A phase I study of a novel MDM2 antagonist APG-115 in patients with advanced solid tumors. Cancer Res. 2019;37:3126. [Google Scholar]
- 49.Tolcher AW, Fang DD, Li Y, et al. A phase Ib/II study of APG-115 in combination with pembrolizumab in patients with unresectable or metastatic melanomas or advanced solid tumors. Ann Onco. 2019;30:i2. doi: 10.1093/annonc/mdz027. [DOI] [Google Scholar]
- 50.Tolcher AW, Reeves JA, McKean M, et al. Preliminary results of a phase II study of alrizomadlin (APG-115), a novel, small-molecule MDM2 inhibitor, in combination with pembrolizumab in patients (pts) with unresectable or metastatic melanoma or advanced solid tumors that have failed immuno-oncologic (IO) drugs. Cancer Res. 2021;39:2506. [Google Scholar]
- 51.Zhang X, Wen X, Yang C, et al. A phase I study of a novel MDM2-P53 antagonist APG-115 in Chinese patients with advanced soft tissue sarcomas. Cancer Res. 2019;37:3124. [Google Scholar]
- 52.Gonzalez-Lopez de Turiso F, Sun D, Rew Y, et al. Rational design and binding mode duality of MDM2-p53 inhibitors. J Med Chem. 2013;56(10):4053–4070. [DOI] [PubMed]
- 53.Rew Y, Sun D, Gonzalez-Lopez De Turiso F, et al. Structure-based design of novel inhibitors of the MDM2-p53 interaction. J Med Chem. 2012;55(11):4936–4954. [DOI] [PubMed]
- 54.Rew Y, Sun D. Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J Med Chem. 2014;57(15):6332–6341. doi: 10.1021/jm500627s. [DOI] [PubMed] [Google Scholar]
- 55.Sun D, Li Z, Rew Y, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57(4):1454–1472. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 56.Canon J, Osgood T, Olson SH, et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther. 2015;14(3):649–658. doi: 10.1158/1535-7163.MCT-14-0710. [DOI] [PubMed] [Google Scholar]
- 57.Yu M, Wang Y, Zhu J, et al. Discovery of potent and simplified piperidinone-based inhibitors of the MDM2-p53 interaction. ACS Med Chem Lett. 2014;5(8):894–899. doi: 10.1021/ml500142b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gonzalez AZ, Eksterowicz J, Bartberger MD, et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J Med Chem. 2014;57(6):2472–2488. doi: 10.1021/jm401767k. [DOI] [PubMed] [Google Scholar]
- 59.Gonzalez AZ, Li Z, Beck HP, et al. Novel inhibitors of the MDM2-p53 interaction featuring hydrogen bond acceptors as carboxylic acid isosteres. J Med Chem. 2014;57(7):2963–2988. doi: 10.1021/jm401911v. [DOI] [PubMed] [Google Scholar]
- 60.Rew Y, Sun D, Yan X, et al. Discovery of AM-7209, a potent and selective 4-amidobenzoic acid inhibitor of the MDM2-p53 interaction. J Med Chem. 2014;57(24):10499–10511. doi: 10.1021/jm501550p. [DOI] [PubMed] [Google Scholar]
- 61.Canon JR, Osgood T, Saiki AY, et al. The MDM2 inhibitor AMG 232 causes tumor regression and potentiates the anti-tumor activity of MEK inhibition and DNA-damaging cytotoxic agents in preclinical models of acute myeloid leukemia. Cancer Res. 2016;76:3761. doi: 10.1158/1538-7445.AM2016-3761. [DOI] [Google Scholar]
- 62.Her NG, Oh JW, Oh YJ, et al. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018;9(8):792. doi: 10.1038/s41419-018-0825-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sahin I, Zhang S, Navaraj A, et al. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Discov. 2020;6:57. doi: 10.1038/s41420-020-0292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gluck WL, Gounder MM, Frank R, et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drug. 2020;38(3):831–843. doi: 10.1007/s10637-019-00840-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moschos SJ, Sandhu SK, Lewis KD, et al. Phase 1 study of the p53-MDM2 inhibitor AMG 232 combined with trametinib plus dabrafenib or trametinib in patients (Pts) with TP53 wild type (TP53WT) metastatic cutaneous melanoma (MCM) Cancer Res. 2017;35:2575. [Google Scholar]
- 66.Erba HP, Becker PS, Shami PJ, et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019;3(13):1939–1949. doi: 10.1182/bloodadvances.2019030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gessier F, Kallen J, Jacoby E, et al. Discovery of dihydroisoquinolinone derivatives as novel inhibitors of the p53-MDM2 interaction with a distinct binding mode. Bioorg Med Chem Lett. 2015;25(17):3621–3625. doi: 10.1016/j.bmcl.2015.06.058. [DOI] [PubMed] [Google Scholar]
- 68.Holzer P, Masuya K, Furet P, et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58(16):6348–6358. doi: 10.1021/acs.jmedchem.5b00810. [DOI] [PubMed] [Google Scholar]
- 69.Townsend EC, DeSouza T, Murakami MA, et al. The MDM2 inhibitor NVP-CGM097 is highly active in a randomized preclinical trial of B-cell acute lymphoblastic leukemia patient derived xenografts. Blood. 2015;126(23):797. doi: 10.1182/blood.V126.23.797.797. [DOI] [Google Scholar]
- 70.Wang HQ, Zubrowski M, Emerson E, et al. The Mdm2 inhibitor, NVP-CGM097, in combination with the BRAF inhibitor NVP-LGX818 elicits synergistic antitumor effects in melanoma. Cancer Res. 2014;74:5466. doi: 10.1158/1538-7445.AM2014-5466. [DOI] [Google Scholar]
- 71.Wang HQ, Battalagine L, Liang J, et al. The Mdm2 inhibitor NVP-CGM097 enhances the anti-tumor activity of NVP-LDK378 in ALK mutant neuroblastoma models. Cancer Res. 2014;74:2929. doi: 10.1158/1538-7445.AM2014-2929. [DOI] [Google Scholar]
- 72.Reuther C, Heinzle V, Nölting S, et al. The HDM2 (MDM2) inhibitor NVP-CGM097 inhibits tumor cell proliferation and shows additive effects with 5-fluorouracil on the p53–p21-Rb-E2F1 cascade in the p53wild type neuroendocrine tumor cell line GOT1. Neuroendocrinology. 2018;106(1):1–19. doi: 10.1159/000453369. [DOI] [PubMed] [Google Scholar]
- 73.Zhang M, Chen XY, Dong XD, et al. NVP-CGM097, an HDM2 inhibitor, antagonizes ATP-binding cassette subfamily B member 1-mediated drug resistance. Front Oncol. 2020;10:1219. doi: 10.3389/fonc.2020.01219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bauer S, Demetri G, Jeay S, et al. A phase I, open-label, multi-center, dose escalation study of oral NVP-CGM097, a p53/HDM2-protein-protein interaction inhibitor, in adult patients with selected advanced solid tumors. Ann Oncol. 2016;27:116. doi: 10.1093/annonc/mdw368.09. [DOI] [Google Scholar]
- 75.Furet P, Masuya K, Kallen J, et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg Med Chem Lett. 2016;26(19):4837–4841. doi: 10.1016/j.bmcl.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 76.Vaupel A, Holzer P, Ferretti S, et al. In vitro and in vivo characterization of a novel, highly potent p53-MDM2 inhibitor. Bioorg Med Chem Lett. 2018;28(20):3404–3408. doi: 10.1016/j.bmcl.2018.08.027. [DOI] [PubMed] [Google Scholar]
- 77.Jeay S, Chène P, Ferretti S, et al. NVP-HDM201: cellular and in vivo profile of a novel highly potent and selective PPI inhibitor of p53-Mdm2. Cancer Res. 2016;76:1225. doi: 10.1158/1538-7445.AM2016-1225. [DOI] [Google Scholar]
- 78.Ferretti S, Rebmann R, Berger M, et al. Insights into the mechanism of action of NVP-HDM201, a differentiated and versatile Next-Generation small-molecule inhibitor of Mdm2, under evaluation in phase I clinical trials. Cancer Res. 2016;76:1224. doi: 10.1158/1538-7445.AM2016-1224. [DOI] [Google Scholar]
- 79.Meille C, Guerreiro N, Jullion A, et al. Abstract CT154: Optimization of the dose and schedule of an HDM2 inhibitor NVP-HDM201 in a first-in-human Phase I study using a mechanism-based PK/PD model. Cancer Res. 2017;77:CT154.
- 80.Jeay S, Ferretti S, Holzer P, et al. Dose and schedule determine distinct molecular mechanisms underlying the efficacy of the p53–MDM2 inhibitor HDM201. Cancer Res. 2018;78(21):6257–6267. doi: 10.1158/0008-5472.CAN-18-0338. [DOI] [PubMed] [Google Scholar]
- 81.Guerreiro N, Jullion A, Ferretti S, Fabre C, Meille C. Translational modeling of anticancer efficacy to predict clinical outcomes in a first-in-human phase 1 study of MDM2 inhibitor HDM201. AAPS J. 2021;23(2):28. doi: 10.1208/s12248-020-00551-z. [DOI] [PubMed] [Google Scholar]
- 82.Seipel K, Marques MAT, Sidler C, Mueller BU, Pabst T. MDM2- and FLT3-inhibitors in the treatment of FLT3-ITD acute myeloid leukemia, specificity and efficacy of NVP-HDM201 and midostaurin. Haematologica. 2018;103(11):1862–1872. doi: 10.3324/haematol.2018.191650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stein EM, DeAngelo DJ, Chromik J, et al. Results from a first-in-human phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin Cancer Res. 2022;28(5):870–881. doi: 10.1158/1078-0432.CCR-21-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stein E, Chromik J, DeAngelo DJ, et al. Abstract CT152: Phase I dose-and regimen-finding study of NVP-HDM201 in pts with advanced TP53 wt acute leukemias. Cancer Res. 2017;77(13 Supplement):CT152.
- 85.Wei AH, Breccia M, Ooi M, et al. Preliminary results from a phase Ib study exploring MDM2 inhibitor siremadlin (HDM201) in combination with B-cell lymphoma-2 (BCL-2) Inhibitor venetoclax in patients with acute myeloid leukemia (AML) or high-risk myelodysplastic syndrome (HR-MDS) Blood. 2021;138:1283. doi: 10.1182/blood-2021-144650. [DOI] [Google Scholar]
- 86.Razak AA, Bauer S, Blay JY, et al. Abstract CT009: Results of a dose-and regimen-finding Phase Ib study of HDM201 in combination with ribociclib in patients with locally advanced or metastatic liposarcoma. Cancer Res. 2018;78(13 Supplement): CT009.
- 87.Stein EM, Chromik J, Carpio C, et al. Siremadlin (HDM201) is well tolerated and demonstrates clinical activity in patients with acute myeloid leukemia who have relapsed after allogeneic stem cell transplantation: a subset analysis of safety and preliminary efficacy. Blood. 2021;138:3417. doi: 10.1182/blood-2021-149751. [DOI] [Google Scholar]
- 88.Yee K, Martinelli G, Vey N, et al. Phase 1/1b study of RG7388, a potent MDM2 antagonist, in acute myelogenous leukemia (AML) patients (Pts) Blood. 2014;124:116. doi: 10.1182/blood.V124.21.116.116. [DOI] [Google Scholar]
- 89.Bian Y, Yang L, Sheng W, et al. Ligustrazine induces the colorectal cancer cells apoptosis via p53-dependent mitochondrial pathway and cell cycle arrest at the G0/G1 phase. Ann Palliat Med. 2021;10(2):1578–1588. doi: 10.21037/apm-20-288. [DOI] [PubMed] [Google Scholar]
- 90.Lan Y, Lou J, Hu J, Yu Z, Lyu W, Zhang B. Downregulation of SNRPG induces cell cycle arrest and sensitizes human glioblastoma cells to temozolomide by targeting Myc through a p53-dependent signaling pathway. Cancer Biol Med. 2020;17(1):112–131. doi: 10.20892/j.issn.2095-3941.2019.0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu YF, Xu XP, Lu XP, et al. SIRT7 activates p53 by enhancing PCAF-mediated MDM2 degradation to arrest the cell cycle. Oncogene. 2020;39(24):4650–4665. doi: 10.1038/s41388-020-1305-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang W, Soares J, Greninger P, et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):D955-D961. [DOI] [PMC free article] [PubMed]
- 93.Kim J, Yu L, Chen W, et al. Wild-type p53 promotes cancer metabolic switch by inducing PUMA-dependent suppression of oxidative phosphorylation. Cancer Cell. 2019;35(2):191–203.e8. doi: 10.1016/j.ccell.2018.12.012. [DOI] [PubMed] [Google Scholar]
- 94.Salomao N, Karakostis K, Hupp T, Vollrath F, Vojtesek B, Fahraeus R. What do we need to know and understand about p53 to improve its clinical value? J Pathol. 2021;254(4):443–453. doi: 10.1002/path.5677. [DOI] [PubMed] [Google Scholar]
- 95.Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018;15(1):13–30. doi: 10.1038/nrclinonc.2017.151. [DOI] [PubMed] [Google Scholar]
- 96.Walerych D, Lisek K, Sommaggio R, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol. 2016;18(8):897–909. doi: 10.1038/ncb3380. [DOI] [PubMed] [Google Scholar]
- 97.Michaelis M, Rothweiler F, Barth S, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011;2(12):e243. doi: 10.1038/cddis.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Melo AN, Eischen CM. Protecting the genome from mdm2 and mdmx. Genes Cancer. 2012;3(3–4):283–290. doi: 10.1177/1947601912454139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haronikova L, Bonczek O, Zatloukalova P, et al. Resistance mechanisms to inhibitors of p53-MDM2 interactions in cancer therapy: Can we overcome them? Cell Mol Biol Lett. 2021;26(1):53. doi: 10.1186/s11658-021-00293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bartel F, Harris LC, Würl P, et al. MDM2 and its splice variant messenger RNAs: expression in tumors and down-regulation using antisense Oligonucleotides11NIH grants CA92401 and CA21765, American Lebanese Syrian Associated Charities (LCH), Deutsche Krebshilfe eV grant 2130-Ta2, Land Sachsen-Anhalt grant 3347A/0021B, and GSGT eV (FB and HT) Mol Cancer Res. 2004;2(1):29–35. doi: 10.1158/1541-7786.29.2.1. [DOI] [PubMed] [Google Scholar]
- 101.Okoro DR, Rosso M, Bargonetti J. Splicing up mdm2 for cancer proteome diversity. Genes Cancer. 2012;3(3–4):311–319. doi: 10.1177/1947601912455323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lim CC, Chan SK, Lim YY, et al. Development and structural characterisation of human scFv targeting MDM2 spliced variant MDM215kDa. Mol Immunol. 2021;135:191–203. doi: 10.1016/j.molimm.2021.04.016. [DOI] [PubMed] [Google Scholar]
- 103.Comiskey DF, Jr, Montes M, Khurshid S, Singh RK, Chandler DS. SRSF2 Regulation of MDM2 reveals splicing as a therapeutic vulnerability of the p53 pathway. Mol Cancer Res. 2020;18(2):194–203. doi: 10.1158/1541-7786.MCR-19-0541. [DOI] [PubMed] [Google Scholar]
- 104.Zhong H, Chen G, Jukofsky L, et al. MDM2 antagonist clinical response association with a gene expression signature in acute myeloid leukaemia. Br J Haematol. 2015;171(3):432–435. doi: 10.1111/bjh.13411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Siu LL, Italiano A, Miller WH, et al. Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors. J Clin Oncol. 2014;32(15):2535–2535. doi: 10.1200/jco.2014.32.15_suppl.2535. [DOI] [Google Scholar]
- 106.Watters JW, Dickson MA, Schwartz GK, et al. TP53 mutations emerge in circulating cell-free DNA obtained from patients undergoing treatment with the HDM2 antagonist SAR405838. J Clin Oncol. 2015;33(15):2515–2515. doi: 10.1200/jco.2015.33.15_suppl.2515. [DOI] [Google Scholar]
- 107.Lam S, Lodder K, Teunisse AF, Rabelink MJ, Schutte M, Jochemsen AG. Role of Mdm4 in drug sensitivity of breast cancer cells. Oncogene. 2010;29(16):2415–2426. doi: 10.1038/onc.2009.522. [DOI] [PubMed] [Google Scholar]
- 108.Graves B, Thompson T, Xia M, et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc Natl Acad Sci USA. 2012;109(29):11788–11793. doi: 10.1073/pnas.1203789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wu D, Prives C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018;25(1):169–179. doi: 10.1038/cdd.2017.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stefaniak J, Lewis AM, Conole D, et al. Chemical instability and promiscuity of arylmethylidenepyrazolinone-based MDMX inhibitors. ACS Chem Biol. 2018;13(10):2849–2854. doi: 10.1021/acschembio.8b00665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Giustiniano M, Daniele S, Pelliccia S, et al. Computer-aided identification and lead optimization of dual murine double minute 2 and 4 binders: structure-activity relationship studies and pharmacological activity. J Med Chem. 2017;60(19):8115–8130. doi: 10.1021/acs.jmedchem.7b00912. [DOI] [PubMed] [Google Scholar]
- 112.Twarda-Clapa A, Krzanik S, Kubica K, et al. 1,4,5-Trisubstituted imidazole-based p53-MDM2/MDMX antagonists with aliphatic linkers for conjugation with biological carriers. J Med Chem. 2017;60(10):4234–4244. doi: 10.1021/acs.jmedchem.7b00104. [DOI] [PubMed] [Google Scholar]
- 113.Merlino F, Daniele S, La Pietra V, et al. Simultaneous targeting of RGD-integrins and dual murine double minute proteins in glioblastoma multiforme. J Med Chem. 2018;61(11):4791–4809. doi: 10.1021/acs.jmedchem.8b00004. [DOI] [PubMed] [Google Scholar]
- 114.Chang YS, Graves B, Guerlavais V, et al. Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci USA. 2013;110(36):E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meric-Bernstam F, Saleh M N, Infante J R, et al. Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX-and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas. J Clin Oncol. 2017;35(15_suppl): 2505.
- 116.Carvajal LA, Neriah DB, Senecal A, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med. 2018;10(436):eaao3003. doi: 10.1126/scitranslmed.aao3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gu L, Zhang H, Liu T, et al. Discovery of dual inhibitors of MDM2 and XIAP for cancer treatment. Cancer Cell. 2016;30(4):623–636. doi: 10.1016/j.ccell.2016.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.He S, Dong G, Wu S, et al. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): discovery of novel multitargeting antitumor agents. J Med Chem. 2018;61(16):7245–7260. doi: 10.1021/acs.jmedchem.8b00664. [DOI] [PubMed] [Google Scholar]
- 119.Daniele S, La Pietra V, Barresi E, et al. Lead optimization of 2-phenylindolylglyoxylyldipeptide murine double minute (MDM)2/translocator protein (TSPO) dual inhibitors for the treatment of gliomas. J Med Chem. 2016;59(10):4526–4538. doi: 10.1021/acs.jmedchem.5b01767. [DOI] [PubMed] [Google Scholar]
- 120.Carita G, Frisch-Dit-Leitz E, Dahmani A, et al. Dual inhibition of protein kinase C and p53-MDM2 or PKC and mTORC1 are novel efficient therapeutic approaches for uveal melanoma. Oncotarget. 2016;7(23):33542–33556. doi: 10.18632/oncotarget.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shirai Y, Shiba H, Iwase R, et al. Dual inhibition of nuclear factor kappa-B and Mdm2 enhance the antitumor effect of radiation therapy for pancreatic cancer. Cancer Lett. 2016;370(2):177–184. doi: 10.1016/j.canlet.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 122.Wang Z, Song T, Feng Y, et al. Bcl-2/MDM2 dual inhibitors based on universal pyramid-like α-helical mimetics. J Med Chem. 2016;59(7):3152–3162. doi: 10.1021/acs.jmedchem.5b01913. [DOI] [PubMed] [Google Scholar]
- 123.Qin JJ, Sarkar S, Voruganti S, Agarwal R, Wang W, Zhang R. Identification of lineariifolianoid A as a novel dual NFAT1 and MDM2 inhibitor for human cancer therapy. J Biomed Res. 2016;30(4):322–333. doi: 10.7555/JBR.30.20160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang J, Li Y, Aguilar A, Liu Z, Yang CY, Wang S. Simple structural modifications converting a bona fide MDM2 PROTAC degrader into a molecular glue molecule: a cautionary tale in the design of PROTAC degraders. J Med Chem. 2019;62(21):9471–9487. doi: 10.1021/acs.jmedchem.9b00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang B, Wu S, Liu J, Yang K, Xie H, Tang W. Development of selective small molecule MDM2 degraders based on nutlin. Eur J Med Chem. 2019;176:476–491. doi: 10.1016/j.ejmech.2019.05.046. [DOI] [PubMed] [Google Scholar]
- 126.Wang B, Liu J, Tandon I, et al. Development of MDM2 degraders based on ligands derived from Ugi reactions: lessons and discoveries. Eur J Med Chem. 2021;219:113425. doi: 10.1016/j.ejmech.2021.113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li Y, Yang J, Aguilar A, et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J Med Chem. 2019;62(2):448–466. doi: 10.1021/acs.jmedchem.8b00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kandarpa M, Peterson L, Potu H, et al. AML-397: preclinical efficacy of a PROTAC-based MDM2 degrader in AML models. Clin Lymph Myelom Leuk. 2020;20:S212. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.