

Abstract
Amyotrophic Lateral Sclerosis (ALS), a late-onset neurodegenerative disorder characterized by the loss of motor neurons in the central nervous system, has no known cure to-date. Disease causing mutations in human Fused in Sarcoma (FUS) leads to aggressive and juvenile onset of ALS. FUS is a well-conserved protein across different species, which plays a crucial role in regulating different aspects of RNA metabolism. Targeted misexpression of FUS in Drosophila model recapitulates several interesting phenotypes relevant to ALS including cytoplasmic mislocalization, defects at the neuromuscular junction and motor dysfunction. We screened for the genetic modifiers of human FUS-mediated neurodegenerative phenotype using molecularly defined deficiencies. We identified hippo (hpo), a component of the evolutionarily conserved Hippo growth regulatory pathway, as a genetic modifier of FUS mediated neurodegeneration. Gain-of-function of hpo triggers cell death whereas its loss-of-function promotes cell proliferation. Downregulation of the Hippo signaling pathway, using mutants of Hippo signaling, exhibit rescue of FUS-mediated neurodegeneration in the Drosophila eye, as evident from reduction in the number of TUNEL positive nuclei as well as rescue of axonal targeting from the retina to the brain. The Hippo pathway activates c-Jun amino-terminal (NH2) Kinase (JNK) mediated cell death. We found that downregulation of JNK signaling is sufficient to rescue FUS-mediated neurodegeneration in the Drosophila eye. Our study elucidates that Hippo signaling and JNK signaling are activated in response to FUS accumulation to induce neurodegeneration. These studies will shed light on the genetic mechanism involved in neurodegeneration observed in ALS and other associated disorders.
Keywords: Neurodegeneration, Amyotrophic Lateral Sclerosis (ALS) FUS, Fused in Sarcoma, TLS, Drosophila eye, Hippo pathway, JNK signaling, Cell death
Introduction
Amyotrophic Lateral Sclerosis (ALS), a fatal late-onset neurodegenerative disorder, is characterized by degeneration of motor neurons in the brain and spinal cord (Brenner and Weishaupt, 2019; Riggs, 1985). One of the hallmarks of ALS is disruption in RNA metabolism, which results in degeneration of motor neurons (Daigle et al., 2013; Donnelly et al., 2013; Ling et al., 2013; Nussbacher et al., 2019; Ortega et al., 2020). Mutations in more than 30 genes including superoxide dismutase1 (SOD1)(Rosen et al., 1993), TAR DNA-binding protein-43 (TDP-43)(Kabashi et al., 2008; Rutherford et al., 2008), C9ORF72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011), ubiquilin-2 (UBQLN2) (Deng et al., 2011), optineurin (OPTN)(Maruyama et al., 2010), and fused in sarcoma (FUS)(Kwiatkowski et al., 2009; Vance et al., 2009) results in ALS (White and Sreedharan, 2016). Of these, FUS is not only involved in causing ALS associated neurodegeneration (Chen et al., 2011), but also serves as a factor in the pathogenesis of other neurodegenerative disorders like polyglutamine diseases (Doi et al., 2008; Kino et al., 2016). FUS is a DNA/RNA binding protein that plays crucial role(s) in mRNA metabolism, nucleocytoplasmic RNA transport, and translation (Coyne et al., 2017; Kwiatkowski et al., 2009; Yang et al., 1998; Zinszner et al., 1997). Normally FUS shuttles between the nucleus and cytoplasm. Pathogenic mutations in FUS result in cytoplasmic mislocalization of a small fraction of the FUS protein. It suggests that cytoplasmic retention of FUS is likely causing cytotoxicity (Lanson et al., 2011; Zinszner et al., 1997). Further, deletion of Nuclear Export Signal (NES) disrupts the ability of this protein to enter the cytoplasm, which likely results in ALS neuropathy (Lanson et al., 2011). The RNA-binding ability of FUS has been suggested to play a major role in FUS mediated neurodegeneration and has been demonstrated by generating mutations in RNA recognition motifs (RRM) like R518K and R521C However, the exact mechanism(s) that can block FUS-mediated neurodegeneration is unclear. Therefore, it is important to understand functional consequences of disease-causing mutations in FUS, which promote cytoplasmic accumulation of mutant FUS, in order to discern the mechanism of FUS-mediated neurodegeneration.
Since the genetic machinery is highly conserved, several animal models including mice, zebrafish, and fruit flies are in use to study the molecular mechanisms of ALS disease (Casci and Pandey, 2015; Pandey and Nichols, 2011; Picher-Martel et al., 2016). Amongst these, Drosophila melanogaster a.k.a fruit fly, serves as a highly versatile and genetically tractable human disease model (McGurk et al., 2015; Olesnicky and Wright, 2018; Pandey and Nichols, 2011; Sarkar et al., 2016; Singh and Irvine, 2012). The Drosophila eye serves as a useful model to study neurodegenerative disorders like ALS (Casci and Pandey, 2015) as the retinal neurons are sensitive to cellular insult(s) and are not crucial for the survival and reproductive ability of the fly (Sarkar et al., 2016; Tare et al., 2011). The adult compound eye of Drosophila develops from an epithelial bi-layer structure, which is housed inside the larva, called an eye-antennal imaginal disc. The larval eye imaginal disc develops into the adult compound eye comprised of 800 unit eyes called ommatidia (Kumar, 2018; Ready et al., 1976; Singh et al., 2005; Singh et al., 2012; Tare et al., 2013). Misexpression of wild-type FUS or mutant FUS, in the differentiating retinal neurons of Drosophila eye, using a GMR-Gal4 driver (Moses and Rubin, 1991) results in severe degeneration of retinal neurons. Targeted misexpression of FUS (FUS) or mutant FUS in motor neurons exhibits locomotor dysfunction (Casci and Pandey, 2015; Coyne et al., 2017; Daigle et al., 2013; Lanson et al., 2011; Pandey and Nichols, 2011; Xia et al., 2012).
We have used a well-characterized fruit fly model to screen for the genetic modifiers of FUS-mediated neurodegeneration using molecularly defined deletions. We identified a deficiency, which uncovers hippo (hpo) and other genes as a suppressor of FUS-mediated neurodegeneration. The Hippo pathway is an evolutionarily conserved growth regulatory pathway (Kango-Singh and Singh, 2009; Ma et al., 2019). The core components of the Hippo pathway are Ste20 family protein kinase hpo (Harvey et al., 2003; Pantalacci et al., 2003; Udan et al., 2003; Wu et al., 2003), the nuclear Dbf-2-related (NDR)-family kinase- warts (wts)(Justice et al., 1995; Xu et al., 1995), and yorkie (yki)- the Drosophila orthologs of the mammalian transcription co-activator yes-associated protein (YAP) (Huang et al., 2005). When Hippo pathway is activated, Hpo binds with and phosphorylates Salvador (Sav), a protein containing WW and coiled-coil domains, and phosphorylates the downstream Wts kinase (Kango-Singh et al., 2002; Kango-Singh and Singh, 2009; Tapon et al., 2002). The active, phosphorylated form of Wts binds with an adaptor protein Mob-as-tumor suppressor (Mats) (Lai et al., 2005) and in turn phosphorylates the transcriptional co-activator Yki. The phosphorylated form of Yki binds with 14-3-3 proteins, and undergoes 14-3-3 mediated proteasomal degradation in the cytoplasm (Dong et al., 2007; Ren et al., 2010; Wei et al., 2007). However, downregulation of the Hippo signaling leads to release of Wts-mediated repression of Yki, and the unphosphorylated Yki goes into the nucleus, binds to its partner(s) such as the TEAD family transcription factor Scalloped (Sd), and regulates expression of the downstream targets of the Hippo pathway like expanded (ex), drosophila inhibitor of apoptosis (diap1), cyclins, bantam (ban), etc. (Fulford et al., 2018; Kango-Singh and Singh, 2009; Yu and Guan, 2013). Loss-of-function of hpo, sav, wts, mats or overexpression of yki activity leads to cell proliferation and over growth, while gain-of-function of the Hpo signaling leads to apoptosis or cell death (Kango-Singh and Singh, 2009).
The Hippo pathway activates JNK, an evolutionarily conserved signaling pathway, to trigger cell death (Ma et al., 2015; Ma et al., 2017). JNK signaling is involved in a wide array of signaling events underlying tumorigenesis and is known to regulate cell proliferation, invasive migration, and cell death (Dhanasekaran and Reddy, 2017). JNK, or stress activated kinase proteins of mitogen-activated protein kinase (MAPK) superfamily (Adachi-Yamada et al., 1999; Adachi-Yamada and O'Connor, 2002), induce cell death due to phosphorylation of transcription factors involved in cell death (Dhanasekaran and Reddy, 2008). JNK signaling pathway in Drosophila gets activated downstream of the Tumor Necrosis Factor (TNF) homolog Eiger (Egr) and its receptor Wengen (Wgn) by a conserved downstream signaling cascade. The core components of this pathway comprises of Tak1 (TGF-β- activating kinase 1); JNK Kinase Kinase (JNKKK), Hemipterous (Hep; JNK Kinase- JNKK, which is closely related to mitogen-activated protein Kinase Kinases (MAPKKs) (Glise et al., 1995; Tournier et al., 1997). Other components are Basket (Bsk; a Jun kinase), D-Jun (Jun), and Puckered (Puc). bsk acts downstream of hep and is activated by constitutive hep phosphorylation (Sluss et al., 1996). Phosphorylation of Jun leads to activation of the JNK signaling pathway that functions as a dual specificity protein phosphatase (Martín-Blanco et al., 1998). puc functions as a transcriptional target of JNK signaling and is known to regulate JNK signaling through a negative feedback loop (Adachi-Yamada et al., 1999; Adachi-Yamada and O'Connor, 2002). Furthermore, ectopic activation of JNK signaling induces cell death during early eye imaginal disc development (Singh et al., 2006). Here we present evidences to show that targeted misexpression of FUS triggers neurodegeneration as seen in ALS models by activating the Hippo signaling pathway, which we identified in a forward genetic screen. We further investigated the genetic mechanism and found that activation of the Hippo signaling pathway due to gain-of-function of FUS in turn triggers the JNK signaling to induce neurodegeneration in the developing Drosophila eye.
Materials and Method
Fly Stocks
The fly stocks used in this study are listed at FlyBase (http://flybase.bio.indiana.edu). We used the transgenic fly strains that carry full length FUS (UAS-FUS)(Lanson et al., 2011) and mutant FUS, UAS-FUS R518K and UAS-FUS R521C(Casci et al., 2019). Other stocks used are UAS-hpo (Udan et al., 2003), yw, hsflp; UAS-hpoRNAisymp19/SM6-TM6B,Tb (Pantalacci et al., 2003), UAS-wts13f (Kwon et al., 2015), UAS-wtsRNAi (Fernández et al., 2011; Rauskolb et al., 2011), UAS-ykiRNAi (N+C) (Zhang et al., 2008), a hyperactivated form that allows yki activation, UAS-yki3SA (Oh and Irvine, 2008), UAS-Djunaspv7(Treier et al., 1995), UAS-bskDN (Adachi-Yamada et al., 1999), UAS-hepAct (Glise et al., 1995), UAS-puc (Martín-Blanco et al., 1998) and diap1-4.3-GFP (Ren et al., 2010). The diap1-4.3- GFP gene, contains a 4.3-kb diap1 genomic fragment from +1.37 kb to +5.68 kb, which contains a Hippo response element that drives the Diap1 expression in response to Hippo pathway activity where GFP is a reporter for Yki activity. We used a molecularly defined deficiency Df(2R)BSC782/+, which is located on the right arm of the second chromosome, and uncovers βTub56D, par-1, CG16926, CG7744, CG15120, mei-W68, oseg6, TBCB, rep and hpo genes (listed in FlyBase). We used the Canton-S (Wild-type) stock of D. melanogaster in this study. Fly stocks used in this study (Supplementary Table 1) were maintained at 25°C on cornmeal, molasses food.
Genetic Crosses
We employed the Gal4/UAS system for targeted misexpression studies (Brand and Perrimon, 1993). The GMR-Gal4 enhancer directs expression of transgenes in the differentiating retinal precursor cells in the developing larval eye-antennal imaginal discs (Moses and Rubin, 1991; Tare et al., 2011). We used GMR-Gal4 to drive expression of transgenes. We used GMR-Gal4/ CyO; UAS-FUS / TM3 Sb e Ser (GMR>FUS), GMR-Gal4/ CyO; UAS-FUS R518K / TM3 Sb e Ser (GMR>FUS R518K), GMR-Gal4/ CyO; UAS-FUS R521K/ TM3Sb e Ser (GMR>FUSR521K) to sample FUS-mediated neurodegeneration in the eye. All fly stocks were maintained at 25°C. All Gal4/UAS crosses were maintained at 18°C, 25°C, or 29°C to sample different induction levels (Singh and Choi, 2003). The third instar larval eye-antennal imaginal discs were used for immunohistochemistry purposes.
Immunohistochemistry
The eye-antennal imaginal discs were dissected from the wandering third-instar Drosophila larvae in 1X PBS (Phosphate Buffered Saline, pH 7.4) following the standard protocol (Sarkar et al., 2018a; Singh et al., 2002). Tissue samples were then fixed in 4% paraformaldehyde for 20 minutes and washed with 1X PBST (1XPBS + 0.2%TritonX-100), 3 times for 10 minutes each. Tissues were incubated with different combinations of primary and secondary antibodies. The primary antibodies used were: rabbit anti-FUS (1:500, Bethyl Laboratories, A300-302A), mouse anti-Dlg (1:100, Developmental Studies Hybridoma Bank-DSHB, 4F3), mouse anti-Wg (1:50, DSHB, 4D4), rat anti-Elav (1:100; DSHB, 7E8A10), mouse anti-Chaoptin, MAb24B10 (1:100, DSHB, 24B10) (Zipursky et al., 1984), and rabbit anti-β-galactosidase (1:100, Promega, Z3781). Secondary antibodies (Jackson Immuno Research) used were donkey anti-rabbit IgG conjugated with FITC (dilution 1:200), donkey anti-mouse IgG conjugated with Cy3 (dilution 1:250), and goat anti-rat IgG conjugated with Cy5 (dilution 1:250). After primary and secondary antibody incubation, tissue samples were washed with 1XPBST (X3) for 10 minutes each to remove excess antibody bound to sample. Following washing, tissue samples were mounted in Vectashield mountant (Vector laboratories-H1000). The images were scanned using Olympus Fluoview-3000 Laser Scanning Confocal Microscope (LSCM) and final figures were made using Adobe Photoshop CS6 software (Sarkar et al., 2018a). The quantification of GFP intensity was done using built-in program in Olympus Fluoview 3000 confocal microscope. The statistical analysis of eye-antennal imaginal discs images was conducted using Microsoft Excel 2017 software, p-values were calculated using a Student’s two-tailed t-test and the error bars represent the values of standard deviation calculated from the mean (Tare et al., 2016).
Detection of Cell Death
The TUNEL assay marks the cells undergoing cell death. In TUNEL staining, the cleavage of double and single-stranded DNA is labeled by a Fluorescent tag (TMR Red) (McCall and Peterson, 2004; White et al., 1994). The fluorescently labeled nucleotides are added to 3’ OH ends in a template-independent manner by Terminal Deoxynucleotidyl transferase (TdT). The dying cell’s fragmented DNA is tagged with a fluorochrome and can be detected using a fluorescence microscope or confocal microscope. Eye antennal discs after secondary antibody staining (Singh et al., 2006) were blocked in 10% normal donkey serum in Phosphate buffered saline with 0.2% Triton X-100 (PBT) and labeled for TUNEL assays using a cell death detection kit from Roche Diagnostics (12156792910). For negative controls, eye-antennal imaginal discs were treated with a cell death detection labeling mix without the TdT enzyme added.
The TUNEL positive nuclei were counted from five sets of eye imaginal discs for each experiment. The cell count number was used for statistical analysis using Microsoft excel 2013. The p-values were calculated using a Student’s two-tailed t- test. The error bars represent the standard deviation from the mean (Steffensmeier et al., 2013).
Adult Eye Imaging
The adult flies were prepared for imaging by freezing them at −20°C for approximately 2 hours. Following this incubation, the flies were mounted on a dissection needle. The needle carrying the fly was placed horizontally over a glass slide using a putty. Images were captured on a MrC5 color camera mounted on an Axioimager.Z1 Zeiss Apotome using a Z-sectioning method (Sarkar et al., 2018a; Wittkorn et al., 2015). The final images were generated by compiling individual stacks from the Z-section using the extended depth of focus function of Axiovision software version 4.6.3.
Scanning Electron Microscopy (SEM)
SEM was carried out using the standard protocol (Tare and Singh, 2009). The flies with phenotype were dehydrated through a series of increasing concentrations of acetone. The dehydrated fly samples were then incubated in 1:1 ratio of acetone and Hexamethyldisilazane (HMDS, Electron Microscopy Sciences) solution for 24 hours, followed by incubation in 100% HMDS alone. The flies were then air dried in the fume hood. The samples were mounted onto the conductive carbon tape attached to stubs (Electron Microscopy Sciences). The flies mounted on the stub were then subjected to sputter coating with gold using a Denton vacuum sputter coater (DV502). The samples were photographed using a Hitachi S-4800 High Resolution Scanning Electron Microscope (HRSEM) (Singh et al., 2019). The images were later analyzed using Adobe Photoshop CS6 software.
Western Blot
The protein samples were extracted from heads of Wild-type, GMR>FUS, GMR> FUS-R518K, and GMR>FUS-R521C adult flies using a standardized protocol (Gogia et al., 2017). The primary antibodies used were Phospho-SAPK/JNK (Thr183/Tyr185) (81E11) (1:3000, Cell Signaling) and anti-FUS (1:1500, A300-302A, Bethyl laboratories). A signal was detected using a Horse Radish Peroxidase (HRP) conjugated goat anti-rabbit IgG (1:5000), and Super Signal West Dura Extended Duration Substrate (Thermo Scientific). Images were captured using the BioSpectrum® 500 imaging system and analyzed using Adobe Photoshop CS6 software. To measure the band intensity, statistical analysis was conducted on western blots using Microsoft Excel 2017 software. The p-values were calculated using Student’s two-tailed t-test and the error bars represent the values of standard deviation calculated from the mean (Sarkar et al., 2018b).
HEK293T cells (ATCC® CRL-3216™) were cultured in Advanced DMEM supplemented with 10% FBS and 1% Glutamax and grown at 37°C and 5% CO2(Casci et al., 2019). HEK293T cells were transiently transfected using Turbofect (Invitrogen) with pCI-neo-HA-FUS WT, pCI-neo-HA-FUS R518K and pCI-neo-HA-FUS R521C. Cells were lysed in RIPA buffer (150 mM NaCl, 1% NP40, 0.1% SDS, 1% sodium deoxycholate, 50 mM NaF, 2 mM EDTA, 1 mM DTT, 2.5 mM Na orthovanadate, 1x protease inhibitor cocktail [Roche 11836170001), pH 7.4] 24 hours post-transfection, and centrifuged down at 12000xg for 10 minutes. The supernatant was boiled in 1X NuPage LDS-Sample buffer (Invitrogen NP0007) at 95°C for 5 minutes. SDS-PAGE was performed using 4-12% NuPage Bis-Tris gels (Invitrogen), and the separated proteins were transferred onto nitrocellulose membranes using the iBlot2 system (Life Technologies 13120134). Nitrocellulose membranes were incubated overnight at 4°C with primary antibody (1:1000 LATS1, SCBT #398560; 1:500 Phospho-LATS1 (Thr1079), CST #8654; 1:8000 α-tubulin, Sigma T5168) followed by secondary antibody at room temperature for 1 hour. Membranes were imaged on Odyssey CLx (LI-COR Biosciences) and quantification of bands was performed using Image Studio (LI-COR Biosciences). Statistical analysis was performed using Graphpad Prism.
Real Time Quantitative Polymerase Chain Reaction
Real time Quantitative Polymerase Chain Reaction (RT-qPCR) was performed according to the standardized protocol (Mehta and Singh, 2017; Mehta et al., 2019). Total RNA was extracted in 500 μl of TRIzol Reagent (Thermo Fisher, Cat. No. # 15596926) from twenty pairs of third instar eye-antennal imaginal discs (n = 40), which were dissected from wild-type (WT), GMR>FUS, GMR>FUS-R518K and GMR>FUS-R521C background larvae. The quality of isolated RNA was determined by using the Nanodrop 2000 spectrophotometer (Thermo Scientific). Good quality samples had A260/A280 ratio greater than 2 and a peak at 260 nm. cDNA was produced from total RNA through RT-PCR using the first- strand cDNA synthesis kit (GE healthcare, Cat# 27926101). RT-qPCR was performed using iQ™ SYBR Green Supermix (Bio-Rad) and Bio-Rad iCycler (Bio-Rad) following the kits protocol for 25 μl. Primers used for Jun-N-terminal kinase (JNK) are: (fwd: CCAACCGTCCGAAACTATGT); rev: CCGGCGGCTATTCTGATTATTA). The expression level of Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control to normalize the results (fwd: CAATGGATTTGGTCGCATCG; rev: CCGTTGACCACCAGGAAACC). The fold change was calculated relative to the expression level of the respective control (WT, Canton-S).
Results
Search for genetic modifier of human-FUS mediated neurodegeneration.
The larval eye imaginal discs develop into an adult compound eye comprised of nearly 800 ommatidia arranged in a stereotypical fashion (Fig. 1 A, F). We stained the eye-antennal imaginal discs with Disc large (Dlg: Green, a membrane specific marker) and pan neural marker Embryonic Lethal Abnormal Vision (Elav: Red), which marks the nuclei of the photoreceptor neurons (Fig. 1A). Targeted misexpression of full length human-FUS (FUS) in differentiating retinal neurons using a Glass Multiple Repeat (GMR) eye specific promoter (GMR-Gal4/+), exhibits a strong retinal degeneration phenotype in the eye imaginal disc (n=10, Fig. 1C) and highly reduced adult eye with loss of eye pigmentation (n=200, Fig S1A, Fig. 1H). The control GMR-Gal4 driver alone does not show any neurodegenerative phenotype as seen in the eye imaginal disc and the adult eye (Fig. 1B, G, S1A). We tested other transgenic alleles of mutant FUS as well. Targeted misexpression of FUS R518K (Fig. 1D, I), and FUS R521C (Fig. 1E, J) also exhibits strong neurodegenerative phenotypes both in eye imaginal discs and the adult eye (Fig. S1A).
Figure 1. Identification of a molecularly defined deletion as a genetic modifier of human FUS mediated neurodegeneration.

Eye imaginal disc and the adult eye of (A, F) Wild-type and (B, G) GMR-Gal4/+ (an eye specific promoter) that serve as control. (A)Third instar larval eye antennal imaginal disc, which develop into (F) the adult compound eye. Note that the eye-antennal imaginal discs are stained with a membrane specific marker Disc large (Dlg: Green) and pan neural marker Embryonic Lethal Abnormal Vision (Elav: Red), which marks the nuclei of the photoreceptor neurons. In comparison to controls, misexpression of (C, H) FUS (GMR-Gal4/CyO; UAS-FUS/ TM3Sb e Ser, GMR>FUS) or mutant FUS (D, I) FUS R518K (Gal4/CyO; UAS-FUS R518K/ TM3Sb e Ser, GMR>FUS R518K), and (E, J) FUS R521C (Gal4/CyO; UAS-FUS R5521C/ TM3Sb e Ser, GMR>FUS R521C) in differentiating retinal neurons using GMR-Gal4 driver results in strong neurodegeneration in retinal neurons as seen in (C, D, E) eye imaginal discs and the (H, I, J) adult eye. Note that phenotype worsens as the (C, D, E) larval eye-antennal imaginal discs transforms into (H, I, J) adult eyes. In a deficiency screen, Df(2R)BSC782 was identified as a genetic modifier of FUS mediated neurodegenerative phenotype. (K) The map shows the region and genes (including hpo) covered by deficiency line (Df(2R)BSC782/+). (L, Q) Df(2R)BSC782/+ (M, R) GMR>Df(2R)BSC782/+ served as controls. In a heterozygous background of (Df(2R)BSC782/+), misexpression of (N, S) FUS (GMR>FUS/+;Df(2R)BSC782/+), or mutant FUS, (O, T) FUS R518K (GMR>FUS R518K/+ ; Df(2R)BSC782/+) and (P, U) FUS R521C (GMR>FUS R521C/+; Df(2R)BSC782/+), results in the significant rescue of FUS mediated neurodegeneration as seen in (N, O, P) the eye-antennal imaginal discs and (S, T, U) the adult compound eyes of Drosophila respectively. The neurodegeneration phenotype in the eye was significantly rescued and pigment cells were restored. The orientation of all imaginal discs in the figure is posterior to left and dorsal up. Magnification of all eye discs is 20X.
We verified if these transgenes were expressing human FUS protein (GMR>FUS, in heterozygous combination) in the Drosophila eye by using an antibody against human FUS protein. We found that FUS expression was restricted to the GMR expression domain of the developing third instar eye imaginal disc of GMR>FUS, GMR>FUS R518K, and GMR>FUS R521C (Fig. S2A-C, G). These results confirmed that the neurodegeneration seen in the eye-antennal imaginal discs and the adult fly eyes is due to misexpression of the human FUS protein. We also calculated the intensity of FUS signals as seen by immunohistochemistry detected by FUS antibody in eye-antennal imaginal disc and found that there was no significant difference of FUS levels in all three alleles (Fig. 1G). The protein extracts from these samples showed a significant accumulation of FUS protein in a semi-quantitative western blot (Fig. S2 H, H’).
In order to understand the genetic basis of the FUS gain-of-function mediated neurodegeneration as seen in ALS, we employed a forward genetic screen using the molecularly defined deficiencies. We identified a deficiency line Df(2R)BSC782, which in heterozygous combination (Df(2R)BSC782/+) can rescue the GMR>FUS mediated neurodegeneration as seen in the eye imaginal disc and the adult eye (Fig. 1N, S). This molecularly defined Df(2R)BSC782/+ deficiency is located on the right arm of the second chromosome, and uncovers βTub56D, par-1, CG16926, CG7744, CG15120, mei-W68, oseg6, TBCB, rep, and hpo genes (Fig. 1K). Targeted misexpression of other FUS transgenes in Df(2R)BSC782/+ background, which includes GMR>FUS R518K+Df(2R)BSC782/+ (Fig. 1O, T) and GMR>FUS R521C + Df(2R)BSC782/+ (Fig. 1P, U), exhibit a stronger rescue with a near complete wild-type phenotype in both the eye-antennal imaginal discs as well as the adult eyes. The heterozygous combination of deficiency Df(2R)BSC782/+ alone (Fig. 1L, Q) and GMR>Df(2R)BSC782/+ (Figure 1M, R) respectively, which served as controls, exhibit a wild-type phenotype in the eye-antennal imaginal disc and the adult eye. We also tested the levels of FUS expression levels in eye-imaginal discs from GMR>FUS, Df(2R)BSC782/+, GMR>FUS R518K+Df(2R)BSC782/+ and GMR>FUS R521C + Df(2R)BSC782/+, where FUS-mediated neurodegenerative phenotypes are rescued (Fig.1). Interestingly, in this background, FUS levels were not significantly affected (Fig.S2D-G).
hpo acts as a genetic modifier of FUS mediated neurodegeneration.
To narrow down the gene, which is responsible for modifying the FUS mediated neurodegeneration phenotype, we individually tested the genes uncovered by Df(2R)BSC782 deficiency (Fig. 1K), using gain-of-function and loss-of-function approaches. Of these, modulation of hippo (hpo), one of the genetic loci uncovered by Df(2R)BSC782, affects the FUS neurodegenerative phenotype (Fig. 2). In comparison to the wild-type adult eye (Fig. 2A), and control GMR-Gal4/+ eye (Fig. 2B), gain-of-function of FUS as seen in GMR>FUS/+ (Fig. 1H, 2C), GMR>FUS R518K/+ (Fig. 1I, 2D), and GMR>FUS R521C/+ (Fig. 1J, 2E) results in a strong neurodegenerative phenotype (Lanson et al., 2011). These eyes are of reduced size and exhibit a rough eye phenotype, with disorganized lenses that clearly marks the onset of neurodegeneration. Gain-of-function of hpo in various backgrounds like FUS (GMR>FUS + hpo) (Fig. 2F), FUS R518K (GMR>FUSR518K+hpo; Fig. 2G), and FUS R521C (GMR>FUS R521C+hpo; Figure 2H) exhibit further enhancement in the severity of neurodegenerative phenotypes of FUS gain-of-function in the eye. On the contrary, loss-of-function of hpo by misexpressing hpoRNAi, with FUS (GMR>FUS+hpoRNAi), FUS R518K (GMR>FUS R518K+hpoRNAi), or FUS 521C (GMR>FUS R521C+hpoRNAi), rescues FUS mediated neurodegenerative phenotype (Fig. 2 I, J, K). These phenotypes were statistically significant (Fig. S1B). Our data strongly suggests that the hpo gene plays a role in triggering FUS mediated neurodegeneration in the Drosophila eye.
Figure 2. hpo is the genetic modifier of FUS mediated neurodegeneration in Drosophila eye.
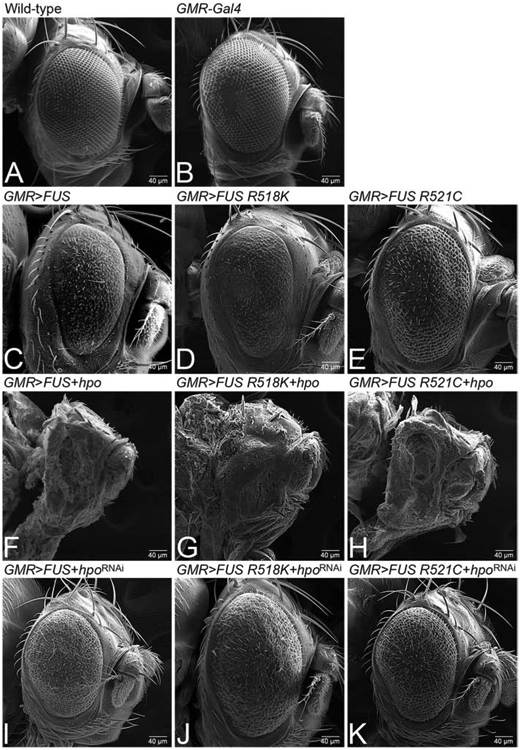
(A) Wild-type and (B) GMR-Gal4/+ adult fly eyes serve as controls. Targeted misexpression of FUS or mutant FUS R518K and FUS R521C in the GMR domain of eye (C) GMR>FUS (D) GMR>FUS R518K (E) GMR>FUS R521C results in reduced and rough eye phenotype due to neurodegeneration. Activation of Hippo signaling by misexpressing hpo along with FUS or mutant FUS (FUS R518K and FUS R521C) in the GMR domain of eye using GMR driver, (F) GMR>FUS + hpo, (G) GMR>FUS R518K+ hpo, (H) GMR>FUS R521C +hpo, further enhances the FUS mediated neurodegeneration phenotype as compared to misexpressing FUS or mutant FUS in GMR domain alone. Downregulation of Hippo signaling by misexpressing hpoRNAi along with FUS (I) GMR>FUS+ hpoRNAi, or mutant FUS (J) GMR>FUS R518K+ hpoRNAi, (K) GMR>FUS R521C+hpoRNAi in the GMR domain rescues the FUS mediated neurodegeneration, as seen in SEM images of the adult eyes of FUS or mutant FUS in GMR domain alone. Note that all transgenes are in heterozygous combination. Magnification of all the scanning electron microscopic images of adult fly eyes is 180X.
Hippo signaling downregulation can rescue FUS mediated neurodegeneration
Hpo, a kinase, is one of the core components of an evolutionarily conserved Hippo signaling pathway. We therefore tested if this neuroprotective function is exclusive to hpo gene or if it is dependent on the Hippo signaling pathway. We therefore tested other members of the Hippo signaling pathway. Ectopic expression of FUS, FUS R518K, and FUS R521C in differentiating retinal neurons using a GMR driver, which results in a neurodegenerative phenotype in the eye-antennal imaginal discs (Fig.1) and the adult fly eye (Fig. 1, 2C, D, E). In comparison to the controls (Fig. 3A, B), activation of Hippo signaling via misexpression of wts and ykiRNAi along with FUS (GMR>FUS+wts, GMR>FUS+ykiRNAi), results in a strong enhancement of neurodegenerative phenotypes (Fig. 3F, I) as compared to the controls (Fig.3A, B, C). Similarly, activation of Hippo signaling in the gain-of-function of FUS R518K (GMR>FUS R518K+wts, Fig. 3G; GMR>FUS R518K+ykiRNAi, Fig. 3J), or FUS R521C (GMR>FUS R521C+wts, Fig. 3H; and GMR>FUS R521C+ykiRNAi, Fig. 3K) backgrounds in the GMR domain of eye worsens the FUS mediated neurodegeneration phenotype (Fig. 3C, D, E).
Figure 3. Downregulation of Hippo signaling rescues FUS mediated neurodegeneration.

(A) Wild-type and (B) GMR-Gal4/+ adult fly eyes as controls. Targeted misexpression of WT FUS or mutant FUS R518K and FUS R521C in the GMR domain of eye (C) GMR>FUS (D) GMR>FUS R518K (E) GMR>FUS R521C results in rough eye or neurodegeneration phenotype. Activation of Hippo signaling by misexpressing wts or ykiRNAi along with full length wild-type FUS or mutant FUS (FUS R518K and FUS R521C) in the GMR domain of eye using GMR driver, (F) GMR>FUS+wts, (G) GMR>FUS R518K+wts, (H) GMR>FUS R521C+wts, (I) GMR>FUS+ykiRNAi, (J) GMR>FUS R518K+ykiRNAi, (K) GMR>FUS R521C+ykiRNAi, further enhances or worsens the FUS or mutant FUS alone mediated neurodegeneration phenotype. In-activation of Hippo signaling by misexpressing wtsRNAi, yki3SA along with full length wild-type FUS (L) GMR>FUS+wtsRNAi, (O) GMR>FUS+yki3SA, or mutant FUS (M) GMR>FUS R518K+wtsRNAi, (N) GMR>FUS R521C+wtsRNAi, (P) GMR>FUS R518K+yki3SA, (Q) GMR>FUS R521C+yki3SA in GMR domain rescues the FUS mediated neurodegeneration, as seen in scanning electron microscopic (SEM) images of adult fly eyes of FUS alone or mutant FUS. Note that all transgenes are in heterozygous combination. Magnification of all the SEM images of the adult fly eyes is 180X.
We further verified our hypothesis by downregulation of Hippo signaling using wts RNAi and yki misexpression in the FUS gain-of-function backgrounds. Downregulation of Hippo signaling in FUS (GMR>FUS+wtsRNAi, Fig. 3L GMR>FUS+yki3SA, Fig. 3O), FUS R518K (GMR>FUS R518K+wtsRNAi, Fig. 3M; GMR>FUS R518K+yki3SA, Fig. 3P), or FUS R521C (GMR>FUS R521C+wtsRNAi, Fig. 3N; and GMR>FUS R521C+yki3SA, Fig. 3Q) results in overgrowth and in the case of wts showed rescue of the neurodegenerative phenotype. These phenotypes were statistically significant (Fig.S2C).
Downregulation of Hippo signaling can block FUS mediated neuronal cell death.
We decided to test if Hippo signaling downregulation rescues FUS mediated neurodegeneration by blocking cell death. We used TUNEL staining, which marks the fragmented DNA, to mark the nuclei of the dying neurons (Cutler et al., 2015; Gogia et al., 2017; McCall and Peterson, 2004; Sarkar et al., 2018b; Tare et al., 2011; White et al., 1994). We performed TUNEL staining in third instar larval eye-antennal imaginal discs of the wild-type larvae (Figure. 4A), GMR>FUS (Fig. 4B). The number of TUNEL positive cells, shown as white dots, were quantified from five eye-antennal imaginal discs (n=5) of each genotype. We took the mean, standard deviation, standard error, and p-values of this data and recorded it on a graph (Fig. 4I). Note that there are fewer TUNEL positive dying cells nuclei in the wild-type eye-antennal imaginal discs, which serves as control, as compared to the experiments (Fig. 4 A, I). Targeted misexpression of FUS (GMR>FUS) exhibits an almost two-fold increase in the number of TUNEL positive cells as compared to the control (Fig. 4 B, I). We also tested other components of the Hippo signaling pathway. Activation of Hippo signaling as seen in GMR>FUS+hpo (Fig. 4C), GMR>FUS+wts (Fig. 4E), GMR>FUS+ykiRNAi (Fig. 4G) results in the enhancement of the neurodegenerative phenotype of GMR>FUS alone (Fig. 4I). Furthermore, activation of Hippo can result in a nearly two-fold increase in cell death as compared to the GMR>FUS alone (Fig. 4I). Inactivation of Hippo signaling in GMR>FUS background as seen in GMR>FUS+hpoRNAi (Fig. 4D), GMR>FUS+wtsRNAi (Fig. 4F), GMR>FUS+yki3SA (Fig. 4H) resulted in the suppression of GMR>FUS mediated neurodegeneration. This suppression is accompanied by a significant decrease in the number of TUNEL positive cells or dying nuclei/ cell death in differentiating eye neurons compared to the wild-type (Fig. 4I). These observations confirm that downregulation of Hippo signaling blocks cell death in the differentiating photoreceptor neurons and thus plays a primary role of rescuing FUS mediated neurodegeneration.
Figure 4. Downregulation of Hippo signaling blocks cell death in the differentiating neurons to rescue FUS mediated neurodegeneration.

TUNEL, marks the fragmented DNA within the nuclei of dying cells (also called as TUNEL positive cells). TUNEL staining was carried out in (A) wild-type third instar larval eye-antennal imaginal disc, displaying randomly distributed TUNEL positive dying cells shown as white dots. The area between the white dotted line and posterior margin of the eye imaginal disc is the domain of GMR-Gal4 driver. The number of TUNEL positive cells were counted from five eye-antennal imaginal discs (n=5) of all the genotypes in the GMR-Gal4 domain. (B) Misexpression of FUS in the differentiating photoreceptor neurons of the eye using GMR-Gal4, eye specific promoter (GMR>FUS), show elevated levels of TUNEL positive cells, indicating increased frequency of cell death in photoreceptor neurons, as compared to the wild-type control (A,I). Note that the number of dying cells (TUNEL positive cells) increase nearly ~0.5 fold in (B) GMR>FUS as compared to the (A) wild-type eye-antennal imaginal discs. Activation of Hippo signaling by misexpressing hpo, wts, ykiRNAi in WT FUS background, in GMR domain of eye in (C) GMR>FUS+hpo, (E) GMR>FUS+wts, and (G) GMR>FUS+ykiRNAi, results in further increase in the number of dying retinal neurons (or TUNEL positive cells). On the other hand, downregulation of Hippo signaling by misexpressing hpoRNAi, wtsRNAi, yki3SA in full length wild-type FUS background in (D) GMR>FUS+hpoRNAi, (F) GMR>FUS+wtsRNAi, and (H) GMR>FUS+yki3SA, results in significant reduction in the dying retinal neurons. (I) A graph representing the Quantitative analysis (statistics). Comparing the number of dying nuclei of neurons, when FUS is ectopically expressed alone, and when FUS is expressed together with components of Hippo pathway in the eye, clearly shows that downregulation of Hippo signaling significantly rescues FUS mediated neurodegeneration by blocking neuronal cell death. The p-values for the estimation of cell death were calculated between GMR>FUS and all the components of Hippo pathway using Student’s two-tailed t-test (n=5) in Microsoft excel software. GMR>FUS phenotype was found to be statistically significant with respect to Wild-type (****, p<0.0001). The genotypes including GMR>hpo+ FUS (****, p<0.0001), GMR>FUS+hpoRNAi (****, p<0.0001), GMR>FUS + wts (****, p<0.0001), GMR>FUS + wtsRNAi (****, p<0.0001) and (G) GMR>FUS + yki, (****, p<0.0001), GMR>FUS + ykiRNAi (*, p<0.05). Note that all transgenes are in heterozygous combination. Magnification of all eye-antennal imaginal discs is 20X.
Loss of Hippo signaling restores impaired axonal targeting due to FUS expression.
During Drosophila eye development, the axonal projections from photoreceptor neurons innervates the medulla and lamina layers of the brain’s optic lobe. We used a sensory neuron marker 24B10 (Chaoptin, DSHB), which marks the photoreceptor neurons and their axons (Fig. 5A) (Zipursky et al., 1984). Axonal targeting studies using Chaoptin staining is a qualitative approach to determine the functionality of the retinal neurons. We asked if the neurodegenerative phenotype seen in GMR>FUS also involves disruption of axonal targeting from the retina to the brain. We reasoned that if the downregulation of Hippo signaling that rescues the GMR>FUS phenotype in larval eye-antennal disc, it also may rescue the axonal targeting from the retina to the brain. In the wild-type eye-antennal imaginal discs, which serve as control, R1-R6 axons of the respective photoreceptors of an ommatidium innervates the lamina of the brain while R7 and R8 axons innervate the medulla (Meinertzhagen and Hanso, 1993; Newsome et al., 2000). It is known that when axonal targeting is disrupted it impairs axonal transport and leads to neurodegeneration observed in neurodegenerative diseases (Sarkar et al., 2018b). We investigated the effect of modulating hpo levels in the full-length FUS background on the axonal projections of photoreceptor retinal axons. In comparison to the wild-type eye-antennal imaginal disc (Fig. 5A), ectopic expression of FUS (GMR>FUS; Fig. 5B), FUSR518K (GMR> FUSR518K; Fig. 5C) and FUSR521C (GMR>FUSR521C; Fig. 5D) in the GMR domain resulted in a disorganized, defective and aberrant axonal targeting. Furthermore, gain-of-function of hpo along with FUS (GMR>FUS+hpo; Fig. 5E), FUS R518K (GMR> FUS R518K+ hpo; Fig. 5F) and FUS R521C (GMR>FUS R521C+ hpo; Fig. 5G) in the GMR domain severely impaired axonal targeting in larval eye-antennal imaginal discs as seen in (Fig. 5B, C, D). The axonal projections significantly decrease, and fail to innervate the lamina and medulla regions of the optic lobe (Fig. 5E, F, G). In contrast, loss-of-function of hpo signaling by misexpressing hpoRNAi in FUS (GMR>FUS+hpoRNAi; Fig. 5H), FUS R518K (GMR> FUS R518K+ hpoRNAi; Fig. 5I) and FUS R521C (GMR>FUS R521C+ hpoRNAi; Fig. 5J) background significantly restores retinal axonal targeting in the developing eye antennal imaginal discs.
Figure 5. Loss of function of Hpo restores axonal projections (impaired by gain of function of FUS) from retina to the brain.

MAb24B10 (Chaoptin), marks the retinal axons that innervate lamina and medulla regions of the brain. Top panel shows, (A) wild-type larval eye-antennal imaginal disc, and (B) GMR>FUS (C) GMR>FUS R518K, and (D) GMR>FUS R521C showing expression of the retinal axons marked by 24B10 in control and when FUS or mutant FUS (FUS R518K and FUS R521C) are misexpressed in the GMR domain respectively. Activation of Hippo signaling by misexpressing hpo, along with FUS or mutant FUS results in impaired targeting of retinal axons to the brain (E) GMR>FUS+hpo, (F) GMR>FUS R518K+hpo, (G) GMR>FUS R521C+hpo, as compared to the wild-type (A). In-activation of Hippo signaling by misexpressing hpoRNAi, along with FUS and mutant FUS results in restoration of targeting of retinal axons to the brain (H) GMR>FUS+hpoRNAi, (I) GMR>FUS R518K+hpoRNAi, (J) GMR>FUS R521C+hpoRNAi. Note that all transgenes are in heterozygous combination. Magnification of all the eye-antennal imaginal discs is 20X.
We further extended our analysis to other members of Hippo signaling pathway to determine their role. Downregulation of Hippo signaling by misexpressing wtsRNAi, yki3SA with FUS or mutant human FUS (GMR>FUS+wtsRNAi, GMR>FUS R518K+wtsRNAi, GMR>FUS R521C+wtsRNAi, GMR>FUS+yki3SA GMR>FUS R518K+yki3SA and GMR>FUS R521C+yki3SA), in the GMR domain of the developing eye also showed rescue of axonal targeting (Fig. S3). In contrast, activation of Hippo signaling by misexpressing wts or ykiRNA with FUS or mutant FUS (GMR>FUS+wts, GMR>FUS R518K+ wts, GMR>FUS R521C+wts, GMR>FUS+ykiRNAi, GMR>FUSR518K+ykiRNAi and GMR>FUS R521C+ykiRNAi), in the GMR domain of the eye showed enhanced loss of the axonal targeting phenotype as compared to the controls (Fig. S3). These results confirm that downregulation of Hippo signaling in the FUS or mutant FUS backgrounds restores axonal targeting from the retinal neuron to the brain.
Misexpression of FUS activates Hippo signaling
A transcriptional target of Yki, death- associated inhibitor of apoptosis (diap1), serves as a functional read out of the Hippo signaling pathway (Ren et al., 2010). Drosophila inhibitor of apoptosis protein 1 (Diap1), is the direct transcriptional target of the Hippo pathway. The diap1-4.3- GFP gene, contains a Hippo response element, which drives the Diap1 expression in response to Hippo pathway activity where GFP is a reporter to study Hpo activity. When Hippo signaling is downregulated, which corresponds to the higher levels of Yki in the nuclei, activation of diap1-4.3-GFP levels (Ren et al., 2010) is triggered. In diap1-4.3- GFP, the GFP reporter expression is used to study Hpo activity.
Thus, when Hippo signaling is activated, diap1-4.3-GFP levels are decreased in the eye disc. We checked for diap1-4.3-GFP expression to further validate if Hippo signaling is activated in GMR>FUS background. The wild-type expression of diap1-4.3- GFP (tagged with a GFP reporter) is mostly restricted to the photoreceptor neurons present towards the posterior end of eye discs, and in the antennal region of eye-antennal imaginal disc (Fig. 6A, A’), as well as serves as the control. The GMR domain extends from the posterior margin of the eye disc to the Morphogenetic furrow (MF) which is marked by a proneural marker, ELAV (Fig. 6A, A’, white arrowheads). Increasing the levels of FUS (GMR>FUS+diap1-4.3-GFP, Fig. 6B, B’), FUS R518K (GMR>FUS R518K+diap1-4.3-GFP, Fig. 6C, C’) and FUS R521C (GMR>FUS R521C+diap1-4.3-GFP; Fig. 6D, D’) in the GMR domain resulted in significant reduction of Diap1 levels (Fig. 6E). These results confirm that an increase in levels of FUS protein does not affect the expression of downstream targets of Hippo signaling.
Figure 6. Targeted misexpression of FUS in the developing eye of Drosophila, transcriptionally activates Hippo signaling.

diap1-4.3-GFP (diap1, tagged with GFP reporter) shows the transcriptional activity of Hippo signaling pathway. Gain-of-function of hpo triggers cell death and result in downregulation of diap1-4.3-GFP levels. Panels shows diap1-4.3-GFP (green) and Elav (red) expression in the eye-antennal imaginal discs from larvae of all the genotypes. (A, A’) Wild-type expression of diap1-4.3-GFP in eye-antennal imaginal discs, which serve as control. The white arrowheads marks the boundary of domain of GMR expression domain on dorsal and ventral margins of the eye disc. Misexpression of FUS or mutant FUS (R518K and FUS R521C) along with diap1-4.3-GFP in the GMR domain of eye (B, B’) GMR>FUS+ diap1-4.3-GFP, (C, C’) GMR>FUS R518K+ diap1-4.3-GFP, (D, D’) GMR>FUS R521C+ diap1-4.3-GFP, exhibits downregulation of diap1-4.3-GFP reporter. (E) Quantitative analysis, comparing the GFP intensity levels in the eye imaginal disc of (A) diap-1-4.3GFP, which serve as control, (B) GMR>FUS, (C) GMR>FUS R518K, and (D) GMR>FUSR521C. The quantification of GFP intensity was done using in built program in Olympus Fluoview laser scanning confocal microscope. The p- values for the estimation of diap1 4.3-GFP intensity levels were calculated from five individual eye-imaginal disc for each genotype using a Student’s t- test in MS excel and a graph was plotted. (A, A’, B’, C’, D’) The diap1 4.3-GFP signal intensity was calculated only in the GMR domain, which extends from the posterior margin of the eye imaginal disc to the MF which is marked by white arrowheads. The data exhibits that GFP levels are reduced in the backgrounds where FUS targeted expression occurs. Genotypes including, GMR>FUS+ diap1-4.3-GFP (***, p<0.001), GMR>FUS R518K+ diap1-4.3-GFP (**, p<0.01) and GMR>FUS R521C+ diap1-4.3-GFP (***, p<0.001) were found to be statistically significant from diap1-4.3-GFP (control). GMR>FUS+ diap1-4.3-GFP was found to be statistically significant from GMR>FUS R518K+ diap1-4.3-GFP (***, p<0.001) while non-significant from GMR>FUS R521C+ diap1-4.3-GFP (N.S., p>0.05). GMR>FUS R518K+ diap1-4.3-GFP was found to be statistically significant as compared to GMR>FUS R521C+ diap1-4.3-GFP (**, P<0.01). Note that all transgenes are in heterozygous combination. The magnification of all the eye-antennal imaginal discs is 20X. (F, F’, F”) Semi-quantitative western blot (the tubulin bands in western blot, serve as controls) and (F’, F”) quantitative analysis of western blot represented in the form of a graph (provides the levels of (F’) p- LATS1 and (F”) LATS1 levels in untransfected HEK293T cells (atcc® crl-3216™) (negative control), HEK293T cells (atcc® crl-3216™) cells transfected with WT FUS, mutant FUS (FUS R518K and FUS R521C). The intensity of the (F’) p-LATS1, and (F”) LATS1 bands was quantified, normalized by image studio ( Li-COR Biosciences), and the graph was made using Graphpad Prism. The p-values for the estimation of p-JNK levels in all combinations in Western Blot were calculated in a set of three (n=3), using one-way ANOVA with Tukey’s multiple comparisons test. The p-values for untransfected versus FUS R518K = 0.0015 and p-value for untransfected versus R521C = 0.0011. Errors bar indicate s.e.m.
To determine if Hippo signaling is activated in mammalian cells, we checked the levels of LATS1 and phosphorylated LATS1, one of the components of Hippo signaling cascade, in HEK293T cells ectopically expressing either FUS-WT or ALS-associated mutants, FUS-R518K and FUS-R521C. We observed that while the total levels of LATS1 protein were unchanged between the groups (Fig. 6F,F”), the levels of phosphorylated LATS1 were increased in the mutant FUS-expressing cells when compared to the untransfected control (Fig. 6F,F’), suggesting a hyper-activation of upstream kinase cascade. Ectopic expression of FUS-WT alone did not change the phosphorylated LATS1 levels (Fig 6F, F’), indicating that activation of Hippo signaling was specific to mutant FUS.
Modulating levels of JNK signaling pathway modulates FUS mediated neurodegeneration
It is known that Hippo regulates JNK signaling (Ma et al., 2015; Ma et al., 2017), which is involved in neurodegeneration and cell death (Flerdegen et al., 1997; Sarkar et al., 2016; Tare et al., 2011). We tested if modulating JNK signaling levels can block FUS-mediated neurodegeneration. JNK signaling comprises of a series of kinases, which in turn regulates expression of puckered (puc) (Fig. 7A). puc, a dual phosphatase, regulates JNK signaling through a negative feedback loop (Martin-Blanco et al., 1998). The phospho-Jun kinase enzyme can phosphorylate its downstream substrate Jun in its N- terminal domain. The phosphorylated Jun is used to quantify levels of JNK activation using an antibody against p-Jun (Tare et al., 2011). Initially we tested JNK activation levels by quantifying the JNK levels with quantitative-PCR (qPCR) (Fig. 7B) and then quantifying levels of the p-JNK protein in semi-quantitative western blot (Fig. 7C, C’). In GMR>FUS background, JNK gene expression levels increased two fold, whereas in other constructs like GMR>FUS R518K or GMR>FUS R521C, the JNK expression levels were 10 and 4 folds respectively (Fig. 7B). We also employed a semi-quantitative approach to compare the amount of phospho-Jun kinase (p-JNK) levels in the wild type to that of FUS (GMR>FUS) and mutant FUS (GMR>FUS R518K and GMR>FUS R521C). In comparison to the wild type, p-JNK levels were upregulated in GMR> FUS, GMR> FUS R518K and GMR>FUS R521C backgrounds. Tubulin bands served as a control in the western blot. We calculated the intensity of the bands and presented them as a graph (Figure 7C, C’).
Figure 7. Modulating the levels of JNK signaling affects FUS mediated neurodegeneration.

Panel shows, (A) Schematic representation of JNK signaling pathway, (B) levels of phospho-JNK (p-JNK) in quantitative-PCR (q-PCR), (C) semi-quantitative western blot (the tubulin bands in western blot, serve as controls) and (C’) quantitative analysis of western blot represented in the form of a graph (provides the levels of JNK in WT FUS and mutant FUS (FUS R518K and FUS R521C) background. (C’) The p-JNK bands intensity was quantified, normalized and the graph was made using Image J software (NIH). The p-values for the estimation of p-JNK levels in all combinations in Western Blot were calculated in a set of three (n=3), using Student’s t-test in MS Excel software. GMR>FUS was found to be statistically significant from 1. Wild-type (p<0.05, *), 2. GMR>FUS R518K (p<0.0001, ****), and 3. GMR>FUS-R521C (P<0.001, ***). Genotypes including GMR>FUS, GMR>FUS-R518K were found to be statistically different (p<0.05, *) and (p<0.001, ***) respectively, while GMR>FUS-R512C was found to be non-significant (p>0.05, N.S.) as compared to Wild-type (Canton-S, Control). (B, C, C’) When compared to wild-type controls, higher levels of p-JNK were observed when FUS or mutant FUS are misexpressed in GMR domain of the eye (GMR>FUS, GMR>FUS R518K and GMR>FUS R521C) in q-PCR and western blot approaches. (D) Wild-type eye-antennal imaginal disc and (D’) adult fly eye, serve as the control. Note the expression of Wg (green) and Elav (red) in the eye-antennal imaginal discs from third instar larvae of all the genotypes. Activation of JNK signaling by misexpressing activated hemipterous (hep; GMR>hepAct), and activated Djun (GMR>junaspv7) alone in GMR domain of the eye, results in few number of dying cells in (I,E) eye imaginal discs and (I’,E’) adult fly eye, respectively. Furthermore, activation of JNK signaling by misexpressing hep and jun in FUS or mutant FUS background using GMR-Gal4 driver, (J,J’) GMR>FUS+hepAct, (K,K’) GMR>FUS R518K+hepAct and (L,L’) GMR>FUS R521C+hepAct, (F,F’) GMR>FUS+junaspv7, (G,G’) GMR>FUS R518K+junaspv7 and (H,H’) GMR>FUS R521C+junaspv7, results in (J,K,L,F,G,H) dramatic increase in dying cell population in the eye-antennal imaginal discs, leading to (J’,K’,L’,F’,G’,H’) no-eye or small eye with loss of pigmentation phenotype in the adult fly eyes. However, downregulation of JNK signaling by misexpression of dominant negative bsk, and puc (a dual phosphatase) alone in GMR domain (GMR>bskDN and GMR>puc), results in near wild-type (M,Q) eye-antennal imaginal discs and (M’,Q’) adult fly eyes respectively. However, downregulating JNK signaling by misexpressing bsk, and puc in FUS or mutant FUS background (N,N’) GMR>FUS+bskDN, (O,O’) GMR>FUS R518K+bskDN, (P,P’) GMR>FUS R521C+bskDN and (R,R’) GMR>FUS+puc, (S,S’) GMR>FUS R518K+puc and (T,T) GMR>FUS R521C+puc, results in significant reduction in the dying cell population in (N,O,P,R,S,T) eye imaginal discs, thereby leading to a strong rescue of the (N’,O’,P’,R’,S’,T’) adult fly eye phenotype. The magnification of all eye-antennal imaginal disc is 20X.
To further test if FUS mediated neurodegeneration can be suppressed by modulating the levels of JNK signaling, we initially activated JNK signaling by misexpressing Djunaspv7 and constitutively active hemipterous (hepAct) in FUS and mutant FUS (FUS R518K and FUS R521C) backgrounds. In comparison to the wild-type eye imaginal disc and the adult eye (Fig. 7D, D’), we analyzed their resultant phenotypes in both eye-antennal imaginal discs (Fig. 7 F, G, H, J, K, L) and the adult fly eyes (Fig. 7 F’, G’, H’ J’,K’,L’). Targeted misexpression of D-jun and hep alone in the GMR domain (GMR>jun and GMR>hep), which served as control, led to a strong neurodegenerative phenotype in eye-antennal imaginal discs (Fig. 7E, I), adult fly eyes (Fig. 7 E’, I’). Moreover, activation of the JNK signaling by misexpressing hepAct along with FUS or mutant FUS in the GMR domain worsens the neurodegeneration phenotype (Fig. 7J-L’) as seen in eye imaginal discs (Fig. 7D) and adult fly eyes (Fig. 7D’). Similarly, misexpression of Djunaspv7 with FUS or mutant FUS in the GMR domain (GMR>FUS+Djunaspv7, GMR>FUS R518K+Djunaspv7 and GMR>FUS R521C+Djunaspv7), resulted in a small, rough eye phenotype with loss of pigment cells (Fig. 7F-H’). We further validated our results by downregulating the JNK signaling in FUS gain-of-function backgrounds. Misexpression of bskDN (GMR>bskDN) or puc (GMR>puc) alone, which serve as controls, exhibits near wild-type eye imaginal discs (Fig. 7M,Q) and adult fly eyes (Fig. 7 M’Q’). Downregulating JNK by misexpressing bskDN with FUS in GMR domain (GMR>FUS+bskDN) or FUS R518K (GMR>FUS R518K+bskDN), GMR>FUS R521C (GMR>FUS R521C+bskDN), exhibits a strong rescue of the FUS mediated neurodegenerative phenotype (Fig. 7N-P’). Similarly, when puc was misexpressed with FUS or mutant FUS (GMR>FUS+puc) or FUS R518K (GMR>FUS R518K+puc), GMR>FUS R521C (GMR>FUS R521C+puc) as seen in the eye imaginal discs (Fig. 7 R, S, T) and adult fly eyes (Fig. 7 R’, S’, T). Thus, activation of JNK signaling is responsible in FUS mediated neurodegeneration.
Discussion
ALS, a progressive neurodegenerative disorder, is caused by mislocalization of FUS in cytoplasm which impairs the RNA metabolism (Lanson et al., 2011). However, the molecular genetic basis of this disease is elusive. It is possible that mislocalization of FUS proteins trigger(s) aberrant signaling, which results in the onset of the progressive neurodegenerative phenotype. Therefore, there is a need to identify the downstream targets of signaling pathways activated by the FUS mislocalization. Generation of animal model systems like Drosophila, which are genetically tractable, have facilitated the use of genetic screens to identify the modifiers of neurodegenerative phenotypes (Casci and Pandey, 2015; Pandey and Nichols, 2011; Sarkar et al., 2016).
Targeted misexpression of FUS and its mutants in the differentiating retinal neurons using transgenic approaches exhibited strong neurodegenerative phenotypes in the Drosophila adult eye (Daigle et al., 2013; Lanson et al., 2011). In a forward genetic screen using the molecularly defined deficiencies, we identified a deficiency line Df(2R)BSC782, which can rescue FUS mediated neurodegeneration. This deficiency uncovers hpo and other genes (Fig.1). In a candidate gene approach, where we tested individual genes uncovered by the deficiency Df(2R)BSC782, we identified hpo as one of the genetic modifiers of ALS phenotypes (Fig. 2). Interestingly, it has been shown that FUS/ LATS1/2 inhibits hepatocellular carcinoma (HCC) via activating Hippo pathway (Bao et al., 2018). Gain-of-function of hpo results in cell death whereas hpo loss-of-function results in over proliferation and overgrowth (Kango-Singh and Singh, 2009; Udan et al., 2003). We found that modulating the levels of hpo can modify FUS mediated neurodegeneration phenotype. Loss-of-function of hpo can significantly rescue the neurodegenerative phenotype of FUS (GMR>FUS) and other mutant (GMR>FUS R518K and GMR>FUS R521C) accumulation (Fig. 2). We further confirmed that the Hippo pathway is activated in FUS overexpressing retinal neurons (GMR>FUS) to trigger neurodegeneration in the Drosophila eye (Fig. 3).
Role of Hippo in neurodegeneration
Studies from ours and other labs have shown that other components of Hippo pathway like Fat (Ft), Teashirt (Tsh), and Crumbs (Crb) induce neurodegeneration in models of Alzheimer’s Disease (Moran et al., 2013; Napoletano et al., 2011; Sarkar et al., 2016; Steffensmeier et al., 2013). In addition, the Hippo pathway may also be involved in neural development (Jiang et al., 2009; Wittkorn et al., 2015). The major function of the Hippo signaling pathway has been in growth regulation and cancer (Kango-Singh and Singh, 2009; Ma et al., 2019; Snigdha et al., 2019). Recently, Hippo signaling has been implicated in many disease models where it plays role in apoptosis, autophagy, regeneration and cell survival (Calamita and Fanto, 2011; Ma et al., 2019). Thus, it is interesting to find a role for Hippo pathway in FUS mediated neurodegeneration.
Our genetic analysis suggested that Hippo signaling acts downstream of FUS accumulation (Fig. S2). In genetic backgrounds where FUS and its mutant proteins were misexpressed in the eye, we found a robust accumulation of FUS protein in the eye imaginal disc. In GMR>FUS, Df(2R)BSC782/+ background, FUS accumulation was unaltered suggesting that the rescue of the neurodegeneration phenotype occurred by a mechanism downstream of FUS accumulation (Fig.S1). Our data shows that FUS accumulation triggers downstream signaling events involving Hippo and JNK signaling. We tested the levels of diap1-4.3-GFP, which serves as a reporter for Hippo signaling, in GMR>FUS and GMR>FUS R518K and GMR>FUS R521C background. When Hippo signaling is activated it triggers cell death and diap1-4.3-GFP levels are downregulated (Zhang et al., 2008). We found that diap1-4.3-GFP levels were downregulated in these backgrounds, which suggests that Hippo signaling is activated (Fig. 6). Earlier studies have shown that Hippo can regulate JNK signaling (Ma et al., 2015; Ma et al., 2017). We also tested the reporters of JNK signaling and found that JNK signaling is activated in gain-of-function of FUS (Fig. 7).
Furthermore, if we block these signaling pathways, even though the neurodegenerative phenotype caused by accumulation of FUS is rescued, it does not affect the accumulation of FUS protein suggesting that JNK signaling doesn’t block expression or turnover of toxic FUS protein. Interestingly, JNK signaling is dependent on timing (temporal scale). We have assayed the effect of JNK signaling in the matrure third instar eye-imaginal disc. Based on our results, we propose a model that accumulation of FUS triggers Hippo and JNK signaling, which acts downstream of FUS accumulation. Activation of Hippo signaling and JNK signaling triggers neurodegeneration (Fig. 8).
Figure 8. Model representing the mechanism by which FUS leads to neurodegeneration and cell death.
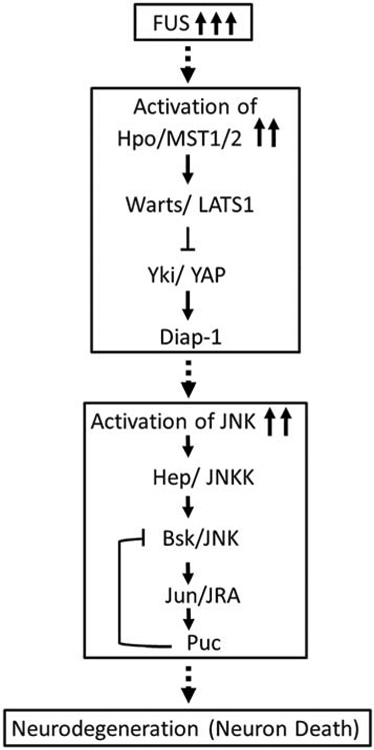
Increase in the levels of human FUS or mutant FUS, FUS R518K and FUS R521C activates Hippo signaling, which in turn leads to the activation of JNK signaling. This aberrant Hippo and JNK signaling due to accumulation of FUS triggers cell death in the retinal neurons of eye. Thus, downregulation of Hippo and JNK signaling in FUS background, rescues FUS-mediated neurodegeneration.
Thus, we demonstrate that inactivation of Hippo and JNK signaling can rescue FUS-mediated neurodegeneration in Drosophila eye. Our data suggests that this neuropathy can be alleviated by modulating levels of Hippo signaling and JNK signaling pathways (Fig. 2, 3, 7). Both pathways are crucial for normal development and in disease backgrounds for onset and progression. The members of these two pathways seem to be interesting therapeutic targets, which can mitigate the onset or rapid progression of neurodegenerative diseases like ALS.
MST, the mammalian homologue of hpo, a kinase, is involved in many biological processes from cell proliferation to cell death, patterning and growth (Kango-Singh and Singh, 2009; Yu and Guan, 2013). There are two hpo homologues MST1 and MST2 in the mammalian genome (Creasy and Chernoff, 1995). In addition, hpo is involved in other neurodegenerative models like the superoxide dismutase SOD1 G93A mouse model of ALS (Lee et al., 2013), and in VAPB models in Drosophila (Sanhueza et al., 2015). The SOD1 G93A mouse model of ALS is linked to decrease in motor neuron survival. It has been shown that downregulation of MST1, fly homolog of hpo, exhibits neuroprotective function in this mouse ALS model (Lee et al., 2013). Hippo signaling dysregulation has been identified in Huntington’s disease brain and neuronal stem cells (Mueller et al., 2018). Thus, MST1/ Hippo can be an interesting link between cancer and neurodegenerative disorder like ALS (Yamamoto et al., 2019). The members of Hippo pathway can serve as potential therapeutic target for ALS as well as other neurodegenerative disorder.
Supplementary Material
Supplementary Figure 1: Quantitative analyses of severity percentage of eye degeneration phenotype. We examined the eye phenotypes of 200 flies per genotype and the phenotypes are represented in the images in Fig1, 2, 3. For quantitative analysis, 200 flies per genotype were randomly selected for scoring according to the criteria where “No-eye” was assigned to category 6, 80% eye degeneration was assigned to category 5, 60-80% eye degeneration was assigned to category 4, 40-60% eye degeneration was assigned to category 3, 20-40% eye degeneration was assigned to category 2, 0-20% eye degeneration was assigned to category 1 and Wild type was assigned to category 0. Comparisons were made using Non-Parametric: Mann Whitney T Test. Data show mean phenotype score ± S.D. ** =p<0.001. (A) Comparison of eye degeneration phenotypes between GMR-Gal4/+ with GMR>FUS, GMR> FUS R518K GMR> FUS R521C. (B) Comparison of eye degeneration phenotypes between GMR>FUS, GMR>FUS+hpo, GMR>FUS+hpo RNAi, GMR> FUS R518K+hpo, GMR>FUS R518K+hpo RNAi, GMR> FUS R521C+hpo, and GMR> FUS R521C+hpo RNAi. (C) Comparison of eye degeneration phenotypes between GMR>FUS, GMR>FUS+wts, GMR>FUS+wts RNAi, GMR> FUS R518K+wts, GMR> FUS R518K+wts RNAi, GMR> FUS R521C+wts, and GMR> FUS R521C+wts RNAi, GMR>FUS+yki3SA, GMR>FUS+ykiRNAi, GMR> FUS R518K+yki3SA, GMR> FUS R518K+ykiRNAi, GMR> FUS R521C+ yki3SA, and GMR> FUS R521C+ ykiRNAi.
Supplementary Figure 2. Accumulation of FUS triggers cell death. FUS protein accumulation and localization was checked in all the genotypes using FUS antibody. The expression of FUS protein was not detected in wild-type eye-antennal imaginal discs. Misexpression of FUS (A, A’) GMR>FUS or mutant FUS (C, C’) GMR>FUS R518K, and (E, E’) GMR>FUS R521C eye, exhibit robust FUS expression in differentiating photoreceptor neurons in the GMR domain of eye imaginal discs. Misexpression of FUS or mutant FUS in heterozygous background of deficiency Df(2R)BSC782/+, which also uncovers hpo gene, in the GMR domain of eye (B,B’) GMR>FUS+Df(2R)BSC782/+, (D,D’) GMR>FUS R518K+Df(2R)BSC782/+ and (F,F’) GMR>FUS R521C+Df(2R)BSC782/+, showed similar expression level of FUS in the eye imaginal discs. (G) Quantitative analysis of images to compare the FUS intensity levels in FUS or FUS mutants (GMR>FUS, GMR>FUS R518K, and GMR>FUS R521C) and when FUS or FUS mutants are misexpressed together with deficiency Df(2R)BSC782/+ in the GMR domain (GMR>FUS+ Df(2R)BSC782/+, GMR>FUS R518K+ Df(2R)BSC782/+ and GMR>FUS R521C+ Df(2R)BSC782/+). The data is represented in the form of a graph. The quantification of levels of FUS expression was done using built-in program in LSM 3000, Laser Scanning Confocal Microscopy and statistical analysis was conducted using Microsoft Excel. The p-values were calculated using Student’s t-Test (n=3) in MS Excel, where the levels of expression of FUS was found to be non-significant in GMR>FUS + Df(2R)BSC782/+ (p>0.05, N.S.), GMR>FUS R518K + Df(2R)BSC782/+ (p>0.05, N.S.) and GMR>FUS R521C + Df(2R)BSC782/+ (p>0.05, N.S.), as compared to GMR>FUS, GMR>FUS R518K, and GMR>FUS R521C respectively. (H) Semi-quantitative western blot was used to detect FUS protein as a 70kDa band in GMR>FUS and mutant FUS (GMR>FUS R518K and GMR>FUS R521C) background. Tubulin with a band size of 55kDa serve as a loading control. Note that 70kDa FUS band was absent in wild-type but was found in all the three FUS transgenic fly lines used in the study. (H’) The FUS bands intensity was quantified in WT FUS and mutant FUS (FUS R518K and FUS R521C backgrounds. It was normalized and the graph was made using Image J software (NIH). The p-values for the estimation of FUS levels in all combinations in Western Blot were calculated in a set of three (n=3), using Student’s t-Test in MS Excel software. Wild-type was found to be statistically significant from GMR>FUS (p<0.0001, ****), GMR>FUS R518K (p0.0001, ****), and GMR>FUS-R521C (p0.0001, ****). Genotypes including GMR>FUS, GMR>FUS-R518K were found to be statistically non-significant (p>0.05, NS) and GMR>FUS, GMR>FUS-R521C were also non-significant (p>0.05, NS) respectively. The genotypes GMR>FUS-R518K and GMR>FUS-R521C was found to be statistically significant (p<0.01, *). Scanning Electron Micrographs (SEM) showing the higher magnification images of (I, I’) Wild-type, (J, J’) GMR>FUS, (K, K’) GMR>FUS R518K and (L, L’) GMR>FUS R521C adult eyes at 180X and 800X respectively.
Supplementary Figure 3. Downregulation of Hippo signaling restores axonal projections (impaired by gain of function of FUS) from retina to the brain. Top panel shows, (A) Wild-type larval eye-antennal imaginal disc, and (B) GMR>FUS (G) GMR>FUS R518K, and (L) GMR>FUS R521C showing expression of the retinal axons marked by 24B10 in control and when WT FUS or mutant FUS (FUS R518K and FUS R521C) are misexpressed in the GMR domain respectively. Activation of Hippo signaling by misexpressing vote, wts, ykiRNAi along with FUS or mutant FUS results in impaired targeting of retinal axons to the brain (C) GMR>FUS+ wts, (H) GMR>FUS R518K+wts, (M) GMR>FUS R521C+wts, (D) GMR>FUS+ ykiRNAi, (I) GMR>FUS R518K+ykiRNAi and (N) GMR>FUS R521C+ykiRNAi as compared to the (A) wild-type. Downregulation of the Hippo signaling by misexpressing wtsRNAi, yki3SA along with WT FUS or mutant FUS results in restoration of targeting of retinal axons to the brain (E) GMR>FUS+wtsRNAi, (J) GMR>FUS R518K+wtsRNAi, (O) GMR>FUS R521C+wtsRNAi, (F) GMR>FUS+yki3SA, (K) GMR>FUS R518K+yki3SA and (P) GMR>FUS R521C+yki3SA. Magnification of all the eye-antennal imaginal discs is 40X.
Acknowledgements
We thank Bloomington Drosophila Stock Center (BDSC) for the Drosophila strains, and Developmental Studies Hybridoma Bank (DSHB) for the antibodies. Confocal Microscopy and Scanning Electron Microscopy were supported by core facility at University of Dayton. N.G. and A.S.M. are supported by the University of Dayton graduate program. MKS is supported by start-up research funds from the University of Dayton, and a subaward from NIH grant R01CA183991 (PI Nakano). N.R. and UBP are supported by _________________ This work is supported by NIH1R15GM124654-01 from NIH, Schuellein Chair Endowment Fund and STEM Catalyst Grant from the University of Dayton to A.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: None
Competing Interests: The authors declare no competing interests.
References –
- Adachi-Yamada T, et al. , 1999. Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature. 400, 166. [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T, O'Connor MB, 2002. Morphogenetic apoptosis: a mechanism for correcting discontinuities in morphogen gradients. Dev Biol. 251, 74–90. [DOI] [PubMed] [Google Scholar]
- Bao L, et al. , 2018. A FUS-LATS1/2 Axis Inhibits Hepatocellular Carcinoma Progression via Activating Hippo Pathway. Cell Physiol Biochem. 50, 437–451. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N, 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118, 401–15. [DOI] [PubMed] [Google Scholar]
- Brenner D, Weishaupt JH, 2019. Update on amyotrophic lateral sclerosis genetics. Curr Opin Neurol. [DOI] [PubMed] [Google Scholar]
- Calamita P, Fanto M, 2011. Slimming down fat makes neuropathic hippo: the Fat/Hippo tumor suppressor pathway protects adult neurons through regulation of autophagy. Autophagy. 7, 907–9. [DOI] [PubMed] [Google Scholar]
- Casci I, et al. , 2019. Muscleblind acts as a modifier of FUS toxicity by modulating stress granule dynamics and SMN localization. Nat Commun. 10, 5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casci I, Pandey UB, 2015. A fruitful endeavor: modeling ALS in the fruit fly. Brain Res. 1607, 47–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, et al. , 2011. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein & cell. 2, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne AN, et al. , 2017. Failure to Deliver and Translate-New Insights into RNA Dysregulation in ALS. Front Cell Neurosci. 11, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasy CL, Chernoff J, 1995. Cloning and characterization of a member of the MST subfamily of Ste20-like kinases. Gene. 167, 303–6. [DOI] [PubMed] [Google Scholar]
- Cutler T, et al. , 2015. Drosophila Eye Model to Study Neuroprotective Role of CREB Binding Protein (CBP) in Alzheimer's Disease. PLoS One. 10, e0137691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle JG, et al. , 2013. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Human Molecular Genetics. 22, 1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, et al. , 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H-X, et al. , 2011. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 477, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Reddy EP, 2008. JNK signaling in apoptosis. Oncogene. 27, 6245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Reddy EP, 2017. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer. 8, 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi H, et al. , 2008. RNA-binding protein TLS is a major nuclear aggregate-interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. J Biol Chem. 283, 6489–500. [DOI] [PubMed] [Google Scholar]
- Dong J, et al. , 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 130, 1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, et al. , 2013. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 80, 415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández BG, et al. , 2011. Actin-Capping Protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development. 138, 2337. [DOI] [PubMed] [Google Scholar]
- Fulford A, et al. , 2018. Upstairs, downstairs: spatial regulation of Hippo signalling. Curr Opin Cell Biol. 51, 22–32. [DOI] [PubMed] [Google Scholar]
- Glise B, et al. , 1995. hemipterous encodes a novel drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell. 83, 451–461. [DOI] [PubMed] [Google Scholar]
- Gogia N, et al. , 2017. An undergraduate cell biology lab: Western Blotting to detect proteins from Drosophila eye. Dros. Inf. Serv. 100, 218–225. [Google Scholar]
- Harvey KF, et al. , 2003. The Drosophila Mst Ortholog, hippo, Restricts Growth and Cell Proliferation and Promotes Apoptosis. Cell. 114, 457–467. [DOI] [PubMed] [Google Scholar]
- Herdegen T, et al. , 1997. The c-Jun transcription factor – bipotential mediator of neuronal death, survival and regeneration. Trends in Neurosciences. 20, 227–231. [DOI] [PubMed] [Google Scholar]
- Huang J, et al. , 2005. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 122, 421–434. [DOI] [PubMed] [Google Scholar]
- Jiang Q, et al. , 2009. yap is required for the development of brain, eyes, and neural crest in zebrafish. Biochem Biophys Res Commun. 384, 114–9. [DOI] [PubMed] [Google Scholar]
- Justice RW, et al. , 1995. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–46. [DOI] [PubMed] [Google Scholar]
- Kabashi E, et al. , 2008. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nature Genetics. 40, 572–574. [DOI] [PubMed] [Google Scholar]
- Kango-Singh M,et al. , 2002. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development. 129, 5719–30. [DOI] [PubMed] [Google Scholar]
- Kango-Singh M, Singh A, 2009. Regulation of organ size: insights from the Drosophila Hippo signaling pathway. Dev Dyn. 238, 1627–37. [DOI] [PubMed] [Google Scholar]
- Kino Y, et al. , 2016. FUS/TLS acts as an aggregation-dependent modifier of polyglutamine disease model mice. Sci Rep. 6, 35236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar JP, 2018. The fly eye: Through the looking glass. Dev Dyn. 247, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, et al. , 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 323, 1205. [DOI] [PubMed] [Google Scholar]
- Kwon HJ, et al. , 2015. Drosophila C-terminal Src kinase regulates growth via the Hippo signaling pathway. Developmental Biology. 397, 67–76. [DOI] [PubMed] [Google Scholar]
- Lai ZC, et al. , 2005. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 120, 675–85. [DOI] [PubMed] [Google Scholar]
- Lanson NA Jr., et al. , 2011. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet. 20, 2510–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, et al. , 2013. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc Natl Acad Sci U S A. 110, 12066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S-C, et al. , 2013. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron. 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, et al. , 2019. The Hippo Pathway: Biology and Pathophysiology. Annu Rev Biochem. 88, 577–604. [DOI] [PubMed] [Google Scholar]
- Ma X, et al. , 2015. Impaired Hippo signaling promotes Rho1–JNK-dependent growth. Proceedings of the National Academy of Sciences. 112, 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, et al. , 2017. Hippo signaling promotes JNK-dependent cell migration. Proceedings of the National Academy of Sciences of the United States of America. 114, 1934–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco E, et al. , 1998. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Blanco E, et al. , 1998. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes & Development. 12, 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, et al. , 2010. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 465, 223. [DOI] [PubMed] [Google Scholar]
- McCall K, Peterson JS, 2004. Detection of apoptosis in Drosophila. Methods Mol Biol. 282, 191–205. [DOI] [PubMed] [Google Scholar]
- McGurk L, et al. , 2015. Drosophila as an In Vivo Model for Human Neurodegenerative Disease. Genetics. 201, 377–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Singh A, 2017. Real time quantitative PCR to demonstrate gene expression in an undergraduate lab. Dros. Inf. Serv . 100, 5. [Google Scholar]
- Mehta AS, et al. , 2019. Comparative transcriptomic analysis and structure prediction of novel Newt proteins. PLoS One. 14, e0220416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinertzhagen IA, Hanso TE, The development of the optic lobe. In: Bate M, Matrtinez-Arias A, (Eds.), The development of Drosophila melanogaster. Cold Spring Harbor Laboratory Press, 1993, pp. 1363–1491. [Google Scholar]
- Moran MT, et al. , 2013. Homeotic Gene teashirt (tsh) has a neuroprotective function in amyloid-beta 42 mediated neurodegeneration. PLoS One. 8, e80829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses K, Rubin GM, 1991. Glass encodes a site-specific DNA-binding protein that is regulated in response to positional signals in the developing Drosophila eye. Genes Dev. 5, 583–93. [DOI] [PubMed] [Google Scholar]
- Mueller KA, et al. , 2018. Hippo Signaling Pathway Dysregulation in Human Huntington's Disease Brain and Neuronal Stem Cells. Sci Rep. 8, 11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoletano F, et al. , 2011. Polyglutamine Atrophin provokes neurodegeneration in Drosophila by repressing fat. EMBO J. 30, 945–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, et al. , 2000. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 127, 851–60. [DOI] [PubMed] [Google Scholar]
- Nussbacher JK, et al. , 2019. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron. 102, 294–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Irvine KD, 2008. In vivo regulation of Yorkie phosphorylation and localization. Development. 135, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesnicky EC, Wright EG, 2018. Drosophila as a Model for Assessing the Function of RNA-Binding Proteins during Neurogenesis and Neurological Disease. J Dev Biol. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega JA, et al. , 2020. Nucleocytoplasmic Proteomic Analysis Uncovers eRF1 and Nonsense-Mediated Decay as Modifiers of ALS/FTD C9orf72 Toxicity. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nichols CD, 2011. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 63, 411–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantalacci S, et al. , 2003. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nature Cell Biology. 5, 921. [DOI] [PubMed] [Google Scholar]
- Picher-Martel V, et al. , 2016. From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathologica Communications. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb C, et al. , 2011. Zyxin Links Fat Signaling to the Hippo Pathway. PLOS Biology. 9, e1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ready DF, et al. , 1976. Development of the Drosophila retina, a neurocrystalline lattice. Dev Biol. 53, 217–40. [DOI] [PubMed] [Google Scholar]
- Ren F, et al. , 2010. Hippo signaling regulates Yorkie nuclear localization and activity through 14-3-3 dependent and independent mechanisms. Dev Biol. 337, 303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, et al. , 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs JE, 1985. Trauma and Amyotrophic Lateral Sclerosis. Archives of Neurology. 42, 205–205. [DOI] [PubMed] [Google Scholar]
- Rosen DR, et al. , 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 362, 59. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, et al. , 2008. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS genetics. 4, e1000193–e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, et al. , 2015. Network analyses reveal novel aspects of ALS pathogenesis. PLoS Genet. 11, e1005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, et al. , 2018a. Characterization of a morphogenetic furrow specific Gal4 driver in the developing Drosophila eye. PLOS ONE. 13, e0196365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, et al. , 2018b. A soy protein Lunasin can ameliorate amyloid-beta 42 mediated neurodegeneration in Drosophila eye. Scientific Reports. 8, 13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, et al. , 2016. Alzheimer's disease: the silver tsunami of the 21(st) century. Neural Regen Res. 11, 693–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Choi KW, 2003. Initial state of the Drosophila eye before dorsoventral specification is equivalent to ventral. Development. 130, 6351–60. [DOI] [PubMed] [Google Scholar]
- Singh A, et al. , 2019. Proximal fate marker homothorax marks the lateral extension of stalk-eyed fly Cyrtodopsis whitei. Genesis. e23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Irvine KD, 2012. Drosophila as a model for understanding development and disease. Dev Dyn. 241, 1–2. [DOI] [PubMed] [Google Scholar]
- Singh A, et al. , 2002. Eye suppression, a novel function of teashirt, requires Wingless signaling. Development. 129, 4271–80. [DOI] [PubMed] [Google Scholar]
- Singh A, et al. , Dorso-ventral boundary is required for organizing growth and planar polarity in the Drosophila eye. In: Mlodzik M, (Ed.), “Planar Cell Polarization during Development: Advances in Developmental Biology and Biochemistry”. Elsevier Science & Technology Books., 2005, pp. 59–91. [Google Scholar]
- Singh A, et al. , 2006. Lobe and Serrate are required for cell survival during early eye development in Drosophila. Development. 133, 4771–81. [DOI] [PubMed] [Google Scholar]
- Singh A, et al. , 2012. A glimpse into dorso-ventral patterning of the Drosophila eye. Dev Dyn. 241, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluss H, et al. , 1996. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. [DOI] [PubMed]
- Snigdha K, et al. , 2019. Hippo Signaling in Cancer: Lessons From Drosophila Models. Front Cell Dev Biol. 7, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensmeier AM, et al. , 2013. Novel neuroprotective function of apical-basal polarity gene crumbs in amyloid beta 42 (abeta42) mediated neurodegeneration. PLoS One. 8, e78717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, et al. , 2002. Salvador Promotes Both Cell Cycle Exit and Apoptosis in Drosophila and Is Mutated in Human Cancer Cell Lines. Cell. 110, 467–478. [DOI] [PubMed] [Google Scholar]
- Tare M, et al. , 2011. Activation of JNK signaling mediates amyloid-ss-dependent cell death. PLoS One. 6, e24361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, et al. , Molecular Genetic Mechanisms of Axial Patterning: Mechanistic Insights into Generation of Axes in the Developing Eye. In: Singh A, et al., (Eds.), Molecular Genetics of Axial Patterning, Growth and Disease in the Drosophila Eye. Springer, Springer NewYork Heidelberg Dordrecht London, 2013, pp. 37–75. [Google Scholar]
- Tare M, et al. , 2016. Cullin-4 regulates Wingless and JNK signaling-mediated cell death in the Drosophila eye. Cell Death Dis. 7, e2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Singh A, 2009. Drosophila adult eye model to teach Scanning Electron Microscopy in an undergraduate cell biology laboratory. Dros Infor Serv 91, 174–180. [Google Scholar]
- Tournier C, et al. , 1997. MAP kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. [DOI] [PMC free article] [PubMed]
- Treier M, et al. , 1995. JUN cooperates with the ETS domain protein pointed to induce photoreceptor R7 fate in the Drosophila eye. Cell. 83, 753–760. [DOI] [PubMed] [Google Scholar]
- Udan RS, et al. , 2003. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nature Cell Biology. 5, 914. [DOI] [PubMed] [Google Scholar]
- Vance C, et al. , 2009. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science. 323, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, et al. , 2007. Mob as tumor suppressor is activated by Hippo kinase for growth inhibition in Drosophila. EMBO J. 26, 1772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, et al. , 1994. Genetic control of programmed cell death in Drosophila. Science. 264, 677–83. [DOI] [PubMed] [Google Scholar]
- White MA, Sreedharan J, 2016. Amyotrophic lateral sclerosis: recent genetic highlights. Curr Opin Neurol. 29, 557–64. [DOI] [PubMed] [Google Scholar]
- Wittkorn E, et al. , 2015. The Hippo pathway effector Yki downregulates Wg signaling to promote retinal differentiation in the Drosophila eye. Development. 142, 2002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, et al. , 2003. hippo Encodes a Ste-20 Family Protein Kinase that Restricts Cell Proliferation and Promotes Apoptosis in Conjunction with salvador and warts. Cell. 114, 445–456. [DOI] [PubMed] [Google Scholar]
- Xia R, et al. , 2012. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol Neurodegener. 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, et al. , 1995. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development. 121, 1053–63. [DOI] [PubMed] [Google Scholar]
- Yamamoto I, et al. , 2019. Cancer-related genes and ALS. Front Biosci (Landmark Ed). 24, 1241–1258. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. , 1998. Oncoprotein TLS Interacts with Serine-Arginine Proteins Involved in RNA Splicing. Journal of Biological Chemistry. 273, 27761–27764. [DOI] [PubMed] [Google Scholar]
- Yu FX, Guan KL, 2013. The Hippo pathway: regulators and regulations. Genes Dev. 27, 355–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, et al. , 2008. The TEAD/TEF Family of Transcription Factor Scalloped Mediates Hippo Signaling in Organ Size Control. Developmental Cell. 14, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, et al. , 1997. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. Journal of Cell Science. 110, 1741. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, et al. , 1984. Neuronal development in the Drosophila retina: monoclonal antibodies as molecular probes. Cell. 36, 15–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Quantitative analyses of severity percentage of eye degeneration phenotype. We examined the eye phenotypes of 200 flies per genotype and the phenotypes are represented in the images in Fig1, 2, 3. For quantitative analysis, 200 flies per genotype were randomly selected for scoring according to the criteria where “No-eye” was assigned to category 6, 80% eye degeneration was assigned to category 5, 60-80% eye degeneration was assigned to category 4, 40-60% eye degeneration was assigned to category 3, 20-40% eye degeneration was assigned to category 2, 0-20% eye degeneration was assigned to category 1 and Wild type was assigned to category 0. Comparisons were made using Non-Parametric: Mann Whitney T Test. Data show mean phenotype score ± S.D. ** =p<0.001. (A) Comparison of eye degeneration phenotypes between GMR-Gal4/+ with GMR>FUS, GMR> FUS R518K GMR> FUS R521C. (B) Comparison of eye degeneration phenotypes between GMR>FUS, GMR>FUS+hpo, GMR>FUS+hpo RNAi, GMR> FUS R518K+hpo, GMR>FUS R518K+hpo RNAi, GMR> FUS R521C+hpo, and GMR> FUS R521C+hpo RNAi. (C) Comparison of eye degeneration phenotypes between GMR>FUS, GMR>FUS+wts, GMR>FUS+wts RNAi, GMR> FUS R518K+wts, GMR> FUS R518K+wts RNAi, GMR> FUS R521C+wts, and GMR> FUS R521C+wts RNAi, GMR>FUS+yki3SA, GMR>FUS+ykiRNAi, GMR> FUS R518K+yki3SA, GMR> FUS R518K+ykiRNAi, GMR> FUS R521C+ yki3SA, and GMR> FUS R521C+ ykiRNAi.
Supplementary Figure 2. Accumulation of FUS triggers cell death. FUS protein accumulation and localization was checked in all the genotypes using FUS antibody. The expression of FUS protein was not detected in wild-type eye-antennal imaginal discs. Misexpression of FUS (A, A’) GMR>FUS or mutant FUS (C, C’) GMR>FUS R518K, and (E, E’) GMR>FUS R521C eye, exhibit robust FUS expression in differentiating photoreceptor neurons in the GMR domain of eye imaginal discs. Misexpression of FUS or mutant FUS in heterozygous background of deficiency Df(2R)BSC782/+, which also uncovers hpo gene, in the GMR domain of eye (B,B’) GMR>FUS+Df(2R)BSC782/+, (D,D’) GMR>FUS R518K+Df(2R)BSC782/+ and (F,F’) GMR>FUS R521C+Df(2R)BSC782/+, showed similar expression level of FUS in the eye imaginal discs. (G) Quantitative analysis of images to compare the FUS intensity levels in FUS or FUS mutants (GMR>FUS, GMR>FUS R518K, and GMR>FUS R521C) and when FUS or FUS mutants are misexpressed together with deficiency Df(2R)BSC782/+ in the GMR domain (GMR>FUS+ Df(2R)BSC782/+, GMR>FUS R518K+ Df(2R)BSC782/+ and GMR>FUS R521C+ Df(2R)BSC782/+). The data is represented in the form of a graph. The quantification of levels of FUS expression was done using built-in program in LSM 3000, Laser Scanning Confocal Microscopy and statistical analysis was conducted using Microsoft Excel. The p-values were calculated using Student’s t-Test (n=3) in MS Excel, where the levels of expression of FUS was found to be non-significant in GMR>FUS + Df(2R)BSC782/+ (p>0.05, N.S.), GMR>FUS R518K + Df(2R)BSC782/+ (p>0.05, N.S.) and GMR>FUS R521C + Df(2R)BSC782/+ (p>0.05, N.S.), as compared to GMR>FUS, GMR>FUS R518K, and GMR>FUS R521C respectively. (H) Semi-quantitative western blot was used to detect FUS protein as a 70kDa band in GMR>FUS and mutant FUS (GMR>FUS R518K and GMR>FUS R521C) background. Tubulin with a band size of 55kDa serve as a loading control. Note that 70kDa FUS band was absent in wild-type but was found in all the three FUS transgenic fly lines used in the study. (H’) The FUS bands intensity was quantified in WT FUS and mutant FUS (FUS R518K and FUS R521C backgrounds. It was normalized and the graph was made using Image J software (NIH). The p-values for the estimation of FUS levels in all combinations in Western Blot were calculated in a set of three (n=3), using Student’s t-Test in MS Excel software. Wild-type was found to be statistically significant from GMR>FUS (p<0.0001, ****), GMR>FUS R518K (p0.0001, ****), and GMR>FUS-R521C (p0.0001, ****). Genotypes including GMR>FUS, GMR>FUS-R518K were found to be statistically non-significant (p>0.05, NS) and GMR>FUS, GMR>FUS-R521C were also non-significant (p>0.05, NS) respectively. The genotypes GMR>FUS-R518K and GMR>FUS-R521C was found to be statistically significant (p<0.01, *). Scanning Electron Micrographs (SEM) showing the higher magnification images of (I, I’) Wild-type, (J, J’) GMR>FUS, (K, K’) GMR>FUS R518K and (L, L’) GMR>FUS R521C adult eyes at 180X and 800X respectively.
Supplementary Figure 3. Downregulation of Hippo signaling restores axonal projections (impaired by gain of function of FUS) from retina to the brain. Top panel shows, (A) Wild-type larval eye-antennal imaginal disc, and (B) GMR>FUS (G) GMR>FUS R518K, and (L) GMR>FUS R521C showing expression of the retinal axons marked by 24B10 in control and when WT FUS or mutant FUS (FUS R518K and FUS R521C) are misexpressed in the GMR domain respectively. Activation of Hippo signaling by misexpressing vote, wts, ykiRNAi along with FUS or mutant FUS results in impaired targeting of retinal axons to the brain (C) GMR>FUS+ wts, (H) GMR>FUS R518K+wts, (M) GMR>FUS R521C+wts, (D) GMR>FUS+ ykiRNAi, (I) GMR>FUS R518K+ykiRNAi and (N) GMR>FUS R521C+ykiRNAi as compared to the (A) wild-type. Downregulation of the Hippo signaling by misexpressing wtsRNAi, yki3SA along with WT FUS or mutant FUS results in restoration of targeting of retinal axons to the brain (E) GMR>FUS+wtsRNAi, (J) GMR>FUS R518K+wtsRNAi, (O) GMR>FUS R521C+wtsRNAi, (F) GMR>FUS+yki3SA, (K) GMR>FUS R518K+yki3SA and (P) GMR>FUS R521C+yki3SA. Magnification of all the eye-antennal imaginal discs is 40X.