

Abstract
The distribution of DNA among bacterioplankton and bacterial isolates was determined by flow cytometry of DAPI (4′,6′-diamidino-2-phenylindole)-stained organisms. Conditions were optimized to minimize error from nonspecific staining, AT bias, DNA packing, changes in ionic strength, and differences in cell permeability. The sensitivity was sufficient to characterize the small 1- to 2-Mb-genome organisms in freshwater and seawater, as well as low-DNA cells (“dims”). The dims could be formed from laboratory cultivars; their apparent DNA content was 0.1 Mb and similar to that of many particles in seawater. Preservation with formaldehyde stabilized samples until analysis. Further permeabilization with Triton X-100 facilitated the penetration of stain into stain-resistant lithotrophs. The amount of DNA per cell determined by flow cytometry agreed with mean values obtained from spectrophotometric analyses of cultures. Correction for the DNA AT bias of the stain was made for bacterial isolates with known G+C contents. The number of chromosome copies per cell was determined with pure cultures, which allowed growth rate analyses based on cell cycle theory. The chromosome ratio was empirically related to the rate of growth, and the rate of growth was related to nutrient concentration through specific affinity theory to obtain a probe for nutrient kinetics. The chromosome size of a Marinobacter arcticus isolate was determined to be 3.0 Mb by this method. In a typical seawater sample the distribution of bacterial DNA revealed two major populations based on DNA content that were not necessarily similar to populations determined by using other stains or protocols. A mean value of 2.5 fg of DNA cell−1 was obtained for a typical seawater sample, and 90% of the population contained more than 1.1 fg of DNA cell−1.
Aquatic heterotrophic bacterioplankton, which are too small for observation by light microscopy, are commonly visualized with fluorescent DNA stains (14). The intensity of stain fluorescence as determined by flow cytometry, together with light scatter data, can help characterize natural populations (10, 11, 43, 70), determine rates of growth (16), locate DNA-deficient organisms (49), provide a cell mass basis for comparative and absolute descriptions of organism affinity for nutrients (5), and identify low-mass particles (49) as bacteria in order to quantify a major component of aquatic living carbon (9).
The mean DNA content of bacterioplankton has been estimated from analysis of filter-retained material and an organism count together with the number of organisms observed (17) and from analysis of images of individual cells (36), but mean values (17, 44) vary more than expected. In early studies, flow cytometry was used to observe differences among cells in monocultures of commonly grown large-cell species (60). Fluorescence from DAPI (4′,6-diamidino-2-phenylindole)-bound DNA was responsible for locating predominant very small oligobacteria (28). DAPI has been used to estimate the genome sizes of Synechococcus (3) and oligobacterial (52) isolates. Stains such as PicoGreen (62), Hoechst 33258, SYBR Green (4, 38), SYTOX Green (66), Syto 13 (18), YOYO, YO-PRO (39), and TOTO (24) have also been used, but the specificity and species dependence of these stains have not been evaluated. Among these stains the in vitro binding of DAPI by DNA is best understood (61). DAPI is bright and stable enough and is minimally affected by DNA conformation (1). To improve the utility of DAPI as a quantitative probe for DNA in individual organisms, we studied binding, salt effects, specificity, staining conditions, and permeation requirements. We show that this stain can be used to measure DNA content, chromosome size, and chromosome stability, as well as the distribution of DNA among various types of oligobacteria or among oligobacteria growing at various rates.
MATERIALS AND METHODS
Cultures and seawater samples.
The marine organisms Cycloclasticus oligotrophus (10), Marinobacter arcticus (10), and Sphingomonas sp. strain RB2256 (53) were grown in synthetic seawater medium containing 1 M Na+ (52) and 1 to 10 μM acetate, mixed amino acids, and glucose, respectively, as carbon sources. Escherichia coli DH1 (ATCC 33849) and Brevundimonas diminuta (formerly Pseudomonas diminuta ATCC 19146) were grown in low-salt (M9) mineral medium (15) containing 100 μM glucose. Methanobacterium thermoautotrophicum ΔH was grown in mineral medium supplemented with a stream of methane gas. Cultures were grown from stock preparations stored in glycerol at −50°C (20). E. coli and C. oligotrophus cultures containing subpopulations of cells with up to five genome copies were produced either by treatment with rifampin or by constitutive chromosome runout (58) following exhaustion of the limiting carbon source. Low-DNA-content cells were produced by 20-fold dilution of a C. oligotrophus batch culture in a medium containing 28 mg of acetate liter−1 to obtain 107 cells ml−1 and incubation at 20°C.
Seawater was collected at a depth of 15 m with a Niskin bottle from the R/V Alpha Helix in Thumb Cove off Resurrection Bay in the Gulf of Alaska. Surface water was collected from East Twin Lake (145 km southwest of Fairbanks, Alaska) by dipping with a baked 3-liter carafe from the front of an aircraft pontoon while another researcher was paddling upwind. Other freshwater samples were collected in a similar manner from a small boat. All samples were preserved with filtered (pore size, 0.2 μm) formalin (0.5% formaldehyde), placed on ice, returned to the laboratory, and stored at 5°C in the dark.
Flow cytometry.
Preserved samples were directly stained, and fresh samples were treated with formaldehyde and refrigerated at least overnight in the dark before staining. Bacterial populations were diluted with basal medium to concentrations of about 106 cells ml−1, filtered through a 1.0-μm-pore-size filter (natural samples only), permeabilized with 0.1% Triton X-100, and stained with freshly diluted DAPI obtained from a frozen stock solution (0.5 μg ml−1) for 60 ± 10 min at 10°C in the dark (7). An internal standard mixture, consisting of 0.6- and 0.9-μm-diameter beads (Polysciences) was added to each sample. The smaller particles were used to normalize fluorescence intensity and correct for instrument drift; the larger particles were added to a concentration of 1 × 105 particles ml−1 (as determined with a Coulter Counter) and used to ratiometrically determine cell population sizes (49).
Measurements were obtained with a modified (47) Ortho Cytofluorograf IIs equipped with a 5-W argon laser. Computerized operation was accomplished with a Cicero system and Cyclops software (Cytomation, Inc.). The laser was tuned to 351.1 and 363.8 nm and was operated with a 100-mW output. Blue fluorescence from DAPI was collected at 90° to the beam through a 424-nm long-pass dichroic filter and a 450- to 490-nm band pass filter. Fluorescence intensity was determined with a calibrated (51) 3.5-decade dynamic-range logarithmic amplifier with analysis triggered by fluorescence.
DNA content.
DAPI-DNA fluorescence intensity was converted from a logarithmic distribution over 256 channels to 103.5 linear channels by Cyclops software. Gains were set with reference to the 0.6-μm-diameter standard beads, and formaldehyde-preserved E. coli (5.12 fg of DNA cell−1) (31, 50) was used at the end of each day to normalize differences in the freshly prepared staining solutions.
To account for differences in the G+C contents of species due to the AT-binding specificity of DAPI (32), the apparent DNA content was adjusted to the E. coli standard content by using the probability (P) of finding n adjacent AT pairs:
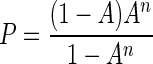 |
1 |
where n is the number needed for binding to DAPI and A is the portion of DNA comprised of adenine plus thymidine. For DAPI, n is at least 3 (69), and the corrected value was considered the product of the apparent value and the ratio of P for E. coli (0.0657) to P for the unknown. When the G+C content was known, P was calculated and related to P for E. coli and the DNA content corrected from the ratio of P for E. coli to P for the unknown. For bacterioplankton a G+C content of 52% was obtained from G+C measurements for the variable region of rRNA-DNA extracted from bulk seawater (L. B. Fandino, personal communication).
Effect of salt and nucleases.
Salt effects were tested by diluting 40‰ salinity filtered seawater, incubating for 1 h, and then staining. To determine the extent of DAPI binding site loss due to nucleic acid hydrolysis, cultures were incubated for 75 min at 37°C with DNase I (2,000 Kunitz units ml−1; Sigma Chemical Co.), RNase A (400 U ml−1; Sigma), and proteinase K (10 μg ml−1; Sigma) and then permeabilized with Triton X-100 and stained with DAPI as described above.
Spectrophotometric analysis of DNA.
The organisms were centrifuged at 5,000 × g for 10 min, resuspended in basal medium at a concentration of 1 × 109 cells ml−1 (as determined with a Coulter Counter ZBI [Coulter Electronics]), diluted with STE buffer (0.1 M NaCl, 50 mM Tris HCl, 1 mM Na2EDTA) to concentrations of 0.5 × 107 to 14 × 107 organisms ml−1, lysed with 0.1% sodium dodecyl sulfate for 10 min at 60°C, and treated with proteinase K (100 μg ml−1, 37°C, 30 min) to reduce aggregation. KCl was added to a concentration of 40 mM, the sample was chilled on ice for 30 min, and the resulting potassium dodecyl sulfate precipitate was removed by centrifugation (10,000 × g, 20 min, 5°C) (45). The supernatant was stained with Hoechst 33258 at a concentration of 0.05 μg ml−1, and its fluorescence was determined by using a spectrofluorimeter (MPF-66; Perkin-Elmer Corp.) with excitation set at 350 nm and emission set at 450 nm (34). The DNA concentration, uncorrected for G+C content, was obtained from a standard curve prepared with calf thymus DNA (type I; Sigma).
Biomass.
Biomass values were obtained from a calibrated Rayleigh-Gans-based forward light scatter standard curve by using formaldehyde-treated cells with a known formaldehyde-free dry mass and an aspect ratio of 3 for all organisms (48, 49).
Cell cycle analysis.
C. oligotrophus was grown in batch cultures at various rates dictated by initial acetate concentrations of 12 to 200 mg liter−1. The rates were determined by measuring the change in total cell mass by flow cytometry. DNA contents were determined as described above. The proportions of cells in the B phase (the period between cell division and the beginning of replication when cells have one chromosome), the C phase (when the chromosome is replicating), and the D phase (when there are two chromosomes [12]) were determined by the peak reflect method (22) and used along with the growth rate determined from the rate of biomass increase to calculate (59) the time spent in each phase.
RESULTS AND DISCUSSION
Binding.
DAPI attains sufficient binding energy near certain regions with 3 or more base pairs, such as the duplex d(-AATT-) of double-stranded DNA and poly[d(A-T)]2, to widen the minor groove, attach, and fluoresce with an improved quantum yield when it is excited (1). For M. arcticus, a DAPI concentration of 0.1 μg ml−1 was sufficient to saturate half the strong binding sites in both one- and two-chromosome cells (Fig. 1). The binding constant (Kb) was 4.0 × 105 mol−1 for intact cells in 0.6 M Na+ at 10°C as determined from a Scatchard plot, and this value was close to 1.8 × 105 mol−1, the value computed from the reported value for pure DNA at an equivalent salinity (69) and corrected for temperature by using an enthalpy change of 15 kJ mol−1, which was obtained from Arrhenius plots of DAPI binding data versus temperature data (Fig. 2).
FIG. 1.

Effect of DAPI concentration on DNA fluorescence intensity (I). The values are means for the first (1n) and second (2n) fluorescence peaks from M. arcticus, such as those shown for C. oligotrophus in Fig. 4A. (Inset) Scatchard transformation for the 2n cells.
FIG. 2.

Effect of staining time and temperature on fluorescence of M. arcticus at different DAPI concentrations.
DAPI binding requires penetration of the cell envelope, but gram-positive and lithotrophic organisms can be particularly impermeant (29, 67). In the absence of Triton X-100, almost all M. thermoautotrophicum cells remained in a low-fluorescence cluster on two-dimensional histograms or dot plots (Table 1). Prolonged treatment with formaldehyde resulted in some increase in fluorescence; one-third of the organisms were repositioned in a moderately bright cluster. Permeabilization with Triton X-100 dramatically increased the fluorescence of the whole population and resulted in a single bright cluster with normal brightness for cultivated bacteria, although individual chromosomes were not resolved. About 35% of Resurrection Bay bacterioplankton were also made more fluorescent by Triton X-100 treatment (Table 1). Staining of these resistant organisms was nearly complete in 10 min and reasonably stable over time during incubation at 10°C in the presence of 0.5 μg of DAPI ml−1 and 0.1% Triton X-100 (Table 2). For E. coli formaldehyde treatment alone was sufficient to optimize staining, and for preserved samples of M. arcticus and C. oligotrophus the DAPI-DNA fluorescence was increased by about 5% (data not shown) by Triton X-100 treatment. Staining for 3 h or more resulted in a loss of chromosome resolution. The organisms belonging to several easily stainable gram-negative species then appeared in a single major cluster that included the low-DNA cells (“dims”) discussed below. Thus, long staining times appeared to result in nonspecific staining of whole organisms that was detrimental to quantitative measurement, and cell envelopes became fluorescent. When a 60-min staining time was used to integrate staining with multiple-sample analysis by flow cytometry, the results were satisfactory.
TABLE 1.
Effect of fixative and permeabilization on the intensities of DAPI-DNA fluorescence of recalcitrant organisms as determined by flow cytometry
| Sample | Treatment
|
Low-fluorescence cluster intensitya | High-fluorescence cluster
|
||
|---|---|---|---|---|---|
| Fixative(s) | No. of days | Intensity | % of total population | ||
| M. thermoautotrophicum | Unfixed | 83 | 385 | 1 | |
| M. thermoautotrophicum | CH2O | 7 | 38 | 300 | 10 |
| M. thermoautotrophicum | CH2O | 50 | 60 | 185 | 35 |
| M. thermoautotrophicum | CH2O and Triton X-100 | 50 | Absent | 425 | 100 |
| Resurrection Bay | CH2O | 63 | 49 | 62 | 49 |
| Resurrection Bay | CH2O and Triton X-100 | 63 | 84 | 242 | 86 |
Intensity of fluorescence expressed as a linear channel number for cells in a distinct low-fluorescence (i.e., lightly stained) cluster having a dry mass similar to that of cells in a high-fluorescence cluster. Data were obtained from dot plots of DNA versus cell mass such as those shown in Fig. 7 for single species as shown in Fig. 8.
TABLE 2.
Stability of DNA content in preserved C. oligotrophus and E. coli samples
| Sample | Length of preservation (days) | DNA content (fg cell−1)
|
|
|---|---|---|---|
| 1n cells | 2n cells | ||
| C. oligotrophus | 0a | 3.36 ± 0.26b | 6.30 ± 0.52 |
| 0.1 | 3.38 ± 0.24 | 6.27 ± 0.50 | |
| 2 | 3.49 ± 0.23 | 6.50 ± 0.47 | |
| 49 | 3.53 ± 0.31 | 6.38 ± 0.66 | |
| 219 | 3.53 ± 0.47 | 6.52 ± 0.93 | |
| E. coli | 0a | 1.09 ± 0.41c | 1.09 ± 0.41c |
| 0 | 0.90 ± 0.33 | 6.57 ± 2.03 | |
| 2 | 5.87 ± 0.46 | 10.45 ± 0.83 | |
| 13 | 6.08 ± 0.54 | 10.82 ± 1.05 | |
| 49.0 | 6.43 ± 0.53 | 11.30 ± 0.86 | |
| 49.1 | 5.38 ± 0.35 | 10.10 ± 0.46 | |
| E. coli (seawater)d | 219 | 5.16 ± 0.29 | 10.06 ± 0.61 |
Before formalin was added.
Mean ± standard deviation.
Only one subpopulation was apparent.
In synthetic seawater medium.
Preservation.
Formaldehyde-treated, refrigerated E. coli and C. oligotrophus cells were indistinguishable after several months from cells that were freshly prepared in either freshwater or saltwater media on the basis of either dry mass (49) or apparent chromosome size (Table 2). For M. arcticus there was a loss of resolution in 1n and 2n cells over 11 years, but Coulter Counter populations remained unchanged (49). For the E. coli used as a DNA standard, the amount of fluorescence per cell changed ±8.6% (n = 27) over 18 months (data not shown). This change in E. coli fluorescence was random, so the small variation was not due to storage and most likely was due to a change in the stain working stock solution.
There have been reports of population losses in stored samples (63, 64) as determined by microscopic methods. Our shipboard DAPI microscopic counts agreed with the flow cytometry counts only within about ± 30%. Intercalibration with results obtained in two other laboratories (unpublished data) by using five independent preserved seawater samples from two locations gave flow cytometry count/DAPI count ratios of 0.782 ± 0.12 and 0.92 ± 0.25. The ratios of flow cytometry cell counts to acridine orange cell counts were 1.42 ± 0.56 and 0.88 ± 0.66. Values obtained by microscopic methods seem to be subjective and to require discrimination among viruses, bacteria, and other fluorescent particles. Examination of heavily stained cells by flow cytometry (Table 3) revealed only fair stability in a population. Samples were normally collected in triplicate and were frequently reanalyzed. These samples consistently gave very stable population and DNA data. However, an 18-day-old culture of C. oligotrophus showed a 38% increase in population in response to extended DAPI–Triton X-100 treatment. Two-thirds of the population was in an off-scale cluster for which the mass-per-cell value was large but the number and level of fluorescence were low. Additional staining time resulted in a normal histogram, and the total cell mass remained unchanged. Stationary-phase organisms sometimes produce clumps, and the data are consistent with formation of aggregates that were stain resistant but dissociated with additional exposure to the Triton X-100–stain mixture. Thus, Triton X-100 may increase the apparent number of organisms in old populations, but the axial-ratio-corrected cell mass should remain unchanged. Longer staining times and higher stain concentrations can increase the apparent sizes of populations, perhaps by bringing debris with nonspecific DAPI binding into the bacterial regions on histograms. Because error is exacerbated at the small end of the size spectrum, where electronic and particle contamination are more prevalent, the variations in total bacterial biomass are more dependable than the variations in cell number. Slow concentration-dependent changes in stain binding (Fig. 2) demonstrated the value of carefully controlled staining conditions for the organisms analyzed, both the standard organisms and the unknown organisms.
TABLE 3.
Changes in apparent organism abundance for preserved natural populations following heavy staininga
| Population | Length of preservation (days) | Population size (106 cells ml−1) | % Loss (% gain) |
|---|---|---|---|
| Port Valdez seawater | 1 | 3.9 | |
| 10 | 4.0 | (2) | |
| 30 | 3.7 | 5 | |
| Resurrection Bay seawater | 1 | 5.8 | |
| 10 | 4.5 | 22 | |
| 30 | 4.1 | 29 | |
| Bering Sea seawater | 1 | 0.76 | |
| 30 | 0.61 | 20 | |
| Harding Lake freshwater | 1 | 3.1 | |
| 10 | 2.9 | 6 | |
| 30 | 3.1 | 0 |
Samples were collected in 1969 and stained with 1.0 μg of DAPI ml−1.
Standard curves.
DAPI-DNA fluorescence intensity increased with chromosome number for both E. coli and C. oligotrophus in laboratory media with near perfect linearity and negligible intercepts (Fig. 3). Changing the salinity from 13 mM sodium in M9 medium to the salinity in seawater medium decreased the fluorescence of E. coli 20%, while diluting the seawater medium used to grow C. oligotrophus so that the sodium concentration was decreased from 700 to 13 mM increased the fluorescence only 8%. Severely (40-fold) diluting the media increased the fluorescence 28 to 42% depending on the number of chromosomes per organism (Table 4). Based on the reduction in the 2n/1n fluorescence ratio, the 2n cells exhibited more of the competitive effects of sodium ions, and the effects were sufficient to reduce the Kb for pure DNA by 103-fold (69). The apparent accessibility of E. coli DNA to salt and the linearity of the C. oligotrophus fluorescence curve over a range of chromosome numbers suggest that the DNA contents of unknown organisms can be estimated by using an E. coli standard that is equilibrated in the media of the unknown organisms, provided that extremely high salt concentrations are avoided.
FIG. 3.

Effect of chromosome number (n) on DAPI-DNA fluorescence intensity in both fresh and saline media. For E. coli n = 1 to 4, y = 741.1x − 48.9 (r2 = 0.995), and y = 569.5x − 3.3 (r2 = 0.998). For C. oligotrophus, n = 1 to 3, y = 439x − 14.9 (r2 = 0.998), and y = 409.1x − 5.4 (r2 = 0.999).
TABLE 4.
Effect of salinity on apparent DNA content of C. oligotrophus
| Salinity (‰) | Apparent DNA content (fg cell−1)
|
2n/1n ratio | |
|---|---|---|---|
| 1n cells | 2n cells | ||
| 40 | 3.01 | 6.02 | 2.0 |
| 20 | 3.22 | 4.10 | 1.94 |
| 10 | 3.35 | 6.27 | 1.94 |
| 5 | 3.50 | 6.60 | 1.89 |
| 1 | 4.28 | 7.80 | 1.82 |
The background fluorescence values for brownwater lakes such as East Twin Lake were small, about 8% of the mean value for the microflora. The background fluorescence was even less in lakes without a golden yellow cast, and the bacterial signals for several such lakes were better separated from the background signals.
Packing.
The concentrations of DNA in bacteria are several times the concentration found in human diploid nuclei (1%, by volume) (25) and are sufficient to give measured attenuation of DAPI-DNA fluorescence in the larger aquatic bacteria (10) of up to 10%. Distances are too great for resonance energy transfer (13), but the complex could absorb reemitted photons. Packing effects were examined by comparing the relative fluorescence values for cells with one and two chromosomes in numerous E. coli subpopulations, but no packing effects were detected (data not shown). For C. oligotrophus the change in the amount of fluorescence per chromosome was also undetectable with cells having one to five chromosomes (Fig. 4). However, the dry mass of the organisms also increased, and without an increase in cell density; the DNA concentration increased only by a factor of 0.5 (Fig. 4C). Fluorescence attenuation due to increased DNA packing could therefore have escaped detection. The corrections for packing appeared to be small for the relatively large commonly cultured bacteria, such as E. coli, due to low DNA concentrations, and the corrections for aquatic forms were small due to the thin cross section associated with small size. Therefore, corrections for fluorescence attenuation due to DNA packing were not incorporated into measurements of cellular DNA content.
FIG. 4.

DNA content of C. oligotrophus. (A) DNA histogram for a stationary-phase culture with chromosome runout. The coefficients of variation for the one- to five-chromosome peaks ranged from 4.28 to 8.73%. (B) Linearity between DNA content determined from the standard curve and modal values for the fluorescence intensity from panel A. (C) Relationships between DNA content and dry mass and between DNA content and DNA concentration. Values were calculated by using a genome size of 3.5 fg cell−1 and a dry weight/wet weight ratio of 0.2; dry mass was determined by using forward light scatter intensity data (49).
DNA from extracted populations versus flow cytometry.
The kinetics of Hoechst staining of pure DNA as determined with a spectrofluorimeter were similar to those of DAPI staining of whole cells as determined by flow cytometry. The standard curve was linear for DNA concentrations between 0.04 and 1.2 μg ml−1, and there was an intercept at 0.08 μg ml−1 (data not shown). The method was applied to three species of bacteria, and the DAPI-DNA fluorescence from each species was linear with the population of added cells (Fig. 5). Mean values for the DNA contents of four species determined by flow cytometry were compared to average DNA content values based on the amount extracted by wet methods and the number of cells determined by flow cytometry (Fig. 6). The values were not G+C corrected since the AT-specific stain DAPI (56) has a binding mechanism similar to that of Hoechst 33258 (37). Despite possible errors due to incomplete pelleting and/or resuspension of the small low-density organisms, there was a good correlation between the two methods.
FIG. 5.

Fluorescence intensity as determined with a spectrophotometer of Hoechst 33258-stained DNA extracted from various dilutions of cultures of three species of bacteria.
FIG. 6.

Comparison of apparent DNA contents of DAPI-stained cells of various species determined by flow cytometry with values determined by spectrophotometric analysis of Hoechst-stained extracts of populations measured by flow cytometry (y = 1.04x + 0.12; r2 = 0.899).
Specificity.
The absence of intercepts in the standard curves (Fig. 3) is consistent with the absence of strongly binding cellular components other than DNA. Staining of cell components that bind DAPI more weakly than DNA binds DAPI should result in a concave-up Scatchard plot. The linearity observed (Fig. 1, inset) suggests that the concentrations were insufficient for nonspecific binding of DAPI to weak sites, such as G+C-rich regions of DNA (2, 69), where binding is by intercalation, or to other macromolecules, such as polyphosphate, slime (30), RNA, and cell wall components (33). Nonspecific staining occurs in the presence of excessive concentrations of DAPI (Fig. 2); however, at the concentration used, 0.5 μg ml−1, no sites with Kb values sufficient to affect DAPI-DNA fluorescence were detected. The large Kb value and the absence of saturation with DAPI (Fig. 2) are consistent with binding primarily at the AT-n ≥ 3 sites when long staining times are avoided.
Effective DNase treatment should eliminate DAPI fluorescence if the stain is specific; the reduction in C. oligotrophus fluorescence was only 88% (data not shown). However, neither E. coli fluorescence nor M. arcticus fluorescence was significantly affected, confirming that substantially intact DNA can persist in Triton X-100-permeabilized DNase-treated cells (26). RNase had no effect on fluorescence, which is also consistent with good specificity for DNA, but its efficacy in intact cells is unknown. Our data call into question the practice of using nuclease treatment to render broad-spectrum dyes DNA or RNA specific.
The DNA measurements for intact cells determined by flow cytometry were similar to those obtained for extracted DNA, as mentioned above. Our data corroborated the thermodynamic measurements that showed that there was undetectable interference from weaker binding sites (Fig. 1, inset) at the concentrations used. In addition, the agreement between values for DNA in intact cells and values for solubilized DNA from these cells suggests that minimal error was attributable to changes in medium chemistry, packing effects, or binding hindrance by supercoiling (61). Thus, under restricted staining time, concentration, and temperature conditions used, DAPI appears to be a specific stain for the DNA of small nonpigmented bacterium-like organisms when it is used with a size parameter such as forward light scatter.
Bacterioplankton DNA.
The DNA content of indigenous seawater bacterioplankton determined by the methods used in this study was less than previously published values, but the concentrations were surprisingly high. According to the histogram for a typical euphotic-zone seawater sample (Fig. 7), the maximum DNA content can be 10% (dry weight basis) with a range of 4 to 16% for the bulk of the population. The DNA content of an extinction culture isolate, C. oligotrophus, was also high, 14%, compared to 2.2% for E. coli, but the low dry weight was mitigated by the dilute cytoplasm of C. oligotrophus (16% solids, compared to 26% solids for E. coli) (10). None of the small-genome bacterioplankton of seawater have been isolated. The maximum fraction that we have been able to cultivate from Alaskan waters by extinction culture techniques is about 10% (46; unpublished data), and all of the organisms have moderate genome sizes. Thus, small genome size and resistance to isolation are covariant. The histogram obtained is typical for both lakewater and seawater in that there is a single loosely defined major cluster that accommodates regions rich in 1n and in 2n cells having fairly small genomes and it is possible that some larger-genome 1n cells are present in the subpopulation labeled 2n.
FIG. 7.

Dot plot of euphotic zone marine bacterioplankton collected from Thumb Cove in Resurrection Bay off the Gulf of Alaska. The locations of dims, a cluster at the likely position of Synechococcus, and 1n cells and a possible location of 2n cells are indicated.
Dims.
A significant population with low fluorescence corresponding to a DNA concentration of about 0.5 fg cell−1 appears in most lakewater (9) and seawater (Fig. 7) samples. This population is usually separated from the normally fluorescent organisms by a particle-free area in scatter-fluorescence (cell mass-DNA) histograms. Starvation has been reported to result in a decrease in the amount of DNA from 30 to 1 fg per cell for isolate ANT-300 (41). Such low-DNA particles have been referred to as dims (57) due to the weak fluorescence when they are stained with DAPI. It has been suggested that these particles are cytoplasmless ghosts and that such corpses can constitute most of the bacterium-sized particles (27). However, cytoplasm is the main contributor of the dipoles that are responsible for light scattering, and the scatter signal is nearly as large as that for most particles in the main bacterial cluster. It is noteworthy that DNA staining of mixed species requires attention to permeabilization; some species are quite resistant, as mentioned above. Also, stain concentrations must be adequate so that the stain occupies the intended sites but does not stain low-affinity sites. The specific affinities for amino acids of the low-mass half of lakewater bacterioplankton sorted from the total population were the same as those of the large-cell bacterial fraction (8), so the small cells can be as active as the large cells, but we have not specifically determined the uptake rate of the dims. The particle mass of most viruses is small for detection by flow cytometry, and our efforts to quantify viruses by using DAPI staining have been unsuccessful. Based on unpublished observations of numerous diverse lake and seawater sites and depths, dims usually account for between 10 and 30% of the total population, values which are less than some much higher values that have been reported (18, 35).
Low-fluorescence organisms have appeared in starved cultures of E. coli (49) and in M. thermoautotrophicum cultures (Table 1). Repositioning of 37% of the in situ Resurrection Bay bacteria into a high-fluorescence cluster after permeabilization showed that these organisms are DAPI-impermeant organisms rather than low-DNA-content organisms, as would be expected for a normal unculturable archaeal population of that size (42). The remaining 14% should represent the true possibly low-DNA-content forms (dims). Formation of dims from acetate-grown C. oligotrophus cells in the laboratory (Fig. 8A) after substrate was withheld (Fig. 8B) was demonstrated by a fourfold decrease in fluorescence and a loss of the 2n population. With longer staining times these dims became indistinguishable from those with a normal DNA content. Since the intensity of scattered light from them, which was largely a measure of cytoplasmic protein content, was reduced only slightly, the DNA signal was preferentially lost, either through hydrolysis or through leakage. The rate (3% day−1) and extent (100%) of transformation to dims is shown in Fig. 8C. Dims in the seawater sample (Fig. 7) contained 0.2 to 0.8 fg of DNA cell−1 and weighed 1 to 8 fg (dry weight).
FIG. 8.

Formation of low-fluorescence dims from C. oligotrophus cells. (A) Histogram of light scatter (cell mass) versus DAPI-DNA fluorescence for a normal population. s, 0.6-μm beads. (B) Population formed after substrate removal. The computed amount of DNA decreased from 2.1 and 4.2 fg cell−1 for the 1n and 2n clusters to 0.2 to 0.6 fg cell−1. (C) Rate of dim formation.
Data have established that dim cells can be generated from normal bacteria by starvation. The moderate rate of dim formation (3% day−1), coupled with doubling times ranging from 1 day to 1 month, is consistent with the higher levels of dims that appear in more active systems, such as warm surface lakewater and seawater, than in deep seawater. One advantage of intact DNA leakage during dim formation, if it occurs, is to maintain the dissolved DNA pool for cross-species genetic exchange.
DNA content and growth rate.
Because in situ growth rates of most aquatic heterotrophic bacteria are difficult to evaluate and the isolation success rate is low and because large-genome representatives do not always behave well in continuous cultures for controlled steady-state growth, the cell cycle of these organisms is not well established. C. oligotrophus has many oligobacterial properties, such as a high affinity for nutrients, large surface-to-dry mass ratio, and low cell mass (10). Mild clumping caused imperfect behavior in continuous cultures. Acetate supported good growth, but the associated specific affinity was low (10), so the rate of growth could be controlled for several days in batch culture by adjusting the acetate concentration (Fig. 9) before significant substrate depletion occurred. The number of genomes was apparent and growth rate dependent. The amount of dry mass increased from 25 to 42 fg cell−1 as the number of multiple-chromosome cells increased, and there was an increase in the amount of cell mass per chromosome (Table 5), which is consistent with a need for additional cytoplasmic enzyme capacity (8). Cell cycle parameters showed that there was a rather constant C phase and a nearly 30-fold decrease in the time spent as single-chromosome organisms. Both the chromosome ratio and the growth rate were linear with acetate concentration up to the maximum specific growth rate (μmax) (Fig. 10). Gradual saturation by acetate was absent, as expected for a substrate that enters by diffusion with truncation at the μmax, as previously observed in a continuous culture (10), due to limitation at some downstream metabolic step. The growth rate(μ) is given directly by the chromosome number:
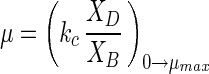 |
2 |
where XB and XD are the masses of one- and two-chromosome organisms, respectively, and kc is the cell cycle constant (0.07 h−1 in this case). kc can be related to the concentration of substrate through specific affinity theory (5):
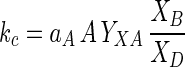 |
3 |
where aA is the specific affinity for a substrate, A is the concentration of that substrate, and YXA is the intervening cell yield. The advantages of this method are that the problems of bottle effect and dissimilar responses to probes common with other methods are minimized, mechanism-sensitive specific affinity theory is used to empirically link the chromosome ratio to nutrient concentrations (5) and to phenomenological treatments for phototrophs (40, 65), and requirements for a defined relationship between μmax and organism affinity as defined by the Michaelis-Menten relationship are eliminated (6). However, this method has not been tested with the abundant small-genome organisms, problems such as the causes of chromosome runout (58), when suddenly starved organisms generate multiple genomes, are not understood, and the distribution of genome sizes among bacterioplankton is not yet known with certainty. Still, the amount of DNA per cell, like cell size (55), provides a potentially species-independent reflection of the rate of growth that can be conveniently measured, and the method seems to reflect general levels of activity in near-arctic lakes, where a large change in specific affinity occurs over the seasons and the specific affinity varies with the amount of DNA per cell (unpublished data). Without an increase in the mean genome size during the spring, which is not anticipated due to an increase in the supply of major substrates, such as amino acids, an increase in the number of copies per cell with growth rate is likely.
FIG. 9.

Growth curves and dry mass-DNA histograms for C. oligotrophus. The histograms show the results obtained after 24 h of growth. The distributions of light scatter and fluorescence intensities associated with the cells at three growth rates are given (lower scales), and these values were converted to dry weights and DNA contents (upper scales) for the main subpopulations.
TABLE 5.
Dependence of cell cycle parameters on the rate of growth of C. oligotrophusa
| Acetate concn (mg liter−1) | Doubling time (h) | B phase
|
C phase
|
D phase
|
Dry mass of cells (fg cell−1)
|
1n/2n mass ratio | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| % of cells | Length (h) | % of cells | Length (h) | % of cells | Length (h) | 1n cells | 2n cells | |||
| 12 | 36.3 | 84.6 | 28.9 | 2.7 | 1.1 | 12.7 | 6.3 | 23.3 | 31.2 | 0.75 |
| 36 | 16 | 68.9 | 9.8 | 7.3 | 1.3 | 23.8 | 5.0 | 22.0 | 31.3 | 0.70 |
| 107 | 4 | 26.6 | 1.0 | 28.3 | 1.0 | 45.1 | 2.2 | 24.9 | 37.0 | 0.67 |
| 193 | 4 | 37.6 | 1.2 | 28.4 | 1.1 | 34.0 | 1.7 | 26.7 | 37.8 | 0.70 |
The values are from a computer analysis of the data shown in Fig. 9.
FIG. 10.

Effect of acetate concentration on the growth rate (●) and the chromosome ratio of C. oligotrophus determined from the 2n and 1n chromosome peak heights (Fig. 9) after 17 h (▵) and 24 h (▿) in batch culture.
Genome sizing.
Since the fluorescence associated with 1n cells can be resolved by flow cytometry, the genome sizes of isolates can be estimated easily. Data suggest a small genome size, approximately 1.5 Mb, for the uncultivatable bulk of the bacterioplankton. This is because slowly growing populations are rich in 1n cells and extra copies of chromosomes are a particular burden in oligotrophic systems.
If the G+C content of an isolate is different from that of E. coli, a correction is required (equation 1). However, a 10% difference from the standard value results in a 40% change in fluorescence, and a value of 3 for n A · T combinations may not be maximal (1). The method was tested with some cultures of organisms with previously reported genome sizes and with some isolates (Table 6). One might question whether flow cytometry underestimates bacterial DNA content due to conformations not accessible to stain, inadequate cell envelope permeabilization, high G+C content, absorption by pigments, exclusion of stain by sodium ions, self-absorption due to packing, or incomplete staining as discussed above. However, our examinations failed to detect significant errors, and the last three possible reasons for underestimates mentioned above are minimized in small organisms. The very large mean value for the dry weight of DNA for natural populations (12 to 16%) compared to the values for commonly cultured organisms, such as E. coli (1.3%) (8), suggests that if the potential errors, which generally favor underestimates, are significant, then the DNA contents would be even higher than the very high values measured. Electrophoretic methods for genome sizing were problematic for C. oligotrophus as well (unpublished), and the values can differ from those obtained by sequence analysis. However, agreement with results of wet methods in which a different probe was used, and failure to find an indication of error aside from differences with previously published genome size determinations are consistent with reasonable cross-species accuracy, and relative intraspecies accuracy should be excellent.
TABLE 6.
Genome sizes of various bacteria as determined by flow cytometry
ACKNOWLEDGMENTS
This work was supported by National Science Foundation Polar and LExEn programs.
Information was provided by Hugh Ducklow, Tim Hollibaugh, and L. B. Fandino.
REFERENCES
- 1.Albert F G, Eckdahl T T, Fitzgerald D J, Anderson J N. Heterogeneity in the actions of drugs that bind in the DNA minor groove. Biochemistry. 1999;38:10135–10146. doi: 10.1021/bi990382p. [DOI] [PubMed] [Google Scholar]
- 2.Barcellona M L, Favilla R, VonBerger J, Avitabile M, Ragusa N, Masotti L. DNA–4′-6-diamidino-2-phenylindole interactions: a comparative study employing fluorescence and ultraviolet spectroscopy. Arch Biochem Biophys. 1986;250:48–53. doi: 10.1016/0003-9861(86)90700-9. [DOI] [PubMed] [Google Scholar]
- 3.Binder B J, Chisholm S W. Cell cycle regulation in marine Synechococcus sp. strains. Appl Environ Microbiol. 1995;61:708–717. doi: 10.1128/aem.61.2.708-717.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binder B J, Liu Y C. Growth rate regulation of rRNA content of a marine Synechococcus (cyanobacterium) strain. Appl Environ Microbiol. 1998;64:3346–3351. doi: 10.1128/aem.64.9.3346-3351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Button D K. Nutrient uptake by microorganisms according to kinetic parameters from theory as related to cytoarchitecture. Microbiol Mol Biol Rev. 1998;62:636–645. doi: 10.1128/mmbr.62.3.636-645.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Button D K. Abandon Michaelis-Menten? ASM News. 2000;66:510. [Google Scholar]
- 7.Button D K, Robertson B R. Use of high-resolution flow cytometry to determine the activity and distribution of aquatic bacteria. In: Kemp P F, Sherr B F, Sherr E B, Cole J J, editors. Handbook of methods in aquatic microbial ecology. Ann Arbor, Mich: Lewis; 1993. pp. 163–173. [Google Scholar]
- 8.Button D K, Robertson B R. Effect of nutrient kinetics and cytoarchitecture on bacterioplankter size. Limnol Oceanogr. 2000;45:499–505. [Google Scholar]
- 9.Button D K, Robertson B R, Jüttner F. Microflora of a subalpine lake: bacterial populations, size, and DNA distributions, and their dependence on phosphate. FEMS Microbiol Ecol. 1996;21:87–101. [Google Scholar]
- 10.Button D K, Robertson B R, Schmidt T, Lepp P. A small, dilute-cytoplasm, high-affinity, novel bacterium isolated by extinction culture that has kinetic constants compatible with growth at measured concentrations of dissolved nutrients in seawater. Appl Environ Microbiol. 1998;64:4467–4476. doi: 10.1128/aem.64.11.4467-4476.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Button D K, Schut F, Quang P, Martin R M, Robertson B. Viability and isolation of typical marine oligobacteria by dilution culture: theory, procedures and initial results. Appl Environ Microbiol. 1993;59:881–891. doi: 10.1128/aem.59.3.881-891.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper S. Bacterial growth and division: biochemistry and regulation of prokaryotic and eukaryotic division cycles. New York, N.Y: Academic Press, Inc.; 1991. [Google Scholar]
- 13.Cramer W A, Knaff D B, editors. Energy transduction in biological membranes. New York, N.Y: Springer-Verlag; 1989. [Google Scholar]
- 14.Daley R J, Hobbie J E. Direct counts of aquatic bacteria by a modified epifluorescent technique. Limnol Oceanogr. 1975;20:875–882. [Google Scholar]
- 15.Eisenstadt E, Carlton B C, Brown B J. Gene mutation. In: Gerhardt P, Murray R G E, Wood W A, Krieg N R, editors. Methods for general and molecular bacteriology. Washington, D.C.: American Society for Microbiology; 1994. pp. 297–316. [Google Scholar]
- 16.Ellenbroek F M, Cappenberg T E. DNA synthesis and tritiated thymidine incorporation by heterotrophic freshwater bacteria in continuous culture. Appl Environ Microbiol. 1991;57:1675–1682. doi: 10.1128/aem.57.6.1675-1682.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuhrman J A, Azam F. Thymidine incorporation as a measure of heterotrophic bacterioplankton production in marine surface waters: evaluation and field results. Mar Biol. 1982;66:109–120. [Google Scholar]
- 18.Gasol J M, Zwiefel U L, Peters F, Fuhrman J A, Hagström A. Significance of size and nucleic acid content heterogeneity as measured by flow cytometry in natural planktonic bacteria. Appl Environ Microbiol. 1999;65:4475–4483. doi: 10.1128/aem.65.10.4475-4483.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gauthier M J, LaFay B, Christen R, Fernandez L, Acquaviva M, Bonin P, Bertrand J-C. Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int J Syst Bacteriol. 1992;42:568–576. doi: 10.1099/00207713-42-4-568. [DOI] [PubMed] [Google Scholar]
- 20.Gherna R L. Culture preservation. In: Gerhardt P, Murray R G E, Wood W A, Krieg N R, editors. Methods for general and molecular bacteriology. Washington, D.C.: American Society for Microbiology; 1994. pp. 278–292. [Google Scholar]
- 21.Gillis M, De Ley J, De Cleene M. The determination of molecular weight of bacterial genome DNA from renaturation rates. Eur J Biochem. 1970;12:143–153. doi: 10.1111/j.1432-1033.1970.tb00831.x. [DOI] [PubMed] [Google Scholar]
- 22.Gray J W, Dolbeare F, Pallavicini M G. Quantitative cell-cycle analysis. In: Melamed M R, Lindmo T, Mendelsohn M L, editors. Flow cytometry and sorting. 2nd ed. New York, N.Y: Wiley-Liss, Inc.; 1990. pp. 445–467. [Google Scholar]
- 23.Grothues D, Tümmler B. New approaches in genome analysis by pulsed-field gel electrophoresis: application to the analysis of Pseudomonas species. Mol Microbiol. 1991;5:2763–2776. doi: 10.1111/j.1365-2958.1991.tb01985.x. [DOI] [PubMed] [Google Scholar]
- 24.Guindulain T, Comas J, Vives-Rego J. Use of nucleic acid dyes SYTO-13, TOTO-1, and YOYO-1 in the study of Escherichia coli and marine prokaryotic populations by flow cytometry. Appl Environ Microbiol. 1997;63:4608–4611. doi: 10.1128/aem.63.11.4608-4611.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamada S, Fujita S. Problem of size dependence in fluorescence DNA cytometry. Cytometry. 1989;10:394–401. doi: 10.1002/cyto.990100406. [DOI] [PubMed] [Google Scholar]
- 26.Helenius A, Simons K. Solubilization of membranes by detergents. Biochim Biophys Acta. 1975;415:29–79. doi: 10.1016/0304-4157(75)90016-7. [DOI] [PubMed] [Google Scholar]
- 27.Hessenberger A, Leppard G G, Herndl G J. Relationship between the intracellular intregrity and the morphology of the capsular envelope in attached and free-living marine bacteria. Appl Environ Microbiol. 1996;62:4521–4528. doi: 10.1128/aem.62.12.4521-4528.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hobbie J E, Daley R J, Jasper S. Use of Nuclepore filters for counting bacteria by fluorescence microscopy. Appl Environ Microbiol. 1977;33:1225–1228. doi: 10.1128/aem.33.5.1225-1228.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hüber H, Hüber G, Stetter K O. A modified DAPI fluorescence staining procedure suitable for the visualization of lithotrophic bacteria. Syst Appl Microbiol. 1985;6:105–106. [Google Scholar]
- 30.Kogure K, Koike I. Particle counter determination of bacterial biomass in seawater. Appl Environ Microbiol. 1987;53:275. doi: 10.1128/aem.53.2.274-277.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohara Y, Akiyame K, Isono K. The physical map of the whole E. coli chromosome: application of a new strategy for rapid analysis and sorting of a large genomic library. Cell. 1987;50:495–508. doi: 10.1016/0092-8674(87)90503-4. [DOI] [PubMed] [Google Scholar]
- 32.Langlois R G, Carrano A V, Gray J W, VanDilla M A. Cytochemical studies of metaphase chromosomes by flow cytometry. Chromosoma. 1980;77:229–251. doi: 10.1007/BF00286050. [DOI] [PubMed] [Google Scholar]
- 33.Lessie T G, Gaffney T. Catabolic potential of Pseudomonas cepacia. Bacteria. 1986;10:439–481. [Google Scholar]
- 34.Li W, Wood A M. Vertical distribution of North Atlantic ultraphytoplankton: analysis by flow cytometry and epifluorescence microscopy. Deep-Sea Res. 1988;35:1615–1638. [Google Scholar]
- 35.Li W K W, Jellett J F, Dickie P M. DNA distributions in planktonic bacteria stained with TOTO or TO-PRO. Limnol Oceanogr. 1995;40:1485–1495. [Google Scholar]
- 36.Loferer-Krössbacher K P, Witzel K-P, Psenner R. DNA content of aquatic bacteria measured by densitometric image analysis. Arch Hydrobiol. 1999;54:185–198. [Google Scholar]
- 37.Maria A A F, Carrondo C T, Coll M, Aymami J, Andrew H J, Wang G A, van der Marel J H, Alexander R. Binding of a Hoechst dye to d(CGCGATATCGCG) and its influence on the conformation of the DNA fragment. Biochemistry. 1989;28:7840. doi: 10.1021/bi00445a047. [DOI] [PubMed] [Google Scholar]
- 38.Marie D, Partensky F, Jacquet S, Vaulot D. Enumeration and cell cycle analysis of natural populations of marine picoplankton by flow cytometry using the nucleic acid stain SYBR Green I. Appl Environ Microbiol. 1997;63:186–193. doi: 10.1128/aem.63.1.186-193.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marie D, Vaulot D, Partensky F. Application of the novel nucleic acid dyes YOYO-1, YO-PRO-1, and PicoGreen for flow cytometric analysis of marine prokaryotes. Appl Environ Microbiol. 1996;62:1649–1655. doi: 10.1128/aem.62.5.1649-1655.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monfort P, Baleux B. Cell cycle characteristics and changes in membrane potential during growth of Escherichia coli as determined by a cyanine fluorescent dye and flow cytometry. J Microbiol Methods. 1996;25:79–86. [Google Scholar]
- 41.Moyer C L, Morita R Y. Effect of growth rate and starvation- survival on cellular DNA, RNA, and protein of a psychrophilic marine bacterium. Appl Environ Microbiol. 1989;55:2710–2716. doi: 10.1128/aem.55.10.2710-2716.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray A E, Massana A, Strawzewski S, Passow U, Alldredge A, DeLong E F. A time series assessment of planktonic archaeal variability in the Santa Barbara Channel. Aquat Microb Ecol. 1999;20:129–145. [Google Scholar]
- 43.Ouverney C C, Fuhrman J A. Combined microautoradiography-16S rRNA probe technique for determination of radioisotope uptake by specific microbial cell types in situ. Appl Environ Microbiol. 1999;65:1746–1752. doi: 10.1128/aem.65.4.1746-1752.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paul J H, Jeffrey W H, DeFlaun M. Particulate DNA in subtropical oceanic and estuarine planktonic environments. Mar Biol. 1985;90:95–101. [Google Scholar]
- 45.Plumley F G, Schmidt G W. Nitrogen-dependent expression of photosynthetic genes. Proc Natl Acad Sci USA. 1989;86:2678–2682. doi: 10.1073/pnas.86.8.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quang P, Button D K, Robertson B R. Use of species distribution data in the determination of bacterial viability by extinction culture of aquatic bacteria. J Microbiol Methods. 1998;33:203–210. [Google Scholar]
- 47.Robertson B R, Button D K. Characterizing aquatic bacteria according to population, cell size and apparent DNA content by flow cytometry. Cytometry. 1989;10:70–76. doi: 10.1002/cyto.990100112. [DOI] [PubMed] [Google Scholar]
- 48.Robertson B R, Button D K. Bacterial biomass from measurements of forward light scatter intensity by flow cytometry. In: Robinson P, editor. Current protocols in cytometry. Vol. 1. New York, N.Y: John Wiley & Sons; 1999. pp. 11.9.1–11.9.11. [DOI] [PubMed] [Google Scholar]
- 49.Robertson B R, Button D K, Koch A L. Determination of the biomasses of small bacteria at low concentration in a mixture of species with forward light scatter measurements by flow cytometry. Appl Environ Microbiol. 1998;64:3900–3909. doi: 10.1128/aem.64.10.3900-3909.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rudd K E, Miller W, Ostell J, Benson D A. Alignment of Escherichia coli K12 DNA sequences to a genomic restriction map. Nucleic Acids Res. 1990;18:313–321. doi: 10.1093/nar/18.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmid I, Schmid P, Giorgi J V. Conversion of logarithmic channel numbers into relative linear fluorescence intensity. Cytometry. 1988;9:533–538. doi: 10.1002/cyto.990090605. [DOI] [PubMed] [Google Scholar]
- 52.Schut F, DeVries E, Gottschal J C, Robertson B R, Harder W, Prins R A, Button D K. Isolation of typical marine bacteria by dilution culture: growth, maintenance, and characteristics of isolates under laboratory conditions. Appl Environ Microbiol. 1993;59:2150–2160. doi: 10.1128/aem.59.7.2150-2160.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schut F, Gottschal J C, Prins R A. Isolation and characterization of the marine ultramicrobacterium Sphingomonas sp. strain RB2256. FEMS Microbiol Rev. 1997;20:363–369. [Google Scholar]
- 54.Segers P, Vancanneyt M, Pot R, Torck U, Hoste B, Dewettinck D, Falsen E, Kersters K, DeVos P. Classification of Pseudomonas diminuta Leifson and Hugh 1954 and Pseudomonas vesicularis Buesing, Doell, and Freytag 1953 in Brevundimonas gen. nov. as Brevundimonas diminuta comb. nov. and Brevundimonas vesicularis comb. nov., respectively. Int J Syst Bacteriol. 1994;44:499–510. doi: 10.1099/00207713-44-3-499. [DOI] [PubMed] [Google Scholar]
- 55.Servais P, Courties C, Lebaron P, Troussellier M. Coupling bacterial activity measurements with cell sorting by flow cytometry. Microb Ecol. 1999;38:180–189. doi: 10.1007/s002489900160. [DOI] [PubMed] [Google Scholar]
- 56.Shapiro H M. Practical flow cytometry. 3rd ed. New York, N.Y: Alan R. Liss; 1995. [Google Scholar]
- 57.Sieracki M, Viles C L. Distributions and fluorochrome-staining properties of sub-micrometer particles and bacteria in the North Atlantic. Deep-Sea Res. 1992;39:1919–1929. [Google Scholar]
- 58.Skarstad K, Bernander R, Wold S, Steen H B, Boye E. Cell cycle analysis of microorganisms. In: Al-Rubeai M, Emery A N, editors. Flow cytometry applications in cell culture. New York, N.Y: Marcel Dekker, Inc; 1996. pp. 241–255. [Google Scholar]
- 59.Slater M L, Sharrow S O, Gart J J. Cell cycle of Saccharomyces cerevisiae in populations growing at different rates. Proc Natl Acad Sci USA. 1977;74:3850–3854. doi: 10.1073/pnas.74.9.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steen H B, Boye E. Bacterial growth studied by flow cytometry. Cytometry. 1980;1:32–36. doi: 10.1002/cyto.990010108. [DOI] [PubMed] [Google Scholar]
- 61.Storl K, Burckhardt G, Lown J W, Zimmer C. Studies on the ability of minor groove binders to induce supercoiling in DNA. FEBS Lett. 1999;334:49–54. doi: 10.1016/0014-5793(93)81678-s. [DOI] [PubMed] [Google Scholar]
- 62.Tranvik L J. Rapid fluorometric assay of bacterial density in lake water and seawater. Limnol Oceanogr. 1997;42:1629–1634. [Google Scholar]
- 63.Troussellier M, Courtiess C, Zettelmaier S. Flow cytometric analysis of coastal lagoon bacterioplankton and picoplankton: fixation and storage effects. Estuarine Coastal Shelf Sci. 1995;40:621–633. [Google Scholar]
- 64.Turley C M, Hughes D J. The effect of storage temperature on the enumeration of epifluorescence-detectable bacterial cells in preserved sea-water samples. J Mar Biol Assoc UK. 1994;74:259–262. [Google Scholar]
- 65.Vaulot D, Marie D, Olson R J, Chisholm S W. Growth of Prochlorococcus, a photosynthetic prokaryote, in the equatorial Pacific Ocean. Science. 1995;268:1480–1482. doi: 10.1126/science.268.5216.1480. [DOI] [PubMed] [Google Scholar]
- 66.Veldhuis M J W, Cucci T L, Sieracki M E. Cellular DNA content of marine phytoplankton using two new fluorochromes: taxonomic and ecological implications. J Phycol. 1997;33:527–531. [Google Scholar]
- 67.Walberg M, Gaustad P, Steen H B. Uptake kinetics of nucleic acid targeting dyes in S. aureus, E. facealis and B. cereus; a flow cytometric study. J Microbiol Methods. 1999;35:167–178. doi: 10.1016/s0167-7012(98)00118-3. [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, Lau P C K, Button D K. A marine oligobacterium harboring genes known to be part of aromatic hydrocarbon degradation pathways of soil pseudomonads. Appl Environ Microbiol. 1996;62:2169–2173. doi: 10.1128/aem.62.6.2169-2173.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilson W D, Tanious F A, Barton H J, Jones R L, Fox K, Wydra R L, Strekowski L. DNA sequence dependent binding modes of 4′,6-diamidino-2-phenylindole (DAPI) Biochemistry. 1990;29:8452–8461. doi: 10.1021/bi00488a036. [DOI] [PubMed] [Google Scholar]
- 70.Zubkov M V, Fuchs B M, Eilers H, Burkill P, Amann R. Determination of total protein content of bacterial cells by SYPRO staining and flow cytometry. Appl Environ Microbiol. 1999;65:3251–3257. doi: 10.1128/aem.65.7.3251-3257.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]