

Abstract
We evaluated mRNA and miRNA in COVID-19 patients and elucidated the pathogenesis of COVID-19, including protein profiles, following mRNA and miRNA integration analysis. mRNA and miRNA sequencing was done on admission with whole blood of 5 and 16 healthy controls (HCs) and 10 and 31 critically ill COVID-19 patients (derivation and validation cohorts, respectively). Interferon (IFN)-α2, IFN-β, IFN-γ, interleukin-27, and IFN-λ1 were measured in COVID-19 patients on admission (day 1, 181 critical/22 non-critical patients) and days 6–8 (168 critical patients) and in 19 HCs. In the derivation cohort, 3,488 mRNA and 31 miRNA expressions were identified among differentially expressed RNA expressions in the patients versus those in HCs, and 2,945 mRNA and 32 miRNA expressions in the validation cohort. Canonical pathway analysis showed the IFN signaling pathway to be most activated. The IFN-β plasma level was elevated in line with increased severity compared with HCs, as were IFN-β downstream proteins, such as interleukin-27. IFN-λ1 was higher in non-critically ill patients versus HCs but lower in critical than non-critical patients. Integration of mRNA and miRNA analysis showed activated IFN signaling. Plasma IFN protein profile revealed that IFN-β (type I) and IFN-λ1 (type III) played important roles in COVID-19 disease progression.
Keywords: MT: Non-coding RNAs, IFN-β, IFN-λ1, IL-27, type I IFN, type III IFN, SARS-CoV-2, coronavirus disease
Graphical abstract
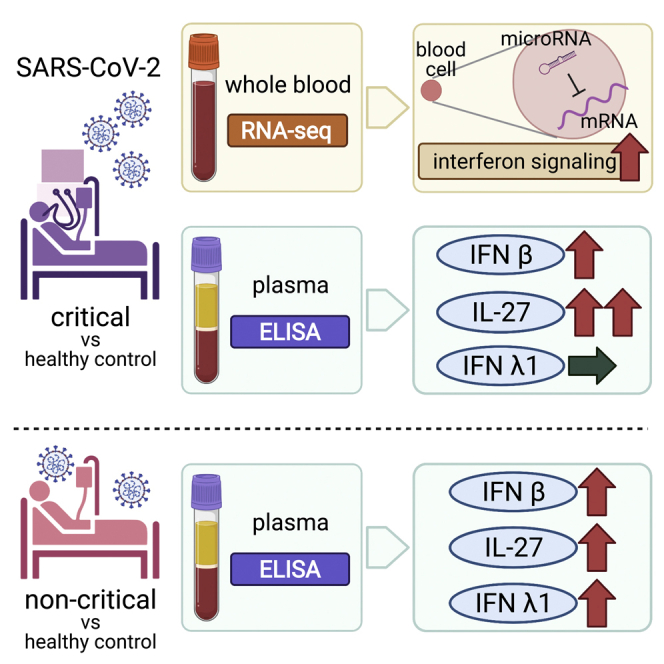
Integration of mRNA and microRNA analysis in critically ill COVID-19 patients showed that the interferon (IFN) signaling pathway was enhanced. Subsequent evaluation of type I, type II, and type III IFN-related proteins in plasma showed that IFN-β (type I) and IFN-λ1 (type III) played a role in disease progression.
Introduction
Coronavirus disease 2019 (COVID-19) is a highly contagious and infectious disease caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) with an enveloped RNA beta coronavirus 2. It was recognized by the World Health Organization as a pandemic in March 2020 and has infected over 490,000,000 people and caused over 6,100,000 deaths as of April 13, 2022.1,2
COVID-19 is characterized by respiratory symptoms; approximately 15% of patients develop pneumonia, and 5% are critically ill, with respiratory failure from acute respiratory distress syndrome, shock, and/or multi-organ dysfunction.3 Upon entering the blood, the virus binds to pattern recognition receptors such as Toll-like receptors on immune cells as pathogen-associated molecular patterns, and activated intracellular transcription factors bind to DNA in the nucleus, resulting in the transcription of messenger RNA (mRNA) and progression of inflammatory responses through translated cytokines and other proteins. Recently, non-coding RNAs (ncRNAs), such as microRNAs (miRNAs), have been reported to be associated with the pathogenesis of inflammation.4 Excessive inflammation can lead to systemic inflammatory response syndrome and multiple organ failure.
With the development of transcriptome technology, it is clear that about 98% of RNAs are ncRNAs, which are not translated into proteins. ncRNAs are now understood to play diverse roles in development, differentiation, and immune response and are essential for life phenomena. The miRNAs, a common short ncRNA, have RNA interference effects and bind to the 3′ end of the UTR of mRNA to inhibit protein translation. About 60% of all human mRNAs are regulated by miRNAs, and they are attracting increased attention.5 miRNAs are related to the pathogenesis of chronic diseases, such as cancer,6 and acute diseases, such as sepsis,7 through RNA interference.
This study aimed to evaluate mRNA and miRNA in whole blood of patients with COVID-19 and to elucidate the pathogenesis of COVID-19, including the subsequent protein profile following mRNA and miRNA integration analysis.
Results
Patient characteristics
We included 10 and 31 critically ill patients with COVID-19 and 5 and 16 healthy control (HCs) in the RNA sequencing (RNA-seq) derivation and validation cohorts and 203 patients with COVID-19 and 19 HCS in the interferon (IFN) protein profile cohort, respectively (Table 1). In the RNA-seq validation cohort, median ages of the HC and COVID-19 groups were 55 and 73 years, and their body mass indexes (BMIs) were 22 and 22.7 kg/m2, respectively. The values were not significantly different except for age in the RNA-seq validation cohort. All patients with COVID-19 were treated in intensive care units (ICUs) and with mechanical ventilation. Mortality rates were 10% and 12.9% in the RNA-seq derivation and validation cohorts, respectively. Among the 203 patients with COVID-19 in the IFN protein profile cohort, 22 were non-critically ill and 181 were critically ill, of whom 144 were male (70.1%), with a mean age of 67 (57–74) years and BMI of 25.0 (22.8–27.8) kg/m2. There were no significant differences between the critical and non-critical COVID-19 groups. BMI was not significantly different between the survivor and non-survivor groups. In both RNA-seq cohorts, the viral strain in cases with known strains was SARS-CoV2 clade B. In the IFN protein profile cohort, some cases were clade 20A or 20I (Alpha, V1). Details of the patient characteristics are shown in Tables 1 and S1.
Table 1.
Patient characteristics of each cohort
| Characteristic | RNA-seq derivation cohort |
RNA-seq validation cohort |
IFN proteins profile cohort |
|||
|---|---|---|---|---|---|---|
| Controls (N = 5) | Patients with COVID-19 (N = 10) | Controls (N = 16) | Patients with COVID-19 (N = 31) | Controls (N = 19) | Patients with COVID-19 (N = 203) | |
| Age, median, IQR | 71 (67–79.5) | 74 (66–78.8) | 55 (34–59.5) | 73 (62–76) | 58 (55–71) | 67 (57–74) |
| Sex, male (%) | 3 (60) | 5 (50) | 8 (50) | 22 (70.1) | 13 (68.4) | 144 (70.9) |
| BMI, median IQR | 21.5 (19.7–26.3) | 24.9 (22.3–26.5) | 22 (20.3–24.6) | 22.7 (21.8–25.2) | 21.7 (20.1–24.8) | 25.0 (22.8–27.8) |
| Comorbidities, n (%) | ||||||
| Diabetes | 0 (0) | 7 (70) | 1 (6.2) | 12 (38.7) | 1 (5.3) | 78 (38.4) |
| Hypertension | 2 (40) | 3 (30) | 2 (12.5) | 16 (51.6) | 4 (21.1) | 99 (48.8) |
| Hyperlipidemia | 3 (60) | 2 (20) | 6 (37.5) | 10 (32.3) | 10 (52.6) | 62 (30.5) |
| Hyperuricemia | 0 (0) | 0 (0) | 1 (6.3) | 6 (19.4) | 1 (5.3) | 23 (11.3) |
| Chronic heart disease | 1 (20) | 1 (10) | 1 (6.3) | 2 (6.5) | 2 (10.5) | 22 (10.8) |
| Chronic lung disease | 0 (0) | 1 (10) | 0 (0) | 6 (19.4) | 0 (0) | 26 (12.8) |
| Chronic kidney disease | 0 (0) | 1 (10) | 0 (0) | 7 (22.6) | 0 (0) | 23 (11.3) |
| Immunocompromised condition | 0 (0) | 0 (0) | 0 (0) | 5 (16.1) | 0 (0) | 6 (3.0) |
| Malignant neoplasm | 0 (0) | 3 (30) | 0 (0) | 0 (0) | 0 (0) | 16 (7.9) |
| Length of hospitalization | 24.5 (6.5–71.8) | 21 (13–41) | 11 (7–17.8) | |||
| Acuity score | ||||||
| 1 = death | 1 (10) | 4 (12.9) | 31 (15.3) | |||
| 2 = intubated/ventilated, survived | 9 (90) | 27 (87.1) | 160 (78.8) | |||
| 3 = hospitalized, O2 required, survived | 0 (0) | 0 (0) | 12 (5.9) | |||
| 4 = hospitalized, no O2 required, survived | 0 (0) | 0 (0) | 0 (0) | |||
| 5 = discharged/not hospitalized, survived | 0 (0) | 0 (0) | 0 (0) | |||
| Intubation required, n (%) | 10 (100) | 31 (100) | 185 (91.1) | |||
| Extracorporeal membrane oxygenation | 2 (20) | 3 (9.7) | 9 (4.8) | |||
| Late recovery, n (%) | 5 (50) | 19 (61.3) | 89 (43.8) | |||
| SOFA score, median, IQR | 5.5 (2.5–6) | 6 (3–6) | 5 (3–6) | |||
| APACHE II score, median, IQR | 16 (15.25–18.8) | 12 (10–17) | 12 (9–15) | |||
| Clade, n (%) | ||||||
| 20A | 0 (0) | 0 (0) | 1 (0.5) | |||
| 20B | 3 (30) | 20 (64.5) | 22 (10.8) | |||
| 20I (Alpha, V1) | 0 (0) | 0 (0) | 14 (6.9) | |||
| Unknown | 7 (70) | 11 (35.5) | 166 (81.8) | |||
IQR, interquartile range; BMI, body mass index; SOFA, Sequential Organ Failure Assessment; APACHE, Acute Physiology and Chronic Health Evaluation.
Data are shown as group number (percentage) or median (IQR).
In the RNA-seq derivation cohort, 21,831 mRNA, 2,024 miRNA, and 1,468 miRNA-targeted mRNA expressions were available for analysis, as were 18,959 mRNA, 2,628 miRNA, and 1,363 miRNA-targeted mRNA expressions in the RNA-seq validation cohort.
In the RNA-seq derivation cohort, 1,747 mRNA and 16 miRNA expressions were upregulated and 1,741 mRNA and 15 miRNA expressions were downregulated (p < 0.05, |log2 fold change| >1) (Figure S1A). In the RNA-seq validation cohort, 1,906 mRNA and 15 miRNA expressions were upregulated and 1,039 mRNA and 17 miRNA expressions were downregulated (false discovery rate [FDR] < 0.1, |log2 fold change| >0.6) (Figure 1A). Multidimensional scaling showed that mRNA, miRNA, and miRNA-targeted mRNA expressions could be used to distinguish between HCs and patients with COVID-19 both in the RNA-seq derivation (Figure S1B) and validation cohorts (Figure 1B). Four miRNAs were commonly upregulated and five miRNAs were commonly downregulated in both the derivation and validation cohorts. Their respective receiver operating characteristic (ROC) curves and areas under the curve (AUCs) are shown in Figure 2. The AUC of hsa.miR.143.3p, which activates the IFN signaling via inhibition of TC-PTP (Figure 4 and S3), was highest among the up- and downregulated miRNAs (AUC = 0.754).
Figure 1.

Volcano plot and multidimensional scaling analyses for mRNA, miRNA, and miRNA-targeted mRNA expressions of the RNA-seq validation cohort
(A) Volcano plot representing the differentially expressed mRNA and miRNA expressions in COVID-19 compared with the healthy control subjects. Among the differentially expressed RNA expressions, 1,906 mRNA and 14 miRNA expressions were upregulated and 1,039 mRNA and 20 miRNA expressions were downregulated. The significant differentially expressed RNA expressions are indicated. The vertical dotted lines represent |log2 fold change| >0.6. The horizontal dotted line represents the threshold for FDR < 0.1. Red dots indicate upregulated RNA expressions, and blue dots indicate downregulated RNA expressions. (B) Multidimensional scaling plot of all mRNA, miRNA, and miRNA-targeted mRNA expressions in COVID-19 compared with the healthy control subjects.
Figure 2.

Receiver operating characteristic (ROC) curve analysis of up- and downregulated miRNAs
(A) The four miRNAs that were commonly upregulated in the RNA-seq derivation cohort and the RNA-seq validation cohort were used for an ROC curve analysis, and the AUC was calculated to evaluate the prognostic accuracy of each marker. (B) The five miRNAs that were commonly downregulated in the RNA-seq derivation cohort and the RNA-seq validation cohort were used for an ROC curve analysis, and the area under the curve was calculated to evaluate the prognostic accuracy of each marker.
Figure 4.

The activated interferon pathway predicted by ingenuity pathway analysis
mRNAs and miRNAs with FDR < 0.1, |log2 fold change| > 0.6 were included in the activated interferon pathway.
Canonical pathway, canonical signaling pathway, and upstream regulators analyses
To investigate signaling pathways significantly involved in COVID-19, outcomes from RNA-seq were subjected to canonical pathway analysis (CPA) in ingenuity pathway analysis (IPA), and the activated canonical signaling pathways in COVID-19 were screened and listed. In the RNA-seq derivation cohort, CPA predicted activation of 24 pathways (the top 15 are shown in Figure S2A) and inhibition of 3 pathways (Table S2). The IFN signaling pathway had the highest p value (Benjamini-Hochberg-adjusted p value = 7.9 × 10−11, Z score = 3.962). Twenty-four mRNAs were involved in the IFN signaling pathway (Figure S2B). CPA of miRNA-targeted mRNA showed that six pathways were activated and no pathway was inhibited (Figure S2C, Table S3). Ten mRNAs involved in the IFN signaling pathway, one of the activated pathways, were shown (Figure S2D). Analysis of upstream regulators identified 393 activated potential upstream regulators and 120 inhibited potential upstream regulators (p value of overlap <0.05). Each of the top 20 is shown in Figures S2E and S2F. The CPA by IPA using mRNAs showed activated IFN pathway as well as that using miRNA-targeted mRNA. The canonical signaling pathway showed the predicted relations between RNAs in the activated IFN pathway (Figure S3). In the RNA-seq validation cohort, CPA prediction showed activation of 55 pathways (the top 15 are shown in Figure 3A) and inhibition of 3 pathways (Table S4). The IFN signaling pathway had the highest p value (Benjamini-Hochberg-adjusted p value = 5.0 × 10−11, Z score = 4.264). Twenty-three mRNAs were involved in the IFN signaling pathway (Figure 3B). CPA showed that miRNA-targeted mRNA were involved in 28 activated pathways (the top 15 are shown in Figure 3C; all pathways, including one inhibited pathway, are shown in Table S5), and 13 mRNAs were involved in the IFN signaling pathway (Figure 3D). Analysis of upstream regulators identified 301 activated potential upstream regulators and 1,059 inhibited potential upstream regulators (p value of overlap <0.05). Each of the top 20 is shown in Figures 3E and 3F. The canonical signaling pathway analysis by IPA showed the predicted relations between RNAs in the activated IFN pathway (Figure 4). miR-5196-3p had predicted activation for IFN-γRα both in the derivation and validation cohorts.
Figure 3.

Canonical pathway and upstream analysis
(A) Top 15 activated canonical signaling pathways in COVID-19 mRNA identified using Ingenuity pathway analysis. (B) Heatmap of gene expression as calculated through RNA-seq involved in the interferon signaling pathway of the samples. (C) Top 15 activated canonical pathways in miRNA-targeted mRNA expressions. (D) Heatmap of gene expression of miRNA-targeted mRNA expressions involved in the interferon signaling pathway of the samples. (E) Top 20 activated upstream regulators. (F) Top 20 inhibited upstream regulators. PD-1, programmed death-1; PD-L1, programmed death-ligand 1; TREM1, triggering receptor expressed on myeloid cells 1; PKR, protein kinase R; STAT, signal transducer and activator of transcription; NAD, nicotinamide adenine dinucleotide; IL, interleukin; iNOS, inducible nitric oxide synthase.
Gene set enrichment analysis
To validate the results of CPA by IPA, we also performed gene set enrichment analysis (GSEA) for mRNAs and miRNA-targeted mRNAs in the RNA-seq validation cohort. GSEA predicted the activation of IFN-related pathways and genes (Figure S4). These results support the activated IFN signaling pathway and IFN-related upstream regulators in IPA.
Correlation engine analysis
BaseSpace Correlation Engine analysis of the mRNA and miRNA-targeted mRNA expressions showed statistically significant correlations between COVID-19 and several diseases, including melioidosis in the RNA-seq derivation and validation cohorts (Figures 5 and S5). The top 5 statistically significant correlated studies in the BaseSpace Correlation Engine analysis using mRNA and miRNA-targeted mRNA expressions in COVID-19 in the RNA-seq derivation and validation cohorts are shown in Tables S6, S7, S8, and S9.
Figure 5.

BaseSpace Correlation Engine analysis of mRNA and miRNA-targeted mRNA genes expressions in the RNA-seq validation cohort compared with the healthy control subjects
The patterns of changes in genes expressions in the patients with COVID-19 in this study ((A) mRNA gene expressions and (B) miRNA-targeted mRNA gene expressions) are compared with the patients with melioidosis. Venn diagrams illustrate the overlap in genome-wide changes in gene expression between the patients with COVID-19 and those with melioidosis. Bar graphs depict the –log of the overlap p values for upregulated (red arrows) or downregulated (green arrows) genes.
RNA expressions of upstream regulators by real-time PCR for validation
qRT-PCR analysis of three selected upstream regulators (STAT1, STAT3, and IL1B) was performed to validate RNA-seq results. Gene expression between RNA-seq and qRT-PCR showed similarity in each of the three upstream regulators (Figure S6A). RNA expressions calculated via RNA-seq were significantly positively correlated to expressions determined via qRT-PCR (Figure S6B): STAT1, R2 = 0.69, p < 0.0001; STAT3, R2 = 0.52, p < 0.0001; IL1B, R2 = 0.52, p < 0.0001).
Enzyme-linked immunosorbent assays
Because the IFN pathway was potently activated in CPA using mRNAs as well as that using miRNA-targeted mRNA expressions, the IFN-related proteins were evaluated to assess their role in the critically ill patients with COVID-19. Compared with those in the HCs, plasma IFN-β levels were significantly higher in the critical and non-critical COVID-19 groups on day 1, but they did not change with the presence of mechanical ventilation. Plasma interleukin (IL)-27 levels were significantly elevated in the critical COVID-19 group compared with those in the non-critical COVID-19 group and the HCs. Significantly increased plasma IFN-λ1 levels were seen in the non-critical COVID-19 group in comparison with those in the HCs which were decreased in the critical COVID-19 group.
Plasma IFN-α2 and IFN-γ levels were not significantly different between the three groups (Figure 6A). In the critical COVID-19 group, when compared over time between day 1 and days 6–8, there was a trend toward lower levels of IFN-α2, IFN-β, IFN-γ, IL-27, and IFN-λ1 in the patients who died, but no significant differences were found (Figure 6B).
Figure 6.

Changes in the interferon protein levels
The interferons were transformed to common logarithm values to normalize the data distribution. All data are expressed as the mean ± SE. (A) IFNα/β, IFN-γ, IL-27, and IFN-λ1 on day 1 in each group. Asterisks indicate a statistically significant difference between the control, no mechanical ventilation, and mechanical ventilation groups. (B) The interferon levels in survivors and non-survivors on each day in the patients on mechanical ventilation due to COVID-19. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. SE, standard error; IFN, interferon; IL, interleukin.
Discussion
Pathogen-associated molecular patterns of the COVID-19 virus components, such as genomic RNA and damage-associated molecular patterns released by damaged cells, are recognized by pattern recognition receptors, including Toll-like receptors.8,9 Subsequently stimulated cell signaling activates transcription factors, such as nuclear factor κB, which bind to DNA. Translated RNAs promote transcription of proteins, such as inflammatory cytokines, which leads to systemic inflammation.10
Several articles have shown whole-blood transcriptome analyses of COVID-19. In the Gill et al. study, transcriptome analysis using blood buffy coat cells showed an enhanced IFN signaling pathway in the COVID-19-positive patients compared with COVID-19-negative patients.11 Arunachalam et al. showed IFN response-enriched clusters of T cells and monocytes in patients with moderate or severe COVID-19 (no ICU stay) compared with control subjects in their single RNA-seq analysis with peripheral blood mononuclear cells.12 The whole-blood RNA-seq analysis in our study showed an activated IFN signaling pathway in the critically ill patients with COVID-19 versus the HCs (Figure 3A). Upstream regulator analysis in IPA in the validation cohort showed IFN signal-associated mRNA expressions of IFN-γ, IFN-α, IFN-α2, IFN- λ1, IRF7, STAT1, and STAT3 (Figure 3E), suggesting that activation of IFN signaling is important in the pathogenesis of COVID-19. Interestingly, the PD-1, the PD-L1 cancer immunotherapy pathway, which is responsible for T cell activation, was activated, and T cell activation-related pathways were downregulated (Figure 3A), indicating that PD-1 signaling is one cause of the inhibition of T cell activation.
miRNA transcription is processed in the nucleus in stimulated cells and in mRNA, and miRNA expressions change in the blood cells of patients with COVID-19.13,14 Li et al. reported that 35 miRNA expressions were upregulated and 38 miRNA expressions were downregulated in patients with mild-to-moderate COVID-19 compared with HCs.14 In this study, we first performed comprehensive analysis of miRNAs in critically ill COVID-19 patients. In the RNA-seq validation study, 15 miRNA expressions were significantly increased and 17 were significantly decreased in the critically ill patients with COVID-19 compared with the HCs. Only a few significantly upregulated and downregulated miRNA expressions were common between the Li et al. study and our study, thus suggesting that differentially expressed miRNA expression may be dependent on the severity of COVID-19.13
MiRNAs are reported to function through transcription of interfering mRNA.5 In this study, we first evaluated miRNAs and performed mRNA-miRNA integration analysis in critically ill COVID-19 patients. In our RNA-seq validation study, CPA enriched by the miRNA-targeted mRNA showed activated pro-inflammation-related pathways, including the IFN signaling pathway (Figure 3A), suggesting that miRNA could contribute to the activated IFN signaling pathway through the regulation of mRNA expressions in critically ill COVID-19 patients. This is supported by the Zheng et al. study in which enrichment analysis using miRNA-targeted mRNA showed a downregulated type I IFN response during recovery from severe COVID-19.13
The IFN signaling pathway has been shown to play a pivotal role in the immune response against COVID-19.15, 16, 17, 18 IFN signalings, which are involved in both innate and acquired immunity, are classified into three groups. Type I, type II, and type III are associated with IFN-α/β, IFN-γ, and IFN-λ, respectively. Type I and type III IFNs are produced in the early phase of infection and contribute to viral growth and suppression.19 Natural killer and T cells mainly secrete IFN-γ, and it is not secreted by virus-infected cells. IFN-γ promotes an antiviral immune response in an indirect manner. In the RNA-seq validation study, CPA and the RNA expressions involved in the IFN signaling pathway (Figures 3B and 4) showed activated type I and type II signals in the whole-blood cells. Also, upstream regulator analysis showed type I (e.g., IFN-α, IFN-α2)-, type II (e.g., IFN-γ)-, and type III (e.g., IFN-λ1)-related RNA expressions (Figure 3E). Following the results of the activated IFN signal through mRNA-miRNA integration analysis, plasma IFN-related proteins profiles were evaluated. IFN-β protein levels (type I) increased in the critically ill patients with COVID-19 compared with those in the non-critically ill patients with COVID-19 and HCs. IL-27 protein levels, which are promoted by type I and type II signalings,20 showed significant increases in the critically ill patients with COVID-19 in comparison with HCs. This suggests that the type I IFN signal was activated via ligands, such as IFN-β protein, and subsequently promoted IL-27 proteins played a role in the pathogenesis of COVID-19.
The relation between dysregulation of IFN signaling and the disease progression of COVID-19 has been reported. Galani et al. showed lower IFN-α and IFN-λ1 protein levels in the serum of patients with moderate-to-severe COVID-19 than those in patients with flu,15 suggesting the functional dysregulation of type I and type III IFNs in COVID-19. In addition, Hadjadj et al. showed decreased IFN-associated mRNA expressions in whole-blood cells and a decrease in type I IFNs, such as IFN-α2, in the plasma of the patients with disease progression.21 In this study, significantly increased IFN-λ1 protein levels (type III) were present in the non-critically ill patients with COVID-19 compared with the HCs but were significantly decreased in the critically ill patients with COVID-19. This indicates that a decrease in IFN-λ1 protein levels might result in a lack of protection against the COVID-19 virus, thus causing disease progression.
During the period in which this study was conducted, SARS-CoV2 clade B was mainly prevalent, with a bias toward viral strain types. In addition, the clade was not identified in all cases. Therefore, this study alone has limitations in showing the association between viral strains and IFN.
In conclusion, in this study, we first evaluated mRNA and miRNA expressions in whole-blood cells in critically ill patients with COVID-19. Integration of mRNA and miRNA analysis showed activation of IFN signaling. The IFN protein profile in plasma revealed that IFN-β (type I) and IFN-λ1 (type III) played an important role in the disease progression of COVID-19.
Materials and methods
Study design and participants
This prospective observational multicenter study was conducted at Osaka University Graduate School of Medicine, Osaka Prefectural Nakakawachi Emergency and Critical Care Center, and Local Independent Administrative Agency Osaka Prefectural Hospital Organization Osaka General Medical Center. All patients were diagnosed as having COVID-19 using SARS-CoV-2 RT-PCR testing and as having pneumonia by chest computed tomography scan.
To elucidate the pathogenesis of COVID-19, mRNA and miRNA in whole blood of these patients were analyzed. In the validation cohort study, an adequate sample size to perform multiple statistical tests was provided, and different sequencing platforms from those of the derivation cohort were used to confirm reproducibility. Patients admitted to the ICU of Osaka University Graduate School of Medicine between August and November 2020 were assigned to the RNA-seq derivation cohort and those between July 2020 and February 2021 were assigned to the RNA-seq validation cohort. The IFN protein profile cohort included patients admitted to the ICUs of Osaka University Graduate School of Medicine and Osaka Prefectural Nakakawachi Emergency and Critical Care Center between August 2020 and April 2021 and to Local Independent Administrative Agency Osaka Prefectural Hospital Organization Osaka General Medical Center between April and June 2021. HCs were also enrolled after providing written informed consent.
Clinical data
Clinical data were collected from the patients’ electronic medical records by the investigators included age, sex, BMI, Acute Physiology and Chronic Health Evaluation II and Sequential Organ Failure Assessment scores, comorbid conditions (hypertension, diabetes, and hyperlipidemia), and clinical variables (day of extubation, day of weaning off mechanical ventilation, and hospital outcome). The SARS-CoV2 clades were based on Nextstrain.22
Disease severity was defined by a modified ordinal scale: 1 = death; 2 = intubated/ventilated, survived; 3 = hospitalized, O2 required, survived; 4 = hospitalized, no O2 required, survived; and 5 = discharged/not hospitalized, survived.23 Patients were respectively divided into two groups based on their use of mechanical ventilation at sample collection: critically ill patients with COVID-19 (intubated on admission) and non-critically ill patients with COVID-19 (not intubated on admission).
Identification of mRNA/miRNA expressions
Total RNA isolation of leukocytes using a PAXgene Blood RNA System (BD Bioscience, San Jose, CA) from 10 to 31 patients with COVID-19 on day 1 (admission day) and from 5 and 16 HCs was performed in the RNA-seq derivation and validation cohorts, respectively. All blood samples for the analyses were collected in collection tubes and stored at −30°C until further analysis. Library preparation was performed using a TruSeq stranded mRNA sample prep kit (Illumina, San Diego, CA) according to manufacturer’s instructions.
Library preparation and RNA sequencing
Total RNA isolation of leukocytes from patients with COVID-19 and HCs was performed using a PAXgene Blood RNA System (BD Bioscience, San Jose, CA). In the derivation cohort, library preparation was performed using a TruSeq stranded mRNA sample prep kit (Illumina) according to the manufacturer’s instructions. Sequencing was performed on an Illumina NovaSeq 6000 platform in 101-bp paired-end mode. In the validation cohort, full-length cDNA was generated using a SMART-seq HT Kit (Takara Bio, Mountain View, CA) according to the manufacturer’s instructions. An Illumina library was prepared using a Nextera DNA Library Preparation Kit (Illumina) according to SMARTer kit instructions. DNA libraries were converted to libraries for DNBSEQ using an MGIEasy Universal Library Conversion Kit (App-A). Sequencing was performed on a DNBSEQ-G400RS platform in 2 × 100-bp paired-end mode.
RNA-seq analysis
The sequenced reads were mapped to the human reference genome sequences (hg19) using TopHat, version 2.0.13, in combination with Bowtie2, version 2.2.3, and SAMtools, version 0.1.19. The fragments per kilobase of exons per million mapped (FPKM) fragments were calculated using Cufflinks, version 2.2.1. Gene-level expression raw read counts were calculated using featureCounts, and relative RNA expression levels were calculated using the DESeq algorithm.
miRNA-seq and miRNA-seq analysis
Small RNA libraries were constructed following the manufacturer’s instructions using the NEBNext Small RNA Library Prep Set for Illumina (New England Biolabs, Ipswich, MA) and sequenced using the HiSeq 2500 platform (Illumina) in 75-bp single-end reads in the derivation cohorts and by the NovaSeq 6000 platform (Illumina) in 101-bp single-end reads in the validation cohorts. miRNA-seq analysis was conducted using Strand NGS, version 3.0, software (Strand Life Sciences, Bengaluru, India) according to the small RNA alignment and small RNA analysis pipeline using default parameters. Before analysis of the small RNA-seq data, reads were trimmed by the adapter sequences and mapped to the human hg19 reference genome. Relatively small RNA expression levels were calculated using the DESeq algorithm.
Statistical analysis of mRNA and miRNA
Multidimensional scaling with the cmdscale command in R24 was performed using log2-normalized mRNA FPKM values and normalized miRNA values to compare expressions between HCs and patients with COVID-19. Volcano plot analysis was conducted to visualize and identify the significant changes in the expression lists. Significance was defined by a |log2 fold change| > 1 and p < 0.05 in the RNA-seq derivation cohort and a |log2 fold change| > 0.6 and FDR < 0.1 the RNA-seq validation cohort. Significantly different mRNA and miRNA expressions were used for further analysis. The upregulated or downregulated miRNAs that were common to both the RNA-seq derivation and RNA-seq validation cohorts were analyzed using ROC curves to determine whether day 1 miRNAs in the RNA-seq validation cohort were associated with prognosis. As there were few deaths, early recovery and late recovery were used as clinical outcomes. We defined early recovery, as in previous studies,25 as the clinical outcome of patients who were treated with invasive mechanical ventilation (IMV) for 12 days or less or who did not receive treatment with IMV. Non-survivors were defined as late recovery. The AUC, accuracy, sensitivity, and specificity were also measured.
To evaluate functional characteristics and upstream regulators of the RNA expressions, the data were analyzed by CPA, canonical signaling pathway analysis, and upstream regulator analysis with calculation of Z scores and p values in IPA (QIAGEN, https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/).26 The Z score predicts the activation state of the upstream regulator by using RNA expression patterns of the downstream state of that regulator. The canonical pathway and upstream regulator were considered activated if the |Z score| was >2 with p < 0.05. To describe specific relations between RNAs, the canonical signaling pathway analysis was performed using significantly different mRNA and miRNA expressions. Correlation of the current RNA-seq to the GEO database was by BaseSpace Correlation Engine (Illumina).
Statistical analysis of integration of mRNA and miRNA analysis
We used Ingenuity’s miRNA Target Filter in silico analysis to identify predicted miRNA and target mRNA interactions. The predicted target mRNA expressions of the miRNAs were obtained from IPA (QIAGEN, https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/features/microrna-target-filter/), which uses TargetScan, miRecords, and TarBase as the databases for miRNA-targeted mRNA expressions. Only significantly up- or downregulated mRNAs were included in the analysis. Subsequently, CPA were conducted using the target mRNA expressions.
GSEA
To confirm reproducibility of the IPA, sensitivity analysis was conducted using GSEA. The expression profiles of mRNAs or miRNA-targeted mRNAs that were significantly changed in the COVID-19 patients compared with HCs (|log2 fold change| >1 and FDR < 0.1) were used to conduct the GSEA. compareCluster functions from the R package ClusterProfiler27 (v3.12.0) were used to determine significant enrichment (q < 0.05) of biological processes. Furthermore, unsupervised hierarchical clustering analysis was performed using mRNAs under the Gene Oncology terms “defense response to virus” and “response to virus.” The clustering and visualization of the heatmap were conducted with Python (v3.9.7) using the Seaborn plotting module28 (v0.11.2).
Analysis of qRT-PCR for validation of upregulated RNA expressions
qRT-PCR analysis on three select upregulated RNAs (STAT1, STAT3, and IL1B) was performed for technical validation. Total RNA samples from 14 patients with COVID-19 and 10 HCs in the RNA-seq derivation and validation cohorts were used. qRT-PCR analysis was performed as described previously29 with some modifications. In brief, total RNA were reverse transcribed to cDNA using a ReverTraAce qPCR RT Master Mix (Toyobo, Osaka, Japan) in accordance with the manufacturer’s protocol. qRT-PCR was performed with THUNDERBIRD SYBR qPCR Mix (Toyobo) on a Step One Plus real-time PCR cycler (Applied Biosystems). Each of the specific primers used is summarized in Table S10. The levels of mRNA expression are expressed relative to the GAPDH levels.
Analysis of IFN-related cytokines
Blood samples were collected from the critically and non-critically ill patients with COVID-19 on day 1, from those critically ill with COVID-19 on days 6–8 (two time points per patient), and from the HCs. Enzyme-linked immunosorbent assay (ELISA, R&D Systems, Minneapolis, MN) was performed to measure plasma levels of IFN-α2, IFN-β, IFN-γ, IL-27, and IFN-λ1. Frozen plasma samples were thawed, and subsequent measurements were conducted according to the manufacturer’s protocol. Absorbance was analyzed using a microplate reader (SH-9000Lab; Corona Electric, Ibaraki, Japan). The minimum detectable levels in pg/mL were: IFN-α2, <3.1; IFN-β, <7.8; IFN-γ, <9.4; IL-27, <156; and IFN-λ1, <62.5.
Continuous values are shown as median and interquartile range, and categorical variables are shown using frequencies and proportions. Comparisons were performed using the nonparametric Mann-Whitney test as appropriate. No imputation was made for missing data. p < 0.05 was considered to indicate statistical significance. The data were analyzed using R version 4.0.2 (R Foundation for Statistical Computing, Vienna, Austria) and JMP Pro version 16.0.0 (SAS Institute, Cary, NC) and are presented using GraphPad Prism, version 8.4.3 (GraphPad Software, LLC, La Jolla, CA).
Data availability
The raw data concerning this study were submitted under Gene Expression Omnibus accession numbers GSE182152 and GSE179850 for future access.
Acknowledgments
We greatly appreciate the patients, families, and healthy volunteers involved in this study. Also, we would like to thank all of the medical staff who cooperated with this study. This study was supported by the Takeda Science Foundation and the Japan Agency for Medical Research and Development (grant no. 20fk0108404h0001). The graphical abstract was created at BioRender.com. This study followed the principles of the Declaration of Helsinki and was approved by the institutional review board of Osaka University Hospital (permit nos. 16109 and 885 [Osaka University Critical Care Consortium Novel Omix Project; Occonomix Project]). Informed consent was obtained from the patients or their relatives and the healthy volunteers for the collection of all blood samples.
Author contributions
Y.T. conceived and designed this study, contributed to acquisition, analysis, and interpretation of the data, and was responsible for drafting, editing, and submission of the manuscript. H. Matsumoto was involved in the conceptualization of this study and contributed significantly to the data analysis, interpretation of the results, and manuscript preparation. J.Y., T.M., T.E., H. Matsuura, Y.M., T.K., and H.H. contributed to acquiring the data. F.S., M.I., and D.O. helped analyze the data. H.O. conducted the literature review. All authors have read and understood the journal’s policies and believe that neither the manuscript nor the study violates any of these. All authors meet the authorship criteria detailed in the submission guidelines, and all authors agree with the content of the manuscript.
Declaration of interests
All authors of this manuscript have no conflicts of interest to disclose.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.07.005.
Supplemental information
References
- 1.WHO coronavirus (COVID-19) dashboard; 2020. https://covid19.who.int.
- 2.CDC . Centers for Disease Control and Prevention; 2020. Coronavirus Disease 2019 (COVID-19)https://www.cdc.gov/coronavirus/2019-ncov/index.html [Google Scholar]
- 3.Rahman S., Montero M.T.V., Rowe K., Kirton R., Kunik F. Epidemiology, pathogenesis, clinical presentations, diagnosis and treatment of COVID-19: a review of current evidence. Expet Rev. Clin. Pharmacol. 2021;14:601–621. doi: 10.1080/17512433.2021.1902303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abu-Izneid T., AlHajri N., Ibrahim A.M., Javed M.N., Salem K.M., Pottoo F.H., Kamal M.A. Micro-RNAs in the regulation of immune response against SARS CoV-2 and other viral infections. J. Adv. Res. 2021;30:133–145. doi: 10.1016/j.jare.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonas S., Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 6.Mehta A., Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016;16:279–294. doi: 10.1038/nri.2016.40. [DOI] [PubMed] [Google Scholar]
- 7.Scicluna B.P., Uhel F., van Vught L.A., Wiewel M.A., Hoogendijk A.J., Baessman I., Franitza M., Nürnberg P., Horn J., Cremer O.L., et al. The leukocyte non-coding RNA landscape in critically ill patients with sepsis. Elife. 2020;9:e58597. doi: 10.7554/eLife.58597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng M., Karki R., Williams E.P., Yang D., Fitzpatrick E., Vogel P., Jonsson C.B., Kanneganti T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021;22:829–838. doi: 10.1038/s41590-021-00937-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Totura A.L., Whitmore A., Agnihothram S., Schäfer A., Katze M.G., Heise M.T., Baric R.S. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. 2015;6 doi: 10.1128/mBio.00638-15. e00638–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Z., Wei Y., Tao C. An enlightening role for cytokine storm in coronavirus infection. Clin. Immunol. 2021;222:108615. doi: 10.1016/j.clim.2020.108615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill S.E., dos Santos C.C., O’Gorman D.B., Carter D.E., Patterson E.K., Slessarev M., Martin C., Daley M., Miller M.R., Cepinskas G., et al. Transcriptional profiling of leukocytes in critically ill COVID19 patients: implications for interferon response and coagulation. Intensive Care Med. Exp. 2020;8:75. doi: 10.1186/s40635-020-00361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O., et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng H.Y., Xu M., Yang C.X., Tian R.R., Zhang M., Li J.J., Wang X.C., Ding Z.L., Li G.-M., Li X.L., et al. Longitudinal transcriptome analyses show robust T cell immunity during recovery from COVID-19. Signal Transduct. Target. Ther. 2020;5:294. doi: 10.1038/s41392-020-00457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C., Hu X., Li L., Li J.H. Differential microRNA expression in the peripheral blood from human patients with COVID-19. J. Clin. Lab. Anal. 2020;34:e23590. doi: 10.1002/jcla.23590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galani I.E., Rovina N., Lampropoulou V., Triantafyllia V., Manioudaki M., Pavlos E., Koukaki E., Fragkou P.C., Panou V., Rapti V., et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021;22:32–40. doi: 10.1038/s41590-020-00840-x. [DOI] [PubMed] [Google Scholar]
- 16.Gadotti A.C., de Castro Deus M., Telles J.P., Wind R., Goes M., Garcia Charello Ossoski R., de Padua A.M., de Noronha L., Moreno-Amaral A., Baena C.P., Tuon F.F. IFN-γ is an independent risk factor associated with mortality in patients with moderate and severe COVID-19 infection. Virus Res. 2020;289:198171. doi: 10.1016/j.virusres.2020.198171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J.S., Shin E.C. The type I interferon response in COVID-19: implications for treatment. Nat. Rev. Immunol. 2020;20:585–586. doi: 10.1038/s41577-020-00429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim Y.M., Shin E.C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021;53:750–760. doi: 10.1038/s12276-021-00592-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crosse K.M., Monson E.A., Beard M.R., Helbig K.J. Interferon-stimulated genes as enhancers of antiviral innate immune signaling. J. Innate Immun. 2018;10:85–93. doi: 10.1159/000484258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida H., Hunter C.A. The immunobiology of interleukin-27. Annu. Rev. Immunol. 2015;33:417–443. doi: 10.1146/annurev-immunol-032414-112134. [DOI] [PubMed] [Google Scholar]
- 21.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nextstrain Genomic epidemiology of SARS-CoV-2 with subsampling focused globally since pandemic start; 2022. https://nextstrain.org/ncov/gisaid/global/all-time
- 23.World Health Organization COVID-19 therapeutic trial synopsis. 2020. https://www.who.int/publications-detail-redirect/covid-19-therapeutic-trial-synopsis
- 24.The R.F. The R project for statistical computing. 1998. https://www.r-project.org/
- 25.Ebihara T., Matsumoto H., Matsubara T., Togami Y., Nakao S., Matsuura H., Kojima T., Sugihara F., Okuzaki D., Hirata H., et al. Cytokine elevation in severe COVID-19 from longitudinal proteomics analysis: comparison with sepsis. Front. Immunol. 2021;12:798338. doi: 10.3389/fimmu.2021.798338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krämer A., Green J., Pollard J., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waskom M., Botvinnik O., O’Kane D., Hobson P., Lukauskas S., Gemperline D.C., Augspurger T., Halchenko Y., Cole J.B., Warmenhoven J., et al. mwaskom/seaborn: v0.8.1 (September 2017) Zenodo. 2017 [Google Scholar]
- 29.Matsuura H., Matsumoto H., Okuzaki D., Shimizu K., Ogura H., Ebihara T., Matsubara T., Hirano S.I., Shimazu T. Hydrogen gas therapy attenuates inflammatory pathway signaling in septic mice. J. Surg. Res. 2021;263:63–70. doi: 10.1016/j.jss.2021.01.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data concerning this study were submitted under Gene Expression Omnibus accession numbers GSE182152 and GSE179850 for future access.