

Abstract
Complex intercellular interactions as well as biomolecular and biomechanical cues from the extracellular matrix (ECM) profoundly affect cellular functions. Traditional transcriptomic and proteomic approaches have provided insight into disease progression by identifying discrete cellular subpopulations or microenvironmental signatures characteristic of normal or pathological tissues, however these techniques do not examine how a given cellular state relates to its interactions with neighboring cells or its surrounding ECM with multiparametric characterization (i.e. ECM alignment, mechanical forces, crosslinking, etc.). Emerging spatial-omic techniques can provide high-resolution mapping of expression profiles similar to scRNA-seq and mass spectroscopy directly within tissues. The ability to preserve the spatial context of cells within samples, their cellular geometry, as well as their surrounding ECM gives spatial-omics the opportunity to interrogate previously unexplored signaling modalities, which has the potential to revolutionize ECM research and our understanding of fibrotic diseases. In this review, we present current spatial transcriptomic and proteomic techniques and discuss how they may be applied to investigate cell-ECM interactions.
Keywords: Spatial transcriptomics, spatial proteomics, matrisome, fibrosis, multiplex imaging
Introduction:
High-throughput or –omic approaches developed over the past decades have expanded studies of individual genes or proteins into the unbiased realm of transcriptomics or proteomics. This has dramatically transformed biomedical research [1] and considerably advanced our understanding of ECM biology and pathophysiology [2–4]. To list a few examples, single-cell RNA sequencing (sc-RNAseq) alone has permitted the classification of fibroblast subpopulations in human skin [5]; the identification of an alveolar macrophage population expressing pro-fibrotic genes in idiopathic pulmonary fibrosis (IPF) patients [6]; the distinction of central vein-associated hepatic stellate cells as the dominant contributor to pathogenic collagen production in a mouse model of centrilobular fibrosis [7]; and the finding that after injury in the murine lung the number of activated fibroblasts, determined by multiple genes, increases despite the total fibroblast population size remaining constant [8]. Furthermore, microarray analyses have demonstrated the alteration and enrichment of developmental pathways in IPF lungs [9]; mass-spectrometry-based proteomics can now profile the ECM composition of normal and fibrotic samples in an unbiased manner and identify signatures distinguishing normal vs diseased states [2, 10–13]; and global gene expression studies have provided insight into the active role of matrix metalloproteases (MMPs) in pulmonary fibrosis [14, 15]. Additionally, and of interest to researchers primarily focused on fibroblasts, sc-RNAseq can be harnessed to identify cell populations with no reliable surface marker [16, 17]. However, techniques capable of achieving truly -omic level information such as bulk RNA sequencing (RNAseq), single-cell RNASeq (sc-RNAseq), or mass spectrometry (MS) require the dissociation of cells from their native context, which results in the loss of any spatial information and other significant cellular information, including but not limited to cell shape, cell-cell, and cell-ECM interactions. Additionally, bulk –omic techniques mask tissue, cellular and sub-cellular heterogeneity by averaging gene expression or protein abundance across whole samples [18]. And yet, these pieces of information are imperative to fully understand biological mechanisms such as signal transduction cascades, differentiation states, and interactions of multiple cell types (e.g. immune-stromal).
Preserving tissue integrity using classic in-situ hybridization (ISH) has demonstrated the importance of transcript localization by facilitating discoveries such as the contribution of RNA gradients on cell polarization during embryogenesis, which ultimately dictates cell differentiation at different developmental stages [19]. Classic microscopy techniques provide spatial resolution for investigating known pathways such as epithelial-to-mesenchymal transition in fibrosis [20], but spectral overlap limits depth for the number of protein/RNA targets that can be simultaneously detected. Previous methods to circumvent this limitation involve overlapping serial sections comprising different markers. However, these methods assume that tissues are homogeneous in the z-direction across the stained sections, which is known to be untrue.
Spatial-omic techniques are now offering a new perspective. By quantify dozens to hundreds of genes, transcripts, or proteins, spatial-omics enable the collection of valuable molecular, cellular and microenvironmental information in the context of native tissue or cellular structures. In this review, we provide an overview of currently available methods for spatial transcriptomics and proteomics and further describe recent studies that have applied such methods to advance our understanding of ECM and fibroblast biology. Given our expertise in pulmonary fibrosis, many of the examples provided within this review will be in the context of the lung, but we will expand to other organ systems where the literature has expanded to demonstrate the relevance of these powerful techniques to ECM research. We propose that these methods can be used to investigate phenotypic traits in the context of cellular heterogeneity, microenvironmental, cellular and sub-cellular dynamics, and eventually help us decipher the fundamental bases of pathologies. We invite our readers to refer to the following reviews for in-depth technical descriptions of spatial transcriptomic and proteomic technics [21–26].
Spatial transcriptomics or RNA imaging:
Recent advances in techniques and technology have enabled the in-situ visualization of RNA transcripts in tissue sections. For example, multiplex transcript technology such as RNAscope has enabled the visualization of up to 12 mRNA molecules commercially helping both clinicians and researchers characterize cells, similarly to the way immunohistochemistry identifies the relative abundance and spatial distribution for protein, even when the mRNA is lowly expressed [27]. Spatial transcriptomic approaches expand multiplex imaging to 100–1000s of transcripts and can be distinguished by whether they measure pre-determined targets or gather global expression data. In this section, we will review current techniques with respect to these categories while providing insight on each technique’s advantages and limitations (Table 1).
Table 1:
Overview of spatial transcriptomic techniques discussed
| Method | Number of Targets | Advantages | Limitations |
|---|---|---|---|
| Single Molecule Fluorescent in situ Hybridization (smFISH) [28] | See derivatives of technique (MERFISH and seqFISH) | Absolute quantification (100% sensitivity); High signal:noise; High sensitivity to image RNA in low abundance | Target must be selected, biasing the results |
| Multiplexed Error Robust FISH (MERFISH) [33–35, 37] | ~10,000 | Detects and corrects errors; Same as smFISH | Requires transcripts longer than 3kb; Special equipment |
| Sequential FISH (seqFISH) [31, 32] | 10,000+ | Single cell resolution; Reduces the number of necessary multiplexing cycles; Same as smFISH | Decreased detection sensitivity with each cycle; Time consuming protocol |
| In-situ sequencing (ISS) [44] | 100+ | Heighten target binding specificity; Can detect Small Nuclear Polymorphisms (SNPs) | Padlock probes are costly; Time consuming protocol |
| Proximity Ligation in situ Hybridization (PLISH) [38] | Unlimited targeted detection 4 transcripts/cycle | High signal to noise; Cost effective; Detects low abundant RNAs | Laborious protocol; Time consuming protocol |
| Spatially-resolved Transcript Amplicon Readout Mapping (STARmap) [45] | 1,000 transcripts/6 imaging cycles | 1,000 genes in only six imaging cycles | Padlock probes are costly |
| Spatial Transcriptomics [50] | Entire transcriptome/non-targeted | Similar protocol as RNA-seq; High transcript count | Lower sensitivity for low abundant transcripts; Not single cell resolution (~100um); Special slide needed; Sequencing data analysis |
| Slide-Seq [52] | Entire transcriptome/non-targeted | Single cell resolution (10um) | Lower sensitivity for low abundant transcripts; Sequencing data analysis |
| Fluorescent in situ RNA Sequencing (FISSEQ) [47] | Entire transcriptome/non-targeted | Unbiased sequencing; Maintains sample integrity | Low target detection; Very time consuming protocol |
Imaging of pre-determined mRNA targets:
The first instance of imaging individual RNA species can be ascribed to single molecule fluorescent in-situ hybridization (smFISH), which provides absolute quantification of the copy number and localization of RNA molecules within cells. In smFISH, a series of fluorescently-conjugated oligonucleotides bind multiple complimentary sites of the same mRNA target to amplify the signal for visualization [28]. Not only can this approach detect nearly all transcripts present, effectively having close to 100% sensitivity, but smFISH also enhances specificity by increasing signal to noise ratio through the requirement of multiple successful binding events (~24 probes/target) to be detected by diffraction-limited fluorescent microscopy [21, 26]. smFISH is beneficial for the imaging of lowly abundant RNAs which require highly sensitive molecular tools to properly quantify their expression and determine their intracellular organization. In a recent study, Dobie et al. used smFISH on fibrotic liver samples and identified TREM2+CD9+ scar-associated macrophages in collagen-positive scar regions [29].
Originally smFISH was limited by the number of available fluorescent channels, but resourceful manipulations, such as combinatorial binding where pre-annotated color patterns are assigned to single targets [30], or temporal barcoding (also known as sequential FISH or seqFISH) where typically three targets are imaged per cycle allowing for the detection of F^n targets (F= fluorophores, n=hybridization cycles) [31], can be used to multiplex the system. Here, we are defining barcodes as a unique sequence (usually in the form of a synthetic DNA oligonucleotide) that is assigned to a specific cell, protein, or transcript of interest when performing multiplex imaging to identify that entity. Barcodes are essential for multiplexing the system in some spatial-omic techniques and therefore are widely deployed in the field of spatial-omics. Different forms of barcodes will be discussed throughout this review as well as visual examples for cells (Fig. 1) and proteins (Fig. 2B). For more depictions of the variations of spatial transcriptomic barcodes we recommend the following reviews [25, 26].
Figure 1: Slide-seq multiplex imaging workflow.
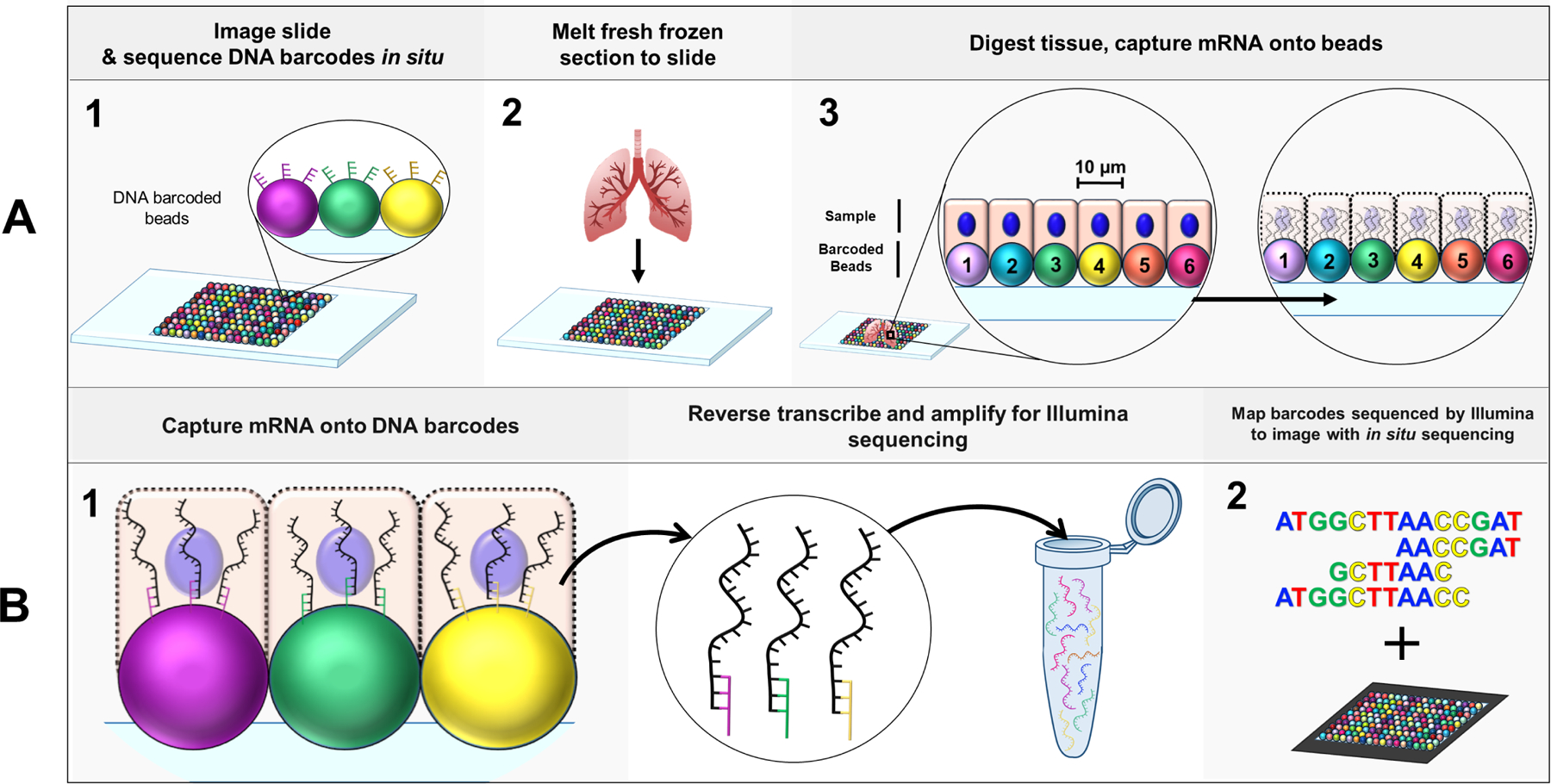
(A) Schematic of initial Slide-seq steps (A1) A rubber coated coverslip is covered in a monolayer of 10µm micro-particles (“beads”) to create a “puck”. Each bead is conjugated to its own unique barcode whose sequence is determined in situ via SOLiD (Sequencing by Oligonucleotide Ligation and Detection) for high resolution localization of each bead on the puck. (A2) Fresh frozen tissue can then be melted onto the puck, (A3) where the cells (approximately one cell/bead) overlap with the DNA barcoded beads. The tissue is digested and the mRNA of each cell is captured onto the DNA barcodes residing directly beneath the respective cell. (B) Schematic of library preparation for Slide-seq. (B1) mRNA are reverse transcribed to incorporate in a 3’-end, barcoded RNA-seq library preparation. Products are then amplified and undergo Illumina sequencing. (B2) Transcript profiles associated with a barcode sequence obtained from Illumina sequencing are mapped to their specific tissue location by matching the Illumina data to the barcode sequence from the initial in situ sequencing step in A1 [52].
Figure 2: Co-Detection by indEXing (CODEX) multiplex imaging workflow.
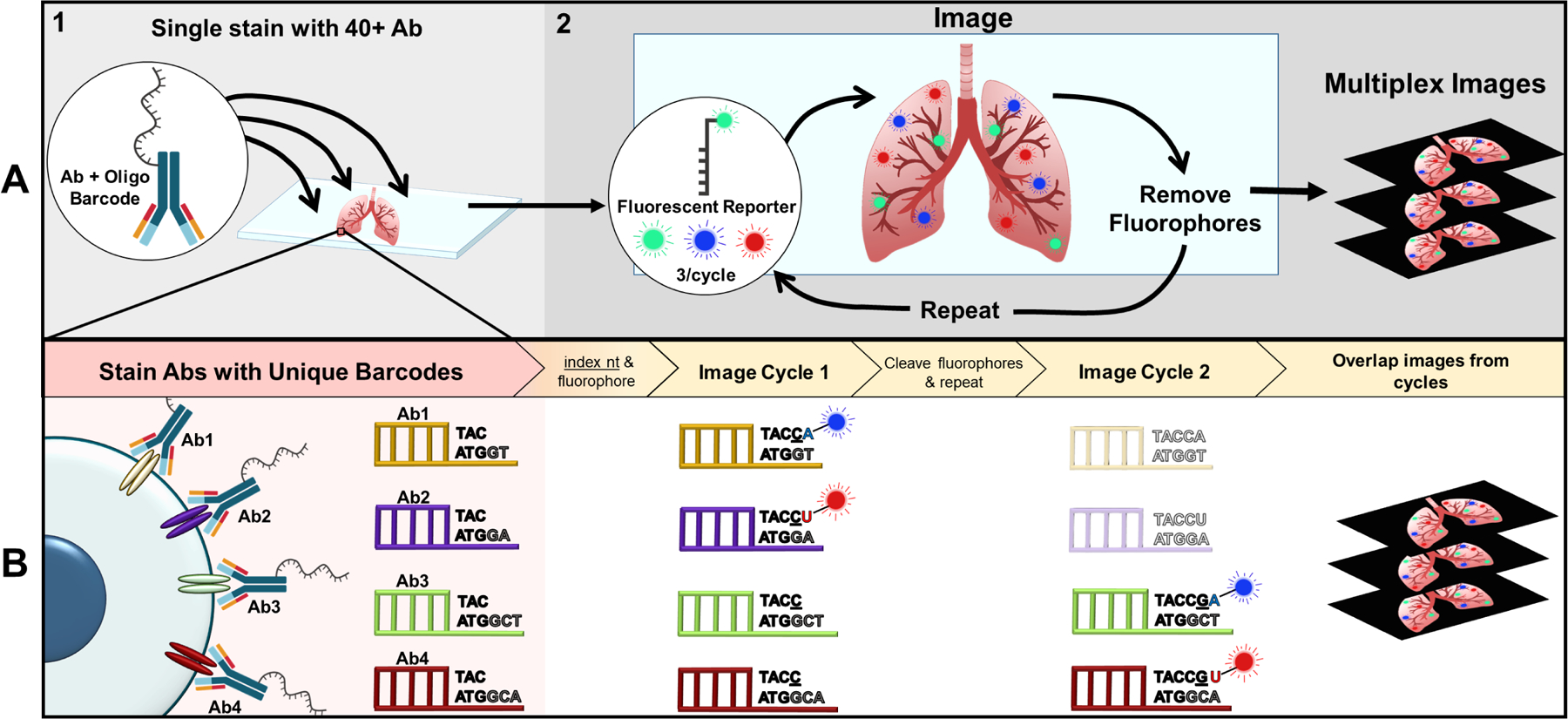
(A) Top left moving right depicting a macro-view of CODEX workflow. (A1) Tissue section (either FFPE or fresh frozen) is simultaneously stained with the entire panel of antibodies tagged with their own unique oligonucleotide barcode by the user. Of note, the commercial system currently provides ~40 barcodes that can be conjugated to any antibody of interest. (A2) The stained section then undergoes several imaging cycles automated within the fluidics device where three fluorophores are imaged and subsequently removed via TCEP cleavage each cycle (14 cycles for 40 antigens). Images are then overlapped to create a ~40 protein multiplexed image from a single tissue section. (B) Bottom depicting cellular-level view of CODEX workflow with two fluorophores (for simplicity) and indexing technology. Each antibody is conjugated to a unique barcode with a 5’ overhang. The indexing nucleotide (denoted by underlining) and two nucleotides bound to fluorophores are added to the section. As shown in cycle 1, the indexing nucleotide is incorporating into all of the barcodes, while the fluorescent nucleotides are only able to bind to their respective pre-designated barcodes to image the antibodies denoted for that cycle. Fluorophores are removed to repeat the process. Barcodes for antibodies in earlier cycles will be shorter than later cycle oligo-tags. After the antigen of interest for that cycle is imaged, the oligonucleotide will no longer be indexed, as denoted by the lighter shading in cycle 2 [70].
Abbreviations: Antibody (Ab); nucleotide (nt); FFPE (Formalin Fixed Paraffin Embedded); Tris(2-carboxyethyl)phosphine hydrochloride (TCEP)
For example, a recent study conducted by Eng et al. expanded the capabilities of seqFISH (seqFISH+) to visualize 10,000+ genes within a single cell by altering the design of the barcodes. Their method vastly reduces the number of necessary multiplexing cycles by replacing standard 3–5 colored barcodes with 60 pseudocolored barcodes per image cycle, resulting in a total of 24,000 imaged genes after four cycles [32]. Combinatorial binding, however, increases the chance for misinterpreting signals due to hybridization error and temporal barcodes decrease detection sensitivity exponentially with each cycle [33]. By incorporating modified hamming codes (linear error-correcting codes previously used in telecommunication to automatically correct one bit errors) into the same workflow as temporal barcoded smFISH probes, multiplexed error robust FISH (MERFISH) can detect and correct errors while maintaining multiplexing to reduce RNA misidentification [23]. Xia et al. recently increased the gene throughput of MERFISH from the original ~1,000 genes [33] to ~10,000 transcripts [34]. This was achieved by applying expansion microscopy (ExM), a technique that expands a polymer to physically enlarge the biological structures and space out molecules within a cell, in combination with several other modifications to bring the field closer to genome-wide imaging (MERFISH and ExM were originally combined in this article [35]). A detailed description of the methods behind expansion microscopy can be found here [36]. Normal MERFISH requires transcripts longer than 3kb to accommodate the error-robust barcodes [33], but a study published in 2019 integrated branched DNA (bDNA) amplification into the MERFISH protocol to increase signal without altering fluorescent spot size, which will enhance shorter RNA imaging and increase imaging throughput [37]. Implementing additional amplification steps is an effective tool that has been previously incorporated into a number of other multiplexed FISH techniques [38–42] to increase signal and are described elsewhere [21, 26]. Amplification techniques must be applied carefully, however, as to avoid biasing the system with too many modifications, but the various derivatives of smFISH are advantageous for quantifying low abundant transcripts that would normally be undetected in methods such as scRNA-seq [43].
Rolling circle amplification (RCA) is used in another spatial transcriptomic technique known as in-situ sequencing (ISS). In ISS, single-stranded DNA oligonucleotides with 5’ and 3’ ends complimentary to the target sequence, also known as padlock probes, first bind cDNA libraries generated in situ from target RNAs [44]. The padlock probe hybridized to the cDNA forms a circle complex which undergoes RCA and sequencing by ligation to visualize over 100 transcripts per sample with single cell resolution [44]. ISS leverages padlock probes to heighten target binding specificity, allowing for the detection of non-coding and microRNA, but are expensive to produce and result in a laborious protocol. For this reason, Nagendran et al. developed a unique method known as proximity ligation in-situ hybridization (PLISH) to lower costs as well as increase scalability by replacing padlock probes with RNA-templated proximity ligation that requires two probes to adjacently bind onto the RNA sequence to form the circle complex [38]. In other instances the advantages of padlock probes can be built upon as demonstrated by spatially-resolved transcript amplicon readout mapping (STARmap), which requires the binding of both a padlock probe and RNA primer to induce RCA, creating a multi-cDNA containing amplicon [45]. After cDNA synthesis, the tissue is converted into an optically transparent 3D hydrogel to anchor DNA amplicons. A modified ISS technique is then applied to visualize and decode pre-encoded five-base barcodes. STARmap can impressively read 1,000 genes in only six imaging cycles [45]. Although the previously described target methods are limited by library size, they are essential for acquiring a high detection efficiency and can be valuable tools for analyzing alterations in specific transcripts of interest within multiple fibroblast subpopulations simultaneously [46].
Genome-scale transcriptome sequencing:
For experiments that require an unbiased gene selection, spatial transcriptomic approaches that evaluate the whole genome were created. ISS is a primary example of a targeted method that has been modified to create an unbiased next generation fluorescent in-situ RNA sequencing (FISSEQ) technique [47]. Instead of using random primers and targeted padlock probes, FISSEQ uses specialized tagged-random primers that allow for RNA to be synthesized into cDNA amplicons in situ. All cDNA amplicons are then cross-linked together within cells for stabilization, followed by RCA and profiled by Sequencing by Oligonucleotide Ligation and Detection (SOLiD), effectively sequencing the whole transcriptome with single-nucleotide resolution [22, 47]. Implementing this method allowed for the localized examination of 8,102 human primary fibroblasts genes in a wound healing model to assess gene alteration in response to injury and further showed that contact-inhibited and migratory fibroblasts differentially express ECM genes [47].
While FISSEQ is capable of directly sequencing a multitude of transcripts within the tissue, other unbiased spatial transcriptomic techniques rely on methods that allow them to employ bulk or single-cell RNA-seq and map the results back to prior images. Notably, the techniques mentioned till this point have z-scanning capabilities that are dependent upon the specifications of the microscope incorporated into the individual lab’s setup, whereas the remainder of the spatial transcriptomic approaches lack z-scanning capabilities. These methods follow a similar workflow, where the tissue is imaged, cells are labeled based on spatial location, but are then disassociated, processed for sequencing, and then the transcripts are mapped back to the original image using computational methods. For example, advanced tools such as laser capture microdissection (LCM) are used to select single cells or entire populations of cells of interest to be sequenced post-imaging [48, 49]. Another technique called Visium by 10x Genomics uses specialized slides containing spatially-distinguishable capture probes within a 100µm diameter chip [50]. Cells are permeabilized within the tissue, releasing the mRNA contents of each cell to locally bind the positional molecular barcodes residing directly under the individual cells on the glass coverslip. RNA-seq data generated from the reverse transcripts of the captured mRNA are aligned to pre-permeabilization images of the tissue via spatial barcodes for 2D positional transcriptome analysis [50]. This method captures twice the amount of transcripts as LCM-seq with few additions to the normal RNA-seq pre-processing workflow [23]. As this method relies on RNA-seq, it is reasonable that the reported sensitivity is comparable to the ability of scRNA-seq to detect single RNA molecules, 5–40% [51], at 6.9±1.5% [23] and below the near 100% sensitivity of smFISH. A method recently developed by the Macosko lab, Slide-seq, reached an even finer resolution by employing coverslips coated in a monolayer of 10 μm microparticles (“beads”) with distinct DNA-barcodes which can be mapped to their spatial address via sequencing of the barcode by oligonucleotide ligation and detection (Fig. 1) [52]. Frozen tissue sections are placed on these slides, imaged, and then processed to release mRNA to be captured by the microparticles for 3’ end RNA sequencing on commercial instruments. Although this method may be less sensitive to very low abundant transcripts, Slide-seq is highly scalable and achieves 10um spatial resolution, yielding the probing of localized gene expression within an individual cell [52].
Spatially-resolved proteomics:
RNAs and their protein counterparts are not expressed in a 1:1 ratio due to RNA degradation, post transcriptional, translational, and post-translational modifications [12, 53, 54], making it imperative to evaluate proteins, their concentration, interactions, and location. This was, for example, captured in the data collected for the atlas of the aging lung, where basement membrane collagen IV mRNA was downregulated, but protein was upregulated [10]. To achieve this objective many techniques have been developed that we have divided into 1) methods that require tissue dissociation and rely on computational mapping techniques or 2) methods that preserve tissue samples, often at the cost of decreased sequencing depth (Table 2).
Table 2:
Overview of spatial proteomic techniques discussed
| Method | Number of Targets | Advantages | Limitations |
|---|---|---|---|
| Imaging Mass Cytometry (IMC) [55, 56] | ~40 antibodies | 1 micron resolution; Single section and staining; Low signal spillover; No autofluorescence | Limited by number of rare metals; Biased by target selection; Special equipment; Costly; Long imaging time; Data analysis |
| Multiplexed Ion Beam Imaging (MIBI) [57] | Capable of ~100 targets, commercially 40+ | 200–300 nm resolution; Parts-per-billion sensitivity; Low signal spillover; No autofluroescence | Biased by target selection; Costly; Special equipment; Long imaging time; Data analysis |
| Spatially Targeted Optical Micro-Proteomics (STOMP) [62] | 1,000s of analytes | 1 micron resolution; Analyzes post translational protein modifications; Unbiased target selection | Special equipment needed; Trouble measuring low abundant proteins; Data analysis |
| Matrix-Assisted Laser Desorption/Ionization (MALDI) [49] | 1,000s of analytes | Analyzes post translational protein modifications; Unbiased target selection | Low resolution; Trouble measuring low abundant proteins; Requires matrix; Data analysis |
| Tissue-based Cyclic ImmunoFluorescence (t-CyCIF) [67, 68] | 60 antibodies | Cost effective; Uses common lab items; Enhanced signal to noise with each cycle | Time consuming |
| Co-Detection by indEXing (CODEX) [70] | ~40 antibodies | Single section and staining; Cost effective; Uses common microscope | Requires special reagents and equipment |
Note: For other comprehensive tables from previous reviews on spatial proteomics please refer to [24].
Dissociative techniques:
Originally presented in 2014, imaging mass cytometry (IMC) is a method capable of dictating protein distributions with 1 micron resolution [55, 56]. Here rare earth metals are conjugated to antibodies rather than fluorochromes to allow for simultaneous imaging of ~40 proteins. Sections undergo a single staining step to reduce technical variability, and then the sample is nebulized one pixel at a time via a laser ablation system. Each ablated pixel is enriched for heavy metal reporter ions and quantified by time of flight (TOF) MS on a per cell basis for single-cell analysis of cell type and state [56]. Noise and channel spillover is reduced in this method as the metal isotopes have distinct detection peaks, unlike fluorophores which may “blead” due to spectral overlap, and these heavy metals are not commonly observed in vivo, ergo eliminating background noise and autofluorescence [24, 56]. Multiplexed Ion Beam Imaging (MIBI) also utilizes mass tagged antibodies to analyze highly multiplexed protein expression patterns with morphological context. Instead of using a laser as in IMC, an ion beam is used to generate secondary ions from the antibodies, which can image up to 100 targets simultaneously after performing secondary ion MS [57]. Recent adaptations of MIBI have shown that the ion beam can precisely image at various depths, unlike the laser used in IMC, to acquire multiple scans in the z direction and create 3D images with ~250 nm resolution in the axial dimension [58]. However, both these techniques require long imaging times in addition to being costly. The area sampled is also confined by the time it takes to process MS data obtained from each pixel and convert it to an image. For more details on these techniques please refer to the following reviews [24, 59–61].
Thousands of molecules, including metabolites, lipids, peptides, proteins, and glycans, can be unbiasedly analyzed through the use of MS. Spatially targeted optical micro-proteomics (STOMP) couples MS and affinity photolabeling to fully harnesses a global spatial proteomic approach [62]. In this method, regions of interest (ROI) are selected in situ, a hexa-histidine photo-tag is then cross-linked to ROI-labeled proteins using two-photon excitation, which enables specific isolation of ROI proteins for mass spectrometry via anti-His bead pull-down. STOMP can identify proteins in any region of the cell with single micron resolution (.67um in xy axis and 1.48um axially), an entire order of magnitude greater than LCM [62]. Hadley et al., using STOMP, identified proteins present in amyloid plaques, fully demonstrating the power of 1µm resolution [62]. Imaging MS can also be paired with matrix-assisted laser desorption/ionization (MALDI) to obtain unbiased, de novo peptide sequencing of 1,000s of analytes from specified, spatial areas [63]. Details on the various ways to implement MALDI-IMS have been reviewed elsewhere [64]. Briefly, a matrix is overlaid onto a tissue section and laser excitation causes absorption of the sample’s ions into the matrix. Mass spectra generated at each xy coordinate are mapped back to the image to visualize xy spatial distribution [63]. MS methods are a valuable tool for spatial proteomics as they allow for the unbiased analysis of multiple analytes as well as post translational protein modifications like the citrullination of fibronectin which was shown to alter focal adhesion stability in fibroblasts [65], bringing us closer to an analysis that encompasses the whole proteome and distinguishes the ECM from cells. However, resolution often suffers with these approaches as compared to optical imaging in order to accommodate MS instrument sensitivity of lowly abundant species from small areas.
Techniques preserving tissue integrity:
In order to preserve morphological context and enhance resolution, a myriad of approaches that modulate standard antibody markers to present high-dimensional data have been developed to create non-destructive methods. Most of these techniques rely on iterative imaging of successive antibody staining cycles, where the sample is stained, followed by fluorophore bleaching, inactivation, or cleaving in preparation for a new batch of markers to be stained. These methods (MELC, SIMPLE, and MultiOmyx) are reviewed in detail elsewhere [24]. Instead, we will focus on improvements to the aforementioned and newer methods. For example, tissue-based cyclic immunofluorescence (t-CyCIF) is an advancement of a previous method developed initially by Gerdes et al. [66] and later adapted for cell culture by Lin et al. [67, 68], where multiplexed images are obtained by oxidizing fluorophores in a high pH hydrogen peroxide solution to erase the signal and image new proteins. In the span of three years the multiplexing capabilities of this technique expanded from imaging 16 channels after fluorescent-protein-based live cell imaging [68], to identifying 60 markers in formalin-fixed, paraffin-embedded (FFPE) samples [67]. Previous versions were limited by computational processing capabilities, but as analytical methods have improved the new constraint has become maintaining tissue integrity at higher cycles [67, 68]. The advantages of t-CyCIF are that it uses commonly available reagents, conventional microscopes that allow z-stacking, and commercial antibodies that can be selected to suite specific researcher interests. Our readers can refer to a recent study from the Hynes lab that reports the validation of ~100 antibodies recognizing ECM proteins [69]. However, the t-CyCIF protocol is time consuming as it requires an additional antibody incubation period for each imaging cycle. Alternatively, the CO-Detection by indEXing (CODEX) workflow facilitates imaging of up to 40 antigens after a single antibody incubation period comprising the entire antibody panel [70]. This method developed in the Nolan lab and now commercialized by Akoya Biosciences, combines in situ iterative indexing polymerization of unique DNA barcodes with automated assay performance via a cyclic fluidic device and automated imaging acquisition. In each cycle, one non-fluorescent nucleotide binds to the 5’ overhang of all oligo barcodes to index the system (Fig. 2). An additional fluorescent nucleotide, particular to that cycle, is incorporated only into the barcode of the antibody to be imaged that cycle [70]. The fluorophore is then cleaved by TCEP and washed away via the fluidics device in preparation for the next imaging cycle. The oligo on the previously imaged antibody is no longer polymerized as it is shorter than the following DNA barcodes. CODEX can also be integrated into any colored epifluorescence microscope, making it a versatile multiplexed imaging platform capable of spatial resolution in the lateral (xy) and axial (z) dimensions to create 3D images [70]. Although some of these techniques are capable of xyz resolution, most notably MIBI, t-CyCIF, and CODEX, they were designed with the intention of evaluating single cells and to our knowledge have not been applied to tissue sections thicker than 10 µm. These tools will be useful for further probing known location dependent protein-protein interactions, such as the decreased nuclear interaction of LEM domain-containing protein 3 and SMAD2/3 correlated to increased ECM stiffness in IPF tissue [71].
Multiplexed spatial-omics:
Few techniques have been developed to multiplex image RNA and protein in parallel to achieve -omics data on multiple molecular levels. Xia et al. demonstrated standard antibody staining in combination with MERFISH to identify localized RNA expression [34]. Techniques such as CITE-seq and REAP-seq have combined proteomic information with RNA-seq, but lack spatial context [72, 73]. Interestingly, recent studies such as those conducted in the Bodenmiller lab have been able to achieve imaging of 3 mRNA and 16 protein simultaneously with single-cell resolution [74]. The GeoMx™ Digital Spatial Profiling (DSP) (Nanostring Technologies, Inc.) system extends its method to accommodate the multiplexing of 40 proteins and up to 900 mRNA probes to be simultaneously analyzed with respect to spatial distribution, which can theoretically be extended to a maximum of 96 antibodies and an unlimited number of RNA readout through next generation sequencing (NGS) [75, 76]. This approach leverages oligo-tags attached to antibodies or RNA probes via a photocleavable linker. In this approach, pre-determined ROIs (identified by standard immunofluorescence) are exposed to photocleaving light directed by an automated digital-micromirror device, releasing the oligonucleotide tags to be aspirated into an automated microfluidics device and subsequently quantified through the nCounter System or NGS [75, 76]. Although DSP can overlay spatial transcriptome and proteome analyses through spatially aligned adjacent tissue sections, its protocol is optimized for interrogating tissue niche’s or ROI, not single cell profiling on whole tissue sections [76]. DSP’s pipeline, however, is compatible with the workflow of other spatial-omic techniques that do quantify expression with single cell resolution. RNA isoforms can also be identified through the integration of NGS in this system [75, 76]. Expanding these techniques will allow for -omic level profiling in various tissue regions, as well as allow for full paneling as transcripts can still provide information on proteins that do not have validated antibodies. In addition, continued progress must be made towards adding epigenetic and post-translational modifications, as well as profiling other biomolecules (lipids, glycans), if we want to truly grasp the complexity, heterogeneity, and dynamics of cellular and pathophysiological processes.
-Omics and Spatial-omics Approaches to Study the Matrisome:
ECM proteins have different biochemical properties than those of intracellular compartments. The highly modified and cross-linked nature of ECM proteins not only confers resilience to tensile or compressive stresses at the tissue and cell level, but also poses challenges in studying these proteins with conventional biochemical methods. In this section, we will first present approaches recently developed to specifically study ECM proteins with high throughput. We will then discuss new emerging ideas that can be implemented to improve the resolution with which we can study the extent of ECM roles on pathophysiology.
The matrisome: a comprehensive classification of ECM proteins:
High-throughput methods require careful annotation of the data generated in order to identify biologically relevant interactions among genes and proteins. Until recently, annotation tools only provided a partial view of the complex compartment that is the ECM [77]. To fill this gap in knowledge, we devised a computational pipeline to predict the “matrisome”, which is the collection of proteins that form ECM scaffolding (termed the core matrisome) but also those that interact with them (termed matrisome-associated components) [78]. This comprehensive collection accompanied by detailed nomenclature has allowed ECM research to enter the –omics era [2].
Indeed, the lists defined above can be used to annotate -omic datasets and map the matrisome of a given tissue or produced by a given type of cells. For example, Etich and collaborators discovered a unique matrisome-gene signature regulated in interferon-γ (IFNγ)- and dexamethasone-primed macrophages in vitro by revisiting previously published transcriptomic data [79]. They further validated the contribution of macrophage-secreted ECM proteins in skin wound repair in vivo [80]. A study conducted by Hamburg-Shields and collaborators highlights another example of how the list of matrisome genes are useful for the study of ECM biology. Here, they induced fibrosis in a mouse model by expressing a non-degradable form of β-catenin, then extracted and sequenced RNA from the dermis [81]. While all the changes observed in mutant skin were increases in gene expression, 36 of 175 (20.6 %) genes were matrisome genes, suggesting that β-catenin is one of the drivers of fibrosis [81]. With increased access to scRNA-seq technology, gene expression profiles of cells from fibrotic tissues have also been investigated. For instance, the analysis of the gene expression profile for different cell populations isolated from lung samples from either healthy control or IPF patients identified a new subtype of macrophages specific to IPF [6]. scRNA-seq of lung mesenchymal cells obtained from healthy donors vs. patients presenting with systemic sclerosis-associated interstitial lung disease identified different subpopulations of fibroblasts (one of them being characterized by a high level of the core matrisome gene MFAP5) and myofibroblasts [82]. However, these studies have left the matrisome largely unexplored, and annotating these datasets may reveal novel or unsuspected correlations that may lead to novel findings on the role of the ECM in pathophysiology.
Mass-spectrometry-based approaches to profile the ECM of tissues
Transcriptomic datasets convey a representative picture of the level of gene expression in a given condition, but do not fully reflect the state of protein abundance, since there is on average a 50% correlation between the two metrics [83]. Proteomic approaches such as those based on MS can be applied to take a snapshot of protein abundance in cells or tissue at a given time point or over time. However, as mentioned previously, ECM proteins present challenges for these methods mostly because of their insolubility [84]. This is further complicated by the quantity of peptides needed to perform an experiment, due to a lower sensitivity in proteomic tools compared to transcriptomic tools. To circumvent these challenges, we and others have developed methods to enrich and solubilize ECM proteins. While nuances between methods exist, they all follow a similar pipeline consisting of: ECM enrichment, protein solubilization and digestion into peptides, followed by MS analysis. For more details, we invite our readers to refer to a review we recently wrote on “matrisomics” [84]. The resulting proteomic data then can be annotated using the matrisome list, which can uncover ECM characteristics of the sample. Although much can be gained from the qualitative comparisons between matrisome profiles, labeled-based quantitative proteomic using isobaric tandem mass tag (TMT), or isobaric tag for relative and absolute quantification (iTRAQ) can provide additional information. In a 2017 study, we employed quantitative proteomics to characterize the changes in ECM composition in bleomycin-induced lung fibrosis and lung adenocarcinoma and found that the two diseases, both characterized by desmoplasia, shared a common ECM signature not observed in healthy lung tissues, but could also be distinguished from one another by the presence of specific ECM components [11]. This has similarly been observed in the context of liver fibrosis and hepatocellular carcinomas [85]. Novel proteomic approaches and bioinformatic tools are now focusing on probing post translational modifications of the ECM. One such study by Merl-Pham et al. investigates the effect of TGF-β on the ECM organization, and found novel sites of prolyl-3-hydroxylation site occupancy and lysine-O-glycosylation along with novel crosslinking enzymes that may help shed light on the process of fibril assembly [86]. The analysis of ECM proteins can be further extended with a technique called Quantitative Detergent Solubility Profiling (QDSP) [12]. As previously mentioned, most of the studies conducted on the ECM involve enrichment of the insoluble ECM by removing more soluble cellular components, yielding multiple fractions containing more soluble proteins in the process. Analysis of all the fractions (and not of only the ECM-enriched fraction) can shed light into mechanisms affecting ECM protein assembly and cross-linking during disease progression [12, 13, 87] or aging [10]. For example, high throughput degradomic and proteomic approaches were able to expand our knowledge of MMP cleavage specificity to establish regulatory roles of MMPs and design more effective therapeutics [88, 89].
Transcriptomic and proteomic characterizations of tissues or cells can be combined in multi-omic studies. This integration can reveal the different levels of regulations (transcriptional, translational, and post-translational) at play in healthy and diseased tissues. The Mann group has pioneered the use of multi-omics to study murine lung tissues in the context of bleomycin-induced injuries [12] and aging [10]. These approaches are particularly useful when genes expression level and protein abundance do not correlate. Together with changes in solubility and abundance seen in proteomic analyses, the transcriptomic analyses of upstream regulators can provide a systems-biology view of the matrisome. Last, recent attempts have been made to integrate miRNA data into transcriptomic and proteomic data. One such study examined the mRNA and miRNAs profiles of pulmonary fibroblasts from IPF and scleroderma patients, and revealed specific fibrotic signatures at both the miRNA, ECM gene, and ECM protein expression levels [90].
Probing the Regional Characteristics of ECM Composition and Architecture:
The studies described above have greatly advanced our understanding of the complexity of the ECM and have shed light on the multiple cell types and subtypes that can contribute to the production and remodeling of the ECM in healthy and diseased tissues. However, because of their destructive natures, the methods mentioned above do not allow researchers to probe sub-tissular heterogeneity of the ECM. To overcome this, approaches have focused on isolating a ROI in order to capture and evaluate smaller groups of cells or a defined microenvironment that are otherwise lost in the bulk processing of the samples. LCM is one of those methods, as it allows for the manual selection of a specific region to be excised from a tissue slide for analysis. LCM allows isolation of a particular compartment, as demonstrated by the isolation of glomeruli for the characterization of the glomerular basement membrane [91].
Another approach to characterize the localization and pattern of ECM protein distribution within tissues, is ECM Image Mass Spectrometry (ECM-IMS) [92]. This technique builds upon MALDI (see above), which records the mass-to-charge (m/z) ratio resulting from each pixel that is ionized. The resulting data can be represented by the distribution of m/z ratios that can be turned into a characteristic signature, allowing for the differentiation of, for example, a pathological tissue from its normal counterpart. One of the earlier studies utilizing the ECM-IMS aimed to find ECM signatures distinguishing young vs. old skin samples. Once age-dependnet m/z signal identified, the authors later identified proteins that were more abundant in young skin samples; these included collagen IV, collagen VII, collagen XVII, and nidogen I [93]. Another study using ECM-IMS compared low grade lung adenocarcinoma (LUAD) samples to normal lung from tissue microarrays by identifying and imaging peptides obtained from collagenase type III digestion. This resulted in a regional distribution tissue map of peptides of specific m/z ratios plotted with respect to their intensity. The authors were thus able to identify a characteristic m/z signature composed of 25 peptides that distinguished LUAD samples from normal tissues. Furthermore, because these spectra were assigned to spatial coordinates, the authors could also identify peptides preferentially found in the tumor core, around blood vessels, or in normal tissues adjacent to tumors [94]. To the best of our knowledge, other spatial proteomic approaches described above have not yet been applied to specifically study the ECM composition of tissues. However, in a recent study, Alföldi and collaborators used cytometry by time-of-flight (CyTOF) to profile protein expression changes in lung cancer cells cultured in 2D, 3D, or grown in vivo and among the 12 markers used in the study, one of them, Galectin-3 is a matrisome-associated proteins [95].
Importantly, to fully capture the complexity of the ECM, one needs, in addition to profiling its biochemical composition, to determine, in a time- and regionally-resolved manner, its architectural organization and biophysical and mechanical properties. These methods do not per se fall under the term of –omic approaches, and will thus not be discussed here, but we are listing for our readers recent and comprehensive reviews illustrating methods (second harmonic generation, atomic force microscopy) that permit these kinds of analysis [96–98].
Future Directions and Challenges:
Multiplex spatial-omic techniques will soon become prevalent, due to their ability to provide quantitative data on dozens of protein or RNA species within the same sample. Multiplex spatial-omics will help resolve the cellular complexities that contribute to overall cell phenotype/state, cell-cell interactions, as well as how these molecular identities link to their respective tissue architecture, and therefore tissue function by obtaining insight from multiple spatial scales [99, 100]. These methods although beneficial, still require advances to increase the resolution and throughput of the system and decrease acquisition time, as well as cost. There is also a need to increase the depth of these systems. At the RNA level, splicing variants are traditionally difficult to detect, and yet determining which splicing variant of a given ECM protein is being actively translated is of paramount importance [101, 102]. In the case of spatial image-based proteomics, construct libraries for each image cycle are limited by the number of variant (either barcodes, fluorescent dyes, or rare metals) available. Furthermore, antibodies must be validated and only rarely detect isoforms.
Interestingly, none of the current spatial-omic approaches presented in this review are capable of performing multiplexed spatial-omics on live cells ex vivo or in vivo. Researchers must be cognizant that these molecular profiles are representative of a single snapshot within the tissue. The observed phenomenon that cells operate through discontinuous transcription dictated by stochastic bursts, or “transcription bursts”, indicates that genetic profiles we observe in fixed samples may simply be an artifact of expression heterogeneity. Bulk sequencing techniques mask heterogeneity, as they are analyzing cells at a single time point; confining cell identification to the cell’s active transcriptional state at that moment, while other cells with similar functional roles are in a transcriptionally dormant state and are not accounted for [103].
What is possibly the most considerable challenge spatial-omics users will encounter is finding proper statistical tools for analyzing the vast amount of data produced by these systems. Furthermore, standardizing the way researchers present and analyze this data will require a community effort where computational algorithms and techniques are made publicly shared. Some of the commercialized spatial-omic techniques described in this review provide helpful software for cell segmentation and identification, but these methods still could benefit from enhancements to accommodate more complex samples with unusual cell shapes and sizes, such as those observed in IPF. Machine learning algorithms, such as CellDissect [104], can be implemented and have the potential to address the aforementioned issue. Visualizing structures that are typically difficult to evaluate through standard procedures due to microscope and affinity reagent limitations, such as lipid rafts, will need creative solutions in order to benefit from spatial-omic techniques as well. To facilitate the movement of spatial-omic platforms into clinical settings, approaches will have to continue to be developed in a cost and time effective manner.
Acknowledgements:
The authors would like to thank Martin Davis (Naba lab) for his critical reading of the manuscript as well as all members of the Barker lab for contributing to the peer review process.
Funding sources:
This work was supported in part by the United States National Institutes of Health, specifically the National Heart, Lung and Blood Institute for funding (R01 HL 127283 and R01 HL 132585) to THB, the NIH Cancer Training Grant T32 CA 009109 to GCB in addition to a start-up fund from the Department of Physiology and Biophysics at the University of Illinois at Chicago and a Catalyst Award from the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust (grant C-088) to AN.
Abbreviations:
- ISH
in-situ hybridization
- sc-RNAseq
single-cell RNA sequencing
- IPF
idiopathic pulmonary fibrosis
- MMPs
matrix metalloproteases
- ECM
extracellular matrix
- RNAseq
RNA sequencing
- MS
mass spectrometry
- smFISH
single molecule fluorescent in situ hybridization
- seqFISH
sequential FISH
- MERFISH
multiplexed error robust FISH
- ExM
expansion microscopy
- ER
endoplasmic reticulum
- bDNA
branched DNA
- ISS
in situ sequencing
- RCA
rolling circle amplification
- PLISH
proximity ligation in situ hybridization
- STARmap
spatially-resolved transcript amplicon readout mapping
- FISSEQ
fluorescent in situ RNA sequencing
- SOLiD
Sequencing by Oligonucleotide Ligation and Detection
- LCM
laser capture microdissection
- EMT
epithelial to mesenchymal transition
- IMC
imaging mass cytometry
- CyTOF
cytometry by time-of-flight
- MIBI
multiplexed ion beam imaging
- STOMP
spatially targeted optical micro-proteomics
- ROI
regions of interest
- MALDI
matrix-assisted laser desorption/ionization
- MELC
multi-epitope-ligand cartography
- SIMPLE
sequential immunoperoxidase labeling and erasing
- t-CyCIF
tissue-based cyclic immunofluorescence
- FP
fluorescent protein
- CODEX
CO-Detection by indEXing
- CITE-seq
Cellular Indexing of Transcriptomes and Epitopes by Sequencing
- REAP-seq
RNA expression and protein sequencing assay
- DSP
Digital Spatial Profiling
- NGS
next generation sequencing
- T1D
Type I Diabetes
- IFNγ
interferon-γ
- TMT
tandem mass tag
- TGF-β
transforming growth factor beta
- QDSP
Detergent Solubility Profiling
- LUAD
low grade lung adenocarcinoma
References:
- [1].Hasin Y, Seldin M, Lusis A, Multi-omics approaches to disease, Genome Biology 18(1) (2017) 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO, The extracellular matrix: Tools and insights for the “omics” era, Matrix biology : journal of the International Society for Matrix Biology 49 (2016) 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Socovich AM, Naba A, The cancer matrisome: From comprehensive characterization to biomarker discovery, Seminars in cell & developmental biology 89 (2019) 157–166. [DOI] [PubMed] [Google Scholar]
- [4].Ricard-Blum S, Miele AE, Omic approaches to decipher the molecular mechanisms of fibrosis, and design new anti-fibrotic strategies, Seminars in Cell & Developmental Biology (2019). [DOI] [PubMed] [Google Scholar]
- [5].Tabib T, Morse C, Wang T, Chen W, Lafyatis R, SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin, J Invest Dermatol 138(4) (2018) 802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen C-I, Ren Z, Verma R, Abdala-Valencia H, Nam K, Chi M, Han S, Gonzalez-Gonzalez FJ, Soberanes S, Watanabe S, Williams KJN, Flozak AS, Nicholson TT, Morgan VK, Winter DR, Hinchcliff M, Hrusch CL, Guzy RD, Bonham CA, Sperling AI, Bag R, Hamanaka RB, Mutlu GM, Yeldandi AV, Marshall SA, Shilatifard A, Amaral LAN, Perlman H, Sznajder JI, Argento AC, Gillespie CT, Dematte J, Jain M, Singer BD, Ridge KM, Lam AP, Bharat A, Bhorade SM, Gottardi CJ, Budinger GRS, Misharin AV, Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis, American journal of respiratory and critical care medicine 199(12) (2019) 1517–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, Picelli S, Zagorska A, Pendem SV, Hudson TE, Wu MM, Budas GR, Breckenridge DG, Harrison EM, Mole DJ, Wigmore SJ, Ramachandran P, Ponting CP, Teichmann SA, Marioni JC, Henderson NC, Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis, Cell Rep 29(7) (2019) 1832–1847.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, Ruan Q, Wei Y, Ni M, Adler C, Zhang W, Devalaraja-Narashimha K, Grindley J, Halasz G, Morton L, Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing, Am J Respir Cell Mol Biol 61(1) (2019) 74–85. [DOI] [PubMed] [Google Scholar]
- [9].Selman M, Pardo A, Kaminski N, Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs?, PLoS Med 5(3) (2008) e62–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, Tsitsiridis G, Ansari M, Graf E, Strom T-M, Nagendran M, Desai T, Eickelberg O, Mann M, Theis FJ, Schiller HB, An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics, Nature communications 10(1) (2019) 963–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gocheva V, Naba A, Bhutkar A, Guardia T, Miller KM, Li CM-C, Dayton TL, Sanchez-Rivera FJ, Kim-Kiselak C, Jailkhani N, Winslow MM, Del Rosario A, Hynes RO, Jacks T, Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival, Proceedings of the National Academy of Sciences of the United States of America 114(28) (2017) E5625–E5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schiller HB, Fernandez IE, Burgstaller G, Schaab C, Scheltema RA, Schwarzmayr T, Strom TM, Eickelberg O, Mann M, Time- and compartment-resolved proteome profiling of the extracellular niche in lung injury and repair, Molecular Systems Biology 11(7) (2015) 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Massey VL, Dolin CE, Poole LG, Hudson SV, Siow DL, Brock GN, Merchant ML, Wilkey DW, Arteel GE, The hepatic “matrisome” responds dynamically to injury: Characterization of transitional changes to the extracellular matrix in mice, Hepatology (Baltimore, Md.) 65(3) (2017) 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pardo A, Selman M, Kaminski N, Approaching the degradome in idiopathic pulmonary fibrosis, Int J Biochem Cell Biol 40(6–7) (2008) 1141–1155. [DOI] [PubMed] [Google Scholar]
- [15].Yu G, Kovkarova-Naumovski E, Jara P, Parwani A, Kass D, Ruiz V, Lopez-Otin C, Rosas IO, Gibson KF, Cabrera S, Ramirez R, Yousem SA, Richards TJ, Chensny LJ, Selman M, Kaminski N, Pardo A, Matrix metalloproteinase-19 is a key regulator of lung fibrosis in mice and humans, American journal of respiratory and critical care medicine 186(8) (2012) 752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors, Science (New York, N.Y.) 356(6335) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Walter JM, Joshi NR, Anekalla KR, Quattie-Pimentel ACM, Chiu S, Fernandez R, Akbarpour M, Chen C-I, Ren Z, Verma R, Abdala-Valencia H, Nam K, Winter DR, Chi M, Han S, Gonzalez FJ, Soberanes S, Watanabe S, Williams KJN, Secunda KE, Argento C, Gillespie CT, Dematte JE, Jain M, Singer BD, Ridge KM, Gottardi CJ, Lam A, Yeldandi AV, Hinchcliff M, Bag R, Hrusch CL, Guzy R, Bonham C, Sperling AI, Basu A, Hamanaka RB, Mutlu GM, Pivarski KL, Pivarski KL, Wang X, Marshall SA, Shilatifard A, Perlman HR, Sznajder JI, Bharat A, Bhorade SM, Budinger GS, Misharin A, Integrated Single-Cell Transcriptomic Analysis of Human Lung Reveals Distinct Patterns of Intercellular Heterogeneity in Idiopathic Pulmonary Fibrosis, A71. THE EPIGENOME, GENOME AND NON-CODING RNAs IN LUNG DISEASE, pp. A2286–A2286.
- [18].Yang J, Tanaka Y, Seay M, Li Z, Jin J, Garmire LX, Zhu X, Taylor A, Li W, Euskirchen G, Halene S, Kluger Y, Snyder MP, Park IH, Pan X, Weissman SM, Single cell transcriptomics reveals unanticipated features of early hematopoietic precursors, Nucleic acids research 45(3) (2017) 1281–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Little SC, Tikhonov M, Gregor T, Precise developmental gene expression arises from globally stochastic transcriptional activity, Cell 154(4) (2013) 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brown AC, Fiore VF, Sulchek TA, Barker TH, Physical and chemical microenvironmental cues orthogonally control the degree and duration of fibrosis-associated epithelial-to-mesenchymal transitions, The Journal of Pathology 229(1) (2013) 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pichon X, Lagha M, Mueller F, Bertrand E, A Growing Toolbox to Image Gene Expression in Single Cells: Sensitive Approaches for Demanding Challenges, Molecular cell 71(3) (2018) 468–480. [DOI] [PubMed] [Google Scholar]
- [22].Strell C, Hilscher MM, Laxman N, Svedlund J, Wu C, Yokota C, Nilsson M, Placing RNA in context and space - methods for spatially resolved transcriptomics, The FEBS journal 286(8) (2019) 1468–1481. [DOI] [PubMed] [Google Scholar]
- [23].Moor AE, Itzkovitz S, Spatial transcriptomics: paving the way for tissue-level systems biology, Current opinion in biotechnology 46 (2017) 126–133. [DOI] [PubMed] [Google Scholar]
- [24].Parra ER, Novel platforms of multiplexed immunofluorescence for study of paraffin tumor tissues, J Cancer Treat Diagnosis 2 (2018) 42–53. [Google Scholar]
- [25].Crosetto N, Bienko M, Van Oudenaarden A, Spatially resolved transcriptomics and beyond, Nature Reviews Genetics 16(1) (2015) 57–66. [DOI] [PubMed] [Google Scholar]
- [26].Lein E, Borm LE, Linnarsson S, The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing, Science (New York, N.Y.) 358(6359) (2017) 64–69. [DOI] [PubMed] [Google Scholar]
- [27].Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y, RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues, J Mol Diagn 14(1) (2012) 22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Femino AM, Fay FS, Fogarty K, Singer RH, Visualization of single RNA transcripts in situ, Science (New York, N.Y.) 280(5363) (1998) 585–90. [DOI] [PubMed] [Google Scholar]
- [29].Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC, Resolving the fibrotic niche of human liver cirrhosis at single-cell level, Nature 575(7783) (2019) 512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lubeck E, Cai L, Single-cell systems biology by super-resolution imaging and combinatorial labeling, Nat Methods 9(7) (2012) 743–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L, Single-cell in situ RNA profiling by sequential hybridization, Nat Methods 11(4) (2014) 360–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC, Cai L, Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH, Nature 568(7751) (2019) 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X, Spatially resolved, highly multiplexed RNA profiling in single cells, Science (New York, N.Y.) 348(6233) (2015) aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xia C, Fan J, Emanuel G, Hao J, Zhuang X, Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression, Proceedings of the National Academy of Sciences 116(39) (2019) 19490–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang G, Moffitt JR, Zhuang X, Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy, Scientific reports 8(1) (2018) 4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wassie AT, Zhao Y, Boyden ES, Expansion microscopy: principles and uses in biological research, Nat Methods 16(1) (2019) 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xia C, Babcock HP, Moffitt JR, Zhuang X, Multiplexed detection of RNA using MERFISH and branched DNA amplification, Sci Rep 9(1) (2019) 7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nagendran M, Riordan DP, Harbury PB, Desai TJ, Automated cell-type classification in intact tissues by single-cell molecular profiling, eLife 7 (2018) e30510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Battich N, Stoeger T, Pelkmans L, Image-based transcriptomics in thousands of single human cells at single-molecule resolution, Nat Methods 10(11) (2013) 1127–33. [DOI] [PubMed] [Google Scholar]
- [40].Choi HMT, Chang JY, Trinh LA, Padilla JE, Fraser SE, Pierce NA, Programmable in situ amplification for multiplexed imaging of mRNA expression, Nat Biotechnol 28(11) (2010) 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Marras SAE, Bushkin Y, Tyagi S, High-fidelity amplified FISH for the detection and allelic discrimination of single mRNA molecules, Proceedings of the National Academy of Sciences 116(28) (2019) 13921–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shah S, Lubeck E, Schwarzkopf M, He TF, Greenbaum A, Sohn CH, Lignell A, Choi HM, Gradinaru V, Pierce NA, Cai L, Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing, Development (Cambridge, England) 143(15) (2016) 2862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kharchenko PV, Silberstein L, Scadden DT, Bayesian approach to single-cell differential expression analysis, Nat Methods 11 (2014) 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wahlby C, Nilsson M, In situ sequencing for RNA analysis in preserved tissue and cells, Nat Methods 10(9) (2013) 857–60. [DOI] [PubMed] [Google Scholar]
- [45].Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, Evans K, Liu C, Ramakrishnan C, Liu J, Nolan GP, Bava F-A, Deisseroth K, Three-dimensional intact-tissue sequencing of single-cell transcriptional states, Science 361(6400) (2018) eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shah S, Lubeck E, Zhou W, Cai L, In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus, Neuron 92(2) (2016) 342–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SS, Li C, Amamoto R, Peters DT, Turczyk BM, Marblestone AH, Inverso SA, Bernard A, Mali P, Rios X, Aach J, Church GM, Highly multiplexed subcellular RNA sequencing in situ, Science (New York, N.Y.) 343(6177) (2014) 1360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nichterwitz S, Chen G, Aguila Benitez J, Yilmaz M, Storvall H, Cao M, Sandberg R, Deng Q, Hedlund E, Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling, Nature communications 7 (2016) 12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gozal YM, Cheng D, Duong DM, Lah JJ, Levey AI, Peng J, Merger of laser capture microdissection and mass spectrometry: a window into the amyloid plaque proteome, Methods in enzymology 412 (2006) 77–93. [DOI] [PubMed] [Google Scholar]
- [50].Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg Å, Pontén F, Costea PI, Sahlén P, Mulder J, Bergmann O, Lundeberg J, Frisén J, Visualization and analysis of gene expression in tissue sections by spatial transcriptomics, Science (New York, N.Y.) 353(6294) (2016) 78–82. [DOI] [PubMed] [Google Scholar]
- [51].Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW, Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells, Cell 161(5) (2015) 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, Macosko EZ, Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution, Science (New York, N.Y.) 363(6434) (2019) 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gygi SP, Rochon Y, Franza BR, Aebersold R, Correlation between protein and mRNA abundance in yeast, Molecular and cellular biology 19(3) (1999) 1720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang K.-l., Lin C, McLellan MD, Yan P, Davies SR, Townsend RR, Skates SJ, Wang J, Zhang B, Kinsinger CR, Mesri M, Rodriguez H, Ding L, Paulovich AG, Fenyö D, Ellis MJ, Carr SA, Nci C, Proteogenomics connects somatic mutations to signalling in breast cancer, Nature 534(7605) (2016) 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang YJ, Traum D, Schug J, Gao L, Liu C, Atkinson MA, Powers AC, Feldman MD, Naji A, Chang K-M, Multiplexed in situ imaging mass cytometry analysis of the human endocrine pancreas and immune system in type 1 diabetes, Cell metabolism 29(3) (2019) 769–783. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schuffler PJ, Grolimund D, Buhmann JM, Brandt S, Varga Z, Wild PJ, Gunther D, Bodenmiller B, Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry, Nat Methods 11(4) (2014) 417–22. [DOI] [PubMed] [Google Scholar]
- [57].Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, Multiplexed ion beam imaging of human breast tumors, Nature medicine 20(4) (2014) 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Keren L, Bosse M, Thompson S, Risom T, Vijayaragavan K, McCaffrey E, Marquez D, Angoshtari R, Greenwald NF, Fienberg H, Wang J, Kambham N, Kirkwood D, Nolan G, Montine TJ, Galli SJ, West R, Bendall SC, Angelo M, MIBI-TOF: A multiplexed imaging platform relates cellular phenotypes and tissue structure, Sci Adv 5(10) (2019) eaax5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chang Q, Ornatsky OI, Siddiqui I, Loboda A, Baranov VI, Hedley DW, Imaging Mass Cytometry, Cytometry Part A 91(2) (2017) 160–169. [DOI] [PubMed] [Google Scholar]
- [60].Baharlou H, Canete NP, Cunningham AL, Harman AN, Patrick E, Mass Cytometry Imaging for the Study of Human Diseases-Applications and Data Analysis Strategies, Front Immunol 10 (2019) 2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bodenmiller B, Multiplexed Epitope-Based Tissue Imaging for Discovery and Healthcare Applications, Cell Syst 2(4) (2016) 225–38. [DOI] [PubMed] [Google Scholar]
- [62].Hadley KC, Rakhit R, Guo H, Sun Y, Jonkman JE, McLaurin J, Hazrati L-N, Emili A, Chakrabartty A, Determining composition of micron-scale protein deposits in neurodegenerative disease by spatially targeted optical microproteomics, eLife 4 (2015) e09579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Caprioli RM, Farmer TB, Gile J, Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS, Analytical chemistry 69(23) (1997) 4751–4760. [DOI] [PubMed] [Google Scholar]
- [64].Ryan DJ, Spraggins JM, Caprioli RM, Protein identification strategies in MALDI imaging mass spectrometry: a brief review, Current opinion in chemical biology 48 (2019) 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Stefanelli VL, Choudhury S, Hu P, Liu Y, Schwenzer A, Yeh C-R, Chambers DM, Pesson K, Li W, Segura T, Midwood KS, Torres M, Barker TH, Citrullination of fibronectin alters integrin clustering and focal adhesion stability promoting stromal cell invasion, Matrix Biology 82 (2019) 86–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F, Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue, Proc Natl Acad Sci U S A 110(29) (2013) 11982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lin J-R, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, Sorger PK, Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes, eLife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lin J-R, Fallahi-Sichani M, Sorger PK, Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method, Nature communications 6 (2015) 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rickelt S, Hynes RO, Antibodies and methods for immunohistochemistry of extracellular matrix proteins, Matrix Biology 71–72 (2018) 10–27. [DOI] [PubMed] [Google Scholar]
- [70].Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, Nolan GP, Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging, Cell 174(4) (2018) 968–981.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chambers DM, Moretti L, Zhang JJ, Cooper SW, Santangelo PJ, Barker TH, LEM domain-containing protein 3 antagonizes TGFbeta-SMAD2/3 signaling in a stiffness-dependent manner in both the nucleus and cytosol, J Biol Chem 293(41) (2018) 15867–15886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P, Simultaneous epitope and transcriptome measurement in single cells, Nat Methods 14(9) (2017) 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, Moore R, McClanahan TK, Sadekova S, Klappenbach JA, Multiplexed quantification of proteins and transcripts in single cells, Nat Biotechnol 35(10) (2017) 936. [DOI] [PubMed] [Google Scholar]
- [74].Schulz D, Zanotelli VRT, Fischer JR, Schapiro D, Engler S, Lun X-K, Jackson HW, Bodenmiller B, Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry, Cell systems 6(1) (2018) 25–36. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Merritt CR, Ong GT, Church S, Barker K, Geiss G, Hoang M, Jung J, Liang Y, McKay-Fleisch J, Nguyen K, High multiplex, digital spatial profiling of proteins and RNA in fixed tissue using genomic detection methods, BioRxiv (2019) 559021. [DOI] [PubMed]
- [76].Van TM, Blank CU, A user’s perspective on GeoMxTM digital spatial profiling, Immuno-Oncology Technology 1 (2019) 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Naba A, Hoersch S, Hynes RO, Towards definition of an ECM parts list: an advance on GO categories, Matrix Biol 31(7–8) (2012) 371–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO, The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices, Molecular & cellular proteomics : MCP 11(4) (2012) M111.014647–M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Steiger J, Stephan A, Inkeles MS, Realegeno S, Bruns H, Kröll P, de Castro Kroner J, Sommer A, Batinica M, Pitzler L, Kalscheuer R, Hartmann P, Plum G, Stenger S, Pellegrini M, Brachvogel B, Modlin RL, Fabri M, Imatinib Triggers Phagolysosome Acidification and Antimicrobial Activity against Mycobacterium bovis Bacille Calmette-Guérin in Glucocorticoid-Treated Human Macrophages, J Immunol 197(1) (2016) 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Etich J, Koch M, Wagener R, Zaucke F, Fabri M, Brachvogel B, Gene Expression Profiling of the Extracellular Matrix Signature in Macrophages of Different Activation Status: Relevance for Skin Wound Healing, International journal of molecular sciences 20(20) (2019) 5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hamburg-Shields E, DiNuoscio GJ, Mullin NK, Lafyatis R, Atit RP, Sustained β-catenin activity in dermal fibroblasts promotes fibrosis by up-regulating expression of extracellular matrix protein-coding genes, J Pathol 235(5) (2015) 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, Rojas M, Lafyatis R, Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease, Ann Rheum Dis 78(10) (2019) 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, Jane-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, Golji J, Porter DA, Rejtar T, Wang YK, Kryukov GV, Stegmeier F, Erickson BK, Garraway LA, Sellers WR, Gygi SP, Quantitative Proteomics of the Cancer Cell Line Encyclopedia, Cell 180(2) (2020) 387–402.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Taha IN, Naba A, Exploring the extracellular matrix in health and disease using proteomics, Essays Biochem 63(3) (2019) 417–432. [DOI] [PubMed] [Google Scholar]
- [85].Arteel GE, Naba A, The liver matrisome, looking beyond collagens, JHEP Reports [DOI] [PMC free article] [PubMed]
- [86].Merl-Pham J, Basak T, Knüppel L, Ramanujam D, Athanason M, Behr J, Engelhardt S, Eickelberg O, Hauck SM, Vanacore R, Staab-Weijnitz CA, Quantitative proteomic profiling of extracellular matrix and site-specific collagen post-translational modifications in an in vitro model of lung fibrosis, Matrix Biology Plus 1 (2019) 100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wierer M, Prestel M, Schiller HB, Yan G, Schaab C, Azghandi S, Werner J, Kessler T, Malik R, Murgia M, Aherrahrou Z, Schunkert H, Dichgans M, Mann M, Compartment-resolved Proteomic Analysis of Mouse Aorta during Atherosclerotic Plaque Formation Reveals Osteoclast-specific Protein Expression, Molecular & cellular proteomics : MCP 17(2) (2018) 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].auf dem Keller U, Prudova A, Eckhard U, Fingleton B, Overall CM, Systems-level analysis of proteolytic events in increased vascular permeability and complement activation in skin inflammation, Sci Signal 6(258) (2013) rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Schlage P, auf dem Keller U, Proteomic approaches to uncover MMP function, Matrix Biol 44–46 (2015) 232–8. [DOI] [PubMed] [Google Scholar]
- [90].Mullenbrock S, Liu F, Szak S, Hronowski X, Gao B, Juhasz P, Sun C, Liu M, McLaughlin H, Xiao Q, Feghali-Bostwick C, Zheng TS, Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts, Genes (Basel) 9(12) (2018) 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hobeika L, Barati MT, Caster DJ, McLeish KR, Merchant ML, Characterization of glomerular extracellular matrix by proteomic analysis of laser-captured microdissected glomeruli, Kidney international 91(2) (2017) 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Angel PM, Schwamborn K, Comte-Walters S, Clift CL, Ball LE, Mehta AS, Drake RR, Extracellular Matrix Imaging of Breast Tissue Pathologies by MALDI-Imaging Mass Spectrometry, Proteomics Clin Appl 13(1) (2019) e1700152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Mondon P, Hillion M, Peschard O, Andre N, Marchand T, Doridot E, Feuilloley MG, Pionneau C, Chardonnet S, Evaluation of dermal extracellular matrix and epidermal-dermal junction modifications using matrix-assisted laser desorption/ionization mass spectrometric imaging, in vivo reflectance confocal microscopy, echography, and histology: effect of age and peptide applications, J Cosmet Dermatol 14(2) (2015) 152–160. [DOI] [PubMed] [Google Scholar]
- [94].Angel PM, Bruner E, Bethard J, Clift CL, Ball L, Drake RR, Feghali-Bostwick C, Extracellular Matrix Alterations in Low Grade Lung Adenocarcinoma Compared to Normal Lung Tissue by Imaging Mass Spectrometry, J Mass Spectrom (2019) 10.1002/jms.4450. [DOI] [PMC free article] [PubMed]
- [95].Alföldi R, Balog JÁ, Faragó N, Halmai M, Kotogány E, Neuperger P, Nagy LI, Fehér LZ, Szebeni GJ, Puskás LG, Single Cell Mass Cytometry of Non-Small Cell Lung Cancer Cells Reveals Complexity of In vivo And Three-Dimensional Models over the Petri-dish, Cells 8(9) (2019) 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jorba I, Uriarte JJ, Campillo N, Farré R, Navajas D, Probing Micromechanical Properties of the Extracellular Matrix of Soft Tissues by Atomic Force Microscopy, Journal of Cellular Physiology 232(1) (2017) 19–26. [DOI] [PubMed] [Google Scholar]
- [97].Alcaraz J, Otero J, Jorba I, Navajas D, Bidirectional mechanobiology between cells and their local extracellular matrix probed by atomic force microscopy, Semin Cell Dev Biol 73 (2018) 71–81. [DOI] [PubMed] [Google Scholar]
- [98].James DS, Jambor AN, Chang HY, Alden Z, Tilbury KB, Sandbo NK, Campagnola PJ, Probing ECM remodeling in idiopathic pulmonary fibrosis via second harmonic generation microscopy analysis of macro/supramolecular collagen structure, Journal of biomedical optics 25(1) (2019) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S, Imaging individual mRNA molecules using multiple singly labeled probes, Nat Methods 5(10) (2008) 877–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Rodriguez AJ, Czaplinski K, Condeelis JS, Singer RH, Mechanisms and cellular roles of local protein synthesis in mammalian cells, Current opinion in cell biology 20(2) (2008) 144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Castelletti F, Donadelli R, Banterla F, Hildebrandt F, Zipfel PF, Bresin E, Otto E, Skerka C, Renieri A, Todeschini M, Caprioli J, Caruso RM, Artuso R, Remuzzi G, Noris M, Mutations in FN1 cause glomerulopathy with fibronectin deposits, Proc Natl Acad Sci U S A 105(7) (2008) 2538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Muro AF, Chauhan AK, Gajovic S, Iaconcig A, Porro F, Stanta G, Baralle FE, Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan, The Journal of cell biology 162(1) (2003) 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Patange S, Girvan M, Larson DR, Single-cell systems biology: Probing the basic unit of information flow, Current Opinion in Systems Biology 8 (2018) 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kesler B, Li G, Thiemicke A, Venkat R, Neuert G, Automated cell boundary and 3D nuclear segmentation of cells in suspension, Scientific reports 9(1) (2019) 10237–10237. [DOI] [PMC free article] [PubMed] [Google Scholar]