

Abstract
Anaerobic methane oxidation was investigated in 6-m-long cores of marine sediment from Aarhus Bay, Denmark. Measured concentration profiles for methane and sulfate, as well as in situ rates determined with isotope tracers, indicated that there was a narrow zone of anaerobic methane oxidation about 150 cm below the sediment surface. Methane could account for 52% of the electron donor requirement for the peak sulfate reduction rate detected in the sulfate-methane transition zone. Molecular signatures of organisms present in the transition zone were detected by using selective PCR primers for sulfate-reducing bacteria and for Archaea. One primer pair amplified the dissimilatory sulfite reductase (DSR) gene of sulfate-reducing bacteria, whereas another primer (ANME) was designed to amplify archaeal sequences found in a recent study of sediments from the Eel River Basin, as these bacteria have been suggested to be anaerobic methane oxidizers (K. U. Hinrichs, J. M. Hayes, S. P. Sylva, P. G. Brewer, and E. F. DeLong, Nature 398:802–805, 1999). Amplification with the primer pairs produced more amplificate of both target genes with samples from the sulfate-methane transition zone than with samples from the surrounding sediment. Phylogenetic analysis of the DSR gene sequences retrieved from the transition zone revealed that they all belonged to a novel deeply branching lineage of diverse DSR gene sequences not related to any previously described DSR gene sequence. In contrast, DSR gene sequences found in the top sediment were related to environmental sequences from other estuarine sediments and to sequences of members of the genera Desulfonema, Desulfococcus, and Desulfosarcina. Phylogenetic analysis of 16S rRNA sequences obtained with the primers targeting the archaeal group of possible anaerobic methane oxidizers revealed two clusters of ANME sequences, both of which were affiliated with sequences from the Eel River Basin.
Anaerobic methane oxidation is a process that effectively controls emission of methane from many anaerobic environments into the atmosphere and thus plays an important role in the global methane budget (2, 35). Three lines of evidence support the hypothesis that biologically mediated methane oxidation occurs under anaerobic conditions: geochemical modeling, rate measurements obtained with radioactive tracers, and changes in stable carbon isotope ratios for methane and carbon dioxide. The geochemical models indicate that a methane-consuming process is required to explain the concave methane concentration profile found in anoxic layers of marine sediments (27, 34, 43). Radioisotope tracer experiments with 14CH4 and 35SO42− have shown that maximum anaerobic methane oxidation rates coincide with local maximum rates of sulfate reduction (2, 7, 13, 16, 17, 19–21) according to the net chemical reaction equation:
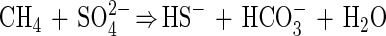 |
1 |
Methane produced by biological methanogenesis is strongly enriched for the light 12C carbon isotope and depleted for the heavy 13C isotope. The stable carbon isotope composition of organic compounds in the methane-sulfate transition zone has been used to determine if the compounds were derived from oxidation of isotopically light methane (i.e., the 12C-enriched methane produced by methanogenic bacteria). Unusually light CO2 was found in the transition zone together with light 13C-depleted lipids that were presumably also formed from light methane (1, 3, 5, 23, 31, 44).
Despite numerous isolation attempts by several scientific research groups, the organisms responsible for anaerobic methane oxidation have not been identified yet, and the mechanism remains unknown. Two scenarios for anaerobic methane oxidation have been proposed: (i) a single sulfate-reducing bacterium and (ii) a consortium of different bacteria. The consortium hypothesis assumes that methane is oxidized by an unknown bacterium in association with a sulfate reducer (7, 10, 13, 15, 17, 46). It is presumed that an unknown compound is used as an electron shuttle between the two organisms to carry electron equivalents from the methane-oxidizing organism to the sulfate-reducing organism. One potential carrier that has frequently been suggested is hydrogen. In this case the equations become:
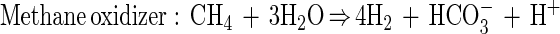 |
2 |
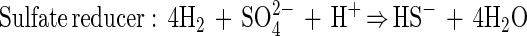 |
3 |
The consortium hypothesis is supported by the results of several studies in which the effects of molybdate, 2-bromoethanesulfonic acid, and fluoroacetate, which inhibit specific groups of bacteria, were monitored (2, 13, 46). Cultures of methanogenic bacteria have been shown to oxidize trace amounts of methane during growth (14, 45). Furthermore, recent studies have shown that 13C in lipid biomarkers for methanogenic archaea was depleted (10, 15, 32). In addition, a molecular investigation using restriction fragment length polymorphism and phylogenetic analysis of 16S rRNA indicated that methane was consumed by a phylogenetically distinct group of archaea (15). For these reasons it has been suggested that methanogenic bacteria are part of the consortium. A recent study by Boetius et al. (4) provided microscopic evidence that a consortium of archaea and sulfate-reducing bacteria is present in gas-hydrate-rich sediment. Laboratory experiments have shown that sulfate-reducing bacterial cultures may cooxidize small amounts of methane (6, 18, 38). However, some of the anaerobic methane oxidation rates measured with 14CH4 prepared biologically by using Methanobacterium thermoautotrophicum may not be accurate as they could include carbon monoxide oxidation rates (14). The problem occurs because M. thermoautotrophicum produces significant amounts of 14CO. If the radioactive methane was not purified and the measured rates were low, the 14CO2 detected may simply have been generated by 14CO oxidation (14). Hopcalite can be used to purify 14CH4 (14). Alternatively, a methanogenic strain that does not produce high concentrations of carbon monooxide could be used (14).
Regardless of possible methodological problems, it is evident that some sulfate-reducing bacteria may cooxidize small quantities of methane (14), but the pure cultures tested so far could not account for the anaerobic methane oxidation rates measured in various environments (18).
The free energy available for a consortium performing anaerobic methane oxidation at in situ concentrations of CO2, sulfide, methane, and sulfate is −22.35 kJ per mol of methane oxidized (14, 45). Such a low energy yield approaches the minimum requirement for even the most frugal bacteria if two species have to share it. Either the bacteria must be well adapted to an extremely penurious existence, or an unidentified energy-yielding reaction is coupled to the anaerobic oxidation of methane (14).
We describe here a study of the anaerobic methane oxidation in a marine sediment from Aarhus Bay, Denmark. The biogeochemical concentration profiles and rate measurements obtained strongly support the hypothesis that anaerobic methane oxidation occurs in a narrow zone about 150 cm below the sediment surface. We tried to evaluate the relative abundance of participating organisms by using minimum cycles for detectable products PCR (MCDP-PCR) and to identify the organisms based on a phylogenetic analysis of molecular sequences retrieved at different depths. We investigated the occurrence of sulfate-reducing bacteria and a distinct group of archaea that was recently proposed to be associated with anaerobic oxidation of methane (15).
The sulfate-reducing bacteria are a polyphyletic group, which limits the usefulness of 16S rRNA-based approaches because physiological inferences can be made only if a molecular isolate is very closely related to known pure cultures of sulfate-reducing bacteria. We instead used a primer set designed by Wagner et al. (42) that targets a key enzyme in sulfate respiration, the dissimilatory sulfate reductase (DSR). This primer set amplifies a 1.9-kb fragment of the α- and β-subunits of the phylogenetically conserved DSR gene. We also designed a primer set which amplifies an 817-bp fragment of the 16S rRNA gene of the specific group of archaea that have been proposed to be involved in anaerobic methane oxidation by Hinrichs et al. (15).
MATERIALS AND METHODS
Sediment sampling and handling.
Sediment cores were collected in June 1999 at station 6 in Aarhus Bay, a semienclosed embayment off the east coast of Jutland, Denmark (56°09′30"N, 10°19′15"E). The salinity of the water just above the sediment at a depth of 16 m was 31‰, and the temperature was 5.4°C. The sediment consisted of very fine sand, silt, and clay, and the sediment was permanently reduced. Cores 7 cm wide and 6 m long were obtained with a piston corer with a removable polyvinyl chloride tube as a liner (20). The cores and polyvinyl chloride tubes were cut into four 1.5-m sections on the ship. The cores were stored in sealed plastic bags and brought to the laboratory. Core A was analyzed within 24 h after sampling. It was split longitudinally, and subsamples were taken at 20-cm intervals to determine the sulfate-methane transition zone. Density and porosity were also measured. Core B was stored at in situ temperature until it was analyzed 1 week after sampling. Ten-centimeter sections were cut off the end of the core, and subsamples were immediately taken from the two exposed surfaces by subcoring with syringes. The core was sampled at 4- to 6-cm intervals from the surface to a depth of 190.5 cm. Sediment from core B was used to quantify sulfate reduction and methane oxidation rates and for DNA extraction (as described below) and also to obtain the same measurements that were obtained with core A.
Porewater analyses. (i) Sulfate and chlorinity.
The porewater used to determine sulfate and chloride concentrations was collected by centrifuging 10 cm3 of sediment and was preserved in 20% (wt/vol) zinc acetate. Porewater samples were filtered, and concentrations of sulfate and chloride were determined by ion chromatography (Sykam, Gilching, Germany). The eluent contained 7.5 mM Na2CO3, 5% ethanol, and 50 mg of 4-hydroxybenzonitrile liter−1 (13).
(ii) Methane.
Ten cubic centimeters of sediment from each depth was incubated in a 100-ml infusion bottle containing 10 ml of 1 M NaOH to trap CO2 and to stop methanogenesis. The bottles were capped immediately with bromobutyl rubber stoppers and shaken vigorously for 1 min. Gas samples were injected into a gas chromatograph equipped with a flame ionization detector (Packard) after separation on a Poropack Q column (13). The methane peak was recorded on a strip chart recorder and quantified by comparison with standards.
(iii) Density and porosity.
The density and porosity of the sediment were measured for every third depth by using 50-cm3 sediment samples. Porosity was determined from the density and the water content; the latter was measured by drying 10 g of sediment overnight at 110°C.
(iv) Steady-state sulfate reduction rates, methane oxidation rates, and methane production rates estimated from sulfate and methane concentration profiles.
Net production and consumption rates were estimated from the curvature of in situ concentration profiles. For the sake of simplicity all metabolic rates pertaining to methane and sulfate were considered positive if the compound was being produced and negative if it was being consumed; i.e., production rates [P(z)] are positive rates, and consumption rates [R(z)] are negative rates, and the resulting net rate of metabolism of methane or sulfate [M(z)] is M(z) = P(z) + R(z). Using this convention, Fick's second law of one-dimensional diffusion becomes:
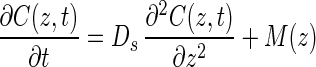 |
4 |
where C(z,t) is the concentration at depth z and time t, Ds is the diffusivity, and M(z) is the net metabolic rate of production and consumption for either methane or sulfate. An assumption of this formula is that Ds does not change in the depth range being investigated. Ds for marine sediments can be estimated from the equation given by Ullman and Aller (41):
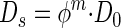 |
5 |
where D0 is the molecular diffusion coefficient and φ is the porosity. The exponent m ranges from 2 to 3 in different sediments depending on porosity. In our study we used a value of 2.5, which is typical for fine-grained silty sediments with a porosity greater than 0.71 and less than 0.86 (41). The molecular diffusion coefficients for methane and sulfate in seawater at 5°C were 0.95 × 10−5 and 0.58 × 10−5 cm2 s−1, respectively (26). Under steady-state conditions equation 4 becomes:
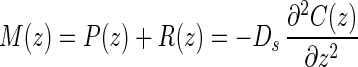 |
6 |
where C(z) is the concentration at depth z.
The net metabolic rate is directly proportional to the second derivative of the concentration with respect to depth (that is, to the curvature of the profile). As measured concentration profiles consist of discrete data points with inherent small variations, differentiation of the raw data often leads to a very noisy pattern. To correct for this, we compared the measured concentration profiles to simulated concentration profiles calculated from activity profiles by integration of equation 6. An activity profile consists of a number of zones with constant activity in each zone. A simplex optimization method was used to find the activity profile that minimized the difference between the theoretically calculated and measured concentration profiles (33). This procedure was used to obtain a set of optimized activity profiles with an increasing number of zones that described the concentration profile best. Each of the solutions was tested by evaluating the sensitivity of the solution to additions of noise to the concentration profile. The activity profile that had the highest number of zones yet was still robust towards noise addition was finally chosen (33).
Rate measurements. (i) Sulfate reduction measurement.
Approximately 10 μl of radiolabelled sulfate (37 kBq of 35SO42−/μl) was injected into 10-cm3 subsamples of sediment. The subsamples were incubated in an anaerobe jar at 5°C for 67 h. Incubation was stopped by mixing the sediment with 10 ml of 20% (wt/vol) zinc acetate. Reduced sulfur compounds (H2S, FeS, FeS2, and S0) were stripped from the sediment as H2S by single-step chromium distillation (12). A flow of N2 was used to carry the H2S from the reaction vessel to tubes containing 10 ml of 2% (wt/vol) zinc acetate. Hydrogen sulfide was trapped as ZnS. A scintillation counter (Tricarb 2200 CA; Packard) was used to measure the radioactivity of the reduced sulfur compounds in the zinc acetate trap and the radioactivity of the nonreduced 35SO42− after addition of scintillation liquid (Ecoscint A; Packard). The sulfate reduction rate was calculated as described by Fossing and Jørgensen (12).
(ii) Methane oxidation rates.
Radioisotope-labelled methane was biosynthesized by Methanococcus deltae and was purified with Hopcalite as described by Harder (14). Approximately 20 μl of radiolabelled methane (0.6 kBq of 14CH4/μl) was injected into 10-cm3 subsamples of sediment, which were incubated at 5°C for 67 h under anaerobic conditions. Incubations was stopped by mixing the sediment with 10 ml of 1 M NaOH. Radiolabelled 14CO2 was collected from the samples as described by Hansen et al. (13). Methane radioactivity was measured by injecting samples of headspace gas into a gas chromatograph equipped with a flame ionization detector. 14CO2 was then produced by combustion and trapped in a scintillation vial containing 1 ml of 1 M NaOH. Finally, 10 ml of HiOnic Fluor scintillation cocktail (Packard) was added to the trap. Methane oxidation rates were calculated from the amount of 14CO2 formed and from the concentration and radioactivity of methane as described by Iversen and Blackburn (19).
Nucleic acid extraction.
Nucleic acids were extracted by using a FastDNA spin kit for soil (Bio 101, Vista, Calif.) according to the manufacturer's instructions. This method relies on mechanical cell lysis by bead beating (FastPrep DNA extractor; Bio 101) followed by selective DNA adsorption to microporous silicate filters. The bound DNA is then washed with ethanol in the presence of chaotropic salts and finally eluted in a low-salt buffer. Nucleic acid extraction was evaluated on a 1% Seakem GTG agarose gel (FMC Bioproducts, Rockland, Maine) electrophoresed with TAE buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA; pH 8.3). The gel was stained with SybrGold (100 ng/ml; Molecular Probes, Leiden, The Netherlands) for 20 min. For analysis and documentation a transilluminator and a digital camera (Gel Doc 2000; Bio-Rad, Hercules, Calif.) were used. Images were acquired and analyzed with the software Quantity One (Bio-Rad). Larger amounts of DNA were recovered from the samples closest to the sediment surface; however, the concentrations of recovered DNA were similar for all samples obtained at depths below 100 cm from the sediment surface. The standard error was less than 10% of the average DNA concentration in each case. To test the quality of the DNA recovered, PCR amplification of ribosomal DNA was performed with universal primers for 24 cycles. Similar amounts of PCR product were obtained with DNA extracts from all depths below 100 cm (standard error, <15%).
MCDP-PCR amplification.
MCDP-PCR is a method that is used to evaluate the relative abundance of template genes present in a sample (9). The principle is to determine the minimum number of PCR cycles necessary to detect a faint amplificate (approximately 0.1 ng) on an agarose gel. The rationale behind the method is that product inhibition reduces amplification efficiency in the last cycles of a PCR when the product concentration is high. Thus, by reducing the number of cycles to the minimum number required for a detectable product, product interference is reduced as much as possible. The number of cycles required to produce a detectable product has been shown to indicate the relative abundance of template molecules (9, 11). An automated version of this technique is widely used in real-time PCR approaches developed by Hoffmann-La Roche, Bio-Rad, and Perkin-Elmer to quantify target DNA in diagnostic medical applications (28). An alternative approach to obtain a barely detectable product is to dilute the sample prior to amplification. However, dilution also reduces the concentration of PCR-inhibiting compounds (e.g., humic acids) present in the sample, which makes it difficult to compare the amplification results obtained for differently diluted samples. It is difficult to get an absolute estimate of gene copies by MCDP-PCR; however, it is possible to evaluate the relative abundance of genes in different samples. In this study, two primer pairs were used. The first primer set was designed by Wagner et al. (42) to specifically amplify a 1.9-kb fragment of the DSR gene; this set consisted of primers DSR1F (5′-ACSCACTGGAAGCACG-3′) and DSR4R (5′-GTGTAGCAGTTACCGCA-3′). A different primer was designed to target the sequences of archaea that were proposed by Hinrichs et al. (15) to be anaerobic methane oxidizers; this primer was primer ANMEF (5′-GGCUCAGUAACACGUGGA-3′). When sequences were subjected to Probe Match analysis with small-subunit rRNAs of the Ribosomal Database Project (Center for Microbial Ecology, University of Michigan), only four sequences of uncultured archaea matched the probe sequence. All the matching sequences were sequences of tentative methane oxidizers from the Eel River Basin study. The highly specific primer was used together with a universal 16S rRNA primer (907R; 5′-CCGTCAATTCCTTTRAGTTT-3′) (25) to detect these archaea in the sediment. The reaction mixture used for PCR amplification contained 36.5 μl of distilled H2O, 5 μl of buffer (100 mM Tris-HCl, 750 mM KCl, 15 mM MgCl2; pH 8.8), 5 μl of a 10× deoxynucleoside triphosphate (dNTP) mixture (containing each dNTP at a concentration of 125 μM), 1 μl of each primer (50 pmol/μl), and 0.5 μl of Taq polymerase (5,000 U/ml; Pharmacia). One microliter (approximately 10 ng) of diluted DNA extract was added. PCR amplification was carried out with a DNA Thermocycler (PT-200 Peltier thermal cycler; MJ Research). Each cycle in the PCR program for DSR gene amplification consisted of 1 min of denaturation at 94°C, 1 min of annealing at 54°C, and 3 min of extension at 72°C. The reaction was completed by a 10-min final extension step at 72°C. PCR with 28, 30, 32, and 34 cycles were performed with the DSR gene primers. The PCR program used for amplification of 16S ribosomal DNA with the ANME primer set consisted of 30 s of denaturation at 92°C, 1 min of annealing at 57°C, and 0.45 s (plus 1 s/cycle) of extension at 72°C. The reaction was completed by a 5-min final extension step at 72°C. With the ANME primer pair 25, 26, 27, 28, and 29 cycles were performed. The PCR products were loaded on a 2% Nusieve 3:1 agarose gel (FMC Bioproducts), and electrophoresis was performed with TAE buffer. The gels were stained with SybrGold (100 ng/ml; Molecular Probes), and the results were evaluated and documented as described above. The detection limit of SybrGold was less than 100 pg of double-stranded DNA per band in routine applications. Band intensities were estimated by using custom macros for the image analysis program NIH Image 1.62b7 written by Wayne Rasband (available from zippy.nimh.nih.gov). The intensity estimates were standardized by using a constant camera aperture and division by exposure time. The reproducibility of the estimates was evaluated by comparing band intensities in marker lanes of the gels analyzed.
Cloning of DSR and 16S rRNA genes.
Fresh PCR amplificates obtained with primers DSR1F and DSR4R and with primers ANMEF and 907R were purified with a QIAquick PCR purification kit (Qiagen GmbH, Hilden, Germany). The amplificates were ligated into a pCR-XL-TOPO vector and transformed into ONE SHOT Escherichia coli cells by following the manufacturer's directions (TOPO XL PCR cloning; Invitrogen, Leek, The Netherlands). Each clone was screened for an insert of the correct length by direct PCR amplification with the original primers, using 16 cycles and the PCR programs mentioned above. Plasmids of randomly selected clones containing the correct-size insert were then recovered with a QIAprep spin miniprep kit (Qiagen GmbH).
Sequencing.
Partial DNA sequences were obtained from extracted plasmid templates with Cy-5-labelled primers targeting the plasmid sequence surrounding the insert (VektorF [5′-TTTGGCCCTCTAGATG-3′] and VektorR [5′-CTATGCATCAAGCTTGG-3′]) (T. R. Thomsen, M. Wagner, K. K. Brandt, K. Ingvorsen, and N. B. Ramsing, unpublished data), using a thermosequenase fluorescent cycle sequencing kit (Pharmacia Biotech, Uppsala, Sweden) and an ALFexpress DNA sequencer (Pharmacia Biotech). The following components were used for each sequencing reaction: 3.375 μl of distilled H2O, 0.375 μl of dimethyl sulfoxide, 1.25 μl of primer (1.25 pmol/μl), 2 μl of a dNTP mixture, and 1 to 2 μl of template (isolated plasmids). Amplification was carried out for 40 cycles, with each cycle consisting of 30 s of denaturation at 94°C, 30 s of annealing at 57°C, and 5 min of extension at 72°C.
Phylogenetic analysis.
DSR gene sequences were aligned and analyzed by using the ARB program package (available from www.mikro.biologie.tu-muenchen.de/Pub/ARB) (39). DNA sequences were translated into amino acid sequences and aligned manually with the Genetic Data Environment (GDE), version 2.2, sequence editor implemented in the ARB software environment. Nucleic acid sequences were subsequently aligned on the basis of the amino acid alignment. Trees based on aligned sequences were constructed by using the FITCH distance matrix program in Phylip 3.53. Only unambiguously aligned amino acid positions from the α- and β-subunits of the DSR gene found in all sequences were used. The final data set consisted of 264 amino acids. Both nucleic acid and amino acid alignments were also evaluated by using PAUP*, version 4.0 (40). The nucleic acid alignment was analyzed by distance matrix and maximum-likelihood approaches, whereas the amino acid alignment was evaluated by using parsimony- and distance matrix-based algorithms. For all types of phylogenetic analysis we used the default settings in PAUP*, version 4.0.
Sequences of archaeal 16S rRNA were manually aligned using SeqPup, version 0.6 (software@bio.indiana.edu), with the general small-subunit alignment used by the Ribosomal Database Project, version 7.1. Phylogenetic trees were generated by using PAUP*, version 4.0. Distance matrix, parsimony, and maximum-likelihood analyses were performed with unambiguously aligned nucleotide positions (631 nucleotides).
Bootstrap analysis was performed with PAUP*, version 4.0, by using 100 resamplings of the nucleotide sequence for both genes.
Nucleotide sequence accession numbers.
The GenBank accession numbers for the small-subunit sequences are as follows: ANME1, AF314243; ANME7, AF314244; ANME8, AF314249; ANME11, AF314247; ANME12, AF314242; ANME14, AF314246; ANME16, AF314245; ANME17, AF314248; ANME19, AF314241; and ANME23, AF314250. The GenBank accession numbers for DSR gene sequences are as follows: a-a, AF316066; a-E, AF316067; a-F, AF316065; a-G, AF316050; a-20, AF316070; a-24, AF316044; a-54, AF316068; a-55, AF316063; a-70, AF316039; a-73, AF316062; a-75, AF316042; b-2, AF316051; b-4, AF316073; b-6, AF316071; b-15, AF316046; b-18, AF316053; b-20, AF316061; b-28, AF31606; b-30, AF316059; b-31, AF316047; b-37, AF316052; c-C, AF316054; c-D, AF316048; c-E, AF316049; c-F, AF316058; c-I, AF316056; c-J, AF316040; c-1, AF316045; c-18, AF316069; c-23, AF316064; c-31, AF316072; c-40, AF316057; c-47, AF316043; c-59, AF316055; c-63, AF316041; Desulfobacter halotolerans, AF521159; Desulfocella halophila, AF321158; KYF135, AF321149; KYF124, AF321156; KYF128, AF321154; KYF136, AF321155; KYF312, AF321153; KYF313, AF321152; KYF314, AF321157; KYF322, AF321151; and KYF 324, AF321150.
RESULTS
Biogeochemical analyses.
The sediment density was about 1.30 g cm−3 at all depths below 50 cm (data not shown). The porosity ranged from 0.71 to 0.86 ml cm−3, as shown in Fig. 1. The sulfate concentration (Fig. 1) was highest at the sediment surface (18.7 mM in porewater) and decreased with depth to less than 2 mM below a depth of 136 cm. The methane concentration (Fig. 1) was less than 0.1 mM in the upper 150 cm but increased gradually below a depth of 150 cm to a maximum value of 1.5 mM at a depth of 281 cm. The cores contained numerous gas bubbles at depths below approximately 290 cm, which probably explains the large variations in the concentrations measured below this depth. Bubbles were formed when the methane core was brought to the surface as the in situ hydrostatic pressure was removed. The upper limit of bubble formation coincided with a methane concentration of about 1.5 mM, which corresponded to a partial pressure of 1 atm. The zone of bubble formation at the in situ hydrostatic pressure was likely to be considerably deeper in the sediment. The depth sounder employed during sampling showed that there was strong reflectance from a sediment layer approximately 3.5 m below the seafloor. The reflection (often referred to as the methane mirror) was most likely caused by small methane bubbles within the sediment. The sulfate-methane transition zone was at a depth of 140 to 160 cm. The methane profile was concave in this depth interval, indicating that there was net consumption. Activities were calculated from the core A profiles because the concentration measurements were made immediately after sampling. Sulfate reduction activity was greatest in the zone ranging from a depth of 138 cm to a depth of 159 cm, and the calculated rate was 1.74 nmol cm−3 day−1 (Fig. 2A). The calculated maximum methane oxidation value was 0.59 nmol cm−3 day at a depth of 140 to 169 cm (Fig. 2B). Net methane production began at a depth of 169 cm but reached a maximum value of 1.14 nmol cm−3 day−1 at a depth of 215 cm (Fig. 2C). The narrow zone of high methane production was most likely an artifact as the inflexion point of the methane profile (Fig. 1) occurred where the partial pressure of methane was 1 atm. Thus, it is likely that methane was vented from the deeper parts of the profile by bubble formation. The measured rates of sulfate reduction and methane oxidation are also shown in Fig. 2A and B, respectively. The highest sulfate reduction rate occurred at the 140.5-cm depth (1.05 nmol cm−3 day−1), whereas the highest methane oxidation rate occurred at the 150.5-cm depth (0.79 nmol cm−3 day−1). The calculated cumulative sulfate reduction rate, based on the sulfate concentration profile, was 365.57 μmol m−2 day−1, whereas the measured rate, obtained by isotopic techniques, was only 84.16 μmol m−2 day−1. The calculated cumulative methane oxidation rate, based on the methane concentration profile, was 170.35 μmol m−2 day−1, whereas the measured rate, obtained by isotopic techniques, was 45.17 μmol m−2 day−1. Sulfate reduction values obtained by Jørgensen et al. (22) at station 6 were integrated to estimate that the sulfate reduction rate in the total core was 4,801.41 μmol m−2 day−1.
FIG. 1.

Concentrations of sulfate and methane in sediment cores A and B. Porosity is indicated by the vertical line without symbols. Depths at which molecular investigations were done are indicated (a, 21.5 cm; b, 81.5 cm; c, 156.5 cm).
FIG. 2.

(A) Depth profiles for measured sulfate reduction rates obtained by tracer techniques and for rates calculated from the sulfate concentration profiles. (B) Depth profiles for measured methane oxidation rates obtained by tracer techniques and for rates calculated from the methane concentration profiles. (C) Depth profile for methane production rates based on the concentration profile for methane. The depths at which molecular investigations were done are indicated (a, 21.5 cm; b, 81.5 cm; c, 156.5 cm).
MCDP-PCR amplification.
MCDP-PCR results obtained for different sampling depths with the ANMEF-907R and DSR gene primer sets are shown in Fig. 3A and C, respectively. The depth profiles for band intensity per gram of sediment analyzed are shown in Fig. 3B, D, and E. Amplifications in which different numbers of cycles were used were scaled to the lowest number of cycles employed by dividing the intensities by 2n where n is the number of additional cycles employed. When amplification results obtained with different numbers of PCR cycles with the ANME primer pair were compared (Fig. 3B), the scaled data curves were all similar and had a peak at and below the sulfate-methane transition zone. The similarity of the scaled curves obtained with different numbers of PCR cycles indicates that the assumed ideal amplification giving rise to a doubling of product for each cycle was reasonable.
FIG. 3.

(A) Agarose gel showing amplificates obtained with primers ANMEF and 907R after 27 PCR cycles. (B) Depth profile for relative gel band intensities of amplificates obtained with the ANMEF-907R primer set. Different symbols correspond to different numbers of PCR cycles, as follows: open circles, 29 cycles; gray circles, 28 cycles; solid circles, 27 cycles. (C) Gel showing amplificates obtained with the DSR gene primer set after 28 PCR cycles. (D and E) Depth profiles for gel band intensities of amplificates obtained with the DSR gene primer set by using different numbers of PCR cycles, as follows: solid circles, 28 cycles; light gray circles, 30 cycles; dark gray circles, 32 cycles; open circles, 34 cycles. The band intensities in panels B, D, and E were scaled by dividing by 2 for every cycle beyond the lowest number used with the primer set (e.g., the measured band intensities of DSR gene amplificates obtained after 30 cycles were divided by 4 to be equivalent to the intensities obtained after 28 cycles). For more information see Results. The samples picked for cloning and sequencing were samples from zone a (depth, 21.5 cm), zone b (depth, 81.5 cm), zone c (depth, 156.5 cm).
Amplifications with the DSR gene primer pair (Fig. 3D and 3E) in which more cycles were used gave more product. However, the amount of amplificate produced did not double for every cycle added. Thus, when the amount of amplificate was divided by 2 for every additional cycle, the lowest number of cycles gave the highest scaled intensity. Thus, it is clear that the amplification efficiency for the DSR gene was substantially less than 100%, at least for the last PCR cycles when the amplification product accumulated. The peak for the DSR gene amplificate in the sulfate-methane transition zone (Fig. 3D and E) appeared most prominently with the lowest number of PCR cycles. It is possible that the peak was affected by product inhibition even with the lowest number of cycles employed in this study, and it may have underestimated the actual abundance. Nevertheless, both primer pairs (the ANME and DSR primer pairs) revealed that a pronounced peak in the amount of amplificate produced (or, equivalently, a reduced number of cycles for a detectable product) occurred 156.5 cm below the sediment surface.
Phylogenetic analysis.
On the basis of the results obtained by MCDP-PCR we decided to clone DSR genes from three zones, zone a (depth, 21.5 cm), zone b, (81.5 cm), and zone c, (156.5 cm). We also cloned the amplificates acquired with the primers specific for 16S rRNA targeting ANME-like archaea in zone c to verify that these sequences were indeed related to the archaeal 16S rRNA targets reported by Hinrichs et al. (15). Phylogenetic trees for the DSR gene based on unambiguously aligned nucleic acid and amino acid data sets were estimated by using distance matrix, parsimony, and maximum-likelihood criteria. The three methods resulted in congruent tree topologies except for the phylogenetic position of Desulfobulbus propionicus. Maximum-likelihood and distance matrix approaches using nucleic acid sequences, as well as parsimony analysis using amino acid sequences, placed D. propionicus in the cluster of phylogenetically related incompletely oxidizing sulfate-reducing bacteria in the δ subgroup of the class Proteobacteria. However, distance matrix analysis based on amino acid sequences placed D. propionicus among the low-G+C-content gram-positive bacteria with Desulfotomaculum as the closest relative. We chose to present distance matrix trees derived with PAUP*, version 4.0, using default settings.
The distance matrix tree based on nucleic acids shown in Fig. 4 is a consensus tree in which only branching orders supported by bootstrap values of more than 50% are included. Branching orders that are not supported by bootstrapping are reduced to multifurcations. The tree topography derived from amino acid sequences (Fig. 5) was likewise subjected to bootstrap analysis. However, as the number of residues used to derive the amino acid phylogeny was limited, parts of the tree are collapsed, and the bootstrap values are generally lower. Still, the trees are very similar in terms of both branching order and branch length.
FIG. 4.

Phylogenetic tree based on nucleic acids. Sequences obtained at different depths (zone a, 21.5 cm; zone b, 81.5 cm; zone c, 156.5 cm) of a station 6 sediment core were compared to pure cultures of sulfate-reducing bacteria. The tree was constructed by using default settings for the distance matrix algorithm in PAUP*, version 4.0, and bootstrapping with 100 replicates was performed. Only nodes supported by bootstrap values greater than 50% are shown. Scale bar = 0.05 substitution per nucleotide position.
FIG. 5.

Phylogenetic tree based on amino acids. Sequences obtained at different depths (zone a, 21.5 cm; zone b, 81.5 cm; zone c, 156.5 cm) of a station 6 sediment core were compared to pure cultures of sulfate-reducing bacteria. The tree was constructed by using default settings for distance matrix analysis in PAUP*, version 4.0. Scale bar = 0.05 substitution per nucleotide position.
Three prominent groups of DSR gene sequences were present in the trees. Group I included seven sequences from zone a (21.5 cm) and four sequences from Kysing Fjord. This group was placed in the group of complete oxidizers in the δ subgroup of the Proteobacteria, and it was affiliated with the genera Desulfosarcina, Desulfococcus, and Desulfonema.
Group II consisted of six sequences recovered from zone b (81.5 cm), four sequences from zone a (21.5 cm), and three sequences from surface sediment from a shallow Danish fjord, Kysing Fjord (Thomsen et al., unpublished). It was deeply rooted in the DSR gene tree, and it had no close relatives.
Group III contained 14 sequences from zone c (156.5 cm) and four sequences from zone b (81.5 cm). This group of DSR gene sequences appeared to be very deeply rooted in the tree. They were not related to any known DSR gene sequences from pure cultures or even to any of the numerous DSR sequences which have been retrieved from various environments. Nevertheless, all members of this cluster showed high homology to DSR gene sequences when a Blast search was performed through the GenBank database. All sequences retrieved from zone c (156.5 cm) belonged to this cluster.
A phylogenetic tree constructed by using distance matrix analysis and 100 bootstrap resamplings for 10 sequences amplified with the ANMEF-907R primer set from zone c (156.5 cm) and their closest phylogenetic relatives are shown in Fig. 6. A comparison of phylogenetic trees obtained by the different methods revealed consistent topologies, and these topologies were supported by maximum-likelihood trees for subsets of these species. Five ANME sequences labelled group I were phylogenetically related to three clones of archaea from a salt marsh (29) and three clones from the Eel River Basin (15). Five other ANME sequences labelled group II clustered together with nine sequences from the Eel River Basin (15). The previously described species that are most closely related to this cluster belong to the orders Methanosarcinales and Methanomicrobiales. The different ANME clones were not very closely related and may represent different species.
FIG. 6.

Phylogenetic tree of archaeal rRNA sequences obtained from station 6 at a depth of 156.5 cm (zone c), sequences of pure cultures of archaea, and environment sequences. The tree was constructed by using default settings for distance matrix analysis in PAUP*, version 4.0, and bootstrapping with 100 replicates was performed. Only nodes supported by bootstrap values greater than 50% are shown. Scale bar = 0.05 substitutions per nucleotide position.
DISCUSSION
Biogeochemical studies.
Based on concentration profiles and rate measurements, the sediment could be divided into three functional zones. The upper zone, zone I, from 0 to 140 cm, is the sulfate-containing upper zone; in this zone the maximum sulfate concentration, present at the surface (18.7 mM) decreases gradually. The sulfate-methane transition zone, zone II, from 140 to 160 cm, is the transition zone in which the two compounds are present simultaneously. Finally, the methanogenic zone, zone III, extends downwards from 160 cm. In this zone net methanogenesis increases and diffusion from below augments the methane concentration until it reaches a maximum value of 1.5 mM at 281 cm, where methane bubble formation starts. The slightly concave methane profile in zone II indicates that net methane oxidation occurs in this zone (27).
The measured methane oxidation rates were low throughout zone I, but a high level of activity was measured at a single point in the sulfate-methane transition zone at a depth of 150.5 cm. Curiously, this activity occurred 10 cm below a similarly prominent peak in the measured sulfate reduction rate at a depth of 140.5 cm. The difference in position is unexplained as the two measurements were made at the same time with subcores from the same spot in a 6-m piston core. The point equidistant between the two peak sites (146.5 cm) gave low rates for both processes. The calculated rates occurred at a greater range of depths than the measured rates, and the maximum methane oxidation rates coincided with the maximum sulfate reduction rates at depths of 140 to 159 cm. The activity results are in agreement with data obtained in previous studies (20, 22, 36).
The integrated measured rates of sulfate reduction and methane oxidation over the area are indicated above. Methane could account for 52% of the electron donors required to sustain the measured sulfate reduction rates in the sulfate-methane transition zone. When the calculated rates based on concentration profiles were used, the methane oxidation rates were 47% of the sulfate reduction rates in the transition zone. Both approaches showed that methane is probably a primary electron donor in the sulfate-methane transition zone. The measured and calculated rates based on concentration profiles revealed similar tendencies, although the calculated rates of sulfate reduction and methane oxidation were 4.3 and 3.8 times higher, respectively, than the measured rates. In a previous study, Devol (7) likewise found that measured rates were lower than calculated rates. Some variability in measured and calculated rates could be explained by spatial heterogeneity in the sample and a distribution of measurement points that was too coarse. Furthermore, the anaerobic methane oxidation rates could have been underestimated because of accidental degassing of radioactive methane during handling.
Other environmental studies of these processes have obtained similar values. Anaerobic methane oxidation in the sulfate-methane transition zone could account for 89% of the electron donor requirement for sulfate reduction in Skagerrak (20), as well as 61% in Kattegat (20), 100% in an upwelling area of Namibia (30), 50 to 85% in Amazon Fan sediment (5), and 10 to 30% in Norsminde Fjord (13).
We estimated that the contribution of methane to the electron donors required for total sulfate reduction per area of seafloor integrated over the whole sediment core was 9.4%. This estimate was made by integrating the sulfate reduction rates obtained in a previous study performed by Jørgensen et al. (22) at station 6 at all depths (see above). The relative importance of methane as an electron donor for total sulfate reduction compares well to the relative importance in sediment cores of different lengths from other habitats; 12% of the total sulfate reduction could be explained by methane oxidation in Scan Bay, 23 to 40% could be explained in Saanich Inlet (8), 10% could be explained in Skagerrak and Kattegat (20), 1.6 to 2.3% could be explained in Big Soda Lake (21), and 0.01 to 0.06% could be explained in Kysing Fjord (19).
Molecular work.
The MCDP-PCR profiles obtained with both the DSR and ANME primer pairs revealed a pronounced peak in the amount of amplificate produced from samples obtained in the sulfate-methane transition zone. The peak could have been due to PCR inhibitors present in surrounding strata. Nevertheless, the homogeneous nature of the sediment that far from the sediment surface makes this an unlikely explanation. Thus, the MCDP-PCR results support the possible occurrence of higher numbers of sulfate-reducing bacteria and special archaea in zone c (156.5 cm below the surface) than in zone b. Still, the largest DSR gene amplificate from sulfate-reducing bacteria was detected in the top sediment. However, the DSR gene of these sulfate-reducing bacteria was apparently phylogenetically different from the genes found in the deep sediment. Most sequences from zone a (depth, 21.5 cm) did cluster with environmental sequences from other estuarine sediments (i.e., Kysing Fjord), and the cluster (group I) was affiliated with DSR genes from known complete oxidizers belonging to the genera Desulfosarcina, Desulfococcus, and Desulfonema.
In the top sediment a broad range of electron donors are probably present, while at a depth of 156.5 cm the most prominent electron donor available is methane. At this depth, a very deeply branching group (group III) of DSR genes was discovered. These genes were not related to any previously characterized DSR gene from sulfate-reducing bacteria. Four sequences from zone b (depth, 81.5 cm) were affiliated with these sequences, but no sequence from the top sediment was represented. This indicated that these deeply branching DSR genes from unknown sulfate-reducing bacteria are present mainly in the deeper sediment. The fact that all 14 DSR gene sequences retrieved from the sulfate-methane transition zone belong to this cluster suggests that these bacteria constitute a prominent part of the sulfate-reducing bacterium community found at this depth and that these organisms may play an important role in anaerobic methane oxidation. In the study of the Eel River Basin (15) sulfate-reducing bacteria were also found in the seep sediment.
The phylogenetic tree based on 16S rRNA sequences obtained with the primers targeting the special group of possibly methane-oxidizing archaea (ANME) from the recent study by Hinrichs et al. (15) contained two clusters of ANME sequences. One cluster consisted of ANME clones related to unknown archaea from a salt marsh and to sequences from a seep sediment from the Eel River Basin (15), while the second group comprised ANME clones affiliated with the ANME sequences from the Eel River Basin. None of the ANME sequences obtained in this study were closely related to each other or to other sequences in the Ribosomal Database Project, and it thus seems likely that the diversity of archaea in the sediment is quite high. However, the two ANME clusters were separate entities in the phylogenetic tree. Elvert and Suess (10) proposed that methanogenic archaea may be able to switch from a methane formation metabolism to a metabolism favoring consumption. However, Hinrichs et al. (15) found that there probably are obligatorily or dominantly methanotrophic archaea. In both studies the authors assumed that the deep-sea ecosystem provides the necessary conditions favoring anaerobic methane oxidation by archaea and sulfate-reducing bacteria. In our study we found that special gas hydrate conditions (very low hydrogen and very high methane concentrations) are not a prerequisite for anaerobic methane oxidation, as the process also occurs in more normal sediments. It is, however, remarkable that the same phylogenetic group of archaea was also found in our study in a normal marine sediment.
Our results support the hypothesis that anaerobic methane oxidation was carried out by a consortium of sulfate-reducing bacteria and a special group of archaea. The spatial separation between the distinct zone in which a peak sulfate reduction rate occurred and the zone in which an equally pronounced peak methane oxidation rate occurred could be explained by the combined action of two distinct groups of bacteria, which are responsible for net anaerobic methane oxidation with sulfate as the ultimate electron acceptor. We also demonstrated that both special archaea and sulfate-reducing bacteria were present in the sulfate-methane transition zone. Sulfate and reduced sulfur compounds are the only possible electron acceptors for anaerobic methane oxidation at that depth (2). It is presumed that the special group of methane oxidizers converts methane to an unknown substrate, which is utilized by the sulfate-reducing bacteria. Different compounds have been proposed as possible interspecies electron carriers; these compounds include hydrogen (2, 14, 16, 17, 24), acetate, methanol (46), and formate (14). However, a theoretical study by Sørensen et al. (37) showed that neither hydrogen, acetate, nor methanol can serve as the elusive interspecies electron carrier. It is impossible for members of a consortium to be located close enough to each other to keep both the generation of the intermediate by methanogens and the consumption of the intermediate by sulfate-reducing bacteria exergonic. If the interspecies electron carrier is methanol or an other alcohol, inhibition of sulfate-reducing bacteria should prevent complete oxidation of methane to carbon dioxide, and this has not been observed in experiments in which the sulfate-reducing bacterium inhibitor molybdate has been added (2, 13). Thus, it appears that the identity of a possible interspecies electron carrier is still unknown.
Another possibility is that a single organism can carry out both anaerobic methane oxidation and sulfate reduction. The deeply branching sulfate-reducing bacteria could be responsible without any participation from archaea such as the new ANME group. The ANME group could also represent a new order of uncultivated methanogens that are not involved at all in anaerobic methane oxidation. Finally, it is also possible that the new group of sulfate-reducing bacteria could belong to the archaea. The novelty of this deeply rooted cluster of sequences unfortunately prevents any qualified guess concerning their phylogenetic position; they may even be the archaea whose sequences were retrieved with the ANME primers.
Great diversity was demonstrated in the archaeal sequences retrieved from a depth of 156.5 cm. The total diversity of sulfate-reducing bacteria demonstrated by the sequences retrieved from all three depths, ranging from top sediment to 156.5 cm, is still unresolved as we never retrieved the same sequence twice. Finally, our results demonstrate the advantage of combining molecular methods and accurate biogeochemical data analysis when complex environments and reactions are studied. Future attempts to isolate bacteria responsible for anaerobic methane oxidation should include both the ANME primers and a new primer pair targeting the novel deeply branching group of sulfate-reducing bacteria retrieved from a depth of 156.5 cm.
ACKNOWLEDGMENTS
This work was supported by Statens Naturvidenskabelige Forskningsråd, Statens Tekniske Videnskabelige Forskningsråd, and the Carlsberg Foundation.
We thank Verner Dam, Leif Flensborg, and Erik Jensen for sampling the piston core and Dorte T. Ganzhorn and Jane Frydenberg for excellent technical assistance. We also thank Michael Wagner, Technische Universität München, and David Stahl, Northwestern University, Evanston, Ill., for helpful advice and access to their DSR gene sequences from pure cultures. Finally, we thank Ketil Sørensen for the radioactive methane used to carry out the experiment and for many inspiring discussions.
REFERENCES
- 1.Alperin M J, Reeburg W S, Whiticar M J. Carbon and hydrogen isotope fractionation resulting from anaerobic methane oxidation. Global Biogeochem Cycles. 1988;2:279–288. [Google Scholar]
- 2.Alperin M J, Reeburgh W S. Inhibition experiments on anaerobic methane oxidation. App Environ Microbiol. 1985;50:940–945. doi: 10.1128/aem.50.4.940-945.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blair E N, Aller R C. Anaerobic methane oxidation on the Amazon shelf. Geochim Cosmochim Acta. 1995;59:3705–3715. [Google Scholar]
- 4.Boetius A, Ravenschlag K, Schubert C J, Rickert D, Widdel F, Gieseke A, Amann R, Jørgensen B B, Witte U, Pfannkuche O. Microscopic identification of a microbial consortium apparently mediating anaerobic methane oxidation above marine gas hydrate. Nature. 2000;407:623–626. doi: 10.1038/35036572. [DOI] [PubMed] [Google Scholar]
- 5.Burns S J. Carbon isotope evidence for coupled sulfate reduction-methane oxidation in Amazon Fan sediments. Geochim Cosmochim Acta. 1998;62:797–804. [Google Scholar]
- 6.Davis J B, Yarbrough H F. Anaerobic oxidation of hydrocarbons by Desulfovibrio desulfuricans. Chem Geol. 1966;1:137–144. [Google Scholar]
- 7.Devol A H. Methane oxidation rates in the anaerobic sediments of Saanich Inlet. Limnol Oceanogr. 1983;28:738–742. [Google Scholar]
- 8.Devol A H, Anderson J J, Kuivila K, Murray J W. A model for coupled sulfate reduction and methane oxidation in the sediment of Saanich Inlet. Geochim Cosmochim Acta. 1984;48:993–1004. [Google Scholar]
- 9.Diaco R. Practical considerations for the design of quantitative PCR assays. In: Innis M A, Gelfand D H, Sninsky J J, editors. PCR strategies. San Diegol, Calif: Academic Press; 1995. pp. 93–95. [Google Scholar]
- 10.Elvert M, Suess E. Anaerobic methane oxidation associated with marine gas hydrates: superlight C-isotopes from saturated and unsaturated C20 and C25 irregular isoprenoids. Naturwissenchaften. 1999;86:295–300. [Google Scholar]
- 11.Ferre F. Quantitative or semi-quantitative PCR: reality versus myth. PCR Methods Applic. 1992;2:1–9. doi: 10.1101/gr.2.1.1. [DOI] [PubMed] [Google Scholar]
- 12.Fossing H, Jørgensen B B. Measurement of bacterial sulfate reduction in sediments: evaluation of a single-step chromium reduction method. Biogeochemistry. 1989;8:205–222. [Google Scholar]
- 13.Hansen L B, Finster K, Fossing H, Iversen N. Anaerobic methane oxidation in sulfate depleted sediments: effects of sulfate and molybdate additions. Aquat Microb Ecol. 1998;14:195–204. [Google Scholar]
- 14.Harder J. Anaerobic methane oxidation by bacteria employing 14C-methane uncontaminated with 14C-carbon monooxide. Mar Geol. 1997;137:13–23. [Google Scholar]
- 15.Hinrichs K U, Hayes J M, Sylva S P, Brewer P G, DeLong E F. Methane-consuming archaebacteria in marine sediments. Nature. 1999;398:802–805. doi: 10.1038/19751. [DOI] [PubMed] [Google Scholar]
- 16.Hoehler T M, Alperin M J. Anaerobic methane oxidation by methanogen-sulfate reducer consortium: geochemical evidence and biochemical evidence. In: Lidstrøm M E, Tabita F R, editors. Microbial growth on C-I compounds. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1996. pp. 326–333. [Google Scholar]
- 17.Hoehler T M, Alperin M J, Albert D B, Martens C S. Field and laboratory studies of methane oxidation in an anoxic marine sediment: evidence for a methanogen-sulfate reducer consortium. Global Biogeochem Cycles. 1994;8:451–463. [Google Scholar]
- 18.Iversen N. Interaktioner mellem fermenteringsprocesser og de terminale processer. Ph. D. thesis. Aarhus, Denmark: Aarhus University; 1984. [Google Scholar]
- 19.Iversen N, Blackburn T H. Seasonal rates of methane oxidation in anoxic marine sediments. Appl Environ Microbiol. 1981;41:1295–1300. doi: 10.1128/aem.41.6.1295-1300.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iversen N, Jørgensen B B. Anaerobic methane oxidation rates at the sulfate-methane transition in marine sediments from Kattegat and Skagerrak (Denmark) Limnol Oceanogr. 1985;30:944–955. [Google Scholar]
- 21.Iversen N, Oremland R S, Klug M J. Big Soda Lake (Nevada). 3. Pelagic methanogenesis and anaerobic methane oxidation. Limnol Oceanogr. 1987;32:804–814. [Google Scholar]
- 22.Jørgensen B B, Bang M, Blackburn T H. Anaerobic mineralization in marine sediments from the Baltic Sea-North Sea transition. Mar Ecol Progr Ser. 1990;59:39–54. [Google Scholar]
- 23.Jørgensen N O. Methane derived carbonate cementation of marine sediments from the Kattegat, Denmark. Mar Geol. 1992;103:1–13. [Google Scholar]
- 24.King G M. Regulation of methane oxidation: contrast between anoxic sediments and oxic soils. In: Lidstrøm M E, Tabita F R, editors. Microbial growth on C-1 compounds. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1996. pp. 318–325. [Google Scholar]
- 25.Lane D J. 16/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. Chichester, United Kingdom: Wiley; 1991. pp. 113–175. [Google Scholar]
- 26.Li Y H. Diffusion of ions in sea water and in deep-sea sediments. Geochim Cosmochim Acta. 1974;38:703–714. [Google Scholar]
- 27.Martens C S, Berner R A. Interstitial water chemistry of anoxic Long Island Sound sediments. Limnol Oceanogr. 1977;22:10–25. [Google Scholar]
- 28.Morrison T B, Weis J J, Wittwer C T. Quantification of low-copy transcripts by continuous SYBR® Green I monitoring during amplification. BioTechniques. 1998;24:954–962. [PubMed] [Google Scholar]
- 29.Munson M A, Nedwell D B, Embley T M. Phylogenetic diversity of Archaea in sediment samples from a coastal salt marsh. Appl Environ Microbiol. 1997;63:4729–4733. doi: 10.1128/aem.63.12.4729-4733.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niewöhner C, Hensen C, Kasten S, Zabel M, Schulz H D. Deep sulfate reduction completely mediated by anaerobic methane oxidation in sediments of the upwelling area off Namibia. Geochim Cosmochim Acta. 1998;62:455–464. [Google Scholar]
- 31.Oremland R S, Marias D J. Distribution, abundance and carbon isotopic composition of gaseous hydrocarbons in Big Soda Lake, Nevada: an alkaline, meromictic lake. Geochim Cosmochim Acta. 1983;47:2107–2114. [Google Scholar]
- 32.Pancost R D, Damsté J S S, Saskia de Lint M, van den Maarel J E C, Gottschal J C. Biomarker evidence for widespread anaerobic methane oxidation in Mediterranean sediments by a consortium of methanogenic archaea and bacteria. Appl Environ Microbiol. 2000;66:1126–1132. doi: 10.1128/aem.66.3.1126-1132.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsing N B, Ferris M J, Ward D M. Highly ordered vertical structure of Synechococcus populations with the one-millimeter-thick photic zone of a hot spring cyanobacterial mat. Appl Environ Microbiol. 2000;66:1038–1049. doi: 10.1128/aem.66.3.1038-1049.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reeburgh W. Methane consumption in Carioco trench waters and sediments. Earth Planet Sci Lett. 1976;28:337–344. [Google Scholar]
- 35.Reeburgh W. Coupling of the carbon and sulphur cycles through anaerobe methane oxidation. New York, N.Y: John Wiley & Sons; 1989. [Google Scholar]
- 36.Reeburgh W S. Anaerobic methane oxidation: rate depth distributions in Skan Bay sediments. Earth Planet Sci Lett. 1980;47:345–352. [Google Scholar]
- 37.Sørensen, K. B., K. Finster, and N. B. Ramsing. Thermodynamic and kinetic requirements in anaerobic methane oxidizing consortia: exclude hydrogen, acetate and methanol as possible electron shuttles, Microb. Ecol., in press. [DOI] [PubMed]
- 38.Sorokin X. On the ability of sulfate-reducing bacteria to utilize methane for the reduction of sulfate. Dokl Akad Nauk SSSR Ser Bio. 1957;115:816–818. [Google Scholar]
- 39.Strunk O, Ludwig W, Gross O, Reichel B, Stuckmann N, May M, Nunhoff B, Lenke M, Ginhart T, Vilbig A, Westran R. ARB-a software environment for sequence data. Technische Universit; 1998. ä München, Munich, Germany. [Google Scholar]
- 40.Swofford, D. L. PAUP∗, version 4.0. Sinauer Associates, Sunderland, Mass.
- 41.Ullman W J, Aller R C. Diffusion coefficients in nearshore marine sediments. Limnol Oceanogr. 1982;27:552–556. [Google Scholar]
- 42.Wagner M, Roger A J, Flax J L, Brusseau G A, Stahl D A. Phylogeny of dissimilatory sulfite reductase supports an early origin of sulfate respiration. J Bacteriol. 1998;180:2975–2982. doi: 10.1128/jb.180.11.2975-2982.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward B B, Kilpartick K A, Novelli P C, Scranton M I. Methane oxidation and methane fluxes in the ocean surface layer and deep anoxic waters. Nature. 1987;327:226–229. [Google Scholar]
- 44.Whiticar M J, Faber E. Methane oxidation in sediments and water column environments—isotope evidence. Org Geochem. 1986;10:759–768. [Google Scholar]
- 45.Zehnder A J B, Brock T D. Methane formation and methane oxidation by methanogenic bacteria. J Bacteriol. 1979;137:420–432. doi: 10.1128/jb.137.1.420-432.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zehnder A J B, Brock T D. Anaerobic methane oxidation: occurrence and ecology. Appl Environ Microbiol. 1980;39:194–204. doi: 10.1128/aem.39.1.194-204.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]