

ABSTRACT
Pseudorabies virus (PRV) is a porcine alphaherpesvirus that belongs to the Herpesviridae family. We showed earlier that infection of porcine epithelial cells with PRV triggers activation of the nuclear factor κB (NF-κB) pathway, a pivotal signaling axis in the early immune response. However, PRV-induced NF-κB activation does not lead to NF-κB-dependent gene expression. Here, using electrophoretic mobility shift assays (EMSAs), we show that PRV does not disrupt the ability of NF-κB to interact with its κB target sites. Assessing basal cellular transcriptional activity in PRV-infected cells by quantitation of prespliced transcripts of constitutively expressed genes uncovered a broad suppression of cellular transcription by PRV, which also affects the inducible expression of NF-κB target genes. Host cell transcription inhibition was rescued when viral genome replication was blocked using phosphonoacetic acid (PAA). Remarkably, we found that host gene expression shutoff in PRV-infected cells correlated with a substantial retention of the NF-κB subunit p65, the TATA box binding protein, and RNA polymerase II—essential factors required for (NF-κB-dependent) gene transcription—in expanding PRV replication centers in the nucleus and thereby away from the host chromatin. This study reveals a potent mechanism used by the alphaherpesvirus PRV to steer the protein production capacity of infected cells to viral proteins by preventing expression of host genes, including inducible genes involved in mounting antiviral responses.
IMPORTANCE Herpesviruses are highly successful pathogens that cause lifelong persistent infections of their host. Modulation of the intracellular environment of infected cells is imperative for the success of virus infections. We reported earlier that a DNA damage response in epithelial cells infected with the alphaherpesvirus pseudorabies virus (PRV) results in activation of the hallmark proinflammatory NF-κB signaling axis but, remarkably, that this activation does not lead to NF-κB-induced (proinflammatory) gene expression. Here, we report that PRV-mediated inhibition of host gene expression stretches beyond NF-κB-dependent gene expression and in fact reflects a broad inhibition of host gene transcription, which correlates with a substantial recruitment of essential host transcription factors in viral replication compartments in the nucleus, away from the host chromatin. These data uncover a potent alphaherpesvirus mechanism to interfere with production of host proteins, including proteins involved in antiviral responses.
KEYWORDS: herpesvirus, host gene expression, pseudorabies virus, shutoff
INTRODUCTION
Virus-infected cells prompt an acute set of responses to limit virus replication and to alert neighbor cells and the immune system about the viral infection (1, 2). An excellent paradigm of these defensive strategies is the activation of the nuclear factor kappa B (NF-κB) pathway, a principal cellular signaling axis in innate immunity (3).
In line with this, we found earlier that the porcine alphaherpesvirus pseudorabies virus (PRV) also triggers activation of the NF-κB pathway (4, 5). Alphaherpesviruses constitute the largest subfamily of the herpesvirus family and include, among others, the human pathogens herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) and varicella-zoster virus (VZV), bovine herpesvirus 1 (BHV-1), equine herpesvirus 1 (EHV-1), and feline herpesvirus 1 (FHV-1) (6, 7).
We identified that infection of epithelial cells with PRV leads to a rapid DNA damage response (DDR) within 2 to 4 h postinoculation (hpi), which in turn leads to activation of NF-κB (4, 5). Although the hallmark aspects of NF-κB activation can be observed in PRV-infected cells (i.e., degradation of the NF-κB inhibitory factor IκBα followed by phosphorylation of the NF-κB p65 subunit at Ser536 and nuclear import of p65 in the infected cell nucleus), this does not lead to NF-κB-dependent gene expression (4, 5). Importantly, the transcriptional activity of NF-κB in PRV-infected cells was rescued by the addition of the viral DNA polymerase inhibitor phosphonoacetic acid (PAA), suggesting that shutoff of NF-κB-dependent gene expression is driven by (processes depending on) viral replication (5).
In this report, we demonstrate that PRV-induced inhibition of NF-κB-dependent gene transcription is a consequence of a broader repression of host cell transcription activity in PRV-infected epithelial cells. NF-κB electrophoretic mobility shift assays (EMSAs) show that PRV does not interfere with the capacity of NF-κB to interact with its κB binding sites. However, PRV infection results in substantial sequestration of the NF-κB p65 subunit along with the TATA box binding protein (TBP) and the RNA polymerase II inside virus replication compartments (VRCs) and thereby away from host chromatin, which may constitute the basis of PRV-induced inhibition of (NF-κB-driven) host gene transcription. Host gene expression and subcellular localization of p65, TBP, and RNA polymerase II at sites of host chromatin were restored by the addition of PAA. In addition, we found that PRV infection has remarkable consequences on nuclear physiology, illustrated by the relocation of the linker histone H1 to PRV VRCs, although the reversible acetylation of the nucleosomal histone H3 associated with the highly compacted host DNA in the infected cell is preserved. Hence, this report reveals a broad host cell transcription inhibition caused by PRV infection of epithelial cells, thereby interfering with (antiviral) host cell responses against the virus.
RESULTS
PRV does not affect the ability of NF-κB to interact with κB sites and does not encode a factor that binds κB sequences.
To investigate how PRV infection inhibits NF-κB-dependent gene expression, we assessed the ability of NF-κB in PRV-infected cells to interact with κB sites, a crucial step leading to NF-κB-induced gene transcription. To this end, NF-κB EMSAs were performed to assess the in vitro complexing capacity of NF-κB from PRV-infected cells with κB-responsive elements. The κB-binding capacity of NF-κB in PRV-infected swine testicle (ST) cells was evaluated at 4, 5, 6, 8, and 12 hpi, time points that correlate with the gradually increased and continuous NF-κB activation observed from 4 hpi onwards (Fig. 1A to C). The EMSA data showed that the κB-binding capacity of NF-κB progressively increases throughout PRV infection from 5 hpi onwards (Fig. 1B and C), which fits with the kinetics of PRV-induced IκBα degradation and nuclear import of NF-κB p65 (Fig. 1A) (4). Of note, the ability of NF-κB to bind κB sequences in PRV-infected cells at 8 hpi is comparable to that observed in mock-infected cells stimulated with tumor necrosis factor alpha (TNF-α) for 30 min or doxorubicin for 6 h, further highlighting that PRV does not disrupt κB-binding fitness of NF-κB transcription factors. Furthermore, the NF-κB EMSA results demonstrate that PAA treatment reduces but does not impair the in vitro interaction of NF-κB with κB sites (Fig. 1B and C). This less powerful complexing of NF-κB with κB sites in the presence of PAA is likely the result of the weaker DDR, which is the trigger for NF-κB activation in PRV-infected cells, in line with our earlier observations (5). As a control, Western blotting (WB) confirmed that complete IκBα degradation was achieved by doxorubicin, TNF-α, and PRV infection (Fig. 1D). In line with previous data (5), in PRV-infected cells, PAA restored NF-κB-dependent gene expression and thereby replenishment of the IκBα protein (Fig. 1D).
FIG 1.

Activated NF-κB in PRV-infected cells contains p65 and p50 and is able to bind κB sites in DNA. (A) Merged confocal microscopy pictures of NF-κB p65 (shown in green) and cell nuclei (shown in blue) in PRV-infected ST cells at 4, 6, and 8 hpi (PRV WT Kaplan, MOI of 10). (B) NF-κB EMSA in PRV-infected ST cells treated or not with 400 μg/mL PAA at 4, 5, 6, 8, and 12 hpi (PRV WT Kaplan, MOI of 10) and in ST cells exposed to 5 μM doxorubicin for 6 h or to 100 ng/mL porcine TNF-α for 30 min. (C) Graph representing the semiquantitative analysis of the intensity of protein bands corresponding to NF-κB-κB complexes in the EMSAs of panel B (set relative to mock-infected cells). Means and standard deviations of the results of three independent repeats of the NF-κB EMSA are shown. (D) Western blot analysis of IκBα in PRV-infected ST cells treated or not with 400 μg/mL PAA at 8 and 12 hpi (PRV WT Kaplan, MOI of 10) and in ST cells stimulated with 100 ng/mL porcine TNF-α for 30 min or 5 μM doxorubicin for 6 h. Detection of the PRV proteins US3 and gB was performed to validate successful PAA treatment. (E) NF-κB supershift EMSAs in mock-infected ST cells and in PRV-infected ST cells at 8 hpi, either treated or not with 400 μg/mL PAA. Supershift assays were performed using anti-p65, anti-p50, or rabbit IgG isotype control antibodies and cold κB probes. Arrows indicate locations of supershifted NF-κB complex bands. (F) Western blot analysis of IκBα in PRV-infected ST cells at 6 hpi, treated or not with 10 μM MG132, 50 μM MLN4924, or 10 μM KU60019. MG132 and MLN4924 inhibitors were added at 2 hpi, and KU60019 was added 30 min before inoculation and kept throughout infection. The PRV US3 protein was detected as infection control. (G) NF-κB EMSA in PRV-infected ST cells at 6 hpi, treated or not with 10 μM MG132, 50 μM MLN4924, or 10 μM KU60019, and in ST cells stimulated with 100 ng/mL porcine TNF-α for 30 min. KU60019 was added 30 min before inoculation and kept throughout infection, while MG132 and MLN4924 were added at 2 hpi. (H) Graph representing the semiquantitative analysis of the intensity of protein bands corresponding to NF-κB-κB complexes in the EMSAs of panel G (set relative to mock-infected cells). Means and standard deviations of the results of three independent repeats of the NF-κB EMSA are shown.
Subsequently, we verified the composition of κB-interacting NF-κB complexes in PRV-infected ST cells at 8 hpi by performing supershift EMSAs, making use of anti-p65 and anti-p50 antibodies (Fig. 1E). The results show that most, if not all, of the NF-κB complexes that interact in vitro with κB sites contain the p65 subunit of NF-κB, confirming that the NF-κB p65 that is imported in the nucleus of PRV-infected ST cells is apt to bind κB sequences. Moreover, a supershifted migration of NF-κB-κB complexes (NF-κB bound to κB sequences) was also observed using the anti-p50 antibody (Fig. 1E), indicating that the p50 subunit of NF-κB also makes part of NF-κB complexes in PRV-infected cells. Hence, the major NF-κB complex induced by PRV infection appears to contain the p65/p50 heterodimer, arguing against the possibility that NF-κB complexes formed in PRV-infected cells would be intrinsically repressive, as such repressive complexes are typically composed of NF-κB dimers that lack the transactivation domain that is present only in Rel proteins like NF-κB p65 (RelA) (8). In addition, the assay further confirmed the NF-κB EMSA results by the elimination of NF-κB-κB complexes using cold probes as competitive inhibitors in PRV-infected cell lysates (Fig. 1E).
These EMSA data indicate that the inability of NF-κB to trigger gene expression in PRV-infected cells cannot be explained by a lack of κB-binding capacity. Alternatively, PRV infection possibly may result in an occupancy of κB sites, e.g., by PRV proteins, thereby interfering with the NF-κB-κB interaction. To assess this hypothesis, we explored the binding of other (viral) factors to κB sites in the absence of NF-κB activation in PRV-infected cells. To this end, NF-κB activation in PRV-infected cells was suppressed by the use of three independent approaches using (i) the proteasome inhibitor MG132 to inhibit IκBα degradation, (ii) the Nedd8 inhibitor MLN4924, which inhibits IκBα ubiquitination and degradation, or (iii) the ATM inhibitor KU60019, which inhibits the ATM-dependent DDR and therefore PRV-induced DDR-dependent NF-κB activation (Fig. 1F). Notably, none of these inhibitors had a deleterious impact on virus replication, as illustrated by similar expression levels of the viral US3 protein in the absence or presence of the inhibitors (Fig. 1F) and similar expression levels of late viral proteins (4, 5). All three inhibitors abolished the κB-binding capacity of NF-κB in PRV-infected cells, and EMSAs showed no alternative proteins interacting with κB sites in the presence of these inhibitors (Fig. 1G and H). Hence, the data do not suggest a possible competitive inhibition by PRV(-induced) proteins for the binding with κB sites.
NF-κB p65, TATA box binding protein (TBP), and RNA polymerase II are largely sequestered inside PRV replication compartments.
The intact κB-binding capacity of NF-κB during PRV infection in combination with the lack of NF-κB-dependent host gene expression suggests that NF-κB may not have efficient access to host target genes in PRV-infected cells. To address this, the relative position of NF-κB p65 with respect to nuclear chromatin and RNA polymerase II, which is indispensable for (NF-κB-induced) gene transcription, was assessed by confocal microscopy. First, the formation of virus replication compartments (VRCs) in the nucleus of PRV-infected cells was evaluated. To this end, we used a PRV strain encoding a viral DNA-binding ICP8 protein fused to enhanced green fluorescent protein (eGFP), which had been used before to visualize the formation of PRV VRCs (9). In line with this earlier report, we observed that from around 4 hpi onwards, PRV infection leads to the formation and expansion of PRV VRCs in the nucleus, which leads to major host cell chromatin marginalization (Fig. 2A). Using host cell chromatin marginalization as a proxy for VRC localization, a PRV strain encoding an eGFP-tagged VP26 capsid protein (10) confirmed that the expanding VRCs at 8 hpi are filled with viral capsid protein (Fig. 2A).
FIG 2.

PRV infection results in substantial sequestration of NF-κB p65, RNA polymerase II, and TATA box binding protein (TBP) in nuclear virus replication compartments (VRCs). (A) Confocal microscopy pictures of ST cells infected with PRV ICP8-GFP (PRV 926, MOI of 10) at 4, 6, and 8 hpi or with PRV VP26-GFP (PRV GS443, MOI of 10) at 8 hpi. Viral proteins fused to GFP are shown in green, and the counterstained cell nuclei are represented in gray (upper row) or blue (lower row). Bar, 10 μm. (B) Confocal microscopy pictures of RNA polymerase II (in green) and NF-κB p65 (in red) in ST cells infected with PRV at 6 hpi (PRV WT Kaplan, MOI of 10) or treated with 100 ng/mL porcine TNF-α for 30 min. Cell nuclei are shown in gray. Bar, 50 μm. (C) Confocal microscopy pictures of RNA polymerase II (in green) in PRV-infected ST, SK-6, and PPK cells (PRV WT Kaplan, MOI of 10). ST and PPK cells were fixed at 6 hpi, while SK-6 cells were fixed at 8 hpi. The counterstained cell nuclei are shown in gray. Bar, 50 μm. (D) Confocal microscopy pictures of PKK cells stained for pancytokeratin (in green). Cell nuclei are shown in blue. Bar, 50 μm. (E) Confocal microscopy pictures of TBP (in green) in PRV-infected PPK, ST, and SK-6 cells at 6 hpi (ST and PPK) and 8 hpi (SK-6) (PRV WT Kaplan, MOI of 10). The counterstained cell nuclei are shown in gray. Bar, 50 μm. (F) Western blot analysis of RNA polymerase II and TBP in mock-infected and PRV-infected ST cells at 8 hpi (PRV WT Kaplan, MOI of 10). Detection of the viral gE protein served as the infection control, whereas H3 and α-tubulin protein levels were used as loading controls for nuclear and cytoplasmic proteins, respectively.
Again by the use of host cell chromatin marginalization as a proxy for VRC formation, nuclear distribution of NF-κB p65 was found to largely correspond with PRV VRCs in PRV-infected ST cells at 6 hpi. In addition, and remarkably, RNA polymerase II also substantially accumulates inside VRCs and is apparently physically largely disconnected from host chromatin (Fig. 2B and C). Importantly, the sequestration of RNA polymerase II in VRCs was confirmed in PRV-infected primary porcine kidney (PPK) cells as well as SK-6 swine kidney cells, indicating that this is not a cell type-dependent phenotype (Fig. 2C). As a control for purity of the primary PPK cell population, the epithelial cell marker cytokeratin was stained (Fig. 2D). Addition of PAA suppressed the formation of noticeable VRCs and restored the colocalization of cellular DNA with RNA polymerase II (Fig. 2C). We also consistently observed an increased cytoplasmic signal for RNA polymerase II in PRV-infected cells compared to mock-infected cells. Although speculative, this possibly may point to disturbed nucleocytoplasmic transport in infected cells. In any case, these findings suggest that PRV infection leads to the inhibition of NF-κB-induced gene transcription by preventing efficient access of NF-κB p65 as well as RNA polymerase II to the host genome. These data also suggest that the inhibition of host gene expression may stretch well beyond NF-κB-dependent gene expression and may involve most host genes expressed via RNA polymerase II.
To assess whether TBP, another key host protein that is involved in transcription and acts upstream of RNA polymerase II, was also recruited to VRCs, and additional immunofluorescence (IF) assays were performed. TBP constitutes a subunit of the general transcription factor TFIID and the most upstream factor of the transcriptional cascade, as it directly interacts with cellular gene promoters and is critical for the subsequent placement of RNA polymerase II on transcription start sites (11–13). Just like RNA polymerase II, TBP was mainly distributed within VRCs in the various cell types infected with PRV. Again, inhibition of VRC formation by PAA rescued the access of TBP to nuclear host DNA (Fig. 2E). As a control, Western blot analyses indicate that protein levels and electrophoretic mobility of RNA polymerase II and TBP are not affected in PRV-infected ST cells at 8 hpi (Fig. 2F). In conclusion, the substantial recruitment of TBP and RNA polymerase II in VRCs point toward a broad appropriation of the cellular transcription machinery by PRV.
PRV infection induces a PAA-sensitive broad shutoff of host transcription.
The mismatch between nuclear distribution of host chromatin and factors that are required for (NF-κB-dependent) transcription suggests that PRV induces a broad repression of host transcription. Taking into consideration that PAA treatment leads to the rescue of both colocalization of RNA polymerase II and TBP with host DNA and NF-κB-dependent gene transcription, we hypothesized that such broad virus-induced inhibition of host transcription would likely be PAA sensitive. To assess this, cellular transcription activity was evaluated by reverse transcription-quantitative PCR (RT-qPCR) analyses for quantification of prespliced transcripts corresponding to different genes. Analyzing prespliced transcripts rather than mature transcripts allows the accurate estimation of transcription efficiency without confounding factors, such as potential differences in transcript degradation. We selected IκBα as a readout of NF-κB-induced gene transcription, and the genes coding for GAPDH (glyceraldehyde-3-phosphate dehydrogenase), B2M (β2-microglobulin), and PGK1 (phosphoglycerate kinase 1) were chosen as constitutively expressed genes for the measurement of basal host gene transcription. These three constitutive genes are typically used as housekeeping genes and are located on different chromosomes. RT-qPCR assays conducted to assess prespliced mRNA levels were based on the generation of amplicons corresponding to sequences in the intron-exon boundaries that are present only in unspliced transcripts. Interestingly, although statistically significant differences were observed only for the GAPDH gene, transcription of all three constitutively expressed genes appeared to be reduced in PRV-infected cells at 8 hpi and, in the case of the GAPDH gene, already at 6 hpi, in line with a general shutoff of host gene expression in PRV-infected cells (Fig. 3A). Importantly, and in agreement with the earlier described PAA-dependent release of NF-κB-induced gene expression, addition of PAA rescued the expression of the different constitutively expressed host genes in PRV-infected cells, without increasing their expression in mock-infected cells. In the case of the B2M and PGK1 genes, addition of PAA to PRV-infected cells even resulted in a markedly (albeit not statistically significant) increased transcription at 6 hpi, suggesting that virus induced increased transcriptional activity at this time point. As a control, addition of the transcription inhibitor actinomycin D from 6 to 8 hpi suppressed the increase of constitutive gene transcription by PAA in this time window in PRV-infected ST cells (Fig. 3A). When transcriptional rates for the different constitutively expressed genes are examined at different time points postinoculation, consistent with our other data, host transcriptional activity drops substantially and significantly for the GAPDH, B2M, and PGK1 genes from 4 hpi onwards (Fig. 3B and C). Independent data sets were used to generate Fig. 3B and C, which accounts for the slight discrepancies at the 6-hpi time point observed in the two figure panels.
FIG 3.
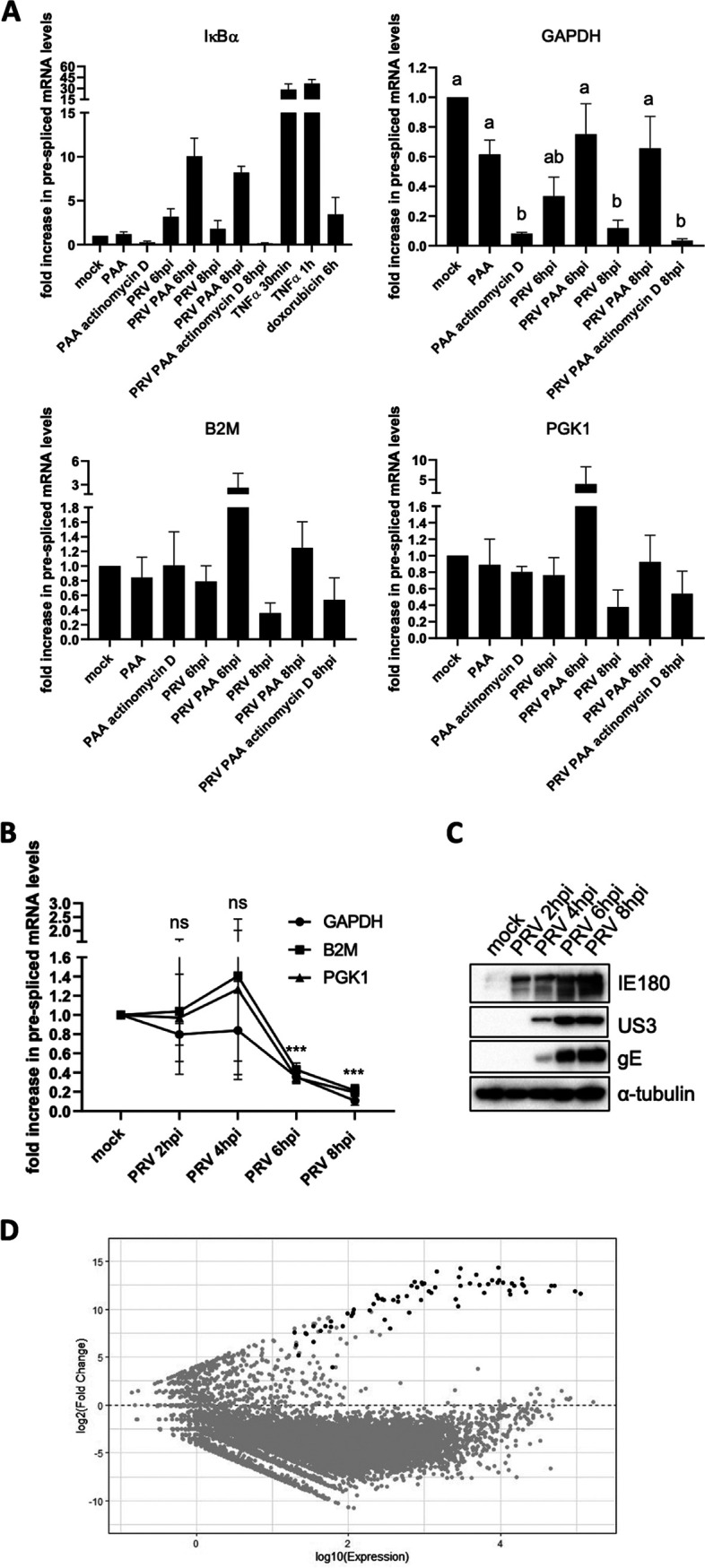
PRV infection results in broad inhibition of host gene expression. (A) Graphs showing the means and standard deviation of results of three independent repeats of quantification of the fold difference in prespliced transcript levels versus the mock condition, determined via RT-qPCR. Results are shown for prespliced transcripts corresponding to the IκBα, GAPDH, B2M, and PGK1 genes. ST cells were either mock infected or PRV infected (6 and 8 hpi with PRV WT Kaplan, MOI of 10) and treated or not with 400 μg/mL PAA or 5 μg/mL actinomycin D. PAA was added 30 min prior to inoculation and maintained throughout infection, while actinomycin D was added from 6 to 8 hpi. In mock-infected cells, the treatment duration was identical to that in infected cells. As positive controls for IκBα gene transcription, ST cells were treated with 100 ng/mL porcine TNF-α for 30 min or 5 μM doxorubicin for 6 h, respectively. Different letters in the GAPDH graph indicate conditions that are statistically significantly different from each other (P < 0.05). (B) Graphs showing mean and standard deviation of results of three independent repeats of RT-qPCR-based quantification of prespliced mRNA levels corresponding to GAPDH, B2M, and PGK1 genes in PRV-infected ST cells at 2, 4, 6, and 8 hpi (PRV WT Kaplan, MOI of 10). Statistically significant differences in results compared to mock-infected cells are indicated (ns, nonsignificant; ***, P < 0.001). (C) Western blot analysis of the PRV proteins IE180, US3, and gE in PRV-infected ST cells at 2, 4, 6, and 8 hpi (PRV WT Kaplan, MOI of 10). (D) MA plot analysis of RNA-seq data sets of ST cells infected with PRV WT NIA3 at 16 hpi (MOI of 10) compared to transcript levels found in mock-infected cells. Gray dots indicate cellular transcripts, whereas black dots correspond to viral transcripts.
To confirm that PRV infection of ST cells causes a broad inhibition of cellular transcription, we analyzed RNA sequencing (RNA-seq) data of PRV-infected ST cells at 16 hpi. Figure 3D confirms that PRV infection of ST cells leads to a broad downregulation of host transcripts, with >75% of cellular transcripts being significantly downregulated, whereas less than 1% of cellular transcripts are upregulated. In agreement with our RT-qPCR data on the corresponding pre-mRNAs (Fig. 3A and B), GAPDH, B2M, and PGK1 mRNAs were significantly reduced in the RNA-seq data set. Further in line with earlier RT-qPCR data (4), the RNA-seq data indicated that also transcripts corresponding to the A20 and IκBα NF-κB-inducible genes were statistically significantly downregulated in PRV-infected ST cells compared to mock-infected cells, Interleukin-6 (IL-6) was downregulated albeit not statistically significantly and TNF-α was not detectably expressed in any sample. Although the RNA-seq data were generated at a late time point in infection (16 hpi) and time course analyses would be needed to carefully assess this, we hypothesize that the small percentage (<1%) of upregulated cellular transcripts may be the result of their enhanced transcription early in infection, prior to the inhibition of host gene transcription by PRV. In conclusion, these data demonstrate that the PAA-sensitive shutoff of NF-κB-dependent gene transcription is at least partially and possibly entirely due to a broad inhibition of host gene transcription in PRV-infected cells, probably driven by the formation and expansion of VRCs and the sequestration of the host transcription complex in these VRCs.
The PRV-induced shutoff of host gene transcription does not appear to depend on the impermeability of cellular chromatin or on the PRV-induced DNA damage response.
The inability of TBP and RNA polymerase II, largely retained inside PRV VRCs, to efficiently access the host chromatin may possibly rely on an impermeability of the densely compacted cellular genome to any type of protein. To evaluate this, the relative positions of histones and the modularity of histone epigenetics were evaluated. As illustrated in Fig. 4A, the core histone H3, composing the nucleosome, localizes mostly at sites of marginalized host chromatin in PRV-infected ST cells at 6 hpi. The linker histone H1 also colocalizes substantially with marginalized cellular DNA but in addition is present in PRV VRCs at 6 hpi and also, although limitedly, at 10 hpi (Fig. 4A). Since H3 does not appear to detach substantially from host chromatin, we analyzed whether enzymes affecting histone H3 acetylation still have access to host chromatin by inducing the accumulation of acetyl-H3 using the broad-spectrum histone deacetylase (HDAC) inhibitor trichostatin A (TSA). Interestingly, TSA was able to trigger H3 acetylation at lysine 27 (H3K27ac) when added to PRV-infected ST cells either before (from 2 to 6 hpi) or after (from 8 to 12 hpi) the appearance of chromatin marginalization and the virus-induced shutoff of host gene transcription (Fig. 4B and C). This suggests that histone acetyltransferases and/or deacetylases are active despite the compression of cellular chromatin, indicating that marginalized host chromatin in PRV-infected cells is not impermeable to nuclear enzymes.
FIG 4.

PRV-induced suppression of host transcription is not caused by HDAC-mediated epigenetic silencing. (A) Confocal microscopy pictures of histone 3 (H3, upper panel) and histone 1 (H1, lower panel) at 6 hpi and 10 hpi in PRV-infected ST cells (PRV WT Kaplan, MOI of 10). Histones are shown in green, and cell nuclei are shown in blue. Pictures in the third column show zoomed regions indicated by white dashed boxes. Bar, 50 μm. (B and C) Western blot analysis of acetylated H3 at lysine 27 (H3K27ac) and total H3 levels in mock-infected or PRV-infected ST cells (PRV WT Kaplan, MOI of 10) that were either exposed or not to 5 μM trichostatin (TSA) for 4 h. In panel B, PRV-infected cells were incubated with TSA from 2 to 6 hpi, while in panel C, TSA was added from 8 to 12 hpi. The levels of PRV proteins IE180 (immediate early protein), US3 (early protein), and gE (late protein) (panel B) or of gE (panel C) were assessed as infection controls. (D) Upper two rows, confocal microscopy images of ST cells at 6 hpi with PRV ICP8-GFP (PRV 926, MOI of 10), treated or not with 5 μM TSA from 2 to 6 hpi. Cell nuclei are shown in blue. Bar, 50 μm. Lower row, confocal microscopy pictures of Hoechst-stained PRV-infected ST cell nuclei (PRV WT Kaplan, MOI of 10) at 6 hpi, treated or not with 5 μM TSA from 2 to 6 hpi. Bar, 50 μm. (E) Western blot analysis of H3K27ac in PRV-infected ST cells (PRV WT Kaplan, MOI of 10 PFU/cell) at 6 and 8 hpi, treated or not with 400 μg/mL PAA. PAA treatment was initiated 30 min before infection and was kept during the entire course of infection. As a positive control for H3 acetylation, treatment of mock-infected ST cells with 5 μM TSA was included. Total H3 proteins were assessed as a loading control, whereas viral US3 and gB proteins served as infection controls and confirmed successful PAA treatment. (F) Western blot analysis of the DDR marker phosphorylated histone H2A.X at serine residue 139 (also termed γH2A.X) in PRV-infected ST cells (PRV WT Kaplan, MOI of 10) at 8 hpi, treated or not with 10 μM ATM inhibitor KU60019. KU60019 treatment was initiated 30 min prior to infection and maintained during the infection. Total H2A.X and PRV gE protein levels were assayed as loading and infection controls, respectively. (G) Graphs showing means and standard deviations of results of three independent repeats of RT-qPCR-based quantification of prespliced mRNA corresponding to GAPDH, B2M, and PGK1 genes in mock- or PRV-infected ST cells (PRV WT Kaplan, MOI of 10 PFU/cell) at 8 hpi, treated or not with 10 μM KU60019 or 5 μM TSA. TSA was added at 2 hpi, while KU60019 was added 30 min before virus inoculation and kept throughout the infection. Different letters indicate conditions that are statistically significantly different from each other (P < 0.05).
The TSA-induced relaxation of host DNA did not noticeably affect (ICP8-GFP-positive) PRV VRC growth or chromatin marginalization (Fig. 4D), suggesting that the marginalization of cellular chromatin likely depends on mechanical forces and not on epigenetic host chromatin compaction. In addition, and in line with this, the release of the PRV-induced shutoff of cellular gene transcription by PAA is not associated with noticeable PAA-mediated effects on H3 K27 acetylation (Fig. 4E). Finally, to assess whether HDACs and/or the PRV-induced DDR contribute to the virus-induced shutoff of host gene expression, we tested whether the repression of host gene transcription could be released by TSA-induced inhibition of HDACs or by blocking the massive PRV-induced DDR using the ATM inhibitor KU60019 (Fig. 4F). As shown in Fig. 4G, neither inhibition of HDACs nor ATM restored transcription levels of GAPDH, B2M, or PGK1 genes in PRV-infected ST cells at 8 hpi. In conclusion, neither histone deacetylation nor the PRV-induced DDR causes the virus-induced shutoff of cellular gene transcription, further pointing toward the substantial sequestration of key host cell transcription factors in PRV VRCs as a possible cause for this suppression.
PRV-induced NF-κB activation does not affect the transcription of viral genes that contain potential κB sites upstream of their start codon.
Although PRV infection triggers nuclear recruitment of p65/p50 NF-κB heterodimers that are able to bind κB sites, at least in vitro (Fig. 1B and E), this does not result in upregulation of the transcription of prototypical NF-κB-driven cellular genes due to the virus-induced shutoff of host gene expression. Taking into consideration that nuclear NF-κB p65 in PRV-infected cell nuclei is distributed mainly within VRCs (Fig. 1A and B), it is possible that NF-κB complexes may interact with viral genomes to enhance transcription of specific PRV genes. To address this hypothesis, the PRV Kaplan genome was analyzed for the presence of possible κB sites by using the consensus sequence 5′-GGGRNWYYCC-3′, in which N represents any base, R represents any purine, Y represents any pyrimidine, and W may be an adenine or a thymine (8, 14). This in silico analysis rendered 36 potential κB sites in the PRV genome, of which only 9 were located within the upstream 600 bp of the start codon of a viral gene, either overlapping with other viral open reading frames (ORFs) or present in noncoding regions of the genome (Table 1). These 9 κB sites were possibly controlling the expression of 6 PRV genes, i.e., US1 (at bp −577 and −405 in the two copies of the gene), US4 (at bp −152), UL24 (at bp −425), UL39 (at bp −41), UL50 (at bp −87), and UL51 (at bp −290 ) (Table 1). Evaluation of the mRNA expression kinetics of these 6 viral genes indicated that the peak in mRNA levels already takes place at 4 hpi for the UL39, UL50, and UL51 genes. Increased transcription from 4 hpi onwards, albeit limited, was observed only for US1, US4, and UL24 genes (Fig. 5A). Since our earlier and current results indicate that noticeable NF-κB activation and NF-κB interaction with κB sites were detected only from 4 hpi onwards (4, 5) (Fig. 1B), this suggests that viral gene transcription prior to 4 hpi may not be directly associated with NF-κB activity. To assess whether PRV-induced NF-κB activation was involved in expression of any of these 6 viral genes, the corresponding mRNA levels were quantified at 4 hpi and 8 hpi in the absence or presence of the ATM inhibitor KU60019, which efficiently blocks PRV-induced DDR-dependent NF-κB activation (Fig. 1F to H and 5B) (5). Figure 5C shows that none of the candidate genes displayed reduced transcription at either 4 hpi or 8 hpi upon treatment with the ATM inhibitor KU60019, suggesting that NF-κB does not contribute to transcription of these genes. To confirm this, we assessed mRNA levels of the selected viral genes in PRV-infected ST cells that were either knocked down or not for NF-κB p65 via small interfering RNA (siRNA). As described above, the NF-κB p65 is an essential component of the NF-κB complexes induced by PRV (Fig. 1E). In agreement with the assays conducted using the ATM inhibitor KU60019, NF-κB p65 knockdown did not affect transcription of the six selected viral genes, either at 4 hpi or 8 hpi (Fig. 5D and E). Although this further indicates that NF-κB does not contribute to PRV gene expression, we cannot exclude at this point that κB-binding sites in the viral genome may interact with the NF-κB complexes located in the VRCs for a purpose(s) other than the transcription of the currently selected viral genes.
TABLE 1.
Potential κB sequences present in the PRV Kaplan genomea
| Sequence (5′–3′) | Strand | Positions | Location |
|---|---|---|---|
| GGGGCTCTCC | − | 698–707 | Noncoding region |
| GGGGCTCCCC | + | 7430–7439 | 290 bp upstream of UL51 ORF |
| GGGACTCCCC | − | 9468–9477 | 87 bp upstream of UL50 ORF |
| GGGGTTTCCC | + | 32711–32720 | Noncoding region |
| GGGGTTTCCC | − | 32711–32720 | Noncoding region |
| GGGGCTCCCC | + | 33949–33958 | Noncoding region |
| GGGGATCTCC | + | 36332–36341 | Noncoding region |
| GGGAGATCCC | − | 36333–36342 | Overlapping with the UL36 ORF |
| GGGGCTCCCC | − | 44025–44034 | Overlapping with the UL37 ORF |
| GGGACTCTCC | + | 46722–46731 | 41 bp upstream of the UL39 ORF |
| GGGGCTCCCC | − | 60095–60104 | 425 bp upstream of the UL24 ORF |
| GGGACTCTCC | + | 64238–64247 | Noncoding region |
| GGGAATCCCC | − | 64324–64333 | Noncoding region |
| GGGAATCCCC | + | 64324–64333 | Noncoding region |
| GGGGGTTTCC | + | 95406–95415 | Noncoding region |
| GGGAAACCCC | − | 95407–95416 | Overlapping with the UL1 ORF |
| GGGGGATTCC | + | 95788–95797 | Noncoding region |
| GGGGGATCCC | + | 96697–96706 | Noncoding region |
| GGGGCTCCCC | + | 97975–97984 | Noncoding region |
| GGGGGTCCCC | + | 100847–100856 | Noncoding region |
| GGGGCTCTCC | − | 104975–104984 | Overlapping with the IE180 ORF |
| GGGAGTCCCC | + | 106180–106187 | Noncoding region |
| GGGGGATTCC | + | 114358–114367 | Internal repeat sequence |
| GGGAATCCCC | − | 114359–114368 | Noncoding region |
| GGGGGATCCC | + | 115604–115613 | Internal repeat sequence, 577 bp upstream of the US1 ORF |
| GGGGGATCCC | − | 115606–115615 | Noncoding region |
| GGGGCTTCCC | + | 115776–115785 | Internal repeat sequence, 405 bp upstream of the US1 ORF |
| GGGGCTTTCC | + | 119313–119322 | Overlapping with the US3 ORF, 152 bp upstream of the US4 ORF |
| GGGGCTCCCC | − | 126209–126218 | Noncoding region |
| GGGGCTTCCC | − | 128954–128963 | 405 bp upstream of the US1 ORF |
| GGGGGATCCC | + | 129124–129333 | Terminal repeat sequence |
| GGGGGATCCC | − | 129126–129135 | 577 bp upstream of the US1 ORF |
| GGGAATCCCC | + | 130371–130380 | Terminal repeat sequence |
| GGGGGATTCC | − | 130372–130381 | Noncoding region |
| GGGAGTCCCC | − | 138550–138559 | Noncoding region |
| GGGGCTCTCC | + | 139755–139764 | Overlapping with the IE180 ORF |
Sequences are indicated from the 5′ to the 3′ end of the corresponding DNA strand. The nucleotide positions are defined based on the positive DNA strand from the 5′ to 3′ end of the PRV Kaplan genome.
FIG 5.

Knockdown of NF-κB p65 or inhibition of ATM does not affect expression of PRV genes that contain potential κB sites upstream of their start codon. (A) Graphs show the accumulation of transcripts corresponding to the UL51, UL50, UL39, UL24, US1, and US4 viral genes via RT-qPCR assays throughout the first 8 h of infection in ST cells (PRV WT Kaplan, MOI of 10). Viral mRNA copy numbers are indicated per 12.5 ng of total RNA. The dotted lines indicate the amount of viral mRNA reached within the first 4 h of infection, before noticeable NF-κB activation. (B) Confocal microscopy images showing NF-κB p65 (green) and PRV gB (red) in mock-infected ST cells and in PRV-infected ST cells at 8 hpi (PRV WT Kaplan, MOI of 10), either treated with 10 μM KU60019 or not. KU60019 was added 30 min before virus inoculation and kept throughout the infection. Cell nuclei are shown in blue. Bar, 50 μm. (C) Graphs representing the mean and standard deviation of results of three independent repeats of quantification of mRNA levels corresponding to the UL51, UL50, UL39, UL24, US1, and US4 viral genes, determined via RT-qPCR in PRV-infected ST cells at 4 hpi and 8 hpi (PRV WT Kaplan, MOI of 10), treated (gray bars) or not (black bars) with 10 μM KU60019. KU60019 was added 30 min before virus inoculation and kept throughout the infection. Viral mRNA copy numbers are indicated per 12.5 ng of total RNA. (D) Western blot analysis of NF-κB p65 at 4 hpi and 8 hpi (PRV WT Kaplan, MOI of 10) in PRV-infected ST cells that were either knocked down or not for NF-κB p65 via siRNA. Infection was monitored by detection of the viral IE180 and gE proteins. (E) Graphs representing the means and standard deviations of results of three independent repeats of viral transcript quantitation via RT-qPCR assays for the UL51, UL50, UL39, UL24, US1, and US4 genes at 4 hpi and 8 hpi (PRV WT Kaplan, MOI of 10) in PRV-infected ST cells that were either knocked down (gray bars) or not (black bars) for NF-κB p65 via siRNA. Viral mRNA copy numbers are indicated per 12.5 ng of total RNA.
DISCUSSION
PRV infection of porcine epithelial cells induces a DDR-dependent activation of the NF-κB signaling pathway that culminates in the nuclear import of the NF-κB p65 subunit but does not result in an upregulated transcription of NF-κB-responsive genes (4, 5). Here, we show that this lack of NF-κB-dependent gene expression is not due to a PRV-induced inability of NF-κB complexes, which contain predominantly p65/p50 heterodimers, to interact with NF-κB-responsive DNA sequences (κB sites) and does not appear to involve virus-encoded competitive inhibitors that bind κB sites. Instead, the shutoff of NF-κB-induced gene transcription is very likely the result of a more general PRV-induced suppression of host gene transcription that we describe here and that also affects constitutively expressed housekeeping genes. This broad shutoff of host gene expression correlates with a spatial sequestration of critical host transcription factors (including TBP, RNA polymerase II, and NF-κB p65) in virus replication compartments in the nucleus, likely preventing their efficient access to host chromatin.
HSV-infected human epithelial cells have been reported to trigger an aberrant and persisting NF-κB activation, similar to what we observe in PRV-infected epithelial cells. Interestingly, EMSAs in these reports also indicated an unaffected ability of activated NF-κB to bind κB sites throughout the course of the infection without leading to NF-κB-dependent gene transcription or to IκBα protein replenishment (15, 16), in line with what we describe for PRV. However, other studies suggest that HSV-1-induced persistent NF-κB activation may be to some extent productive, since it has been reported to prevent virus-induced apoptosis and to facilitate virus egress from HSV-1-infected epithelial cells (17, 18). The differences in NF-κB transcription activity may possibly be cell type dependent, as the pathway seems to be productive in HSV-1-infected monocytic cells (19–22). Amici et al. explained the lack of IκBα protein replenishment in HSV-1-infected epithelial cells by the redirection of NF-κB factors from cellular to viral gene promoters, in particular that of the ICP0 gene, to enhance virus replication (23). Other studies suggest that the proviral effect of HSV-1-induced NF-κB activation may be a consequence of improved virus release without affecting viral protein expression (15, 18, 24). In any case, our data indicate that in PRV-infected epithelial cells, the DDR/NF-κB signaling axis activation does not affect virus replication efficiency or infectious virus production and does not seem to affect expression of viral genes that harbor potential κB sites in the region upstream of their start codon, at least in cell culture (4, 5).
The suppression of NF-κB transcriptional activity in PRV-infected cells correlates with a substantially reduced constitutive expression of the GAPDH, B2M, and PGK1 housekeeping genes. HSV-1 has also been reported to induce a shutoff of cellular gene transcription starting very early in infection and mediated by diverse immediate early proteins such as ICP0, ICP4, and ICP27 (25). In PRV-infected ST cells, the onset of inhibition of host gene transcription coincides with the initiation of exponential viral DNA replication and L protein expression and is prevented by the addition of the DNA polymerase inhibitor phosphonoacetic acid (PAA). In PRV infection, the protein expression of the HSV-1 ICP4 homolog, the IE180 protein, is not substantially reduced by PAA treatment, in line with its expression kinetics as an immediate early gene (4, 5). Moreover, PAA treatment also does not affect transcription of the two early EP0 and UL54 genes, which encode the protein homologs of ICP0 and ICP27 of HSV-1, respectively (data not shown), indicating that the mechanisms underlying host gene transcription repression differ in PRV and HSV-1. In agreement with our data in PRV-infected cells, Jenkins and Spencer showed that TBP, RNA polymerase II, and also different subunits of TFIIE (p56E) and TFIIH (p62H) were concentrated inside (ICP4 positive) HSV-1 VRCs (26). In addition, the retention of TBP and TBP-associated factors (hsTAF1 and hsTAF4) within HSV-1 replication factories was reversed by PAA-dependent abrogation of VRC growth (27). Hence, although there may be (possibly cell type-dependent) differences in this respect, both PRV and HSV-1 have the capacity to trigger a PAA-sensitive sequestration of critical host cell transcription factors in VRCs, thereby complicating their access to host chromatin.
Several HSV-1 proteins affect the activity of RNA polymerase II (e.g., phosphorylation status of the large subunit of RNA polymerase II) (25–33) and general transcription factors (GTFs) involved in viral gene expression, such as ICP4 (26, 30, 34–42), VP16 (43, 44), ICP22, UL13 (30–33), ICP0 (26), ICP27, and ICP8 (26, 45). The IE62 protein of VZV also cooperatively works with the cellular transcription machinery to regulate viral gene expression (46–51). For PRV, it is known that the IE180 protein promotes RNA polymerase II-dependent gene transcription by modulating the activity of other (cellular) transcription factors, particularly via formation of the preinitiation complex (52, 53). However, as discussed above, a potential complexing of IE180 protein or viral E proteins with factors of the host cell transcription complex does not explain the shutoff of cellular gene transcription in PRV-infected cells, as this shutoff is effectively prevented by PAA treatment, which does not affect viral IE and E protein expression. Therefore, we hypothesize that, instead, the PAA-sensitive virus-induced sequestration of RNA polymerase II and GTFs within the growing PRV VRCs may be the main reason for the PRV-induced suppression of host gene transcription.
Our data suggest that the permeability of marginalized cellular DNA toward DNA-targeting enzymes, like histone deacetylases, does not seem to be an obstacle and therefore likely does not explain the apparent lack of association of RNA polymerase II and GTPs with host chromatin. Therefore, we speculate that the preferential transcription of viral genes rather than cellular genes may rely on an the association of PRV factors with the transcription machinery, thereby leading to retention of these host factors inside the PAA-sensitive VRCs.
Interestingly, earlier microarray-based transcriptome studies of PRV-infected porcine PK-15 epithelial cells also indicated a broad shutoff of host gene expression between 4 hpi and 8 hpi (54, 55), which is in line with our current results. In addition, microarray-based transcriptome analyses of PRV-infected cells of other species (i.e., rat embryonic fibroblasts and human HEK-293 cells) also pointed to virus-induced suppression of host gene expression at later stages of infection (56–58). Our RNA-seq analysis indicated that at late stages of infection, <1% of host genes showed statistically increased expression. Although the RNA-seq data were generated at a late time point in infection (16 hpi) and additional time course experiments are needed to assess this carefully, it is possible that this very low percentage of upregulated cellular transcripts may in fact represent transcripts that were upregulated earlier in infection, prior to the inhibition of host gene transcription by PRV, and/or whether some host genes escape the broad inhibition of host gene transcription from 4 hpi onwards in PRV-infected porcine epithelial cells. Of note, some of the upregulated host genes that were identified in other, microarray-based transcriptome studies of PRV-infected cells and tissues were also found to be upregulated in our RNA-seq data set, such as the COX1, ARC, and SLC4A1 genes (56, 59). Ray et al. have demonstrated earlier that the COX1 gene, which encodes cyclooxygenase 1, contributes to PRV replication in rat embryo fibroblasts (60). Hence, it might be interesting in future assays to carefully identify host genes that are consistently upregulated in several of these transcriptome studies and determine their putative biological role in PRV infection. Of note, HSV-1 infection has been reported to impair transcription termination (61). It will be interesting in future research to assess whether transcription termination is also suppressed in PRV-infected cells, as this may further add to host shutoff in addition to the reduced levels of prespliced host mRNA levels that we observe here.
In conclusion, we report that PRV infection of porcine epithelial cells triggers a broad shutoff of cellular gene transcription. Since this broad inhibition of host gene expression appears to be highly efficient at repressing NF-κB-dependent proinflammatory gene transcription in PRV-infected epithelial cells, we assume that other (antiviral) inducible pathways may be similarly hindered from 4 hpi onwards. Hence, this broad inhibition of host gene expression in infected epithelial cells may serve as a wide-scope mechanism to inhibit innate immune responses at mucosal epithelia that represent the primary site of PRV infection.
MATERIALS AND METHODS
Cells and viruses.
Swine testicle (ST) cells (ATCC CRL-1746) were cultured in modified Eagle’s medium (MEM) supplemented with 10% inactivated fetal bovine serum (FBS), 100 U/mL penicillin, 0.1 mg/mL streptomycin, 50 μg/mL gentamicin, and 1 mM sodium pyruvate (all from Gibco, Thermo Fisher Scientific) (ST medium). A swine kidney (SK-6) cell line (62) was cultivated in MEM supplemented with 10% FBS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 50 μg/mL gentamicin (SK medium).
Porcine primary epithelial kidney (PPK) cells were isolated from 18-week-old piglets (Sus scrofa). In brief, after the removal of the renal capsule and the outer layer of the kidney cortex, the intermediate layer of the kidney cortex was fragmented in small pieces and sequentially digested with trypsin at 2.5 mg/mL, resulting in a single-cell suspension. The primary kidney cells collected from several rounds of trypsinization were cultured in PPK medium consisting of MEM (with l-glutamine) supplemented with 10% FBS, 100U/mL penicillin, 0.1 mg/mL streptomycin, 50 μg/mL gentamicin, and 0.05% (vol/wt) lactoalbumin hydrolysate until full cell confluence was achieved. A total of 800,000 PPK cells were seeded on each well of a 24 well plate well, and the next day, the confluent cell monolayer was used to perform the corresponding assay.
The wild-type (WT) PRV strain used was PRV WT Kaplan (63). The PRV GS443 strain expresses PRV VP26 capsid protein fused to GFP (VP26-GFP) by coupling the PRV UL35 open reading frame to that of GFP in the PRV WT Becker backbone (10). The PRV 926 strain was also originated from PRV WT Becker and encodes the PRV ICP8-GFP protein (and VP26-mRFP), in which the PRV UL29 and GFP open reading frames were fused (9). PRV GS443 virus was kindly provided by G. A. Smith (Northwestern University, IL, USA), and PRV 926 was a generous gift from J. B. Bosse (Centre for Structural Systems Biology, Hamburg, Germany) and L. W. Enquist (Princeton University, NJ, USA). Virus stocks were grown and titrated by serial dilution on ST cells. Every infection was performed on confluent cell monolayers and always using a multiplicity of infection (MOI) of 10 PFU/cell.
Chemicals and cytokines.
Purified recombinant porcine TNF-α was purchased from R&D Systems (catalog number 690-T). Doxorubicin hydrochloride was obtained from Thermo Fisher Scientific (catalog number BP25161). The viral DNA polymerase phosphonoacetic acid (PAA) was purchased from Sigma-Aldrich (catalog number 284270). The 26S proteasome inhibitor MG132 was obtained from Sigma-Aldrich (catalog number M7449). The Nedd8 inhibitor MLN4924 (pevonedistat) was purchased from Selleckchem (catalog number S7109). The ATM kinase inhibitor KU60019 was also purchased from Selleckchem (catalog number S1570). The broad-spectrum histone deacetylase inhibitor trichostatin A (catalog number T1952) and the gene transcription inhibitor actinomycin D (catalog number A9415) were purchased from Sigma-Aldrich.
NF-κB EMSA.
Five million cells were harvested per condition and resuspended in ice-cold electrophoretic mobility shift assay (EMSA) lysis buffer composed of 20 mM HEPES (pH 7.9), 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.15 mM EGTA, 20% glycerol, 1% Nonidet P-40 (NP-40), 0.5 mM dithiothreitol (DTT), and protease inhibitor cocktail (1 tablet/10 mL of EMSA lysis buffer, cOmplete mini EDTA free; catalog number 11836170001, Roche). Cell lysates were incubated for 30 min on ice and later centrifuged at 14,000 × g for 10 min. The supernatant was collected and frozen at −20°C. Cell extracts (10 μg) were incubated with 1 μg of poly(dI-dC) (catalog number P4929, Sigma-Aldrich) and EMSA binding buffer (75 mM NaCl, 15 mM Tris HCl [pH 7.5], 1.5 mM EDTA, 1.5 mM DTT, 7.5% glycerol, 0.3% NP-40, 20 μg/mL bovine serum albumin [BSA]) in a total reaction volume of 9 μL for 20 min at 4°C. For supershift assays, 1 μL (~0.2 μg) of the following antibodies were added to the above reaction mixture: anti-NF-κB P65 (Cell Signaling; clone D14E12, catalog number 8242), anti-NF-κB P50 (Upstate; catalog number 06–886), or control normal rabbit IgG (Cell Signaling; catalog number 2729). To demonstrate the specificity of the NF-κB complexes, 0.5 μL of unlabeled probe (see below) was added to the above reaction mixture in a 25-fold molar excess. One microliter of [32P]-labeled double-stranded oligonucleotides (5′-TCAACAGAGGGGACTTTCCGAGAGGCC-3′) containing the intronic κB site (underlined) from the Igκ gene was added to the reaction mixture and incubated for 20 min at room temperature. The samples were separated on a 4% native polyacrylamide gel, which was then dried and exposed to film or a phosphor screen, and the NF-κB binding bands were quantified by Image Quant software (64).
Antibodies for the detection of PRV proteins.
The mouse monoclonal antibodies used for the detection of PRV proteins were anti-gB (clone 1C11; 1:100 for Western blotting [WB]), anti-gE (clone 18E8, 1:100 for WB) (65), and anti-US3 (1:100 for WB) (66) antibodies. The anti-IE180 was a rabbit polyclonal antibody (1:1,000 for WB) (67). The anti-US3 antibody was generously provided by L. W. Enquist (Princeton University, NJ, USA), and the anti-IE180 antiserum was a gift of E. Tabares (Autonoma University of Madrid, Madrid, Spain).
Antibodies for the detection of cellular proteins.
The antibodies purchased from Cell Signaling Technology (CST) were the mouse monoclonal antibodies anti-IκBα (clone L35A5, 1:1,000 for WB; catalog number 4814) and anti-NF-κB p65 (clone L8F6, 1:400 for IF and 1:1,000 for WB; catalog number 6956) and the rabbit antibodies anti-H3 (clone D1H2, 1:2,000 for WB; catalog number 4499), anti-H3K27ac (clone D5E4, 1:1,000 for WB; catalog number 8173), anti-H2A.X (clone D17A3, 1:1,000 for WB; catalog number 7631), and anti-phospho-H2A.X at Ser139 (γH2A.X) (1:1,000 for WB; catalog number 2577). From Santa Cruz Biotechnology, we obtained the mouse monoclonal antibodies anti-H1 (clone AE4, 1:100 for IF; catalog number sc-8030), anti-H3 (clone 1G1, 1:100 for IF; catalog number sc-517576), and anti-RNA polymerase II (clone CDT4H8, 1:50 for IF and 1:500 for WB; catalog number sc-47701). The mouse monoclonal anti-TBP antibody was purchased from Thermo Fisher Scientific (clone 3TF1-3G3, 1:1,000 for IF and WB; catalog number MA3-080). The rabbit polyclonal anti-pancytokeratin antibody (1:200 for IF; catalog number ab9377) and the horseradish peroxidase (HRP)-conjugated mouse monoclonal antibody anti-α-tubulin were obtained from Abcam (clone DM1A; catalog number ab40742).
Western blot assays.
Samples were harvested in ice-cold 1× radioimmunoprecipitation (RIPA) buffer, made from 10× RIPA buffer (catalog number ab156034; Abcam) diluted in ultrapure water and containing protease inhibitor cocktail (cOmplete mini EDTA free; catalog number 11836170001, Roche). Cell lysates were kept at 4°C for 20 min before storage at −20°C. SDS-PAGE and Western blot (WB) procedures were extensively described previously (68). The blocking solution was composed of 5% (wt/vol) nonfat dry milk diluted in 0.1% PBS-Tween 20 (PBS-T). However, for the detection with determined antibodies, the blocking buffer used consisted of 5% (wt/vol) BSA (fraction V; catalog number 1120180100, Sigma-Aldrich) diluted in PBS-T. Blotted polyvinylidene difluoride (PVDF) membranes were blocked for 1 h at room temperature. Primary antibodies diluted in the corresponding blocking buffer were incubated with gentle shaking overnight at 4°C. Prior to the incubation with the secondary antibodies, three washing steps with PBS-T of 10 min each were carried out. HRP-linked secondary antibodies, goat anti-IgG mouse-HRP (1:2,000; catalog number P0447, Dako) and goat anti-IgG rabbit-HRP (1:3,000; catalog number P0448, Dako), were diluted in the corresponding blocking solution and incubated at room temperature for 1 h. Subsequently to the three washing steps after incubation with the secondary antibodies, protein bands were visualized by chemiluminescence using a ChemiDoc imaging device (Bio-Rad). Depending on the levels of the targeted protein and the antibody sensitivity, the use of ECL Plus substrate (GE Health Care) or SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific) was required.
Immunofluorescence assays.
Cell monolayers were rinsed with sterile phosphate-buffered saline (PBS) containing calcium and magnesium prior to fixation using 4% paraformaldehyde for 15 min at room temperature. After the total removal of paraformaldehyde, fixed cells were incubated with the blocking/permeabilizing buffer consisting of 0.3% Triton X-100 and 5% inactivated FBS diluted in PBS for 1 h at 37°C. Next, primary antibodies were diluted in the incubation buffer (0.3% Triton X-100 and 1% [wt/vol] BSA diluted in PBS) and incubated overnight at 4°C. Once the fixed cells were washed three times to eliminate the unbound primary antibody, fluorochrome-conjugated goat anti-IgG mouse secondary antibodies (1:200; Invitrogen, Thermo Fisher Scientific) were incubated for 1 h at 37°C. Finally, cell nuclei were counterstained by using Hoechst 33342 (1:200; catalog number H3570, Invitrogen, Thermo Fisher Scientific) diluted in PBS for 10 min at room temperature. Most of the pictures shown were taken with a Leica SPE confocal microscope, except those shown in Fig. 4A, which were captured with a Thunder imaging system (Leica). The resulting images were analyzed using ImageJ imaging software (NIH, USA).
RNA isolation and reverse transcription.
An RNeasy minikit (catalog number 74104, Qiagen) was used to isolate RNA according to the procedure provided by the manufacturer. Total RNA was treated with DNase I (RNase free; catalog number M0303S, New England Biolabs) for 10 min at 37°C. To stop the DNase I activity, up to 5 mM EDTA (catalog number AM9260G, Invitrogen) was added for the later incubation at 75°C for 10 min. Reverse transcription (RT) was performed on 500 ng of DNase I-treated RNA, by making use of the iScript cDNA synthesis kit (catalog number 1708891, Bio-Rad), in a final volume of 20 μL, in accordance with the guidelines provided by the manufacturer. The single-step reverse transcription consisted of 5 min at 25°C (priming), 20 min at 46°C (reverse transcription), and 1 min at 95°C (inactivation).
qPCR for analysis of unspliced transcript levels and viral mRNAs.
Primer DNA oligonucleotides were produced by Integrative DNA Technologies (IDT). For the analyses of unspliced mRNA levels, primer sequences were obtained via Primer-BLAST software (NIH, USA) from manually established sequences placed on exon-intron boundaries of approximately 200 bp, without considering the first exon. Primers were selected based on their capacity to generate an amplicon that comprises intron and exon sequences. The primer sequences are shown in Table 2. The complete sequences of IκBα (NFKBIA, gene ID 406188; range, positions 64509779 to 64509964, chromosome 7), GAPDH (gene ID 396823; range, positions 64130601 to 64130829, chromosome 5), B2M (gene ID 397033; range, positions 126867515 to 126867898, chromosome 1), and PGK1 (gene ID 407608; range, positions 62203312 to 62203548, chromosome X) genes in Sus scrofa were drawn from the NCBI Gene database. The quantitative PCR (qPCR) assays were performed on MicroAmp Fast optical 96-well reaction plates (catalog number 4346906, Thermo Fisher Scientific) in a total volume of 20 μL and using SYBR green PCR master mix (catalog number 4309155, Thermo Fisher Scientific). qPCR assays were launched in a StepOnePlus real-time PCR system (catalog number 4376600, Thermo Fisher Scientific). Every condition was assessed in duplicate, and the qPCR data analysis was done by the double delta threshold cycle method. Data were normalized with the levels of the rRNA 28S. Importantly, the qPCR assays performed using RNA samples that were not subjected to reverse transcription gave negative results for detection of any of the prespliced transcripts.
TABLE 2.
Sequences of primers used for detection of prespliced transcriptsa
| Primer | Forward sequence (5′–3′) | Reverse sequence (5′–3′) |
|---|---|---|
| Prespliced IκBα | AGG GTG GCA ACA AGT GCT TA | AGC ACC CAA AGA CAC CAA CA |
| Prespliced GAPDH | CAT CCA CAG TCT TCT GGG TGG | TTC ACT CCC CCT CCT TGT CTC |
| Prespliced B2M | GGC GGA TGG AAC CCA GAT AC | CGG TGA AAT CCT CTG GCG TT |
| Prespliced PGK1 | GGA AGA ATT GCT AGG GAC TGA | TCC AGC CTT CTT TGG CAG ATT |
| rRNA 28S (60) | GGG CCG AAA CGA TCT CAA CC | GCC GGG CTT CTT ACC CAT T |
The forward sequence is that closer to the 5′ end of the chromosome in which the gene is located.
For the quantification of viral transcripts, a standard curve was created by 1/10 serial dilution of viral genome isolated virus particles from infectious supernatants by using the DNeasy blood and tissue kit (catalog number 69405, Qiagen) in accordance with the procedure given by the manufacturer. The standard curve included viral genome concentrations ranging from 5 × 107 to 5 copies, considering that 1 PFU corresponds to one viral genome, from which an equation with an R2 of ≥0.99 was generated for the calculation of the absolute number of mRNA copies (per 12.5 ng total yield of RNA subjected to reverse transcription reaction) by linear regression, which was normalized to the relative amount of rRNA 28S. This procedure was adapted from that described earlier (69). All the primer sequences (Table 3) were specific for PRV strain Kaplan (GenBank accession number JQ809328) and were obtained from Tombácz et al. (70).
TABLE 3.
Sequences of primers used for detection of viral transcripts
| Primer | Forward sequence (5′–3′) | Reverse sequence (5′–3′) |
|---|---|---|
| US1 | AGC TCA ACG AGC GCG ACG TCT A | CGG AAG CTA AAC TCG GAC GCG A |
| UL24 | TGT GCT TCG TCA TCG AGC TC | TGG GCG TGT TGA GGT TCC |
| UL39 | GCT GGC CAA GTT CAA GAC G | CGC ACA TGT CGA TGA GCA G |
| UL50 | CTT CTT CGA GGT CTT TGC GC | ATG TCG TAT CCG GCG TCC T |
| UL51 | GCT CAT GCA CCT GTA CCT CTC G | ACG TCG GAC ATC ACC ACG TTG C |
| US4 | ACC TCG ATC TAC ATC TGC GTC G | GGC CCT GGT GAT CGC CAT |
RNA-seq analysis.
RNA isolations were performed using the RNeasy minikit (Qiagen) according to the manufacturer’s procedure. DNase treatment was performed on-column to eliminate DNA contamination (catalog number 79254, Qiagen). The concentration and quality of the total extracted RNA were checked via the Quant-it RiboGreen RNA assay (Life Technologies) and the RNA 6000 nano chip (Agilent Technologies). The QuantSeq 3′ mRNA library prep FWD kit (Lexogen) was used for library preparation. Library quality control (QC) was performed using the high-sensitivity DNA chip (Agilent Technologies). Sequencing was performed on the NextSeq 500 SR 76 high-output system (Illumina). Data from mock-infected ST cells or ST cells infected with WT PRV NIA3 (71) (MOI of 10, 16 hpi) were downloaded and used for analysis in the present study. A combined genome annotation containing the porcine genome (Sus scrofa) (GCF_000003025.6) and the Suid herpesvirus 1 (PRV) reference genome (GenBank accession number NC_006151.1) was generated. Reads were aligned using HISAT2 in the QuasR package in R software (72), and they were subsequently counted via qCount. Differential gene expression was determined using DESeq2 (73). Transcripts with an adjusted P value of less than 0.05 were considered significantly up- or downregulated. Noncoding transcripts were discarded from these analyses. Size factors were determined based on total library sizes. Results are represented as MA plots.
In silico analysis of potential κB sequences in the PRV genome.
The κB binding sequences present in the PRV genome were determined using R software. To that end, the consensus sequence 5′-GGGRNWYYCC-3′ in which N represents any base, R represents purine, W represents A or T, and Y represents pyrimidine, that is 5′-GGG(AG)(GATC)(TA)(TC)(TC)CC-3′, was identified in the PRV Kaplan genome (GenBank accession number JQ809328) and in the reverse complement DNA strand. The resulting κB sequences were exported as a bed file and visualized with the Integrative Genomics Viewer (IGV) genome browser. Viral genes that contain potential κB sites 600 bp upstream of the coding DNA sequence were selected for further study.
NF-κB p65 gene expression knockdown via siRNA.
The procedure for silencing RNA-mediated knockdown of porcine p65 gene expression was described in detail previously (4).
Statistics.
Unpaired t tests were performed for statistical analyses of the data shown in Fig. 3B. Tukey’s test for multiple comparisons was applied to data shown in Fig. 3A and 5G.
Data availability.
All sequencing data were deposited in GEO under accession number GSE201012.
ACKNOWLEDGMENTS
We thank Thomas C. Mettenleiter (Friedrich-Loeffler Institute, Insel Reims, Germany) for supplying the PRV WT Kaplan strain, Gregory A. Smith (Northwestern University, IL, USA) for generously sharing the PRV VP26-GFP virus strain (PRV GS443), Lynn W. Enquist (Princeton University, NJ, USA) and Jens B. Bosse (Centre for Structural Systems Biology, Hamburg, Germany) for kindly providing the PRV ICP8-GFP mutant virus (PRV 926), and the ID-DLO (the Netherlands) for providing the NIA3 strain. We also thank our colleagues Jan Clement, Leen Hermans and Sofie Denaeghel for help with animal caretaking and/or preparation of PPK cells and Ann Machtelinckx for excellent assistance. N.R. and A.T. supported by Ph.D. grants from the Research Foundation Flanders (F.W.O.-Vlaanderen) (https://www.fwo.be). This research was supported by grants to H.W.F. from the F.W.O.-Vlaanderen (grant numbers G017615 and G.019617N) (https://www.fwo.be) and the Special Research Fund of Ghent University (G.O.A. grant 01G01317 and grant BOFBAS2018000301) (https://www.ugent.be). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Herman W. Favoreel, Email: herman.favoreel@ugent.be.
Felicia Goodrum, University of Arizona.
REFERENCES
- 1.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Sun L, Liu S, Chen ZJ. 2010. SnapShot: pathways of antiviral innate immunity. Cell 140:436–436.e2. 10.1016/j.cell.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayden MS, West AP, Ghosh S. 2006. NF-kappaB and the immune response. Oncogene 25:6758–6780. 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 4.Romero N, Van Waesberghe C, Favoreel HW. 2020. Pseudorabies virus infection of epithelial cells leads to persistent but aberrant activation of the NF-κB pathway, inhibiting hallmark NF-κB-induced proinflammatory gene expression. J Virol 94:e00196-20. 10.1128/JVI.00196-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romero N, Favoreel HW. 2021. Pseudorabies virus infection triggers NF-κB activation via the DNA damage response but actively inhibits NF-κB-dependent gene expression. J Virol 95:e01666-21. 10.1128/JVI.01666-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roizmann B, Desrosiers RC, Fleckenstein B, Lopez C, Minson AC, Studdert MJ. 1992. The family Herpesviridae: an update. The Herpesvirus Study Group of the International Committee on Taxonomy of Viruses. Arch Virol 123:425–449. 10.1007/BF01317276. [DOI] [PubMed] [Google Scholar]
- 7.Pomeranz LE, Reynolds AE, Hengartner CJ. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69:462–500. 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132:344–362. 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 9.Bosse JB, Hogue IB, Feric M, Thiberge SY, Sodeik B, Brangwynne CP, Enquist LW. 2015. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc Natl Acad Sci USA 112:E5725–E5733. 10.1073/pnas.1513876112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci USA 98:3466–3470. 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodrich JA, Cutler G, Tjian R. 1996. Contacts in context: promoter specificity and macromolecular interactions in transcription. Cell 84:825–830. 10.1016/s0092-8674(00)81061-2. [DOI] [PubMed] [Google Scholar]
- 12.Burley SK, Roeder RG. 1996. Biochemistry and structural biology of transcription factor IID (TFIID). Annu Rev Biochem 65:769–799. 10.1146/annurev.bi.65.070196.004005. [DOI] [PubMed] [Google Scholar]
- 13.Burley SK. 1996. The TATA box binding protein. Curr Opin Struct Biol 6:69–75. 10.1016/s0959-440x(96)80097-2. [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann A, Natoli G, Ghosh G. 2006. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 25:6706–6716. 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 15.Patel A, Hanson J, McLean TI, Olgiate J, Hilton M, Miller WE, Bachenheimer SL. 1998. Herpes simplex type 1 induction of persistent NF-kappa B nuclear translocation increases the efficiency of virus replication. Virology 247:212–222. 10.1006/viro.1998.9243. [DOI] [PubMed] [Google Scholar]
- 16.Amici C, Belardo G, Rossi A, Santoro MG. 2001. Activation of I kappa b kinase by herpes simplex virus type 1. A novel target for anti-herpetic therapy. J Biol Chem 276:28759–28766. 10.1074/jbc.M103408200. [DOI] [PubMed] [Google Scholar]
- 17.Goodkin ML, Ting AT, Blaho JA. 2003. NF-kappaB is required for apoptosis prevention during herpes simplex virus type 1 infection. J Virol 77:7261–7280. 10.1128/jvi.77.13.7261-7280.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramani VC, Djalilian AR, Sanderson RD, Shukla D. 2015. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat Commun 6:6985. 10.1038/ncomms7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sciortino MT, Medici MA, Marino-Merlo F, Zaccaria D, Giuffrè-Cuculletto M, Venuti A, Grelli S, Bramanti P, Mastino A. 2008. Involvement of gD/HVEM interaction in NF-kB-dependent inhibition of apoptosis by HSV-1 gD. Biochem Pharmacol 76:1522–1532. 10.1016/j.bcp.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 20.Marino-Merlo F, Papaianni E, Medici MA, Macchi B, Grelli S, Mosca C, Borner C, Mastino A. 2016. HSV-1-induced activation of NF-κB protects U937 monocytic cells against both virus replication and apoptosis. Cell Death Dis 7:e2354. 10.1038/cddis.2016.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marino-Merlo F, Papaianni E, Frezza C, Pedatella S, De Nisco M, Macchi B, Grelli S, Mastino A. 2019. NF-κB-dependent production of ROS and restriction of HSV-1 infection in U937 monocytic cells. Viruses 11:428. 10.3390/v11050428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venuti A, Musarra-Pizzo M, Pennisi R, Tankov S, Medici MA, Mastino A, Rebane A, Sciortino MT. 2019. HSV-1\EGFP stimulates miR-146a expression in a NF-κB-dependent manner in monocytic THP-1 cells. Sci Rep 9:5157. 10.1038/s41598-019-41530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amici C, Rossi A, Costanzo A, Ciafrè S, Marinari B, Balsamo M, Levrero M, Santoro MG. 2006. Herpes simplex virus disrupts NF-kappaB regulation by blocking its recruitment on the IkappaBalpha promoter and directing the factor on viral genes. J Biol Chem 281:7110–7117. 10.1074/jbc.M512366200. [DOI] [PubMed] [Google Scholar]
- 24.Taddeo B, Zhang W, Lakeman F, Roizman B. 2004. Cells lacking NF-kappaB or in which NF-kappaB is not activated vary with respect to ability to sustain herpes simplex virus 1 replication and are not susceptible to apoptosis induced by a replication-incompetent mutant virus. J Virol 78:11615–11621. 10.1128/JVI.78.21.11615-11621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spencer CA, Dahmus ME, Rice SA. 1997. Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J Virol 71:2031–2040. 10.1128/JVI.71.3.2031-2040.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jenkins HL, Spencer CA. 2001. RNA polymerase II holoenzyme modifications accompany transcription reprogramming in herpes simplex virus type 1-infected cells. J Virol 75:9872–9884. 10.1128/JVI.75.20.9872-9884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quadt I, Günther AK, Voss D, Schelhaas M, Knebel-Mörsdorf D. 2006. TATA-binding protein and TBP-associated factors during herpes simplex virus type 1 infection: localization at viral DNA replication sites. Virus Res 115:207–213. 10.1016/j.virusres.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Rice SA, Long MC, Lam V, Spencer CA. 1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J Virol 68:988–1001. 10.1128/JVI.68.2.988-1001.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rice SA, Long MC, Lam V, Schaffer PA, Spencer CA. 1995. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J Virol 69:5550–5559. 10.1128/JVI.69.9.5550-5559.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leopardi R, Ward PL, Ogle WO, Roizman B. 1997. Association of herpes simplex virus regulatory protein ICP22 with transcriptional complexes containing EAP, ICP4, RNA polymerase II, and viral DNA requires posttranslational modification by the U(L)13 protein kinase. J Virol 71:1133–1139. 10.1128/JVI.71.2.1133-1139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long MC, Leong V, Schaffer PA, Spencer CA, Rice SA. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J Virol 73:5593–5604. 10.1128/JVI.73.7.5593-5604.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fraser KA, Rice SA. 2005. Herpes simplex virus type 1 infection leads to loss of serine-2 phosphorylation on the carboxyl-terminal domain of RNA polymerase II. J Virol 79:11323–11334. 10.1128/JVI.79.17.11323-11334.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraser KA, Rice SA. 2007. Herpes simplex virus immediate-early protein ICP22 triggers loss of serine 2-phosphorylated RNA polymerase II. J Virol 81:5091–5101. 10.1128/JVI.00184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith CA, Bates P, Rivera-Gonzalez R, Gu B, DeLuca NA. 1993. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J Virol 67:4676–4687. 10.1128/JVI.67.8.4676-4687.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu B, DeLuca N. 1994. Requirements for activation of the herpes simplex virus glycoprotein C promoter in vitro by the viral regulatory protein ICP4. J Virol 68:7953–7965. 10.1128/JVI.68.12.7953-7965.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuddus R, Gu B, DeLuca NA. 1995. Relationship between TATA-binding protein and herpes simplex virus type 1 ICP4 DNA-binding sites in complex formation and repression of transcription. J Virol 69:5568–5575. 10.1128/JVI.69.9.5568-5575.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu B, Kuddus R, DeLuca NA. 1995. Repression of activator-mediated transcription by herpes simplex virus ICP4 via a mechanism involving interactions with the basal transcription factors TATA-binding protein and TFIIB. Mol Cell Biol 15:3618–3626. 10.1128/MCB.15.7.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carrozza MJ, DeLuca NA. 1996. Interaction of the viral activator protein ICP4 with TFIID through TAF250. Mol Cell Biol 16:3085–3093. 10.1128/MCB.16.6.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grondin B, DeLuca N. 2000. Herpes simplex virus type 1 ICP4 promotes transcription preinitiation complex formation by enhancing the binding of TFIID to DNA. J Virol 74:11504–11510. 10.1128/jvi.74.24.11504-11510.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zabierowski SE, Deluca NA. 2008. Stabilized binding of TBP to the TATA box of herpes simplex virus type 1 early (tk) and late (gC) promoters by TFIIA and ICP4. J Virol 82:3546–3554. 10.1128/JVI.02560-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sampath P, Deluca NA. 2008. Binding of ICP4, TATA-binding protein, and RNA polymerase II to herpes simplex virus type 1 immediate-early, early, and late promoters in virus-infected cells. J Virol 82:2339–2349. 10.1128/JVI.02459-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lester JT, DeLuca NA. 2011. Herpes simplex virus 1 ICP4 forms complexes with TFIID and mediator in virus-infected cells. J Virol 85:5733–5744. 10.1128/JVI.00385-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stringer KF, Ingles CJ, Greenblatt J. 1990. Direct and selective binding of an acidic transcriptional activation domain to the TATA-box factor TFIID. Nature 345:783–786. 10.1038/345783a0. [DOI] [PubMed] [Google Scholar]
- 44.Goodrich JA, Hoey T, Thut CJ, Admon A, Tjian R. 1993. Drosophila TAFII40 interacts with both a VP16 activation domain and the basal transcription factor TFIIB. Cell 75:519–530. 10.1016/0092-8674(93)90386-5. [DOI] [PubMed] [Google Scholar]
- 45.Zhou C, Knipe DM. 2002. Association of herpes simplex virus type 1 ICP8 and ICP27 proteins with cellular RNA polymerase II holoenzyme. J Virol 76:5893–5904. 10.1128/jvi.76.12.5893-5904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim SK, Shakya AK, Kim S, O'Callaghan DJ. 2016. Functional characterization of the serine-rich tract of varicella-zoster virus IE62. J Virol 90:959–971. 10.1128/JVI.02096-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng H, He H, Hay J, Ruyechan WT. 2003. Interaction between the varicella zoster virus IE62 major transactivator and cellular transcription factor Sp1. J Biol Chem 278:38068–38075. 10.1074/jbc.M302259200. [DOI] [PubMed] [Google Scholar]
- 48.Rahaus M, Desloges N, Yang M, Ruyechan WT, Wolff MH. 2003. Transcription factor USF, expressed during the entire phase of varicella-zoster virus infection, interacts physically with the major viral transactivator IE62 and plays a significant role in virus replication. J Gen Virol 84:2957–2967. 10.1099/vir.0.19335-0. [DOI] [PubMed] [Google Scholar]
- 49.Ruyechan WT, Peng H, Yang M, Hay J. 2003. Cellular factors and IE62 activation of VZV promoters. J Med Virol 70:S90–S94. 10.1002/jmv.10328. [DOI] [PubMed] [Google Scholar]
- 50.Yang M, Peng H, Hay J, Ruyechan WT. 2006. Promoter activation by the varicella-zoster virus major transactivator IE62 and the cellular transcription factor USF. J Virol 80:7339–7353. 10.1128/JVI.00309-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Folster JM, Jensen NJ, Ruyechan WT, Inoue N, Schmid DS. 2011. Regulation of the expression of the varicella-zoster virus open reading frame 66 gene. Virus Res 155:334–342. 10.1016/j.virusres.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Abmayr SM, Feldman LD, Roeder RG. 1985. In vitro stimulation of specific RNA polymerase II-mediated transcription by the pseudorabies virus immediate early protein. Cell 43:821–829. 10.1016/0092-8674(85)90255-7. [DOI] [PubMed] [Google Scholar]
- 53.Abmayr SM, Workman JL, Roeder RG. 1988. The pseudorabies immediate early protein stimulates in vitro transcription by facilitating TFIID: promoter interactions. Genes Dev 2:542–553. 10.1101/gad.2.5.542. [DOI] [PubMed] [Google Scholar]
- 54.Flori L, Rogel-Gaillard C, Cochet M, Lemonnier G, Hugot K, Chardon P, Robin S, Lefèvre F. 2008. Transcriptomic analysis of the dialogue between Pseudorabies virus and porcine epithelial cells during infection. BMC Genomics 9:123. 10.1186/1471-2164-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flori L, Rogel-Gaillard C, Mariani V, Lemonnier G, Cochet M, Hugot K, Chardon P, Robin S, Lefèvre F. 2008. A combined transcriptomic approach to analyse the dialogue between pseudorabies virus and porcine cells. Dev Biol (Basel) 132:99–104. 10.1159/000317149. [DOI] [PubMed] [Google Scholar]
- 56.Ray N, Enquist LW. 2004. Transcriptional response of a common permissive cell type to infection by two diverse alphaherpesviruses. J Virol 78:3489–3501. 10.1128/jvi.78.7.3489-3501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brukman A, Enquist LW. 2006. Suppression of the interferon-mediated innate immune response by pseudorabies virus. J Virol 80:6345–6356. 10.1128/JVI.00554-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blanchard Y, Le MN, Le CM, Blanchard P, Leger J, Jestin A. 2006. Cellular gene expression survey of PseudoRabies Virus (PRV) infected human embryonic kidney cells (HEK-293). Vet Res 37:705–723. 10.1051/vetres:2006027. [DOI] [PubMed] [Google Scholar]
- 59.Paulus C, Sollars PJ, Pickard GE, Enquist LW. 2006. Transcriptome signature of virulent and attenuated pseudorabies virus-infected rodent brain. J Virol 80:1773–1786. 10.1128/JVI.80.4.1773-1786.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ray N, Bisher ME, Enquist LW. 2004. Cyclooxygenase-1 and -2 are required for production of infectious pseudorabies virus. J Virol 78:12964–12974. 10.1128/JVI.78.23.12964-12974.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rutkowski AJ, Erhard F, L'Hernault A, Bonfert T, Schilhabel M, Crump C, Rosenstiel P, Efstathiou S, Zimmer R, Friedel CC, Dölken L. 2015. Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun 6:7126. 10.1038/ncomms8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grauwet K, Cantoni C, Parodi M, De Maria A, Devriendt B, Pende D, Moretta L, Vitale M, Favoreel HW. 2014. Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc Natl Acad Sci USA 111:16118–16123. 10.1073/pnas.1409485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaplan AS, Vatter AE. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394–407. 10.1016/0042-6822(59)90068-6. [DOI] [PubMed] [Google Scholar]
- 64.Jackson SS, Miyamoto S. 2015. Dissecting NF-κB signaling induced by genotoxic agents via genetic complementation of NEMO-deficient 1.3E2 cells. Methods Mol Biol 1280:197–215. 10.1007/978-1-4939-2422-6_11. [DOI] [PubMed] [Google Scholar]
- 65.Nauwynck HJ, Pensaert MB. 1995. Effect of specific antibodies on the cell-associated spread of pseudorabies virus in monolayers of different cell types. Arch Virol 140:1137–1146. 10.1007/BF01315422. [DOI] [PubMed] [Google Scholar]
- 66.Olsen LM, Ch'ng TH, Card JP, Enquist LW. 2006. Role of pseudorabies virus Us3 protein kinase during neuronal infection. J Virol 80:6387–6398. 10.1128/JVI.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gómez-Sebastián S, Tabarés E. 2004. Negative regulation of herpes simplex virus type 1 ICP4 promoter by IE180 protein of pseudorabies virus. J Gen Virol 85:2125–2130. 10.1099/vir.0.80119-0. [DOI] [PubMed] [Google Scholar]
- 68.Deruelle M, Geenen K, Nauwynck HJ, Favoreel HW. 2007. A point mutation in the putative ATP binding site of the pseudorabies virus US3 protein kinase prevents Bad phosphorylation and cell survival following apoptosis induction. Virus Res 128:65–70. 10.1016/j.virusres.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 69.Broto L, Romero N, Méndez F, Diaz-Beneitez E, Candelas-Rivera O, Fuentes D, Cubas-Gaona LL, Courtillon C, Eterradossi N, Soubies SM, Rodríguez JR, Rodríguez D, Rodríguez JF. 2021. Type I Interferon acts as a major barrier to the establishment of infectious bursal disease virus (IBDV) persistent infections. J Virol 95:e02017-20. 10.1128/JVI.02017-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tombácz D, Tóth JS, Petrovszki P, Boldogkoi Z. 2009. Whole-genome analysis of pseudorabies virus gene expression by real-time quantitative RT-PCR assay. BMC Genomics 10:491. 10.1186/1471-2164-10-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McFerran JB, Dow C, McCracken RM. 1979. Experimental studies in weaned pigs with three vaccines against Aujeszky’s disease. Comp Immunol Microbiol Infect Dis 2:327–334. 10.1016/0147-9571(79)90020-1. [DOI] [PubMed] [Google Scholar]
- 72.Gaidatzis D, Lerch A, Hahne F, Stadler MB. 2015. QuasR: quantification and annotation of short reads in R. Bioinformatics 31:1130–1132. 10.1093/bioinformatics/btu781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All sequencing data were deposited in GEO under accession number GSE201012.