

Summary
Autism spectrum disorder and intellectual disability are comorbid neurodevelopmental disorders with complex genetic architectures. Despite large-scale sequencing studies, only a fraction of the risk genes was identified for both. We present a network-based gene risk prioritization algorithm, DeepND, that performs cross-disorder analysis to improve prediction by exploiting the comorbidity of autism spectrum disorder (ASD) and intellectual disability (ID) via multitask learning. Our model leverages information from human brain gene co-expression networks using graph convolutional networks, learning which spatiotemporal neurodevelopmental windows are important for disorder etiologies and improving the state-of-the-art prediction in single- and cross-disorder settings. DeepND identifies the prefrontal and motor-somatosensory cortex (PFC-MFC) brain region and periods from early- to mid-fetal and from early childhood to young adulthood as the highest neurodevelopmental risk windows for ASD and ID. We investigate ASD- and ID-associated copy-number variation (CNV) regions and report our findings for several susceptibility gene candidates. DeepND can be generalized to analyze any combinations of comorbid disorders.
Keywords: autism, deep learning, genome-wide association, comorbidity, intellectual disability, graph convolution, node classification, semisupervised learning
Graphical abstract
Highlights
-
•
DeepND can co-analyze comorbid neurodevelopmental disorders to discover risk genes
-
•
The approach employs multitask learning to learn shared and disorder-specific weights
-
•
DeepND uses graph convolution to process gene interactions in multiple networks
-
•
The model includes a mixture-of-experts model to detect informative networks
The bigger picture
While risk-gene-discovery algorithms have complemented exome/genome-sequencing studies of neurodevelopmental disorders, they are not capable of co-analyzing multiple comorbid conditions like autism and intellectual disability. A common approach is analyzing disorders one by one and comparing the outcomes. With this approach, the method does not utilize cross-disorder interactions and is bound by limited evidence per disorder. We address this gap with a technique, Deep Neurodevelopmental Disorders (DeepND), that uses multitask learning to co-analyze data from multiple disorders to learn shared and disorder-specific patterns. DeepND includes graph convolutional neural networks that process gene-interaction information from multiple networks. DeepND also learns which networks are important for disorder etiologies. Based on this, we propose an interpretable risk-gene-discovery algorithm for neuropsychiatric disorders.
We propose a neural-network-based model that can simultaneously analyze multiple comorbid neurodevelopmental disorders to discover risk genes. DeepND learns the shared and disorder-specific interaction patterns of a risk gene in multiple disorders. It can also extract which gene-interaction networks are the most informative for the gene risk assessment and the etiology of each disorder. Our method can also work with any combination of comorbid conditions and is not limited to neurodevelopmental disorders.
Introduction
Autism spectrum Disorder (ASD) is a common neurodevelopmental disorder with a complex genetic architecture in which around a thousand risk genes have a role.1 Large consortia efforts have been paving the way for understanding the genetic, functional, and cellular aspects of this complex disorder via large-scale exome-2, 3, 4, 5, 6, 7, 8 and genome-9, 10, 11, 12, 13, 14 sequencing studies. The latest and also most comprehensive study to date analyzed ∼36,000 samples (6,430 trios) to pinpoint 102 risk genes (false discovery rate [FDR] ≤ 0.1).15 Overwhelming evidence suggests that genetic architectures of neuropsychiatric disorders overlap.16, 17, 18 For instance, out of the twenty five SFARI category I ASD risk genes (i.e., highest risk), only five are solely associated with ASD. Genes like chromodomain helicase DNA-binding protein 8 (CHD2), SCN2A, and ARID1B are associated with six neurodevelopmental disorders. Intellectual disability (ID) is one of such comorbid disorders that manifests itself with impaired mental capabilities. Reminiscent of ASD, ID also has a complex genetic background with hundreds of risk genes involved and is identified by rare de novo disruptive mutations observed in whole-exome- and genome-sequencing studies.19, 20, 21, 22, 23, 24, 25 ASD and ID are frequently observed together.26 In 2018, the CDC reported that of children with ASD were also diagnosed with ID and were borderline.27 They also share a large number of risk genes.28 Despite these similarities, Robinson et al. also point to differences in genetic architectures and report that intelligence quotient (IQ) positively correlates with family history of psychiatric disease and negatively correlates with de novo disruptive mutations.29 Yet, the shared functional circuitry behind is mostly unknown.
The current lack of understanding on how comorbid neuropsychiatric disorders relate mostly stems from the incomplete profiling of individual genetic architectures. Statistical methods have been used to assess gene risk using excess genetic burden from case-control and family studies,1 which have recently been extended to work with multiple traits.30 Yet, these tools work with genes with observed disruptive mutations (mainly de novo). It is often of interest to use these as prior risks and obtain a posterior gene-interaction-network-adjusted risk that can also assess risk for genes with no prior signal. Network-based computational gene risk-prediction methods come in handy for (1) imputing the insufficient statistical signal and providing a genome-wide risk ranking and (2) finding out the affected cellular circuitries such as pathways and networks of genes.1,31, 32, 33, 34, 35, 36, 37, 38, 39 While these methods have helped unravel the underlying mechanisms, they have several limitations. First, by design, they are limited to work with a single disorder. In order to compare and contrast comorbid disorders such as ASD and ID using these tools, one approach is to bag the mutational burden observed for each disorder, assuming two are the same. However, disorder-specific features are lost as a consequence.31 The more common approach is to perform independent analyses per disorder and intersect the results. Unfortunately, this approach ignores valuable source of information coming from the shared genetic architecture and loses prediction power as per-disorder analyses have less input (i.e., samples, mutation counts) and less statistical power.15,33,40 Second, current network-based gene-discovery methods can work with one or two integrated gene-interaction networks.33,35,38,41,42 This means that numerous functional interaction networks (e.g., co-expression, protein interaction, etc.) are disregarded, which limits and biases the predictions. Gene co-expression networks that model brain development are a promising source of diverse information regarding gene risk but currently cannot be fully utilized, as the signal coming from different networks cannot be deconvoluted. Usually, investigating such risky neurodevelopmental windows is an independent downstream analysis.42,43 Should this process be integrated within the risk-assessment framework, it has the potential to provide valuable biological insights and also to improve the performance of the genome-wide risk assessment task.
Here, we address these challenges with a cross-disorder gene-discovery algorithm (Deep Neurodevelopmental Disorders [DeepND]). For the first time, DeepND analyzes comorbid neurodevelopmental disorders simultaneously over multiple gene co-expression networks and explicitly learns the shared and disorder-specific features using multitask learning. Thus, the predictions for the disorders depend on each other’s genetic architecture.
The proposed DeepND architecture uses graph convolution to extract associations between genes from gene co-expression networks. This information is processed by a mixture-of-experts model that can self learn critical neurodevelopmental time windows and brain regions for each disorder etiology, which makes the model interpretable. We provide a genome-wide risk ranking for each disorder and show that the prediction power is improved in both single- (single disorder) and multitask settings. DeepND identifies the prefrontal cortex brain region and from early- to late-mid-fetal and from mid-childhood to young adulthood periods as the highest neurodevelopmental risk windows for both disorders.
Finally, we investigate frequent ASD and ID associated copy-number-variation regions and pinpoint risk genes that are disregarded by other algorithms. The software has been released at http://github.com/ciceklab/deepnd. This neural network architecture can easily be generalized to other disorders with a shared genetic component and can be used to prioritize focused functional studies and possible drug targets.
Results
Using a deep-learning framework that combines graph convolutional neural networks with a mixture-of-experts model, we perform a genome-wide risk assessment for ASD and ID simultaneously in a multitask learning setting and detect neurodevelopmental windows that are informative for risk assessment for each disorder. Our results point to the shared disrupted functionalities and novel risk genes that provide a road map to researchers who would like to understand the ties between these two comorbid disorders.
Genome-wide risk prediction for ASD and ID
To have a model of evolving gene interactions throughout brain development, we extracted 52 gene co-expression networks from the BrainSpan dataset44,45 using hierarchical clustering of brain regions and a sliding-window approach.43 These networks are used to assess the network-adjusted posterior risk for each gene for which disease agnostic the prior risk features such as the probability of loss-of-function intolerance (pLI) is given. Note that one can use disease-specific features such as mutation burden in case-control or family studies. However, current knowledge on ground-truth risk genes mostly rely on these data, and using these as features would cause a leak in the training procedure.
The deep neural network architecture (DeepND) performs a semi-supervised learning task, which means a set of positively and negatively labeled genes are required as ground truth per disorder. As also done in Krishnan et al.,42 we obtained 594 ASD-positive genes (Table S1) from the SFARI dataset (http://gene.sfari.org),46 which are categorized into three evidence levels based on the strength of evidence in the literature. For ID, we curated a ground-truth ID risk-gene set of 580 genes using landmark review studies on ID gene risk21,47, 48, 49, 50, 51 (Table S2). We generated three evidence level sets similar to the ASD counterpart: E1, E2, and E3 sets where each set includes genes that are recurrently indicated in multiple studies. As for the negatively labeled genes, we used 1,074 non-mental-health-related genes for both disorders, which were curated by Krishnan et al., after discarding 181 of them that are shown to be related to other neurodevelopmental disorders, such as epilepsy, schizophrenia, or bipolar disorder (Table S1).
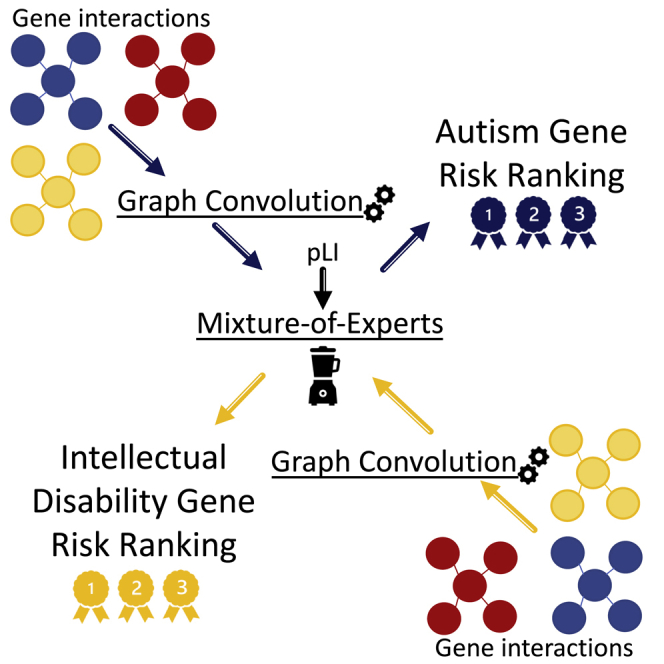
DeepND uses the multitask-learning paradigm where multiple tasks are solved concurrently (i.e., genome wide risk assessment for ASD and ID). Thus, the network learns a shared set of weights for both disorders and also disorder-specific set of weights (Figure 1). First, the model inputs pLI for each gene and, using fully connected layers, produces an embedded feature set per gene. The set of weights learned in these layers are affected by the ground-truth labels for both disorders and, thus, are shared. Then, the architecture branches out to two single-task layers, with one per disorder (Figure 1, blue for ASD and yellow for ID). For each single-task branch, these embeddings are input to 52 graph convolutional neural networks (GCNs). Each GCN processes a co-expression network that represents a neurodevelopmental window and extracts network-adjusted gene risk signatures (i.e., embeddings)15,37 (experimental procedures). Finally, these embeddings are fed into a fully connected gating network along with the prior risk features. The gating network assigns a weight to each GCN that is proportional to the informativeness of the embedding coming from each neurodevelopmental window. Thus, the model also learns which windows are important for prediction of ASD/ID risk genes. This also means that they are important in the etiology of the disorder. In the end, each disorder-specific subnetwork produces a genome-wide ranking of genes being associated with that disorder along with risk probabilities. To quantify the contribution of the co-analyzing comorbid disorders (i.e., multitask) as opposed to individual analysis (i.e., single task), we also present our results of DeepND when it is run on a single-task mode (DeepND-ST). In this mode, the fully connected layers that generate embeddings are removed, and the feature (i.e., pLI) is directly fed into the GCNs (experimental procedures). We show that the genome-wide risk assessment of DeepND is robust, precise, and sensitive and substantially improves the state of the art both in terms of performance and interpretability of the predictions.
Figure 1.

System model
System model of the proposed deep-learning architecture for genome-wide cross-disorder risk assessment (DeepND). The algorithm takes the following information as inputs: (1) gene-specific features such as pLI, (2) disorder-specific ground-truth genes that are labeled as positive with varying levels of evidence based on a literature search, and (3) non-psychiatric genes that are labeled as negative. The unique gene identifiers are passed through fully connected multitask layers that learn shared weights for both disorders and produces a new feature representation affected by both losses. This new representation is then input to the starting point of the single-task layers. Graph convolutional neural networks (GCNs) each process one of fifty-two gene co-expression networks that represent different brain regions and neurodevelopmental time windows. The outputs of GCNs are then weighted by the gating network using gene pLIs to learn which networks are informative for the gene risk assessment (shade of the network indicates importance). Thus, DeepND learns which neurodevelopmental windows confer more risk for each disorder’s etiology using the gating network (mixture of experts). The final output is a genome-wide risk-probability ranking per disorder, which are then used for various downstream analyses to understand the underlying functional mechanisms and to compare/contrast both disorders. The single-task layers are exclusively trained with the ground-truth genes of the disorder they belong. Thus, they learn only disorder-specific parameters and disorder-specific networks that implicate risk.
We benchmark the performances of DeepND and other gene-discovery algorithms for neurodevelopmental disorders. Note that these tools are mainly designed for ASD. However, to the best of our knowledge, there are no such algorithms designed for ID or for other neurodevelopmental disorders. Thus, we compare them against DeepND in terms of both ASD and ID. However, one can primarily focus on their ASD performance. First, we compare DeepND and the state-of-the-art algorithm by Krishnan et al. using their experimental settings. Evaluating performances of the algorithms using E1 genes through 5-fold cross shows that DeepND achieves a median area under receiver operating curve (AUC) of and a median area under the precision recall (AUPR) of for ASD, which correspond to and improvements over the evidence-weighted support vector machine (SVM) algorithm of Krishnan et al., respectively (Figures 2A and 2B). Similarly for ID, DeepND achieves a median AUC of and a median AUPR of , which correspond to improvements of and , respectively. We observe that even the single-task mode DeepND-ST performs better than Krishnan et al.’s algorithm in all settings other than ID’s AUPR. As expected, the multitask setting of DeepND performs better than DeepND-ST and leads to an improved AUC (up to ) and AUPR (up to ) for both disorders.
Figure 2.

Evaluation of DeepND genome-wide risk assessment for ASD and ID
(A and B) The AUC distributions of Krishnan et al., DeepND, and DeepND-ST for ASD and ID genome-wide risk assessments. Every point corresponds to the performance on a test fold in the repeated cross validation setting (B). The AUPR curve distributions are for the same methods as in (A). Center line: median; box limits: upper and lower quartiles; whiskers: 1.5× interquartile range; points: outliers.
(C) Matthews correlation coefficient (MCC) between the rankings of each method and the ground-truth genes are shown for varying rank-percentage-threshold values (x axis). This ranking threshold p sets the top p% of a ranking as positive predictions. Methods compared are DeepND, DeepND - ST, evidence-weighted SVM of Krishnan et al., DAWN (only compared for ASD due to low intersection ratio with ground-truth gene set of ID), DAMAGES score of Zhang and Shen, and random-forest-based method of Duda et al. Results for ASD and ID shown when (1) E1 genes are used as the true risk genes and (2) E1 + E2 genes are considered as the true risk genes.
(D) PR curves to compare the same set of methods in (C).
We also compare the ranking performance DeepND with the following methods from the literature using a rank-percentage-threshold-based method: Krishnan et al.,42 DAWN,41 DAMAGES score,52 and the random forest-based method of Duda et al.39 That is, we obtain the final ranking of each method and mark p% of the top genes predicted as risk genes. We calculate the Matthews correlation coefficient of these marked lists with the ground truth. We vary p by and obtain the plots in Figure 2C. Results show that for varying threshold levels, the DeepND curve always dominates all others, indicating that it achieves the highest correlation with the ground truth both when we use E1 and E1 + E2 genes as positive ground truth. Comparing the precision-recall performance of the same set of methods shows that the PR curve of DeepND dominates all others in all settings and for both disorders (Figure 2D).
Finally, we compare DeepND with an ensemble learner, forecASD.38 This method uses the outputs of other methods including the ones we compared above and learns to predict ASD/ID risk genes in a semi-supervised setting. However, labels used by forecASD are also used by the voter methods whose outputs are used as features, which suggests information leakage during the training process and that the performance might be overly optimistic. Comparing DeepND and forecASD shows that despite this advantage of forecASD, DeepND performs slightly worse or on par with respect to rank-percentage-threshold curve and PR curve comparisons in ASD (see Figure S1). DeepND performs better than forecASD in all settings for ID. We also considered adding DeepND as an additional voter into the ensemble of forecASD to check if it benefits the final performance. For both disorders and in all settings, forecASD that uses DeepND as a voter outperforms both DeepND and original forecASD. This shows the informativeness of DeepND predictions.
Critical neurodevelopmental windows for ASD and ID risk
We observe that the top percentile genes in the DeepND ASD and ID rankings have a high overlap with a Jaccard index of . The top 3 deciles also have relatively high overlap with Jaccard indices 0.66, and 0.69, respectively (Figure S2).
To further investigate the shared genetic component, we focus on the spatiotemporal neurodevelopmental windows that are deemed important by DeepND for an accurate ranking of risk genes for both disorders. The neural network analyzes co-expression networks that represent 13 neurodevelopmental time windows (from embryonic period to late adulthood; Figures 3A and 4) and brain-region clusters (prefrontal and motor-somatosensory cortex [PFC-MSC]; mediodorsal nucleus of the thalamus and cerebellar cortex [MDCBC]; primary visual and superior temporal cortex [V1C-STC]; striatum, hippocampus, amygdala [SHA]) generated using Brainspan RNA-Seq dataset45 in accordance with Willsey et al.43 (experimental procedures; Figure 3B).
Figure 3.

Spatiotemporal brain regions and heatmaps for the probabilities assigned to each spatiotemporal window by DeepND
The BrainSpan dataset, which models the spatiotemporal gene expression of human neurodevelopment,45 is used to obtain gene co-expression networks. This dataset contains samples from 12 brain regions and spans 15 time points from early fetal period to late adulthood.
(A) We generate 13 neurodevelopmental time windows using a sliding window of length 3, which provides sufficient data in each window as was also done by Willsey et al.43
(B) We obtain 4 brain-region clusters based on transcriptional similarity during fetal development that also reflect topographical closeness and functional segregation. The brain regions considered are as follows. HIP, hippocampal anlage (for (1)–(2)), hippocampus (for (3)–(15)); OFC, orbital prefrontal cortex; DFC, dorsal prefrontal cortex; VFC, ventral prefrontal cortex; MFC, medial prefrontal cortex; M1C, primary motor cortex; S1C, primary somatosensory cortex; IPC, posterior inferior parietal cortex; A1C, primary auditory cortex; STC, superior temporal cortex; ITC, inferior temporal cortex; V1C, primary visual cortex; AMY, amygdala; STR, striatum; MD, mediodorsal nucleus of the thalamus; CBC, cerebellar cortex.
(C) Heatmaps show which spatiotemporal windows lead to assignment of higher risk probabilities to top-percentile genes for ASD (left) and ID (right). The numbers in boxes are softmaxed outputs of each respective GCN, averaged for top-percentile genes and then normalized. The weights assigned to each co-expression network by the MoE lets each GCN learn to make a better prediction.
We investigate which neurodevelopmental windows more confidently distinguish disorder risk genes. Figure 3C shows normalized average probabilities assigned to top-percentile risk genes (experimental procedures). We observe that the networks of the PFC-MSC brain region, spanning several time windows from early- to mid-fetal periods and from early childhood to young adulthood periods consistently are better predictors for ASD and ID risk. The periods 3–5 and 4–6 were also previously indicated as two of the highest risk regions and were subject to network analyses for ASD gene discovery.41,43 DeepND also captures strong signals from these windows, in addition to period 11–13, for both ASD and ID. We see that V1C-STC brain regions are second to PFC-MSC and MDCBC regions, providing the most subtle signal. We assess the empirical of significance of these findings via a permutation test. That is, we run DeepND in the exact same settings but shuffle the labels of the risk genes, keeping the same number of positively and negatively labeled genes. For each of the squares in Figure 3C, we obtain a distribution of average probabilities (background distribution) and check if the actual informativeness of that window is higher than the average background value. See these distributions in Figures S4 and S5 for ASD and ID, respectively. We observe that informativeness of PFC-MSC windows are the most significant compared with other brain regions and that MD-CBC results are consistently insignificant. Overall, we observe that not only earlier time windows but also later time windows are important for both etiologies. Figure S3 shows the heatmaps of DeepND-ST for ASD and ID.
The weakest source of information is the MDCBC 2-4 network. We observe roughly 12,500 links between top-percentile ASD and ID genes. The network contains close to 37.5 m links. On the other hand, PFC-MSC 4–6 contains close to 785,000 edges. Yet, there exists close to 13,000 links between top-percentile ASD and ID genes, which is the reason behind DeepND focusing on this window as its top predictor. Visualization of the top 30 genes for each disorder in the PFC-MSC 4–6 network with only very high co-expression links () is provided in Figure 4A.
Figure 4.

Network analyses of the risk genes
(A) The co-expression relationships between top 30 ASD and ID risk genes in PFC-MSC 4–6 region. This is found as the informative co-expression network for DeepND and is also indicated in the literature as an important window for ASD.43 Only links for at least 0.95 absolute correlation and genes with at least one connection are shown.
(B) The protein-protein interactions between the top-percentile risk genes are shown. The protein-protein interactions (PPIs) are obtained from the tissue-specific PPI network of frontal cortex in the DifferentialNet database.53 Only interactions between the top-percentile risk genes are shown.
Enrichment analysis of the predicted risk genes
In addition to the prediction performance benchmark above, we also evaluate the enrichment of our ASD and ID gene risk rankings in gene lists, which are shown to be related to these disorders. That is, while these gene sets are not ground-truth sets, they have been implicated as being associated with the etiology of either disorder. Thus, enrichment of members of these sets in the higher deciles of the genome-wide risk ranking of DeepND is an indication of the wellness of the ranking and also provides a means of comparing and contrasting the disrupted circuitries affected by ASD and ID. These lists are (1) targets of transcription regulators like CHD8,54 FMRP,2,55,56 RBFOX,2,57,58 and TOP1,59 (2) susceptibility pathways like WNT signaling60 and MAPK signaling,61 and (3) affected biological processes and molecular functions like post-synaptic density complex62,63 and histone modification.2,64
The first decile of ASD risk genes has the highest enrichment in all categories. All enrichments are significant with respect to Binomial test (Figure 5; experimental procedures). We observe the same trend for ID that the top decile of the genome-wide risk ranking is the most enriched in most categories but the p values are more subtle. CHD8 is the highest ASD risk gene known to date with the highest mutation burden in large ASD cohorts2,15 and with downstream functional analysis.54 Accordingly, DeepND ranks CHD8 as the 37th top risk gene whereas Krishnan et al. ranks it 1,943th genome wide. While it has a solid association to ASD etiology, its ties to ID are not well established. Bernier et al. reports that 9 out of 15 ASD probands with mutations in CHD8 also have ID. In accordance, DeepND places CHD8 as the 53rd highest risk gene for ID.
Figure 5.

The enrichment of our ASD and ID gene risk rankings in various disease-related gene lists
The enrichment of our ASD and ID gene risk rankings in various disease-related gene lists (i.e., each panel) are shown: (1) ASD- and/or ID-related transcription regulators, (2) ASD- and/or ID-related pathways, and (3) ASD- and/or ID-related biological functions or protein complexes. While these do not fully contain ground-truth genes, they have been indicated in the literature as being enriched with risk genes for either disorder. Percentage of genes in the corresponding gene set (x axis) that occurred within each decile of the genome-wide risk ranking per ASD (blue) and ID (yellow) are shown. The gene sets used are as follows: (1) targets of RBFOX (splice); (2) targets of RBFOX (splice target); (3) targets of FMRP (all peak); (4) targets of CHD8; (5) targets of TOP1; (6) WNT pathway; (7) MAPK signaling pathway; (8) GTPase regulator activity; (9) post-synaptic density complex genes; (10) synaptic genes; and (11) histone-modifier genes.
We also perform an untargeted enrichment analysis of the top-percentile predictions using the Enrichr system.65,66 Unsurprisingly, we find that for both disorders, nervous system development is the top enriched Biological Process GO term (Fisher exact test, p = 1.48 × 10-12 for ASD and p – 3.95 × 10-14 for ID), which indicates that it is a shared function that is disrupted in the etiology of both disorders (Table S3). As for the GO Molecular Function enrichment, two disorders overall have similar terms, but their top terms are different. The top function affected for ASD is histone-lysine N-methyltransferase activity, indicating a disruption in the transcription-factor activity, whereas for ID, the top affected function is voltage-gated cation channel activity, which indicates a disruption in synaptic activity.
As transcription-factor activity is highly affected in both disorders, we further investigate if there are any master transcription regulators upstream that regulate the high-risk genes for ASD and ID in the ChEA database.67 We find that SMARCD1 is the top transcription factor, with 82 of its targets coinciding with the top-percentile DeepND-predicted ASD risk genes (Fisher’s exact test, p = 1.14 × 10-20) and 99 of its targets coinciding with the top-percentile DeepND-predicted ID risk genes (Fisher’s exact test, p = 7.18 × 10-18). Sixty-two targets are shared among ASD and ID. Note that 204 genes overlap in top-percentile ASD and ID risk genes. This means that 30% of the shared set of genes among two disorders are targeted by SMARCD1 (Table S3). When the union set of the top-percentile genes for ASD and ID are considered (312 genes), we find that the top transcription factor targeting these is again SMARCD1 (82 out of 2,071 targets; Fisher’s exact test, p = 1.24 × 10-21). These results indicate that SMARCD1 might be playing a role in the convergent etiologies of these comorbid disorders as a shared upstream chromatin modeler.
Other interactions between ASD and ID genes
We further investigate the protein-protein interactions (PPIs) between the top-percentile risk genes in tissue-specific PPI networks for frontal cortex obtained from the DifferentialNet database53 using the NetworkAnalyst system.68 This analysis reveals several hub proteins such as HECW2, EP300, and CREBBP1, as shown in Figure 4B. In this list, the HECW2 gene stands out as it has the highest degree in this network and has very low prior risk for both ASD (TADA p = 0.95) and ID (extTADA Q = 0.97). Yet, it is in the top percentile for ID risk. Note that DeepND did not use any PPI network information in its reasoning and yet was able to identify this hub protein, which has been linked with ASD3 and ID69 via de novo disruptive mutations in simplex families.
Evaluation of novel predictions and identification of candidates within ASD- and ID-associated CNV regions
Recurrent copy-number variations in several regions of the genome are associated with ASD and ID etiology. However, these are large regions, and it is not clear which genes in particular are driver genes. We investigate the DeepND risk ranking of genes within (1) six regions that frequently harbor ASD-related copy-number variations (CNVs) (16p11.2, 15q11-13, 15q13.3, 1q21.1, and 22q11)42 and (2) six regions that were reported to harbor mental-retardation-related CNVs (16p11.2, 1q21.1, 22q11.21, 22q11.22, 16p13, and 17p11.2).70 Note that these CNVs in these regions might confer risk for both disorders (Table S4).
DeepND highly ranks several candidate genes for ASD and ID that (1) are within these CNV regions, (2) have low prior risk (e.g., E2 or lower, low TADA p value, etc.), and (3) low posterior risk assigned by other algorithms (Table S4). We discuss some of them below.
NIPA2 is an ASD E3-E4 gene that encodes a magnesium transporter and is located on 15q11-13. It does not participate in any of the relevant gene sets (e.g., risk pathways), and it has a low TADA (p = 0.768). Although it is ranked in the 5th decile for ASD, it is listed in the top decile by DeepND for ID. Its linkage to Prader-Willi syndrome by Goytain et al.71 also suggests that NIPA2 might is an important candidate for ID.
DeepND links MICAL3, which is in the 22q11.21–22 CNV region, to ASD and ID. It is a gene related to actin and Rab GTPase binding, ranked as the 191st for ASD risk and 147th for ID risk despite having ASD-TADA and not being in relevant risk-gene groups. Krishnan et al. rank it 11,665th, and DAMAGES score ranks it 1,916th. DAWN provides no ranking as it is not co-expressed with other networks of interest for DAWN. While disruption in the Rab GTPase cycle was shown to cause ID,72 there are no established ties between MICAL3 and ASD/ID. Moreover, MICAL family genes are related to cytoskeletal organization, which recently has been deemed an important molecular function in ASD etiology.15,34
It is also an important task to pinpoint genes that might seem related to a neurodevelopmental disorder but actually are not. This also corresponds to untangling the genetic architectures of comorbid disorders. One example that DeepND pinpoints is ZBTB20. Although ZBTB20 is not located within investigated CNV regions, it is regarded as an important risk gene for ASD. It is a CHD8 target and is listed as an E1 gene for ASD with TADA . DAWN and Krishnan et al. rank it as ∼500th ASD risk gene. However, DeepND consistently ranks it in the last decile with 0.252 probability of being an ASD risk gene. DeepND predicts that there is somewhat a higher chance for ZBTB20 to be a candidate risk gene for ID, though it is still in the 5th decile. This gene acts as a transcriptional repressor; it plays a role in various processes such as postnatal growth. Its shown relation to Primrose syndrome, which is specifically characterized by ID rather than other symptoms of ASD,73,74 also suggests that ZBTB20 has a closer relation to ID. Yet, more mutational evidence is required for a more concrete assessment.
Finally, DeepND ranks LMTK2 as the 2nd and 7th highest risk gene for ASD and ID, respectively, while other algorithms do not even list it in the top 1,000 (Table S1). It has very low TADA Q values for both disorders. However, it is a target of CHD8 and FMRP and is also a post-synaptic gene. It is a gene that plays a role in nerve growth factor (NGF)-TrkA signaling and in spermatogenesis. With this background in the literature, we think this could be a novel new candidate for both disorders identified by DeepND.
Generalization of DeepND’s performance for other diseases and disorders
We investigated whether DeepND generalizes to conditions other than ASD and ID. We investigate whether gene prediction performance increases in the multitask setting. That is, for the disease/disorder pairs mentioned below, we compared the genome-wide gene risk assessment performance of DeepND (1) in the single-task setting (DeepND-ST—separately for each disease/disorder type) and (2) in the multitask setting (DeepND).
First, we co-analyzed ID and attention-deficit/hyperactivity disorder (ADHD). As stated by Faraone et al., in contrast to the rich literature on familial co-transmission of ADHD and several other disorders, such is lacking for ID and ADHD.75 Only a few studies suggest comorbidity: an increased risk of ADHD for people with ID, and lower IQs in people with ADHD.76, 77, 78 We use the exact same setup we used to co-analyze ASD and ID for this analysis, as described in the experimental procedures. The sole difference is the positive ground-truth risk genes of ADHD, which is compiled from the following review.75 We find 32 × 101 genes (see Table S6). We observe that in the multitask setting, the median AUC and AUPR values increase by 15% and 0.2% for ADHD, respectively. While the median AUPR for ID increases substantially by 17%, the median AUC slightly decreases by 5%. Thus, we see improvement in three out of four metrics (see Table S7). The consistent improvements in ADHD results are important, as the number of known risk genes for ADHD is relatively small. DeepND bringing the median AUC value from close to random 0.49 up to 0.64 is a leap forward in gene risk assessment of ADHD.
Second, we investigated whether DeepND can be useful outside the domain of neurodevelopmental disorders. We co-analyzed breast and ovarian cancers. In addition to the risk created by the environmental factors, both breast and ovarian cancers are known to be hereditary with a large number of common risk genes/factors.79 The positive ground-truth gene sets for each cancer are curated from the following sources.80, 81, 82 We ended up with 142 and 58 positive risk genes (E1) for breast and ovarian cancers, respectively. We selected 300 random genes to generate a common negative risk gene set. The lists of these genes are given in Table S5. We used the following PPI networks: Human Protein Reference Database (HPRD),83 BioGRID,84 and DifferentialNet.53 All other settings (e.g., early stop condition, features, learning rates) are same as in the ASD-ID analysis (see learning and the cross-validation setting of DeepND). We observed that the results of DeepND improved the median DeepND-ST performance by ∼20% in terms of AUC and ∼12% in terms of AUPR for both cancer types, as shown in Table S6’s AUC and AUPR distributions. See Table S5 for gene-wise risk scores. These results show that DeepND can work with diseases/disorders other than neurodevelopmental disorders. It was able to capture the information from the overlapping genetic risk architecture and improve the gene risk prediction for both cancers.
Discussion
Neurodevelopmental disorders have been challenging geneticists and neuroscientists for decades with complex genetic architectures harboring hundreds of risk genes. Tracing inherited rare and de novo variation burdens has been the main driver of risk-gene discovery. However, overlapping genetic components and confounding clinical phenotypes make it hard to pinpoint disorder-specific susceptible genes and to understand differences. For instance, Satterstrom et al. pinpoint 102 ASD risk genes with FDR <10% with the largest ASD cohort to date covering nearly 6,500 trios.15 Yet, they still need to manually segregate these risk genes into two groups: (1) 53 ASD predominant risk genes that are distributed across a spectrum of ASD phenotypes and (2) 49 neurodevelopmental delay risk genes causing impaired cognitive, social, and motor skills. Thus, comorbidity is a further obstacle to be reckoned with in addition to identifying individual susceptible genes. Nevertheless, the shared risk genes and biological pathways offer opportunities for computational risk-assessment methods that were not explored before. So far, only disorder-specific analyses were possible by design of the network-based gene-discovery algorithms. These are limited in power due to distinct datasets that lead to limited cohort sizes. Here, we proposed a novel approach that can co-analyze comorbid neurodevelopmental disorders for gene risk assessment. The method is able to leverage the shared information using a multitask learning paradigm for the first time for this task. DeepND learns both a shared and a disorder-specific set of weights to calculate the genome-wide risk for each disorder. DeepND is a multitask deep learner that uses state-of-the-art techniques such as graph convolution and mixture of experts to learn non-linear relationships of genes on 52 brain co-expression networks. The model is also interpretable, as it is able to learn which neurodevelopmental windows (i.e., networks) provide more information for distinguishing high-risk genes and thus are important for understanding disease etiology. Our benchmarks show these techniques enable DeepND to outperform existing gene-discovery methods.
We focus on ASD and ID in this study and identify similarities such as shared affected pathways and neurodevelopmental windows and differences such as regulatory relationships and novel risk genes. We think the findings in this paper will help guiding neuroscientists researching ASD and ID in prioritizing downstream functional studies. DeepND is not an algorithm specific to these disorders, though. It can easily be extended to consider other comorbid disorders such as schizophrenia and epilepsy by adding similar single-task layers for each disorder. It can also be used for other comorbid disorders that are not related to neurodevelopment at all.
We demonstrated the advantage of being able to employ multiple co-expression networks and pointed to cases where, for instance, DAWN was not able to capture relationships between genes as they are limited with a single co-expression network. For the clarity of discussion and interpretability, we focused on only networks produced from the BrainSpan dataset. However, DeepND can also employ any combination of other types of gene-interaction networks such as protein-interaction networks, as also explained in generalization of DeepND’s performance for other diseases and disorders.
While the BrainSpan dataset and co-expression network-based analyses have been frequently used to understand the relationships between genes in the context of ASD,33, 34, 35, 36,38,41,43,43,85, 86, 87 the dataset has several limitations: it contains post-mortem tissue samples from distinct individuals of different ages. The time intervals we use to generate the co-expression networks are not equal, and there is an inherent sample variability. Moreover, these samples belong to neurotypical individuals and do not convey the biology (i.e., gene relationships) of an individual with autism. These factors potentially limit the prediction power of DeepND. Yet, as of today, BrainSpan is one of the most useful resources with which to base our analyses. One alternative to BrainSpan is the BrainVar dataset. This dataset also contains healthy samples from various time points in the neurodevelopment. Yet, it contains only samples from the dorsolateral prefrontal cortex region. We foresee that with the ongoing interest in single-cell RNA sequencing (RNA-seq) studies, we are going to have access to better models of gene interactions during neurodevelopment with autism and other comorbid disorders in the near future.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources should be directed to the lead contact, A. Ercument Cicek (cicek@cs.bilkent.edu.tr).
Materials availability
This study did not generate any new unique materials.
Ground-truth risk-gene sets
Our genome-wide gene risk-prediction algorithm is based on a semi-supervised learning framework in which some of the samples (i.e., genes) are labeled (as ASD/ID risk gene or not) and are used to learn a ranking for the unlabeled samples.
We obtain the labels for ASD from SFARI dataset.46 These genes are classified into three evidence levels indicating the quality of the evidence (E1–E3, E1 indicating the highest risk). The list contains 185 E1 genes (from SFARI category I), 200 E2 genes (from SFARI category II), and 470 E3 genes (from SFARI category III) as positively labeled ASD risk genes. We also obtain 893 non-mental-health-related genes from OMIM and Krishnan et al. as negatively labeled genes. The list and corresponding evidence levels are listed in Table S1. In the loss calculations during training of the model, genes in these categories are assigned the following weights: E1 (1.0), E2 (0.50), E3/E4 (0.25) and negative (1.0) genes. The performance is evaluated only on E1 genes for the sake of compatibility with other methods.
For ID, we rely on review studies from the literature that provide in-depth analyses and lists of ID risk genes. We considered the known-gene lists from 2 landmark review studies as our base ground-truth ID risk-gene list.21,47 We divide this set into 3 parts (i.e., E1, E2, and E3) with respect to evidence obtained from 4 other studies.48, 49, 50, 51 The genes that are indicated by 5 or more studies are assigned to the highest-risk class, E1, and the genes that are indicated by 4 studies are assigned to the second-highest-risk class, E2. The remaining genes are assigned to the third risk-gene class, E3. See Table S2 for a detailed breakdown of evidence for each gene. We use the same set of negative genes as ASD, which are non-mental-health-related genes. Consequently, we have 123 × 101, 224 × 102, and 232 × 103 genes and 893 negative genes for ID. See Table S1 for a complete list of ground-truth genes for both disorders. The weights we use per gene class are as follows: E1 (1.0), E2 (0.50), E3 (0.25), and negative (1.0) genes. For the sake of consistency, we also report the performance of the model on E1 genes for ID.
Gene risk features
The only gene risk feature we use is the gene pLI, for both disorders. See Table S1 for the pLI of each gene obtained from Satterstrom et al.15 Note that neurodevelopmental risk genes are known to be conserved15 and that pLI has been used to quantify autism risk in the literature.52 Yet, the ground-truth labels described in ground-truth risk-gene sets are assigned independently and have no ties to this information.
Gene co-expression networks
We used the BrainSpan dataset of the Allen Brain Atlas44,45 in order to model gene interactions through neurodevelopment and generated a spatiotemporal system of gene co-expression networks. This dataset contains 57 post-mortem brains (16 regions) that span 15 consecutive neurodevelopmental periods from 8 post-conception weeks to 40 years. To partition the dataset into developmental periods and clusters of brain regions, we follow the practice in Willsey et al.43 and refer the reader to this paper for details. Note that this is the same grouping also employed by Norman and Cicek34 and Liu et al.41,85 Brain regions were hierarchically clustered according to their transcriptional similarity, and four clusters were obtained (Figure 3B): (1) V1C-STC, (2) PFC-MSC, (3) SHA, and (4) MDCBC. In the temporal dimension, 13 neurodevelopmental windows (Figure 3A) were obtained using a sliding-window approach (i.e., [1–3], [3–5], , [13–15]). A spatiotemporal window of neurodevelopment and its corresponding co-expression network are denoted by the abbreviation for its brain-region cluster followed by the time window of interest, e.g. “PFC-MSC(1–3)” represents interactions among genes in the region PFC-MSC during the time interval [1–3].
Using the above-mentioned partitioning, we obtained 52 spatiotemporal gene co-expression networks, each of which contain 25,825 nodes representing genes. An undirected unweighted edge between two nodes is created if their absolute Pearson correlation coefficient is greater than or equal to 0.8 in the related partition of BrainSpan data.
The reason for picking 0.8 as our threshold is as follows: we observed that, overall, a lower threshold results in an AUC and AUPR performance increase, and we found out that 0.7 and 0.8 had very close median performances and that 0.8 was the smallest threshold that let us fit all 52 gene co-expression networks into the eight graphics processing units (GPUs) we used (total memory of 114 GB).
Instead of using binary co-expression networks, we also tested using weighted co-expression networks where we use the co-expression values as the edge weights. We found that binary edges provide slightly better median performance and opted to use binary co-expression networks.
DeepND model
Problem formulation
Each 52 co-expression network j described in gene co-expression networks is represented as a graph , where the vertex set contains genes in the human genome and denotes the binary adjacency matrix. Note that n = 25,825. Let be the feature matrix for disorder D where each row is a list of d features associated with gene (and node) . In our application, is a one-hot-encoded vector, uniquely representing the gene i. We also use pLI as a feature that represents how constrained the gene i is (i.e., ) an . Let be an l dimensional vector, where [i] = 1 if the the node i is a risk gene for ASD, and [i] = 0 if the gene is non-mental-health related, , l < n. Note that, in this semi-supervised learning task, only the first l genes out of n have labels. is defined similarly for ID, using its ground-truth risk and non-risk gene sets. The goal of the algorithm is to learn a function . denotes , and denotes .
GCN model
CNNs have revolutionized the computer-vision field by significantly improving the state of the art by extracting local patterns on grid-structured data.88 Applying the same principle on arbitrarily structured graph data has also been a success.89, 90, 91 While all these spectral approaches have proven useful, they are computationally expensive. Kipf and Welling have proposed an approach (GCN) to approximate the filters as a Chebyshev expansion of the graph Laplacian92 and let them operate on the 1-hop neighborhood of each node.93 This fast and scalable approach extracts a network-adjusted feature vector for each node (i.e., embedding) that incorporates high-dimensional information about each node’s neighborhood in the network. The convolution operation of DeepND is based on this method. Given a gene co-expression network , GCN inputs the normalized adjacency matrix with self loops (i.e., ) and the feature vector , for gene i and for disorder D. d = 1 in this application and is shared among ASD and ID. Then, the first layer embedding produced by GCN is computed using the following propagation rule: = , where is the weight matrix at the input layer to be learned, is the rectified linear unit function, and is the normalized version of a diagonal matrix where . We pick = 4 in this application. Each subsequent layer k is defined similarly as follows: = . Thus, the output of a k-layered GCN, for gene i on co-expression network j, is denoted as follows: = . In this application, we use two GCN layers and . The final layer is softmax to produce probabilities for the positive class. That is, the output of a GCN model j, for a gene i, is = .
Mixture-of-experts model
Mixture-of-experts (MoE) model is an ensemble machine-learning approach that aims to find out informative models (experts) among a collection.94 Specifically, MoE inputs the features and assigns weights to the outputs of the experts. In DeepND architecture, individual experts are the 52 GCNs that operate on 52 gene co-expression networks, as explained above (). For every gene i, MoE also inputs . It produces a weight of length 52, which is passed through a softmax layer (i.e., . The outputs of the GCNs are weighted by this network, and the weighted sum is used to produce a risk probability for every gene i using softmax. That is, . The GCNs produce 52 values (one per co-expression network), which are concatenated to produce . Finally, the following dot product is used to produce the risk probability of gene i for disorder D: .
Multitask learning model
The above-mentioned GCN and MoE cascade inputs , , and the co-expression networks along with labels for a single disorder to predict the risk probability for every gene. Thus, it corresponds to the single-task version of DeepND (i.e., DeepND-ST.) On the other hand, DeepND is designed to work concurrently with multiple disorders (i.e., ASD and ID.) DeepND employs one DeepND-ST cascade per disorder and puts a multilayer perceptron (MLP) as a precursor to two DeepND-ST subnetworks. The weights learned on these subnetworks are only affected by the back-propagated loss of the corresponding disorder, and hence, these are single-task parts of the architecture. On the contrary, the weights learned on the MLP part are affected by the loss back propagated from both subnetworks. Thus, this part corresponds to the multitask component of the DeepND architecture. The MLP layer inputs and passes it through a fully connected layer followed by Leaky ReLU activation (negative slope = -1.5) to learn a weight matrix and output a dimensional embedding to be input DeepND-ST instead of . That is, .
Learning and the cross-validation setting of DeepND
To evaluate both DeepND-ST and DeepND approaches, we use a 5-fold cross-validation scheme. All labeled genes are uniformly and randomly distributed to the folds. At each training iteration, we leave one fold for validation and one fold for testing. We train the model on the remaining three folds of the labeled genes. For all the genes in the left-out fold, their feature vectors are nullified, and their labels are masked and not included in the training loss calculation when input to training in order to prevent information leakage.
The model is trained up to 1,000 epochs with early stop, which is determined with respect to the loss calculated on the validation fold using only E1 genes as the positives and all negative genes. Once the model converges, the test performance is reported on the test fold in the same manner as in the validation fold. The model uses cross-entropy loss (evidence weighted) and ADAM optimizer95 for updating the weights, which are initialized using Xavier initialization.96 DeepND-ST uses a fixed learning rate of 1 × 10-4 for both disorders. DeepND uses the learning rates of 7 × 10-4, 1 × 10-4, and 1 × 10-3 for the shared layer, ASD single-task layer, and ID single-task layer, respectively. To ensure proper convergence of DeepND, should one of the single-task subnetworks converge, the learning rate of that subnetwork and the shared layer are cut down twenty folds. This lets the yet-underfit subnetwork keep learning until early stop or the epoch limit.
The above-mentioned procedure produces four results as for every left-out test fold as all remaining 4 folds are used as validation. We repeat this process 10 times with random initialization to obtain a total of 200 results for performance comparison (Figure 2). For ASD, positively labeled genes’ weights equal 1.00 (E1), 0.50 (E2), 0.25 (E3), and 0.25 (E4). For ID, positively labeled genes’ weights equal 1.00 (E1) and 0.50 (E2). For both disorders, negatively labeled genes have a weight of 1.00. Note that this procedure is in line with the setting of Krishnan et al. for fairness of comparison.
Genome-wide risk prediction for ASD and ID and comparison with other methods
We compare the performance of DeepND-ST and DeepND with other state-of-the-art network-based neurodevelopmental disorder gene risk-assessment methods from the literature that output a genome-wide risk ranking: Krishnan et al.,42 DAWN,85 DAMAGES score,52 random-forest-based method of Duda et al.,39 and forecASD.38
We run Krishnan et al.’s approach as described in their manuscript.42 This is an evidence-weighted SVM classifier that identifies risk genes based on similarity of network features. They use a human brain-specific functional interaction network to generate features as input.97 Note that the ground-truth gene set is the same as ours as well as the evidence weights for ASD. We perform a 5-fold cross-validation. That is, for each iteration, we train their SVM model on 80% of the labeled genes and evaluate the model on E1 genes and all negative genes in the left-out 20% of the labeled genes as suggested. We repeat this procedure 10 times. We post-process SVM outputs to produce risk probabilities using isotonic regression, which ensures that the gene ranking is preserved. We use the pLI value of each gene as the dependent variable. In a 10-fold cross-validation setting, we detect knots on the left-out fold and fit another isotonic-regression line to interpolate the knots. We use SVM output for all genes to produce gene risk-probability values for the corresponding disorder and produce the genome-wide risk ranking. We compare this method and DeepND-ST/DeepND with respect to (1) the AUC and AUPR distributions calculated on the left-out fold at each cross-validation iteration (Figures 2A and 2B) and (2) the Matthews correlation coefficient of the final predictions with the ground-truth (using a rank percentage threshold; Figures 2C and 2Ciii) PR curves (Figure 2D).
DAWN is a hidden Markov-random-field-based approach that assigns a posterior, network-adjusted disorder risk score to every gene based on the guilt-by-association principle. It inputs transmission and de novo association analysis (TADA) p values as prior features along with a partial co-expression network to assess connectivity. We input the TADA p values to DAWN.15,37 The method also uses partial co-expression networks. We use two networks that the authors suggest as the most useful for this task in their manuscript.85 These represent prefrontal cortex/mid-fetal period (i.e., PFC-MSC 3–5 and PFC-MSC 4–6). We generate these networks using the RNA-seq data in the BrainSpan dataset. Note that DeepND utilizes the same dataset and uses these networks and 50 others. Instead of partial co-expression networks, DeepND uses co-expression networks. The DAMAGES score is a principal-component-analysis-based technique that assess the risk of genes based on (1) the similarity of their expression profiles in 24 specific mouse CNS cell types, (2) the enrichment of mutations in cases as opposed to controls, and (3) the pLI score of the gene. We directly obtained the risk ranking from Shen and Zhang.52 We also benchmark the random-forest-based method presented by Duda et al.39 The method inputs a combination of (1) gene-expression data from several microarray studies, (2) PPIs from multiple databases, (3) a quantitative physical interaction (PI) score for all protein isoform pairs in mouse, and (4) phenotype annotations from the Mouse Genome Informatics (MGI) database. They combine these modalities by a Bayesian network, and the final probabilities of functional interactions between gene pairs are represented with a matrix. Since this method does not support evidence weights for the ground-truth set, in the experimental setting, we used our ground-truth gene set for ASD after discarding the evidence information to obtain results. We compare all above mentioned methods with respect to (1) the Matthews correlation coefficient of the final predictions with the ground-truth (using a rank percentage threshold; Figures 2C and 2Cii) PR curves (Figure 2D).
Apart from the previously mentioned methods, we also consider forecASD, which is a random-forest-based ensemble method. It inputs features derived from (1) BrainSpan transcriptome, (2) the STRING PPI network, and (3) the outputs of several previously published ASD gene prediction methods: Krishnan et al.,42 DAWN,85 DAMAGES score,52 and TADA.98 These features are combined in a two-layer architecture. In the first layer, each derived feature set is used to train a random-forest branch. Then, in the second step, the results from the first layer are combined and are used to train the final ensemble classifier. We trained the original forecASD architecture with our ground-truth sets for ASD and ID and obtained their results. Then, in a separate run, we also added the final ranking of DeepND as an extra feature and retrained the model. We compared these two results with our final ranking using (1) the Matthews correlation coefficient of the final predictions with the ground-truth (using a rank percentage threshold) and (2) PR curves (Figure S1).
Enrichment analyses
We evaluate the enrichment of DeepND’s ASD and ID gene risk rankings in gene lists, which are known to be enriched in disorder risk genes in the literature. These lists are (1) targets of transcription regulators like CHD8,54 FMRP,55,99 RBFOX1,57,58 and TOP1,59 (2) susceptibility pathways like WNT signaling60 and MAPK signaling,61 and (3) affected biological processes and molecular functions like post-synaptic density complex62,63 and histone modification.2,64 We use the binomial test to determine whether the top decile in the corresponding ranking significantly deviates from uniform enrichment (Figure 5).
We perform a Gene Ontology term enrichment analysis of the top-percentile predictions using the Enrichr system65,66 with respect to Biological Process and Molecular Function terms. As both analyses point to transcription-factor regulation, we investigate the connectivity of the high-risk genes in the ChEA database,67 which is a large repository that lists experimentally validated transcriptional regulation relationships in various organisms.
Spatiotemporal network analyses
We investigate which GCNs (i.e., neurodevelopmental windows) are better predictors of gene risk for each disorder. For each gene i, the average risk probability assigned by the corresponding GCN is calculated over all iterations such that i is in the test fold. The average values for top-percentile genes for each disorder are shown in Figure 3.
To check whether obtained average risk probabilities per neurodevelopmental window that are assigned by that GCN are empirically significant, we performed a permutation test. First, we randomly redistribute the labels of the ground-truth genes while keeping the counts same. We obtain 100 randomized ground-truth sets. Keeping all other settings same, we train 100 DeepND models and obtain 100 ASD and 100 ID risk rankings. Using these rankings, we calculate the average risk probability assigned by each GCN and obtain two background distributions for each GCN: one for ASD and one for ID (Figures S4 and S5). We then compare the original average probability assigned by that GCN with the corresponding background distribution to assess significance.
Experimental setup
All DeepND models are trained and tested on a Super-Micro Super-Server 4029GP-TRT with 2 Intel Xeon Gold 6140 Processors (2.3 GHz, 24.75 M cache), 251 GB RAM, 6 NVIDIA GeForce RTX 2080 Ti (11 GB, 352 bit), and 2 NVIDIA TITAN RTX GPUs (24 GB, 384 bit). For DeepND-ST, we used 32,080 RTX and 1 TITAN RTX cards. For DeepND, we used 5 RTX 2080 and one TITAN RTX cards.
The running time of DeepND depends on the number, size (number of nodes), and density (number of edges) of the networks used. Note that the number of input genes and the features are constant for each condition so that it is independent of the considered disease. When we used the 3 PPI networks to co-analyze breast and ovarian cancers, DeepND took approximately 3 h. When we used the 52 co-expression networks, it took approximately 16 h.
Acknowledgments
We thank all families who participated in the studies and enabled this study with data. The authors would like to acknowledge Autism Sequencing Consortium PIs and contributors. This work was supported by a pilot grant from the Simons Foundation (SFARI 640935, A.E.C.). We also acknowledge support by TUBA GEBIP 2017 and Bilim Akademisi BAGEP 2020 Awards to A.E.C.
Author contributions
A.E.C. conceived, designed, and supervised the study. I.B. and O.K. implemented the DeepND software. I.B., O.K., and A.E.C. performed the computational experiments and wrote the manuscript.
Declaration of interests
The authors declare no competing financial interests.
Published: June 2, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.patter.2022.100524.
Supplemental information
The table marks the gold standard genes: 855 positively and 893 negatively labeled genes for ASD; and 579 positively and 893 negatively labeled genes for ID are given along with their evidence levels (E1 - E4, E1 indicating the highest risk). The table also provides the rankings from other gene discovery algorithms from the literature. For each gene, participation in disorder-related gene sets are provided (e.g., WNT pathway, CHD8 targets etc.). We also provide 29 gene specific details compiled from the literature.
values
The table provides information about the studies used to generate the ground truth labels for ID. For each gene, the base studies which indicates it as a risk gene are provided.
The table lists (i) the GO enrichment analysis for the top percentile risk genes for both disorders (Biological Process and Molecular Function). It also provides lists of top transcription factor (TF) regulators whose targets are enriched with the top percentile ASD and/or ID risk genes, respectively. The TF enrichment results are based on the experimentally validated TF-gene relationships in ChEA 2016 database. All results are obtained using the EnrichR system.
DeepND risk rankings for the genes within 6 ASD related (16p11.2, 15q11-13, 15q13.3, 1q21.1 and 22q11) (Krishnan et al., 2016) and 6 mental retardation related CNV regions (16p11.2, 1q21.1, 22q11.21, 22q11.22, 16p13 and 17p11.2). Frequency of these CNV regions within ASD and ID individuals are provided when available.
The table marks the gold standard genes: 142 positively labeled gene for breast cancer, 58 positively labeled gene for ovarian cancer and 300 negatively labeled genes for both diseases. It also provides information about the studies used to generate the ground truth labels for both diseases.
The table provides the genome-wide risk probability predictions and rankings of DeepND for ADHD and also marks the 31 positive and 893 negative gold standard gene for ADHD.
Data and code availability
All datasets, which are referenced in the relevant experimental procedures subsections, used in this study are publicly available. The processed data that were used to obtain results presented in this paper are also publicly available at Zenodo: https://doi.org/10.5281/zenodo.3892979. All data supporting the key findings such as gene risk evidence levels and gene risk predictions are available within the article and corresponding supplemental tables.
DeepND has been implemented and released at http://github.com/ciceklab/deepnd (Zenodo: https://doi.org/10.5281/zenodo.6501496). We provide the environment, which contains all dependencies for an easy setup. We give a small example to train and test both DeepND-ST/DeepND models. Finally, we provide the code and links to the full set of datasets to reproduce the results (genome-wide risk rankings and data for heatmaps) presented in this manuscript for ASD and ID.
References
- 1.He X., Sanders S.J., Liu L., Rubeis S.D., Lim E.T., Sutcliffe J.S., Schellenberg G.D., Gibbs R.A., Daly M.J., Buxbaum J.D., et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013 doi: 10.1371/journal.pgen.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Rubeis S., He X., Goldberg A.P., Poultney C.S., Samocha K., Cicek A.E., Kou Y., Liu L., Fromer M., Walker S., et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014 doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E., et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014 doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012 doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D., et al. Sporadic autism exomes reveal a highly interconnected protein network ofde novomutations. Nature. 2012 doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K., et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012 doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V., et al. Patterns and rates of exonicde novomutations in autism spectrum disorders. Nature. 2012 doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A., et al. De novo gene disruptions in children on the autistic spectrum. Neuro. 2012 doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grove J., Ripke S., Als T.D., Mattheisen M., Walters R.K., Won H., Pallesen J., Agerbo E., Andreassen O.A., Anney R., et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019 doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feliciano P., Daniels A.M., Snyder L.G., Beaumont A., Camba A., Esler A., Gulsrud A.G., Mason A., Gutierrez A., Nicholson A., et al. Spark: a us cohort of 50,000 families to accelerate autism research. Neuron. 2018 doi: 10.1016/j.neuron.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devlin B., Melhem N., Roeder K. Do common variants play a role in risk for autism? evidence and theoretical musings. Brain Res. 2011 doi: 10.1016/j.brainres.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma D., Salyakina D., Jaworski J.M., Konidari I., Whitehead P.L., Andersen A.N., Hoffman J.D., Slifer S.H., Hedges D.J., Cukier H.N., et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14. 1. Ann. Hum. Genet. 2009 doi: 10.1111/j.1469-1809.2009.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meta-analysis of gwas of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24. 32 and a significant overlap with schizophrenia. Mol. Autism. 2017 doi: 10.1186/s13229-017-0137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anney R., Klei L., Pinto D., Almeida J., Bacchelli E., Baird G., Bolshakova N., Bolte S., Bolton P.F., Bourgeron T., et al. Individual common variants exert weak effects on risk for autism spectrum disorders. Hum. Mol. Genet. 2012 doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satterstrom F.K., Kosmicki J.A., Wang J., Breen M.S., De Rubeis S., An J.-Y., Peng M., Collins R., Grove J., Klei L., et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020 doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell K.J. The genetics of neurodevelopmental disease. Curr. Opin. Neurobiol. 2011 doi: 10.1016/j.conb.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Roeper J. Closing gaps in brain disease — from overlapping genetic architecture to common motifs of synapse dysfunction. Curr. Opin. Neurobiol. 2018 doi: 10.1016/j.conb.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 18.Lee P.H., Anttila V., Won H., Feng Y.-C.A., Rosenthal J., Zhu Z., Tucker-Drob E.M., Nivard M.G., Grotzinger A.D., Posthuma D., et al. 2019. Genome Wide Meta-Analysis Identifies Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Ligt J., Willemsen M.H., Van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., De Vries P., Gilissen C., et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012 doi: 10.1097/01.ogx.0000428160.59063.a6. [DOI] [PubMed] [Google Scholar]
- 20.Firth H.V., Wright C.F., study D. The deciphering developmental disorders (ddd) study. Dev. Med. Child Neurol. 2011 doi: 10.1111/j.1469-8749.2011.04032.x. [DOI] [PubMed] [Google Scholar]
- 21.Gilissen C., Hehir-Kwa J.Y., Thung D.T., van de Vorst M., van Bon B.W., Willemsen M.H., Kwint M., Janssen I.M., Hoischen A., Schenck A., et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014 doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 22.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N., et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012:61480–61489. doi: 10.1016/S0140-6736. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava S., Cohen J.S., Vernon H., Barañano K., McClellan R., Jamal L., Naidu S., Fatemi A. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 2014 doi: 10.1002/ana.24251. [DOI] [PubMed] [Google Scholar]
- 24.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M., et al. A de novo paradigm for mental retardation. Nat. Genet. 2010 doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X., Petrovski S., Xie P., Ruzzo E.K., Lu Y.-F., McSweeney K.M., Ben-Zeev B., Nissenkorn A., Anikster Y., Oz-Levi D., et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet. Med. 2015 doi: 10.1038/gim.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno-De-Luca A., Myers S.M., Challman T.D., Moreno-De-Luca D., Evans D.W., Ledbetter D.H. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013 doi: 10.1016/S1474-4422-70011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baio J., Wiggins L., Christensen D.L., Maenner M.J., Daniels J., Warren Z., Kurzius-Spencer M., Zahorodny W., Rosenberg C.R., White T., et al. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveillance Summaries. 2018 doi: 10.15585/mmwr.ss6706a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J., Cai T., Jiang Y., Chen H., He X., Chen C., Li X., Shao Q., Ran X., Li Z., et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from npdenovo database. Mol. Psychiatr. 2016 doi: 10.1038/mp.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson E.B., Samocha K.E., Kosmicki J.A., McGrath L., Neale B.M., Perlis R.H., Daly M.J. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl. Acad. Sci. Unit. States Am. 2014 doi: 10.1073/pnas.1409204111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen T.-H., Dobbyn A., Brown R.C., Riley B.P., Buxbaum J.D., Pinto D., Purcell S.M., Sullivan P.F., He X., Stahl E.A. Mtada is a framework for identifying risk genes from de novo mutations in multiple traits. Nat. Commun. 2020 doi: 10.1038/s41467-020-16487-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Mantilla A.J., Moreno-De-Luca A., Ledbetter D.H., Martin C.L. A cross-disorder method to identify novel candidate genes for developmental brain disorders. JAMA Psychiatr. 2016 doi: 10.1001/jamapsychiatry.2015.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gandal M.J., Haney J.R., Parikshak N.N., Leppa V., Ramaswami G., Hartl C., Schork A.J., Appadurai V., Buil A., Werge T.M., et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 2018 doi: 10.1126/science.aad6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hormozdiari F., Penn O., Borenstein E., Eichler E.E. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2014 doi: 10.1101/gr.178855.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Norman U., Cicek A.E. St-steiner: a spatio-temporal gene discovery algorithm. Bioinformatics. 2019 doi: 10.1093/bioinformatics/btz110. [DOI] [PubMed] [Google Scholar]
- 35.Gilman S.R., Iossifov I., Levy D., Ronemus M., Wigler M., Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011 doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilman S.R., Chang J., Xu B., Bawa T.S., Gogos J.A., Karayiorgou M., Vitkup D. Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat. Neurosci. 2012 doi: 10.1038/nn.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen H.T., Bryois J., Kim A., Dobbyn A., Huckins L.M., Munoz-Manchado A.B., Ruderfer D.M., Genovese G., Fromer M., Xu X., et al. Integrated bayesian analysis of rare exonic variants to identify risk genes for schizophrenia and neurodevelopmental disorders. Genome Med. 2017 doi: 10.1186/s13073-017-0497-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brueggeman L., Koomar T., Michaelson J.J. Forecasting risk gene discovery in autism with machine learning and genome-scale data. Sci. Rep. 2020 doi: 10.1038/s41598-020-61288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duda M., Zhang H., Li H.-D., Wall D.P., Burmeister M., Guan Y. Brain-specific functional relationship networks inform autism spectrum disorder gene prediction. Transl. Psychiatry. 2018 doi: 10.1038/s41398-018-0098-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buxbaum J., Cicek E., Devlin B., Klei L., Roeder K., De Rubeis S. Combining autism and intellectual disability exome data implicates disruption of neocortical development in both disorders. Eur. Neuropsychopharmacol. 2017 doi: 10.1016/j.euroneuro.2016.09.497. [DOI] [Google Scholar]
- 41.Liu L., Lei J., Sanders S.J., Willsey A.J., Kou Y., Cicek A.E., Klei L., Lu C., He X., Li M., et al. Dawn: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol. Autism. 2014 doi: 10.1186/2040-2392-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishnan A., Zhang R., Yao V., Theesfeld C.L., Wong A.K., Tadych A., Volfovsky N., Packer A., Lash A., Troyanskaya O.G. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 2016 doi: 10.1038/nn.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willsey A.J., Sanders S.J., Li M., Dong S., Tebbenkamp A.T., Muhle R., Reilly S.K., Lin L., Fertuzinhos S., Miller J.A., et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013 doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunkin S.M., Ng L., Lau C., Dolbeare T., Gilbert T.L., Thompson C.L., Hawrylycz M., Dang C. Allen brain atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic acids research. 2012 doi: 10.1093/nar/gks1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G., et al. Spatio-temporal transcriptome of the human brain. Nature. 2011 doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abrahams B.S., Arking D.E., Campbell D.B., Mefford H.C., Morrow E.M., Weiss L.A., Menashe I., Wadkins T., Banerjee-Basu S., Packer A. Sfari gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (asds) Mol. Autism. 2013 doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grozeva D., Carss K., Spasic-Boskovic O., Tejada M.-I., Gecz J., Shaw M., Corbett M., Haan E., Thompson E., Friend K., et al. Targeted next-generation sequencing analysis of 1,000 individuals with intellectual disability. Hum. Mutat. 2015 doi: 10.1002/humu.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vissers L.E., Gilissen C., Veltman J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016 doi: 10.1038/nrg3999. [DOI] [PubMed] [Google Scholar]
- 49.Inlow J.K., Restifo L.L. Molecular and comparative genetics of mental retardation. Genetics. 2004 doi: 10.1534/genetics.166.2.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiurazzi P., Pirozzi F. Advances in Understanding–Genetic Basis of Intellectual Disability. F1000Research. 2016 doi: 10.12688/f1000research.7134.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaplanis J., Samocha K.E., Wiel L., Zhang Z., Arvai K.J., Eberhardt R.Y., Gallone G., Lelieveld S.H., Martin H.C., McRae J.F., et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020 doi: 10.1038/s41586-020-2832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang C., Shen Y. A cell type-specific expression signature predicts haploinsufficient autism-susceptibility genes. Hum. Mutat. 2017 doi: 10.1002/humu.23147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basha O., Shpringer R., Argov C.M., Yeger-Lotem E. The differentialnet database of differential protein–protein interactions in human tissues. Nucleic acids research. 2018 doi: 10.1093/nar/gkx981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cotney J., Muhle R.A., Sanders S.J., Liu L., Willsey A.J., Niu W., Liu W., Klei L., Lei J., Yin J., et al. The autism-associated chromatin modifier chd8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 2015 doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y.S., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W., et al. Fmrp stalls ribosomal translocation on mrnas linked to synaptic function and autism. Cell. 2011 doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ascano M., Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L., Langlois C., Munschauer M., Dewell S., Hafner M., et al. Fmrp targets distinct mrna sequence elements to regulate protein expression. Nature. 2012 doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S., Mill J., Cantor R.M., Blencowe B.J., Geschwind D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011 doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weyn-Vanhentenryck S.M., Mele A., Yan Q., Sun S., Farny N., Zhang Z., Xue C., Herre M., Silver P.A., Zhang M.Q., et al. Hits-clip and integrative modeling define the rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014 doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.King I.F., Yandava C.N., Mabb A.M., Hsiao J.S., Huang H.-S., Pearson B.L., Calabrese J.M., Starmer J., Parker J.S., Magnuson T., et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013 doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalkman H.O. A review of the evidence for the canonical wnt pathway in autism spectrum disorders. Mol. Autism. 2012 doi: 10.1186/2040-2392-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinto D., Delaby E., Merico D., Barbosa M., Merikangas A., Klei L., Thiruvahindrapuram B., Xu X., Ziman R., Wang Z., et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014 doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bayés À., Van De Lagemaat L.N., Collins M.O., Croning M.D., Whittle I.R., Choudhary J.S., Grant S.G. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011 doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zoghbi H.Y., Bear M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harbor Perspect. Biol. 2012 doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iwase S., Bérubé N.G., Zhou Z., Kasri N.N., Battaglioli E., Scandaglia M., Barco A. Epigenetic etiology of intellectual disability. J. Neurosci. 2017 doi: 10.1523/JNEUROSCI.1840-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative html5 gene list enrichment analysis tool. BMC Bioinf. 2013 doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids Research. 2016 doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lachmann A., Xu H., Krishnan J., Berger S.I., Mazloom A.R., Ma’ayan A. Chea: transcription factor regulation inferred from integrating genome-wide chip-x experiments. Bioinformatics. 2010 doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou G., Soufan O., Ewald J., Hancock R.E., Basu N., Xia J. Networkanalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic acids research. 2019 doi: 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halvardson J., Zhao J.J., Zaghlool A., Wentzel C., Georgii-Hemming P., Månsson E., Ederth Sävmarker H., Brandberg G., Soussi Zander C., Thuresson A.-C., et al. Mutations in hecw2 are associated with intellectual disability and epilepsy. J. Med. Genet. 2016 doi: 10.1136/jmedgenet-2016-103814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mefford H.C., Cooper G.M., Zerr T., Smith J.D., Baker C., Shafer N., Thorland E.C., Skinner C., Schwartz C.E., Nickerson D.A., et al. A method for rapid, targeted cnv genotyping identifies rare variants associated with neurocognitive disease. Genome Res. 2009 doi: 10.1101/gr.094987.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goytain A., Hines R.M., Quamme G.A. 2008. Functional Characterization of Nipa2, a Selective Mg2+ Transporter. [DOI] [PubMed] [Google Scholar]
- 72.Seabra M.C., Mules E.H., Hume A.N. Rab gtpases, intracellular traffic and disease. Trends Mol. Med. 2002 doi: 10.1016/s1471-4914(01)02227-4. [DOI] [PubMed] [Google Scholar]
- 73.Cordeddu V., Redeker B., Stellacci E., Jongejan A., Fragale A., Bradley T.E., Anselmi M., Ciolfi A., Cecchetti S., Muto V., et al. Mutations in zbtb20 cause primrose syndrome. Nat. Genet. 2014 doi: 10.1038/ng.3035. [DOI] [PubMed] [Google Scholar]
- 74.Cleaver R., Berg J., Craft E., Foster A., Gibbons R.J., Hobson E., Lachlan K., Naik S., Sampson J.R., Sharif S., et al. Refining the primrose syndrome phenotype: a study of five patients with zbtb20 de novo variants and a review of the literature. Am. J. Med. Genet. 2019 doi: 10.1002/ajmg.a.61024. [DOI] [PubMed] [Google Scholar]
- 75.Faraone S.V., Larsson H. Genetics of attention deficit hyperactivity disorder. Mol. Psychiatr. 2019 doi: 10.1038/s41380-018-0070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frazier T.W., Demaree H.A., Youngstrom E.A. Meta-analysis of intellectual and neuropsychological test performance in attention-deficit/hyperactivity disorder. Neuropsychology. 2004 doi: 10.1037/0894-4105.18.3.543. [DOI] [PubMed] [Google Scholar]
- 77.Antshel K.M., Phillips M.H., Gordon M., Barkley R., Faraone S.V. Is adhd a valid disorder in children with intellectual delays? Clin. Psychol. Rev. 2006 doi: 10.1016/j.cpr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 78.Faraone S.V., Ghirardi L., Kuja-Halkola R., Lichtenstein P., Larsson H. The familial co-aggregation of attention-deficit/hyperactivity disorder and intellectual disability: a register-based family study. Journal of the American Academy of Child & Adolescent Psychiatry. 2017 doi: 10.1016/j.jaac.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 79.Zografos G.C., Panou M., Panou N. Common risk factors of breast and ovarian cancer: recent view. International Journal of Gynecologic Cancer. 2004 doi: 10.1136/ijgc-00009577-200409000-00002. [DOI] [PubMed] [Google Scholar]
- 80.Becker K.G., Barnes K.C., Bright T.J., Wang S.A. The genetic association database. Nat. Genet. 2004 doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- 81.Liu S.-H., Shen P.-C., Chen C.-Y., Hsu A.-N., Cho Y.-C., Lai Y.-L., Chen F.-H., Li C.-Y., Wang S.-C., Chen M., et al. Driverdbv3: a multi-omics database for cancer driver gene research. Nucleic acids research. 2020 doi: 10.1093/nar/gkz964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang T., Ruan S., Zhao X., Shi X., Teng H., Zhong J., You M., Xia K., Sun Z., Mao F. Oncovar: an integrated database and analysis platform for oncogenic driver variants in cancers. Nucleic acids research. 2021 doi: 10.1093/nar/gkaa1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keshava Prasad T., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., et al. Human protein reference database-2009 update. Nucleic acids research. 2008 doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stark C., Breitkreutz B.J., Reguly T., Boucher L., Breitkreutz A. Biogrid: a general repository for interaction datasets. Nucleic Acids Res. 2006 doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu L., Lei J., Roeder K. Network assisted analysis to reveal the genetic basis of autism. Ann. Appl. Stat. 2015 doi: 10.1214/15-AOAS844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gulsuner S., Walsh T., Watts A.C., Lee M.K., Thornton A.M., Casadei S., Rippey C., Shahin H., Braff D., Cadenhead K.S., et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013 doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parikshak N.N., Luo R., Zhang A., Won H., Lowe J.K., Chandran V., Horvath S., Geschwind D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013 doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Krizhevsky A., Sutskever I., Hinton G.E. Advances in neural information processing systems. 2012. Imagenet classification with deep convolutional neural networks; pp. 1097–1105. [DOI] [Google Scholar]
- 89.Bruna J., Zaremba W., Szlam A., LeCun Y. Spectral Networks and Locally Connected Networks on Graphs. arXiv. 2013 doi: 10.48550/arXiv.1312.6203. Preprint at. [DOI] [Google Scholar]
- 90.Duvenaud D.K., Maclaurin D., Iparraguirre J., Bombarell R., Hirzel T., Aspuru-Guzik A., Adams R.P. Advances in neural information processing systems. 2015. Convolutional networks on graphs for learning molecular fingerprints; pp. 2224–2232. [DOI] [Google Scholar]
- 91.Henaff M., Bruna J., LeCun Y. Deep Convolutional Networks on Graph-Structured Data. arXiv. 2015 doi: 10.48550/arXiv.1506.05163. Preprint at. [DOI] [Google Scholar]
- 92.Defferrard M., Bresson X., Vandergheynst P. Advances in neural information processing systems. 2016. Convolutional neural networks on graphs with fast localized spectral filtering; pp. 3844–3852. [DOI] [Google Scholar]
- 93.Kipf T.N., Welling M. Semi-supervised classification with graph convolutional networks. arXiv. 2016 doi: 10.48550/arXiv.1609.02907. Preprint at. [DOI] [Google Scholar]
- 94.Masoudnia S., Ebrahimpour R. Artificial Intelligence Review. 2014. Mixture of experts: a literature survey. [DOI] [Google Scholar]
- 95.Kingma D.P., Ba J. Adam: a method for stochastic optimization. arXiv. 2014 doi: 10.48550/arXiv.1412.6980. Preprint at. [DOI] [Google Scholar]
- 96.Glorot X., Bengio Y. Proceedings of the thirteenth international conference on artificial intelligence and statistics. 2010. Understanding the difficulty of training deep feedforward neural networks; pp. 249–256. [Google Scholar]
- 97.Greene C.S., Krishnan A., Wong A.K., Ricciotti E., Zelaya R.A., Himmelstein D.S., Zhang R., Hartmann B.M., Zaslavsky E., Sealfon S.C., et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015 doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanders S.J., He X., Willsey A.J., Ercan-Sencicek A.G., Samocha K.E., Cicek A.E., Murtha M.T., Bal V.H., Bishop S.L., Dong S., et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015 doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rosina E., Battan B., Siracusano M., Di Criscio L., Hollis F., Pacini L., Curatolo P., Bagni C. Disruption of mtor and mapk pathways correlates with severity in idiopathic autism. Transl. Psychiatry. 2019 doi: 10.1038/s41398-018-0335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table marks the gold standard genes: 855 positively and 893 negatively labeled genes for ASD; and 579 positively and 893 negatively labeled genes for ID are given along with their evidence levels (E1 - E4, E1 indicating the highest risk). The table also provides the rankings from other gene discovery algorithms from the literature. For each gene, participation in disorder-related gene sets are provided (e.g., WNT pathway, CHD8 targets etc.). We also provide 29 gene specific details compiled from the literature.
values
The table provides information about the studies used to generate the ground truth labels for ID. For each gene, the base studies which indicates it as a risk gene are provided.
The table lists (i) the GO enrichment analysis for the top percentile risk genes for both disorders (Biological Process and Molecular Function). It also provides lists of top transcription factor (TF) regulators whose targets are enriched with the top percentile ASD and/or ID risk genes, respectively. The TF enrichment results are based on the experimentally validated TF-gene relationships in ChEA 2016 database. All results are obtained using the EnrichR system.
DeepND risk rankings for the genes within 6 ASD related (16p11.2, 15q11-13, 15q13.3, 1q21.1 and 22q11) (Krishnan et al., 2016) and 6 mental retardation related CNV regions (16p11.2, 1q21.1, 22q11.21, 22q11.22, 16p13 and 17p11.2). Frequency of these CNV regions within ASD and ID individuals are provided when available.
The table marks the gold standard genes: 142 positively labeled gene for breast cancer, 58 positively labeled gene for ovarian cancer and 300 negatively labeled genes for both diseases. It also provides information about the studies used to generate the ground truth labels for both diseases.
The table provides the genome-wide risk probability predictions and rankings of DeepND for ADHD and also marks the 31 positive and 893 negative gold standard gene for ADHD.
Data Availability Statement
All datasets, which are referenced in the relevant experimental procedures subsections, used in this study are publicly available. The processed data that were used to obtain results presented in this paper are also publicly available at Zenodo: https://doi.org/10.5281/zenodo.3892979. All data supporting the key findings such as gene risk evidence levels and gene risk predictions are available within the article and corresponding supplemental tables.
DeepND has been implemented and released at http://github.com/ciceklab/deepnd (Zenodo: https://doi.org/10.5281/zenodo.6501496). We provide the environment, which contains all dependencies for an easy setup. We give a small example to train and test both DeepND-ST/DeepND models. Finally, we provide the code and links to the full set of datasets to reproduce the results (genome-wide risk rankings and data for heatmaps) presented in this manuscript for ASD and ID.