

Abstract
Enterovirus A71 (EV-A71) is a significant human pathogen, especially in children. EV-A71 infection is one of the leading causes of hand, foot, and mouth diseases (HFMD), and can lead to neurological complications such as acute flaccid myelitis (AFM) in severe cases. Although three EV-A71 vaccines are available in China, they are not broadly protective and have reduced efficacy against emerging strains. There is currently no approved antiviral for EV-A71. Significant progress has been made in developing antivirals against EV-A71 by targeting both viral proteins and host factors. However, viral capsid inhibitors and protease inhibitors failed in clinical trials of human rhinovirus infection due to limited efficacy or side effects. This review discusses major discoveries in EV-A71 antiviral development, analyzes the advantages and limitations of each drug target, and highlights the knowledge gaps that need to be addressed to advance the field forward.
KEY WORDS: Enterovirus A71, EV-A71, Antivirals, Acute flaccid myelitis, Hand, Foot and mouth disease (HFMD), Picornavirus, 2C protein
Graphical abstract
The EV-A71 replication cycle provides multiple targets for drug design, and the promising ones include the capsid protein, the 2A and 3C proteases, 2C protein, and the 3D polymerase.

1. Introduction
Enteroviruses (EVs) belong to the Enterovirus genus of the Piconaviridae family. Enteroviruses contain a large variety of serotypes, including more than 100 non-polio enteroviruses and more than 150 rhinoviruses. The Enterovirus genus comprises many important human pathogens, including poliovirus (PV), coxsackievirus, enterovirus D68 (EV-D68), enterovirus A71 (EV-A71), and rhinovirus1. Enteroviruses are non-enveloped, positive-sense, single-stranded RNA viruses with ~7500 nucleotides2. The viral particles are made of icosahedral shaped capsids with approximately 30 nm in diameter. Infection with enteroviruses is generally mild and self-limiting. However, it can progress to life-threatening diseases, such as neonatal sepsis, paralysis, and death. EV-A71 belongs to enterovirus species A and is a major etiological agent of the hand, foot, and mouth disease (HFMD), which mainly affects children. There have been several outbreaks of HFMD in the Asia–Pacific regions3,4. EV-A71 transmits through both the fecal-oral route and the direct contact with virus-contaminated surface or patients’ respiratory droplets5. The basic reproduction number R0 for EV-A71 is 5.066. The mutation rate of EV-A71 was estimated to be 3.1 × 10−3 nucleotide substitutions per site per year7. The symptoms of HFMD include fever, sore throat, and vesicular eruptions on the hands, feet, and oral mucosa8. In addition to EV-A71, coxsackievirus A16 (CV-A16) and CV-A6 are also the main causative agents for HFMD9,10. A recent study showed that the clinical severities of HFMD had a positive correlation with the viral genomic loads of EV-A71 in the throat swabs, suggesting that antivirals should be exploited to reduce viral load and alleviate the clinical outcomes11.
In addition to HFMD, EV-A71 causes various symptoms and diseases ranging from herpangina, rashes, and diarrhea to various neurological complications, including aseptic meningitis, brainstem encephalitis, Guillain–Barré syndrome, acute flaccid paralysis (AFP), neurogenic pulmonary edema, delayed neurodevelopment, and reduced cognitive function12, 13, 14. EV-A71 was the most prevalent AFP-associated virus as assessed from cerebrospinal fluid samples15. Thus, the neurological complications caused by severe EV-A71 infection make it one of the most significant neurotropic viruses known16, 17, 18, 19.
Since its first isolation in 1969 in California, USA20, several outbreaks of EV-A71 have been reported worldwide10, rendering it a major public health concern. However, currently there is no antiviral available for the prevention or treatment of EV-A71 infection21. Three vaccines using inactivated whole viruses have been approved in China22, 23, 24. However, as these vaccines were generated using a single subgenotype, they do not offer broad-spectrum protection against all EV-A71 strains25,26. EV-A71 consists of three genotypes A, B, and C, and genotypes B and C are further divided into subgenotypes B1 to B5 and C1 to C5. In addition, EV-A71 virus constantly mutates, leading to reduced vaccine efficacy. For example, the recently identified EV-A71 C1-GD2019 genotype could not be neutralized by antibodies produced against the EV-A71 C4a genotype, which is the predominant circulating strain in China since 200827. Similarly, the sera from patients infected with genotype B5- or C4 EV-A71 viruses showed reduced neutralization against the 2018–2019 genotype C1 viruses compared to the earlier genotype B5- or C4 viruses28. The EV-A71 C1 genotype is an emerging strain that led to an outbreak of HFMD in Taiwan (China) between 2018 and early 2019. A recent study showed a newly emerged EV-A71 C4 sublineage is more virulent than the B5 lineage that caused 2015–2016 HFMD outbreak in Vietnam29. In addition, EV-A71 viruses carrying naturally occurring mutations have been associated with more severe diseases30. Overall, it is challenging to develop a vaccine against all enteroviruses. As such, there is an urgent need to develop effective broad-spectrum antivirals.
Understanding the viral life cycle is essential in identifying viral proteins and host factors as potential drug targets to prevent EV-A71 infection31. EV-A71 first attaches to the host cell surface by binding of the viral capsid proteins to the cell surface receptors and entering the host cell through endocytosis (Fig. 1). As of most current research, at least six cell receptors have been identified for EV-A7132: the human scavenger receptor class B member 2 (SCARB2)33; the human P-selectin glycoprotein ligand-1 (PSGL-1)34; the sialic-acid-linked glycan35; human annexin 2 protein36; heparan sulfate glycosaminoglycan37; and human tryptophanyl-tRNA synthetase (hWARS)38. The uncoating of EV-A71 is triggered upon its binding to the host cell receptor through a distinctive hydrophobic pocket in the capsid39,40. A lipid molecule (“pocket factor”) naturally occupies this pocket and stabilizes the capsid. The binding of the receptor replaces the lipid, which causes structural rearrangement of capsids and forms the expanded intermediate (“A-particle”) for viral genome RNA release41. Next, the viruses undergo uncoating and release of the viral genome RNA in the cytoplasm. The released positive viral genome RNA is used as a template for the replication of viral genome RNA and translation of the viral polyprotein. The polyprotein is then proteolytically cleaved into structural proteins (VP0, VP1 and VP3) and nonstructural proteins (2A, 2B, 2C, 3A, 3AB, 3C, 3CD) by viral proteases 2A and 3C42. The viral protein VPg (3B) serves as a primer for viral genome replication, which takes place on the surface of membranous vesicles43. Genome replication is catalyzed by the viral RNA-dependent RNA polymerase (3Dpol), and a negative-strand RNA is first synthesized, which serves as a template for synthesis of positive-sense viral genome RNA42,43. Nascent positive RNA molecules either serve as templates for viral polyprotein translation or negative-strand RNA synthesis, or are encapsidated into progeny virions. Virion assembly starts with one copy of each VP0, VP1 and VP3 self-organizing into protomers, with five copies of protomers forming a pentamer44. Together with nascent viral RNA, twelve copies of pentamer form the provirion, which are converted into mature virion upon the cleavage of VP0 into VP4 and VP2, a process that is induced by viral genome RNA45,46. Mature virions are released afterwards and start a new cycle of infection.
Figure 1.

Schematic overview of the life cycle of EV-A71. EV-A71 viral particles attach to the host cell surface by binding to its specific receptor and entering the host cell through endocytosis. Upon uncoating, the viral genome RNA is released and serves as a template for translation into viral polyprotein or the synthesis of negative-strand RNA, which is further used as a template for viral genome RNA replication. Viral polyprotein is cleaved into structural and non-structural proteins by 2Apro and 3Cpro. Viral capsid proteins VP0, VP1 and VP3 first self-organize into a protomer, five of which assemble into a pentamer. Twelve pentamer and viral genome RNA assemble into a provirion, which mature into progeny virion upon the cleavage of VP0 into VP2 and VP4, a process induced by viral genome RNA. Mature virions release and exit from host cells.
While poliovirus has been largely eradicated worldwide with successful vaccination47, non-polio enteroviruses including EV-A71 and EV-D68 that cause polio-like paralytic diseases continue to emerge48. Therefore, it is critical to understand the neurotropic mechanism of these viruses, which might inform the development of effective therapeutic intervention strategies. Enteroviruses gain access to the central nervous system (CNS) mainly through three pathways (Fig. 2): direct infection, Trojan horse invasion, and retrograde axonal transport19. In the direct infection pathway, enterovirus infects the brain microvascular endothelial cells (BMECs) that comprise the blood brain barrier (BBB), allowing invasion into the CNS via the BBB (Fig. 2A)49,50. Mouse transferrin receptor 1 was reported as a poliovirus receptor on BMECs51. Also, cytokines produced during viral infection can modulate BBB integrity, which further facilitate virus invasion52. The second pathway is known as “Trojan horse route”, in which enterovirus-infected peripheral circulating leukocytes acting as carriers to transport virus into the CNS (Fig. 2B). A membrane protein primarily expresses on leukocytes called P-selectin glycoprotein ligand-1 (PSGL-1) was reported as an EV-A71 receptor34. In the third pathway, enteroviruses including EV-D68 and EV-A71 are able to hijack the retrograde axonal transport to enter the CNS (Fig. 2C)53,54.
Figure 2.

Three CNS penetration pathways exploited by enteroviruses. (A) Enterovirus can cross the BBB and reach the CNS by directly infecting BMECs. (B) Enterovirus can infect leukocytes, which act as carriers to transport virus into the CNS. (C) Enteroviruses hijack the retrograde axonal transport to enter the CNS. Viruses first infect muscles, then motor neurons, and finally reach the spinal cord.
This review focuses on small molecule antiviral drug candidates against EV-A71 with either a confirmed mechanism of action or in vivo antiviral efficacy in animal models. Compounds with an unknown or a promiscuous mechanism of action will not be discussed in detail. The sections are organized based on the drug targets. For each drug target, the function of the protein will be introduced42, and prominent examples of antivirals will be discussed alongside with their mechanism of action. The translational potential of each class of drugs will then be compared. New techniques and assays that can facilitate EV-A71 antiviral drug discovery will also be highlighted. Since EV-A71 and EV71 are used interchangeably in the literature, we will use EV-A71 throughout this review to be consistent. For compounds cited in this review, we use the same name as in the original publication. To avoid duplications, the drug target is placed in front of the compound name. Selected examples with translational potential are listed in Table 155, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100.
Table 1.
Selected EV-A71 antivirals with translational potential.
| Inhibitor | Remark | Inhibited | Not inhibited | Ref. |
|---|---|---|---|---|
| Capsid inhibitors | ||||
| Pleconaril | Activity against EV-A71 is controversial Dropped out of clinical trial for rhinovirus infection |
EV-D68, RV-87, RV-A, RV-B | PV-1, RV-B17, RV-A45, RV-C | 56,58,59,64 |
| Vapendavir | Dropped out of clinical trial for rhinovirus infection | EV-A71. EV-C, RV-A, RV-B | EV-D68 | 55,57,65 |
| Pirodavir | Dropped out of clinical trial for rhinovirus infection | EV-A71, EV-D68, EV-A, EV-B, EV-C, EV-D, RV-A, RV-B | RV-A8, RV-A25, RV-A45, RV-87, RV-C | 57,64, 65, 66 |
| PR66 | In vivo antiviral efficacy against EV-A71 in mice | EV-A71 | EV-D68, CV-A16, CV-B1, CV-B2, CV-B3, Echovirus 9, rhinovirus | 60 |
| NLD-22 | Favorable PK properties In vivo antiviral efficacy against EV-A71 in mice |
EV-A71 | – | 61 |
| VP1-14 | Favorable PK properties In vivo antiviral efficacy against EV-A71 in mice |
EV-A71 | – | 62 |
| ICA135 | In vivo antiviral efficacy against CV-A10 in mice | CV-A10, EV-A71, CV-A16, CV-B3, PV-1, EV-D68 | – | 63 |
| CB-30 | Binds to the five-fold axis of the EV-A71 capsid | EV-A71, HIV-1, HIV-2 | – | 67 |
| Suramin | Polypharmacology In vivo antiviral efficacy against EV-A71 in mice and rhesus monkeys |
EV-A71, CV-A2, CV-A3, CV-A10, CV-A12, CV-A16, CV-A9, ECHO25 | ECHO20, PV1-3, EV-D68 | 68, 69, 70, 71 |
| Brilliant black BN (E151) | Binds to the five-fold axis of the EV-A71 capsid In vivo antiviral efficacy against EV-A71 in mice |
EV-A71, CV-A16, CV-A6 | CV-A4, CV-A10 | 72 |
| 2C inhibitor | ||||
| (S)-Fluoxetine | Lack of efficacy against EV-D68 in clinical trials (S)-enantiomer is more active | CV-B3, EV-D68, HRV-A2, HRV-B14 | EV-A71, PV-1 | 73, 74, 75 |
| 2C-12b | Analog of fluoxetine | EV-A71, EV-D68, CV-B3, PV-1, CV-A24, HRV-A2, HRV-B14 | – | 76 |
| Dibucaine | Identified through drug repurposing | EV-A71, EV-D68, CV-B3 | PV-11, RV-A2, RV-B14 | 77 |
| 2C-12a | Dibucaine analog; Did not inhibit Na+ channel |
EV-D68 | – | 78 |
| JX040 | Derived from a HTS hit | CV-B3, EV-A71, PV-1 | – | 79 |
| 2C-7d | Derived from a HTS hit | EV-A71, EV-D68, CV-B3 | – | 80 |
| 3A protein inhibitor | ||||
| Enviroxime | Failed in phase II trial for rhinovirus infection; PI4KIIIβ was also suggested as the drug target |
PV1-3, RV14, RV16, CA21, CB3, RV-87, EV-A71, EV-D68 | – | 64,65,81, 82, 83 |
| Itraconazole | Mutations in 3A confer drug resistance; OSBP was also suggested as the drug target |
EV-A71, CV-A16, CV-B3, PV-1, HRV14, EV-D68 | Equine rhinitis A virus | 84,85 |
| 3Cpro inhibitor | ||||
| Rupintrivir | Failed in phase II trial for rhinovirus infection | EV-A71, CV-A16, EV-D68, RV-87, norovirus, human rhinoviruses | SARS-CoV-2 | 64,65,86, 87, 88, 89 |
| GC-376 | Veterinary drug candidate for FIPV infection in cats | EV-A71, EV-D68, HRV18, HRV51, HRV68 | – | 88 |
| 3Dpol inhibitor | ||||
| Gemcitabine | Identified through drug repurposing; Synergistic effect with interferon-β |
EV-A71, EV-D68, CV-B3, CV-A6, CV-A16, E-7, PV-1, DENV, CHIKV | – | 90 |
| Sofosbuvir | In vivo antiviral efficacy against EV-A71 in mice | EV-A71 | – | 90 |
| FNC | HIV clinical candidate | EV-A71, CV-A16, CV-A6, EV-D68, CV-B3 | – | 91 |
| Favipiravir (T-705) | Synergistic effect with suramin | EV-A71, EV-D68 (not all subtypes) | – | 65,92 |
| NITD008 | Failed in clinical trial for flavivirus infection | EV-A71 | – | 93 |
| IRES inhibitor | ||||
| DMA-135 | Binds to the IRES stem loop 2 | EV-A71 | – | 94 |
| Prunin | Mutations in IRES confer drug resistance; In vivo antiviral efficacy against EV-A71 infection in mice |
EV-A71 | – | 95 |
| Emetine | In vivo antiviral efficacy against EV-A71 infection in mice | EV-A71, CV-A16, CV-B1, EV-D68, Echovirus 6 | – | 96 |
| Host-targeting antivirals | ||||
| OSW-1 | Decreases OSBP level | EV-A71, CV-B3, CV-A21, RV-B14 | – | 97 |
| MDL-860 | Allosteric inhibitor of PI4KB | RV-1, RV-2, RV-8, RV-64, PV-2, EV-A71, EV-D68 | – | 98,99 |
| RYL-634 | DHODH inhibitor | HCV, DENV, ZIKV, CHIV, EV-A71, HIV, RSV | – | 100 |
–Not available.
2. Direct-acting EV-A71 antivirals
2.1. Viral capsid inhibitors
The EV-A71 icosahedral viral capsid is composed of 60 copies of a pentameric subunit comprising VP1, VP2, VP3, and VP4. VP1, VP2, and VP3 are exposed on the viral surface and mediate host cell receptor recognition, and VP4 is present underneath the capsid. The structures of the viral capsid with and without inhibitors or antibodies bound have been solved by X-ray crystallography and cryo-EM42, rendering it feasible for structure-based drug design. Specifically, the VP1 protein contains a hydrophobic pocket located on the surface of the capsid canyon. A “pocket factor”, typically a lipid, binds to this pocket and stabilizes the virion (Fig. 3A)101. Upon receptor binding, the pocket factor is released, leading to particle destabilization and subsequent viral genome release. It has been found that small molecule inhibitors that compete with the pocket factor for the same binding site can regulate virus stability and infectivity (Fig. 3B)55. In contrast to the lipid pocket factor, inhibitors generally bind to the hydrophobic pocket of VP1 with higher affinity and therefore cannot be released upon receptor binding102. In other words, they act as a molecular glue to prevent the uncoating of the viral capsid proteins. In addition to this hydrophobic pocket in VP1, the five-fold axis of the capsid proteins has also been shown to be a viable drug-binding site.
Figure 3.

EV-A71 capsid inhibitors targeting the VP1 hydrophobic pocket. (A) Cryo-EM structure of EV-A71 capsid proteins in complex with sphingosine (PDB: 6UH6) and (B) NLD-22 (PDB: 6LQD). VP1, VP2, VP3, and NLD-22 were colored in gray, tint, yellow, and magenta, respectively. (C) Chemical structures of EV-A71 capsid inhibitors targeting the VP1 hydrophobic pocket.
2.1.1. EV-A71 antivirals targeting the VP1 hydrophobic pocket
The most advanced EV-A71 capsid inhibitors are derived from the WIN series of compounds such as WIN 51711 (Fig. 3C)101. Pleconaril is a representative example of the WIN series of viral capsid inhibitors. Pleconaril treatment protected mice from lethal EV-A71 infection, while ribavirin had no effect56. However, in another study, pleconaril showed no antiviral activity against EV-A71 (EC50 > 262 μmol/L). In contrast, vapendavir and pirodavir inhibited the in vitro replication of 21 EV-A71 strains from genogroups A, B2, B5, C2, and C4 with EC50 values ranging from 0.361 to 0.957 μmol/L57. In our study, pleconaril was also not active against two EV-A71 strains tested, the Tainan/4643/1998 and MP4 (EC50 > 5 μmol/L)58. Vapendavir dropped out of clinical trials of rhinovirus infection because of its significant side effects including headache and drug–drug interactions55. Pleconaril was rejected by FDA as a drug candidate to treat rhinoviruses, citing concerning side effects of menstrual irregularity and drug resistance59.
An imidazolidinone derivative PR66 was identified as a potent EV-A71 antiviral through structure–activity relationship studies60. PR66 inhibits EV-A71 2231 virus with an IC50 of 0.019 ± 0.001 μmol/L as well as several other EV-A71 strains with IC50 values at the nanomolar range. PR66 was not active against EV-D68 (IC50 > 10 μmol/L), coxsackieviruses A16, B1, B2, and B3 (IC50 > 25 μmol/L), echovirus 9 (IC50 > 25 μmol/L), or human rhinovirus (IC50 > 25 μmol/L). Serial viral passage experiments yielded a resistant virus with mutations mapped to the viral capsid VP1 protein (V179F). The recombinant EV-A71 virus with VP1-V179F mutant was not stabilized by PR66 against thermal denaturation. In the in vivo study, PR66 treatment by either oral gavage or intraperitoneal injection significantly improved the disease score and survival rate.
NLD and ALD are structural analogs to PR66 and inhibited EV-A71 replication with IC50 values of 0.025 and 8.54 nmol/L, making them the most potent EV-A71 inhibitors reported so far103. The X-ray crystal structures of EV-A71 VP1 in complex with NLD and ALD have been solved (PDB: 4CEY and 4CEW). Recently, a similar compound NLD-22 was reported by the same group to inhibit EV-A71 with an EC50 value of 5.056 nmol/L in cell culture with improved cellular selectivity (CC50 > 100 μmol/L)61. NLD-22 has favorable pharmacokinetic properties and provided 100% protection for mice infected with a lethal dose of EV-A71, suggesting that NLD-22 might be a promising antiviral drug candidate for HFMD. High-resolution cryo-electron structure showed that NLD-22 binds to the hydrophobic pocket in VP1 (PDB: 6LQD).
Compound VP1-14 (14 in the original publication), an aminopyridyl 1,2,5-thiadiazolidine 1,1-dioxide analog, showed potent antiviral activity against EV-A71 with an EC50 of 4 nmol/L. Compound VP1-14 is highly similar to NLD and contains the 1,2,5-thiadiazolidine 1,1-dioxide replacing the imidazolidinone linker. This compound contains a ketoxime ether and had favorable in vivo PK properties. When dosed at 10 mg/kg either before or after the lethal dose of EV-A71 infection, VP1-14 showed 100% survival protection in mice62.
A virtual screening was conducted to identify compounds that fit in the hydrophobic pocket in the VP1 protein of coxsackievirus A10 (CV-A10)63. Four compounds were identified to inhibit CV-A10 replication in cell culture with EC50 values ranging from 9.11 to 36.36 μmol/L. Among them, ICA135 had broad-spectrum antiviral activity against EV-A71 (IC50 = 3.047 μmol/L), CV-A16 (IC50 = 0.566 μmol/L), CV-B3 (IC50 = 9.68 μmol/L), PV-1 (IC50 = 1.533 μmol/L), and EV-D68 (IC50 = 1.425 μmol/L). The docking study suggested that ICA135 might similarly bind to the hydrophobic pocket in their VP1 proteins. In the CV-A10 infected mouse model study, ICA135 treatment led to improved survival rate and decreased clinical score. Although the in vitro antiviral activity of ICA135 is relatively weak compared to other capsid inhibitors (μmol/L vs. nmol/L), the broad-spectrum antiviral activity and the in vivo antiviral efficacy of ICA135 suggest that this class of compounds warrant further development.
A series of diarylhydrazides were reported as potent EV-A71 capsid inhibitors104. The most potent lead compound VP1-15 (15 in the original publication) inhibited EV-A71 with an EC50 value of 0.02 μmol/L and a high selectivity index (SI > 10,000). Fluorescence-based thermal stability assay showed that compound binding increased the EV-A71 virion stability against thermal denaturation. The in vivo antiviral efficacy for this series of compounds has not been reported.
A natural product isolated from avocado, named avoenin, was found to inhibit EV-D68 replication in cell culture with an EC50 of 2 μmol/L105. It has a weak inhibition against EV-A71 at 38 μmol/L. Mechanistic studies showed that mutations at the capsid protein VP3 confers drug resistance against avoenin. However, further studies are needed to identify the drug-binding site of aveonin.
Three natural products, auraptene, formononetin and yangonin were identified as EV-A71 antivirals106. Auraptene inhibited EV-A71 and CV-A16, while the antiviral activities of formononetin and yangonin were limited to EV-A71. Resistance selection experiment showed that mutations mapped at VP1 and VP4 conferred drug resistance against these three compounds.
A series of benzothiophene derivatives have been found to inhibit human rhinovirus A and B strains107. The most potent compound VP1-6g (6g in the original publication) inhibited EV-A71 with an EC50 value of 15 nmol/L. The time-of-addition experiment and resistance selection suggest that compound 6g binds to the same binding site as pleconaril in the viral capsid protein VP1.
2.1.2. EV-A71 antivirals targeting the five-fold axis of the capsid
MADL385 is a dendrimer consisting of 12 tryptophan residues and has antiviral activity against EV-A71 BrCr strain in cell culture with an EC50 value of 0.28 μmol/L (Fig. 4A)108. Cryo-EM structure showed that MADAL385 binds at the 5-fold axis of the vial capsid. This binding mode was supported by the drug-resistant mutants in VP1, S184T and P246S. Recombinant viruses encoding the VP1-S184T, VP1-P246S, and the VP1-S184T/P246S led to reduced drug sensitivity by 7-, 16-, 31-fold. Subsequently, simplified dendrimers consisting of three or four tryptophan residues were designed to similarly target the five-fold axis of the EV-A71 capsid (Fig. 4B)67. They also inhibit HIV by binding to the glycoprotein gp120. One of the most potent compounds is CB-30, which inhibits multiple strains of EV-A71 with EC50 values ranging from 0.2 to 353 nmol/L (Fig. 4B). Furthermore, recombinant viruses carrying mutations at the viral capsid protein VP1, VP1-S184T, VP1-P246S, VP1-S184T/P246S, showed partial resistance against CB-30, suggesting that CB-30 might bind to the same pocket as MADL385. This was further supported by a model generated from the cryo EM structure, showing that CB-30 might fit around the five-fold axis of the EV-A71 capsid (PDB: 6UH7) (Fig. 4C).
Figure 4.

EV-A71 antivirals targeting the five-fold axis of the capsid proteins. (A) Chemical structure of EV-A71 capsid inhibitors targeting the five-fold axis of the capsid proteins. (B) Chemical structure of CB-30. (C) Binding pose of EV-A71 capsid inhibitor CB-30 (compound 30 in the original publication)67. Reprinted with permission from Ref. 68. Copyright © 2020, American Chemical Society.
A natural product rosmarinic acid (RA) was found to inhibit multiple strains of EV-A71 in cell culture with EC50 values from 31.57 to 114 μmol/L, but not the EV-D68 strains (EC50 > 100)109. Resistant selection identified the VP1-N104K mutation, which was confirmed by reverse genetics to confer drug resistance. The N104 residue was shown to be critical in allowing the virus to bind to heparan sulfate but not the PSGL-1. Interestingly, despite its relatively weak in vitro antiviral activity, RA showed in vivo antiviral efficacy in EV-A71 infected mouse model study. It is likely that the in vivo antiviral efficacy of RA might involve other mechanisms.
Suramin, an FDA-approved pediatric antiparasitic drug, was found to inhibit EV-A71 by blocking the viral entry step68. Several sulfonated and sulfated analogs similarly showed potent antiviral activity against EV-A71. However, the exact mechanism of action or the drug target remains elusive. Nevertheless, the in vivo antiviral activity of suramin was confirmed in two animal models using mice and rhesus monkeys. In mice challenged with a lethal dose of EV-A71, suramin treatment by intraperitoneal injection at 20 or 50 mg/kg improved the survival rate. In adult monkeys challenged with 1 × 106.5 CCID50 dose of EV-A71 FY-23 strain, suramin treatment at −1, 1, 3, 5 dpi by i.v. injection at 50 mg/kg significantly lowered the viral genome copy in the serum. Although these results were promising, the translational potential of suramin remains to be validated in human clinical trials. One potential concern might be its promiscuous mechanism of action since suramin is a known pan-assay interference (PANIS) compound. The binding site of suramin was proposed to be a positively charged region surrounding the 5-fold axis of the capsid, and drug binding leads to the blockage of viral attachment70. Specifically, mutations at residue 145 of the VP1 protein, Q145G and Q145E, resulted in approximately 30-fold loss of antiviral potency. Moreover, the antiviral spectrum of suramin is limited to enterovirus A species.
Sumarin analogs NF449, NF110 and NM16, were found to inhibit EV-A71 by blocking the viral attachment to the host cell heparan sulfate and PSGL-1 receptors, both of which are sulfated molecules71. Residues located at the 5-fold vertex of the EV-A71 capsid were shown to be critical for NF449 binding, and mutations E98Q and K244R reduced the drug sensitivity. Consistent with the proposed mechanism of action, NF449 and NF110 prevented the binding of the monoclonal antibody MA28-7, which recognizes the same epitope as these small molecules.
Brilliant Black BN (E151), a sulfonated food azo dye, was identified to inhibit EV-A71, CV-A16, and CV-A6 replication in cell culture72. Mechanistic studies suggested that E151 binds to the vertex of the 5-fold axis of EV-A71 and prevents viral attachment to the host cell. When 14-day old AG129 mice were challenged with 10 × LD50 of either the EV-A71-B4 or the EV-A71-C1 strains, E151 treatment by i.p. injection at 200 mg/kg/day from 0 dpi to 3 dpi provided 100% survival protection. Mice treated with E151 also had significantly reduced viral titer in the brain and hind limb muscle.
Overall, viral capsid proteins are validated antiviral drug targets. The advantage of capsid inhibitors is their high potency (nmol/L to pmol/L). However, capsid inhibitors generally have a low genetic barrier to drug resistance and their antiviral activity is often limited to certain subtypes of enteroviruses. No broad-spectrum capsid inhibitors have been reported thus far55. Nevertheless, the capsid inhibitors are promising candidates for combination therapy. Further studies need to focus on optimizing the pharmacokinetic properties, improving selectivity, and broadening the antiviral spectrum.
2.2. 2A protease (2Apro)
2A is a viral cysteine protease that cleaves the viral polyprotein between P1 and P2 segments, whereas the viral protease 3C cleaves between P2 and P3. In addition, 2A protease (2Apro) suppresses host protein translation by cleaving the initiation factor eIF4G, thereby inhibiting the host cap-dependent mRNA translation110. The EV-A71 2Apro remains a relatively unexplored drug target and no potent and specific EV-A71 2Apro inhibitor has been reported. In one study, a peptide based inhibitor Z-LVLQTM-FMK showed weak inhibition against EV-A71 replication in cell culture at a high concentration of 200 μmol/L (Fig. 5A)111. In another study, CW33 was found to be a weak inhibitor of EV-A71 2Apro and inhibited the enzymatic activity with an IC50 value of 53.1 μmol/L112. CV33 inhibited EV-A71 replication in cell culture with an EC50 close to 200 μmol/L and its antiviral activity is synergistic with IFN-β. Given their weak inhibitory activities, these two compounds might not be classified as specific 2Apro inhibitors. Further studies are needed to identify more potent and drug-like 2Apro inhibitors. The X-ray crystal structure of the EV-A71 2A-C110A mutant was solved (PDB: 4FVB)113, paving the way for structure-based drug design or virtual screening (Fig. 5B).
Figure 5.

EV-A71 2A protease inhibitors. (A) Chemical structure of the EV-A71 2Apro inhibitors. (B) Modeling of the substrate peptide on the X-ray crystal structure of EV-A71 2Apro C110A mutant (PDB: 4FVB)113. Reprinted with permission from Ref. 115. Copyright © 2013, American Society for Microbiology.
In another study, a library of 32 lycorine derivatives was screened against EV-A71. 1-acetyllycorine was found to be the most potent hit and inhibited multiple strains of EV-A71 in several cell lines114. Resistance selection identified the 2A-F76L mutation that reduced the drug sensitivity by ~10-fold. The F76 residue was located at the conserved zinc-binding motif and the EV-A71 2A-F76L mutant protein had reduced enzymatic activity. However, the exact mechanism of action of 1-acetyllycorine remains elusive.
Overall, EV-A71 2Apro remains a relatively unexplored novel drug target and potent and specific inhibitors are needed for in vitro and in vivo target validation.
2.3. 2B inhibitor
When expressed in xenopus oocyte, the EV-A71 viral 2B protein forms an ion channel that selectively conducts chloride ion115, rendering it a putative member of the viroporin family116. A chloride channel inhibitor 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS) was found to inhibit the EV-A71 replication in RD cells (Fig. 6). However, given the structural similarities of DIDS with many other capsid inhibitors such as suramin and Brilliant Black, the cellular antiviral activity of DIDS might not solely arise from the inhibition of chloride conductance through the 2B ion channel. To further delineate the mechanism of action of DIDS, resistance selection experiment should be performed. Similarly, more potent and specific 2B channel blockers need to be developed, which can be used as chemical probes for target validation. Moreover, the structure of EV-A71 2B protein has not been determined. It remains unknown whether the chloride ion channel function of 2B protein is essential for the viral replication. One study found that EV-A71 2B recruited the proapoptotic protein BAX to the mitochondria and induced cell apoptosis117.
Figure 6.
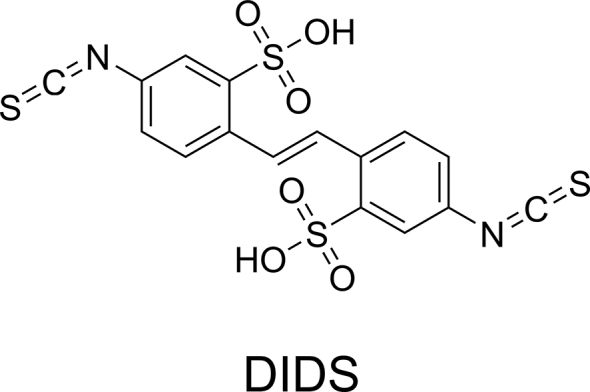
EV-A71 2B inhibitor.
2.4. 2C inhibitor
The 2C protein is a multifunctional protein and its known functions include viral uncoating, membrane remodeling, RNA binding and replication, and encapsidation118. The EV-A71 2C protein was reported to have ATP-dependent RNA helicase and ATP-independent chaperoning activities119. The 2C protein was also involved in modulating the innate host immune response upon viral infection. The detailed mechanism of 2C protein in virus replication and packaging is yet to be elucidated. However, 2C is a high-profile antiviral drug target and structurally diverse small molecule compounds have been identified as 2C-targeting inhibitors.
The X-ray crystal structure of a soluble fragment of EV-A71 2C (residues 116−329) was solved at 2.5 Å resolution (Fig. 7A). The 2C protein contains an adenosine triphosphatase (ATPase) domain, a cysteine-rich zinc finger, and a C-terminal helical domain. The C-terminal helical domain mediates the oligomerization with a neighboring monomer by binding to a concave pocket formed between the ATPase and the zinc finger domains (Fig. 7A). Functional studies showed that the oligomerization is critical to the 2C ATPase activity and the EV-A71 viral replication.
Figure 7.

EV-A71 2C inhibitors. (A) X-ray crystal structure of EV-A71 2C protein (PDB: 5GRB). One monomer is colored in cyan, and another monomer is colored in gray and shown in surface. ATP is shown in sticks. (B) X-ray crystal structure of CV-B3 2C (117-329) in complex with (S)-fluoxetine (PDB: 6T3W). (C) Cryo-electron micrographs of CV-B3 2C with (S)-fluoxetine (SFX) and ATP. Reprinted from Ref. 132. (D) Chemical structure of EV-A71 2C inhibitors.
Fluoxetine was advanced to clinical trials in treating EV-D68 infection, but was found not effective for patients with proven or presumptive EV-D68 associated AFM74,75. A recent study discovered that (S)-fluoxetine is more potent than the racemic fluoxetine mixture in inhibiting CV-B3 and EV-D68 (Fig. 7B). Mechanistic studies showed that (S)-fluoxetine binds to CV-B3 2C protein with higher affinity than the (R)-fluoxetine73. However, neither the racemic mixture nor the individual enantiomer of fluoxetine was active against EV-A71120, suggesting that not all 2C inhibitors have broad-spectrum antiviral activity against enteroviruses. Nevertheless, in a following study, several substituted amides were synthesized and tested against enteroviruses76. The most potent compound 2C-12b (12b in the original publication) inhibited multiple coronaviruses including EV-A71, EV-D68, CV-B3, PV-1, CV-A24, HRV-A2 and HRV-B14 with EC50 values ranging from 0.0029 to 1.39 μmol/L. Compound 2C-12b is structurally similar to fluoxetine, both of which containing three aromatic substitutions connected with a linker. However, unlike fluoxetine, compound 12C-2b is not neuroactive and did not inhibit the serotonin transporter (SERT), dopamine transporter (DAT), or the norepinephrine transporter (NET). The mechanism of action was elucidated through resistance selection using CV-B3, EV-A71, and EV-D68 viruses. In each case, the resistant mutants were mapped to the viral 2C protein. The resistance was confirmed using recombinant viruses encoding the identified 2C mutants as well as the differential scanning fluorimetry binding assay. The mutations were located at the α2 helix of 2C, which is close to the solvent accessible tunnel that the inhibitor might bind. The drug–bound complex structure is needed to fully explain their mechanism of action.
Dibucaine was identified as a CV-B3 antiviral through a drug repurposing screening77. Resistance selection mapped several mutations at the viral 2C protein. A secondary assay showed that dibucaine also inhibited EV-A71 and EV-D68 with EC50 values of 7.59 and 3.03 μmol/L. Given its convenient synthesis, structure–activity relationship studies were conducted, yielding dibucaine analogs such as 2C-12a (12a in the original publication) with a significantly improved antiviral activity and selectivity index against EV-D6878. One potential issue with repurposing dibucaine as an EV-A71 antiviral is its analgesic effect. Dibucaine was clinically used as a local analgesic drug through inhibiting the Na+ channel, and the optimized lead compounds such as 2C-12a had no inhibition against Na+ channel, thereby alleviating the concern of potential side effects78. Follow up studies led to the discovery of 2C-6aw (6aw in the original publication) with favorable in vitro PK properties and a broader spectrum of antiviral activity121, especially against EV-A71. In parallel, another study similarly identified dibucaine analogs with improved antiviral activity and cellular selectivity index through structure–activity relationship (SAR) study122. The most potent compound 2C-6i (6i in the original publication) had in vivo antiviral efficacy in the EV-A71 infected mouse model study and the antiviral activity was synergistic with emetine. However, the in vitro and in vivo PK properties of these compounds have not been reported.
An aryl substituted amide R523062 was identified to inhibit EV-D68 from a HTS123. In the secondary hit validation, R523062 was found to inhibit multiple EV-D68 strains with EC50 values ranging from 2.3 to 6.4 μmol/L. It also inhibited EV-A71 viruses, but with lower potency (EC50 = 9.6–55.7 μmol/L). Serial viral passage experiment led to the identification of resistant EV-D68 viruses with several mutations VP1- G178S, 2A-V112I, 2C-I227L and Q322R, and 3A-V54A. The VP1-G178S and 2A-V112I were eliminated as possible contributors of drug resistance by time-of-addition and enzymatic assays, respectively. Next, to confirm whether the 2C or the 3A mutant confers the phenotypic drug resistance, corresponding recombinant viruses were generated. It was found that the rMO 3A-V54V maintained similar drug sensitivity against R523062 as the rMO, suggesting that the 3A mutant did not contribute to the drug resistance. Furthermore, the rMO 2C-Q322R also demonstrated similar drug sensitivity against R523062 as the rMO. In contrast, the rMO 2C-I227L/Q332R double-mutant virus was resistant with an EC50 shift by more than 10-fold from the CPE assay, suggesting that the I227L might be the drug resistant mutant. However, the rMO 2C-I227L could not be generated, most likely due to the loss of function caused by the mutation. Nevertheless, the binding assay with purified 2C-I227L protein provided confirmative evidence that this single mutation confers drug resistance against R523062. Although R523062 had relatively weak antiviral activity against EV-A71, its simple structure makes it convenient for lead optimization.
A series of pyrazolopyridine derivatives were identified as CV-B3 antivirals through a phenotypic screening124. These compounds also had broad-spectrum antiviral activity against EV-A71, coxsackieviruses, echoviruses, and poliovirus. Mechanistic studies suggest that viral 2C protein is the drug target. Subsequent SAR studies led to more potent and selective inhibitors including JX040 which inhibited EV-A71 with an EC50 value of 0.5 μmol/L79. The in vitro and in vivo pharmacokinetic properties of this series of compounds remain to be profiled.
Our recent study identified a series of pyrazolopyridine analogs with broad-spectrum antiviral activity against EV-D68, EV-A71, and CV-B3 in several cells lines including RD cells and SH-SY5Y neuronal cells80. Compound 2C-7d (7d in the original publication) was used as a chemical probe for the mechanistic studies, which revealed that the viral 2C protein is the drug target. 2C-7d was shown to bind to the 2C proteins from EV-D68, EV-A71, and CV-B3 in the differential scanning fluorimetry (DSF) assay. Resistance selection identified mutations in the 2C protein (D183V/D323G) that confer resistance to EV-D68, which was further confirmed in the antiviral assay using recombinant viruses generated from reverse genetics, and the DSF binding assay. Although the recombinant EV-D68 2C-D183V/D323G virus (rD183V/D323G) had reduced drug sensitivity, competition growth assay showed that it has a compromised fitness of replication compared to the WT virus.
Other reported 2C inhibitors include guanidine, TBZE-029, HBB, MRL-1237, pirlindole, zuclopenthixol, metrifudil, and N6-benzyladenosine (Fig. 7D)64,77,124, 125, 126, among which metrifudil and N6-benzyladenosine were reported to inhibit EV-A71 while the rest have not been tested against EV-A71127. All these compounds were identified from drug repurposing screening and had resistant mutations mapped to the viral 2C protein. Although mutations in the viral 2C protein conferred drug resistance against metrifludil, and N6-benzyladenosine, no direct binding assay experiments were performed.
The broad-spectrum antiviral activity of 2C inhibitors makes them promising drug candidates, but the detailed mechanism of action needs further investigation. It is intriguing that structurally diverse compounds all target the same protein. As of latest research, only the EV-A71 2C, CV-B3 2C, and PV 2C structures have been solved128, 129, 130, and efforts should be made to solve the 2C structures from other enteroviruses including EV-D68, CV-A16, CV-A6, and CV-A10. More importantly, the complex structures of 2C in complex with inhibitors can contribute significantly to the understanding of the mechanism of action of existing 2C inhibitors and guide the development of more potent and selective 2C inhibitors. It is noted that the X-ray crystal structure of CV-B3 2C (117-329) in complex with (S)-fluoxetine was recently reported in a preprint (PDB: 6T3W) (Fig. 7B and C)130. X-ray crystal structure showed that (S)-fluoxetine binds to the allosteric site in the CV-B3 2C (Fig. 7B and C), and drug binding stabilizes the 2C hexamer. Drug-resistant mutations are mapped to both the (S)-fluoxetine binding site (C179F, C179Y and F190L) and the 224-AGSINA-229 loop (A224V, I227V, and A229V). The 224-AGSINA-229 loop locates downstream of the Walker C motif and does not directly interact with (S)-fluoxetine. It was proposed that the 224-AGSINA-229 loop stabilizes the Walker B-containing loop in an “open conformation”, therefore suitable for SFX binding. Giving the success of crystalizing CV-B3 with fluoxetine, it is promising to solve the co-crystal structures of EV-D68 and EV-A71 2Cs with structurally diverse 2C-targeting inhibitors. The structures will help guide the lead optimization and rationalization of the drug resistance mechanism.
2.5. 3A protein
The enterovirus replication complex resides in the replication organelles, the formation of which requires the hijacking of host lipid homeostasis pathways. The viral 3A protein and the host phosphatidylinositol 4-kinase IIIβ (PI4KB), acyl-coenzyme A binding domain containing 3 (ACBD3), and oxysterol-binding protein (OSBP) are known to be essential for the formation of the replication organelles (Fig. 8A)131,132. Overexpression of 3A protein or EV-A71 infection led to the increased interactions of PI4KB and ACBD3133. Furthermore, viral 3A, PI4KB and ACBD3 co-localize at the replication organelles where viral RNA replication takes place, and 3A interacts directly with ACBD3 (Fig. 8A). Mutations in the 3A protein I44A and H54Y interrupts the PI4KB–ACBD3 interaction. A recent study also showed that ACBD3 is essential in mediating the 3A-PI4KB interactions134. The X-ray crystal structures of the C-terminal Golgi-dynamics domain (GOLD) of ACBD3 in complex with the 3A protein from several enteroviruses including EV-A71, PV1, EV-D68, and RVB14 were solved (PDBs: 6HLW, 6HLV, 6HLN, and 6HLT) (Fig. 8B)135. The structures of all GOLD:3A complexes are highly similar, implying that diverse enteroviruses use a conserved mechanism to recruit the host ACBD3 protein to the Golgi. The 3A protein has been shown to stimulate the ACBD3:PI4KB interaction and subsequent enrichment of PI4KB at the replication organelle. It is likely that upon binding to viral 3A protein, ACBD3 undergoes a conformational change that is essential for its binding with PI4KB. However, the exact binding mode of these proteins in the replication organelles is yet to be determined by high-resolution structures.
Figure 8.

EV-A71 3A structure and inhibitors. (A) Model of the EV-A71 3A protein and the associated proteins in the replication organelle. (B) X-ray crystal structure of the C-terminal Golgi-dynamics domain (GOLD) of ACBD3 in complex with the cytoplasmic domain of the EV-A71 3A protein (PDB: 6HLW). The ACBD3 and 3A proteins are colored in grey and rainbow, respectively. (C) Chemical structure of EV-A71 3A inhibitors.
Enviroxime is a broad-spectrum enterovirus antiviral targeting the viral 3A protein (Fig. 8C). Enviroxime was evaluated in clinical trials for rhinovirus infection, but the clinical development was halted due to the lack of therapeutic benefits and gastrointestinal side effects81,82. Other compounds that inhibit EV-A71 through a similar mechanism of action as enviroxime include AN-12-H5 and GW5074127,136,137. These compounds demonstrated cross-resistance with enviroxime and had resistant mutations mapped to the same region in the viral 3A protein. Interestingly, AN-12-H5 had an additional mechanism of action by targeting the viral capsid protein VP1, which inhibited the early stage of EV-A71 replication136. AN-12-H5 and GW5074 had no structural similarities to enviroxime, raising the question of whether they target the viral 3A protein directly or indirectly.
TTP-8307 was found to inhibit CV-B3 by directly inhibiting OSBP-mediated lipid shuttling (Fig. 8C)138,139. Mutations in the CV-B3 3A protein (V45A, I54F, and H57Y) resulted in phenotypic drug resistance. TTP-8307 had cross-resistance with another 3A-targeting enterovirus inhibitor enviroxime. Similarly, mutations in the 3A proteins from RV14 and PV1 viruses conferred drug resistance against enviroxime83. Although resistant mutants were mapped to the viral 3A protein, the exact mechanism of action of TTP-8307 or enviroxime remains unknown. There is no experimental evidence to support that TTP-8307 and enviroxime bind directly to the viral 3A protein. Another possibility might be that mutations in 3A disrupt the interactions between 3A and other viral or host factors that are required for the viral replication. Although TTP-8307 has not been tested against EV-A71, it is expected that it will be active against EV-A71 accordingly to the proposed mechanism of action.
Itraconazole (ITZ) was identified as an EV-A71 antiviral with an EC50 value of 1.15 μmol/L through a drug repurposing screening of the FDA-approved drug library84. In addition, ITZ also had antiviral effect against CV-A16, CV-B3, PV1, and EV-D68, suggesting that it might be a promising broad-spectrum antiviral candidate against picornaviruses. Drug resistance selection coupled with reverse genetics identified two mutations located at the 3A protein V51L and V75A that conferred drug resistance. Interestingly, ITZ-resistant viruses do not show cross-resistance to the other two 3A-targeting antivirals posaconazole and GW5074, implying that these compounds might target different regions of the 3A protein or target 3A indirectly. The difficulty in pursuing 3A as a drug target is the lack of a biochemical assay to access the direct effect of the inhibitors against 3A.
A recent study showed that itraconazole and posaconazole were also potent antivirals against Parechovirus A3 (PeV-A3)140. Similar to EV-A71, PeV-A3 primarily infects children and causes sepsis-like illness, meningitis, and encephalitis. Resistant viruses identified from serial viral passage experiments had mutations mapped to viral capsid proteins VP0, VP3, VP1 as well as nonstructural proteins 2A and 3A. All single mutations led to drug resistance against posaconazole, but the 3A-T1L was the predominant mutate and was 119-fold more resistant to posaconazole. This study suggests that itraconazole and posaconazole might have a polypharmacology and their antiviral mechanism of action might involve multiple drug targets including the viral 3A and VP1 proteins as well as the host OSBP and ORP4.
In summary, although structurally diverse compounds have been identified as putative viral 3A inhibitors, there is a lack of evidence that these compounds interact directly with the 3A protein. They may target the 3A-interacting proteins such as ACBD3 and OSBP. In addition, no 3A inhibitor has advanced to in vivo mouse model study. Nevertheless, the multifunctional roles of 3A render it a high-profile antiviral drug target.
2.6. 3C protease
The EV-A71 3C protease catalyzes the cleavage of the viral polyprotein at 8 different sites86. Similar to the 2A protease, the EV-A71 3C protease also plays a role in inhibiting host cap-dependent translation by cleaving the eukaryotic initiation factor 4A (eIF4A)141 and eukaryotic initiation factor 5B (eIF5B)142. For more details regarding the functions of 3C protease in viral life cycle please refer to the reference143.
The 3C or 3CL protease has a high substrate preference for glutamine at the P1 position, therefore the majority of the 3C protease inhibitors are designed to have a pyrrolidone at the P1 position as a substrate mimetic. The general structure of 3C protease inhibitors is shown in Fig. 9A. The 3C inhibitors are either di-, tri- or tetra-peptide conjugated with a reactive warhead. Commonly used reactive warheads include aldehyde, ketoamide and α,β-unsaturated ester. The aldehyde warhead can also be converted to bisulfite and cyanohydrin144 prodrugs. Recent studies also identified novel reactive warheads including 4-iminooxazolidin-2-one145, which is a bioisostere of the cyanohydrin, and the dually activated Michael acceptor146. The 4-iminooxazolidin-2-one-based inhibitors 3C-4e and 3C-4g (4e and 4g in the original publication) inhibited EV-A71 with EC50 values of 0.21 and 0.10 μmol/L, respectively (Fig. 9A)145. More importantly, 4e and 4g had drastically improved stability in human plasma and moderate stability in mouse and human microsomes. The EV-A71 inhibitors with the dually activated Michael acceptor such as 3C-30 (30 in original publication) showed improved selectivity against host proteases cathepsin K and calpain I (Fig. 9A). Non-covalent EV-A71 3C protease inhibitors such as DC07090 have also been reported147. DC07090 was identified through an in silico docking and had an IC50 of 21.72 μmol/L in the enzymatic assay. It also inhibited the EV-A71 viral replication with an EC50 of 22.09 μmol/L. Although the enzymatic inhibition and antiviral activity of DC07090 were moderate, DC07090 represents a novel chemotype that can be further optimized as a more potent and specific EV-A71 antiviral.
Figure 9.

EV-A71 3C inhibitors. (A) Chemical structures of EV-A71 3Cpro inhibitors. (B) X-ray crystal structure of EV-A71 3Cpro in complex with rupintrivir (AG7088) (PDB: 3SJO). (C) X-ray crystal structure of EV-A71 3Cpro in complex with 3C-18p (PDB: 7DNC). Reprinted with permission from Ref. 157. Copyright © 2021, American Chemical Society. (D) X-ray crystal structure of EV-A71 3Cpro in complex with macrocyclic inhibitor 3C-4 (PDB: 6LKA).
Rupintrivir (AG7088) was developed as a specific human rhinovirus 3C protease inhibitor. Although rupintrivir was well tolerated148, its phase II trial for rhinovirus infection induced common cold was halted due to a lack of significant therapeutic benifits31. As the 3C or 3C-like protease of enteroviruses, noroviruses, coronaviruses share a high sequence similarity, rupintrivir was also shown to have broad-spectrum antiviral activity against EV-A71, CV-A16, EV-D68, norovirus, and human rhinoviruses86,88. However, rupintrivir was not active against the SARS-CoV-2 main protease89. The X-ray crystal structure of EV-A71 3C protease in complex with rupintrivir was solved (Fig. 9B), revealing a covalent complex formation between the catalytic C147 with the Michael acceptor from rupintrivir86,149.
Novel phenylthiomethyl ketone-based inhibitors such as 3C-7a (7a in original publication) were identified from fragment-based screening as irreversible inhibitors of CV-B3 and EV-D68 3Cpro with Kinact/KI of 54.4 and 160 L/mol∙s, respectively150. However, these compounds have not been tested in the antiviral assay, and it is unknown whether they can inhibit EV-A71 3Cpro.
GC373, GC375, and GC376 were reported to inhibit EV-A71 viral replication with IC50 values of 11.1, 15.2, and 10.3 μmol/L88. NK-1.8k inhibits EV-A71 3C protease with an IC50 of 0.11 μmol/L and multiple strains of EV-A71 in RD cells with EC50 values from 0.093 to 0.105 μmol/L151. The antiviral activity of NK-1.8k is consistent between RD and 293T cells. However, NK-1.8k was less active in Vero cells with an EC50 of 2.41 μmol/L. The cell-type dependent difference might be due to the drug efflux in Vero cells. Recent studies have shown that similar compounds are P-gp substrates and that co-treatment with P-gp inhibitor CP-100356 can boost the cellular antiviral activity152,153. Resistance selection identified the 3C-N69S mutation that conferred drug resistance. The FRET assay also showed that the 3C-N69S mutant had reduced protease activity compared to the WT protein. The IC50 of NK-1.8k against the 3C-N69S mutant was 1.15 μmol/L, which was a 10-fold increase compared to WT (IC50 = 0.11 μmol/L)154. The X-ray crystal structures of EV-A71 3C and 3C-N69S in complex with NK-1.8k were solved, showing that the mutation destabilized the S2 pocket and negatively impacted drug binding.
A series of α-ketoamides were designed to target the viral Mpro or 3CLpro and were found to have broad-spectrum antiviral activity against coronaviruses and enteroviruses155. However, these compounds were not specifically optimized for the EV-A71 and the most potent compound was 3C-11r with an EC50 of 3.7 μmol/L. Recently, a follow up study from the same group revealed compound 3C-18p (18p in the original publication) as a broad-spectrum antiviral against a panel of enteroviruses, rhinoviruses, and coronaviruses including EV-A71 and SARS-CoV-2. 3C-18p inhibited EV-A71 3Cpro with an IC50 of 2.36 μmol/L. Despite its relatively weak enzymatic inhibition, 3C-18p had surprisingly potent antiviral activity against EV-A71 with an EC50 of 0.03 μmol/L. X-ray crystal structure of EV-A71 3Cpro in complex with 3C-18p showed that the aldehyde formed a covalent bond with the catalytic C147 (PDB: 7DNC) (Fig. 9C). 3C-18p also inhibited additional enteroviruses including EV-D68, CV-A21, CV-B3, RV-B14, and RV-A02-WT with EC50 values of 0.03, 0.43, 4.19, 0.81, and 1.62 μmol/L, respectively. In addition, 3C-18p was also highly active against SARS-CoV-2 3CLpro with an IC50 of 0.034 μmol/L and inhibited viral replication at an EC50 of 0.29 μmol/L. In vivo PK profiling revealed that 3C-18p had a long half-life of 5.85 h following intravenous administration, suggesting it might be a potential candidate for the in vivo antiviral efficacy study.
A macrocyclic 3C protease inhibitor, compound 3C-4 (4 in the original publication), was recently designed as the EV-A71 3C protease inhibitor (Fig. 9D)156. Compound 3C-4 inhibited EV-A71 in cell culture with an EC50 of 4.5 μmol/L. The X-ray crystal structure of 3C in complex with compound 4 was solved (PDB: 6LKA).
Using rupintrivir derived activity-based chemical probe, several host proteins were pulled out, among which the host cysteine protease autophagy-related protein 4 homolog B (ATG4B) was found to hydrolyze the viral polyprotein with comparable activity as the viral 3C protease157. ATG4B knockdown by short hairpin RNAs (shRNAs) inhibited EV-A71 replication in cell culture, suggesting the enzymatic activity of ATG4B is essential for viral replication. However, given that ATG4B and 3Cpro process the same substrate, it remains unclear why the function of ATG4B is essential for EV-A71 replication.
For additional examples of EV-A71 3C inhibitors please refer to Refs. 57,158, 159, 160, 161, 162.
The advantages of the 3Cpro or 3CLpro inhibitors include the broad-spectrum and potent antiviral activity. However, the in vivo antiviral efficacy of 3Cpro inhibitors in EV-A71 infection animal models has not been demonstrated. Another concern with covalent 3Cpro inhibitors is the potential off-target effects. GC376 and its analogs are known to inhibit host cysteine proteases including cathepsin L, calpain 1, and cathepsin K152,163,164.
2.7. 3D polymerase (3Dpol) inhibitor
EV-A71 3Dpol is an RNA-dependent RNA polymerase that mediates the viral RNA synthesis. Similar to other viral polymerase inhibitors, majority of the reported EV-A71 3Dpol inhibitors are nucleoside or nucleotide analogs. Nevertheless, non-nucleoside 3Dpol have also been reported.
Screening of the FDA-approved drug library identified three nucleoside drugs with potent antiviral activity against EV-A71: gemcitabine, LY2334737, and sofosbuvir (Fig. 10A)90. Gemcitabine also had synergistic antiviral effect with interferon-β in inhibiting EV-A71. Significantly, LY2334737 and sofosbuvir protected mice from lethal dose of EV-A71 infection and significantly reduced the viral titers and viral infection-induced damage in the limb muscle tissue of mice.
Figure 10.

EV-A71 3D polymerase inhibitors. (A) Chemical structures of EV-A71 3D polymerase inhibitors. (B) X-ray crystal structure of CV-B3 3Dpol in complex with BPR-3P0128 (PDB: 4Y2A).
A pyrimidine analog, 2′-deoxy-2′-β-fluoro-4′-azidocytidine (FNC), was identified to have broad-spectrum antiviral activity against enteroviruses including EV-A71, CV-A16, CV-A6, EV-D68, and CV-B3 at nanomolar range91. FNC is a clinical candidate for HIV treatment and is currently in a phase II trial in China. In vitro 3Dpol assay, coupled with isothermal titration calorimetry (ITC) binding assay, showed that FNC targets the EV-A71 viral RNA-dependent RNA polymerase (3Dpol). However, it remains unknown whether the metabolites of FNC were incorporated in the viral RNA or act as a chain terminator. In the EV-A71 and CV-A16 infection neonatal mouse model studies, mice treated with FNC at 1 mg/kg every two days were protected from virus-induced death and had reduced viral loads in various tissues.
A broad-spectrum antiviral favipiravir was shown to inhibit EV-A71 replication in cell culture by targeting the viral 3Dpol. The mechanism of action was supported by the viral passage experiment in which the S121N mutation in the 3D polymerase was identified. Recombinant viruses generated through reverse genetics showed a single S121N mutation confers drug resistance against favipavir92.
Through screening a library of nucleoside analogues, NITD008 was identified as a potent inhibitor of EV-A7193. NITD008 is an adenosine analog that was originally developed as a flavivirus antiviral but dropped out of clinical trial due to its side effects165. NITD008 inhibited multiple EV1 strains in several cell lines with EC50 values ranging from 0.108 to 4.951 μmol/L. Mechanistic studies showed that the triphosphate derivative of NITD008 (ppp-NITD008) was a chain terminator in the in vitro primer extension-based RdRp assay with an IC50 value of 0.6 μmol/L. Synergistic antiviral effect was observed when NITD008 was combined with either the 3C protease inhibitor AG7088 or the capsid inhibitor GPP3. In EV-A71 infection mouse model study, NITD008 protected viral induced death, alleviated clinical symptoms, and reduced viral loads in various organs when dosed at 5 mg/kg166. Scape mutants were mapped to the viral 3A and 3D polymerase proteins. Reverse genetics showed that either the 3A or the 3D mutations confers drug resistance, and that a combination of 3A and 3D mutations led to higher resistance.
MRS7704 was reported to inhibit EV-A71 with an EC50 value of 3–4 μmol/L167. However, the mechanism of action was not investigated. Remdesivir is the only FDA approved SARS-CoV-2 antiviral. It is a phosphoramidate prodrug that is converted to the triphosphate active drug, which is subsequently incorporated in the RNA chain where it acts as a chain terminator168. Given the similarities in viral polymerase, it was speculated that remdesivir might inhibit EV-A71. Indeed, remdesivir inhibits EV-A71 with an EC50 of 0.991 μmol/L. Drug time-of-addition experiment showed that remdesivir inhibited EV-A71 at the post-viral entry stage. Remdesivir treatment reduced both viral cRNA and vRNA synthesis in EV-A71-infected cells. In addition, remdesivir also inhibited CV-B3 and EV-D68 with EC50 values of 0.097 and 0.026 μmol/L.
DTriP-22 is a non-nucleoside analog that was identified to have broad-spectrum antiviral activity against multiple strains of EV-A71 and coxsackieviruses with submicromolar EC50 values169. Serial viral passage experiment identified mutations in the viral 2C, 3D, and VP1 proteins. Corresponding recombinant EV-A71 viruses were generated, and it was confirmed that the 3D-R163K mutation conferred drug resistance against DTrip-22. The mechanism of action was further supported by the in vitro polymerase assay in which DTrip-22 inhibited the poly(U) elongation activity of EV-A71 3Dpol, but not the VPg uridylylation activity.
Another nonnucleoside EV-A71 3Dpol inhibitor is aurintricarboxylic acid. Aurintricarboxylic acid was found to inhibit EV-A71 RNA synthesis of the in vitro 3D RdRp activity. However, no resistance selection experiment was performed to further support the proposed mechanism of action170.
GPC-N114 (2,2′-[(4-chloro-1,2-phenylene)bis(oxy)]bis(5-nitro-benzonitrile)) was reported as one of the most potent non-nucleoside inhibitors with broad-spectrum antiviral activity against enteroviruses and cardioviruses171. GPC-N114 inhibits EV-A71 with an EC50 value of 0.13 μmol/L. The mechanism of action was confirmed using the in vitro polymerase assay which showed that GPC-N114 inhibited the RNA elongation catalyzed by CV-B3 3Dpol. The co-crystal structure of CV-B3 3Dpol in complex with GPC-N114 showed that GPC-N1114 binds to the RNA-binding channel in the 3Dpol (PDB: 4Y2A) (Fig. 10B).
BPR-3P0128 is a highly potent antiviral against EV-A71 with an EC50 value of 0.0029 μmol/L172. BPR-3P0128 inhibited the in vitro EV-A71 RNA-dependent RNA polymerase activity and VPg uridyllylation synthesis. No resistant virus was selected in the passage experiment, and BPR-3P0128 was also active against the DTrip-22 resistant EV-A71 virus carrying the 3D-R163K mutant.
Viral polymerase is a high-profile antiviral drug target but also a challenging one. The advantages of targeting the polymerase include the broad-spectrum antiviral activity and high potency. Majority of FDA approved viral polymerase inhibitors are nucleoside or nucleotide analogs, which act as either polymerase inhibitors, chain terminator or mutagens once incorporated in viral RNA or DNA173,174. For this reason, the major obstacle in developing nucleoside/nucleotide polymerase inhibitors is the off-target effects against host polymerase. In addition, certain nucleoside/nucleotide drugs have shown to have immunomodulating activities. Overall, targeting the viral polymerase is a high risk and high award strategy.
2.8. IRES inhibitors
Like other eukaryotic viruses, EV-A71 hijacks cellular translational machinery to synthesize viral proteins. In contrast to the host cap-dependent cellular translation, the EV-A71 viral genome lacks the 5′ cap, and thus initiates viral RNA translation through an internal ribosomal entry site (IRES)-mediated mechanism that is cap-independent175. The IRES is located at the 5′ UTR of EV-A71 mRNA and consists of four domains II to IV. The stem loop II structure located at the EV-A71 internal ribosomal entry site is vital for viral replication and represents a novel drug target. Screening of a focused library of RNA-targeting compounds using the peptide-displacement assay revealed DMA-135 as a potent IRES inhibitor (Fig. 11A)94. DMA-135 inhibited EV-A71 replication with an IC50 of 7.54 μmol/L in SF268 cells. Solution NMR structure of IRES stem loop 2 (SLII) in complex with DMA-135 was solved (PDB: 6XB7) (Fig. 11B). It was proposed that upon binding to DMA-135, SLII undergoes a conformational change that stabilizes the ternary complex with the AUF1 protein, thereby suppressing translation.
Figure 11.

EV-A71 IRES inhibitors. (A) Chemical structures of EV-A71 IRES inhibitors. (B) Solution NMR structure of EV-A71 IRES in complex with DMA-135 (PDB: 6XB7).
Screening of a flavonoid library consisting of 502 compounds against EV-A71 identified prunin as one of the most potent hits (Fig. 11A). Prunin inhibited EV-A71 with an EC50 of 115.3 nmol/L. Serial viral passage experiment with prunin yielded drug-resistant viruses with mutations located at the viral internal ribosome entry site (IRES). In vivo study showed that prunin can effectively reduce the mortality rate and clinical symptoms induced by viral infection in HEVA71-infected BALB/c mice95. Other flavonoids including apigenin, luteolin, kaempferol, formononetin, penduletin, and isorhamnetin have been shown to significantly improve the survival rate of mice infected with lethal dose of EV-A71 virus. Among them, isorhamnetin was the most potent and provided 100% survival protection at a dose of 10 mg/kg176. However, the detailed molecular mechanism of action of these flavonoids remains elusive.
An antiprotozoal drug, emetine, was found to inhibit EV-A71 with an EC50 value of 0.04 μmol/L and a CC50 value of 10 μmol/L in RD cell96. In addition, emetine also showed broad-spectrum antiviral activity against CV-A16, CV-B1, EV-D68, Echov-6 with EC50 values in the nanomolar range. Using a dual reporter assay in which the EV-A71 5′ UTR region was flanked by Renilla and Firefly luciferase, emetine was shown to inhibit viral IRES-mediated translation. Significantly, when dosed by oral gavage at as low as 0.2 mg/kg, emetine completely prevented EV-A71 infection induced death in mice and reduced viral loads in the front and hind limbs, brains, and spleens.
Idarubicin (IDR), an antitumor drug, was identified as a broad-spectrum antiviral against enteroviruses including EV-A71177. IDR is a topoisomerase II inhibitor and is used as an anticancer drug to treat myeloid leukemia. IDR was shown to inhibit EV-A71 IRES-mediated translation of viral proteins, but not the host P53 IRES activity, suggesting that IDR might be selective for viral IRES. IDR blocked the binding between EV-A71 IRES and the host IRES trans-acting factor hnRNP A1.
Overall, IRES represents a novel drug target for the broad-spectrum anti-enteroviral inhibitors. Most of the IRES-targeting inhibitors are natural products and some are used in human, making them ideal candidates for the combination therapy. Expect DMA-135, the detailed mechanism of action of flavonoids, emetine, and idarubicin needs to be further characterized. The results of such characterization are expected to help with the lead optimization.
3. Host-targeting antivirals
Viruses including EV-A71 are obligate intracellular parasites and exploit host proteins/signaling pathways for viral replication. Targeting of host factors provides another opportunity for the development of antivirals. The HIV drug maraviroc is a prominent example of a host-targeting antiviral. Maraviroc is an antagonist of the host CCR5 receptor, which is a co-receptor for HIV-1 viral entry178. Advantages of host-target antivirals include the broad-spectrum antiviral activity and a high genetic barrier to drug resistance. However, the cytotoxicity presents a potential issue with host-targeting antivirals.
Several methods including siRNA, proteomics and insertional mutagenesis have been used to map critical host factors and pathways that are important for EV-A71 replication179,180. The host-targeting antivirals against EV-A71 are discussed below.
The PI4KB–PI4P–OSBP pathway was exploited by enteroviruses to direct cholesterol to the replication organelles. PI4KB is essential for the formation of replication organelles by all enteroviruses. Therefore, proteins in the PI4KB–PI4P–OSBP pathway are critical host-targeting antiviral drug targets. OSBP is a PI4P-binding protein and is located in PI4P-enriched replication organelle membranes. It mediates the exchange of PI4P from the replication organelle membrane with cholesterol from the ER, leading to increased cholesterol content in the replication organelle membrane181,182. OSW-1 is a natural product and was found to interact with OSBP (Fig. 12)183. It has broad-spectrum antiviral activity against enteroviruses including EV-A71 with EC50 values at the nanomolar range97. When cells were treated with the OSW-1 for a short period of time (1–6 h), the cellular OSBP protein level decreased by ~90% in multiple cell lines with no apparent toxicity184. Significantly, the reduction was persistent in multiple generations of cells. Although the specific mechanism for the persistent reduction of OSBP level upon OSW-1 treatment remains unknown, the OSW-1 treated cells become refractory to enterovirus infection, rendering it a prophylactic strategy to prevent multiple enterovirus infection. In another study, itraconazole (ITZ) was proposed to exert its antiviral activity by targeting the oxysterol-binding protein (OSBP) and OSBP-related protein 4 (ORP4)85, although other studies have shown that ITZ targets the viral 3A protein. The role of OSBP and ORP4 in viral replication was shown by knockdown or overexpression experiments in which OSBP knockdown led to inhibition of viral replication, while overexpression of either OSBP or ORP4 diminished the antiviral effect of antiviral effects of ITZ and OSW-1. Binding of ITZ to OSBP might impair its function in shutting cholesterol and phosphatidylinositol-4-phosphate between membranes, which is essential for the viral replication organelle formation.
Figure 12.

Host-targeting EV-A71 inhibitors.
The target of MDL-860 was identified to be the host phosphatidylinositol-4 kinase III beta (PI4KB)98. Specifically, MDL-860 was shown to be a covalent inhibitor by irreversibly modifying C646, which is located at the bottom of a surface pocket that is far apart from the active site. The C646S mutant did not affect the enzymatic activity of PI4KB, suggesting that targeting the C646-located allosteric site might not result in unwanted side effects associated with inhibiting the enzymatic activity of PI4KB. This study unveiled a novel drug target that could be further explored to develop more potent and specific host-targeting antivirals.
Through a phenotypic screening and subsequent SAR studies, RYL-634 was identified as a broad-spectrum antiviral candidate against hepatitis C virus, dengue virus, Zika virus, chikungunya virus, EV-A71, human immunodeficiency virus, respiratory syncytial virus, and others100. The EC50 value of RYL-634 against EV-A71 is 4 nmol/L. To identify the drug target of RYL-634, an activity-based probe containing alkyne pull down tag and a diazirine reactive group was designed and used for cell-based target pull down. Human dihydroorotate dehydrogenase (DHODH) was identified as a putative drug target. DHODH is a well-known host factor for viral replication and structurally diverse DHODH inhibitors have been reported as potent antivirals against both RNA and DNA viruses185,186. The antiviral activity of RYL-634 against HCV was antagonized by uridine, supporting the proposed mechanism of action by targeting DHODH. However, the in vivo antiviral activity of RYL-634 remains to be validated. Another DHODH inhibitor FA-613 was also found to exhibit low micromolar broad-spectrum antiviral activity against multiple viruses including influenza virus, EV-A71, RSV, rhinoviruses, and coronaviruses187.
Heparan sulfate proteoglycans (HSPGs) have also been shown to be an important attachment factor for EV-A7137. A library of sulfated HS disaccharides was designed as decoy receptors to inhibit the cell attachment of EV-A71. Compound HTA-22 (22 in the original publication), a per-sulfated GlcN-α(1,4)-Glc synthetic HS mimetic, was identified to be the most potent inhibitor with an IC50 of 7.9 μmol/L in inhibiting EV-A71 replication188. A time-of-addition experiment showed that HTA-22 inhibited the virus adsorption to cells. The mechanism of action of HTA-22 was further supported by the results that no IC50 shift was observed when HTA-22 was present only during the viral attachment at 4 °C compared to when HTA-22 was present during the entire experiment throughout infection.
A number of heparan sulfate (HS) mimetics including heparin, heparan sulfate, and pentosan polysulfate were tested against EV-A71189. Heparin was found to be the most potent inhibitor and inhibited viral replication by more than 90% at 7.81 μg/mL. Mechanistic studies showed that heparin inhibited the early stage of viral replication by blocking viral attachment to the cells.
Human N-myristoyltransferases 1 (hNMT1), but not hNMT2, has been shown to be an important host factor for EV-A71 replication190. Enteroviruses have a conserved N-terminal myristoylation signal (MGXXXS) in the viral capsid protein VP4, suggesting that it might be a potential antiviral drug target. Genetic knockdown of hNMT1 using siRNA or inhibiting the enzymatic activity of hNMT1 by small molecule inhibitor 4O led to the inhibition of viral replication. It was suggested that the myristate moiety in EV-A71 is important for VP0 cleavage, and inhibition of hNMT1 led to the accumulation of the precursors VP0, VP4-2-3, and P1.
Lactoferrin was reported to inhibit EV-A71 replaiton in both RD and the human neuronal SK-N-SH cells191. Bovine lactoferrin was more potent than the human lactoferrin. Immunofluorescence and ELISA-assay showed that lactoferrin binds to the surface of both RD and SK-N-SH cells. Lactoferrin was found to bind to VP1 and the binding was inhibited by anti-VP1 antibody. When co-administered with the virus, lactoferrin improved the survival rate in EV-A71 infected mouse model. However, the binding between lactoferrin and the host heparan sulfate proteoglycans was not examined. The antiviral activity of lactoferrin in inhibiting EV-A71 might also involve blocking viral attachment to the HSPG attachment factor.
A recent study discovered a conserved drug target that can be exploited for the development of broad-spectrum antivirals192. Specifically, the host AP2M1 protein was found to interact with the YxxØ-motif from multiple viruses and this interaction is critical for the intracellular trafficking of viral proteins. For enteroviruses including EV-A71 and EV-D68, the YxxØ-motif has been found to be highly conserved among the viral 2C protein. AP2M1 facilitates EV-A71 2C protein localization in the ER. A screening identified N-(p-amylcinnamoyl)anthranilic acid (ACA) as an inhibitor of the AP2M1/YxxØ interaction that does not affect the AP2M1 phosphorylation. ACA treatment reduced the rate of colocalization of 2C and ER (63% vs. 21%) and had antiviral activity against EV-A71. In addition, ACA also exhibited broad-spectrum antiviral activity both in vitro and in vivo against influenza, Zika virus, and MERS-CoV infection.
Torin2 is an ATP competitive mTOR kinase and has potent anti-EV-A71 activity with an IC50 of 0.01 μmol/L. A library of Torin2 derivatives were designed, among which compound HTA-11e (11e in the original publication) was the most potent EV-A71 antiviral with an IC50 of 0.027 μmol/L193. However, the role of mTOR in EV-A71 replication was not fully elucidated.
In EV-A71 infected mouse model, GS-9620 was found to improve the survival rates and alleviate clinical symptoms194. It was proposed that the antiviral mechanism of action of GS-9620 might be mediated through activation of the NF-κB and PI3K signaling pathways, and inhibition of the NF-κB and PI3K activities antagonized the antiviral activity of GS-9620. GS-9602 treatment significantly reduced the proinflammatory cytokines/chemokines including IFN-α, IFN-γ and MCP-1 as compared to the control group.
Cyclophilin A (CypA), an immunosuppresssor with the peptidyl-prolyl cis–trans isomerase activity, is a high-profile drug target for host-targeting antivirals195,196. CypA has been shown to be essential for the replication of HCV, HIV-1, vesicular stomatitis virus (VSV), influenza virus, and others197, 198, 199. For EV-A71, a study showed that CypA interacts with viral capsid VP1 and regulates the uncoating of the virus200. Short hairpin RNA (shRNA) knockdown of endogenous CypA expression led to reduced EV-A71 replication. CypA inhibitors HL051001P2 and cyclosporine A inhibited EV-A71 replication at EC50 values of 0.78 and 3.38 μmol/L, respectively. Follow up lead optimization led to the discovery of CypA-11 with an EC50 of 0.37 against EV-A71201.
Host-targeting antivirals are valuable chemical probes to study the viral replication mechanism. The general concern with host-targeting antivirals is the off-target side effects. This can be alleviated through either targeting host factors that are upregulated upon viral replication or combining host-targeting antivirals with direct-acting antivirals.
4. Unknown mechanism of action
Two macrolide antibiotics spiramycin (SPM) and azithromycin (AZM) were found to inhibit both EV-A71 and CV-A16 in cell culture (Fig. 13)202. These two compounds might share a similar mechanism of action and have overlapping resistance profiles. Serial viral passage experiments selected multiple mutants including VP1-N102S, 2A-S7F, 2A-G8R, 2B-I47T, and 2C-M108V. Intriguingly, recombinant EV-A71 viruses carrying each of the single mutant showed resistance against SPM. Therefore, it remains elusive which viral protein is the drug target. In vivo study showed that AZM is more efficacious than SPM in protecting neonatal BALB/c mice from EV-A71 infection.
Figure 13.

EV-A71 inhibitors with unknown mechanism of action.
A lysosomotropic agent chloroquine was found to inhibit EV-A71 replication in cell culture203. Significantly, chloroquine also showed in vivo antiviral efficacy in protecting neonatal mice from a lethal challenge of EV-A71, and chloroquine treatment led to improved survival rate and decreased severity of clinical symptoms.
A natural product chebulagic acid was reported to inhibit EV-A71 replication in RD cells with an IC50 of 12.5 μg/mL. In the EV-A71 infection mouse model study, chebulagic acid treatment improved the survival rate and reduced viral titers in the blood, brain, and muscle. However, the molecular mechanism of action of chebulagic acid is unknown204.
A food additive, sodium copper chlorophyllin (CHL), was identified to inhibit multiple strains of EV-A71 and CV-A16 with IC50 values ranging from 14.73 to 50.83 μmol/L205. Mechanistic studies suggest that CHL might target the viral postattachment stage, but the drug target and the binding site remain elusive. Nevertheless, in vivo study showed that CHL-treated mice had significantly reduced viral titers in the lung and muscle.
Simeprevir, an FDA approved HCV drug, was found to inhibit EV-A71, Zika virus, and HSV-1 in cell culture206. However, its antiviral mechanism of action was independent of the simeprevir-induced transcription of IFN-β and ISG15. Further studies are needed to elucidate the mechanism of action for its broad-spectrum antiviral activity.
A natural product geraniin inhibits EV-A71 both in vitro and in vivo207. Geraniin inhibited EV-A71 replication in RD cells with an IC50 of 10 μg/mL and improved the survival rate of mice infected with a lethal dose of EV-A71. However, the mechanism of action was not studied. Punicalagin, which shares similar structural features with geraniin, was also reported to inhibit EV-A71 both in vitro and in vivo208.
Two alkaloids lycorine and matrine were reported to inhibit EV-A71 replication in cell culture and mouse model studies209,210. Compound treatment in mice that were challenged with a lethal dose of EV-A71 showed an improved survival rate. A lycorine derivative LY-55 had inhibitory activity against both EV-A71 and CV-A16211. LY-55 treatment improved the survival rate, decreased viral loads in muscles and clinical scores in mice challenged with a lethal dose of EV-A71. Mechanistic studies suggest that LY-55 is not virucidal and might inhibit EV-A71 and CV-A16 replication through downregulating autophagy. A similar compound 1-acetyllycorine was also reported to inhibit EV-A71 through targeting the viral 2A protease114.
Genetic knockdown the heat shock protein 90 (HSP90) by siRNA or treatment with HSP90 inhibitor geldanamycin (GA) resulted in the inhibition of EV-A71 replication212. In EV-A71-infected mouse model study, 17-allyamino-17-demethoxygeldanamycin (17-AAG), which is a less cytotoxic HSP90 inhibitor, increased the survival rate of mice challenged with a lethal dose of EV-A71 viruses from several genotypes including C2, C4 and B4.
Prohibitin has been shown to be important in EV-A71 viral replication in both the cell entry and replication steps. The cell surface prohibitin is involved in EV-A71 cell entry into neuronal cells, and the mitochondrial prohibitin contributes to viral replication213. The prohibitin inhibitor rocaglamide inhibited EV-A71 replication in NSC-34 neuronal cells. In the EV-A71 infected mouse model study, rocaglamide treatment prolonged the survival and decreased viral titers in the spinal cord and brain in the infected mice compared to the controls.
Curcumin was proposed to inhibit EV-A71 through down regulating GBF1 and PI4KB, but further studies are needed to dissect the roles of these two proteins in viral replication214. Licorice, a traditional Chinese medicine, was reported to inhibit EV-A71 at a step during viral entry215.
Among the list of compounds with an unknown mechanism of action against EV-A71, many are also reported to inhibit other unrelated viruses, suggesting that they might either target a host factor or have a polypharmacology by interacting with multiple targets.
5. Assay development
To increase the throughput of antiviral screening, reporter viruses are advantageous as they can bypass the additional steps of cell staining. An infectious cDNA clone with a stable eGFP reporter was generated based on an epidemic strain of EV-A71216. The eGFP gene was inserted between the 5′ untranslated region and VP4 gene of the EV-A71 genome. The infectious cDNA clone produced high titers (>106 pfu/mL) of eGFP reporter viruses when transfected into Vero cells. Passage experiments showed that the eGFP reporter virus is stable, suggesting that it is suitable for antiviral drug screening as well as studying the mechanism of viral replication. Subsequently, a similar reporter EV-A71 virus with Gaussia luciferase was created using the same strategy and was shown to be suitable for antiviral drug screening217.
In addition to the screening of antivirals, reporter viruses are also useful in studying viral replication in animal models. For example, a mouse-adapted EV-A71 construct with a bioluminescent reporter (mEV-A71-NLuc) was created and used to study viral spread and tissue tropism in two different mouse strains: the BALB/c mice and AG129 mice218. Infection of BALB/c mice resulted a high viral replication as shown by the strong luminescence for 2 wpi. However, no clinical sings of disease were observed. In contrast, the alpha/beta and gamma interferon receptor deficient AG129 mice showed rapid spread of the virus to the brain with the luminescence signal lasting as long as 8 weeks post infection. This EV-A71 infected mouse model might be useful in testing the in vivo antiviral efficacy of EV-A71 antivirals.
One potential pitfall in drug discovery is the failure of transition from in vitro cell culture to in vivo animal model study. To improve the success rate, it is critical to apply physiologically relevant cell culture models that are close mimetics of the virus targeting organs. Specifically, EV-A71 primarily infects the intestine, and intestine organoids have seen increasing use in studying viral replication and drug testing.
Using stem cell-derived enteroids from human small intestines, it was found that EV-A71, CVB, and echovirus 11 infect specific cell populations in the human intestine and induced virus-specific antiviral and inflammatory signaling pathways219.
In another study, human small intestinal organoids have been found to support productive replication of EV-A71, CV-B2, and PV3, rendering it a physiologically relevant model for studying viral replication kinetics and testing antiviral drug candidates220. In contrast, EV-D68 did not replicate significantly in this organoid, an observation that is consistent with its respiratory tract infection. Using itraconazole as a control, it was shown that the antiviral efficacy obtained from organoids was highly reproducible compared to the standard cell lines Caco-2 and RD cells. Significantly, organoids offered an additional advantage for studying EV-A71 infection induced inflammatory response.
A recent study showed that the optimized human intestinal organoids enabled higher replication of EV-A71 and CV-A16 as well as a more rigorous cellular response upon viral infection221.
Overall, the reporter virus and the organoid model are expected to facilitate EV-A71 drug discovery and might help reduce the triage rate of lead progression.
6. Future perspectives
Significant progress has been made in discovering EV-A71 antiviral drug candidates with both in vitro and in vivo antiviral efficacy. However, there are still several hurdles that need to overcome before moving the drug candidates to human clinical trials. First, there is a need of broad-spectrum antivirals that target not only EV-A71, but also related virus strains CV-A16, and CV-A10, both of which are significant HFMD pathogens. Ideally, the antiviral spectrum should also be extended to EV-D68, rhinoviruses, and other members of the enterovirus family. In this regard, viral 2C, 3C, 3Dpol and IRES inhibitors are more prominent drug candidates. In contrast, the capsid inhibitors and 2A inhibitors generally have a narrower spectrum of antiviral activity. Second, a high safety profile is needed for EV-A71 antivirals. Capsid inhibitors dropped out of clinical trials due to side effects. For example, vapendavir failed because of side effects including headaches and drug–drug interactions, and pleconaril treatment led to menstrual irregularities in women using oral contraceptives222. As the susceptible populations of EV-A71 virus are mainly children and infants, and the infection is generally not lethal, there is a more stringent safety requirement for EV-A71 antivirals. Host-targeting antivirals have been reported to have broad-spectrum antiviral activity against enteroviruses; however, the intrinsic issue is the potential side effects and toxicity, as exemplified by in vivo mouse model study with PI4KB inhibitors223. Third, there is a need for EV-A71 antivirals with a high genetic barrier to drug resistance. Resistant mutants have been selected for almost all direct-targeting antivirals in cell culture, raising the concern that resistance might quickly emerge when EV-A71 antivirals are advanced to the market. Another related issue needs to be addressed is the consequences of resistance including the fitness and transmission of the mutant viruses. Although resistance quickly emerges under the drug selection pressure of 2C inhibitors, the resulting mutants have been shown to have compromised fitness of replication80,123. Nevertheless, it is plausible that viruses can continue mutating and evolving compensatory mutations to rescue the replication and transmission processes.
One strategy that can potentially overcome the spectrum of antiviral activity, selectivity or cytotoxicity, and drug resistance is combination therapy. Combination therapy experiments revealed that rupintrivir plus ITZ or favipiravir was synergistic, rupintrivir plus suramin was additive, and suramin plus favipiravir was synergistic against EV-A71 infection in cell culture. Moreover, rupintrivir in combination with ITZ showed a higher genetic barrier to drug resistance than ITZ alone. Combination of 3C protease inhibitor rupintrivir with interferon-α showed synergistic effect in inhibiting EV-A71224. Combination of the protease inhibitor NK-1.8k with either the polymerase inhibitor NITD008 or the capsid inhibitor GPP3 exhibited strong synergistic antiviral effect against EV-A71151. Overall, these promising results showed that combination therapy can not only achieve a higher antiviral potency, but also a higher resistance barrier92.
Previous studies have provided valuable insights of the viral life cycle; however, many unknowns remain to be addressed. For example, the components and the organization of the replication organelles have not been determined by high-resolution structures. The tissue tropism and neuronal infection by EV-A71 remain to be elucidated. Along this line, receptor usage in different tissues needs to be studied. In addition, identifying host factors that are involved in the replication of different subtypes of EVs will help explain the pathogenicity and discover common host factors as drug targets for broad-spectrum antivirals.
Overall, antiviral drug discovery for EV-A71 is the most advanced compared to other enteroviruses. Modern drug discovery techniques and strategies such as high-throughput screening, artificial intelligence, CRISPER, cryo-EM, proteomics, and molecular dynamics simulations might be instrumental in uncovering additional viral and host factors as drug targets225,226. Further development might yield clinical candidates that might eventually benefit thousands to millions of patients. The expertise gained through studying EV-A71 can be similarly applied to other enteroviruses including EV-D68.
Acknowledgments
This research was supported by the National Institute of Allergy and Infectious Diseasess of Health (NIH, USA; grants AI147325 and AI157046) and the Arizona Biomedical Research Commission Centre Young Investigator grant (ADHS18-198859, USA) to Jun Wang. Yanmei Hu was supported by the NIH training grant T32 GM008804 (USA).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Author contributions
Jun Wang, Yanmei Hu, and Madeleine Zheng wrote the draft manuscript; Jun Wang submitted this manuscript on behalf of other authors.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Tuthill T.J., Groppelli E., Hogle J.M., Rowlands D.J. Picornaviruses. Curr Top Microbiol Immunol. 2010;343:43–89. doi: 10.1007/82_2010_37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lugo D., Krogstad P. Enteroviruses in the early 21st century: new manifestations and challenges. Curr Opin Pediatr. 2016;28:107–113. doi: 10.1097/MOP.0000000000000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ooi M.H., Wong S.C., Lewthwaite P., Cardosa M.J., Solomon T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010;9:1097–1105. doi: 10.1016/S1474-4422(10)70209-X. [DOI] [PubMed] [Google Scholar]
- 4.Tyring S.K. Hand foot and mouth disease: enteroviral load and disease severity. EBioMedicine. 2020;62:103115. doi: 10.1016/j.ebiom.2020.103115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phyu W.K., Ong K.C., Wong K.T. Modelling person-to-person transmission in an enterovirus A71 orally infected hamster model of hand-foot-and-mouth disease and encephalomyelitis. Emerg Microb Infect. 2017;6 doi: 10.1038/emi.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z., Liu Y., Liu F., Ren M., Nie T., Cui J., et al. Basic reproduction number of enterovirus 71 coxsackievirus A16 and A6: evidence from outbreaks of hand, foot and mouth disease in China between 2011 and 2018. Clin Infect Dis. 2020;73:e2552–e2559. doi: 10.1093/cid/ciaa1853. [DOI] [PubMed] [Google Scholar]
- 7.Yip C.C., Lau S.K., Lo J.Y., Chan K.H., Woo P.C., Yuen K.Y. Genetic characterization of EV71 isolates from 2004 to 2010 reveals predominance and persistent circulation of the newly proposed genotype D and recent emergence of a distinct lineage of subgenotype C2 in Hong Kong. Virol J. 2013;10:222. doi: 10.1186/1743-422X-10-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solomon T., Lewthwaite P., Perera D., Cardosa M.J., McMinn P., Ooi M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis. 2010;10:778–790. doi: 10.1016/S1473-3099(10)70194-8. [DOI] [PubMed] [Google Scholar]
- 9.Qiu J., Yan H., Cheng N., Lu X., Hu X., Liang L., et al. The clinical and epidemiological study of children with hand, foot, and mouth disease in Hunan, China from 2013 to 2017. Sci Rep. 2019;9:11662. doi: 10.1038/s41598-019-48259-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puenpa J., Wanlapakorn N., Vongpunsawad S., Poovorawan Y. The history of enterovirus A71 outbreaks and molecular epidemiology in the Asia-Pacific region. J Biomed Sci. 2019;26:75. doi: 10.1186/s12929-019-0573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song C., Li Y., Zhou Y., Liang L., Turtle L., Wang F., et al. Enterovirus genomic load and disease severity among children hospitalised with hand, foot and mouth disease. EBioMedicine. 2020;62:103078. doi: 10.1016/j.ebiom.2020.103078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang L.Y., Lin H.Y., Gau S.S., Lu C.Y., Hsia S.H., Huang Y.C., et al. Enterovirus A71 neurologic complications and long-term sequelae. J Biomed Sci. 2019;26:57. doi: 10.1186/s12929-019-0552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messacar K., Spence-Davizon E., Osborne C., Press C., Schreiner T.L., Martin J., et al. Clinical characteristics of enterovirus A71 neurological disease during an outbreak in children in Colorado, USA, in 2018: an observational cohort study. Lancet Infect Dis. 2020;20:230–239. doi: 10.1016/S1473-3099(19)30632-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ong K.C., Badmanathan M., Devi S., Leong K.L., Cardosa M.J., Wong K.T. Pathologic characterization of a murine model of human enterovirus 71 encephalomyelitis. J Neuropathol Exp Neurol. 2008;67:532–542. doi: 10.1097/NEN.0b013e31817713e7. [DOI] [PubMed] [Google Scholar]
- 15.Suresh S., Rawlinson W.D., Andrews P.I., Stelzer-Braid S. Global epidemiology of nonpolio enteroviruses causing severe neurological complications: a systematic review and meta-analysis. Rev Med Virol. 2020;30 doi: 10.1002/rmv.2082. [DOI] [PubMed] [Google Scholar]
- 16.Ong K.C., Wong K.T. Understanding enterovirus 71 neuropathogenesis and its impact on other neurotropic enteroviruses. Brain Pathol. 2015;25:614–624. doi: 10.1111/bpa.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long L., Xu L., Xiao Z., Hu S., Luo R., Wang H., et al. Neurological complications and risk factors of cardiopulmonary failure of EV-A71-related hand, foot and mouth disease. Sci Rep. 2016;6:23444. doi: 10.1038/srep23444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z., Nicholls J.M., Liu F., Wang J., Feng Z., Liu D., et al. Pulmonary and central nervous system pathology in fatal cases of hand foot and mouth disease caused by enterovirus A71 infection. Pathology. 2016;48:267–274. doi: 10.1016/j.pathol.2015.12.450. [DOI] [PubMed] [Google Scholar]
- 19.Tee H.K., Zainol M.I., Sam I.C., Chan Y.F. Recent advances in the understanding of enterovirus A71 infection: a focus on neuropathogenesis. Expert Rev Anti Infect Ther. 2021;19:733–747. doi: 10.1080/14787210.2021.1851194. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt N.J., Lennette E.H., Ho H.H. An apparently new enterovirus isolated from patients with disease of the central nervous system. J Infect Dis. 1974;129:304–309. doi: 10.1093/infdis/129.3.304. [DOI] [PubMed] [Google Scholar]
- 21.Lin J.Y., Kung Y.A., Shih S.R. Antivirals and vaccines for enterovirus A71. J Biomed Sci. 2019;26:65. doi: 10.1186/s12929-019-0560-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li R., Liu L., Mo Z., Wang X., Xia J., Liang Z., et al. An inactivated enterovirus 71 vaccine in healthy children. N Engl J Med. 2014;370:829–837. doi: 10.1056/NEJMoa1303224. [DOI] [PubMed] [Google Scholar]
- 23.Wei M., Meng F., Wang S., Li J., Zhang Y., Mao Q., et al. 2-Year efficacy, immunogenicity, and safety of Vigoo enterovirus 71 vaccine in healthy Chinese children: a randomized open-label study. J Infect Dis. 2017;215:56–63. doi: 10.1093/infdis/jiw502. [DOI] [PubMed] [Google Scholar]
- 24.Zhu F., Xu W., Xia J., Liang Z., Liu Y., Zhang X., et al. Efficacy, safety, and immunogenicity of an enterovirus 71 vaccine in China. N Engl J Med. 2014;370:818–828. doi: 10.1056/NEJMoa1304923. [DOI] [PubMed] [Google Scholar]
- 25.Fang C.Y., Liu C.C. Recent development of enterovirus A vaccine candidates for the prevention of hand, foot, and mouth disease. Expert Rev Vaccines. 2018;17:819–831. doi: 10.1080/14760584.2018.1510326. [DOI] [PubMed] [Google Scholar]
- 26.Mao Q., Wang Y., Bian L., Xu M., Liang Z. EV-A71 vaccine licensure: a first step for multivalent enterovirus vaccine to control HFMD and other severe diseases. Emerg Microb Infect. 2016;5 doi: 10.1038/emi.2016.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng H., Yi L., Chen X., Zhou H., Zheng H., Lu J., et al. Emergence of a non vaccine-cognate enterovirus A71 genotype C1 in mainland China. J Infect. 2021;82:407–413. doi: 10.1016/j.jinf.2020.12.020. [DOI] [PubMed] [Google Scholar]
- 28.Huang K.A., Huang P.N., Huang Y.C., Yang S.L., Tsao K.C., Chiu C.H., et al. Emergence of genotype C1 Enterovirus A71 and its link with antigenic variation of virus in Taiwan. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1008857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu S.T., Kobayashi K., Bi X., Ishizaki A., Tran T.T., Phung T.T.B., et al. Newly emerged enterovirus-A71 C4 sublineage may be more virulent than B5 in the 2015−2016 hand-foot-and-mouth disease outbreak in northern Vietnam. Sci Rep. 2020;10:159. doi: 10.1038/s41598-019-56703-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang C.S., Liao C.C., Liou A.T., Chou Y.C., Yu Y.Y., Lin C.Y., et al. Novel naturally occurring mutations of enterovirus 71 associated with disease severity. Front Microbiol. 2020;11:610568. doi: 10.3389/fmicb.2020.610568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baggen J., Thibaut H.J., Strating J., van Kuppeveld F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol. 2018;16:368–381. doi: 10.1038/s41579-018-0005-4. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi K., Koike S. Cellular receptors for enterovirus A71. J Biomed Sci. 2020;27:23. doi: 10.1186/s12929-020-0615-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamayoshi S., Yamashita Y., Li J., Hanagata N., Minowa T., Takemura T., et al. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med. 2009;15:798–801. doi: 10.1038/nm.1992. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura Y., Shimojima M., Tano Y., Miyamura T., Wakita T., Shimizu H. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med. 2009;15:794–797. doi: 10.1038/nm.1961. [DOI] [PubMed] [Google Scholar]
- 35.Yang B., Chuang H., Yang K.D. Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol J. 2009;6:141. doi: 10.1186/1743-422X-6-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang S.L., Chou Y.T., Wu C.N., Ho M.S. Annexin II binds to capsid protein VP1 of enterovirus 71 and enhances viral infectivity. J Virol. 2011;85:11809–11820. doi: 10.1128/JVI.00297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan C.W., Poh C.L., Sam I.C., Chan Y.F. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J Virol. 2013;87:611–620. doi: 10.1128/JVI.02226-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeung M.L., Jia L., Yip C.C.Y., Chan J.F.W., Teng J.L.L., Chan K.H., et al. Human tryptophanyl-tRNA synthetase is an IFN-gamma-inducible entry factor for Enterovirus. J Clin Invest. 2018;128:5163–5177. doi: 10.1172/JCI99411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X., Peng W., Ren J., Hu Z., Xu J., Lou Z., et al. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat Struct Mol Biol. 2012;19:424–429. doi: 10.1038/nsmb.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyu K., Ding J., Han J.F., Zhang Y., Wu X.Y., He Y.L., et al. Human enterovirus 71 uncoating captured at atomic resolution. J Virol. 2014;88:3114–3126. doi: 10.1128/JVI.03029-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shingler K.L., Yoder J.L., Carnegie M.S., Ashley R.E., Makhov A.M., Conway J.F., et al. The enterovirus 71 A-particle forms a gateway to allow genome release: a cryoEM study of picornavirus uncoating. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan J., Shen L., Wu J., Zou X., Gu J., Chen J., et al. Enterovirus A71 proteins: structure and function. Front Microbiol. 2018;9:286. doi: 10.3389/fmicb.2018.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melia C.E., Peddie C.J., de Jong A.W.M., Snijder E.J., Collinson L.M., Koster A.J., et al. Origins of enterovirus replication organelles established by whole-cell electron microscopy. mBio. 2019;10 doi: 10.1128/mBio.00951-19. e00951-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang P., Liu Y., Ma H.C., Paul A.V., Wimmer E. Picornavirus morphogenesis. Microbiol Mol Biol Rev. 2014;78:418–437. doi: 10.1128/MMBR.00012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basavappa R., Syed R., Flore O., Icenogle J.P., Filman D.J., Hogle J.M. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9 A resolution. Protein Sci. 1994;3:1651–1669. doi: 10.1002/pro.5560031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curry S., Fry E., Blakemore W., Abu-Ghazaleh R., Jackson T., King A., et al. Dissecting the roles of VP0 cleavage and RNA packaging in picornavirus capsid stabilization: the structure of empty capsids of foot-and-mouth disease virus. J Virol. 1997;71:9743–9752. doi: 10.1128/jvi.71.12.9743-9752.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moffett D.B., Llewellyn A., Singh H., Saxentoff E., Partridge J., Boualam L., et al. Progress toward poliovirus containment implementation—worldwide, 2019–2020. MMWR Morb Mortal Wkly Rep. 2020;69:1330–1333. doi: 10.15585/mmwr.mm6937a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uprety P., Graf E.H. Enterovirus infection and acute flaccid myelitis. Curr Opin Virol. 2020;40:55–60. doi: 10.1016/j.coviro.2020.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Rhoades R.E., Tabor-Godwin J.M., Tsueng G., Feuer R. Enterovirus infections of the central nervous system. Virology. 2011;411:288–305. doi: 10.1016/j.virol.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang H.I., Shih S.R. Neurotropic enterovirus infections in the central nervous system. Viruses. 2015;7:6051–6066. doi: 10.3390/v7112920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mizutani T., Ishizaka A., Nihei C. Transferrin receptor 1 facilitates poliovirus permeation of mouse brain capillary endothelial cells. J Biol Chem. 2016;291:2829–2836. doi: 10.1074/jbc.M115.690941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klein R.S., Garber C., Funk K.E., Salimi H., Soung A., Kanmogne M., et al. Neuroinflammation during RNA viral infections. Annu Rev Immunol. 2019;37:73–95. doi: 10.1146/annurev-immunol-042718-041417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen C.S., Yao Y.C., Lin S.C., Lee Y.P., Wang Y.F., Wang J.R., et al. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. J Virol. 2007;81:8996–9003. doi: 10.1128/JVI.00236-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan S.H., Ong K.C., Wong K.T. Enterovirus 71 can directly infect the brainstem via cranial nerves and infection can be ameliorated by passive immunization. J Neuropathol Exp Neurol. 2014;73:999–1008. doi: 10.1097/NEN.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 55.Egorova A., Ekins S., Schmidtke M., Makarov V. Back to the future: advances in development of broad-spectrum capsid-binding inhibitors of enteroviruses. Eur J Med Chem. 2019;178:606–622. doi: 10.1016/j.ejmech.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang G., Zhou F., Gu B., Ding C., Feng D., Xie F., et al. In vitro and in vivo evaluation of ribavirin and pleconaril antiviral activity against enterovirus 71 infection. Arch Virol. 2012;157:669–679. doi: 10.1007/s00705-011-1222-6. [DOI] [PubMed] [Google Scholar]
- 57.Tijsma A., Franco D., Tucker S., Hilgenfeld R., Froeyen M., Leyssen P., et al. The capsid binder vapendavir and the novel protease inhibitor SG85 inhibit enterovirus 71 replication. Antimicrob Agents Chemother. 2014;58:6990–6992. doi: 10.1128/AAC.03328-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma C., Hu Y., Zhang J., Musharrafieh R., Wang J. A novel capsid binding inhibitor displays potent antiviral activity against enterovirus D68. ACS Infect Dis. 2019;5:1952–1962. doi: 10.1021/acsinfecdis.9b00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Senior K. FDA panel rejects common cold treatment. Lancet Infect Dis. 2002;2:264. doi: 10.1016/s1473-3099(02)00277-3. [DOI] [PubMed] [Google Scholar]
- 60.Ho J.Y., Chern J.H., Hsieh C.F., Liu S.T., Liu C.J., Wang Y.S., et al. In vitro and in vivo studies of a potent capsid-binding inhibitor of enterovirus 71. J Antimicrob Chemother. 2016;71:1922–1932. doi: 10.1093/jac/dkw101. [DOI] [PubMed] [Google Scholar]
- 61.Zhang M., Wang Y., He W., Sun Y., Guo Y., Zhong W., et al. Design, synthesis, and evaluation of novel enterovirus 71 inhibitors as therapeutic drug leads for the treatment of human hand, foot, and mouth disease. J Med Chem. 2020;63:1233–1244. doi: 10.1021/acs.jmedchem.9b01414. [DOI] [PubMed] [Google Scholar]
- 62.Li P., Yu J., Hao F., He H., Shi X., Hu J., et al. Discovery of potent EV71 capsid inhibitors for treatment of HFMD. ACS Med Chem Lett. 2017;8:841–846. doi: 10.1021/acsmedchemlett.7b00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen J., Ye X., Zhang X.Y., Zhu Z., Zhang X., Xu Z., et al. Coxsackievirus A10 atomic structure facilitating the discovery of a broad-spectrum inhibitor against human enteroviruses. Cell Discov. 2019;5:4. doi: 10.1038/s41421-018-0073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smee D.F., Evans W.J., Nicolaou K.C., Tarbet E.B., Day C.W. Susceptibilities of enterovirus D68, enterovirus 71, and rhinovirus 87 strains to various antiviral compounds. Antivir Res. 2016;131:61–65. doi: 10.1016/j.antiviral.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun L., Meijer A., Froeyen M., Zhang L., Thibaut H.J., Baggen J., et al. Antiviral activity of broad-spectrum and enterovirus-specific inhibitors against clinical isolates of enterovirus D68. Antimicrob Agents Chemother. 2015;59:7782–7785. doi: 10.1128/AAC.01375-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lacroix C., Laconi S., Angius F., Coluccia A., Silvestri R., Pompei R., et al. In vitro characterisation of a pleconaril/pirodavir-like compound with potent activity against rhinoviruses. Virol J. 2015;12:106. doi: 10.1186/s12985-015-0330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martinez-Gualda B., Sun L., Marti-Mari O., Noppen S., Abdelnabi R., Bator C.M., et al. Scaffold simplification strategy leads to a novel generation of dual human immunodeficiency virus and enterovirus-A71 entry inhibitors. J Med Chem. 2020;63:349–368. doi: 10.1021/acs.jmedchem.9b01737. [DOI] [PubMed] [Google Scholar]
- 68.Ren P., Zou G., Bailly B., Xu S., Zeng M., Chen X., et al. The approved pediatric drug suramin identified as a clinical candidate for the treatment of EV71 infection-suramin inhibits EV71 infection in vitro and in vivo. Emerg Microb Infect. 2014;3 doi: 10.1038/emi.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiedemar N., Hauser D.A., Maser P. 100 years of suramin. Antimicrob Agents Chemother. 2020;64:e01168-19. doi: 10.1128/AAC.01168-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ren P., Zheng Y., Wang W., Hong L., Delpeyroux F., Arenzana-Seisdedos F., et al. Suramin interacts with the positively charged region surrounding the 5-fold axis of the EV-A71 capsid and inhibits multiple enterovirus A. Sci Rep. 2017;7:42902. doi: 10.1038/srep42902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishimura Y., McLaughlin N.P., Pan J., Goldstein S., Hafenstein S., Shimizu H., et al. The suramin derivative NF449 interacts with the 5-fold vertex of the enterovirus A71 capsid to prevent virus attachment to PSGL-1 and heparan sulfate. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1005184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meng T., Jia Q., Wong S.M., Chua K.B. In vitro and in vivo inhibition of the infectivity of human enterovirus 71 by a sulfonated food azo dye, brilliant black BN. J Virol. 2019;93 doi: 10.1128/JVI.00061-19. e00061-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bauer L., Manganaro R., Zonsics B., Strating J., El Kazzi P., Lorenzo Lopez M., et al. Fluoxetine inhibits enterovirus replication by targeting the viral 2C protein in a stereospecific manner. ACS Infect Dis. 2019;5:1609–1623. doi: 10.1021/acsinfecdis.9b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Messacar K., Sillau S., Hopkins S.E., Otten C., Wilson-Murphy M., Wong B., et al. Safety, tolerability, and efficacy of fluoxetine as an antiviral for acute flaccid myelitis. Neurology. 2019;92:e2118–e2126. doi: 10.1212/WNL.0000000000006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tyler K.L. Rationale for the evaluation of fluoxetine in the treatment of enterovirus D68-associated acute flaccid myelitis. JAMA Neurol. 2015;72:493–494. doi: 10.1001/jamaneurol.2014.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bauer L., Manganaro R., Zonsics B., Hurdiss D.L., Zwaagstra M., Donselaar T., et al. Rational design of highly potent broad-spectrum enterovirus inhibitors targeting the nonstructural protein 2C. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ulferts R., de Boer S.M., van der Linden L., Bauer L., Lyoo H.R., Mate M.J., et al. Screening of a library of FDA-approved drugs identifies several enterovirus replication inhibitors that target viral protein 2C. Antimicrob Agents Chemother. 2016;60:2627–2638. doi: 10.1128/AAC.02182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Musharrafieh R., Zhang J., Tuohy P., Kitamura N., Bellampalli S.S., Hu Y., et al. Discovery of quinoline analogues as potent antivirals against enterovirus D68 (EV-D68) J Med Chem. 2019;62:4074–4090. doi: 10.1021/acs.jmedchem.9b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xing Y., Zuo J., Krogstad P., Jung M.E. Synthesis and structure–activity relationship (SAR) studies of novel pyrazolopyridine derivatives as inhibitors of enterovirus replication. J Med Chem. 2018;61:1688–1703. doi: 10.1021/acs.jmedchem.7b01863. [DOI] [PubMed] [Google Scholar]
- 80.Hu Y., Kitamura N., Musharrafieh R., Wang J. Discovery of potent and broad-spectrum pyrazolopyridine-containing antivirals against enteroviruses D68, A71, and coxsackievirus B3 by targeting the viral 2C protein. J Med Chem. 2021;64:8755–8774. doi: 10.1021/acs.jmedchem.1c00758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Phillpotts R.J., Jones R.W., Delong D.C., Reed S.E., Wallace J., Tyrrell D.A. The activity of enviroxime against rhinovirus infection in man. Lancet. 1981;1:1342–1344. doi: 10.1016/s0140-6736(81)92520-4. [DOI] [PubMed] [Google Scholar]
- 82.Miller F.D., Monto A.S., DeLong D.C., Exelby A., Bryan E.R., Srivastava S. Controlled trial of enviroxime against natural rhinovirus infections in a community. Antimicrob Agents Chemother. 1985;27:102–106. doi: 10.1128/aac.27.1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heinz B.A., Vance L.M. The antiviral compound enviroxime targets the 3A coding region of rhinovirus and poliovirus. J Virol. 1995;69:4189–4197. doi: 10.1128/jvi.69.7.4189-4197.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gao Q., Yuan S., Zhang C., Wang Y., Wang Y., He G., et al. Discovery of itraconazole with broad-spectrum in vitro antienterovirus activity that targets nonstructural protein 3A. Antimicrob Agents Chemother. 2015;59:2654–2665. doi: 10.1128/AAC.05108-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Strating J.R., van der Linden L., Albulescu L., Bigay J., Arita M., Delang L., et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015;10:600–615. doi: 10.1016/j.celrep.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu G., Qi J., Chen Z., Xu X., Gao F., Lin D., et al. Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design. J Virol. 2011;85:10319–10331. doi: 10.1128/JVI.00787-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Musharrafieh R., Ma C., Zhang J., Hu Y., Diesing J.M., Marty M.T., et al. Validating enterovirus D68-2Apro as an antiviral drug target and the discovery of telaprevir as a potent D68-2Apro inhibitor. J Virol. 2019;93 doi: 10.1128/JVI.02221-18. e02221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim Y., Lovell S., Tiew K.C., Mandadapu S.R., Alliston K.R., Battaile K.P., et al. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol. 2012;86:11754–11762. doi: 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu Y., Ma C., Szeto T., Hurst B., Tarbet B., Wang J. Boceprevir, calpain inhibitors II and XII, and GC-376 have broad-spectrum antiviral activity against coronaviruses in cell culture. ACS Infect Dis. 2021;7:586–597. doi: 10.1021/acsinfecdis.0c00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun J., Yogarajah T., Lee R.C.H., Kaur P., Inoue M., Tan Y.W., et al. Drug repurposing of pyrimidine analogs as potent antiviral compounds against human enterovirus A71 infection with potential clinical applications. Sci Rep. 2020;10:8159. doi: 10.1038/s41598-020-65152-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu N., Yang J., Zheng B., Zhang Y., Cao Y., Huan C., et al. The pyrimidine analog FNC potently inhibits the replication of multiple enteroviruses. J Virol. 2020;94:e00204–e00220. doi: 10.1128/JVI.00204-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y., Li G., Yuan S., Gao Q., Lan K., Altmeyer R., et al. In vitro assessment of combinations of enterovirus inhibitors against enterovirus 71. Antimicrob Agents Chemother. 2016;60:5357–5367. doi: 10.1128/AAC.01073-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shang L., Wang Y., Qing J., Shu B., Cao L., Lou Z., et al. An adenosine nucleoside analogue NITD008 inhibits EV71 proliferation. Antivir Res. 2014;112:47–58. doi: 10.1016/j.antiviral.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 94.Davila-Calderon J., Patwardhan N.N., Chiu L.Y., Sugarman A., Cai Z., Penutmutchu S.R., et al. IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat Commun. 2020;11:4775. doi: 10.1038/s41467-020-18594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gunaseelan S., Wong K.Z., Min N., Sun J., Ismail N., Tan Y.J., et al. Prunin suppresses viral IRES activity and is a potential candidate for treating enterovirus A71 infection. Sci Transl Med. 2019;11 doi: 10.1126/scitranslmed.aar5759. [DOI] [PubMed] [Google Scholar]
- 96.Tang Q., Li S., Du L., Chen S., Gao J., Cai Y., et al. Emetine protects mice from enterovirus infection by inhibiting viral translation. Antivir Res. 2020;173:104650. doi: 10.1016/j.antiviral.2019.104650. [DOI] [PubMed] [Google Scholar]
- 97.Albulescu L., Strating J.R., Thibaut H.J., van der Linden L., Shair M.D., Neyts J., et al. Broad-range inhibition of enterovirus replication by OSW-1, a natural compound targeting OSBP. Antivir Res. 2015;117:110–114. doi: 10.1016/j.antiviral.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 98.Arita M., Dobrikov G., Purstinger G., Galabov A.S. Allosteric regulation of phosphatidylinositol 4-kinase III beta by an antipicornavirus compound MDL-860. ACS Infect Dis. 2017;3:585–594. doi: 10.1021/acsinfecdis.7b00053. [DOI] [PubMed] [Google Scholar]
- 99.Torney H.L., Dulworth J.K., Steward D.L. Antiviral activity and mechanism of action of 2-(3,4-dichlorophenoxy)-5-nitrobenzonitrile (MDL-860) Antimicrob Agents Chemother. 1982;22:635–638. doi: 10.1128/aac.22.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Y., Cao L., Gao H., Wu Y., Wang Y., Fang F., et al. Discovery, optimization, and target identification of novel potent broad-spectrum antiviral inhibitors. J Med Chem. 2019;62:4056–4073. doi: 10.1021/acs.jmedchem.9b00091. [DOI] [PubMed] [Google Scholar]
- 101.Plevka P., Perera R., Yap M.L., Cardosa J., Kuhn R.J., Rossmann M.G. Structure of human enterovirus 71 in complex with a capsid-binding inhibitor. Proc Natl Acad Sci U S A. 2013;110:5463–5467. doi: 10.1073/pnas.1222379110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anasir M.I., Zarif F., Poh C.L. Antivirals blocking entry of enteroviruses and therapeutic potential. J Biomed Sci. 2021;28:10. doi: 10.1186/s12929-021-00708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.De Colibus L., Wang X., Spyrou J.A.B., Kelly J., Ren J., Grimes J., et al. More-powerful virus inhibitors from structure-based analysis of HEV71 capsid-binding molecules. Nat Struct Mol Biol. 2014;21:282–288. doi: 10.1038/nsmb.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Han X., Sun N., Wu H., Guo D., Tien P., Dong C., et al. Identification and structure–activity relationships of diarylhydrazides as novel potent and selective human enterovirus inhibitors. J Med Chem. 2016;59:2139–2150. doi: 10.1021/acs.jmedchem.5b01803. [DOI] [PubMed] [Google Scholar]
- 105.Arita M., Fuchino H., Kawakami H., Ezaki M., Kawahara N. Characterization of a new antienterovirus D68 compound purified from avocado. ACS Infect Dis. 2020;6:2291–2300. doi: 10.1021/acsinfecdis.0c00404. [DOI] [PubMed] [Google Scholar]
- 106.Li G., Gao Q., Yuan S., Wang L., Altmeyer R., Lan K., et al. Characterization of three small molecule inhibitors of enterovirus 71 identified from screening of a library of natural products. Antivir Res. 2017;143:85–96. doi: 10.1016/j.antiviral.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 107.Kim J., Jung Y.K., Kim C., Shin J.S., Scheers E., Lee J.Y., et al. A novel series of highly potent small molecule inhibitors of rhinovirus replication. J Med Chem. 2017;60:5472–5492. doi: 10.1021/acs.jmedchem.7b00175. [DOI] [PubMed] [Google Scholar]
- 108.Sun L., Lee H., Thibaut H.J., Lanko K., Rivero-Buceta E., Bator C., et al. Viral engagement with host receptors blocked by a novel class of tryptophan dendrimers that targets the 5-fold-axis of the enterovirus-A71 capsid. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hsieh C.F., Jheng J.R., Lin G.H., Chen Y.L., Ho J.Y., Liu C.J., et al. Rosmarinic acid exhibits broad anti-enterovirus A71 activity by inhibiting the interaction between the five-fold axis of capsid VP1 and cognate sulfated receptors. Emerg Microb Infect. 2020;9:1194–1205. doi: 10.1080/22221751.2020.1767512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuo R.L., Kung S.H., Hsu Y.Y., Liu W.T. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J Gen Virol. 2002;83:1367–1376. doi: 10.1099/0022-1317-83-6-1367. [DOI] [PubMed] [Google Scholar]
- 111.Falah N., Montserret R., Lelogeais V., Schuffenecker I., Lina B., Cortay J.C., et al. Blocking human enterovirus 71 replication by targeting viral 2A protease. J Antimicrob Chemother. 2012;67:2865–2869. doi: 10.1093/jac/dks304. [DOI] [PubMed] [Google Scholar]
- 112.Wang C.Y., Huang A.C., Hour M.J., Huang S.H., Kung S.H., Chen C.H., et al. Antiviral potential of a novel compound CW-33 against enterovirus A71 via inhibition of viral 2A protease. Viruses. 2015;7:3155–3171. doi: 10.3390/v7062764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cai Q., Yameen M., Liu W., Gao Z., Li Y., Peng X., et al. Conformational plasticity of the 2A proteinase from enterovirus 71. J Virol. 2013;87:7348–7356. doi: 10.1128/JVI.03541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guo Y., Wang Y., Cao L., Wang P., Qing J., Zheng Q., et al. A conserved inhibitory mechanism of a lycorine derivative against enterovirus and hepatitis C virus. Antimicrob Agents Chemother. 2016;60:913–924. doi: 10.1128/AAC.02274-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xie S., Wang K., Yu W., Lu W., Xu K., Wang J., et al. DIDS blocks a chloride-dependent current that is mediated by the 2B protein of enterovirus 71. Cell Res. 2011;21:1271–1275. doi: 10.1038/cr.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nieva J.L., Madan V., Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10:563–574. doi: 10.1038/nrmicro2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cong H., Du N., Yang Y., Song L., Zhang W., Tien P. Enterovirus 71 2B induces cell apoptosis by directly inducing the conformational activation of the proapoptotic protein Bax. J Virol. 2016;90:9862–9877. doi: 10.1128/JVI.01499-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang S.H., Wang K., Zhao K., Hua S.C., Du J. The structure, function, and mechanisms of action of enterovirus non-structural protein 2C. Front Microbiol. 2020;11:615965. doi: 10.3389/fmicb.2020.615965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xia H., Wang P., Wang G.C., Yang J., Sun X., Wu W., et al. Human enterovirus nonstructural protein 2CATPase functions as both an RNA helicase and ATP-independent RNA chaperone. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Manganaro R., Zonsics B., Bauer L., Lorenzo Lopez M., Donselaar T., Zwaagstra M., et al. Synthesis and antiviral effect of novel fluoxetine analogues as enterovirus 2C inhibitors. Antivir Res. 2020;178:104781. doi: 10.1016/j.antiviral.2020.104781. [DOI] [PubMed] [Google Scholar]
- 121.Musharrafieh R., Kitamura N., Hu Y., Wang J. Development of broad-spectrum enterovirus antivirals based on quinoline scaffold. Bioorg Chem. 2020;101:103981. doi: 10.1016/j.bioorg.2020.103981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tang Q., Xu Z., Jin M., Shu T., Chen Y., Feng L., et al. Identification of dibucaine derivatives as novel potent enterovirus 2C helicase inhibitors: in vitro, in vivo, and combination therapy study. Eur J Med Chem. 2020;202:112310. doi: 10.1016/j.ejmech.2020.112310. [DOI] [PubMed] [Google Scholar]
- 123.Ma C., Hu Y., Zhang J., Wang J. Pharmacological characterization of the mechanism of action of R523062, a promising antiviral for enterovirus D68. ACS Infect Dis. 2020;6:2260–2270. doi: 10.1021/acsinfecdis.0c00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zuo J., Kye S., Quinn K.K., Cooper P., Damoiseaux R., Krogstad P. Discovery of structurally diverse small-molecule compounds with broad antiviral activity against enteroviruses. Antimicrob Agents Chemother. 2015;60:1615–1626. doi: 10.1128/AAC.02646-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.De Palma A.M., Heggermont W., Lanke K., Coutard B., Bergmann M., Monforte A.M., et al. The thiazolobenzimidazole TBZE-029 inhibits enterovirus replication by targeting a short region immediately downstream from motif C in the nonstructural protein 2C. J Virol. 2008;82:4720–4730. doi: 10.1128/JVI.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ulferts R., van der Linden L., Thibaut H.J., Lanke K.H., Leyssen P., Coutard B., et al. Selective serotonin reuptake inhibitor fluoxetine inhibits replication of human enteroviruses B and D by targeting viral protein 2C. Antimicrob Agents Chemother. 2013;57:1952–1956. doi: 10.1128/AAC.02084-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Arita M., Wakita T., Shimizu H. Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J Gen Virol. 2008;89:2518–2530. doi: 10.1099/vir.0.2008/002915-0. [DOI] [PubMed] [Google Scholar]
- 128.Guan H., Tian J., Zhang C., Qin B., Cui S. Crystal structure of a soluble fragment of poliovirus 2CATPase. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1007304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guan H., Tian J., Qin B., Wojdyla J.A., Wang B., Zhao Z., et al. Crystal structure of 2C helicase from enterovirus 71. Sci Adv. 2017;3 doi: 10.1126/sciadv.1602573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hurdiss D.L., El Kazzi P., Bauer L., Papageorgiou N., Ferron F.P., Donselaar T., et al. Fluoxetine targets an allosteric site in the enterovirus 2C AAA+ ATPase and stabilizes the hexameric complex. bioRxiv. 2021 doi: 10.1101/2021.04.26.440876. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li X., Wang M., Cheng A., Wen X., Ou X., Mao S., et al. Enterovirus replication organelles and inhibitors of their formation. Front Microbiol. 2020;11:1817. doi: 10.3389/fmicb.2020.01817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nagy P.D., Strating J.R., van Kuppeveld F.J. Building viral replication organelles: close encounters of the membrane types. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xiao X., Lei X., Zhang Z., Ma Y., Qi J., Wu C., et al. Enterovirus 3A facilitates viral replication by promoting phosphatidylinositol 4-kinase IIIbeta-ACBD3 interaction. J Virol. 2017;91 doi: 10.1128/JVI.00791-17. e00791-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lyoo H., van der Schaar H.M., Dorobantu C.M., Rabouw H.H., Strating J., van Kuppeveld F.J.M. ACBD3 is an essential pan-enterovirus host factor that mediates the interaction between viral 3A protein and cellular protein PI4KB. mBio. 2019;10 doi: 10.1128/mBio.02742-18. e02742-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Horova V., Lyoo H., Rozycki B., Chalupska D., Smola M., Humpolickova J., et al. Convergent evolution in the mechanisms of ACBD3 recruitment to picornavirus replication sites. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Arita M., Takebe Y., Wakita T., Shimizu H. A bifunctional anti-enterovirus compound that inhibits replication and the early stage of enterovirus 71 infection. J Gen Virol. 2010;91:2734–2744. doi: 10.1099/vir.0.023374-0. [DOI] [PubMed] [Google Scholar]
- 137.Arita M., Wakita T., Shimizu H. Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J Gen Virol. 2009;90:1869–1879. doi: 10.1099/vir.0.012096-0. [DOI] [PubMed] [Google Scholar]
- 138.De Palma A.M., Thibaut H.J., van der Linden L., Lanke K., Heggermont W., Ireland S., et al. Mutations in the nonstructural protein 3A confer resistance to the novel enterovirus replication inhibitor TTP-8307. Antimicrob Agents Chemother. 2009;53:1850–1857. doi: 10.1128/AAC.00934-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Albulescu L., Bigay J., Biswas B., Weber-Boyvat M., Dorobantu C.M., Delang L., et al. Uncovering oxysterol-binding protein (OSBP) as a target of the anti-enteroviral compound TTP-8307. Antivir Res. 2017;140:37–44. doi: 10.1016/j.antiviral.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 140.Rhoden E., Ng T.F.F., Campagnoli R., Nix W.A., Konopka-Anstadt J., Selvarangan R., et al. Antifungal triazole posaconazole targets an early stage of the parechovirus A3 life cycle. Antimicrob Agents Chemother. 2020;64 doi: 10.1128/AAC.02372-19. e02372-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li W., Ross-Smith N., Proud C.G., Belsham G.J. Cleavage of translation initiation factor 4AI (eIF4AI) but not eIF4AII by foot-and-mouth disease virus 3C protease: identification of the eIF4AI cleavage site. FEBS Lett. 2001;507:1–5. doi: 10.1016/s0014-5793(01)02885-x. [DOI] [PubMed] [Google Scholar]
- 142.de Breyne S., Bonderoff J.M., Chumakov K.M., Lloyd R.E., Hellen C.U. Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology. 2008;378:118–122. doi: 10.1016/j.virol.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sun D., Chen S., Cheng A., Wang M. Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses. 2016;8:82. doi: 10.3390/v8030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhai Y., Zhao X., Cui Z., Wang M., Wang Y., Li L., et al. Cyanohydrin as an anchoring group for potent and selective inhibitors of enterovirus 71 3C protease. J Med Chem. 2015;58:9414–9420. doi: 10.1021/acs.jmedchem.5b01013. [DOI] [PubMed] [Google Scholar]
- 145.Ma Y., Shang C., Yang P., Li L., Zhai Y., Yin Z., et al. 4-Iminooxazolidin-2-one as a bioisostere of the cyanohydrin moiety: inhibitors of enterovirus 71 3C protease. J Med Chem. 2018;61:10333–10339. doi: 10.1021/acs.jmedchem.8b01335. [DOI] [PubMed] [Google Scholar]
- 146.Ma Y., Li L., He S., Shang C., Sun Y., Liu N., et al. Application of dually activated michael acceptor to the rational design of reversible covalent inhibitor for enterovirus 71 3C protease. J Med Chem. 2019;62:6146–6162. doi: 10.1021/acs.jmedchem.9b00387. [DOI] [PubMed] [Google Scholar]
- 147.Ma G.H., Ye Y., Zhang D., Xu X., Si P., Peng J.L., et al. Identification and biochemical characterization of DC07090 as a novel potent small molecule inhibitor against human enterovirus 71 3C protease by structure-based virtual screening. Eur J Med Chem. 2016;124:981–991. doi: 10.1016/j.ejmech.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 148.Hayden F.G., Turner R.B., Gwaltney J.M., Chi-Burris K., Gersten M., Hsyu P., et al. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47:3907–3916. doi: 10.1128/AAC.47.12.3907-3916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang J., Fan T., Yao X., Wu Z., Guo L., Lei X., et al. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol. 2011;85:10021–10030. doi: 10.1128/JVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schulz R., Atef A., Becker D., Gottschalk F., Tauber C., Wagner S., et al. Phenylthiomethyl ketone-based fragments show selective and irreversible inhibition of enteroviral 3C proteases. J Med Chem. 2018;61:1218–1230. doi: 10.1021/acs.jmedchem.7b01440. [DOI] [PubMed] [Google Scholar]
- 151.Wang Y., Yang B., Zhai Y., Yin Z., Sun Y., Rao Z. Peptidyl aldehyde NK-1.8k suppresses enterovirus 71 and enterovirus 68 infection by targeting protease 3C. Antimicrob Agents Chemother. 2015;59:2636–2646. doi: 10.1128/AAC.00049-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hu Y., Ma C., Szeto T., Hurst B., Tarbet B., Wang J. Boceprevir, calpain inhibitors II and XII, and GC-376 have broad-spectrum antiviral activity against coronaviruses. ACS Infect Dis. 2021;7:586–597. doi: 10.1021/acsinfecdis.0c00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., et al. Discovery of a novel inhibitor of coronavirus 3CL protease as a clinical candidate for the potential treatment of COVID-19. bioRxiv. 2020 http://doi:10.1101/2020.09.12.293498 Available from: [Google Scholar]
- 154.Wang Y., Cao L., Zhai Y., Yin Z., Sun Y., Shang L. Structure of the enterovirus 71 3C protease in complex with NK-1.8k and indications for the development of antienterovirus protease inhibitor. Antimicrob Agents Chemother. 2017;61 doi: 10.1128/AAC.00298-17. e00298-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., et al. alpha-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 156.Li P., Wu S., Xiao T., Li Y., Su Z., Wei W., et al. Design, synthesis, and evaluation of a novel macrocyclic anti-EV71 agent. Bioorg Med Chem. 2020;28:115551. doi: 10.1016/j.bmc.2020.115551. [DOI] [PubMed] [Google Scholar]
- 157.Sun Y., Zheng Q., Wang Y., Pang Z., Liu J., Yin Z., et al. Activity-based protein profiling identifies ATG4B as a key host factor for enterovirus 71 proliferation. J Virol. 2019;93 doi: 10.1128/JVI.01092-19. e01092-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tan J., George S., Kusov Y., Perbandt M., Anemuller S., Mesters J.R., et al. 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J Virol. 2013;87:4339–4351. doi: 10.1128/JVI.01123-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tan Y.W., Ang M.J., Lau Q.Y., Poulsen A., Ng F.M., Then S.W., et al. Antiviral activities of peptide-based covalent inhibitors of the Enterovirus 71 3C protease. Sci Rep. 2016;6:33663. doi: 10.1038/srep33663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li P., Yang B., Hao F., Wang P., He H., Huang L., et al. Design, synthesis, and biological evaluation of anti-EV71 agents. Bioorg Med Chem Lett. 2016;26:3346–3350. doi: 10.1016/j.bmcl.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 161.Zeng D., Ma Y., Zhang R., Nie Q., Cui Z., Wang Y., et al. Synthesis and structure–activity relationship of alpha-keto amides as enterovirus 71 3C protease inhibitors. Bioorg Med Chem Lett. 2016;26:1762–1766. doi: 10.1016/j.bmcl.2016.02.039. [DOI] [PubMed] [Google Scholar]
- 162.Zhai Y., Ma Y., Ma F., Nie Q., Ren X., Wang Y., et al. Structure–activity relationship study of peptidomimetic aldehydes as enterovirus 71 3C protease inhibitors. Eur J Med Chem. 2016;124:559–573. doi: 10.1016/j.ejmech.2016.08.064. [DOI] [PubMed] [Google Scholar]
- 163.Steuten K., Kim H., Widen J.C., Babin B.M., Onguka O., Lovell S., et al. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect Dis. 2021;7:1457–1468. doi: 10.1021/acsinfecdis.0c00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X., et al. An expedited approach towards the rationale design of non-covalent SARS-CoV-2 main protease inhibitors with in vitro antiviral activity. J Med Chem. 2022;65:2848–2865. doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yin Z., Chen Y.L., Schul W., Wang Q.Y., Gu F., Duraiswamy J., et al. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A. 2009;106:20435–20439. doi: 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Deng C.L., Yeo H., Ye H.Q., Liu S.Q., Shang B.D., Gong P., et al. Inhibition of enterovirus 71 by adenosine analog NITD008. J Virol. 2014;88:11915–11923. doi: 10.1128/JVI.01207-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tosh D.K., Toti K.S., Hurst B.L., Julander J.G., Jacobson K.A. Structure activity relationship of novel antiviral nucleosides against Enterovirus A71. Bioorg Med Chem Lett. 2020;30:127599. doi: 10.1016/j.bmcl.2020.127599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yin W., Mao C., Luan X., Shen D.D., Shen Q., Su H., et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368:1499–1504. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen T.C., Chang H.Y., Lin P.F., Chern J.H., Hsu J.T., Chang C.Y., et al. Novel antiviral agent DTriP-22 targets RNA-dependent RNA polymerase of enterovirus 71. Antimicrob Agents Chemother. 2009;53:2740–2747. doi: 10.1128/AAC.00101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hung H.C., Chen T.C., Fang M.Y., Yen K.J., Shih S.R., Hsu J.T., et al. Inhibition of enterovirus 71 replication and the viral 3D polymerase by aurintricarboxylic acid. J Antimicrob Chemother. 2010;65:676–683. doi: 10.1093/jac/dkp502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.van der Linden L., Vives-Adrian L., Selisko B., Ferrer-Orta C., Liu X., Lanke K., et al. The RNA template channel of the RNA-dependent RNA polymerase as a target for development of antiviral therapy of multiple genera within a virus family. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1004733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Velu A.B., Chen G.W., Hsieh P.T., Horng J.T., Hsu J.T., Hsieh H.P., et al. BPR-3P0128 inhibits RNA-dependent RNA polymerase elongation and VPg uridylylation activities of Enterovirus 71. Antivir Res. 2014;112:18–25. doi: 10.1016/j.antiviral.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 173.Yates M.K., Seley-Radtke K.L. The evolution of antiviral nucleoside analogues: a review for chemists and non-chemists. Part II: complex modifications to the nucleoside scaffold. Antivir Res. 2019;162:5–21. doi: 10.1016/j.antiviral.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Seley-Radtke K.L., Yates M.K. The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antivir Res. 2018;154:66–86. doi: 10.1016/j.antiviral.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lai M.C., Chen H.H., Xu P., Wang R.Y.L. Translation control of Enterovirus A71 gene expression. J Biomed Sci. 2020;27:22. doi: 10.1186/s12929-019-0607-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dai W., Bi J., Li F., Wang S., Huang X., Meng X., et al. Antiviral efficacy of flavonoids against enterovirus 71 infection in vitro and in newborn mice. Viruses. 2019;11:625. doi: 10.3390/v11070625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hou H.Y., Lu W.W., Wu K.Y., Lin C.W., Kung S.H. Idarubicin is a broad-spectrum enterovirus replication inhibitor that selectively targets the virus internal ribosomal entry site. J Gen Virol. 2016;97:1122–1133. doi: 10.1099/jgv.0.000431. [DOI] [PubMed] [Google Scholar]
- 178.Tan Q., Zhu Y., Li J., Chen Z., Han G.W., Kufareva I., et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shih S.R., Stollar V., Li M.L. Host factors in enterovirus 71 replication. J Virol. 2011;85:9658–9666. doi: 10.1128/JVI.05063-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lim Z.Q., Ng Q.Y., Ng J.W.Q., Mahendran V., Alonso S. Recent progress and challenges in drug development to fight hand, foot and mouth disease. Expet Opin Drug Discov. 2020;15:359–371. doi: 10.1080/17460441.2019.1659241. [DOI] [PubMed] [Google Scholar]
- 181.Arita M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol Immunol. 2014;58:239–256. doi: 10.1111/1348-0421.12144. [DOI] [PubMed] [Google Scholar]
- 182.Roulin P.S., Lotzerich M., Torta F., Tanner L.B., van Kuppeveld F.J., Wenk M.R., et al. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER–Golgi interface. Cell Host Microbe. 2014;16:677–690. doi: 10.1016/j.chom.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 183.Burgett A.W., Poulsen T.B., Wangkanont K., Anderson D.R., Kikuchi C., Shimada K., et al. Natural products reveal cancer cell dependence on oxysterol-binding proteins. Nat Chem Biol. 2011;7:639–647. doi: 10.1038/nchembio.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Roberts B.L., Severance Z.C., Bensen R.C., Le A.T., Kothapalli N.R., Nunez J.I., et al. Transient compound treatment induces a multigenerational reduction of oxysterol-binding protein (OSBP) levels and prophylactic antiviral activity. ACS Chem Biol. 2019;14:276–287. doi: 10.1021/acschembio.8b00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang L., Das P., Schmolke M., Manicassamy B., Wang Y., Deng X., et al. Inhibition of pyrimidine synthesis reverses viral virulence factor-mediated block of mRNA nuclear export. J Cell Biol. 2012;196:315–326. doi: 10.1083/jcb.201107058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hoffmann H.H., Kunz A., Simon V.A., Palese P., Shaw M.L. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci U S A. 2011;108:5777–5782. doi: 10.1073/pnas.1101143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cheung N.N., Lai K.K., Dai J., Kok K.H., Chen H., Chan K.H., et al. Broad-spectrum inhibition of common respiratory RNA viruses by a pyrimidine synthesis inhibitor with involvement of the host antiviral response. J Gen Virol. 2017;98:946–954. doi: 10.1099/jgv.0.000758. [DOI] [PubMed] [Google Scholar]
- 188.Earley D.F., Bailly B., Maggioni A., Kundur A.R., Thomson R.J., Chang C.W., et al. Efficient blocking of enterovirus 71 infection by heparan sulfate analogues acting as decoy receptors. ACS Infect Dis. 2019;5:1708–1717. doi: 10.1021/acsinfecdis.9b00070. [DOI] [PubMed] [Google Scholar]
- 189.Pourianfar H.R., Poh C.L., Fecondo J., Grollo L. In vitro evaluation of the antiviral activity of heparan sulfate mimetic compounds against Enterovirus 71. Virus Res. 2012;169:22–29. doi: 10.1016/j.virusres.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 190.Tan Y.W., Hong W.J., Chu J.J. Inhibition of enterovirus VP4 myristoylation is a potential antiviral strategy for hand, foot and mouth disease. Antivir Res. 2016;133:191–195. doi: 10.1016/j.antiviral.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 191.Weng T.Y., Chen L.C., Shyu H.W., Chen S.H., Wang J.R., Yu C.K., et al. Lactoferrin inhibits enterovirus 71 infection by binding to VP1 protein and host cells. Antivir Res. 2005;67:31–37. doi: 10.1016/j.antiviral.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 192.Yuan S., Chu H., Huang J., Zhao X., Ye Z.W., Lai P.M., et al. Viruses harness YxxO motif to interact with host AP2M1 for replication: a vulnerable broad-spectrum antiviral target. Sci Adv. 2020;6 doi: 10.1126/sciadv.aba7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hao T., Li Y., Fan S., Li W., Wang S., Li S., et al. Design, synthesis and pharmacological evaluation of a novel mTOR-targeted anti-EV71 agent. Eur J Med Chem. 2019;175:172–186. doi: 10.1016/j.ejmech.2019.04.048. [DOI] [PubMed] [Google Scholar]
- 194.Zhang Q., Zhao B., Chen X., Song N., Wu J., Li G., et al. GS-9620 inhibits enterovirus 71 replication mainly through the NF-kappaB and PI3K–AKT signaling pathways. Antivir Res. 2018;153:39–48. doi: 10.1016/j.antiviral.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 195.Frausto S.D., Lee E., Tang H. Cyclophilins as modulators of viral replication. Viruses. 2013;5:1684–1701. doi: 10.3390/v5071684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Peel M., Scribner A. Cyclophilin inhibitors as antiviral agents. Bioorg Med Chem Lett. 2013;23:4485–4492. doi: 10.1016/j.bmcl.2013.05.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhou D., Mei Q., Li J., He H. Cyclophilin A and viral infections. Biochem Biophys Res Commun. 2012;424:647–650. doi: 10.1016/j.bbrc.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bienkowska-Haba M., Patel H.D., Sapp M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ma C., Li F., Musharrafieh R.G., Wang J. Discovery of cyclosporine A and its analogs as broad-spectrum anti-influenza drugs with a high in vitro genetic barrier of drug resistance. Antivir Res. 2016;133:62–72. doi: 10.1016/j.antiviral.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Qing J., Wang Y., Sun Y., Huang J., Yan W., Wang J., et al. Cyclophilin A associates with enterovirus-71 virus capsid and plays an essential role in viral infection as an uncoating regulator. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yan W., Qing J., Mei H., Nong J., Huang J., Zhu J., et al. Identification, synthesis and pharmacological evaluation of novel anti-EV71 agents via cyclophilin A inhibition. Bioorg Med Chem Lett. 2015;25:5682–5686. doi: 10.1016/j.bmcl.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 202.Zeng S., Meng X., Huang Q., Lei N., Zeng L., Jiang X., et al. Spiramycin and azithromycin, safe for administration to children, exert antiviral activity against enterovirus A71 in vitro and in vivo. Int J Antimicrob Agents. 2019;53:362–369. doi: 10.1016/j.ijantimicag.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 203.Tan Y.W., Yam W.K., Sun J., Chu J.J.H. An evaluation of chloroquine as a broad-acting antiviral against hand, foot and mouth disease. Antivir Res. 2018;149:143–149. doi: 10.1016/j.antiviral.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 204.Yang Y., Xiu J., Liu J., Zhang L., Li X., Xu Y., et al. Chebulagic acid, a hydrolyzable tannin, exhibited antiviral activity in vitro and in vivo against human enterovirus 71. Int J Mol Sci. 2013;14:9618–9627. doi: 10.3390/ijms14059618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Liu Z., Xia S., Wang X., Lan Q., Li P., Xu W., et al. Sodium copper chlorophyllin is highly effective against enterovirus (EV) A71 infection by blocking its entry into the host cell. ACS Infect Dis. 2020;6:882–890. doi: 10.1021/acsinfecdis.0c00096. [DOI] [PubMed] [Google Scholar]
- 206.Li Z., Yao F., Xue G., Xu Y., Niu J., Cui M., et al. Antiviral effects of simeprevir on multiple viruses. Antivir Res. 2019;172:104607. doi: 10.1016/j.antiviral.2019.104607. [DOI] [PubMed] [Google Scholar]
- 207.Yang Y., Zhang L., Fan X., Qin C., Liu J. Antiviral effect of geraniin on human enterovirus 71 in vitro and in vivo. Bioorg Med Chem Lett. 2012;22:2209–2211. doi: 10.1016/j.bmcl.2012.01.102. [DOI] [PubMed] [Google Scholar]
- 208.Yang Y., Xiu J., Zhang L., Qin C., Liu J. Antiviral activity of punicalagin toward human enterovirus 71 in vitro and in vivo. Phytomedicine. 2012;20:67–70. doi: 10.1016/j.phymed.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 209.Liu J., Yang Y., Xu Y., Ma C., Qin C., Zhang L. Lycorine reduces mortality of human enterovirus 71-infected mice by inhibiting virus replication. Virol J. 2011;8:483. doi: 10.1186/1743-422X-8-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yang Y., Xiu J., Zhang X., Zhang L., Yan K., Qin C., et al. Antiviral effect of matrine against human enterovirus 71. Molecules. 2012;17:10370–10376. doi: 10.3390/molecules170910370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang H., Guo T., Yang Y., Yu L., Pan X., Li Y. Lycorine derivative LY-55 inhibits EV71 and CVA16 replication through downregulating autophagy. Front Cell Infect Microbiol. 2019;9:277. doi: 10.3389/fcimb.2019.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tsou Y.L., Lin Y.W., Chang H.W., Lin H.Y., Shao H.Y., Yu S.L., et al. Heat shock protein 90: role in enterovirus 71 entry and assembly and potential target for therapy. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Too I.H.K., Bonne I., Tan E.L., Chu J.J.H., Alonso S. Prohibitin plays a critical role in Enterovirus 71 neuropathogenesis. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1006778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Qin Y., Lin L., Chen Y., Wu S., Si X., Wu H., et al. Curcumin inhibits the replication of enterovirus 71 in vitro. Acta Pharm Sin B. 2014;4:284–294. doi: 10.1016/j.apsb.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wang L., Yang R., Yuan B., Liu Y., Liu C. The antiviral and antimicrobial activities of licorice, a widely-used Chinese herb. Acta Pharm Sin B. 2015;5:310–315. doi: 10.1016/j.apsb.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Shang B., Deng C., Ye H., Xu W., Yuan Z., Shi P.Y., et al. Development and characterization of a stable eGFP enterovirus 71 for antiviral screening. Antivir Res. 2013;97:198–205. doi: 10.1016/j.antiviral.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 217.Xu L.L., Shan C., Deng C.L., Li X.D., Shang B.D., Ye H.Q., et al. Development of a stable Gaussia luciferase enterovirus 71 reporter virus. J Virol Methods. 2015;219:62–66. doi: 10.1016/j.jviromet.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 218.Caine E.A., Osorio J.E. In vivo imaging with bioluminescent enterovirus 71 allows for real-time visualization of tissue tropism and viral spread. J Virol. 2017;91 doi: 10.1128/JVI.01759-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Drummond C.G., Bolock A.M., Ma C., Luke C.J., Good M., Coyne C.B. Enteroviruses infect human enteroids and induce antiviral signaling in a cell lineage-specific manner. Proc Natl Acad Sci U S A. 2017;114:1672–1677. doi: 10.1073/pnas.1617363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tsang J.O.-L., Zhou J., Zhao X., Li C., Zou Z., Yin F., et al. Development of three-dimensional human intestinal organoids as a physiologically relevant model for characterizing the viral replication kinetics and antiviral susceptibility of enteroviruses. Biomedicines. 2021;9:88. doi: 10.3390/biomedicines9010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhao X., Li C., Liu X., Chiu M.C., Wang D., Wei Y., et al. Human intestinal organoids recapitulate enteric infections of enterovirus and coronavirus. Stem Cell Reports. 2021;16:493–504. doi: 10.1016/j.stemcr.2021.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hayden F.G., Herrington D.T., Coats T.L., Kim K., Cooper E.C., Villano S.A., et al. Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: results of 2 double-blind, randomized, placebo-controlled trials. Clin Infect Dis. 2003;36:1523–1532. doi: 10.1086/375069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lamarche M.J., Borawski J., Bose A., Capacci-Daniel C., Colvin R., Dennehy M., et al. Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob Agents Chemother. 2012;56:5149–5156. doi: 10.1128/AAC.00946-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hung H.C., Wang H.C., Shih S.R., Teng I.F., Tseng C.P., Hsu J.T. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis. 2011;203:1784–1790. doi: 10.1093/infdis/jir174. [DOI] [PubMed] [Google Scholar]
- 225.Zhan P., Pannecouque C., De Clercq E., Liu X. Anti-HIV drug discovery and development: current innovations and future trends. J Med Chem. 2016;59:2849–2878. doi: 10.1021/acs.jmedchem.5b00497. [DOI] [PubMed] [Google Scholar]
- 226.Diep J., Ooi Y.S., Wilkinson A.W., Peters C.E., Foy E., Johnson J.R., et al. Enterovirus pathogenesis requires the host methyltransferase SETD3. Nat Microbiol. 2019;4:2523–2537. doi: 10.1038/s41564-019-0551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]