

Abstract
The mechanism of sphingosine-1-phosphate (S1P)-mediated phagocytosis remains unknown. Here, we found that S1P or FTY720 (an analog of S1P) promoted microglial phagocytosis in stroke independent of S1PRs. First, we used computer simulation of molecular docking to predict that S1P might be a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). Next, microscale thermophoresis (MST), surface plasmon resonance (SPR) and liquid chromatography–tandem mass spectrometry (LC–MS/MS) were performed to reveal that S1P was a novel TREM2 ligand. Then, we confirmed the pro-phagocytosis of S1P targeting in Trem2-Dap12 transfected CHO cells and TREM2 knockdown microglia. Point mutation analysis showed that D104 was the critical binding residue. Trem2−/− mice were used to demonstrate the role of S1P-induced phagocytosis targeting on TREM2 in protecting against ischemic brain injury. Finally, further studies revealed that apolipoprotein E (APOE) loaded with S1P was released by microglia and bound to apoptotic neurons via LDL receptor related protein 1B (LRP1B) and thereby induced microglia to phagocytose apoptotic neurons. Overall, the present work reveals for the first time that S1P acts as a novel endogenous ligand of TREM2 to effectively promote microglial phagocytosis. Our findings provide a new lead compound for developing immunomodulator targeting on TREM2.
KEY WORDS: S1P, TREM2, Microglia, Phagocytosis, Stroke, APOE, LRP1B, Apoptotic neurons
Graphical abstract
S1P interacts with TREM2 to increase microglial phagocytosis and exerts neuroprotective effect in ischemic stroke. Apoptotic neurons showed increased membrane LRP1B expression to mediate S1P-TREM2 induced clearance.
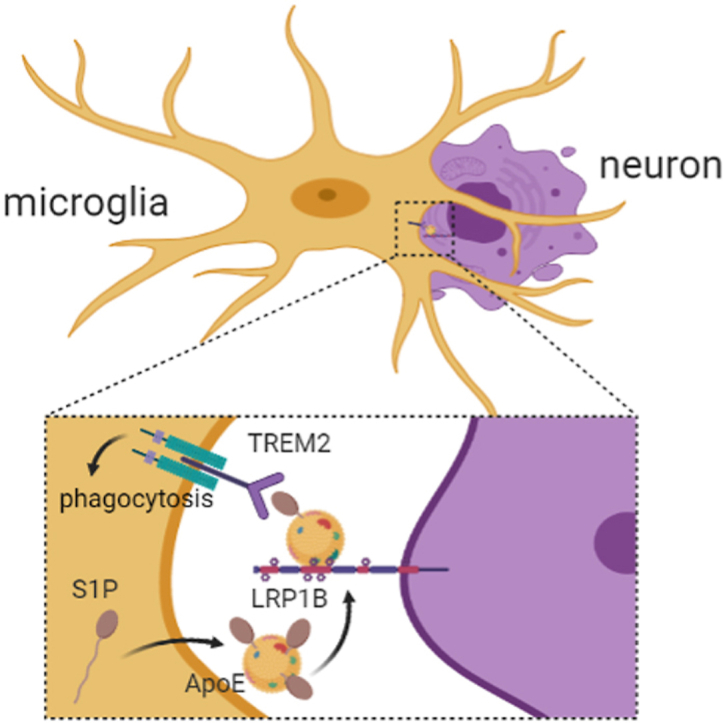
1. Introduction
Sphingosine is one of the most important sphingolipid metabolites, named after the Sphinx for its mysterious features. Phosphorylation of sphingosine forms the pleiotropic and bioactive lipid sphingosine-1-phosphate (S1P, Fig. 1A). Traditionally, S1P acts not only as an intracellular second messenger, but also as an extracellular first messenger in both an autocrine and paracrine manner, via binding with S1P receptors (S1PRs) of which there are currently five known subtypes (S1PR1–5)1, 2, 3. It has been revealed that S1P has a wide range of biological functions including regulating cell differentiation, survival, proliferation, angiogenesis and immune modulation4,5. A few studies have suggested that S1P might regulate microglial phagocytosis6,7. However, the involved mechanisms remain unknown.
Figure 1.

FTY720 promoted microglia phagocytosing neuronal debris in the penumbra of MCAO rats. (A) Chemical structure of S1P, FTY720 and FTY720P. (B) Immunofluorescence staining of NeuN+neuronal debris (green) and IBA1+microglia (red) in Sham, MCAO group or MCAO+FTY720 group. Arrows indicate neurons enwrapping microglia. The percentage of phagocytic microglia (C) and phagocytosed neuronal debris (D) were analyzed; n = 4 for each group, ∗P < 0.05 vs. corresponding group at 24 h after MCAO; #P < 0.05 vs. MCAO group. (E) Immunofluorescence staining of CD68 (green) and IBA1+microglia (red) in Sham, MCAO group and MCAO + FTY720 group. (F) Quantification of CD68 expression, indicated by ratio of CD68+ area to IBA1+ area; n = 4–5, ∗P < 0.05 and ∗∗∗P < 0.001 vs. corresponding group at 24 h after MCAO; ##P < 0.01 and ###P < 0.001 vs. MCAO group. Values are presented as mean ± SD and analyzed by two-way ANOVA and Bonferroni's test. (G) Microglial phagocytosis observed via two-photon microscopy in the penumbra at 48 h after MCAO; n = 3 for each group. (H) TTC staining of brain sections from MCAO or MCAO + FTY720 rats. (I) Quantification analysis of infarct volume; n = 8, #P < 0.05 vs. MCAO group. (J) Neurological deficits via Bederson's scale scores were analyzed in MCAO or MCAO+FTY720 rats (J, n = 8). Values are expressed as mean ± SD and analyzed by two-way ANOVA and Bonferroni's test.
As an important phagocytosis mediator, triggering receptor expressed on myeloid cells 2 (TREM2) is a cell surface receptor of the immunoglobin superfamily. It consists of an ectodomain, a transmembrane region and a short cytoplasmic tail, which transmit downstream signal by coupling with DNAX-activating protein of 12 kDa (DAP12)8,9. Expressed on osteoclast, macrophage, dendritic cell, and exclusively on microglia in the brain, TREM2 primarily participates in phagocytosis10, 11, 12, 13. Till now, the endogenous ligand of TREM2 has not been found. However, a diverse set of potential TREM2 ligands have been proposed, such as bacteria, poly-anionic14,15, apolipoprotein E (APOE) and phospholipids16. ApoE participates in the transition to disease associated microglia (DAM)17, which is TREM2 dependent in phagocytosis enhancement. Moreover, lipidated APOE shows higher affinity to interact with TREM2 and facilitates amyloid β (Aβ) clearance18. As a member of sphingolipid, S1P may be involved in the APOE–TREM2 interaction to regulate microglial phagocytosis.
Ischemic stroke is a leading cause of mortality and disability. In addition to thrombolysis and thrombectomy in the acute stage, there is no effective therapeutic strategy19,20. Disruption of the regional blood supply initiates ischemic cascade leading to neuronal dysfunction and subsequent cell death21,22. Brain edema and inflammatory response in the sub-acute phase contribute to the secondary injury21,23. The damaged and dead neurons could release nucleic acids, proteins and lipids, which induce neuroinflammation and exacerbate brain damage24. In the intracerebral hemorrhagic stroke, blood products introduced from hematoma such as hemoglobin and iron can exacerbate neuronal death25,26. Thus, promoting phagocytic clearance of neurotoxic cellular debris is beneficial to recovery after stroke and could serve as a promising therapeutic strategy.
In the present work, we found that the pro-phagocytic function of S1P and a S1P analog FTY720 was not dependent on S1PRs. Since S1P belongs to phospholipid and can regulate microglial phagocytosis, we speculated and used computer simulation of molecular docking to predict that S1P might target on TREM2 to exert pro-phagocytosis effect. We revealed that S1P and FTY720P could bind to TREM2, promote microglial phagocytosis, and thereby exert neuroprotection in ischemic and hemorrhagic stroke.
2. Materials and methods
2.1. Cells, media and reagents
Primary microglia were cultured with Dulbecco's minimum essential medium (ThermoFisher Scientific, NY, USA) containing 10% fetal bovine serum (Gibco, ThermoFisher Scientific), penicillin at 100 U/mL (Gibco) and streptomycin at 100 μg/mL (Gibco) at 37 °C in a humidified 5% CO2–95% air atmosphere. Primary neurons were cultured in Neuralbasal (Gibco) supplemented with 2% B27 (Invitrogen, ThermoFisher Scientific, CA, USA). S1P and sphingosine ELISA kits were used to detect the expression of S1P and sphingosine, which were purchased from Cloud-Clone Corp. (Wuhan, China). Ceramide ELISA kit used to detect the expression of ceramide, which was purchased from Enzyme-Linked Biotechnology (Shanghai, China). Rabbit anti-IBA1 monoclonal antibody, mouse anti-NeuN monoclonal antibody and mouse anti-CD68 monoclonal antibody were obtained from Wako (1:500, Osaka, Japan), Millipore (1:400, Billerica, MA, USA) and Dako (1:200, Glostrup, Denmark), respectively. Alexa Fluor-488-conjugated goat anti-mouse secondary antibody, Alexa Fluor-546-conjugated donkey anti-rabbit secondary antibody, Alexa Fluor-647-conjugated goat anti-rabbit secondary antibody were obtained from Invitrogen/Thermo Fisher Scientific (NY, USA). The plasmid encoding TREM2 and APOE were purchased from Public Protein/Plasmid Library (Nanjing, China). Small interfering RNA (siRNA) against TREM2 and its negative control (siRNA) were synthesized by Genepharma (Shanghai, China). The TUNEL Apoptosis Assay Kit was purchased from KeyGEN bioTECH (Jiangsu, China).
2.2. Animal model and experimental protocol
Male Sprague–Dawley (SD) rats (260 ± 10 g, Animal Core Facility of Nanjing Medical University, Nanjing, China), C57BL/6J mice (20–25 g, Animal Core Facility of Nanjing Medical University) and Trem2−/− mice (20–25 g, Cyagen Biosciences) were maintained with ad libitum access to standard fodder and water in a well-ventilated environment with approximately 25 °C, 50%–60% humidity and a standard 12 h light/dark cycle. Animals were assigned to Sham group, Model group and FTY720-treated group, randomly. Focal cerebral ischemic stroke was induced as described previously27 and rats were intraperitoneally injected with FTY720 (2 mg/kg, Selleck chemicals) or normal saline at 1 h after MCAO, once a day for two days. The protocols were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University.
2.3. TTC staining
Brains were removed and cut into cerebral coronary slices of 2 mm at 48 h after surgery, which were incubated in TTC (2,3,5-triphenyltetrazolium chloride, 1%, sigma) for 5 min at 37 °C and then, placed in 4% paraformaldehyde solution (pH 7.4) at 4 °C to fix up overnight. Photographies were taken and infarct size was analyzed by ImageJ software.
2.4. Neurological deficit scoring
Neurological deficit of MCAO rats was classified and scored referring to Bederson's scale scores at 24 and 48 h after surgery. When following conditions occurred, corresponding rats were excluded and supplemented: 0 score, massive bleeding during surgery, postoperative respiratory abnormality, early mortality and subarachnoid hemorrhage.
2.5. Immunofluorescence and two-photon microscope
After anesthetizing, rats were transcardially perfused with 37 °C saline followed by 4% paraformaldehyde (PFA). The brains were removed and postfixed in 4% PFA for 24–48 h at 4 °C. Then, they were embedded in paraffin and sectioned coronally at an interval of 5 mm. Coronal sections were processed subsequently as following steps: paraffin melting for 60 min at 60 °C, deparaffinage and rehydration. 0.01 mol/L citrate buffer solution was heated to 92–98 °C with microwave oven simultaneously, slices were placed into the solution for retrieval for 15 min and taken out to cool down naturally to room temperature. For the sake of blocking non-specific antigen, slides were incubated with 10% normal goat serum for 1 h at room temperature. Primary antibodies were incubated overnight at 4 °C at the following dilutions: rabbit anti-IBA1 monoclonal antibody (1:500), mouse anti-NeuN monoclonal antibody (1:400) and mouse anti-CD68 monoclonal antibody (1:200), followed by incubation with secondary antibody: Alexa Fluor-488-conjugated goat anti-mouse, Alexa Fluor-546-conjugated donkey anti-rabbit, Alexa Fluor-647-conjugated goat anti-rabbit secondary antibody at a 1:1000 dilution for 1 h. After washing, coronal sections or cells were counterstained with Hoechst 33342 for 15 min and washed thrice with PBS for 5 min each time. The stained sections or cells were visualized and photographed using fluorescent microscope (Zeiss Axio Vert A1) or two-photon microscope (Zeiss LSM880 with NLO & Airyscan).
2.6. Primary microglia and neuron co-culture system
Primary microglial and neuron cultures were performed as previously described and were isolated from 1- to 2-day-old postnatal Sprague–Dawley rats. All of the animal operational procedures were performed in accordance with the Institution for Animal Care and Use Committee and approved by Animal Core Facility of Nanjing Medical University (Nanjing, China). Briefly, primary cultures of glial cells were obtained from the cerebral cortices, which were earlier digested by 0.25% trypsin/EDTA (Gibco) at 37 °C for 10 min and seeded into poly-d-lysine-coated (0.1 mg/mL; Sigma Chemical, St. Louis, MO, USA) 25-cm2 culture flasks. The microglia cultures were maintained for 7 days.
For in vitro experiments, microglial cells were separated from the mixed primary culture by flapped for 15 min and then plated (2 × 104 cells per well) in neuron (2 × 105 cells per well in a 24-well plate) culture vessels with DMEM containing 10% FBS:Neuralbasal = 1:3. The cells were co-cultured for further treatment the following day.
2.7. Transfection: Knock-down of TREM2
Microglia cells in the co-culture system or in culture vessels (6 × 105 cells per well in a 6-well plate) were transfected using siRNA-mate (Genepharma, Shanghai, China) according to the manufacturer's instructions.
2.8. Phagocytosis assay
Trem2-Dap12 cDNA was generated as previously described13. CHO cells were transfected with the construct to generate a stable cell line that express TREM2-DAP12 chimera. The parental cells or transfected CHO cells were seeded in 24-well plates at the density of 1 × 104 cells per well and incubated overnight. After two washes with PBS, cells were dyed with 5 mmol/L Cell Tracker™ Green (ThermoFisher Scientific, NY, USA) for 20 min, followed by washing and incubation in Opti-MEM medium containing 3 μL/100 μL of pHrodo Red zymosan bioparticles and/or 20 μmol/L S1P and/or 10 μg/mL LPS, or S1P and 2 μmol/L CytoD. The treated cells were examined at 2 and 4 h by fluorescent microscope.
2.9. Oxygen and glucose deprivation/reperfusion (OGD/R)
To initiate OGD, the culture medium was removed, rinsed with phosphate buffered saline (PBS) and replaced with Opti-MEM (Gibco). The cultured cells were placed into the hypoxia chamber (Mitsubishi Gas Chemical Company, Japan) with a premixed gas (1% O2, 94% N2, 5% CO2) at 37 °C for 3 h. After OGD, the cells were perfused by 10% FBS-DMEM medium or 10% FBS-DMEM medium:neurobasal = 1:3 and transferred to a 5% CO2–95% O2 air incubator for relative time. Control cells were incubated under normal conditions throughout the procedure.
2.10. LC–MS/MS
BV2 cells were grown to 80%–90% confluency in two dishes and treated with 20 μmol/L S1P or not. After 2 h, the cells were washed thrice with PBS and collected with 400 μL homogenate buffer per dish (250 mmol/L sucrose, 10 mmol/L HEPES, 1 mmol/L EDTA, 1 mmol/L DTT, NaOH, pH to 7.4). The collected cells were frozen at −80 °C and underwent 5 freezing and thaw cycles to facilitate lysis. Then the buffer was homogenized further with bead mill. After centrifugation at 12,000 rpm for 15 min at 4 °C (D3024R, high speed refrigerated micro centrifuge, Scilogex, USA), the protein of cell lysates was acquired and incubated with 3 μL anti-TREM2 (Abcam, ab125117) overnight at 4 °C on a rotating device, followed by adding 100 μL proteinA+G beads/mL lysate overnight to capture the conjugated polymers at 4 °C on a rotating device. Immunoprecipitates were collected by centrifugation at 8000 rpm for 2 min at 4 °C (D3024R, high speed refrigerated micro centrifuge, Scilogex, USA) and washed thrice with 1 mL homogenate buffer, then resuspended in 50 mmol/L NH4HCO3 twice the volume of beads. After boiling, the supernatant was added 200 μL chromatographic grade ethanol, blended and centrifugated at 12,000 rpm, 4 °C for 30 min to discard precipitation (D3024R, high speed refrigerated micro centrifuge, Scilogex, USA). The solution obtained was purified and concentrated with Amicon Ultra–0.5 mL Centrifugal Filter Units (Millipore, Merck KGaA, Darmstadt, Germany), and detected by Analysis and Testing Center of Nanjing Medical University.
2.11. Preparation of recombinant TREM2 and ApoE
TREM2 expression for binding assay was performed as described previously28. In brief, TREM2 was expressed in freestyle 293F cells and purified using Ni-NTA resin and AKTA for further purification. The protein was dissolved in PBS and immediately used to measure binding affinity rapidly. The pET-24a(+)-human APOE was purchased from Public Protein/Plasmid Library. The constructs were transformed into Escherichia coli BL21(DE3) and the fusion proteins were expressed by addition of IPTG till final concentration of 1 mmol/L for 8 h, followed by extraction and purification with His-tag Protein Purification Kit (Beyotime Biotechnology, Shanghai, China) and concentration with Amicon Ultra–15 mL Centrifugal Filter Units (Millipore, Merck KGaA, Darmstadt, Germany).
2.12. Microscale thermophoresis (MST)
The above-mentioned obtained fusion proteins were labelled using Monolith His-Tag Labeling Kit (NanoTemper Technologies, Munich, Germany). The recombinant TREM2 and S1P were prepared at the concentration of 250 nmol/L and 2 mmol/L, respectively. The binding affinity was detected with Monolith NT.115 (NanoTemper Technologies, Munich, Germany).
2.13. Surface plasmon resonance (SPR)
The obtained fusion proteins and S1P were prepared as described above. The fusion proteins were attached to Sensor chip NTA (Biacore). The binding affinity was detected with GE Biacore T200 (GE, USA).
2.14. Apoptosis assay
Apoptosis was evaluated using TUNEL Apoptosis Assay Kit and performed following the manufacturer's instructions.
2.15. Pull-down assay
2 × 106 primary neurons were adjusted to 400 μL using membrane protein extraction kit (ThermoFisher Scientific). Approximately 200 μL recombinant ApoE conjugated to agarose beads (Cube Biotech, Germany) was washed thrice with PBS. The membrane protein extract or solvent was incubated with beads overnight at 4 °C with constant gentle rotation. Protein bound beads were washed by washing buffer (10 mmol/L HEPES pH 7.4, 150 mmol/L NaCl, 0.25% NP40) for five times. Washed beads were added to solvent and 6 × loading buffer, followed by heating at 100 °C for 10 min. Beads were centrifuged at 5000 rpm for 5 min (D3024R, high speed refrigerated micro centrifuge, Scilogex, USA) and supernatants were separated by SDS-polyacrylamide gel electrophoresis.
2.16. Statistical analysis
The obtained data are presented as mean ± standard error of mean (SEM) of at least two independent experiments. The relationship between two factors was analyzed using Pearson correlation analysis and groups were compared using a two-way ANOVA with post hoc Bonferroni's multiple comparisons test. All of the data were analyzed with GraphPad Prism 6.0 software. A value of P < 0.05 indicated that the difference was statistically significant.
3. Results
3.1. FTY720 promoted microglial phagocytosis in MCAO rats
We first investigated the pro-phagocytic effects of FTY720 in MCAO rats via colabeling with NeuN to identify neuron and with IBA1 to identify microglia. As a marker of macrophage-specific lysosome, expression of CD68 can reflect the phagocytic ability of microglia. We found that supplementing with FTY720, an analog of S1P after phosphorylation, could promote microglia to phagocytose neuronal debris indicated by the increased proportions of (IBA1++NeuN+) cells/total IBA1+ cells (Fig. 1B and C), (IBA1++NeuN+) cells/total NeuN+ cells (Fig. 1B and D) and CD68+ area per microglia (Fig. 1E and F) compared with MCAO group. We observed an increase in the number of microglia that phagocytosing neuronal debris in the penumbra at 48 h after FTY720 treatment via two-photon microscope, confirming that FTY720 significantly enhanced microglial phagocytosis after MCAO (Fig. 1G). These data demonstrate the pro-phagocytic function of FTY720.
To determine the neuroprotective effect of enhanced microglial phagocytosis, we detected the infarct volume and neurological deficit. As shown in Fig. 1H and I, FTY720 treatment significantly decreased infarct volume in MCAO rats. Neurological deficit (Fig. 1J) was also alleviated in MCAO + FTY720 group. These data suggested that microglial phagocytosis enhancement reduced ischemic injury.
3.2. S1P or FTY720P enhanced phagocytosis via TREM2 rather than S1PRs
In order to determine whether S1PRs mediate the pro-phagocytic effect of S1P and FTY720 on microglia, we knocked down S1PR2 and S1PR4, the two major subtypes expressed on the microglia (Supporting Information Fig. S1A). Neither S1PR2 nor S1PR4 knock-down reduced the CD68+ area enhanced by S1P. The result shows that knockdown of S1PR2 or S1PR4 did not affect the pro-phagocytic effect of S1P (Fig. S1B–S1D), indicating certain receptor other than S1PRs mediated microglial phagocytosis.
Since S1P shares structural similarities with phospholipids, we speculated that S1P might be an endogenous ligand for TREM2. Hence, we used computer simulation of molecular docking to predict the potential interactions between S1P or FTY720P (FTY720P is structurally similar to S1P) and TREM2. The predicated results show that D104 of human TREM2 (hTREM2) could form critical hydrogen bond with S1P and FTY720P (Fig. 2A), which is the key of the interaction. S81, T82, Y108 and Y50 also participate in the hydrogen bond network formation.
Figure 2.

S1P is an endogenous TREM2 ligand binding at D104. (A) Molecular docking results showing the potential interaction between TREM2 and S1P or FTY720P. (B) LC–MS/MS spectra of TREM2 immunoprecipitated S1P of BV2 cell extracts after complete medium or 1 h 20 μmol/L S1P treatment. The binding of fluorescently labelled hTREM2 (C), rTREM2 (E), mTREM2 (F) to S1P was analyzed with MST. S1P was titrated from 30.5 μmol/L to 1 mmol/L. The change in the thermophoretic signal led to a KD = 62.59 ± 11.93, 56.80 ± 13.96, 64.62 ± 16.64 μmol/L, respectively. (D) The binding profiles of S1P to different concentrations of hTREM2 were generated by SPR assay. (G)–(I) The binding of fluorescently labelled hTREM2, rTREM2, mTREM2 to FTY720p was analyzed with MST. FTY720P was titrated from 1.53 nmol/L to 50 μmol/L. The change in the thermophoretic signal leads to a KD = 6.75 ± 1.80, 7.17 ± 2.21, 72.60 ± 21.02 μmol/L, respectively. (J) MST result showing no interaction between hTREM2 and FTY720. The binding of fluorescently labelled TREM2 D104A to S1P (K) or FTY720P (L) was analyzed with MST. S1P was titrated from 30.5 nmol/L to 1 mmol/L and FTY720P was titrated from 1.53 nmol/L to 500 μmol/L. The change in the thermophoretic signal led to a KD = 593.23 ± 97.58 μmol/L for S1P binding to TREM2 D104A. The binding of FTY720P to TREM2 D104A was undetected.
Then, we investigated the interaction between S1P/FTY720P and TREM2 via LC–MS/MS (Fig. 2B), microscale thermophoresis (MST, Fig. 2C, E, and F) and surface plasmon resonance (SPR, Fig. 2D). S1P treatment resulted in dramatically more S1P binding at TREM2 detected by LC–MS/MS, and normal cultured cells without S1P treatment showed much less binding of S1P (Fig. 2B), demonstrating the S1P–TREM2 interaction. The interaction was further confirmed via MST and S1P-binding affinities of S1P to human TREM2 (hTREM2), rat TREM2 (rTREM2) and mouse TREM2 (mTREM2) were 62.59 ± 11.93, 56.80 ± 13.96 and 64.62 ± 16.64 μmol/L, respectively. Simultaneously, we examined the binding affinities of FTY720P to hTREM2, rTREM2 and mTREM2, which showed that FTY720P had higher binding affinity to hTREM2, rTREM2 (6.75 ± 1.80, 7.17 ± 2.21 μmol/L, respectively), and relative lower affinity to mTREM2 (72.60 ± 21.02 μmol/L) (Fig. 2G–I). However, the results from MST showed that FTY720 failed to bind to hTREM2, suggesting phosphorylation was the precondition for FTY720 to bind to TREM2 (Fig. 2J). To further determine the binding site of S1P to TREM2, we induced the point mutation according to computer prediction data (Fig. 2A). D104A variants caused significantly lower affinity of S1P to hTREM2 (593.23 ± 97.58 μmol/L, Fig. 2K). The interaction between FTY720P and D104A TREM2 was even undetected (Fig. 2L). These data confirm the prediction results by computer simulation of molecular docking, and revealed that D104 residue was the key binding site of S1P to TREM2.
To determine whether the pro-phagocytic function of S1P is dependent on TREM2, we constructed CHO cell expressing TREM2-DAP12 (T/D CHO cells, Fig. 3A). As shown in Fig. 3B, CHO cells did not phagocytose pHrodo Red zymosan bioparticles with or without S1P or LPS treatment. However, S1P or LPS treatment increased phagocytosis of T/D CHO cells in a time-dependent manner (Fig. 3B and C), which was cancelled by phagocytosis inhibitor CytoD. Collectively, our data demonstrate that S1P or FTY720P functions as a TREM2 ligand to enhance phagocytosis.
Figure 3.

TREM2/DAP12 transfected CHO cells confirm that S1P increases phagocytosis via TREM2. (A) Immunofluorescence image confirming Trem2/Dap12 transfected CHO cells was successfully constructed. (B) Phagocytosis of pHrodo by transfected cells analyzed by fluorescence microscopy. CHO cells or TREM2/DAP12 cells were challenged with complete medium, 20 μmol/L S1P, 20 μmol/L S1P + 2 μmol/L cytoD or 10 μg/mL LPS, respectively. (C) Quantification of phagocytosed pHrodo in each cell (50 cells analyzed in each group); ∗P < 0.05, ∗∗P < 0.001 and ∗∗∗P < 0.0001. TREM2/DAP12 CHO group. The graph represents the mean ± SD and analyzed by one-way ANOVA and Tukey's test.
3.3. S1P and FTY720 exerted pro-phagocytic and neuroprotective effects depending on TREM2 in stroke
In order to further confirm the crucial roles of TREM2 in S1P/FTY720-induced microglial phagocytosis, we used Trem2 knockout (Trem2−/−) mice to investigate the effect of FTY720. Consistent with previous results, FTY720 significantly promoted phagocytosis in WT mice, as indicated by increased proportions of phagocytic microglia (Fig. 4A and B) and CD68+ area per microglia (Fig. 4A and C). However, the pro-phagocytosis effect of FTY720 was abolished in Trem2−/− mice (Fig. 4A–C). In vitro data show similar results. We co-cultured negative control (NC) or TREM2 knockdown microglia (Supporting Information Fig. S2A) with neurons, followed by OGD/R treatment. Phagocytosis by NC microglia significantly increased at 5 h after reperfusion, peaked at 7 h and decreased thereafter (Fig. S2B–S2E). S1P or FTY720 treatment dramatically promoted phagocytosis at 3 h, which was significantly inhibited by TREM2 knock-down. These data highlight the importance of TREM2 in S1P/FTY720-induced pro-phagocytic effect.
Figure 4.

TREM2 deficiency abolished the pro-phagocytic neuroprotective functions of FTY720. (A) Immunofluorescence staining of neuronal debris (green) enwrapped by microglia (red, upper row) or of microglial CD68 level (lower row) in WT or Trem2−/− MCAO mice with saline or FTY720 (1 mg/kg) treatment for 48 h. The percentage of phagocytic microglia (B) and CD68 expression (C) were analyzed; n = 4 for each group. (D) Dynamic changes of S1P level in the penumbra of MCAO rats measured by ELISA; n = 6, ∗P < 0.05 vs. Sham group, #P < 0.05 and ##P < 0.01 vs. MCAO group. (E) TTC staining of brain sections from WT mice and Trem2−/− mice with MCAO or MCAO + FTY720 treatment. (F) Quantification analysis of infarct volume; n ≥ 3, ∗∗P < 0.01 vs. MCAO group and ##P < 0.01 vs. MCAO + FTY720 group. (G) Neurological deficits via Bederson's scale scores were analyzed in WT or Trem2−/− mice (G, n ≥ 5). Values are expressed as mean ± SD and analyzed by two-way ANOVA and Bonferroni's test.
The expression of S1P in the penumbra of ischemic stroke rats were decreased at 24 and 48 h after MCAO (Fig. 4D), indicating the lack of S1P in acute phase of cerebral ischemia. FTY720 treatment supplemented the shortage of S1P (Fig. 4D), promoted the clearance of cellular debris (Fig. 1) and thereby exerted the neuroprotective effects, as indicated by the decreased infarct volume (Fig. 4E and F) and alleviated neurological deficits (Fig. 4G). To determine the participation of TREM2 mediated phagocytosis enhancement in neuroprotection, we investigated the infarct volume and neurologic deficit in Trem2−/− mice. The results show that TREM2 deficiency significantly reduced the protective effects of FTY720 indicated by the infarct volume (Fig. 4E and F) and neurologic deficits (Fig. 4G). We further assessed the protective effects of S1P/FTY720 via microglia-neuron co-culture system. At 24 h after OGD/R, S1P or FTY720 treatment maintained the length of longest neurite of neurons, which was significantly shortened when TREM2 in microglia was knocked down without affecting the number of neurites (Supporting Information Fig. S3A–S3C). Consistently, S1P or FTY720 dramatically decreased apoptotic cells, which was abolished by TREM2 knockdown or CytoD (Fig. S3D and S3E). These data show that TREM2 is pivotal in FTY720/S1P induced phagocytosis and protection.
Considering the importance of phagocytosis in hemorrhagic stroke, which is characterized by accumulated blood products like hemoglobin (Hb) in the central nervous system (CNS), we first investigated the effect of S1P on the microglial clearance of Hb. After 4 h of Hb treatment, primary microglia showed nearly no Hb clearance. Microglia barely phagocytosed Hb when treated with low dose of S1P (250 nmol/L), which is able to activate S1PRs29. However, high dose of S1P (5 μmol/L) induced obviously enhanced phagocytosis of Hb, which was almost completely abolished by cytoD (Supporting Information Fig. S4A). In the hemorrhagic stroke mice, FTY720 treatment dramatically increased Hb clearance at 72 h after ICH (Fig. S4B), ameliorated injury as indicated by reduced hematoma volume (Fig. S4C and S4D) and lower the neurological score (Fig. S4E). Taken together, our data suggest that S1P/FTY720 could promote phagocytosis and alleviated stroke-induced injury via TREM2.
3.4. APOE mediated the microglial recognition and phagocytosis of apoptotic neurons
To investigate the mechanism involved in S1P-mediated microglial phagocytosis of neurons, we first explored the distribution of APOE among neurons, astrocytes and microglia. Slices from sham group showed minimal expression. At 24 h after MCAO, APOE expression remained unchanged and FTY720 failed to affect its expression (Fig. 5A–F). However, the colocalization of APOE to neurons, astrocytes and microglia dramatically increased at 48 h after MCAO, which was further enhanced by FTY720 treatment (Fig. 5A–F). Interestingly, FTY720 significantly increased the number of ApoE positive cells enwrapped by microglia (Fig. 5E and G), implying the potential role of ApoE in mediating microglial phagocytosis of other damaged cells. As reported, APOE is not synthesized in neuron after focal ischemia30. To reveal the source of APOE localized in neurons after MCAO, we cultured neurons, astrocytes and microglia to detect the APOE expression after OGD/R treatment. At 48 h after reoxygenation, neuronal APOE expression decreased and was not affected by S1P (Supporting Information Fig. S5A). APOE expression in astrocyte remained unchanged after OGD/R, but was slightly increased by FTY720 (Fig. S5B). The APOE expression in microglia was dramatically increased after OGD/R, which was reduced after FTY720 treatment (Fig. S5C), suggesting the possibility of APOE release. Microglia transfected with GFP-labelled APOE were co-cultured with neuron in a non-contact system, followed by normal culture or OGD/R treatment for 3 h. Microglia without S1P treatment showed scarcely detected APOE expression and none green fluorescence colocalized with co-cultured neuron. OGD/R dramatically increased APOE expression in microglia, but only a little was observed to attach to neuron. S1P significantly increased the colocalization of APOE with neuron (Fig. 5H), indicating the transmit of microglia derived APOE to neuron under ischemic circumstances. When co-cultured with neuron in the non-contact system, astrocyte showed more APOE expression with nearly no transmit to neuron. However, OGD/R treatment significantly increased its expression, and resulted in a little ApoE attached to neuron, which were not influenced by S1P (Fig. S5D). These results indicate that under the circumstances of S1P treatment, microglia were the main cells to release APOE to neuron.
Figure 5.

Microglia secreted APOE to neurons. (A) Immunofluorescence staining of neurons (red) enwrapped by APOE (green) in MCAO group or MCAO + FTY720 group at 24 and 48 h after MCAO. The percentage of ApoE enwrapped neuron (B) were analyzed. (C) Representative images of APOE expression (green) in astrocyte (red) in the penumbra. APOE (green) was colocalized with GFAP (red). Nuclei counterstained with Hoechst (blue) in MCAO group and MCAO + FTY720 group at 24 and 48 h after MCAO. (D) Quantification of ApoE expression, indicated by ratio of APOE+ area to GFAP+ area. (E) Immunofluorescence staining of APOE (green) expressed in microglia (red) in MCAO group or MCAO + FTY720 group at 24 and 48 h. Quantification of APOE expression, indicated by ratio of APOE+ area to Iba1+ area (F). The percentage of microglia enwrapping APOE + cells to total microglia was analyzed (G). n = 4 for each group, ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001 vs. corresponding group at 24 h, ##P < 0.01 and ###P < 0.001 vs. MCAO group. Values are expressed as mean ± SD and analyzed by two-way ANOVA and Bonferroni's test. (H) ApoE-GFP fluorescence observed at 3 h after OGD/R in microglia-neuron non-contact co-culture system.
Apoptosis reaches a peak at 24–48 h after reperfusion. To test the possibility that released APOE bound to apoptotic neuron, we co-stained NeuN, APOE with TUNEL at 48 h after MCAO. In the penumbra, plentiful apoptotic neurons were observed in the penumbra in both MCAO group and MCAO + FTY720 group (Fig. 6A). Unlike weak APOE fluorescence in MCAO group, FTY720 significantly increased APOE labelling on the surface of apoptotic neurons (Fig. 6A). Since APOE receptors like LDLR and LRP1 mediate the uptake of APOE, and this process can be antagonized by LRP1B because of its slow rate of endocytosis31, we hypothesized that apoptotic neurons may increase LRP1B expression to bind microglia-derived APOE. The expression of LRP1B in cultured neuron following OGD/R was detected via Western blot. The results show that OGD/R treatment significantly increased LRP1B expression, which was not affected by S1P (Fig. 6B). Immunofluorescence labelling of slices of 48 h after reperfusion also showed enhanced LRP1B expression in apoptotic neurons, which was nearly absent in sham group (Fig. 6C). FTY720 treatment had no effect on the expression of neuronal APOE, but increased the APOE attachment (Fig. 6C). To further confirm the binding of APOE via LRP1B, we performed pull-down assay. His-tag labelled APOE was purified and immobilized to Ni-NTA agarose, followed by incubation with membrane protein extracted from OGD/R treated neurons. As expected, negative control showed no bands of APOE receptor. Bands indicating APOE bound LDLR and LRP1 were almost invisible. On the contrary, the band of bound LRP1B was extremely dark (Fig. 6D), indicating that APOE primarily binds to LRP1B.
Figure 6.

APOE mediated S1P–TREM2 interaction-induced apoptotic neuron clearance. (A) Immunofluorescence staining of APOE (green), TUNEL (red), NeuN (white) and Hoechst (blue) in the penumbra of slices from MCAO or MCAO+FTY720 group at 48 h after MCAO. (B) LRP1B protein level identified by Western blot in neuron with or without S1P (10 nmol/L) treatment for 3 h after OGD or normal culture. n = 4 for each group, ∗∗P < 0.01 vs. Con group, ##P < 0.01 vs. Con+S1P group. (C) Immunofluorescence staining of neurons (green), TUNEL (red), APOE (white) and LRP1B (blue) in Sham group, MCAO group and MCAO+FTY720 group at 48 h after MCAO. (D) Pull-down of receptors extracted from the membrane of OGD/R treated neurons by APOE agarose. The binding of fluorescently labelled APOE to S1P and FTY720P (E) was analyzed with MST. S1P was titrated from 30.5 nmol/L to 1 mmol/L. The change in the thermophoretic signal leads to a KD = 6.64 ± 1.68 and 15.94 ± 7.41 μmol/L, respectively. (F) Immunofluorescence staining of CD68 (green), IBA1 (red) and Hoechst (blue) in WT or APOE KD microglia–neuron coculture system. (G) Quantification of CD68 expression as ratio of CD68 area to IBA1 area. (H) Immunofluorescence staining of NeuN (green), IBA1 (red) and Hoechst (blue) in WT or APOE KO microglia–neuron coculture system. Primary cells were treated with complete medium or 10 nmol/L S1P following OGD. The experiment underwent three independent replicates. (I) shows quantification of phagocytosing microglia; ∗∗P < 0.01, ∗∗∗P < 0.001 vs. corresponding Con group, #P < 0.05 and ###P < 0.001 vs. WT group following OGD/R. Values are expressed as mean ± SD and analyzed by two-way ANOVA and Bonferroni's test.
As reported, APOE lipidation correlates with its interaction with TREM2 and subsequent phagocytosis18. As a member of sphingolipid, S1P may lipidate APOE and facilitate the activation of TREM2. MST were used to investigate the interaction between S1P or FTY720P and APOE, which showed that both S1P and FTY720P could bind to APOE (Fig. 6E). The equilibrium dissociation constants are 6.64 ± 1.68 and 15.94 ± 7.41 μmol/L, respectively. To further confirm the importance of APOE in phagocytosis, we co-cultured WT or APOE–/– microglia with neuron, followed by OGD/R. Consistent with previous results, S1P dramatically increased CD68 expression (Fig. 6F and G) and phagocytosis of neuron (Fig. 6H and I) by WT microglia at 3 h after OGD. However, the enhanced CD68 expression was significantly reduced in APOE–/– microglia-neuron co-culture system, and the pro-phagocytosis effect of S1P was abolished (Fig. 6F–I), indicating the importance of APOE in S1P mediated phagocytosis.
4. Discussion
Recent findings have revealed the importance of rapid clearance of cellular debris after ischemic stroke32,33. In the present study, we reveal for the first time that S1P acts as a novel endogenous ligand of TREM2 to effectively promote microglial phagocytosis, and thereby exert neuroprotective effects in stroke.
As is known, S1P can be secreted outside cells and activate S1PRs to regulate cell differentiation, survival, proliferation, angiogenesis and immune modulation4,5. A few studies have suggested that S1P could regulate microglial phagocytosis6,7 via unknown mechanisms. We found that S1P and its analog FTY720 could enhance microglial phagocytosis. However, S1PR2 and S1PR4, two main S1PRs expressed on microglia, did not participated in the pro-phagocytic function, indicating certain receptor instead of S1PRs mediates the effect.
TREM2 is exclusively expressed on microglia in the brain whose endogenous ligand is still unknown. Since S1P structurally resembles phospholipids, we speculated the potential interaction between S1P and TREM2 to promote phagocytosis. Firstly, computer simulation of molecular docking predicated that S1P or FTY720P could bind to TREM2. For verification, LC–MS/MS, MST and SPR were performed and identified the affinity. The results show the binding affinities of S1P to hTREM2, rTREM2 and mTREM2 were 62.59 ± 11.93, 56.80 ± 13.96 and 64.62 ± 16.64 μmol/L, respectively. FTY720P has higher affinities to hTREM2 and rTREM2, but lower affinity to mTREM2. Trem2-Dap12 transfected CHO cells were used to further demonstrate the pro-phagocytic function of S1P via acting on TREM2. Furthermore, the point mutation analysis suggests that D104 is the crucial residue for the binding of S1P to TREM2. Overall, our results reveal that S1P is a novel endogenous ligand for TREM2 to promote phagocytosis, and that S1P can be used as a lead compound for modification to increase its affinity and effects.
Emerging evidence suggest that TREM2 is essential for microglial transition to disease associated microglia (DAM)17,34 and microglial neurodegenerative phenotype (MGnD)35. Whether S1P activates TREM2 to facilitate microglia transition to these phenotypes, and then promote phagocytosis deserves further investigation. The neuroprotection of FTY720 in MCAO models has been proved36,37. However, the contribution of FTY720 induced cellular debris clearance to its protective effects has not been reported. We found that S1P in the penumbra decreased in the acute phase of ischemic stroke, accompanied by dysfunction of microglial phagocytosis. Supplementing with FTY720 could significantly enhance microglial phagocytosis. We further investigated the crucial role of TREM2 in mediating phagocytosis using Trem2−/− mice and TREM2 knockdown microglia. As expected, the pro-phagocytic function of FTY720 and S1P was abolished in Trem2−/− mice and TREM2-deficient microglia, resulting in reduced protective effect. TREM2 deficiency resulted in a slight but no significant infarct volume enlargement, and had nearly no influence on neurologic deficit. As reported, ischemia insulted Trem2 KO mice showed no exacerbated injury at 24 h after reperfusion, but exert dramatically increased injury at 14 days38, indicating no obvious influence of TREM2 on ischemic injury at acute phase. However, the increased phagocytosis and peak of apoptosis39,40 occurred at 48–72 h after MCAO, suggesting that enhancing microglial phagocytosis via TREM2 at this time could be protective. Under pathological circumstances, dead cells-released substances like nucleic acid, proteoglycan, heat shock protein 60, could also activate TREM2 and mediate downstream signaling24. Phagocytosis enhanced by S1P may promote the clearance of these DAMPs. These data confirm that S1P or FTY720 acts on TREM2 to induce phagocytosis and protection.
In addition to cerebral ischemia, phagocytosis also exerts a critical role in hemorrhagic stroke. Rapid clearance of Hb should be protective in the hemorrhagic stroke. However, increased activity and expression of S1P-lyase occur after hemorrhage resulted in a 60% reduction in S1P level41. FTY720 treatment significantly reduced the hematoma volume and neurological deficit42, 43, 44 which could not be abolished by blockage of central S1PRs45. Previous studies have uncovered that S1P forms a complex with Hb and promotes deoxy-Hb to anchor on the membrane29. On the basis of our findings, we supposed that FTY720 may function as a S1P analog to mediate the recognition of Hb by TREM2 for phagocytosis. To prove the point, we investigated the Hb clearance by isolated microglia after Hb treatment. Hb was not cleared by microglia cultured in normal culture medium or low dose of S1P (250 nmol/L), but was dramatically phagocytized by microglia treated with high dose of S1P (5 μmol/L). These data also suggest S1P-induced phagocytosis was mediated by TREM2 rather than S1PRs. The pro-phagocytosis effect of FTY720 was further observed in the peri-hematoma tissue following ICH.
APOE has been shown to regulate microglial phagocytosis via TREM235,46. During ischemic stroke, APOE is not synthesized by neuron and its expression in astrocytes and microglia increases30. However, we found obvious co-localization of APOE with neurons after MCAO treatment, which was enhanced dramatically by FTY720 at 48 h after reperfusion. Moreover, microglia showed more APOE co-localization and enwrapped more APOE positive cells in FTY720 treated group at the same time, implying that microglia may synthesize and release more APOE to mediate phagocytosis of injured neuron. Following Apoe-GFP transfection, microglia was co-cultured with neuron in a non-contact system and treated with OGD/R. APOE expression in microglia and transmission to neuron increased dramatically after S1P treatment. At the same time, we co-cultured astrocyte with neuron after Apoe-GFP transfection. However, S1P treatment showed no influence on the APOE transmit, suggesting the important role of microglia in the process. Further investigation showed that neurons co-localized with APOE underwent apoptosis, and 48 h after ischemia was the peak of apoptosis39,40. As reported, LRP1B antagonizes APOE uptake because of its slow rate of endocytosis31. We observed that apoptotic neuron bound APOE distributed on the surface, implying the interaction via LRP1B. Increased expression of LRP1B by apoptotic neurons was observed and pull-down assay was performed to demonstrate that APOE attached to apoptotic neuron via LRP1B. Our findings suggest the labelling of apoptotic neurons by attaching APOE to induce phagocytosis.
APOE lipidation is positively correlated with microglia phagocytosis18. We found that S1P and FTY720P could bind to APOE, indicating its participation in the APOE mediated clearance of apoptotic neurons. APOE knock-out abolished the pro-phagocytic function of S1P in microglia-neuron co-culture system, while TREM2 knock-out or knock-down had no effect on it, demonstrating the vital role of APOE in TREM2 mediated phagocytosis.
Collectively, our results reveal for the first time that S1P is a novel endogenous ligand for TREM2. S1P and its analog FTY720P targeting on TREM2, promotes microglial phagocytosis and exerts neuroprotection in stroke. Our findings provide a promising strategy for modulating microglial functions and a new lead compound for developing immunomodulator targeting on TREM2.
5. Conclusions
In summary, we reveal that S1P is a novel endogenous TREM2 ligand that promotes microglial phagocytosis and debris clearance. APOE loaded with S1P was released by microglia and bound to apoptotic neurons via LRP1B to mediate the clearance. Targeting TREM2–S1P interaction is a promising strategy to alleviate damaged and dead neurons induced damage, and to exert neuroprotection. These findings provide a new lead compound for developing TREM2 modulator.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Nos. 81973301, 82003732 and 81773701), the Medical Research Project of Jiangsu Commission of Health (No. ZDA2020006, China), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 18KJA310004), the Major Project of Nanjing Medical University (No. NMUD2018008, China), the Postgraduate Research and Practice Innovation Program of Jiangsu Province (Nos. KYCX19_1121 and KYCX20_1417, China) and Priority Academic Program Development of Jiangsu Higer Education Institutions (China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.10.012.
Author contributions
Xiulan Sun conceived the project and wrote the manuscript. Tengfei Xue and Juan Ji assisted with study design, performed in vivo experiments and contributed to data analysis. Yuqin Sun and Xinxin Huang performed all in vitro experiments. Zhenyu Cai and Jin Yang synthesized and purified the TREM2 protein. Wei Guo and Ruobing Guo performed binding assays. Hong Cheng assisted with experiments. All authors critically read and approved the final manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Spiegel S., Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang L., Dong Y., Wang Y., Hu W., Dong S., Chen Y. Sphingosine-1-phosphate (S1P) receptors: promising drug targets for treating bone-related diseases. J Cell Mol Med. 2020;24:4389–4401. doi: 10.1111/jcmm.15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Z., Zhang H. Molecular mechanism of S1P binding and activation of the S1P1 receptor. J Chem Inf Model. 2019;59:4402–4412. doi: 10.1021/acs.jcim.9b00642. [DOI] [PubMed] [Google Scholar]
- 4.Mihanfar A., Nejabati H.R., Fattahi A., Latifi Z., Pezeshkian M., Afrasiabi A., et al. The role of sphingosine 1 phosphate in coronary artery disease and ischemia reperfusion injury. J Cell Physiol. 2019;234:2083–2094. doi: 10.1002/jcp.27353. [DOI] [PubMed] [Google Scholar]
- 5.Raza Z., Saleem U., Naureen Z. Sphingosine 1-phosphate signaling in ischemia and reperfusion injury. Prostag Other Lipid Mediat. 2020;149:106436. doi: 10.1016/j.prostaglandins.2020.106436. [DOI] [PubMed] [Google Scholar]
- 6.Luo B., Gan W., Liu Z., Shen Z., Wang J., Shi R., et al. Erythropoeitin signaling in macrophages promotes dying cell clearance and immune tolerance. Immunity. 2016;44:287–302. doi: 10.1016/j.immuni.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Tran H.B., Jersmann H., Truong T.T., Hamon R., Roscioli E., Ween M., et al. Disrupted epithelial/macrophage crosstalk via spinster homologue 2-mediated S1P signaling may drive defective macrophage phagocytic function in COPD. PLoS One. 2017;12 doi: 10.1371/journal.pone.0179577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paradowska-Gorycka A., Jurkowska M. Structure, expression pattern and biological activity of molecular complex TREM-2/DAP12. Hum Immunol. 2013;74:730–737. doi: 10.1016/j.humimm.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Sudom A., Talreja S., Danao J., Bragg E., Kegel R., Min X.S., et al. Molecular basis for the loss-of-function effects of the Alzheimer's disease-associated R47H variant of the immune receptor TREM2. J Biol Chem. 2018;293:12634–12646. doi: 10.1074/jbc.RA118.002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mecca C., Giambanco I., Donato R., Arcuri C. Microglia and aging: the role of the TREM2–DAP12 and CX3CL1–CX3CR1 axes. Int J Mol Sci. 2018;19:318. doi: 10.3390/ijms19010318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Painter M.M., Atagi Y., Liu C.C., Rademakers R., Xu H., Fryer J.D., et al. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener. 2015;10:43. doi: 10.1186/s13024-015-0040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poliani P.L., Wang Y., Fontana E., Robinette M.L., Yamanishi Y., Gilfillan S., et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 2015;125:2161–2170. doi: 10.1172/JCI77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.N'Diaye E., Branda C.S., Branda S.S., Nevarez L., Colonna M., Lowell C., et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. 2009;184:215–223. doi: 10.1083/jcb.200808080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daws M.R., Sullam P.M., Niemi E.C., Chen T.T., Tchao N.K., Seaman W.E. Pattern recognition by TREM-2: binding of anionic ligands. J Immunol. 2003;171:594–599. doi: 10.4049/jimmunol.171.2.594. [DOI] [PubMed] [Google Scholar]
- 15.Quan D.N., Cooper M.D., Potter J.L., Roberts M.H., Cheng H., Jarvis G.A. TREM-2 binds to lipooligosaccharides of Neisseria gonorrhoeae and is expressed on reproductive tract epithelial cells. Mucosal Immunol. 2008;1:229–238. doi: 10.1038/mi.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Cella M., Mallinson K., Ulrich J.D., Young K.L., Robinette M.L., et al. TREM2 lipid sensing sustains microglia response in an Alzheimer's disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deczkowska A., Keren-Shaul H., Weiner A., Colonna M., Schwartz M., Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173:1073–1081. doi: 10.1016/j.cell.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Yeh F.L., Wang Y., Tom I., Gonzalez L.C., Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 19.Benjamin E.J., Blaha M.J., Chiuve S.E., Cushman M., Das S.R., Deo R., et al. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim A.S., Johnston S.C. Temporal and geographic trends in the global stroke epidemic. Stroke. 2013;44:S123–S125. doi: 10.1161/STROKEAHA.111.000067. [DOI] [PubMed] [Google Scholar]
- 21.Dirnagl U., Iadecola C., Moskowitz M.A. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 22.Mehta S.L., Manhas N., Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Geissmann F., Manz M.G., Jung S., Sieweke M.H., Merad M., Ley K. Development of monocytes, macrophages and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuyama J., Nakamura A., Ooboshi H., Yoshimura A., Shichita T. Pivotal role of innate myeloid cells in cerebral post-ischemic sterile inflammation. Semin Immunopathol. 2018;40:523–538. doi: 10.1007/s00281-018-0707-8. [DOI] [PubMed] [Google Scholar]
- 25.Hankey G.J. Stroke. Lancet. 2017;389:641–654. doi: 10.1016/S0140-6736(16)30962-X. [DOI] [PubMed] [Google Scholar]
- 26.Xi G., Keep R.F., Hoff J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 27.Ji J., Wang J., Yang J., Wang X.P., Huang J.J., Xue T.F., et al. The intra-nuclear SphK2–S1P axis facilitates M1-to-M2 shift of microglia via suppressing HDAC1-mediated KLF4 deacetylation. Front Immunol. 2019;10:1241. doi: 10.3389/fimmu.2019.01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kober D.L., Wanhainen K.M., Johnson B.M., Randolph D.T., Holtzman M.J., Brett T.J. Preparation, crystallization, and preliminary crystallographic analysis of wild-type and mutant human TREM-2 ectodomains linked to neurodegenerative and inflammatory diseases. Protein Expr Purif. 2014;96:32–38. doi: 10.1016/j.pep.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun K., Zhang Y., D'Alessandro A., Nemkov T., Song A., Wu H., et al. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat Commun. 2016;7 doi: 10.1038/ncomms12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez C.G., Hamby M.E., McReynolds M.L., Ray W.J. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer's disease. Front Aging Neurosci. 2019;11:14. doi: 10.3389/fnagi.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winfree L.M., Speese S.D., Logan M.A. Protein phosphatase 4 coordinates glial membrane recruitment and phagocytic clearance of degenerating axons in Drosophila. Cell Death Dis. 2017;8:e2623. doi: 10.1038/cddis.2017.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamaguchi A., Jitsuishi T., Hozumi T. Temporal expression profiling of DAMPs-related genes revealed the biphasic post-ischemic inflammation in the experimental stroke model. Mol Brain. 2020;13:57. doi: 10.1186/s13041-020-00598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;169:1276–1290. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., Fatimy R.E., et al. The TREM2–APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nazaria M., Keshavarza S., Rafati A., Namavar M.R., Haghani M. Fingolimod (FTY720) improves hippocampal synaptic plasticity and memory deficit in rats following focal cerebral ischemia. Brain Res Bull. 2016;124:95–102. doi: 10.1016/j.brainresbull.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Hasegawa Y., Suzuki H., Altay O., Rollanda W., Zhang J.H. Role of the sphingosine metabolism pathway on neurons against experimental cerebral ischemia in rats. Transl Stroke Res. 2013;4:524–532. doi: 10.1007/s12975-013-0260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawabori M., Kacimi R., Kauppinen T., Calosing C., Kim J.Y., Hsieh C.L., et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. 2015;35:3384–3396. doi: 10.1523/JNEUROSCI.2620-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia M., Yang X., Yang T., Deng X., Liang J., Bi J., et al. β-1,3-Galactosyltransferase 2 deficiency exacerbates brain injury after transient focal cerebral ischemia in mice. Brain Res Bull. 2021;169:104–111. doi: 10.1016/j.brainresbull.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 40.Wang S.D., Fu Y.Y., Han X.Y., Yong Z.J., Li Q., Hu Z., et al. Hyperbaric oxygen preconditioning protects against cerebral ischemia/reperfusion injury by inhibiting mitochondrial apoptosis and energy metabolism disturbance. Neurochem Res. 2021;46:866–877. doi: 10.1007/s11064-020-03219-4. [DOI] [PubMed] [Google Scholar]
- 41.Testai F.D., Xu H.L., Kilkus J., Suryadevara V., Gorshkova I., Berdyshev E., et al. Changes in the metabolism of sphingolipids after subarachnoid hemorrhage. J Neurosci Res. 2015;93:796–805. doi: 10.1002/jnr.23542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y.J., Chang G.Q., Liu Y., Gong Y., Yang C., Wood K., et al. Fingolimod alters inflammatory mediators and vascular permeability in intracerebral hemorrhage. Neurosci Bull. 2015;31:755–762. doi: 10.1007/s12264-015-1532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lia R., Venkatb P., Chopp M. RP001 hydrochloride improves neurological outcome after subarachnoid hemorrhage. J Neurol Sci. 2019;399:6–14. doi: 10.1016/j.jns.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Yang Z., Dong S., Zheng Q., Zhang L., Tan X., Zou J., et al. FTY720 attenuates iron deposition and glial responses in improving delayed lesion and long-term outcomes of collagenase-induced intracerebral hemorrhage. Brain Res. 2019;1718:91–102. doi: 10.1016/j.brainres.2019.04.031. [DOI] [PubMed] [Google Scholar]
- 45.Hasegawa Y., Uekawa K., Kawano T., Suzuki H., Kim-Mitsuyama S. Blockage of central sphingosine-1-phosphate receptor does not abolish the protective effect of FTY720 in early brain injury after experimental subarachnoid hemorrhage. Curr Drug Deliv. 2017;14:861–866. doi: 10.2174/1567201813666160907094401. [DOI] [PubMed] [Google Scholar]
- 46.Chen M., Xie M., Peng C., Long S. The absorption of apolipoprotein E by damaged neurons facilitates neuronal repair. Cell Biol Int. 2019;43:623–633. doi: 10.1002/cbin.11135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.