

ABSTRACT
Hereditary hemorrhagic telangiectasia (HHT) is a genetic disorder in which there is faulty development of the arteries. There is a high incidence of pulmonary hypertension (PH) in these patients, the pathophysiology of which is not fully known. An increase in cardiac output, causing high-output cardiac failure, and increased pulmonary vascular resistance secondary to genetic mutations are the main reasons. We report a 25-year-old male with HHT who presented with right heart failure secondary to PH in whom both the above mechanisms were operating. The coexistence of giant pulmonary arteriovenous malformations with severe PH is a rare scenario influencing management decisions that are discussed. In addition, this patient highlights the classical visceral vascular malformations in this rare disorder.
Keywords: Epistaxis, genetic disorder, vascular malformations
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT) (Osler–Weber–Rendu disease), an autosomal dominant disorder with a prevalence of 1 per 10,000, is characterized by mucocutaneous and visceral vascular malformations. Epistaxis due to nasal mucosal telangiectasia is the most common manifestation seen in over 90% of patients. Pulmonary hypertension (PH) manifests in up to 5% of patients,[1] which occurs due to high-output heart failure from excessive shunting through visceral vascular malformations. This group of patients generally present in adulthood after the onset of left ventricular dysfunction. However, few HHT patients manifest a syndrome of heritable pulmonary arterial hypertension (PAH), whose clinical features are similar to idiopathic PAH and present in the first two decades of life.
Our patient presented with severe PH at a younger age with large hepatic and pulmonary vascular malformations, with probably both the etiologies of PH involved. The differentiation of the type of PH is vital in management and prognostification. These issues, including the management of pulmonary arteriovenous malformations (PAVMs) in the presence of PH, are discussed below.
CLINICAL SUMMARY.
A 25-year-old male presented to us with a history of progressive dyspnea of Class 4 severity. There were episodes of epistaxis for the past 5 years. No family members had similar complaints except that his elder brother died suddenly due to an unexplained cause. On examination, he had pallor, pandigital clubbing, bilateral pitting type pedal edema, and telangiectasia on finger pads and abdomen. Saturation on room air was 40%, with partial improvement to 75% on inhaled oxygen. Cardiac examination revealed a right ventricular (RV) apex, a loud pulmonary component of the second heart sound and a continuous murmur in the right parasternal area.
The blood parameters were abnormal with a hemoglobin of 9.5 g/dL, serum creatinine of 1.7 mg/dl, and elevated liver enzymes (more than ten times the upper limit). The electrocardiogram (ECG) had a right-axis deviation and RV hypertrophy with strain pattern [Figure 1a]. Transthoracic echocardiogram demonstrated enlargement of the right atrium and right ventricle with normal function, mild tricuspid regurgitation, and dilated pulmonary arteries [Figure 1b]. The left ventricular (LV) size and function were normal. The estimated PA systolic pressure was elevated at 80 mmHg [Figure 1c]. Computed tomography (CT) angiography showed dilated main PA, dilated right-sided chambers, and large PAVM in the right middle lobe and left lower lobe of the lung [Figure 2a-d]. The abdominal CT demonstrated a dilated celiac artery and its branches with multiple tiny arteriovenous malformations, confluent vascular masses, and a few cysts [Figure 3a and b].
Figure 1.

(a) Electrocardiogram with prominent R in the anterior chest leads with ST-segment depression suggestive of right ventricular strain. (b) Parasternal short-axis view on echocardiogram showing dilated right ventricle with a flattened interventricular septum. (c) Spectral Doppler showing right ventricular systolic pressure gradient of 70 mm of Hg
Figure 2.
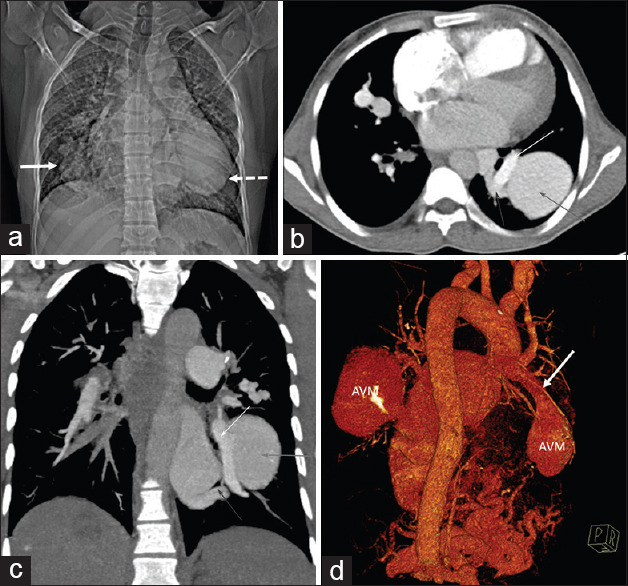
(a) Computed tomography topogram image showing lobulated oval density in the right lower zone paracardiac region (white arrow). Another larger ovoid density is seen in the left retrocardiac region (dashed arrow). (b) Computed tomography pulmonary angiogram image showing the typical appearance of arteriovenous malformation, with a large feeding vessel arising from the pulmonary artery (yellow arrow) and an adjacent draining vein (blue arrow), entering and exiting the malformation arteriovenous malformation (red arrow). (c) Coronal maximum intensity projection contrast-enhanced computed tomography images depicting large simple arteriovenous malformations (red arrows) with feeding artery from the right lower lobe segmental pulmonary artery (yellow arrow) and draining through the right inferior pulmonary vein (blue arrow). (d) Computed tomography volume-rendered image showing the bilateral arteriovenous malformation, with a large feeding vessel arising from the pulmonary artery (white arrow) entering the malformation arteriovenous malformation
Figure 3.
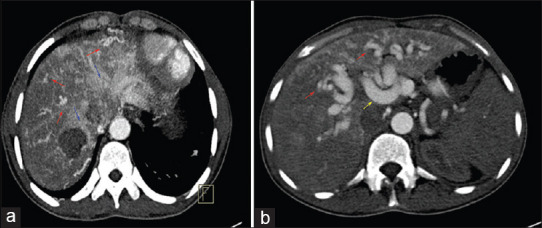
(a) Axial contrast-enhanced early arterial phase computed tomography abdomen image depicts inhomogeneous attenuating pattern within the liver parenchyma with multiple telangiectases (red arrow), large confluent vascular mass (blue arrow). (b) Computed tomography abdomen depicting multiple telangiectases (red arrow) and dilated hepatic artery (yellow arrow)
A clinical diagnosis of respiratory infection with right heart failure, congestive hepatopathy, and acute kidney injury was made and managed with antibiotics. The patient was discharged on oral sildenafil. He had an inadequate response to pulmonary vasodilator therapy requiring a repeat admission after 4 months for similar complaints.
DISCUSSION
HHT is a genetically transmitted disorder. Clinical diagnosis is based on (Curacao criteria) the presence of at least three of the following criteria: epistaxis, mucocutaneous telangiectasias, visceral vascular malformations, and a first-degree relative who meets the criteria for the diagnosis of HHT.[2] Our patient had all the criteria except the family history. The genetic abnormality in the receptors of the transforming growth factor-beta explains its manifestations. Genetic testing, as was not affordable by the patient, was not done.
PH in patients with HHT occurs predominantly by two mechanisms.[3] Firstly, visceral malformations lead to increased cardiac output induced heart failure which can cause pulmonary hypertension. Second, a small number of patients have pulmonary arteriopathy whose clinical features are identical to those of idiopathic PAH. Patients in the former category have vascular malformations with evidence of LV dysfunction and usually present late, unlike those with the hereditary component. Even though echocardiography is helpful, right heart catheterization (RHC) is definitive in differentiating the two mechanisms.[1] PH due to vascular shunting has increased cardiac output, increased pulmonary capillary wedge pressure, and low pulmonary vascular resistance (PVR). In our patient, RHC was not done as the patient was unstable with cyanosis, and the echo was suggestive of suprasystemic RV pressure with a diminutive LV. A recent study showed a higher prevalence of PH in genetically confirmed HHT.[4]
PAVMs manifest as either cyanosis, hemoptysis, high-output failure, or a complication due to right-to-left shunt. Simple PAVMs appear as well-defined rounded nodules on imaging. The connection of the nodule to the draining artery and vein confirms the diagnosis. Endovascular therapy is the preferred management strategy when they are symptomatic, which relieves the volume overload. However, in the presence of severe PH, occlusion of these malformations may increase the PVR by the exclusion of low-pressure pathways and worsen the PA pressure.[5] In this case, PAVM closure was contemplated, but due to severe PAH, we refrained from it.
Hepatic manifestations of HHT are the most common, with the estimated prevalence of hepatic involvement ranging from about 74%–79%.[6] Definitive treatment of hepatic malformations is either by surgical ligation or banding or by transcatheter coil embolization. Transcatheter embolization of hepatic AVMs effectively decreases shunting and improves symptoms, but the rate of serious complications is unacceptably high such as biliary necrosis and death after embolization. For these reasons, the procedure is not routinely recommended. However, liver transplantation is the definitive line of management.
Our patient's unique feature is that he presented with young-onset PH causing right heart failure, which is probably due to pulmonary arteriopathy but with coexisting large visceral vascular malformations. How much each component contributed was challenging to ascertain, leading to a dilemma in management. However, significant RVH on ECG and features of cor pulmonale on echo were suggestive of severe PH, which precluded further interventions for vascular malformations.
Management of PH varies according to the mechanism of its cause. Therapy for PH from high-output heart failure includes diuretics, salt restriction, and correction of anemia. Pulmonary vasodilator therapy may have detrimental hemodynamic effects by increasing pulmonary blood flow. PAH in HHT is treated similarly to other idiopathic PAH. However, there are insufficient data to determine the efficacy of vasodilator therapy.
PH, due to increased cardiac output, has a better prognosis with a dramatic response to interventions. In comparison, PAH caused by obstructive pulmonary vascular disease has a poor prognosis with 1- and 3-year survival rates of 77.8% and 53.3%, respectively, which were much lower than idiopathic PAH.[7] Presentation with right heart failure, suprasystemic RV pressure, and large unmanageable vascular malformations portends a grave prognosis in our patient.
CONCLUSION
HHT has a high prevalence of PH and carries a worse prognosis. It can be due to either due to arteriopathy or the associated vascular malformations.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to thank Dr. Sravanthi Yerram, Consultant Radiologist, Sagar Hospitals, Bengaluru.
REFERENCES
- 1.Lyle MA, Fenstad ER, McGoon MD, Frantz RP, Krowka MJ, Kane GC, et al. Pulmonary hypertension in hereditary hemorrhagic telangiectasia. Chest. 2016;149:362–71. doi: 10.1378/chest.15-0535. [DOI] [PubMed] [Google Scholar]
- 2.Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48:73–87. doi: 10.1136/jmg.2009.069013. [DOI] [PubMed] [Google Scholar]
- 3.Battaile JT, Ochoa CD. Distinct forms of pulmonary hypertension complicate hereditary hemorrhagic telangiectasia. Adv Pulm Hypertens. 2015;14:145–9. [Google Scholar]
- 4.Vorselaars V, Velthuis S, Van Gent M, Westermann C, Snijder R, Mager J, et al. Pulmonary hypertension in a large cohort with hereditary hemorrhagic telangiectasia. Respiration. 2017;94:242–50. doi: 10.1159/000458447. [DOI] [PubMed] [Google Scholar]
- 5.Revuz S, Decullier E, Ginon I, Lamblin N, Hatron PY, Kaminsky P, et al. Pulmonary hypertension subtypes associated with hereditary haemorrhagic telangiectasia: Haemodynamic profiles and survival probability. PLoS One. 2017;12:e0184227. doi: 10.1371/journal.pone.0184227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tortora A, Riccioni ME, Gaetani E, Ojetti V, Holleran G, Gasbarrini A. Rendu-Osler-Weber disease: A gastroenterologist's perspective. Orphanet J Rare Dis. 2019;14:130. doi: 10.1186/s13023-019-1107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W, Xiong CM, Gu Q, Wang XT, Cheng XL, Huang L, et al. The clinical characteristics and long-term prognosis of pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Pulm Circ. 2018;8:2045894018759918. doi: 10.1177/2045894018759918. [DOI] [PMC free article] [PubMed] [Google Scholar]