

This study shows that CD38 is a new member of the IgM-BCR coreceptor; it associates with CD19 in unstimulated B cells and with CD19 and IgM after BCR stimulation. Targeting CD38 with an antibody impairs B cell proliferation and survival, and formation of IgM:CD19 synapses.
Abstract
CD38 is a multifunctional protein expressed on the surface of B cells in healthy individuals but also in B cell malignancies. Previous studies have suggested a connection between CD38 and components of the IgM class B cell antigen receptor (IgM-BCR) and its coreceptor complex. Here, we provide evidence that CD38 is closely associated with CD19 in resting B cells and with the IgM-BCR upon engagement. We show that targeting CD38 with an antibody, or removing this molecule with CRISPR/Cas9, inhibits the association of CD19 with the IgM-BCR, impairing BCR signaling in normal and malignant B cells. Together, our data suggest that CD38 is a new member of the BCR coreceptor complex, where it exerts a modulatory effect on B cell activation upon antigen recognition by regulating CD19. Our study also reveals a new mechanism where α-CD38 antibodies could be a valuable option in therapeutic approaches to B cell malignancies driven by aberrant BCR signaling.
Graphical Abstract

Introduction
CD38 is a 45-kD transmembrane glycoprotein with a short cytoplasmic tail without tyrosine residues or identified activation motifs (Jackson and Bell, 1990). It is an ectoenzyme with multiple functions; it catalyzes the formation of second messengers that regulate Ca2+ mobilization from intracellular stores, and can also mediate cell-to-cell interaction by binding to its ligand, CD31 (PECAM-1; Aarhus et al., 1995; De Flora et al., 2004; Deaglio et al., 1998; Malavasi et al., 2008). Moreover, depending on the orientation of the catalytic domain, whether it faces outside (type II) or inside (type III) of the membrane, CD38 exerts distinct enzymatic functions (Lee et al., 2022). CD38 is expressed on the cell surface of a range of normal and malignant leukocytes and it is particularly highly expressed in terminally differentiated plasma cells and their pathological counterparts, e.g., multiple myeloma (MM), a B cell neoplasm characterized by the proliferation of plasma cells in the bone marrow (Kumar et al., 2010; Malavasi et al., 2008). Daratumumab (trade name Darzalex), a human IgG1κ mAb that recognizes CD38, is approved for treating MM (Sanchez et al., 2016).
In a study of patients’ B cell chronic lymphocytic leukemia cells, CD38 was found to colocalize with components of the IgM class B cell antigen receptor (IgM-BCR), i.e., membrane-bound IgM, Igα, and Igβ as well as with CD81 that is member of the BCR coreceptor (Deaglio et al., 2003). In another study investigating the modalities of CD38-mediated signaling on activated human tonsillar B cells and a Burkitt lymphoma (BL) cell line, Raji, CD38 was found to be associated with CD21, which is also a component of the BCR coreceptor (Funaro et al., 1993). Furthermore, upon engagement via antibodies and cross-linkers or via CD31, CD38 relocalizes in cap areas with the CD19/CD81 complex (Deaglio et al., 2007). During the last few years, it has been uncovered that the BCRs are organized in nanoclusters on the B cell surface, and not as individual freely moving entities (Lee et al., 2017; Maity et al., 2015; Mattila et al., 2013). Upon maturation, naive B cells coexpress two BCR isotypes, IgM and IgD, which localize in different nanoclusters on the cell surface. In resting B cells, IgD-BCRs are organized together with CD19 and other proteins of the BCR coreceptor, whereas IgM-BCRs gain proximity with CD19 only upon activation, forming IgM:CD19 synapses (Kläsener et al., 2014).
Herein, we set out to determine the functional role of CD38 on human B cells, and in particular its interactions with CD19 and the IgM-BCR. We demonstrate that CD38 is fundamental for B cell activation via the IgM-BCR, and that it is essential for the nanoscale reorganization of the BCR and its coreceptor complex and the formation of IgM:CD19 synapses upon BCR engagement.
Results
Targeting CD38 with a mAb impairs B cell proliferation and survival
Human peripheral blood (PB) B cells express CD38 on their surface as shown by flow cytometry (Fig. 1 A). To investigate whether CD38 plays a role in B cell proliferation, we cultured PB B cells in the presence of mAbs recognizing CD38, i.e., α-CD38 clone HB-7 and daratumumab, hereafter referred to as DARA. We analyzed the proliferation after 5 and 7 d in culture with a combination of mitogenic stimuli, i.e., α-IgM/G/A (hereafter α-Ig) and IL-2 combined with TLR9 agonist (cytosine–phosphate–guanine [CpG]; Fig. 1, B and C). The cells cultured with α-CD38 mAbs showed a decreased proliferative response compared to the control B cells without α-CD38 mAbs, and DARA was the most effective inhibitor of B cell proliferation.
Figure 1.

B cell proliferation and survival are impaired by CD38 mAbs. (A) CD38 expression on B cells (gray filled) compared to other lymphocytes from PB (black line). (B and C) Isolated untouched PB B cells were labeled with CTV, and then cultured for 5 and 7 d with a stimuli combination containing F(ab′)2-fragments of goat α-human Ig M/G/A (here indicated as α-Ig) plus IL-2 and CpG, in the presence of two different α-CD38 antibody treatments, i.e., HB-7 or DARA, or no antibodies. Proliferation was calculated as expansion index, which determines the fold-expansion of the overall culture. (D) CD38 and CD20 expression levels on Namalwa cells (gray filled) compared to unstained negative control (black line). (E) Cell number and percentage of dead cells (7AAD+) in Namalwa cells, cultured for 4 d without or with Rituximab (RTX) or DARA. (F) CD38 expression levels on two additional BL cell lines, unstained negative control (black line) and stained (gray filled). (G) Cell number and percentage of dead cells (7AAD+) in the two additional BL cell lines, cultured for 4 d without or with DARA. (H) IgM expression levels on the three BL cell lines. Light gray indicates a negative control for IgM expression, which in this case was the B cell line RS4;11, a cell line derived from B cell precursors. One-way ANOVA with Tukey’s multiple comparisons (C and E) and unpaired two-tailed t tests (G) were used (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). All experiments were repeated at least three times. For each experiment n = 3; error bars show mean ± SD.
This outcome prompted us to investigate the influence of DARA on proliferation and survival of human B cell malignancies. Firstly, we used the BL B cell line Namalwa that expresses high amounts of surface CD38 but is negative for CD20 (Fig. 1 D). We analyzed proliferation and survival of Namalwa cells cultured for 4 d in the presence of either the α-CD20 mAb Rituximab or DARA (Fig. 1 E). In line with the experiment performed on primary PB B cells, we found that Namalwa cells displayed a decrease in proliferation and increase in cell death only in the presence of DARA. Raji and DG75, two other BL B cell lines and positive for CD38 (Fig. 1 F), showed a similar response to treatment with DARA (Fig. 1 G). These BL cell lines represent a mature B cell stage and express an IgM-BCR (Fig. 1 H). Further, although the Namalwa and Raji cells lines are EBV positive, the DG75 BL is not, and hence the effect is not linked to viral proteins, e.g., LMP2A (Portis and Longnecker, 2004). Overall, these results suggest that targeting CD38 has an effect on mature B cell proliferation and survival.
Targeting CD38 with DARA reduces ERK phosphorylation in PB B cells
ERK1/2 are members of the mitogen-activated protein kinase super family that mediate cell proliferation and survival (Wellbrock et al., 2004). Thus, inhibition of ERK phosphorylation would result in reduced cell proliferation. As ERK is located in the signaling pathways downstream of both the BCR and TLR9, we investigated the effect of α-CD38 antibody exposure on pERK induced by the previously used stimuli, i.e., α-Ig, CpG, and IL-2. In the following experiments, the cells were pretreated with DARA such that the cells were incubated for 30 min at 37°C with the mAb and then washed before further procedures. PB mononuclear cells (PBMCs) were stimulated 10 min with α-Ig or 30 min with CpG, and pERK was assessed in CD19+ PB B cells with or without pretreatment with DARA. These time points were the most effective for optimal pERK using these stimuli, while IL-2 did not induce pERK at the investigated time points (Fig. S1). Levels of pERK protein expression were significantly reduced both in B cells pretreated with DARA when stimulated with α-Ig but not CpG (Fig. 2, A and B). We thereafter performed an additional experiment, this time investigating B cells as IgM+ and IgM− by intracellularly staining for IgM and IgD (Fig. 2 C). This allowed us to distinguish the B cells bearing an IgM-BCR or other isotypes, as α-Ig stimulation leads to BCR downmodulation from the cell surface. Both IgM+ and IgM− B cells pretreated with DARA showed significantly reduced levels of pERK protein expression compared to the untreated cells, with the IgM+ cells being more affected than the IgM− (Fig. 2, C and D). These results suggest that targeting CD38 with the mAb DARA inhibits ERK phosphorylation downstream of the BCR.
Figure S1.
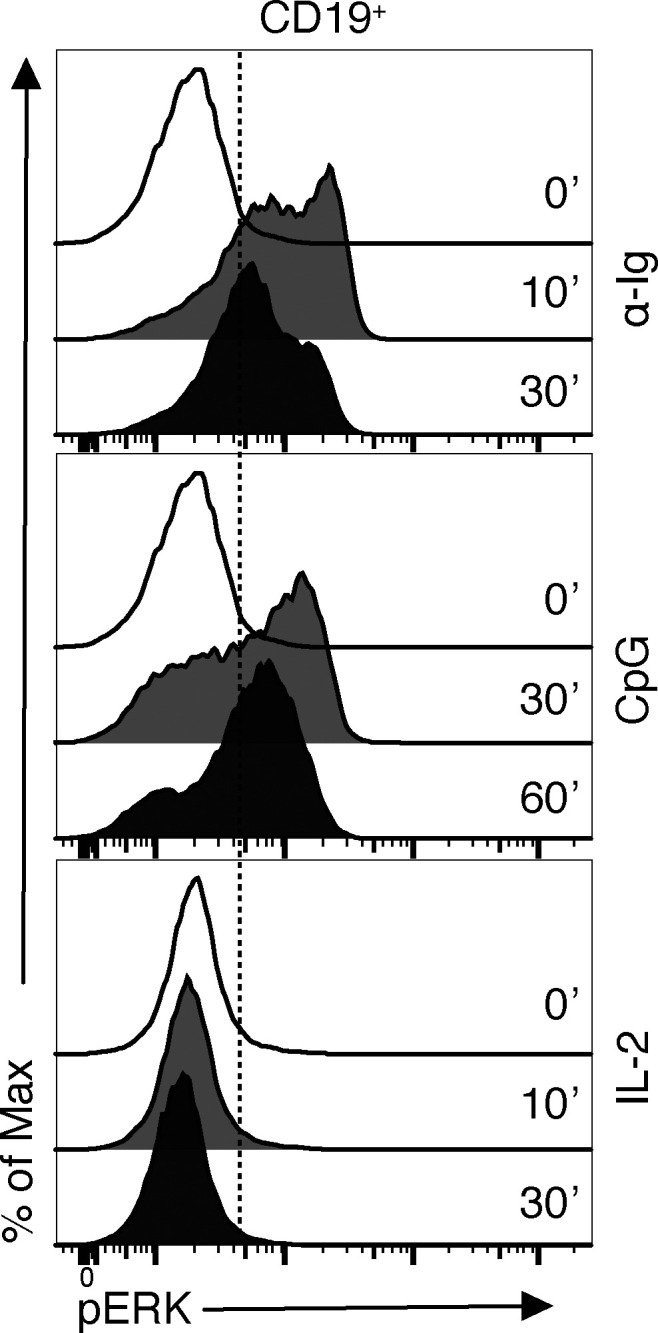
pERK expression kinesis with different stimulations in PB B cells. FACS histogram shows pERK expression in PB B cells in unstimulated (0 min) state and after different stimulations at different time points: α-Ig (10 and 30 min); CpG (30 and 60 min); IL-2 (10 and 30 min).
Figure 2.

Exposure to DARA reduces ERK phosphorylation and affects IgM-BCR trafficking in B cells. (A and B) pERK on CD19+ B cells was measured after stimulation for 10 min with F(ab′)2-fragments α-IgM/G/A (here indicated as α-Ig) or 30 min with CpG, with and without exposure to DARA. (C and D) CD19+ B cells were stimulated for 10 min with α-Ig with and without exposure to DARA. The cells were then stained intracellularly for IgM and IgD and pERK was measured on B cells gated as IgM+ and IgM−. Experiments in A–D were repeated at least twice. For each experiment, n = 5. Error bars show mean ± SD; paired two-tailed t tests were used (*, P < 0.05; **, P < 0.01). (E) Phosphoproteomic comparison of isolated PB IgM+ B cells under four different treatment conditions: unstimulated (−; green), stimulated with an α-IgM for 5 min with and without (light blue) exposure to either a control IgG1 (ctrl; dark blue) or DARA (red). The heatmap shows significantly changing phosphopeptides between treatment groups (ANOVA, P < 0.01, n = 4). (F) Protein phosphorylation in the BCR pathway induced by stimulation with an α-IgM for 5 min (t test, P < 0.05). (G) Phosphopeptides significantly affected by DARA exposure versus stimulation with and without exposure to control IgG1 (t test, P < 0.01). (H) Enrichment map shows GO pathways significantly enriched (P < 0.05) for all significantly changed phosphopeptides identified in G. Nodes in the network represent pathways, and similar pathways with many common genes are connected. Groups of similar pathways are indicated. Pathways without connection were omitted. Nodes are colored according to P value, and the size of the nodes reflects the number of proteins in the node as indicated in the figure.
Phosphoproteomics suggests that exposure to DARA affects IgM-BCR trafficking
In an attempt to identify cellular processes affected by treatment of the cells with DARA, we compared the phosphoproteome of PB IgM+ B cells under four different conditions: unstimulated or α-IgM stimulated for 5 min, the latter with or without pretreatment with an IgG1 control antibody or DARA (Fig. 2 E). Two of the four sample groups, namely those stimulated with α-IgM either pretreated or not with the IgG1 ctrl, clustered close to each other, hereafter referred to as stimulated control groups, and thus support the validity of our approach. To examine the phosphorylation profile at this time point, we compared the stimulated control groups with that of the unstimulated and searched for the phosphorylation status of proteins in the signaling pathway downstream of the BCR (Table S1). We found that in the stimulated samples several of these proteins were phosphorylated, in some cases at more than one site, e.g., SYK and CD20 (MS4A1) as previously described, confirming activation through BCR engagement (Fig. 2 F; Kurosaki et al., 1994; Valentine et al., 1989). In fact, we found that 93 phosphopeptides in α-IgM–stimulated B cells were significantly affected by DARA pretreatment, of which 48 showed a decrease and 45 an increase in phosphorylation (Fig. 2 G and Table S2). Thereafter, we carried out a Gene Ontology (GO) term analysis of all significantly changed phosphopeptides identified. We saw a striking enrichment for GO terms related to actin polymerization and depolymerization, plasma membrane–bounded cell projection, cortical cytoskeleton, endocytic vesicle, spliceosome, and chromosome (Fig. 2 H and Table S3). Whilst it remains unclear if the changes in the phosphorylation state of these proteins are directly responsible for the broader effects of DARA pretreatment, we hypothesized that DARA may be affecting events associated with actin remodeling and endocytic trafficking in the context of IgM-BCR stimulation.
Exposure to DARA prevents IgM-BCR downmodulation
These results pointed towards a role for CD38 in IgM-BCR trafficking, which directed our interest to the effect of DARA on the surface organization of the IgM-BCR upon activation. We sought, therefore, to investigate the kinetics of IgM-BCR downregulation after inducing activation with α-IgM. For these experiments, we used an antibody for IgM detection that recognized an epitope that was not occupied by the α-IgM used for stimulation (Table S4).
Observing the proportion of cells still expressing IgM on the cell surface after activation with α-IgM, we found that IgM downmodulation was prevented in B cells pretreated with DARA (Fig. 3 A). We followed up the kinetics of IgM downmodulation at 5, 15, and 30 min after α-IgM stimulation in B cells with or without DARA. IgM expression on the unstimulated cells was used as reference and set to 100%. The cells pretreated with DARA showed a higher percentage of surface IgM than the control (Fig. 3, A and B). We conclude that pretreatment of PB B cells with DARA impairs IgM-BCR downmodulation.
Figure 3.

IgM-BCR downmodulation is prevented on B cells exposed to DARA. (A) Representative FACS plots showing IgM expression on PB B cells before (0 min) and after inducing IgM downregulation with α-IgM for 5, 15, and 30 min with and without exposure to DARA. (B) IgM expression kinetics measured as frequency of IgM still detectable on the cell surface upon α-IgM stimulation after 5, 15, and 30 min compared to the starting point (0 min) = 100% of IgM present on the surface; no DARA = black circles; DARA = open circles. All experiments were repeated at least three times. For each experiment n = 3; error bars show mean ± SD. Paired two-tailed t tests were used (*, P < 0.05; **, P < 0.01).
DARA inhibits IgM-BCR polarization upon stimulation
To further our analysis on the role of CD38 in the reorganization of the IgM-BCR upon stimulation, we visualized the polarization process, in which activated BCRs aggregate on the cell surface and polarize into caps, a process also known as BCR capping. In unstimulated cells, the IgM-BCR did not aggregate, whereas α-IgM stimulation of the BCR for 5 or 15 min induced capping (Fig. 4 A), as expected. However, in B cells pretreated with DARA, capping of the BCR was inhibited, and at both time points, after addition of α-IgM. Also, CD19 seemed to partially polarize with the IgM in the stimulated cells, while this phenomenon was not recorded in the unstimulated or DARA pretreated cells. We quantified the extent of IgM capping on the B cells, which showed that the inhibition of capping by DARA was significant both at 5 and 15 min after addition of α-IgM (Fig. 4 B). This indicates that DARA prevents IgM-BCR polarization and thus the formation of an immunological synapse, which would normally lead to the activation of downstream signaling pathways.
Figure 4.

CD38 regulates IgM-BCR polarization and the formation of CD19:IgM synapses upon IgM-BCR engagement. (A) IgM capping analyzed by fluorescence microscopy on B cells with and without exposure to DARA in unstimulated (0 min) PB B cells and after 5- and 15-min stimulation with α-IgM. Hoechst (blue), CD19 (red), and IgM (green). Scale bar, 7 µm. (B) Scatter dot plot represents the Δ centroid XY analysis of the IgM capping on the surface of B cells. The higher the value on the y-axis, the more capping is present on the cell surface. (C–E) Fab-PLA study of the proximity of IgM to CD19 on unstimulated (0 min; C), or 5- and 15-min α-IgM–stimulated PB B cells without and with exposure to DARA (D). PLA signals are shown in red and nuclei in blue. Scale bar, 5 µm. (E) Scatter dot plot represents the mean of PLA signals for IgM:CD19 interaction (signal counts). The experiments were repeated twice (A and B) and three (C–E) times. Shown are representative microscope images (B and E). Two-tailed Mann-Whitney t test was used (****, P < 0.0001). In these graphs, every data point is one cell; error bars show mean ± SD.
CD38 regulates the nanoscale reorganization of CD19 and IgM upon BCR engagement
We have previously shown that on unstimulated B cells the IgM-BCR and CD19 are organized in different membrane compartments and that after BCR stimulation form IgM:CD19 synapses (Kläsener et al., 2014). The formation of these synapses initiates a signaling cascade that leads to B cell activation, proliferation, and survival. Here, we sought to determine whether this process was influenced by pretreatment with DARA. To this end, we performed a set of Fab-based proximity ligation assays (Fab-PLA) on PB IgM+ B cells, either unstimulated or stimulated with α-IgM for 5 and 15 min, with or without pretreatment with DARA. We first confirmed that CD19 and IgM are well separated on resting B cells and that after BCR engagement with α-IgM, the IgM:CD19 proximity increases significantly, with the highest number of interactions at 5 min after stimulation (Fig. 4, C–E). The binding of DARA to CD38 inhibits the CD19/IgM-BCR conjugation and this may be the reason for its negative effect on BCR signaling (Fig. 4 E). Of note, we could not determine the reorganization of the CD38 molecule itself in the B cells treated with DARA, because the epitopes on CD38 recognized by the anti-CD38 antibodies used in the Fab-PLA assay were already occupied by DARA (Fig. S2).
Figure S2.

DARA occupies the epitopes recognized by the α-CD38 antibodies used in Fab-PLA. FACS histograms show CD38 expression on PBMCs with (solid black line) and without (gray filled) treatment with DARA by using two antibodies against CD38 (clones: HB-7 and HIT2).
CD38 is in close proximity to the IgM-BCR complex upon stimulation
The previous Fab-PLA experiment did not tell us whether CD38 interacts with the IgM-BCR and, if it does, whether this interaction is evident already before or only after BCR stimulation. We sought, therefore, to address whether CD38 is in close proximity to the IgM-BCR on unstimulated PB IgM+ B cells but found that this is not the case (Fig. 5 A). However, 5 min after addition of the α-IgM stimulus, CD38 was found to be in close proximity to IgM-BCR and, after 15 min, the effect was even more pronounced (Fig. 5, B and C). Based on this observation and previous data finding that CD38 colocalizes with the BCR complex upon B cell activation, we investigated whether CD38 itself polarizes into IgM-like caps upon BCR activation. We found that after 5 min activation with α-IgM, also CD38 molecules undergo reorganization into caps colocalized with those of IgM-BCR (Fig. 5 D; Deaglio et al., 2003; Funaro et al., 1993).
Figure 5.

The IgM-BCR complex and CD38 form synapses upon stimulation. (A–C) Fab-PLA study of the proximity of IgM to CD38 on unstimulated (0 min; A), or 5- and 15-min α-IgM–stimulated PB B cells (B). PLA signals are shown in red and nuclei in blue. Scale bar, 5 µm. (C) Scatter dot plot represents the mean of PLA signals for IgM:CD38 interaction (signal counts). (D) IgM and CD38 capping analyzed by confocal microscopy in unstimulated (0 min) PB B cells and after 5 min stimulation with α-IgM. Hoechst (blue), IgM (green), CD38 (magenta), and CD19 (red). Scale bar, 5 µm. (E) Fab-PLA study of the proximity of CD38 with cyt-Igα and the intracellular SYK on unstimulated (0 min), or 5- and 15-min α-IgM–stimulated PB B cells. Scatter dot plots represent the mean of PLA signals for the interactions (signal counts). (F) IgM, CD38, and CD19 capping analyzed by confocal microscopy in PB B cells and after 5-min stimulation with a cross-linker for CD38. Hoechst (blue), IgM (green), CD38 (magenta), CD19 (red). Scale bar, 5 µm. The experiments were repeated four (A–C), two (D), four (E), and three (F) times. Shown are representative microscope images. (C and E) Kruskal-Wallis test corrected for Dunn’s multiple comparisons was used (****, P < 0.0001). In these graphs, every data point is one cell; error bars show mean ± SD.
We further investigated the interaction between CD38 and the IgM-BCR components or interactors by analyzing the proximity of CD38 with the cytoplasmic region of Igα (cyt-Igα) and the spleen tyrosine kinase (SYK), both necessary for BCR signaling upon stimulation. Because we did not have antibodies that recognize intracellular CD38 epitopes, we detected CD38 with a secondary antibody, thereby covering a larger interaction range (10–60 nm) than with Fab-fragments (10–20 nm). This allowed us to determine the proximity of the extracellular CD38 with intracellular targets, e.g., cyt-Igα and SYK. We found that CD38 was not in close proximity of cytosolic-Igα or SYK in unstimulated B cells but it was upon BCR engagement with α-IgM, corroborating the association of CD38 with the BCR complex after stimulation (Fig. 5 E). Thus, CD38 interacts with the IgM-BCR only on activated B cells.
We then used a different approach in order to establish the interaction between CD38 and the IgM-BCR by inducing CD38 capping with a combination of primary antibodies against CD38 and F(ab′)2 fragments used as cross-linkers for the primary antibodies. After 5-min stimulation with the cross-linkers, CD38 aggregated on the cell surface and polarized into caps (Fig. 5 F). Both IgM and CD19 cocapped with CD38, supporting the interaction of CD38 with both molecules.
The colocalization of CD38 with CD19 is blocked by DARA
The similar interaction behavior of CD19 and CD38 with the IgM-BCR on unstimulated and stimulated B cells suggested that the two molecules might be located in the same membrane compartment. To test for this, we performed CD19:CD38 Fab-PLA on PB IgM+ B cells, either unstimulated or stimulated with α-IgM for 5 and 15 min. We found that CD19 and CD38 molecules are in close proximity already in unstimulated B cells, which indicates that indeed the two molecules reside in the same membrane compartment (Fig. 6 A). In addition, the proximity of CD19 and CD38 increased markedly 5 min after BCR stimulation but decreased again after 15 min, suggesting a higher but transient association of CD38 with CD19 on stimulated B cells (Fig. 6, B and C). Conceivably, the binding of DARA blocks the association of CD38 with CD19 and the IgM-BCR (Fig. 6 D). The transient CD19:CD38 association is seemingly not due to downregulation of either CD19 or CD38 after 15 min of stimulation since both molecules maintained the same expression levels before and after stimulation (Fig. S3), an observation that will have to await further studies in due course as it is not the focus of this study.
Figure 6.

CD19 and CD38 form synapses upon stimulation and migrate together to the IgM-BCR. (A–C) Fab-PLA study of the proximity of CD19 to CD38 on unstimulated (0 min; A), or 5- and 15-min α-IgM–stimulated PB B cells (B). PLA signals are shown in red and nuclei in blue. Scale bar, 5 µm. (C) Scatter dot plot represents the mean of PLA signals for CD19:CD38 interaction (signal counts). (D) Schematic drawing of the proposed localization and interaction of IgM, CD19, and CD38 in unstimulated and α-IgM–stimulated B cells without and with exposure to DARA. The experiment was repeated four times. Shown are representative microscope images. (C) Kruskal-Wallis test corrected for Dunn’s multiple comparisons was used (****, P < 0.0001). In these graphs, every data point is one cell; error bars show mean ± SD.
Figure S3.

CD19 and CD38 are not downregulated during IgM-BCR stimulation. The graphs show CD19, CD38, and IgM expression on unstimulated (0 min), or 5-, 15-, and 30-min α-IgM–stimulated PB B cells.
Loss of CD38 expression is associated with a decreased formation of IgM:CD19 synapses
To corroborate the importance of CD38 for the biology of the IgM-BCR, we used Ramos, an EBV-negative BL cell line that expresses IgM, CD19, and CD38, and generated a CD38-deficient counterpart using CRISPR/Cas9 (Fig. 7 A). The BCR expressed by these two cell lines is specific for the antigen NIP (4-Hydroxy-3-iodo-5-nitrophenylacetyl; He et al., 2018). We analyzed proliferation and survival of Ramos cell lines IgM-NIP and IgM-NIP CD38KO, cultured for 4 d, with and without DARA. We found that IgM-NIP cells displayed a decrease in proliferation and increase in cell death in the presence of DARA, while the IgM-NIP CD38KO Ramos B cells were not affected by this treatment (Fig. 7 B). We then performed Fab-PLA to detect IgM:CD19, IgM:CD38, and CD19:CD38 interactions in these two cell lines, either unstimulated or stimulated for 5 min with α-IgM. Already in the unstimulated IgM-NIP Ramos B cells, we observed some interactions between IgM and CD19, as well as CD38 with IgM and CD19 (Fig. 7 C). Nevertheless, all three interactions increased after stimulation with α-IgM. In the IgM-NIP CD38KO, the IgM:CD19 interactions were still observed albeit at lower levels, in both unstimulated and stimulated cells. This demonstrates that CD38 augments the interaction between IgM-BCR and CD19 in Ramos B cells. All interactions involving CD38 were not observed in the CD38KO cells, as expected. In parallel, cells were stimulated with the cognate antigen namely NIP conjugated to NIP-BSA, which resulted in a similar interaction pattern as that when the cells were stimulated with α-IgM (Fig. 7 D; Bothwell et al., 1981). All together, these experiments provide strong additional evidence to the importance of CD38 in the regulation of IgM:CD19 synapses and, ultimately, in the normal function of the IgM-BCR.
Figure 7.

CD38 deletion impairs proliferation in Ramos B cells and the interaction between IgM-BCR and CD19. (A) Histograms show the expression of IgM, CD19, and CD38 on Ramos cell lines, IgM-NIP and IgM-NIP CD38KO. (B) Cell number and percentage of dead cells (7AAD+) in Ramos cell lines, IgM-NIP and IgM-NIP CD38KO, cultured for 4 d without or with DARA. (C and D) Fab-PLA study of the proximity of IgM, CD19, and CD38 on Ramos cell lines, IgM-NIP and IgM-NIP CD38KO, unstimulated or stimulated for 5 min with either α-IgM (C) or NIP-BSA (D). (E and F) Fab-PLA study of the proximity of IgM:CD19 and cyt-Igα:SYK on IgM-NIP Ramos cells, untreated or treated with DARA, stimulated for 8 min with either α-IgM (E), α-CD40 antibody, CXCL12, or left unstimulated. (F) Scatter dot plot represents the mean of PLA signals for IgM:CD19, IgM:CD38, CD19:CD38, and cyt-Igα:SYK interactions (signal counts). The experiments were repeated twice (A and B) and at least three (C–F) times. (B) For these experiments, n = 4. Unpaired two-tailed t tests were used (***, P < 0.001; ****, P < 0.0001); error bars show mean ± SD. (C) Kruskal-Wallis test corrected for Dunn’s multiple comparisons was used (*, P < 0.05; ****, P < 0.0001). (D–F) Two-tailed Mann-Whitney t test was used (**, P < 0.01; ****, P < 0.0001). In these graphs, every data point is one cell; error bars show mean ± SD.
DARA inhibits IgM:CD19 and Igα:SYK interactions upon IgM-BCR, CD40, and CXCR4 engagement
CD19 and SYK phosphorylation and reorganization of the actin cytoskeleton are not only triggered by engagement of the BCR but also through other receptors, e.g., CD40 and CXCR4 (Keppler et al., 2015). To investigate and compare the effect of other stimuli on IgM:CD19 and cyt-Igα:SYK interactions after DARA pretreatment, we studied this in IgM-NIP Ramos B cells. As control, we determined that DARA inhibits both interactions upon α-IgM stimulation (Fig. 7 E), as might be expected based on our results above. Thereafter, we investigated the effect of DARA on these same interactions in unstimulated cells and after stimulation with either an α-CD40 antibody or the chemokine CXCL12, ligand of CXCR4. When treated with DARA, the unstimulated cells showed a significant decrease in cyt-Igα:SYK interactions, whereas it reduced also IgM:CD19 interactions after engagement of CD40 and CXCR4 (Fig. 7 F). Thus, DARA treatment affects the arrangement of BCR components also after engagement of receptors other than the IgM-BCR itself.
Discussion
On resting B cells, the IgM-BCR and the BCR coreceptor complex are localized inside different nanoscale membrane compartments. Antigen encounter through the BCR results in B cell activation and reorganization of the cell membrane. This process brings together several surface proteins, such as IgM-BCR and CD19, forming an immunological synapse and inducing the activation of intracellular pathways (Kläsener et al., 2014). CD19 is the major stimulatory coreceptor of B cells, and possesses a long cytoplasmic tail with tyrosine residues, of which most are phosphorylated upon BCR stimulation (Del Nagro et al., 2005; Fujimoto et al., 1998). This provides docking sites for downstream proteins and proximity to the BCR signaling components Igα and Igβ and lowers the threshold for B cell activation (Carter and Fearon, 1992).
In this study, we provide evidence for a regulatory role of CD38 in the IgM:CD19 synapse formation and BCR activation. Specifically, we found that the binding of CD38 with an anti-CD38 antibody (DARA) prevents formation of these synapses by hindering the migration of CD19 to the IgM-BCR. Our results are supported by previous studies that indicated a connection between CD38 and components of the BCR and its coreceptor (Deaglio et al., 2003; Deaglio et al., 2007; Funaro et al., 1993). Our findings also suggest that CD38 is, in fact, a component of the BCR coreceptor complex, being in close contact with CD19 not only in stimulated but also in unstimulated B cells. In the latter, CD38 might exert a modulatory effect on the B cell activation threshold by controlling CD19 localization. In fact, BCR-antigen recognition triggers alteration of the actin cytoskeleton, which enables mobility of cell surface molecules and allows the formation of IgM:CD19 synapses (Maity et al., 2015; Mattila et al., 2013; Treanor et al., 2010). As we show here, targeting CD38 affects proteins involved in actin cytoskeleton remodeling that seemingly prevents IgM:CD19 synapse formation. A recent study investigating membrane dynamics of MM cells upon targeting CD38 reported that ligation of CD38 with an antibody led to polar aggregation of the cell membrane, which was followed by the formation of microvesicles that eventually reached the circulation (Malavasi et al., 2021). This further supports the notion that CD38 is an important player in actin cytoskeleton remodeling.
Based on our results, CD38 appears to have a broad role in regulating the molecular dynamics on the B cell surface as DARA treatment affects the arrangement of BCR components not only through engagement of the IgM-BCR but also other receptors, i.e., CD40 and CXCR4. Since CD40 is important for B–T cell interactions, via its ligand on T cells, and CXCR4 is involved in cell migration, CD38 might play a role also in these processes.
The Ig loci are often a target of chromosomal abnormalities that render these loci inactive. Despite this, malignant B cells selectively retain BCR expression, suggesting that this receptor is essential for their survival (Küppers, 2005). Indeed, deregulated BCR signaling is a central mechanism in diverse forms of human B cell lymphoma (Davis et al., 2010; Young et al., 2019). As is the case for normal mature B cells, in BL tumors CD19 plays a major role in BCR signaling via the PI3K pathway (Schmitz et al., 2012). Our results herein show that also CD38 is central in IgM-BCR bearing BL B cell lines: targeting CD38 using DARA affects both proliferation and survival, and deleting CD38 impairs the formation of IgM:CD19 synapses. As we show here, targeting of CD38 with DARA impairs proliferation and survival in both EBV-positive and -negative BL cell lines, which suggest that the role of CD38 in IgM-BCR signaling is EBV independent. This is of importance, as the fraction of EBV-associated BL cases in the US and Europe, 20–30%, is low compared to Sub-Saharan Africa, where it is nearly 100% (Plummer et al., 2016). As most BL are IgM-positive, targeting CD38 would affect BLs whether EBV-positive or not (Magrath, 1990).
Additionally, CD38 has a complex and not fully understood enzymatic activity (Chini et al., 2021). This was highlighted using Isatuximab (ISA, trade name Sarclisa), a human IgG1κ mAb that recognizes CD38 and is also approved for MM treatment. In vitro studies performed on MM cell lines demonstrated that ISA was more efficient than DARA in inhibiting the enzymatic activity of CD38 (Martin et al., 2019). Whether the engagement of CD38 using ISA would be different from that of DARA on preventing IgM-BCR organization in normal and malignant B cells is currently unknown.
Our study not only reveals that CD38 is a major player in human B cell biology but also endorses its importance as a therapeutic target for B cell malignancies, particularly those expressing an IgM-BCR. Moreover, this is extending our understanding of the consequences of using DARA therapeutically.
Materials and methods
Human samples
PBMCs were obtained from anonymized buffy coats. As no personal information or identity was recorded, no written consent or approval by the Human Research Ethics Committee was needed (Swedish law 2003: 460, paragraphs 4 and 13). PB B cells were purified by negative selection in immunomagnetic sorting (Dynabeads Untouched Human B Cells Kit; Invitrogen). IgM+ B cells were purified with the same kit with the addition of biotinylated α-IgG and α-IgA antibodies (BioSite) in the antibody mix provided by the manufacturer, in order to remove class-switched B cells.
Flow cytometry
Cells were stained with directly labelled antibodies (Table S4) and data were acquired on FACSVerse or FACSLyric (BD Biosciences) flow cytometers, and analyzed using FlowJo software version 10 (TreeStar Inc.).
Proliferation of primary B cells
Isolated PB B cells were labelled using the CellTrace Violet (CTV) Cell Proliferation Kit (Life Technologies), according to manufacturer’s instructions, and cell proliferation was measured by CTV dilution. The cells were cultured in 96-well plates at 1 × 104 cells/well in complete medium (Table S5) and stimulated with 2.5 µg/ml goat α-human IgA/IgG/IgM F(ab′)2 fragments (α-Ig; Jackson ImmunoResearch Laboratories) in combination with 20 ng/ml human recombinant IL-2 (R&D Systems) and 2.5 µg/ml TLR9 agonist, CpG-B ODN 2006 (InvivoGen) for 5/7 d with or without antibodies recognizing CD38: α-CD38 mouse clone HB-7 (Ultra-LEAF Purified; Biolegend) and daratumumab (trade name Darzalex, human, IgG1κ, HuMax-CD38, Genmab/Janssen) both at a concentration of 1 µg/ml (de Weers et al., 2011; Laubach et al., 2014; Matas-Céspedes et al., 2017). The proliferation was assessed as expansion index. Data were acquired by flow cytometry.
Proliferation and survival of B cell lines
Namalwa cells (ATCC) were cultured in 96-well plates in complete medium (Table S5) at 2 × 104 cells/well without or with 1 µg/ml Rituximab (Chimeric human, IgG1κ; Roche) or 1 µg/ml DARA. After 4 d, cells were harvested and incubated with 7AAD (BD Biosciences) for 5 min to identify late apoptotic cells. Proliferation was determined via cell counting. The same experiment was performed without or with 1 µg/ml DARA on the following cell lines (all from ATCC): Raji, DG75, and two cell lines derived from Ramos (NIP-IgM and NIP-IgM CD38-KO). Data were acquired by flow cytometry.
Generation of Ramos NIP-IgM and NIP-IgM CD38KO
NIP-IgM and NIP-IgM CD38-KO Ramos cells were generated from H and L chains-KO, activation-induced cytidine deaminase (AID)-KO (MDL-AID-KO) Ramos cell lines as previously described (He et al., 2018). CD38-KO Ramos cells were generated using double-strand break CRISPR/Cas9 with gRNA CD38.2: 5′-TGAGTTCCCAACTTCATTAGTGG-3′ (target intron 1) and CD38.3: 5′-ACAACCCTGTTTCAGTATTC TGG-3′ (target intron 3). For the generation of NIP-IgM Ramos cells, the H and L chains were re-expressed in MDL-AID-KO Ramos cells by retroviral transduction with a construct encoding NIP–specific mouse μ-chain-P2A sequence-NIP–specific L chain.
Phosphoflow assay
The phosphorylation of ERK1/2 was analyzed using the BD PhosFlow Protocol for Human PBMCs. After isolation, PBMCs were incubated for 30 min in complete medium (Table S5) without or with DARA at 1 µg/ml. Afterwards, the samples were washed and resuspended in complete medium and left to equilibrate at 37°C for 20 min. An equal volume of prewarmed complete medium containing either α-Ig, CpG-B, or IL-2 (as described in Materials and methods) were added and the samples were incubated for 10, 30, or 60 min at 37°C, or left unstimulated (control). The samples were then fixed with BD Cytofix Fixation Buffer (BD Biosciences) for 10 min at 37°C, washed twice in Phosflow Perm/Wash Buffer I (BD Biosciences), and stained for pERK1/2 (Table S4) for 60 min at room temperature. Surface and intracellular stainings with other mAbs were performed as required by the experimental design. Samples were washed and resuspended in Phosflow Perm/Wash Buffer I for flow cytometric analysis.
Proteomic and phosphoproteomic
IgM+ PB B cells were isolated from four healthy individuals as described above. Cells were incubated without or with either a control human IgG1 (Ultra-LEAF Purified; Biolegend) or DARA, both at a concentration of 1 µg/ml for 30 min. Cells were either unstimulated or stimulated with 2.5 µg/ml goat α-human IgM F(ab′)2 fragment (α-IgM; Jackson ImmunoResearch Laboratories) for 5 min. Cell lysates were then prepared and subjected to high-resolution mass-spectrometric analysis of protein and phosphopeptides abundance on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific), which was normalized to total protein abundance. Analysis of processed phosphoproteomic data, hierarchical clustering, and heat-map production were performed with Qlucore Omics Explorer 3.5 (Qlucore AB) and followed by GO functional enrichment analysis using the web-based toolset g:Profiler (https://biit.cs.ut.ee/gprofiler). GO results with terms containing a number of genes between 5 and 350 and with a P ≤ 0.05 were included. The terms collected with g:Profiler were then analyzed with Cytoscape App 3.8.2 (http://www.cytoscape.org) and EnrichmentMap (http://www.baderlab.org).
BCR capping and downregulation
PBMCs were incubated for 30 min without (untreated control) and with 1 µg/ml DARA and then washed. IgM-BCR capping and downregulation were induced with 2.5 µg/ml α-IgM for 5, 15, and 30 min. IgM-BCR capping images were acquired on an ImageStream X Mark II imaging flow cytometer (Amnis) and analyzed using the Δ Centroid XY feature in the IDEAS software (Amnis).
Proximity ligation assay (PLA)
PLA experiments were performed as previously described (Kläsener et al., 2018). To generate PLA probes against specific targets, the following unlabeled antibodies were used: α-IgM, α-CD19, α-CD38, α-Igα, and α-SYK (Table S4). To generate Fab-fragments, the buffer of the antibodies was exchanged to PBS with Tween using a Microcon-30 kD centrifugal filter unit (MRCF0R030; Merck Millipore). All Fab fragments were prepared with Pierce Fab Micro preparation kit (Thermo Fisher Scientific) using immobilized papain. For the detection of intracellular interactions of membrane CD38 with Fab α-Igα or α-SYK, we used the secondary α-rabbit antibody for CD38 detection. In brief, after desalting (Zeba spin desalting columns; Thermo Fisher Scientific), all antibodies were coupled with PLA Probemaker Plus or Minus oligonucleotides (Sigma-Aldrich) to generate PLA probes. IgM+ PB B cells were isolated and incubated without or with DARA, as described above. IgM+ PB B cells and cells from two cell lines derived from the Ramos B cell line (NIP-IgM and NIP-IgM CD38-KO) were settled on polytetrafluoroethylene (PTFE) slides (Thermo Fisher Scientific) for 30 min at 37°C. Cells were stimulated for 5 or 15 min with 2.5 or 10 µg/ml α-IgM or left unstimulated and fixed for 20 min with 4% paraformaldehyde. Further experiments on NIP-IgM Ramos B cells were performed where we incubated the cells without or with DARA, as described above, and stimulated for 8 min with either α-IgM (10 µg/ml), α-CD40 antibody (5 µg/ml), CXCL12 (100 ng/ml), or left unstimulated, and then fixed for 20 min with 4% paraformaldehyde. For intracellular PLA, cells were permeabilized with 0.5% saponin for 30 min at room temperature, and blocked for 30 min with blocking buffer (25 μg/ml sonicated salmon sperm DNA, 250 μg/ml BSA, 1 M glycine). PLA was performed with Duolink In Situ Orange (Sigma-Aldrich). Samples were directly mounted on slides with DAPI Fluoromount-G (Southern Biotech) to visualize the PLA signals in relationship to the nuclei. Microscopy images were acquired with a Leica DMi8 microscope (Leica-microsystems). For each experiment, a minimum of 500 primary B cells or Ramos B cells were analyzed with CellProfiler-3.0.0 (https://cellprofiler.org/).
Confocal microscopy
IgM+ PB B cells (isolated as above) were settled on PTFE slides for 30 min at 37°C. Cells were stimulated with 2.5 µg/ml α-IgM for 5 min or left unstimulated and fixed for 20 min with 4% paraformaldehyde. The cells were stained for 1 h at 4°C with primary antibodies and then incubated for 30 min at 4°C with secondary antibodies and Hoechst 34580 (Life Technologies). For the CD38 cross-linking experiment, cells were settled on PTFE slides and then stained with a mouse α-human-CD38 antibody for 1 h at 4°C and then incubated 30 min at 4°C with AF555 labeled α-mouse-IgG F(ab′)2 fragments as cross-linkers. The cells were then washed and incubated 5 min at 37°C in order to induce cross-linking and then fixed and stained as described above. Antibodies and dilutions are listed in Table S4. Cells were mounted with ProLong Gold Antifade Mountant (Life Technologies) and acquired using a LSM700 (Carl Zeiss AG). Image analysis was performed with ImageJ software (National Institutes of Health).
Statistical analysis
Biological replicates and presentation in each figure are shown as mean ± SD as mentioned in the figure legends. No statistical methods were used to predetermine sample size. Data distribution was assumed to be normal. No data exclusion was performed. Mean values and SDs are displayed in the plots. The statistical significance of differences between samples and treatments was determined using unpaired or paired two-tailed t tests or ordinary one-way ANOVA corrected for Tukey’s multiple comparisons, as appropriate. The statistical significance of differences between samples with unequal size, i.e., experiments where every data point is one cell, was determined using two-tailed Mann-Whitney t test or Kruskal-Wallis test corrected for Dunn’s multiple comparisons, as appropriate. All the statistical analyses, with the exception of the phosphoproteomic, were performed with Graph-Pad Prism version 8 (La Jolla). Levels of statistical significance are designated with asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Nonstatistical significant differences were not reported in figures.
Online supplemental material
Fig. S1 shows pERK expression kinesis with different stimulations in PB B cells. Fig. S2 shows that DARA occupies the epitopes recognized by the α-CD38 antibodies used in Fab-PLA. Fig. S3 shows that CD19 and CD38 are not downregulated during IgM-BCR stimulation. Table S1 shows a list of the proteins that are present downstream the BCR signaling pathway, including name of the protein and protein ID. Table S2 shows a list of those phosphopeptides in the α-IgM–stimulated B cells that were significantly affected by DARA pretreatment. Table S3 shows the GO enrichment analysis of all significantly changed phosphopeptides identified in Table S2. Table S4 shows a list of antibodies used in the study, including fluorochrome, clone, name of the company where it was purchased, and dilution. Table S5 shows the complete media composition used in the study, including company and concentration of each reagent.
Supplementary Material
shows a list of the proteins that are present downstream the BCR signaling pathway, including name of the protein and protein ID.
shows a list of those phosphopeptides in the α-IgM–stimulated B cells that were significantly affected by DARA pretreatment.
shows the GO enrichment analysis of all significantly changed phosphopeptides identified in Table S2.
shows a list of antibodies used in the study, including fluorochrome, clone, name of the company where it was purchased, and dilution.
shows the complete media composition used in the study, including company and concentration of each reagent.
Acknowledgments
We are thankful to the Proteomics Core Facility of Sahlgrenska Academy, University of Gothenburg that performed the quantitative proteomic analysis and in particular Evelin Berger and Egor Vorontsov.
This work was supported by the Swedish Research Council (2018-03128), the Swedish Cancer Foundation (19 0464), the Swedish Childhood Cancer Fund (PR2018-0170, PR2020-0147, TJ2019-0098), King Gustav V’s 80-year Foundation (SGI-2018-0510, FAI-2019-0618, FAI-2020-0706), Assar Gabrielsson’s Foundation (FB19-66, FB20-83, FB21-104), Elisabeth Bollan Lindén-stipendiet, Wenner-Gren Foundations, Reumatikerförbundet, ALF (agreement between the Swedish government and the county council), Kungl. Vetenskaps- och Vitterhets-Samhället, Adlerbertska stiftelsen, Stiftelsen Apotekare Hedbergs fond för medicinsk forskning, Stiftelsen Samariten, Amlövs Stiftelser, Lundgrens Stiftelse, Göteborgsregionens Stiftelse för Reumatologisk Forskning, Ingabritt och Arne Lundbergs Forskningsstiftelse, the German Research Foundation (Deutsche Forschungsgemeinschaft) through TRR130 to M. Reth, and Roche Innovation Center Zurich.
Author contributions: A. Camponeschi, K. Kläsener, M. Reth, and I-L. Mårtensson designed the study. A. Camponeschi, K. Kläsener, T. Sundell, C. Lundqvist, M. Sundqvist, N. Ayoubzadeh, M. Gatto, and K. Önnheim contributed to the procurement and processing of samples and acquired and analyzed flow cytometry and microscopy data. A. Camponeschi and P.T. Manna analyzed the proteomic and phosphoproteomic data. P.T. Manna provided statistical expertise. O. Ekwall, H. Forsman, K. Thorarinsdottir, K. Önnheim, I. Gjertsson, M. Reth, and I-L. Mårtensson carried out or contributed essential reagents and materials for the experiments. P.T. Manna, M. Visentini, A. Aranburu, and L. Fogelstrand contributed substantially to the discussions. A. Camponeschi, K. Kläsener, M. Reth, and I-L. Mårtensson wrote the manuscript with contributions from all the authors.
Data availability
All raw and processed data of the 32 datasets from the proteomic and phosphoproteomic experiments are available from the corresponding authors upon reasonable request.
References
- Aarhus, R., Graeff R.M., Dickey D.M., Walseth T.F., and Lee H.C.. 1995. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 270:30327–30333. 10.1074/jbc.270.51.30327 [DOI] [PubMed] [Google Scholar]
- Bothwell, A.L., Paskind M., Reth M., Imanishi-Kari T., Rajewsky K., and Baltimore D.. 1981. Heavy chain variable region contribution to the NPb family of antibodies: Somatic mutation evident in a gamma 2a variable region. Cell. 24:625–637. 10.1016/0092-8674(81)90089-1 [DOI] [PubMed] [Google Scholar]
- Carter, R.H., and Fearon D.T.. 1992. CD19: Lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 256:105–107. 10.1126/science.1373518 [DOI] [PubMed] [Google Scholar]
- Chini, C.C.S., Zeidler J.D., Kashyap S., Warner G., and Chini E.N.. 2021. Evolving concepts in NAD+ metabolism. Cell Metabol. 33:1076–1087. 10.1016/j.cmet.2021.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, R.E., Ngo V.N., Lenz G., Tolar P., Young R.M., Romesser P.B., Kohlhammer H., Lamy L., Zhao H., Yang Y., et al. 2010. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 463:88–92. 10.1038/nature08638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora, A., Zocchi E., Guida L., Franco L., and Bruzzone S.. 2004. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N. Y Acad. Sci. 1028:176–191. 10.1196/annals.1322.021 [DOI] [PubMed] [Google Scholar]
- de Weers, M., Tai Y.T., van der Veer M.S., Bakker J.M., Vink T., Jacobs D.C.H., Oomen L.A., Peipp M., Valerius T., Slootstra J.W., et al. 2011. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 186:1840–1848. 10.4049/jimmunol.1003032 [DOI] [PubMed] [Google Scholar]
- Deaglio, S., Capobianco A., Bergui L., Dürig J., Morabito F., Dührsen U., and Malavasi F.. 2003. CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood. 102:2146–2155. 10.1182/blood-2003-03-0989 [DOI] [PubMed] [Google Scholar]
- Deaglio, S., Morra M., Mallone R., Ausiello C.M., Prager E., Garbarino G., Dianzani U., Stockinger H., and Malavasi F.. 1998. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 160:395–402 [PubMed] [Google Scholar]
- Deaglio, S., Vaisitti T., Billington R., Bergui L., Omede’ P., Genazzani A.A., and Malavasi F.. 2007. CD38/CD19: A lipid raft-dependent signaling complex in human B cells. Blood. 109:5390–5398. 10.1182/blood-2006-12-061812 [DOI] [PubMed] [Google Scholar]
- Del Nagro, C.J., Otero D.C., Anzelon A.N., Omori S.A., Kolla R.V., and Rickert R.C.. 2005. CD19 function in central and peripheral B-cell development. Immunol. Res. 31:119–131. 10.1385/IR:31:2:119 [DOI] [PubMed] [Google Scholar]
- Fujimoto, M., Poe J.C., Inaoki M., and Tedder T.F.. 1998. CD19 regulates B lymphocyte responses to transmembrane signals. Semin. Immunol. 10:267–277. 10.1006/smim.1998.9999 [DOI] [PubMed] [Google Scholar]
- Funaro, A., De Monte L.B., Dianzani U., Forni M., and Malavasi F.. 1993. Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. Eur. J. Immunol. 23:2407–2411. 10.1002/eji.1830231005 [DOI] [PubMed] [Google Scholar]
- He, X., Kläsener K., Iype J.M., Becker M., Maity P.C., Cavallari M., Nielsen P.J., Yang J., and Reth M.. 2018. Continuous signaling of CD79b and CD19 is required for the fitness of Burkitt lymphoma B cells. EMBO J. 37:e97980. 10.15252/embj.201797980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, D.G., and Bell J.I.. 1990. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J. Immunol. 144:2811–2815 [PubMed] [Google Scholar]
- Keppler, S.J., Gasparrini F., Burbage M., Aggarwal S., Frederico B., Geha R.S., Way M., Bruckbauer A., and Batista F.D.. 2015. Wiskott-Aldrich syndrome interacting protein deficiency uncovers the role of the co-receptor CD19 as a generic hub for PI3 kinase signaling in B cells. Immunity. 43:660–673. 10.1016/j.immuni.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kläsener, K., Maity P.C., Hobeika E., Yang J., and Reth M.. 2014. B cell activation involves nanoscale receptor reorganizations and inside-out signaling by Syk. Elife. 3:e02069. 10.7554/eLife.02069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kläsener, K., Yang J., and Reth M.. 2018. Study B cell antigen receptor nano-scale organization by in situ fab proximity ligation assay. Methods Mol. Biol. 1707:171–181. 10.1007/978-1-4939-7474-0_12 [DOI] [PubMed] [Google Scholar]
- Kumar, S., Kimlinger T., and Morice W.. 2010. Immunophenotyping in multiple myeloma and related plasma cell disorders. Best Pract. Res. Clin. Haematol. 23:433–451. 10.1016/j.beha.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki, T., Takata M., Yamanashi Y., Inazu T., Taniguchi T., Yamamoto T., and Yamamura H.. 1994. Syk activation by the Src-family tyrosine kinase in the B cell receptor signaling. J. Exp. Med. 179:1725–1729. 10.1084/jem.179.5.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küppers, R. 2005. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer. 5:251–262. 10.1038/nrc1589 [DOI] [PubMed] [Google Scholar]
- Laubach, J.P., Tai Y.T., Richardson P.G., and Anderson K.C.. 2014. Daratumumab granted breakthrough drug status. Expert Opin. Investig. Drugs. 23:445–452. 10.1517/13543784.2014.889681 [DOI] [PubMed] [Google Scholar]
- Lee, H.C., Deng Q.W., and Zhao Y.J.. 2022. The calcium signaling enzyme CD38: A paradigm for membrane topology defining distinct protein functions. Cell Calcium. 101:102514. 10.1016/j.ceca.2021.102514 [DOI] [PubMed] [Google Scholar]
- Lee, J., Sengupta P., Brzostowski J., Lippincott-Schwartz J., and Pierce S.K.. 2017. The nanoscale spatial organization of B-cell receptors on immunoglobulin M- and G-expressing human B-cells. Mol. Biol. Cell. 28:511–523. 10.1091/mbc.e16-06-0452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrath, I. 1990. The pathogenesis of Burkitt’s lymphoma. Adv. Cancer Res. 55:133–270. 10.1016/s0065-230x(08)60470-4 [DOI] [PubMed] [Google Scholar]
- Maity, P.C., Yang J., Klaesener K., and Reth M.. 2015. The nanoscale organization of the B lymphocyte membrane. Biochim. Biophys. Acta. 1853:830–840. 10.1016/j.bbamcr.2014.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavasi, F., Deaglio S., Funaro A., Ferrero E., Horenstein A.L., Ortolan E., Vaisitti T., and Aydin S.. 2008. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 88:841–886. 10.1152/physrev.00035.2007 [DOI] [PubMed] [Google Scholar]
- Malavasi, F., Faini A.C., Morandi F., Castella B., Incarnato D., Oliviero S., Horenstein A.L., Massaia M., van de Donk N.W.C.J., and Richardson P.G.. 2021. Molecular dynamics of targeting CD38 in multiple myeloma. Br. J. Haematol. 193:581–591. 10.1111/bjh.17329 [DOI] [PubMed] [Google Scholar]
- Martin, T.G., Corzo K., Chiron M., Velde H.V., Abbadessa G., Campana F., Solanki M., Meng R., Lee H., Wiederschain D., et al. 2019. Therapeutic opportunities with pharmacological inhibition of CD38 with Isatuximab. Cells. 8:1522. 10.3390/cells8121522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matas-Céspedes, A., Vidal-Crespo A., Rodriguez V., Villamor N., Delgado J., Giné E., Roca-Ho H., Menéndez P., Campo E., López-Guillermo A., et al. 2017. The human CD38 monoclonal antibody daratumumab shows antitumor activity and hampers leukemia-microenvironment interactions in chronic lymphocytic leukemia. Clin. Cancer Res. 23:1493–1505. 10.1158/1078-0432.CCR-15-2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila, P.K., Feest C., Depoil D., Treanor B., Montaner B., Otipoby K.L., Carter R., Justement L.B., Bruckbauer A., and Batista F.D.. 2013. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity. 38:461–474. 10.1016/j.immuni.2012.11.019 [DOI] [PubMed] [Google Scholar]
- Plummer, M., de Martel C., Vignat J., Ferlay J., Bray F., and Franceschi S.. 2016. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health. 4:e609–e616. 10.1016/S2214-109X(16)30143-7 [DOI] [PubMed] [Google Scholar]
- Portis, T., and Longnecker R.. 2004. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene. 23:8619–8628. 10.1038/sj.onc.1207905 [DOI] [PubMed] [Google Scholar]
- Sanchez, L., Wang Y., Siegel D.S., and Wang M.L.. 2016. Daratumumab: A first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol. 9:51. 10.1186/s13045-016-0283-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz, R., Young R.M., Ceribelli M., Jhavar S., Xiao W., Zhang M., Wright G., Shaffer A.L., Hodson D.J., Buras E., et al. 2012. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 490:116–120. 10.1038/nature11378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treanor, B., Depoil D., Gonzalez-Granja A., Barral P., Weber M., Dushek O., Bruckbauer A., and Batista F.D.. 2010. The membrane skeleton controls diffusion dynamics and signaling through the B cell receptor. Immunity. 32:187–199. 10.1016/j.immuni.2009.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine, M.A., Meier K.E., Rossie S., and Clark E.A.. 1989. Phosphorylation of the CD20 phosphoprotein in resting B lymphocytes. Regulation by protein kinase C. J. Biol. Chem. 264:11282–11287. 10.1016/s0021-9258(18)60461-2 [DOI] [PubMed] [Google Scholar]
- Wellbrock, C., Karasarides M., and Marais R.. 2004. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5:875–885. 10.1038/nrm1498 [DOI] [PubMed] [Google Scholar]
- Young, R.M., Phelan J.D., Wilson W.H., and Staudt L.M.. 2019. Pathogenic B-cell receptor signaling in lymphoid malignancies: New insights to improve treatment. Immunol. Rev. 291:190–213. 10.1111/imr.12792 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
shows a list of the proteins that are present downstream the BCR signaling pathway, including name of the protein and protein ID.
shows a list of those phosphopeptides in the α-IgM–stimulated B cells that were significantly affected by DARA pretreatment.
shows the GO enrichment analysis of all significantly changed phosphopeptides identified in Table S2.
shows a list of antibodies used in the study, including fluorochrome, clone, name of the company where it was purchased, and dilution.
shows the complete media composition used in the study, including company and concentration of each reagent.
Data Availability Statement
All raw and processed data of the 32 datasets from the proteomic and phosphoproteomic experiments are available from the corresponding authors upon reasonable request.