

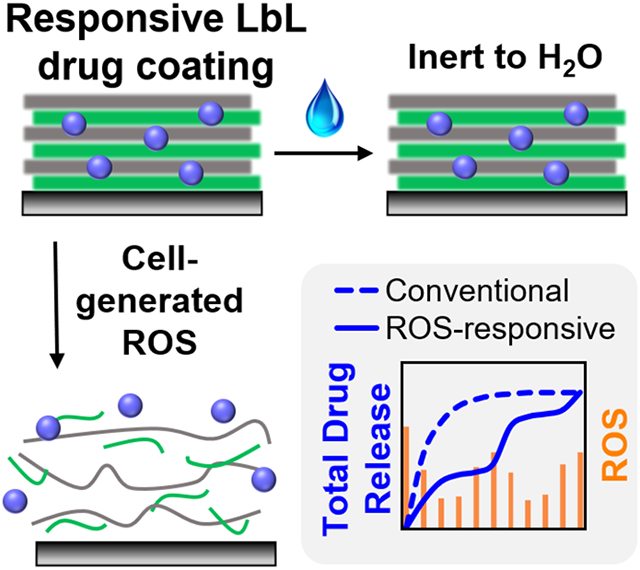
Abstract
Polyelectrolyte multilayer (PEM) coatings, constructed on the surfaces of tissue engineering scaffolds using layer-by-layer assembly (LbL), promote sustained release of therapeutic molecules and have enabled regeneration of large-scale, pre-clinical bone defects. However, these systems primarily rely on non-specific hydrolysis of PEM components to foster drug release, and their pre-determined drug delivery schedules potentially limit future translation into innately heterogeneous patient populations. To trigger therapeutic delivery directly in response to local environmental stimuli, an LbL-compatible polycation solely degraded by cell-generated reactive oxygen species (ROS) was synthesized. These thioketal-based polymers were selectively cleaved by physiologic doses of ROS, stably incorporated into PEM films alongside growth factors, and facilitated tunable release of therapeutic bone morphogenetic protein-2 (BMP-2) upon oxidation. These coatings' sensitivity to oxidation was also dependent on the polyanions used in film construction, providing a simple method for enhancing ROS-mediated protein delivery in vitro. Correspondingly, when implanted in critically-sized rat calvarial defects, the most sensitive ROS-responsive coatings generated a 50% increase in bone regeneration compared with less sensitive formulations and demonstrated a nearly three-fold extension in BMP-2 delivery half-life over conventional hydrolytically-sensitive coatings. These combined results highlight the potential of environmentally-responsive PEM coatings as tunable drug delivery systems for regenerative medicine.
Keywords: layer-by-layer, reactive oxygen species, BMP-2, responsive drug delivery, bone
Graphical Abstract
Polyelectrolyte multilayer coatings constructed with selectively ROS-degradable polymers and the osteogenic growth factor BMP-2 are sensitive to physiologic levels of oxidation, feature robust layer-by-layer film construction with adjustable drug loading, and promote tunable, responsive protein release. Scaffolds coated with the most sensitive ROS-responsive BMP-2 coatings correspondingly promote sustained drug delivery and enhance bone regeneration in critically-sized rat calvarial defects, validating oxidation-mediated drug release in vivo.
1. Introduction
Over half a million bone grafting procedures are performed annually in the United States,[1] and despite the resultant need for bone-substituting materials, regenerating large-scale orthopedic defects in the clinic remains challenging. The current gold-standard treatment involves harvesting autologous bone and surgically integrating the extracted tissue into the defect site; however, this approach is constrained by both donor-site morbidity and the limited amount of harvestable donor bone available in a given patient.[2] Accordingly, considerable research effort has been expended to develop generalizable, synthetic bone grafts comprising biodegradable scaffolds that locally deliver regenerative therapeutics. Natural bone healing relies on cell signaling pathways that are primarily directed by secreted growth factors, including the highly osteoinductive bone morphogenetic protein-2 (BMP-2).[3] BMP-2 has been identified as the most potent member of the BMP family due to its ability to promote cellular proliferation of progenitor cells and spur stem cell differentiation into bone-forming osteoblasts.[4] Moreover, this therapeutic protein has been heavily investigated in drug delivery applications,[5] and was introduced in 2002 in an FDA-approved clinical formulation (INFUSE® Bone Graft from Medtronic[6]) that releases BMP-2 from an absorbable collagen sponge for certain spinal, dental, and maxillofacial indications.
However, many of these biomaterial-based BMP-2 delivery systems discharge the majority of their drug payload in a short-duration bolus over the first several minutes to hours,[7] thereby necessitating that these materials contain high protein doses to compensate for the poorly-controlled release kinetics and rapid clearance.[8] The physiologically-inappropriate BMP-2 dosing facilitated by these carriers not only causes damaging side-effects,[9] but also prematurely depletes the encapsulated growth factor reservoir and can lead to incomplete bone regeneration.[10] Motivated by these persistent shortcomings, many groups have explored the impact of BMP-2 dosing[7a, 11] and release kinetics[7b] from biomaterial implants alongside methodologies for tuning these parameters. In particular, polyelectrolyte multilayer (PEM) thin film deposition represents a promising technology for facilitating localized, highly tunable therapeutic delivery from implanted biomaterials.[12]
Previous work has demonstrated that PEM assemblies can form nanolayered surface coatings on a variety of scaffold materials and are easily fabricated under mild aqueous conditions from a layer-by-layer (LbL) iterative adsorption[13] of charged species. Moreover, the incorporation of therapeutic compounds, including nucleic acids,[14] small molecules,[15] or proteins,[16] into the PEM architecture alongside a biodegradable polymer allows for the pre-programmed release of drug cargo from the film coating.[17] These films have primarily utilized hydrolytically-degradable, cationic poly(β-amino ester) (PBAE) polymers that break down over time in the presence of water via ester hydrolysis[18] to discharge the film-encapsulated payloads. Modulation strategies for tuning PEM drug release kinetics include modifying the hydrophobicity of the PBAE,[19] creating covalent cross-links between layers,[20] incorporating barrier layers in the film architecture,[21] or staggering depositions of different therapeutics to foster sequential release of multiple payloads.[22] In our previous orthopedic work, we have demonstrated extended BMP-2 release from LbL thin film coatings to generate bone both around a non-degradable tibial implant[23] and within a critically-sized cranial defect.[22a]
However, these hydrolytically degradable PEM systems are minimally-responsive to the local tissue environment and are primarily dependent on previously determined drug delivery kinetics for achieving tissue regeneration. Therefore, this work sought to develop a PEM coating that delivers therapeutics directly and selectively in response to local tissue repair signaling and healing-specific environmental stimuli. Many recent drug delivery technologies have employed materials that respond to external triggers, though most promising are “smart” systems[24] that enable on-demand drug release in response to tissue-produced signals, including pH levels,[25] enzymatic activity, or the presence of reactive oxygen species (ROS). ROS are particularly attractive for facilitating material responsiveness since these cell-specific stimuli are highly associated with bone growth and remodeling. ROS are important mediators in many biological processes and the body’s immune response,[26] though ROS levels are also elevated across a number of pathologies[27] and at sites of inflammation and tissue injury.[28] In orthopedics specifically, mature osteocytes actively remodeling bone tissue have been shown to generate increased ROS as part of the natural healing cycle,[29] further motivating the targeting of ROS as a selective mediator of therapeutic release from PEM-coated implants.
Several polymer-based technologies that utilize cellular oxidation as a selective trigger for biomaterial responsiveness have recently emerged,[30] including ROS-degradable tissue engineering constructs[31] and oxidation-sensitive drug delivery vehicles. These triggerable therapeutic carriers include nanoparticles,[32] microparticles,[33] and hydrogels[31e, 31l] that specifically deliver payloads to intracellular or extracellular targets featuring high levels of oxidative stress. Though some groups have employed positively charged, oxidation-sensitive polymers for nanoparticle fabrication,[34] to our knowledge ROS-degradable polymers have never been employed in the construction of LbL coatings. To confer ROS-responsiveness to different biomaterials, many groups have employed thioketal-based polymers due to their ease of synthesis,[35] sensitivity to many different ROS,[34a] and oxidation-specific degradation.[31b] Herein, we report the development of a thioketal-based polycation that forms stable PEM coatings with growth factor proteins upon LbL assembly, selectively releases these therapeutics upon oxidation, and features tunable sensitivity to ROS-mediated protein delivery. Most importantly, the oxidation-responsive drug release kinetics from PEM-coated implants directly correlate with bone growth outcomes in critically-sized calvarial defects. These findings highlight the utility of environmentally-responsive therapeutic delivery in orthopedic regeneration, and the potential to incorporate this principle for LbL-generated delivery systems.
2. Results and Discussion
2.1. Synthesis and Characterization of an ROS-Degradable, LbL-Compatible Polycation
To synthesize the LbL-compatible poly(thioketal β-amino amide) (PTK-BAA) polycation, the terminal amine group in a cysteamine monomer was successfully protected with ethyl trifluoroacetate before further reacting with 2,2-dimethoxypropane to form a protected thioketal diamine monomer as demonstrated in Figure 1A. Following deprotection, the thioketal diamine was further functionalized with reactive acrylamide groups and then polymerized via Michael addition condensation with 4,4′-trimethylene dipiperidine to generate the PTK-BAA polycation (Figure 1A). A conventional PBAE polymer was similarly synthesized as shown in Figure 1B. These Michael addition polymerizations leave both polycations with ionizable tertiary amines in the polymer backbone, though the PTK-BAA replaces the PBAE’s hydrolytically-cleaved esters with hydrolytically-inert amides and introduces ROS-sensitive thioketal units in the main polymer sequence to channel chain scission exclusively through oxidative degradation.
Figure 1.

Synthetic scheme for LbL-compatible polycations, (A) the oxidation-sensitive poly(thioketal β-amino amide) (PTK-BAA) and (B) the hydrolytically-degradable poly(β-amino ester) (PBAE). Both polycations are polymerized using Michael addition condensation and contain ionizable tertiary amines in the polymer backbone. However, the PTK-BAA features ROS-sensitive thioketal units and minimally-degradable amide linkers while the PBAE is degraded through the hydrolytically-labile ester bonds. Successful PTK-BAA polymer synthesis was confirmed by (C) 1H NMR spectroscopy and by (D) MALDI-TOF mass spectrometry. The presence of predicted polymerization products in the polymer bulk further validates the successful step-growth synthesis.
The PTK-BAA structure was confirmed with 1H NMR (Figure 1C), while MALDI-TOF analysis also confirmed the presence of predicted polymerization products and established the bulk polymer’s number average molecular weight (Mn) as 2267 g mol−1 with a polydispersity index (PDI) of 1.36 (Figure 1D). 1H NMR also indicated the successful formation of the PBAE polycation, while GPC analysis showed a polymer Mn of 6621 g mol−1 with a PDI of 1.48. As indicated in previous reports,[34a] the PTK-BAA Michael addition condensation of diacrylamide monomers yielded relatively low molecular weight polymers compared with PBAEs polymerized with diacrylate precursors (~2.3kDa vs ~6.6kDa). The diacrylamide polymerization, carried out for 24h at room temperature, was also relatively insensitive to longer reaction times or higher temperatures while converting to the highest molecular weight when synthesized in a solvent mix of ethanol and water[36] instead of conventionally used tetrahydrofuran (THF) or dimethylformamide (DMF). To serve as a direct non-degradable control for the PTK-BAA, the thioketal-less polycation hexane-poly(β-amino amide) (hexane-PBAA) was also successfully synthesized (Figure S1).
To demonstrate the selective ROS-sensitivity of the thioketal-containing PTK-BAA polycations, polymer samples were incubated in escalating doses of hydrogen peroxide (H2O2) and evaluated for thioketal degradation by 1H NMR. As shown in Figure 2A, the thioketal is unchanged after incubation in pure water but demonstrates dose-dependent degradation with increasing ROS treatment. MALDI-TOF analysis of the 10 mM H2O2-degraded PTK-BAA also demonstrated the elimination of higher molecular weight polymerization products and identified low molecular weight species that corresponded well with predicted oxidized degradation products (Figure 2B). Selective thioketal sensitivity to oxidation is well documented,[30b] though encouragingly PTK polymer degradation could be further amplified in vivo by more highly reactive compounds (particularly hydroxyl radicals)[31c, 34a] that are commonly produced in regenerating tissues. Cytotoxicity of the PTK-BAA polymer and its degradation products with MC3T3-E1 pre-osteoblast cells was also determined in comparison to no treatment, treatment with a cytotoxic control 25kDa LPEI polymer, and treatment with the conventional PBAE polycation used in previous PEM film formulations (Figure S2). As measured by the Cell Titer Glo assay of relative viable cell number, all treatments exhibited significantly higher viability than LPEI treatment as expected while cells incubated with the parent and degraded PTK-BAA polymers were not statistically different from no treatment. Though these results only featured cells treated with film constituents rather than intact films, previous work conclusively demonstrated robust cell attachment to similarly constructed LbL assemblies[37] and motivated the continued exploration of these novel coatings.
Figure 2.

In vitro PTK-BAA degradation. Polymer samples were incubated for 72h in escalating doses of H2O2 and evaluated by (A) NMR, indicating that the characteristic thioketal peak underwent dose-dependent degradation. (B) Polymer samples incubated in 10 mM H2O2 and evaluated by MALDI-TOF also demonstrated the complete disappearance of higher molecular weight signal and generated species corresponding with predicted polymer degradation products.
2.2. LbL Film Construction, Characterization, and In Vitro Drug Release
To first demonstrate the LbL-compatibility of PTK-BAA polycations, model silicon substrates were alternatively dipped in aqueous baths of polyelectrolytes at pH 5.0 to form repeating tetralayer units. The initial layer was the ROS-degradable PTK-BAA polycation, which was followed by the polyanion 450 kDa poly(acrylic acid) (PAA), the cationic model protein lysozyme (fluorescently tagged for quantitation), and then finally PAA again; these tetralayer cycles were repeated 30 times to construct the complete LbL films on model silicon substrates as shown in Figure S3A. PAA is minimally cytotoxic and effectively sequesters cationic growth factors into PEM films due to its high density of ionizable carboxylate groups,[22b] while the highly charged lysozyme (isoelectric point 11.35) has been previously employed as a low-cost model protein for encapsulation studies in LbL assembly.[16a, 38] Film-coated silicon samples were incubated in phosphate buffered saline (PBS, pH 7.4) with escalating quantities of hydrogen peroxide and demonstrated significant dose-dependent protein release as shown in Figure S3B. Though a minor amount of lysozyme is released during incubation in pure PBS, likely initiated by weakened ionic bonds and film swelling during the transition from pH 5.0 to 7.4,[39] the substantial increase in protein liberation seen with peroxide treatment strongly implicates oxidation as the chief driver of drug delivery from PTK-BAA films.
With ROS-triggered protein delivery from PEM constructs successfully validated, PTK-BAA film coatings were fabricated with the therapeutic growth factor protein BMP-2 and characterized. PEM films created on silicon with PTK-BAA, 450 kDa PAA, and BMP-2 displayed a linear increase in film thickness as a function of tetralayer depositions (Figure 3A), indicating robust LbL assembly. Next, films were similarly constructed with fluorescently tagged BMP-2 on 1 mm-thick, microporous poly(lactic-co-glycolic) acid (PLGA) scaffolds fabricated by solvent phase-separation.[22a] Importantly, the LbL dip-coating process formed PEM films on both the top and bottom PLGA surfaces to double the allowable drug loading per sample, and measured BMP-2 loadings (totaled from both sides of each coated scaffold, giving a cumulative surface area of ~1 cm2 for an 8.0 mm diameter sample) displayed a linear increase with increasing tetralayer additions (Figure 3B-C). This tunable therapeutic encapsulation is therefore easily controlled by varying the number of layer depositions to target a specific drug dosing per surface area of coated substrate.
Figure 3.

Characterization of PTK-BAA films fabricated with BMP-2 and 450 kDa PAA. (A) Profilometry measurements of film thickness vs. number of tetralayer depositions indicates controlled, linear film growth. Tunability of total BMP-2 loading in PTK-BAA films via number of tetralayer depositions is demonstrated by (B) IVIS imaging of Cy5-labeled BMP-2, and (C) ELISA measurements of total BMP-2 recovered from degraded films.
In vitro BMP-2 release kinetics from the oxidation-sensitive films were next determined and compared against drug discharge from conventional hydrolytically-degradable PEM films fabricated with PBAE polycations. Film-coated PLGA scaffolds (30 tetralayers) were incubated in PBS or 1 mM H2O2-doped PBS and evaluated for BMP-2 release into the supernatant. As shown in Figure 4A, PBAE film coatings released the majority of their complexed growth factor over the first 24h and demonstrated no difference in release kinetics between PBS or ROS-treated samples. However, PTK-BAA film coatings released relatively little BMP-2 in saline alone but expelled significantly more protein when treated with 1 mM H2O2 (Figure 4A). Polyester-based materials have previously demonstrated minimal responsiveness to ROS-mediated degradation,[31b] and the exhibited initial bolus release of growth factor from PEM films is in line with prior reports.[22a]
Figure 4.

Oxidation-responsive BMP-2 release from PTK-BAA films in vitro. (A) Conventional PBAE films primarily release their BMP-2 payload in a bolus over the first 24h regardless of 1 mM H2O2 treatment, while PTK-BAA films release significantly more BMP-2 in the peroxide media compared against release in pure PBS. (B) PTK-BAA / BMP-2 film-coated samples incubated in constant doses of 1 mM H2O2 released protein consistently over time, whereas samples intermittently incubated in PBS or peroxide released significantly less protein in PBS but equivalent BMP-2 amounts when incubated in H2O2. During the later cycles, the “on-off” responsiveness behavior is delayed but still apparent. All PEM films were constructed with 30 tetralayers on PLGA scaffolds and utilized 450 kDa PAA as the polyanion (n=3 per treatment, *p<0.05).
To further assess the ROS-responsiveness of the PTK-BAA / BMP-2 coatings, drug release from similarly fabricated samples was evaluated with either a “constant” 1 mM H2O2 medium or with a “pulsed” medium that was switched back and forth between PBS and 1 mM H2O2 every 4 days as shown in Figure 4B. Following the initial 24h burst release, samples incubated in the “constant” ROS media released significantly more BMP-2 than the “pulsed” samples incubated in PBS over the first 4-day cycle. However, upon switching the “pulsed” medium to 1 mM H2O2 over days 5-8, the release kinetics were statistically indifferent between the two sample sets (Figure 4B). The oscillating protein release behavior from “pulsed” samples was generally continued until day 20, though with some hysteresis in the responsiveness; the expected PBS “pulse” response during days 9-12 was delayed by 2 days, while the anticipated ROS media response during days 13-16 was also shortly postponed as shown in Figure 4B.
Interestingly, at later time points the pulsed samples do not deliver extra BMP-2 despite possessing more of their initial drug reservoir, though this can potentially be explained by the pulsed samples simply not releasing their entire payload over 20 days due to the reduced oxidative stimulation. Additionally, PEM interlayer diffusion and film rearrangement[21d] that could occur in the non-oxidative cycles may have caused repartitioning of BMP-2 in the film such that less protein was readily exposed when the PTK-BAA component was degraded in ROS media. While this experiment confirmed triggerable BMP-2 delivery based on a global stimulus, these data also indicate this system’s potential for selective drug release to areas of cellular growth and tissue regeneration. Bone remodeling osteoclasts have been previously shown to localize at the periphery of bone injuries[40] and produce high levels of ROS[41] during the healing process. With PTK-BAA coated implants, osteogenic cellular activity could directly liberate BMP-2 at the regenerative defect margin, thereby limiting off-target effects, while preserving the implant’s undisturbed drug reservoir until sufficient new tissue growth reaches the defect’s central regions and releases more growth factor.
2.3. In Vitro Bioactivity of Film-Released BMP-2
The bioactivity of BMP-2 released from PTK-BAA films was evaluated by incubating MC3T3-E1 pre-osteoblasts with releaseate from PBS or 1 mM H2O2-treated film samples. BMP-2-containing releaseate from 30 tetralayer films constructed with PTK-BAA and 450 kDa PAA was first combined into discrete pools across the time course, and then a constant volume of releaseate from each pooled sample (to normalize the time-dependent dose of BMP-2) was lyophilized for 48h to both concentrate the growth factor and remove residual cytotoxic peroxide as previously reported.[42] The dried releaseate was then re-suspended into cell culture media and treated onto the pre-osteoblasts for 48h before allowing further osteogenic cellular differentiation over an additional 4 days. Alkaline phosphatase production by cells was then quantified after normalization to total cellular protein levels, and as demonstrated in Figure 5, confirmed the bioactivity of film-released BMP-2. Moreover, cells treated with releaseate from ROS-treated films significantly increased their alkaline phosphatase levels compared with cells incubated in PBS releaseate, particularly at the later time points despite some variability potentially caused by osmotic stress on the cells from residual releaseate salts (Figure 5). These data further validate the connection between ROS-mediated drug release and regenerative cellular activity.
Figure 5.

In vitro cellular osteogenesis corresponds with oxidation-mediated BMP-2 release from PTK-BAA films. MC3T3-E1 pre-osteoblasts treated with releaseate from 1 mM H2O2-incubated films generated significantly more alkaline phosphatase than cells treated with releaseate from PBS-incubated films (n=3 per treatment, *p<0.05). 30 tetralayer films constructed with 450 kDa PAA.
2.4. Effects of PEM Film Components on In Vitro BMP-2 Release
While the results discussed above establish the initial validation of oxidation-sensitive PEM coatings for responsive therapeutic delivery, interestingly the ROS-mediated discharge of lysozyme (Figure S3) and BMP-2 (Figure 4A) were markedly different despite similar film formulations. It was hypothesized that faster-releasing lysozyme’s lower molecular weight (14.3 kDa vs. 26 kDa for BMP-2) was creating less stable LbL assemblies that were more prone to disassociation upon ROS-mediated PTK-BAA degradation. Previous work has demonstrated that lower molecular weight polyelectrolytes can more easily diffuse through a multilayer assembly;[43] since the BMP-2 size is not easily alterable and the PTK-BAA already features a relatively low molecular weight (~2.3 kDa), the impact of varyingly sized PAA polyanions in PEM films was explored to further adapt the LbL assembly stability. PTK-BAA / BMP-2 films were constructed with 450 kDa, 5 kDa, or 1.8 kDa PAA on PLGA scaffolds, and crucially did not display appreciable differences in total drug loading across formulations (Figure 6A). Additionally, all three formulations displayed limited protein release under non-oxidative conditions while delivering significantly more BMP-2 after treatment with 1 mM H2O2 as expected (Figure 6B). Further validating PTK-BAA films’ fundamental ROS-specific release mechanism, BMP-2 films constructed with hexane-PBAA (no ROS-degradable groups) or conventional PBAE polycations did not significantly increase their drug release in oxidative conditions (Figure S4).
Figure 6.

Varying the PAA polyanion molecular weight (450, 5, or 1.8 kDa) in BMP-2 / PTK-BAA films (A) does not appreciably change the total BMP-2 loading as visualized by fluorescence imaging of films with Cy5-labeled protein. However, PAA1.8 samples (B) display significantly more sensitivity to oxidation via BMP-2 release at both 1 mM and 0.1 mM H2O2 compared against higher molecular weight PAA formulations (n=3 per treatment; *p<0.05, **p<0.005, ***p<0.0005 for 1 mM H2O2 vs PBS; #p<0.05 for 0.1 mM H2O2 vs PBS). All samples featured 30 tetralayer films constructed on PLGA scaffolds.
Although ROS-mediated BMP-2 release from 450 kDa and 5 kDa PAA films was nearly indistinguishable in oxidative media, the 1.8 kDa PAA samples displayed a comparatively significant increase (greater than 40%) in drug release at both 1 and 0.1 mM H2O2 (Figure 6B). These BMP-2 liberation kinetics reasonably support the initial hypothesis that reducing PEM component molecular weight will decrease overall film stability and increase film disassociation (e.g. BMP-2 release) upon PTK-BAA degradation. However, these findings do not generate a strict molecular weight-dependent pattern given that films constructed with 5 kDa PAA behave more similarly to those with 450 kDa PAA than films containing 1.8 kDa PAA. Preliminary investigations also explored films made with 50 kDa PAA and found they did not substantially differ from those made with 450 or 5 kDa PAA (data not shown), further separating the performance of the 1.8 kDa formulation.
A possible explanation arises from the relationship between the PTK-BAA polycation and PAA polyanion; when the PAA’s molecular weight exceeds that of the degradable polymer, the highly-charged yet non-degradable PAA fosters more robust film stability. Conversely, if the PAA component’s molecular weight approaches or falls below that of the PTK-BAA (1.8 < 2.3 kDa), the degradable polycation chains more strongly impact the film architecture and confer additional sensitivity to ROS-mediated multilayer disassembly. Though LbL assemblies are considered effective ionic networks with significant amounts of interpenetration and entanglement,[43] here it would appear that the network stability is highly dependent on the higher molecular weight component in the film. It should also be noted that while this work sought to improve sensitivity to a specific film-disrupting stimulus, many drug-releasing PEM assemblies are designed to maximize film stability to prolong delivery windows.[19b] However, for this environmentally-responsive system, these findings present an additional strategy for enhancing stimuli-triggered therapeutic delivery from PEM assemblies.
2.5. In Vivo Bone Regeneration and Drug Release Kinetics from PTK-BAA / BMP-2 Coated Implants
To explore the efficacy of ROS-responsive, PEM film-mediated growth factor delivery in vivo, PLGA scaffolds coated with PEM films comprising PTK-BAA, BMP-2, and the varyingly sized PAA polyanions (450, 5, or 1.8 kDa) were implanted in large-scale rat calvarial defects to evaluate in vivo bone regeneration. The surgically created 8.0mm-diameter bone injuries are generally accepted as “critically-sized” in that the animal cannot completely heal over their natural lifetime[44] and have been extensively used to evaluate regenerative biomaterial and bone tissue engineering strategies.[11c, 22a, 45] As visualized in computed tomography (CT) micrographs in Figure 7A, new bone development (identified by its lower CT density compared with mature bone) is present at the defect edges for all three film formulations at week 4. This bone growth into the defect margins was quantified using an 8 mm-diameter region of interest (ROI) to selectively measure new bone as seen in Figure S5. The 1.8 kDa PAA PEM coatings, featuring the highest sensitivity to ROS-mediated BMP-2 release (Figure 6B), promoted a greater than 50% increase in bone volume (Figure 7B) and significantly improved tissue mineralization (Figure 7C) within the defect margins when compared against the notably less responsive 450 and 5 kDa PAA film formulations. These significant results further supported the structure-function relationship between PEM components and BMP-2 release as explored in Figure 6. Moreover, histology of bone defects treated with the 1.8 kDa PAA films further demonstrated developing bone formation, though not fully mature bone coverage, in the calvarial defects at week 4 as shown in Figure S6.
Figure 7.

In vivo bone regeneration in critically-sized rat calvarial defects from oxidation-sensitive, BMP-2-coated implants. BMP-2 / PTK-BAA LbL films (30 tetralayers) with varying PAA polyanion molecular weights (450, 5, or 1.8 kDa) were constructed on PLGA scaffolds and implanted into 8 mm diameter rat calvarial bone defects for 4 weeks. (A) Non-invasive microcomputed tomography images of the calvarial bone were collected at week 4 post-surgery (original defect margins in red, new bone growth tinted blue, scale bar 5 mm, n=4 animals per group). Quantitation of (B) bone volume and (C) tissue mineral density inside the defect margins demonstrated that the PAA1.8 films facilitated significantly more bone growth and mineralization than higher molecular weight PAA films (*p<0.05), corresponding to the PAA1.8 samples’ increased BMP-2 release rates following oxidation.
Importantly, the bone regeneration mediated by these oxidation-sensitive drug coatings indicated that tissue-generated ROS levels at the calvarial defect site were sufficient to trigger BMP-2 release from PTK-BAA films. To further confirm and quantify localized in vivo BMP-2 delivery kinetics from the responsive PEM films, lead-candidate PTK-BAA coated implants fabricated with 1.8 kDa PAA and fluorescently-tagged growth factor were directly compared against analogous PBAE-coated samples in the rat calvarial defect model. As visualized in Figure 8A, IVIS imaging of the Cy7-labeled protein indicated a decreased bolus release of BMP-2 in the responsive film formulations and significantly enhanced drug retention over three weeks in vivo (Figure 8B). Moreover, the calculated half-life values for the respective release curves indicated a nearly three-fold increase in BMP-2 retention as quantified in Figure 8C. Given prior reports of bolus growth factor release from hydrolytically-sensitive PBAE films[20a, 22a] and the in vitro release kinetics demonstrated in Figure 4A, the significantly prolonged in vivo BMP-2 release kinetics from PTK-BAA coated implants is well-supported.
Figure 8.

In vivo BMP-2 release from ROS-sensitive PTK-BAA and conventional PBAE-coated implants in rat calvarial defects. As quantified from (A) fluorescence imaging of Cy7-labeled BMP-2, growth factor delivery from the PTK-coated scaffolds (B) exhibits a lower initial bolus discharge and is significantly extended compared against PBAE films. Importantly though, over 95% of the PTK film’s drug payload is released over a three-week time course. (C) Quantification of half-life values for the respective release profiles (displayed as a 95% confidence interval) further demonstrates a nearly three-fold increase in drug retention for PTK-BAA coatings. N=3 animals per treatment, *p<0.05.
Crucially, this extended protein release did not appear to encourage indefinite BMP-2 retention in the PTK-BAA films as all samples discharged ~80% of their BMP-2 by day 14 and 95% of their payload by day 21 (Figure 8B). These findings indicate that local ROS concentrations, quantified at less than 0.1 mM in most tissues,[46] were sufficient to continuously degrade the oxidation-sensitive coatings and promote sustained payload delivery. However, it should be noted that the PTK-BAA / BMP-2 coatings appeared to elicit less bone regeneration than previously reported PBAE / BMP-2 LbL film formulations over the same 4-week time periods.[22a] Importantly, the PTK-BAA polymer and its degradation products did not cause significant toxicity in vitro (Figure S2) or elicit an excessive local inflammatory response or necrosis in vivo[47] as visualized in high-magnification hematoxylin and eosin-stained histology images (Figure S6). Moreover, specific immunohistochemical staining for CD68-expressing macrophages[48] in 4-week tissue sections did not indicate an abundant macrophage presence in these healing bone defects with blank or PTK-BAA coated implants (Figure S7), further confirming the non-inflammatory nature of these degradable films.
Since toxicity or excess inflammation does not appear to cause the decreased calvarial bone regeneration achieved with these ROS-responsive films, the interplay between BMP-2 dose and release kinetics was additionally explored. Rapid BMP-2 release from a collagen substrate (as utilized in the commercial INFUSE® system[6]) requires BMP-2 doses between 1.25 – 2.5 μg to achieve full rat calvarial defect bridging,[11c] and rapid delivery of nanogram-scale BMP-2 doses is insufficient to completely heal these rat bone injuries.[11a] However, previous work has also demonstrated negligible bone formation in rats when defects receive extremely slow rates of BMP-2 delivery.[7b] Conceivably, the strong electrostatic links in the PTK-BAA / BMP-2 assemblies which limited early film decomplexation and prevented a corresponding initial bolus of discharged protein both in vitro and in vivo (Figure 6B and Figure 8B) could have similarly hindered bone regeneration with these responsive films. Importantly, reports of robust rat bone formation at relatively low BMP-2 doses have indicated that growth factor release profiles featuring some initial release coupled with sustained BMP-2 delivery achieved superior bone regeneration outcomes.[7b, 22a]
To evaluate the hypothesis that a burst release of BMP-2 followed by more sustained growth factor delivery would improve bone healing, PLGA scaffolds were coated with “stacked” LbL films (constructed with lead-candidate 1.8 kDa PAA) featuring an inner 15 tetralayers of PTK-BAA / BMP-2 and an outer 15 tetralayers of PBAE / BMP-2 (Figure S8A) and implanted in critically-sized rat calvarial defects for 4 weeks (Figure S8B). Mediated by the PBAE components in the outer portion of the PEM assemblies, these combination film samples expectedly elicited a faster initial burst release of BMP-2 than films solely constructed with PTK-BAA polymers as confirmed by in vivo fluorescent tracking data in Figure S8C. Interestingly, bone growth with these combination films was substantially greater in some of the treated animals but did not elicit a statistically significant increase in bone volumes across the entire treatment cohort when compared to animals treated with the single PTK-BAA film formulations (Figure S8D). Though these findings do not exclude the possibility of some other PTK-BAA film variable inhibiting bone growth, they potentially give some indication of bone healing improvements that can be achieved by increasing initial in vivo BMP-2 delivery rates from these responsive systems.
3. Conclusions
Reactive oxygen species, ubiquitous biological signals in both healthy and diseased tissues, represent a promising cell-specific stimulus for promoting therapeutic delivery from PEM-coated implants. Here, newly synthesized PTK-BAA polycations were selectively degraded via oxidation and built robust multilayered PEM films upon LbL assembly with appropriate polyanions. When constructed with the therapeutic protein BMP-2, the PTK-BAA films produced ROS dose-dependent delivery of the bioactive growth factor while promoting in vitro cellular osteogenesis at levels corresponding with BMP-2 release kinetics. Moreover, these films’ in vitro sensitivity to oxidation could be further modulated by altering the polyanion used in LbL assembly to promote significantly increased rates of ROS-mediated protein liberation. This connection between tunable LbL components and enhanced BMP-2 delivery was further exhibited in critically-sized rat calvarial defects as implants coated in the most sensitive oxidation-responsive PEM films correspondingly generated a greater than 50% increase in new bone formation compared against less responsive film formulations. Moreover, this responsive BMP-2 release was evaluated against conventional hydrolytically-sensitive PEM coatings in vivo and demonstrated a nearly three-fold extension in delivery half-life.
In expanding the future application space for these environmentally-responsive drug coatings, their ability to sustainably deliver therapeutics upon oxidative triggering is potentially useful for a number of indications. Inflammation-triggered delivery of anti-inflammatory drugs to improve tissue repair,[49] ROS-stimulated release of chemotherapeutics to target cancer cells with high levels of oxidative stress,[50] or immune-activated antibiotic discharge to help fight bacterial infections[51] all represent promising future areas of exploration with these intelligent delivery systems. For continued development as a bone regenerative technology, the findings presented here further motivate the prospective development of ROS-responsive coatings with enhanced sensitivity that can more rapidly deliver payload upon local cellular activation to improve performance. In all, these collective data highlight the utility of PTK-BAA polymers as environmentally-responsive PEM film constituents and highlight the potential of tunable drug delivery systems for regenerating large-scale bone defects.
4. Experimental Section
Materials:
All materials and reagents were obtained from MilliporeSigma (Burlington, MA) or Alfa Aesar (Ward Hill, MA) unless otherwise specified. Silicon wafers were bought from Silicon Quest International (Santa Clara, CA). Linear polyethylenimine (LPEI, 25 kDa) was purchased from Polysciences (Warrington, PA). Cy5 and Cy7 NHS ester dyes were procured from Lumiprobe (Hunt Valley, MD). The alkaline phosphatase assay kit was bought from Abcam (Cambridge, United Kingdom). Recombinant human bone morphogenetic protein-2 (BMP-2) was generously donated by Bioventus LLC (Durham, NC) while the Human/Murine /Rat BMP-2 Standard ABTS ELISA Kit was procured from Peprotech (Rocky Hill, NJ). MC3T3-E1 mouse pre-osteoblasts were obtained from American Type Culture Collection (Manassas, VA) and cultured in Gibco Alpha Minimum Essential Medium (containing ribonucleosides, deoxyribonucleosides, 2 mM L-glutamine and 1 mM sodium pyruvate, but without ascorbic acid) with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Lonza). Cell Titer Glo assay kit was acquired from Promega (Madison, WI).
Polycation Synthesis and Characterization:
Detailed experimental procedures for polymer synthesis and chemical characterization are provided in the Supporting Information. In brief, a thioketal diacrylamide monomer was synthesized using methods adapted from previous reports,[34a] polymerized by Michael addition with 4,4′-trimethylene dipiperidine for 24h at room temperature in a 3:1 v/v solvent mix of ethanol:water, and then purified by 3x precipitation into cold THF. The generated PTK-BAA polymer structure was confirmed by 1H NMR, and molecular weight was determined with MALDI-TOF using a 2,5-dihydroxybenzoic acid matrix. The PBAE polymer was synthesized using previously established protocols[19b, 52] and characterized by 1H NMR and gel permeation chromatography (GPC, Waters system with Stryagel Columns, THF mobile phase, linear polystyrene molecular weight standards). To synthesize the hexane-PBAA, a hexane diacrylamide monomer was generated from 1,6 hexane diamine and then polymerized with 4,4′-trimethylene dipiperidine as described above.
Quantification of ROS-Triggered Thioketal Degradation:
PTK-BAA polymers were dissolved at 1 mg mL−1 in deionized water and doped with 0, 0.1, 1, and 10 mM doses of H2O2. Samples were incubated for 72h at 37°C before being lyophilized and then reconstituted in deuterated water to analyze with 1H NMR. The characteristic thioketal peak (δ 1.59) was then normalized against the internal control peaks from the polymer’s dipiperidine units (δ 1.2-1.3). MALDI-TOF analysis of the 10 mM H2O2-incubated polymer was also used to determine degradation product molecular weight.
In Vitro Polymer Cytotoxicity:
MC3T3-E1 mouse pre-osteoblast cells were seeded in a black-walled 96-well plate (5000 cells per well) for 24h before treating with 100 μL of fresh media or media doped with 10 μg mL−1 of 25 kDa LPEI, PBAE, PTK-BAA, or 10 mM H2O2-degraded PTK-BAA polymers (n=3 to 6 biological replicates for each treatment). After 24h of incubation, the plate was equilibrated to room temperature for 30 min before adding 50 μL of room-temperature Cell Titer Glo reagent to each well, shaking for 10 min, and then measuring luminescent signal using a Tecan Infinite 200 PRO microplate reader. All viable cell signals were normalized to no treatment wells.
LbL Film Construction and In Vitro Drug Release with the Model Protein Lysozyme:
Complete procedures for protein labeling, film construction, and drug release quantification can be found in the Supporting Information. Briefly, polyelectrolyte solutions were formulated in 100 mM sodium acetate buffer (pH 5.0), with PTK-BAA dissolved at 0.5 mg mL−1 and the 450 kDa PAA and fluorescein isothiocyanate (FITC)-labeled lysozyme (10% labeled protein by mass) being dissolved at 1 mg mL−1. Silicon substrates were plasma treated and then dipped into the polyelectrolyte baths using a Carl Zeiss HMS Series Programmable Slide Stainer, alternatively immersing in PTK-BAA (5 min), PAA (5 min), lysozyme (5 min), and PAA (5 min) with two 30s rinses in deionized water between dips. Fabricated films were incubated in PBS, PBS with 0.1 mM H2O2, or PBS with 1 mM H2O2 before collecting releaseate samples over time to quantify fluorescence from the labeled lysozyme.
PLGA Scaffold Fabrication:
Poly(lactic-co-glycolic) acid (PLGA: acid terminated, lactide:glycolide 50:50, molecular weight 38 – 54 kDa) was formed into a degradable scaffold using a diffusion-induced phase-separation protocol adapted from previously described methods.[22a, 53] Raw polymer was fully dissolved at 25 wt% in dimethylformamide (DMF), and the solution was deposited on a glass slide. Using a film-casting doctor blade dragged across the dissolved PLGA, a 1 mm-thick polymer solution was cast on the glass before being immediately submerged in a large excess of deionized water to induce phase separation. The rinse water was changed multiple times over 24h before drying the membrane under vacuum.
LbL Film Construction and Characterization with BMP-2:
Complete procedures for protein labeling, film construction, PEM characterization can be found in the Supporting Information. LbL film construction with BMP-2 was carried out similarly to the methods described above for lysozyme samples. All polyelectrolyte solutions were prepared in 100 mM sodium acetate buffer; cationic polymers (PTK-BAA, PBAE, or hexane-PBAA) were prepared at 0.5 mg mL−1 (pH 5.0), PAA polyanions (450, 5, or 1.8 kDa) were dissolved at 1 mg mL−1 (pH 5.0), and BMP-2 (5% Cy5-labeled protein by mass for drug loading quantification samples, 40% Cy7-labeled protein for in vivo release samples) was formulated at 0.04 mg mL−1 (pH 4.1). Plasma-treated substrates were immersed in the initial cationic polymer solution for 30 min before initiating the dipping program: PTK-BAA (5 min), PAA (5 min), BMP-2 (10 min), and PAA (5 min) with two 30s rinses in deionized water between dips. This dipping protocol allowed for the coating of both the top and bottom scaffold surfaces. For PTK-BAA / Cy5-BMP-2 / 450 kDa PAA film samples with increasing numbers of tetralayer depositions, PEM thickness on silicon was quantified using a Dektak Stylus profilometer (Veeco Instruments Inc., Plainville, NY) while BMP-2 loading was quantified by fluorescence imaging with an IVIS Spectrum (PerkinElmer, Waltham, MA) and ELISA.
In Vitro BMP-2 Release from LbL Films:
Additional experimental details can be found in the Supporting Information. Briefly, LbL films comprising BMP-2, 450 kDa PAA, and either PTK-BAA or PBAE degradable polycations were deposited on both top and bottom surfaces of PLGA scaffolds with 30 tetralayer repeats, incubated at 37°C in either PBS or PBS with 1 mM H2O2, and quantified for BMP-2 release using ELISA. For measuring responsive BMP-2 release from these same PTK-BAA films, coated PLGA samples were incubated at 37°C in either a continuous dose of 1 mM H2O2 or alternating between PBS and 1 mM H2O2 every 4 days. For both the “constant” and “pulsed” ROS-treated samples, releaseate was collected every two days and then quantified for BMP-2 amounts by ELISA. In determining the effects of PAA molecular weight on PEM protein, 30 tetralayer PTK-BAA / Cy5-BMP-2 films were fabricated with either 450, 5, or 1.8 kDa PAA on PLGA substrates, fluorescently imaged by IVIS to visualize and quantify relative BMP-2 loading, and finally incubated at 37°C in PBS, PBS with 0.1 mM H2O2, or PBS with 1 mM H2O2 before quantifying BMP-2 release using ELISA. Control 30 tetralayer PEM films were also constructed with Cy5 BMP-2, 1.8 kDa PAA, and either the hydrolytically-sensitive PBAE or non-degradable hexane-PBAA polycations on PLGA substrates. Release experiments and quantifications were performed with the same methodologies as used for PTK-BAA samples.
In Vitro Osteogenic Bioactivity of Film-Released BMP-2:
Additional experimental details can be found in the Supporting Information. Briefly, releaseate from PTK-BAA / BMP-2 / 450 kDa PAA films incubated in PBS or 1 mM H2O2 was pooled across discrete time periods, titrated to equal volumes, and then lyophilized before re-suspending in culture media. MC3T3-E1 pre-osteoblasts were treated with the releaseate-doped media for 48h with an additional 4-day incubation in osteogenic differentiation media before quantifying alkaline phosphatase activity as normalized to total cell protein content.
In Vivo Rat Calvarial Defect Procedures:
All animal work was approved by the Committee on Animal Care at the Massachusetts Institute of Technology (protocol 0718-057-21, PI Dr. Paula Hammond), and adult male Sprague-Dawley rats (300-350g, Taconic) were used in all procedures. Critically-sized calvarial defect studies were carried out using well-established methods.[22a, 44] First, anesthetized animals were cranially shaved, disinfected using betadine and alcohol, and given pre-operative meloxicam (anti-inflammatory) and buprenorphine sustained release (analgesic). Next, an incision through the scalp was used to expose the soft tissue/calvarium before using blunt dissection to displace the periosteum covering the bony surface. With the bone exposed, an 8.0 mm-diameter dental trephine drill with intermittent sterile saline irrigation was used to create a circular defect in the calvarial bone. Without disturbing the dura or superior sagittal sinus, the calvarium was excised before press-fitting the respective PEM-coated PLGA scaffold sample (also 8.0 mm-diameter) into the surgical bone defect. Finally, the periosteum was closed over the bone and implant using 5-0 monofilament absorbable sutures before also suturing the skin over the periosteum along the initial incision. Animals were given post-operative anti-inflammatories and analgesics until recovery. N=4 animal cohort sizes were chosen per treatment group based on power analyses from previous reports with a calvarial defect model.[22a] In vivo studies employing unlabeled BMP-2 films (Figure 7 and Figure S8D) featured n=4 animals per treatment group, though the subsequent studies tracking in vivo release of fluorescently-tagged BMP-2 (Figure 8 and Figure S8C) ultimately only featured n=3 animals per treatment group due to humane euthanasia of animal subjects following surgical complications from excessive blood loss. Microcomputed tomography, IVIS fluorescence imaging, and histological sample preparation details are described in the Supporting Information. Whole slide imaging analysis and visualization were performed using QuPath software[54].
Statistical Analysis:
All data are presented as the mean and standard deviation unless otherwise indicated. Statistical analyses were performed using Student’s t-test for single comparisons or single factor analysis of variance (ANOVA) with Tukey post-hoc tests for multiple comparisons. P-values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
Funding for this work was provided by the National Institutes of Health (NIH) through grants R01DE024747 and F32DE027877 (J.R.M. individual fellowship). P.T.H. also acknowledges support from the David H. Koch (1962) Chair Professorship in Engineering and discloses her competing interests as cofounder of LayerBio Inc. and service on the advisory boards of Performance Indicator and Moderna Therapeutics. The authors would like to thank Dr. Howard Seeherman and Bioventus LLC for their generous donation of BMP-2. The authors also thank the Koch Institute Swanson Biotechnology Center’s core facilities for Preclinical Imaging & Testing, Biopolymers & Proteomics, and Histology. Additionally, the authors thank the MIT Department of Chemistry Instrumentation Facility and the Langer Lab at MIT for use of equipment. Special thanks also go to Dr. Jennifer Haupt for surgical training and Ms. Kathleen Cormier for assistance with histology.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
John R. Martin, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139
MayLin T. Howard, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139
Sheryl Wang, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139.
Adam G. Berger, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139
Paula T. Hammond, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139
References
- [1].Greenwald AS, Boden SD, Goldberg VM, Khan Y, Laurencin CT, Rosier RN, JBJS 2001, 83, S98. [DOI] [PubMed] [Google Scholar]
- [2].Finkemeier CG, The Journal of Bone & Joint Surgery 2002, 84. [DOI] [PubMed] [Google Scholar]
- [3].Schmitt JM, Hwang K, Winn SR, Hollinger JO, Journal of Orthopaedic Research 1999, 17, 269. [DOI] [PubMed] [Google Scholar]
- [4].Yamaguchi A, Katagiri T, Ikeda T, Wozney JM, Rosen V, Wang EA, Kahn AJ, Suda T, Yoshiki S, The Journal of Cell Biology 1991, 113, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Agrawal V, Sinha M, Journal of Biomedical Materials Research Part B: Applied Biomaterials 2017, 105, 904. [DOI] [PubMed] [Google Scholar]
- [6].U. F. a. D. Administration, (Ed.: U. F. a. D. Administration; ), 2002. [Google Scholar]
- [7].a Boerckel JD, Kolambkar YM, Dupont KM, Uhrig BA, Phelps EA, Stevens HY, García AJ, Guldberg RE, Biomaterials 2011, 32, 5241; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Brown KV, Li B, Guda T, Perrien DS, Guelcher SA, Wenke JC, Tissue Engineering Part A 2011, 17, 1735; [DOI] [PubMed] [Google Scholar]; c Carlisle P, Guda T, Silliman DT, Burdette AJ, Talley AD, Alvarez R, Tucker D, Hale RG, Guelcher SA, BrownBaer PR, Journal of Biomedical Materials Research Part B: Applied Biomaterials 2018; [DOI] [PubMed] [Google Scholar]; d Uludag H, D'Augusta D, Palmer R, Timony G, Wozney J, Journal of Biomedical Materials Research 1999, 46, 193. [DOI] [PubMed] [Google Scholar]
- [8].Poynton AR, Lane JM, Spine 2002, 27. [DOI] [PubMed] [Google Scholar]
- [9].a Zara JN, Siu RK, Zhang X, Shen J, Ngo R, Lee M, Li W, Chiang M, Chung J, Kwak J, Wu BM, Ting K, Soo C, Tissue Engineering Part A 2011, 17, 1389; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Carragee EJ, Hurwitz EL, Weiner BK, The Spine Journal 2011, 11, 471; [DOI] [PubMed] [Google Scholar]; c James AW, LaChaud G, Shen J, Asatrian G, Nguyen V, Zhang X, Ting K, Soo C, Tissue Engineering Part B: Reviews 2016, 22, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Uludag H, D'Augusta D, Golden J, Li J, Timony G, Riedel R, Wozney JM, Journal of Biomedical Materials Research 2000, 50, 227. [DOI] [PubMed] [Google Scholar]
- [11].a Cowan CM, Aghaloo T, Chou Y-F, Walder B, Zhang X, Soo C, Ting K, Wu B, Tissue Engineering 2007, 13, 501; [DOI] [PubMed] [Google Scholar]; b Wei L, Yu D, Wang M, Deng L, Wu G, Liu Y, Tissue Engineering Part A 2019; [DOI] [PubMed] [Google Scholar]; c Pelaez M, Susin C, Lee J, Fiorini T, Bisch FC, Dixon DR, McPherson JC Iii, Buxton AN, Wikesjö UME, Journal of Clinical Periodontology 2014, 41, 827. [DOI] [PubMed] [Google Scholar]
- [12].Alkekhia D, Hammond PT, Shukla A, Annual Review of Biomedical Engineering 2020, 22. [DOI] [PubMed] [Google Scholar]
- [13].Decher G, Hong J-D, Makromolekulare Chemie. Macromolecular Symposia 1991, 46, 321. [Google Scholar]
- [14].a DeMuth PC, Su X, Samuel RE, Hammond PT, Irvine DJ, Advanced Materials 2010, 22, 4851; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Castleberry SA, Almquist BD, Li W, Reis T, Chow J, Mayner S, Hammond PT, Advanced Materials 2015. [DOI] [PubMed] [Google Scholar]
- [15].a Smith RC, Riollano M, Leung A, Hammond PT, Angewandte Chemie International Edition 2009, 48, 8974; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shukla A, Avadhany SN, Fang JC, Hammond PT, Small 2010, 6, 2392; [DOI] [PubMed] [Google Scholar]; c Shukla A, Fang JC, Puranam S, Hammond PT, Journal of Controlled Release 2012, 157, 64. [DOI] [PubMed] [Google Scholar]
- [16].a Macdonald M, Rodriguez NM, Smith R, Hammond PT, Journal of Controlled Release 2008, 131, 228; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Macdonald ML, Rodriguez NM, Shah NJ, Hammond PT, Biomacromolecules 2010, 11, 2053; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Macdonald ML, Samuel RE, Shah NJ, Padera RF, Beben YM, Hammond PT, Biomaterials 2011, 32, 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wood KC, Boedicker JQ, Lynn DM, Hammond PT, Langmuir 2005, 21, 1603. [DOI] [PubMed] [Google Scholar]
- [18].Vázquez E, Dewitt DM, Hammond PT, Lynn DM, Journal of the American Chemical Society 2002, 124, 13992. [DOI] [PubMed] [Google Scholar]
- [19].a Zhang J, Fredin NJ, Janz JF, Sun B, Lynn DM, Langmuir 2006, 22, 239; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith RC, Leung A, Kim B-S, Hammond PT, Chemistry of Materials 2009, 21, 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a Almquist BD, Castleberry SA, Sun JB, Lu AY, Hammond PT, Advanced Healthcare Materials 2015, 4, 2090; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bouyer M, Guillot R, Lavaud J, Plettinx C, Olivier C, Curry V, Boutonnat J, Coll J-L, Peyrin F, Josserand V, Bettega G, Picart C, Biomaterials 2016, 104, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a Min Y, Hammond PT, Chemistry of Materials 2011, 23, 5349; [Google Scholar]; b Min J, Braatz RD, Hammond PT, Biomaterials 2014, 35, 2507; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Min J, Choi KY, Dreaden EC, Padera RF, Braatz RD, Spector M, Hammond PT, ACS Nano 2016, 10, 4441; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wood KC, Chuang HF, Batten RD, Lynn DM, Hammond PT, Proceedings of the National Academy of Sciences 2006, 103, 10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a Shah NJ, Hyder MN, Quadir MA, Dorval Courchesne N-M, Seeherman HJ, Nevins M, Spector M, Hammond PT, Proceedings of the National Academy of Sciences 2014, 111, 12847; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shah NJ, Macdonald ML, Beben YM, Padera RF, Samuel RE, Hammond PT, Biomaterials 2011, 32, 6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shah NJ, Hyder MN, Moskowitz JS, Quadir MA, Morton SW, Seeherman HJ, Padera RF, Spector M, Hammond PT, Science Translational Medicine 2013, 5, 191ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Qu M, Jiang X, Zhou X, Wang C, Wu Q, Ren L, Zhu J, Zhu S, Tebon P, Sun W, Khademhosseini A, Advanced Healthcare Materials 2020, 9, 1901714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guo X, Carter MC, Appadoo V, Lynn DM, Biomacromolecules 2019, 20, 3464. [DOI] [PubMed] [Google Scholar]
- [26].Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA, Free Radical Biology and Medicine 2000, 28, 1456. [DOI] [PubMed] [Google Scholar]
- [27].Pacher P, Beckman JS, Liaudet L, Physiological Reviews 2007, 87, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB, Antioxidants & redox signaling 2014, 20, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a Key L, Wolf W, Gundberg C, Ries W, Bone 1994, 15, 431; [DOI] [PubMed] [Google Scholar]; b Garrett IR, Boyce BF, Oreffo R, Bonewald L, Poser J, Mundy GR, Journal of Clinical Investigation 1990, 85, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a Martin JR, Duvall CL, in Oxidative Stress and Biomaterials, Vol. 1 (Eds.: Dziubla T, Butterfield DA), Academic Press, Cambridge, MA, 2016, pp. 225; [Google Scholar]; b El-Mohtadi F, d’Arcy R, Tirelli N, Macromolecular Rapid Communications 2018, 0, 1800699; [DOI] [PubMed] [Google Scholar]; c Song C-C, Du F-S, Li Z-C, Journal of Materials Chemistry B 2014, 2, 3413. [DOI] [PubMed] [Google Scholar]
- [31].a Yu SS, Koblin RL, Zachman AL, Perrien DS, Hofmeister LH, Giorgio TD, Sung H-J, Biomacromolecules 2011, 12, 4357; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martin JR, Gupta MK, Page JM, Yu F, Davidson JM, Guelcher SA, Duvall CL, Biomaterials 2014, 35, 3766; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Martin JR, Nelson CE, Gupta MK, Yu F, Sarett SM, Hocking KM, Pollins AC, Nanney LB, Davidson JM, Guelcher SA, Duvall CL, Advanced Healthcare Materials 2016, 5, 2751; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Patil P, Martin JR, Sarett SM, Pollins AC, Cardwell NL, Davidson JM, Guelcher SA, Nanney LB, Duvall CL, Tissue Engineering Part C: Methods 2017, 23, 754; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gupta MK, Martin JR, Werfel TA, Shen T, Page JM, Duvall CL, Journal of the American Chemical Society 2014, 136, 14896; [DOI] [PubMed] [Google Scholar]; f Dollinger BR, Gupta MK, Martin JR, Duvall CL, Tissue Engineering Part A 2017, 23, 1120; [DOI] [PMC free article] [PubMed] [Google Scholar]; g Gupta MK, Martin JR, Dollinger BR, Hattaway ME, Duvall CL, Advanced Functional Materials 2017, 27, 1704107; [DOI] [PMC free article] [PubMed] [Google Scholar]; h McEnery MAP, Lu S, Gupta MK, Zienkiewicz KJ, Wenke JC, Kalpakci KN, Shimko DA, Duvall C, Guelcher SA, RSC Advances 2016, 6, 109414; [DOI] [PMC free article] [PubMed] [Google Scholar]; i McGough MA, Shiels SM, Boller LA, Zienkiewicz K, Duvall C, Wenke JC, Guelcher S, Tissue Engineering 2018, 25, 949; [DOI] [PMC free article] [PubMed] [Google Scholar]; j McGough MAP, Boller LA, Groff DM, Schoenecker JG, Nyman JS, Wenke JC, Rhodes C, Shimko D, Duvall CL, Guelcher SA, ACS Biomaterials Science & Engineering 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]; k Yao Y, Ding J, Wang Z, Zhang H, Xie J, Wang Y, Hong L, Mao Z, Gao J, Gao C, Biomaterials 2020, 232, 119726; [DOI] [PubMed] [Google Scholar]; l Martin JR, Patil P, Yu F, Gupta MK, Duvall CL, Biomaterials 2020, 263, 120377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a Gupta MK, Meyer TA, Nelson CE, Duvall CL, Journal of Controlled Release 2012, 162, 591; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pu H-L, Chiang W-L, Maiti B, Liao Z-X, Ho Y-C, Shim MS, Chuang E-Y, Xia Y, Sung H-W, ACS Nano 2014, 8, 1213; [DOI] [PubMed] [Google Scholar]; c Broaders KE, Grandhe S, Fréchet JMJ, Journal of the American Chemical Society 2010, 133, 756; [DOI] [PubMed] [Google Scholar]; d El Mohtadi F, d’Arcy R, Burke J, Rios De La Rosa JM, Gennari A, Marotta R, Francini N, Donno R, Tirelli N, Biomacromolecules 2020, 21, 305. [DOI] [PubMed] [Google Scholar]
- [33].a Poole KM, Nelson CE, Joshi RV, Martin JR, Gupta MK, Haws SC, Kavanaugh TE, Skala MC, Duvall CL, Biomaterials 2015, 41, 166; [DOI] [PMC free article] [PubMed] [Google Scholar]; b O'Grady KP, Kavanaugh TE, Cho H, Ye H, Gupta MK, Madonna MC, Lee J, O'Brien CM, Skala MC, Hasty KA, Duvall CL, ACS Biomaterials Science & Engineering 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]; c DeJulius C, Bernardo-Colón A, Naguib S, Backstrom J, Kavanaugh T, Gupta M, Duvall C, Rex T, Journal of Controlled Release 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].a Shim MS, Xia Y, Angewandte Chemie International Edition 2013, 52, 6926; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Song C-C, Ji R, Du F-S, Li Z-C, Macromolecules 2013, 46, 8416. [Google Scholar]
- [35].Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N, Nature Materials 2010, 9, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ferruti P, Marchisio MA, Barbucci R, Polymer 1985, 26, 1336. [Google Scholar]
- [37].Castleberry SA, Li W, Deng D, Mayner S, Hammond PT, ACS Nano 2014, 8, 6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hsu BB, Hagerman SR, Jamieson K, Veselinovic J, O’Neill N, Holler E, Ljubimova JY, Hammond PT, Biomacromolecules 2014, 15, 2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dubas ST, Schlenoff JB, Macromolecules 2001, 34, 3736. [Google Scholar]
- [40].Gouron R, Petit L, Boudot C, Six I, Brazier M, Kamel S, Mentaverri R, Journal of Tissue Engineering and Regenerative Medicine 2017, 11, 382. [DOI] [PubMed] [Google Scholar]
- [41].Lee NK, Choi YG, Baik JY, Han SY, Jeong D.-w., Bae YS, Kim N, Lee SY, Blood 2005, 106, 852. [DOI] [PubMed] [Google Scholar]
- [42].Cheng W, Zheng X, Yang M, Journal of pharmaceutical sciences 2016, 105, 1837. [DOI] [PubMed] [Google Scholar]
- [43].Porcel C, Lavalle P, Decher G, Senger B, Voegel J-C, Schaaf P, Langmuir 2007, 23, 1898. [DOI] [PubMed] [Google Scholar]
- [44].Spicer PP, Kretlow JD, Young S, Jansen JA, Kasper FK, Mikos AG, Nature Protocols 2012, 7, 1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Patel ZS, Young S, Tabata Y, Jansen JA, Wong ME, Mikos AG, Bone 2008, 43, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Weinstain R, Savariar EN, Felsen CN, Tsien RY, Journal of the American Chemical Society 2014, 136, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lin HC, Lee HS, Chiueh TS, Lin YC, Lin HA, Lin YC, Cha TL, Meng E, Molecular medicine reports 2015, 11, 2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Murray PJ, Wynn TA, Nature Reviews Immunology 2011, 11, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brandt CJ, Kammer D, Fiebeler A, Klinge U, Journal of Biomedical Materials Research Part A 2011, 99, 335. [DOI] [PubMed] [Google Scholar]
- [50].Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK, Cancer Research 2008, 68, 1777. [DOI] [PubMed] [Google Scholar]
- [51].Slauch JM, Molecular Microbiology 2011, 80, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lynn DM, Langer R, Journal of the American Chemical Society 2000, 122, 10761. [Google Scholar]
- [53].Mikos AG, Thorsen AJ, Czerwonka LA, Bao Y, Langer R, Winslow DN, Vacanti JP, Polymer 1994, 35, 1068. [Google Scholar]
- [54].Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, Scientific Reports 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.