

Abstract
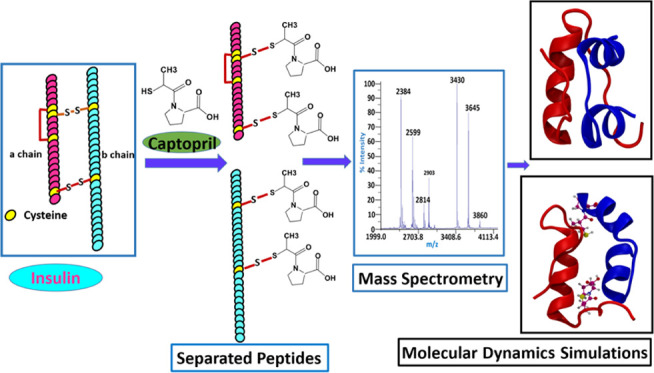
Post-translational modifications remarkably regulate proteins’ biological function. Small molecules such as reactive thiols, metabolites, and drugs may covalently modify the proteins and cause structural changes. This study reports the covalent modification and noncovalent interaction of insulin and captopril, an FDA-approved antihypertensive drug, through mass spectrometric and computation-based approaches. Mass spectrometric analysis shows that captopril modifies intact insulin, reduces it into its “A” and “B” chains, and covalently modifies them by forming adducts. Since captopril has a reactive thiol group, it might reduce the insulin dimer or modify it by reacting with cysteine residues. This was proven with dithiothreitol treatment, which reduced the abundance of captopril adducts of insulin A and B chains and intact Insulin. Liquid chromatography tandem mass spectrometric analysis identified the modification of a total of four cysteine residues, two in each of the A and B chains of insulin. These modifications were identified to be Cys6 and Cys7 of the A chain and Cys7 and Cys19 of the B chain. Mass spectrometric analysis indicated that captopril may simultaneously modify the cysteine residues of intact insulin or its subunits A and B chains. Biophysical studies involving light scattering and thioflavin T assay suggested that the binding of captopril to the protein leads to the formation of aggregates. Docking and molecular dynamics studies provided insights into the noncovalent interactions and associated structural changes in insulin. This work is a maiden attempt to understand the detailed molecular interactions between captopril and insulin. These findings suggest that further investigations are required to understand the long-term effect of drugs like captopril.
Introduction
The protein structure and function are affected by various enzymatic and nonenzymatic post-translational modifications. Small molecules such as metabolites and drugs covalently modify the proteins and affect their function. Previous studies have shown that metabolites such as glucose, methylglyoxal,1 HNE,2 etc. nonenzymatically modify the proteins in vivo. Similarly, various drugs such as aspirin3,4 bisacodyl, benzylpenicillin,5 amoxicillin,6 etc. also modify the proteins covalently. Among such molecules, drugs containing reactive thiol groups could potentially modify the proteins. One such drug molecule that modifies the proteins nonenzymatically is captopril.7,8 It is the first US Food and Drug Administration (FDA)-approved angiotensin-converting enzyme (ACE) inhibitor, which is primarily used for the treatment of hypertension. Previous studies have shown that captopril treatment leads to the formation of captopril–protein conjugates in the plasma.8,9 Captopril is a small-molecular weight compound (217.29 Da), and it contains two asymmetric centers; one is associated with the (S)-proline, and the other is associated with the 3-mercapto-2-methylpropionic acid side chain. The presence of the thiol (−SH) group makes the molecule a potent oxidant scavenger.10,11 The reactive thiol (−SH) group of captopril is involved in the spontaneous thiol–disulfide exchange process during its metabolism, which is a characteristic feature of sulfhydryl compounds.7,9,12,13 The metabolism of captopril involves the formation of mixed disulfide bonds with plasma proteins cysteine and glutathione (GSH) forming their respective conjugates.7,9,14,15
Under the conditions of glutathione (GSH) or cysteine depletion and impaired kidney function, captopril accumulates in the plasma,9,12,13,15,16 perhaps leading to an increased amount of captopril-bound plasma proteins.16 Under disease conditions such as diabetic albuminuria and nephropathy, depletion of plasma albumin and retention of captopril due to kidney insufficiency result in more exposure of captopril to the low-abundant plasma proteins such as insulin.
Although captopril is primarily used to treat hypertension,17,18 congestive heart failure,19 cardiovascular diseases20−22 and demonstrated protective effects on diabetic23 and nondiabetic chronic renal ailments,24,25 some adverse effects such as proteinuria, glomerulonephritis, serum sickness, agranulocytosis, and ulcers have also been reported.9 The clinical conditions for which captopril is used as a therapeutic drug are primarily associated with diabetes and related complications, where insulin plays a key role. Insulin is a low-abundant plasma protein, crucial for maintaining blood glucose homeostasis. Previous studies show that the biological activity of insulin is significantly compromised due to chemical modifications and associated structural changes.26−29 Similarly, during long-term treatment of hypertension using captopril, especially under diabetic conditions, it is possible that insulin will be modified by captopril, which further may alter its molecular conformation, eventually impacting its biological function and stability, leading to undesirable side effects and the onset of diseases.
So far, research has been focused mainly on investigating the therapeutic effects of captopril,19,23 the significance of covalent modification by small molecules,1−3 or the structural change of insulin in various diseases.26,30,31 Previous studies have demonstrated that thiol-containing small molecules such as cysteine and homocysteine cause thiolation of multiple cysteines in HSA,32 and the thiol/disulfide state of the protein is key for its conformation or stability.32,33 Since insulin also contains cysteine disulfide bonds, its biological efficiency may be impacted by captopril-induced alteration in the thiol–disulfide state. To the best of our knowledge, no study reports the interaction between captopril and low-abundant plasma proteins such as insulin. To this end, we have performed a comprehensive study to understand the effect of captopril on insulin. Since thiol/disulfide interchange plays an essential role in stabilizing the protein structure, cross-linking, etc., we first studied the effect of captopril on protein modification and thiolation of cysteine disulfide bonds, peptide separation of dimeric protein insulin and multimeric large protein IgG, and protein aggregation, using a combination of mass spectrometric, electrophoretic, and biophysical assays. Furthermore, docking and molecular dynamics studies have been performed to understand the noncovalent interactions and structural change upon binding with one and two captopril molecules. This work is a maiden attempt to understand the detailed molecular interaction and effects of captopril on insulin as a model protein. Thus, it enlightens a specific area for further investigation that may help predict the impact of long-term captopril treatment in patients.
Results and Discussion
Captopril Reduces and Modifies the Cysteine Residues of Proteins by Disulfide–Thiol Exchange
Insulin is a key protein in regulating blood glucose homeostasis34 and plays a significant role in diabetes and related complications. It consists of A and B chains linked together by two interchain and one intrachain disulfide bond between cysteines, giving the protein an α helical globular structure.35 It is a model amyloidogenic protein used to study a broad class of amyloidogenic protein-associated diseases.30,36,37
Captopril’s protein modifying ability was studied by incubating insulin (106.92 μM) with captopril (5 mM) for 30 min, and the formation of the captopril-induced modifications by was analyzed LC-ESI-MS, using a UPLC-Synapt-XS (Waters). In Figure 1A, the unmodified control insulin showed clusters of [M + 6H]+, [M + 5H]+, and [M + 4H]+ peaks at m/z 968.7544, m/z 1162.3053, and m/z 1452.6439, respectively (Figure 1A). Captopril treatment on insulin led to the formation of singly and doubly captopril-modified intact insulin peaks and the formation of a predominant B chain and its modified adducts. Singly captopril-modified intact insulin showed clusters of [M + 6H + 1C]+, [M + 5H + 1C]+, and [M + 4H + 1C]+ peaks at m/z 1005.0556, m/z 1206.0654, and m/z 1507.0995, respectively (Figure 1B). Similarly, doubly captopril-modified intact insulin showed peaks of [M + 6H + 2C]+ and [M + 5H + 2C]+ at m/z 1041.0952 and m/z 1249.0790, respectively. The intensity of the [M + 4H + 2C]+ peak was significantly less or undetectable.
Figure 1.

Captopril modification of insulin studied by liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI MS). (A) Control insulin (106.92 μM) incubated for 30 min and (B) insulin (106.92 μM) incubated with 5 mM captopril for 30 min. The peaks indicate the different charge states of intact insulin and its respective A and B chains. Single and double captopril adducts at different residues of intact insulin or its chains are indicated by 1C or 2C, respectively.
Furthermore, multiply charged B chain peaks were observed at m/z 686.7099 (M + 5H) and 858.1472 (M + 4H), and its adduct peaks modified with one captopril were found at m/z 729.8962 (M + 5H + C) and 912.1182 (M + 4H + C) and those modified with two captopril were found at m/z 772.9 (M + 5H + 2C) (inset, Figure 1B). A low-intensity peak of the A chain was also observed at m/z 794.2761 (M + 3H) (inset Figure 1B).
The formation of multiply charged peaks corresponding to the A and B chains of insulin suggests that captopril reduces the interchain cysteine disulfide bonds. On the other hand, the increase in the mass of A and B chains by 215.06 Da indicates that captopril (MW 217.07 Da) forms a disulfide bond by reacting with the thiol group (−SH) of reduced cysteine residues of the A or B chain; during this process of disulfide bond formation between the thiol group of captopril and cysteine, two hydrogen atoms are released, and thus, the mass increases by 215.06 Da.
Further captopril-induced modification of insulin was studied by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). The molecular mass of unmodified Insulin is 5808 Da (Figure 2A). However, the treatment of insulin (106.92 μM) with captopril (5 mM) (Figure 2B) led to the formation of A and B chains with an average mass of 2384 and 3430 Da, respectively. Notably, both A and B chains showed captopril modification with an average mass increase of each chain by 215 Da. The addition of a single captopril molecule to the A and B chains led to the formation of peaks corresponding to 2599 and 3645 Da, respectively, while the addition of two captopril molecules to the A and B chains led to the formation of peaks corresponding to 2814 and 3860 Da, respectively. We have also observed a doubly charged species of intact insulin and single captopril-modified intact insulin at m/z 2903 and m/z 3010, respectively. Typically doubly charged ions are rarely observed in MALDI-MS; however, high-molecular weight peptides and proteins sometimes produce doubly charged ions in MALDI-MS.
Figure 2.

Modification of insulin by captopril, cysteine is the probable site of modification, investigated by MALDI-TOF MS. (A) Control insulin (106.92 μM) was incubated for 30 min. (B) Insulin (106.92 μM) was incubated with 5 mM captopril for 30 min. (C) Captopril-modified insulin (106.92 μM) was treated with 5 mM dithiothreitol (DTT). Peaks labeled (1) and (2) indicate the single and double adduct/(s) of captopril in intact insulin or in A and B chains at different residues, respectively.
In both LC-ESI-MS and MALDI-MS, we observed that captopril treatment led to the formation of A and B chains, although the intensity of the A chain was less in the case of ESI-MS. This difference may be attributed to differences in the ionization methods. Both the experiments suggested that captopril may react with the cysteine residues of insulin, reducing disulfide bridges. To validate that captopril modification occurs at the cysteine residues of insulin, captopril-modified insulin was further treated with DTT. DTT is routinely used as a reducing agent, as it is a thiol and known to reduce the disulfide bridges of proteins without forming any protein adduct.38,39 Therefore, it is expected that the treatment of DTT can influence the captopril-induced modification of insulin. In this experiment (Figure 2C), treatment of captopril-modified insulin with DTT (Figure 2C) led to the suppression of peaks significantly, which represent captopril adducts at single residues in both the A and B chains. Interestingly, the peaks corresponding to double captopril adducts completely disappeared; only the peaks representing unmodified A and B chains were prominent. Also, the peak representing the single captopril adduct of doubly charged intact insulin disappeared. This result further strengthened the LC-ESI-MS and MALDI-MS observations that captopril modification possibly occurs at cysteine residues.
To confirm this, we further performed the LC-MS/MS analysis of captopril-modified insulin by two different approaches. In the first approach, LC-MSE (UPLC-Synapt-HDMS, Waters) was used to perform tandem mass spectrometry of control insulin and captopril-modified insulin. This analysis led to the identification of only one modified cysteine residue on the B chain of insulin, which is Cys7 of the B chain. The annotated MS/MS spectrum of the same is provided in the Supporting Information, Figure S1A,B (magnified image of S1A). Since we could not identify other modified cysteine residues, we digested the insulin with thermolysin. In the second approach, the thermolysin-digested peptides of control insulin and captopril-modified insulin were used for LC-MS/MS analysis using a Triple TOF (microLC-Sciex5600). This analysis led to the identification of a total of four modified cysteine residues, on both the A and B chains. On the A chain, Cys6 and Cys7, while on the B chain, Cys7 and Cys19 were found to be modified. The details of the peptide sequence, their m/z, and retention time (RT) are provided in Table 1. Also, an annotated MS/MS spectrum of a modified B chain peptide, VcGERGF, is depicted in Figure 3. The b1, b2, and b3 ions of unmodified VCGERGF (Figure 3A) were compared to those of captopril-modified VcGERGF (Figure 3B). Figure 3 illustrates that b2 and b3 ions and thereafter other b ions of modified VcGERGF show an increase in mass (215.06) corresponding to captopril modification. The annotated MS/MS spectra of the other peptides mentioned in Table 1 are provided in the Supporting Information (Figures S2–S6). Tandem mass spectrometry confirms that formation of the captopril adducts occurs at the cysteine residues.
Table 1. Captopril-Modified Peptides of A and B Chains of Insulin Identified by LC-MS/MS.
| chain | modified peptide | site of modification | charge state | modified m/z | Δ ppm | RT [min] |
|---|---|---|---|---|---|---|
| B chain | VNQHLcGSH | Cys7 | 2 | 1209.52214 | 6.77 | 5.18 |
| B chain | VcGERGF | Cys19 | 2 | 982.418 | 5.98 | 6.01 |
| B chain | LVcGERG | Cys19 | 2 | 948.43039 | 2.75 | 6.75 |
| B chain | VcGERG | Cys19 | 2 | 835.34808 | 5.23 | 3.77 |
| A chain | GIVEQCcTS | Cys7 | 2 | 1154.45989 | 6.25 | 8.48 |
| A chain | VEQcCTS | Cys6 | 2 | 984.3401 | 7.14 | 7.78 |
Figure 3.

Annotated MS/MS spectrum of captopril-modified cysteine residues of insulin peptide VCGERGF, by LC-ESI-MS/MS. (A) Unmodified VCGERGF peptide from control insulin (106.92 μM) incubated for 30 min and (B) captopril-modified VcGERGF peptide from 5 mM captopril-treated insulin (106.92 μM) incubated for 30 min. The yellow bars indicate b ions.
The probable mechanism of the insulin and captopril reaction is depicted in Figure 4. The A and B chains of insulin are connected to each other by two cysteine disulfide bonds between Cys7 (A chain)-Cys7 (B chain) and Cys20 (A chain)-Cys19 (B chain). Also, Cys6 and Cys11 residues of the A chain are connected by an intrachain cysteine disulfide bond. Together, MALDI-MS and LC-ESI-MS indicated that captopril treatment on insulin led to the formation of peaks corresponding to both the A chain and B chain and the single and double captopril adducts of the respective chains, which are pictorially represented in Figure 4A–C, respectively. Furthermore, it indicates that captopril modifies the cysteine residues of insulin involved in interchain disulfide bonding. In the LC-ESI MS/MS analysis, we observed that captopril modifies the Cys6 residue of the A chain, which is involved in the intrachain disulfide bonding. Other cysteine residues engaged in interchain disulfide bond formation were also found to be modified by captopril. Thus, captopril being a reactive thiol facilitates the disulfide–thiol exchange reaction in any disulfide bond of insulin and might not have any preference for intra/interchain disulfide cleavage.
Figure 4.

Probable mechanism of covalent modification of insulin: (A) captopril-induced separation of A and B chains of insulin and/or modification of one cysteine residue by captopril in intact insulin, (B) covalent modification of cysteine residues of chain A, and (C) covalent modification of cysteine residues of chain B by the captopril molecule(s). Image A depicting the modification of intact insulin of a single cysteine residue (Cys7) in chain B is just a pictorial representation and does not reflect all other modifications.
Single and double captopril adduct formation in intact insulin (Figure 1A,B) indicates that captopril modification may occur at any one and two cysteine residues involved in either the interchain or intrachain cysteine disulfide bonds, respectively. Notably, in the case of captopril modification at two cysteine residues of the intact insulin, when the A and B chains are not separated, the formation of two captopril adducts may occur either on both the cysteine residues participating in the same disulfide bond or two cysteine residues participating in two different disulfide bonds, but atleast one interchain disulfide bond should remain intact. In the case of captopril modification at a single cysteine residue or two cysteine residues participating in two formation of different disulfide bonds, the cysteine residue which is not forming captopril adducts would be in the thiol form (Figure 4A-iii). We observed captopril adducts of intact insulin and both the A and B chains in the MALDI-MS spectrum and LC-ESI-MS spectrum, which indicates that captopril may modify the cysteine residues of intact insulin and the A or B chains simultaneously. Since two captopril adducts in each A and B chain were observed, the formation of these chain-specific adducts may occur sequentially or simultaneously. However, we did not observe three and four cysteine-modified A chain. This may be either because of their absence or that they were not observed, possibly due to the lower ionization efficiency of the A chain in its heavily modified state. The scheme indicated in Figure 4 is a pictorial representation of the captopril–insulin interaction and does not indicate a sequential reaction.
As captopril separated the A and B chains of insulin, we investigated whether captopril has similar effects on other multimeric proteins such as IgG. IgG was treated with (0.5–25 mM) captopril. The molecular mass of IgG is about 150 kDa, which consists of two heavy chains (each approximately 50 kDa) and two light chains (each approximately 25 kDa). Each heavy chain is connected to a different light chain by two cysteine disulfide bonds, and the heavy chains are connected to each other by two cysteine disulfide bonds. Nonreducing SDS-PAGE analysis was performed without adding β-mercaptoethanol. Figure S7 lane C displays a single band of about 150 kDa of unmodified IgG, which indicates the intact mass of IgG, whereas treatment of captopril (0.5–25 mM) on IgG resulted in the separation of the heavy and light chains of IgG. Treatment of 0.5 mM captopril on IgG led to the formation of two bands around 25 and 125 kDa, indicating the separation of the light chain from the intact IgG and formation of a trimer consisting of two heavy chains and one light chain corresponding to 125 kDa, whereas treatment at 5 and 10 mM led to an additional band around 100 kDa and 50 kDa, indicating the formation of a dimer of heavy chains and light chains, respectively. At 25 mM concentration of captopril, the separation of IgG subunits was evident. The density of the low-molecular weight bands increased with a higher concentration of captopril. These experiments with insulin and IgG suggest that captopril reduces the disulfide bridges and modulates their quaternary structure. IgGs play an essential role in the defense mechanism against various pathogens. The chronic use of captopril may have an adverse effect on the IgG structure and function, and thus, it may affect the immune response.
The previous studies have reported that insulin is modified into the A and B chains by endogenous thiols such as cysteine, similar to captopril in this study. Furthermore, the stability and unfolding pathway of insulin in the presence of thiols are completely different from its reversible denaturation observed in the absence of thiol, where the disulfide bonds remain intact.40
Captopril Modification Leads to Aggregate Formation in the Candidate Proteins
We have observed in the above-mentioned studies that captopril modifies and reduces insulin and IgG. Such effects of captopril on the protein structure may lead to aggregation of these proteins. To investigate whether captopril causes aggregation in protein, first, thioflavin T assay was performed using the insulin (106.92 μM) and captopril concentration (5 mM); these concentrations were used for all the mass spectrometry-based experiments described above. In the presence of captopril, the fluorescence intensity of insulin was not significantly increased (Figure 5A), whereas in our previous experiment with the captopril–IgG interaction, we have found that the light and heavy chains of IgG are separated increasingly by captopril in the concentration range of 0.5–25 mM. Therefore, we have studied the effects of this range of captopril concentration on the IgG aggregation by thioflavin T assay. An increase in the fluorescence intensity in the presence of captopril was observed in IgG. Furthermore, we have studied the effect of the same range of captopril concentration (0.5–25 mM) on insulin aggregation. A similar increase in the fluorescence intensity was observed for captopril-treated insulin for all the captopril concentrations used (Figure 5B). In the above-mentioned thioflavin T assay, the increased fluorescence intensity of insulin and IgG indicates the formation of fibrillar aggregates in the presence of captopril. Since there was a significant increase in the fibrillar aggregates of insulin when a broad range of captopril concentration was used, to further confirm whether the fibrillar aggregates are insoluble in nature, we have performed light scattering study. A considerable increase in the intensity of the scattered light was observed in captopril-treated insulin (Figure 5C). Since the extent of the scattered light is directly dependent on the particle size, increased scattered light indicates the presence of insoluble aggregates in captopril-treated insulin, involving major structural change-associated irreversible aggregate formation41−43 Thioflavin T (a benzothiazole dye) is a molecular probe used extensively to study fibril formation in various amyloidogenic proteins.44,45 As Thio-T binds to the β sheet-rich structure of the amyloid fibrils, it increases the fluorescence emission signal at 482 nm, when excited at 440 nm.46 Thus, the results of the thioflavin T experiment indicate fibril formation in insulin and IgG by captopril modification. As a whole, all the above-mentioned results suggest that captopril has the ability to induce aggregation in insulin and IgG.
Figure 5.

Captopril modification causes aggregation in insulin. (A) Thioflavin T assay to study fibrillar aggregate formation of insulin (106.92 μM) in the presence or absence of 5 mM captopril incubated for 30 min. (B) ThioflavinT assay to study formation of fibrillar aggregates in insulin (23.76 μM) in the presence (0.5–25 mM) or absence of captopril at 24 h of incubation. (C) Light scattering study to detect the insoluble aggregates of insulin (23.76 μM) in the presence of (0.5–25 mM) or absence of captopril at 13 h of incubation. P indicates the probability value.
Molecular Dynamics Simulations Support that Captopril Induces Structural Changes in Insulin
During the covalent interaction between captopril and the cysteine residues of insulin, noncovalent interactions also take place between captopril and the other amino acids. To understand these interactions and their effect on the insulin structure, docking and molecular dynamics studies were performed. A site-specific docking was performed under two conditions: (i) one molecule of captopril bound close to one of the cysteine disulfide bonds (site 1: Cys20 of chain A and Cys19 of chain B) involved in insulin dimer formation and (ii) two molecules of captopril bound to both the cysteine disulfide bonds (Cys20 of chain A, Cys19 of chain B (site 1); and Cys7 of chain A, Cys7 of chain B (site 2)) involved in insulin dimer formation. The binding energy for the best-docked structure for the insulin complex bound with one and two molecules of captopril is −5.2 and −6.4 kcal/mol, respectively.
The interaction pattern and orientation of captopril bound to both cysteine disulfide bonds of insulin are depicted in Figure 6. The residues that participate in hydrophobic interactions are Glu17 (site 2) for two-bound captopril and Val3 (site 1) for one-bound captopril systems. At (site 2), hydrogen bonds are observed at the oxygen of captopril interacting with the hydrogen of Asn21 and at (site 1) oxygen of Gly8 (site 2) interacting with the hydrogen of captopril. The salt bridge was formed between the carboxylate group of captopril and Arg22 at the site 2.
Figure 6.

Investigation of the noncovalent interaction of insulin and captopril by docking study. The docked captopril at two sites on insulin is shown. Site 1 has residues Gly8 and Val3 interacting with one captopril, and at site 2, residues Asn21, Arg22, and Glu17 are shown to interact with captopril. The cysteine residues are shown in orange at both the sites.
Since biophysical studies suggested the formation of fibrillar aggregates by captopril modification, which is based primarily on the structural change, molecular dynamics studies were performed for systems bound with one molecule of captopril and two molecules of captopril to understand the effect of captopril on the insulin structure in a given time. A control simulation was performed without any captopril for better comparison. To explore the effect of captopril on chain A and chain B, we calculated the center of the mass distance between them, as shown in Figure 7A.
Figure 7.

Interchain distances between chain A and chain B by molecular dynamics study. (A) Time series of interchain distance for control (blue), one-bound captopril (red), and two-bound captopril (yellow) systems. (B,C) Interchain distance at the maximum for one-bound and two-bound systems. Chain A is given in blue, chain B is given in red, and captopril is shown in a ball-and-stick model.
For the first 30 ns, the distance between the chains was ∼0.6 nm for the systems with two bound molecules of captopril and 0.8 nm for the system with one bound molecule of captopril. However, large fluctuations were observed between the interchain distances of insulin under both conditions. The maximum distance of about 1.2 nm between chain A and chain B was observed at 360 ns for one-bound and two-bound systems; the maximum distance was about 1.2 nm at 520 ns, as shown in Figure 7B,C. The control insulin structure was stable throughout the simulation when compared to that of the captopril-bound insulin.
As can be seen clearly from the interchain distances, large conformational changes occur in the presence of captopril. Furthermore, to characterize the changes observed in insulin, secondary structural analysis was performed (Figure 8). The results highlighted that secondary structural elements are intact in control insulin simulations. Interestingly, the insulin secondary structure began to lose its helical content in two-bound simulations at residues 8–11 in chain B, as shown in Figure 8C, while one-bound captopril simulations unwind the helical structure at residues 8–10 in chain A (Figure 8B). Transient β-bridges and β sheets are observed in captopril-bound insulin, especially at residues 11-12 in chain A and residues 4–5 in chain B in the one-bound system (Figure 8B). Such a structural representation is observed at the start of the simulation and remains as such for 150 ns and then becomes a coil-like structure (Figure 8D). For the two-bound system (Figure 8C), β-bridges were observed at residues 11–12 in chain A and residues 9–10 in chain B for longer stretches of time. In the control simulation, insulin forms 55% helix and 32% coil, whereas the helical content was significantly reduced when the captopril was bound (one-bound: 48% helix and 35% coil and two-bound: 43% helix and 38% coil). The percentage of the secondary structural content reported is averaged over the entire simulation timescale.
Figure 8.

Time evolution of the secondary structure elements in insulin by molecular dynamics study (A), control (B), one-bound, and (C) two-bound systems. The X-axis represents the molecular dynamics trajectory time in nanoseconds, while the residue numbers are shown on the Y-axis. For better clarity, the first half of the y-axis 0–21 residues refers to the chain A and second half 0–30 residues refers to chain B. (D) Snapshots for the time points showing a large difference in the secondary structure. Chain A has been highlighted in blue and chain B has been highlighted in red. Distinct conformational changes were observed in two-bound chain B (highlighted in the dotted box); the helical content rapidly converted to a more disordered coil.
Previous studies have suggested that the B chain plays an important role in the initiation of insulin aggregation,47,48 and it is well-established that the decreased helical content and increased β sheets are associated with insulin fibrillation.49 In our simulation study, a large structural change was observed in chain B for both one- and two-bound captopril systems (Figure 8), and second, the helical content was found to be decreased and coil content was found to be increased. These two prominent findings in this simulation study favor the conditions that captopril-bound states initiate the aggregation process in insulin, which is also supported by our experimental study of aggregation (Figure 5B,C).
In this work, we have investigated the role of the captopril interaction with human insulin by molecular docking and simulations. The interchain distance calculations have highlighted that captopril has a large influence on the chain conformation. The distances calculated between the two chains show that captopril causes the insulin molecule to open into a wide conformation. The secondary structural analysis revealed that insulin’s secondary structure did not change in the control simulation. The captopril decreases the helical content and increases the coil of the insulin especially of the chain B for both the one- and two-bound captopril systems.
Conclusions
Captopril contains a reactive thiol as a functional group, which is known to modify proteins by the thiol–disulfide exchange. This study demonstrates that captopril modifies intact insulin and separates it into A and B chains and forms their adducts. A series of mass spectrometric experiments suggested that captopril-modified cysteine residues involved in both interchain and intrachain disulfide bond formation. The drug also initiates aggregation and fibril formation. Interestingly, docking and molecular dynamics studies suggested that captopril affects the insulin structure, corroborating our experimental data. The long-term use of captopril in humans possibly affects the structure and function of some of its target proteins. Thus, our findings warrant further investigation into the long-term effect of drugs like captopril.
Materials and Methods
Most of the chemicals were purchased from Sigma Aldrich (IgG, Thioflavin T, etc.); captopril was purchased from MPI, and insulin was purchased from either Sigma or Biocon.
Captopril–Protein Reaction
For captopril–insulin interaction studies, 5 mM captopril was incubated with insulin (106.92 μM) for 30 min at 37°C. For captopril–IgG interaction studies, 0.5–25 mM captopril was incubated with IgG (6.75 μM) for 24 h at 37°C. Insulin (23.76 μM) was incubated in the presence (0.5–25 mM) or absence of captopril for 13 and 24 h, respectively, for the aggregation study. Reactions were carried out in 50 mM sodium phosphate buffer, pH 7.4, containing sodium azide. Captopril was filtered using a 0.2 micron filter before adding to reaction tubes for incubation. All the reactions were performed in triplicate.
Furthermore, before performing mass spectrometry studies of insulin, captopril was removed using a Sep-Pak cartridge (M/s Waters) (as per the vendor’s instructions), followed by drying the elute using a speed vacuum concentrator and reconstituting it in 3% acetonitrile (ACN).
Effect of DTT on Captopril-Modified Insulin
In this reaction, captopril-treated insulin was incubated with or without 5 mM DTT for 20 min at 37 °C. Control insulin was kept without the addition of captopril or DTT at 37 °C. The captopril-induced insulin modifications, followed by DTT treatment, were studied by MALDI-TOF-MS analysis.
MALDI-TOF-MS
Insulin or captopril-modified Insulin was acquired on a MALDI-TOF-MS system (SCIEX5800) in a range of 2000–6000 Da in the positive reflector mode with an acceleration voltage of 25 kV. The analyte was premixed with sinapic acid in 1:30 ratio for insulin and spotted by the dried droplet method. For all spectral acquisition, the laser power was set just above the ion generation threshold to obtain peaks with highest possible signal-to-noise ratio. All spectra were acquired with 250 shots in three replications. The spectra were processed using Data Explorer for advanced baseline correction, noise removal, and mass calibration.
Insulin Digestion Using Thermolysin
Both unmodified and captopril-modified insulin were digested with thermolysin using the manufacturer’s instructions. Briefly, the digestion was initiated by diluting the reaction solution using a suitable dilution buffer. Thermolysin was added to the reaction mixture, and the reaction was allowed to proceed for 3 h at 37 °C. The digestion reaction was stopped by adding 0.1% formic acid. Later, the digested protein was centrifuged at 14,000 rpm for 20 min at 4 °C, and the supernatant was collected and desalted using a Sep-Pak cartridge (M/s Waters) (as per the vendors instructions), and the eluate was subsequently dried using a speed vacuum concentrator. Peptides were stored at −80 °C until further analysis.
Liquid Chromatography Mass Spectrometry
-
(i)
Insulin or captopril-modified insulin was analyzed on a SYNAPT XS (Waters Corp) mass spectrometer equipped with an ACQUITY UPLC I-Class PLUS system. The mass spectrometer was calibrated using sodium iodide (MS grade, Waters Corp) over 400–4500 m/z for intact and subunit mass analysis and over 50–2000 m/z for peptide mapping. Additionally, 50 fmol/ul leucine enkephalin (Waters Corp) at a flow rate of 5 μL/min is used as the LockMass (556.2771 m/z, singly charged, positive mode) during the data acquisition, and mass correction is applied during data processing. Leucine enkephalin was infused through an independent port (LockSpray), and the LockMass is recorded once in every 40 s.
-
(a)
Intact mass analysis: 1.0 μL of insulin (1μg) was loaded onto an ACQUITY UPLC BEH 300 C4 column (300 Å pore size, 1.7 μm particle size; 2.1 mm × 50 mm, Waters Corp) and resolved using a water/acetonitrile gradient (solvent A: 0.1% formic acid in water, solvent B: 0.1% formic acid in acetonitrile) and at a flow rate of 0.2 mL/min. The column was maintained at 80 °C pre-equilibrated in 5% solvent B. The SYNAPT XS is operated in the positive ion mode. The ionization of the protein is performed using an electrospray ionization (ESI) probe. The m/z spectra were analyzed using MassLynx software, and the peak annotation was performed manually. The mass difference for captopril modification was estimated to be 215.0616 Da.
-
(b)
Tandem mass spectrometry by LC-MSE: LC-MSE (MS at elevated energy) was performed on a SYNAPT XS (Waters Corp) mass spectrometer equipped with an ACQUITY UPLC I-Class PLUS system. A total of 5 μL of insulin (5 μg) was separated on an ACQUITY UPLC peptide BEH C18 column, (130 Å pore size, 1.7 μm particle size, 2.1 mm × 100 mm, Waters Corp) using the following water/acetonitrile gradient (solvent A: 0.1% formic acid in water, solvent B: 0.1% formic acid in acetonitrile). The column was equilibrated at 1% B at a flow rate of 0.2 mL/min, and the column temperature was set at 60 °C. The MSE method for data acquisition for the peptide mapping was used on a Waters SYNAPT XS mass spectrometer. The m/z spectra were analyzed using UNIFI software using the amino acid sequence for chain A and chain B of human insulin. The captopril modification at the cysteine residue increases the mass of the peptide by 215.0616 Da.
-
(ii)
Tandem mass spectrometry by DDA: Peptides were reconstituted in 3% ACN + 0.1% formic acid. A total of 1.5 μg of peptide digest was loaded onto a C18 reverse-phase column (dimensions: 100 mm × 0.3 mm, 3 μm, 120 Å) of a microLC 200 liquid chromatography system (Eksigent Technologies) coupled to a Triple TOF 5600 mass spectrometer (SCIEX). Peptides were separated over a 15 min gradient of 3–40% acetonitrile in water with 0.1% formic acid at a flow rate of 7 μL/min.
-
(c)
Raw files obtained from the Triple-TOF-MS (*.WIFF files) were converted to *.mzml using msConvert and then analyzed using Proteome discoverer 2.4 (Thermo Scientific) software for the identification of unmodified and captopril-modified peptides. The mass spectral data were searched again in the insulin database (P01308, Uniprot) using the SEQUEST algorithm with trypsin thermolysin as protease, with a minimum of one missed cleavage allowed. Captopril modification of cysteine residues was selected as a variable modification (215.0616 Da.).
Nonreducing SDS-PAGE of IgG
A total of 20 μg of the control and modified IgG was reconstituted in the Laemmli sample buffer (without β-mercaptoethanol). Then, these samples were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS PAGE). Proteins on the gel were visualized by Coomassie brilliant blue (CBBR-250) staining and destaining.
Thioflavin T Assay Preparation
Thio-T (50 μM) was added to captopril-modified Insulin or IgG. Thioflavin T fluorescence was measured immediately in black 96-well plates with excitation at 440 nm and emission at 482 nm using a Thermo Scientific Varioskan flash fluorescence spectrometer. The background fluorescence of the corresponding blank control (consisting of different concentrations of captopril in addition to the buffer) was subtracted to obtain the actual fluorescence of the protein.
Light Scattering
Light scattering was recorded at 400 nm using a PerkinElmer LS 55 fluorescence spectrometer. The excitation and emission slit widths were set at 2.5 and 10 nm, respectively, to follow the aggregation of insulin after 13 h of incubation with captopril.
Computational Methods
Protein and ligand preparation: The insulin protein X-ray crystallographic structure (resolution 1.47 Å) was obtained from the Protein Data Bank. The protein has two chains: chain A with 21 amino acid residues and chain B with 30 amino acid residues. Water molecules and other heteroatom ligands were excluded from the protein molecule. The ligand molecule captopril was obtained from the ChEMBL database50 in the SMILES format. The SMILES format was further converted into the PDB format using OpenBabel,51 with all hydrogens added. Molecular Docking: The protein and ligand docking studies were performed using the Autodock suite (Autodock 4.2.6).52 The grid-based docking was performed, which uses a grid box to define the search space. Ligand-centered grid maps were generated using the Autogrid program with a grid box size of 26 Å × 26 Å × 34 Å. Two docked systems were generated based on the site at which the ligand is bound: one captopril bound and two captopril bound are referred to as 1-bound and 2-bound, respectively. Based on docking results, the lowest energy and best-docked structure for each system were further considered for molecular dynamics simulations. Molecular dynamics simulations: The molecular dynamics simulations were performed for both the 1-bound and 2-bound docked structures and a control system without a ligand. The Gromacs version 4.5.5 package53 was used to perform the simulations using the CHARMM36 force field.54 The parameters of ligands for the CHARMM force field were obtained from the swissparam server.54 The system energy is minimized via 10,000 steps with a constant temperature (300 K). The system was coupled to Berendson’s thermostat method and a standard pressure of 1.0 bar.55 A time step of 2 fs was used, and the trajectory was written out every 100 ps. The protein–ligand complex and control system were subjected to production run for 640 ns. The simulations were analyzed using Gromacs utilities and in-house scripts.
Acknowledgments
A.G. would like to acknowledge the Department of Science and Technology, New Delhi, Government of India, for Women Scientist Fellowship (SR/WOS-A/LS-1165/2014).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00660.
MS/MS spectra of captopril-modified undigested insulin, annotated MS/MS spectra of captopril-modified cysteine residues of insulin peptides, peptide segregation study of different concentrations of captopril-modified IgG, and aggregation study of different concentrations of captopril-modified IgG (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Rabbani N.; Thornalley P. J. Protein Glycation—Biomarkers of Metabolic Dysfunction and Early-Stage Decline in Health in the Era of Precision Medicine. Redox Biol. 2021, 42, 101920 10.1016/j.redox.2021.101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Forman H. J. Signaling by 4-Hydroxy-2-Nonenal: Exposure Protocols, Target Selectivity and Degradation. Arch. Biochem. Biophys. 2017, 617, 145–154. 10.1016/j.abb.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannuruswamy G.; Jagadeeshaprasad M. G.; Kashinath K.; Kesavan S. K.; Bhat S.; Korwar A. M.; Chougale A. D.; Boppana R.; Reddy D. S.; Kulkarni M. J. Molecules with O-Acetyl Group Protect Protein Glycation by Acetylating Lysine Residues. RSC Adv. 2016, 6, 65572–65578. 10.1039/C6RA11313C. [DOI] [Google Scholar]
- Tatham M. H.; Cole C.; Scullion P.; Wilkie R.; Westwood N. J.; Stark L. A.; Hay R. T. A Proteomic Approach to Analyze the Aspirin-Mediated Lysine Acetylome. Mol. Cell. Proteomics 2017, 16, 310–326. 10.1074/mcp.O116.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.; Jenkins R. E.; Berry N. G.; Maggs J. L.; Farrell J.; Lane C. S.; Stachulski A. V.; French N. S.; Naisbitt D. J.; Pirmohamed M.; Park B. K. Direct Evidence for the Formation of Diastereoisomeric Benzylpenicilloyl Haptens from Benzylpenicillin and Benzylpenicillenic Acid in Patients. J. Pharmacol. Exp. Ther. 2011, 338, 841–849. 10.1124/jpet.111.183871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariza A.; Collado D.; Vida Y.; Montañez M. I.; Pérez-Inestrosa E.; Blanca M.; Torres M. J.; Cañada F. J.; Pérez-Sala D. Study of Protein Haptenation by Amoxicillin through the Use of a Biotinylated Antibiotic. PLoS One 2014, 9, e90891 10.1371/journal.pone.0090891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai T.; Ikeda T.; Kawai K.; Kameyama E.; Shindo H. In Vitro Studies on the Metabolic Pathway of SQ 14225 (Captopril) and Mechanism of Mixed Disulfide Formation. J. Pharmacobio-Dyn. 1981, 4, 677–684. 10.1248/bpb1978.4.677. [DOI] [PubMed] [Google Scholar]
- Coleman J.; Yeung J.; Roberts D.; Breckenridge A.; Park B. Drug-specific Antibodies in Patients Receiving Captopril. Br. J. Clin. Pharmacol. 1986, 22, 161–165. 10.1111/j.1365-2125.1986.tb05244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. K.; Grabowski P. S.; Yeung J. H. K.; Breckenridge A. M. Drug Protein Conjugates—I. A Study of the Covalent Binding of [14C]Captopril to Plasma Proteins in the Rat. Biochem. Pharmacol. 1982, 31, 1755–1760. 10.1016/0006-2952(82)90680-3. [DOI] [PubMed] [Google Scholar]
- Chopra M.; Scott N.; McMurray J.; McLay J.; Bridges A.; Smith W.; Belch J. Captopril: A Free Radical Scavenger. Br. J. Clin. Pharmacol. 1989, 27, 396–399. 10.1111/j.1365-2125.1989.tb05384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Tang Y.; Rong W.; Zhang X.; Zhao W.; Zi Y. Analysis of Binding Interaction between Captopril and Human Serum Albumin. Am. J. Anal. Chem. 2011, 2, 250–257. 10.4236/ajac.2011.22030. [DOI] [Google Scholar]
- Migdalof B. H.; Antonaccio M. J.; Mc Kinstry D. N.; Singhvi S. M.; Lan S.-J.; Egli P.; Kripalani K. J. Captopril: Pharmacology, Metabolism, and Disposition. Drug Metab. Rev. 1984, 15, 841–869. 10.3109/03602538409041080. [DOI] [PubMed] [Google Scholar]
- Yeung J. H. K.; Breckenridge A. M.; Park B. K. Drug Protein Conjugates-VI. Role of Glutathione in the Metabolism of Captopril and Captopril Plasma Protein Conjugates. Biochem. Pharmacol. 1983, 32, 3619–3625. 10.1016/0006-2952(83)90313-1. [DOI] [PubMed] [Google Scholar]
- Duchin K. L.; McKinstry D. N.; Cohen A. I.; Migdalof B. H. Pharmacokinetics of Captopril in Healthy Subjects and in Patients with Cardiovascular Diseases. Clin. Pharmacokinet. 1988, 14, 241–259. 10.2165/00003088-198814040-00002. [DOI] [PubMed] [Google Scholar]
- Kubo S. H.; Cody R. J. Clinical Pharmacokinetics of the Angiotensin Converting Enzyme Inhibitors. A Review. Clin. Pharmacokinet. 1985, 10, 377–391. 10.2165/00003088-198510050-00001. [DOI] [PubMed] [Google Scholar]
- Yeung J. H. K.; Breckenridge A. M.; Kevin Park B. Drug-Protein Conjugates—IV: The Effect of Acute Renal Failure on the Disposition of [14C]Captopril in the Rat. Biochem. Pharmacol. 1983, 32, 2467–2472. 10.1016/0006-2952(83)90003-5. [DOI] [PubMed] [Google Scholar]
- Sullivan J. M.; Ginsburg B. A.; Ratts T. E.; Johnson J. G.; Barton B. R.; Kraus D. H.; McKinstry D. N.; Muirhead E. E. Hemodynamic and Antihypertensive Effects of Captopril, an Orally Active Angiotensin Converting Enzyme Inhibitor. Hypertension 1979, 1, 397–401. 10.1161/01.hyp.1.4.397. [DOI] [PubMed] [Google Scholar]
- Posadas C.; Sánchez G.; Boyer J. L.; Delmar M.; Serrano P. A. [Effect of inhibition of converting enzyme by captopril on arterial pressure, renin and aldosterone in essential hypertension]. Arch. Inst. Cardiol. Mex. 1982, 52, 295–300. [PubMed] [Google Scholar]
- Levine T. B.; Franciosa J. A.; Cohn J. N. Acute and Long-Term Response to an Oral Converting-Enzyme Inhibitor, Captopril, in Congestive Heart Failure. Circulation 1980, 62, 35–41. 10.1161/01.cir.62.1.35. [DOI] [PubMed] [Google Scholar]
- Pfeffer M. A.; Braunwald E.; Moyé L. A.; Basta L.; Brown E. J.; Cuddy T. E.; Davis B. R.; Geltman E. M.; Goldman S.; Flaker G. C.; Klein M.; Lamas G. A.; Packer M.; Rouleau J.; Rouleau J. L.; Rutherford J.; Wertheimer J. H.; Hawkins C. M. Effect of Captopril on Mortality and Morbidity in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 1992, 327, 669–677. 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- St John Sutton M.; Pfeffer M. A.; Plappert T.; Rouleau J. L.; Moyé L. A.; Dagenais G. R.; Lamas G. A.; Klein M.; Sussex B.; Goldman S. Quantitative Two-Dimensional Echocardiographic Measurements Are Major Predictors of Adverse Cardiovascular Events after Acute Myocardial Infarction. The Protective Effects of Captopril. Circulation 1994, 89, 68–75. 10.1161/01.CIR.89.1.68. [DOI] [PubMed] [Google Scholar]
- Plosker G. L.; McTavish D. Captopril. Drugs Aging 1995, 7, 226–253. 10.2165/00002512-199507030-00007. [DOI] [PubMed] [Google Scholar]
- Fouad A. A.; Al-Mulhim A. S.; Jresat I.; Morsy M. A. Protective Effects of Captopril in Diabetic Rats Exposed to Ischemia/Reperfusion Renal Injury. J. Pharm. Pharmacol. 2012, 65, 243–252. 10.1111/j.2042-7158.2012.01585.x. [DOI] [PubMed] [Google Scholar]
- Nagasawa T.; Hye Khan M. A.; Imig J. D. Captopril Attenuates Hypertension and Renal Injury Induced by the Vascular Endothelial Growth Factor Inhibitor Sorafenib. Clin. Exp. Pharmacol. Physiol. 2012, 39, 454–461. 10.1111/j.1440-1681.2012.05699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z.; Huang D.; Jiang J.; Li Y.; Li H.; Ke Y. Captopril Alleviates Hypertension-Induced Renal Damage, Inflammation, and NF-KB Activation. Braz. J. Med. Biol. Res. 2018, 51, 20187338 10.1590/1414-431x20187338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Yang C.; Huang L.; Chen Y.; Li Y.; Cheng C.; Cheng B.; Zheng L.; Huang K. Glycated Insulin Exacerbates the Cytotoxicity of Human Islet Amyloid Polypeptides: A Vicious Cycle in Type 2 Diabetes. ACS Chem. Biol. 2019, 14, 486–496. 10.1021/acschembio.8b01128. [DOI] [PubMed] [Google Scholar]
- Jia X.; Olson D. J. H.; Ross A. R. S.; Wu L.; et al. Structural and Functional Changes in Human Insulin Induced by Methylglyoxal. FASEB J. 2006, 20, 1555–1557. 10.1096/fj.05-5478fje. [DOI] [PubMed] [Google Scholar]
- Ku M. C.; Fang C. M.; Cheng J. T.; Liang H. C.; Wang T. F.; Wu C. H.; Chen C. C.; Tai J. H.; Chen S. H. Site-Specific Covalent Modifications of Human Insulin by Catechol Estrogens: Reactivity and Induced Structural and Functional Changes. Sci. Rep. 2016, 6, 28804 10.1038/srep28804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S. J.; Boyd A. C.; O’harte F. P. M.; Mckillop A. M.; Wiggam M. I.; Mooney M. H.; Mccluskey J. T.; Lindsay J. R.; Ennis C. N.; Gamble R.; Sheridan B.; Barnett C. R.; Mcnulty H.; Bell P. M.; Flatt P. R. Demonstration of Glycated Insulin in Human Diabetic Plasma and Decreased Biological Activity Assessed by Euglycemic-HyperInsulinemic Clamp Technique in Humans. Diabetes 2003, 52, 492–498. 10.2337/diabetes.52.2.492. [DOI] [PubMed] [Google Scholar]
- Abedini A.; Schmidt A. M. Mechanisms of Islet Amyloidosis Toxicity in Type 2 Diabetes. FEBS Lett. 2013, 587, 1119–1127. 10.1016/j.febslet.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm K. R.; Yanamandra K.; Gruden M. A.; Zamotin V.; Malisauskas M.; Casaite V.; Darinskas A.; Forsgren L.; Morozova-Roche L. A. Immune Reactivity towards Insulin, Its Amyloid and Protein S100B in Blood Sera of Parkinson’s Disease Patients. Eur. J. Neurol. 2007, 14, 327–334. 10.1111/j.1468-1331.2006.01667.x. [DOI] [PubMed] [Google Scholar]
- Nakashima F.; Shibata T.; Kamiya K.; Yoshitake J.; Kikuchi R.; Matsushita T.; Ishii I.; Giménez-Bastida J. A.; Schneider C.; Uchida K. Structural and Functional Insights into S-Thiolation of Human Serum Albumins. Sci. Rep. 2018, 8, 932 10.1038/s41598-018-19610-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert H. F.Thiol/Disulfide Exchange and Redox Potentials of Proteins. In Bioelectrochemistry of Biomacromolecules; Lenaz G.; Milazzo G., Eds.; Birkhäuser Basel: Basel, 1997; pp 256–324. 10.1007/978-3-0348-9179-0_5. [DOI] [Google Scholar]
- Röder P. V.; Wu B.; Liu Y.; Han W. Pancreatic Regulation of Glucose Homeostasis. Exp. Mol. Med. 2016, 48, e219 10.1038/emm.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M.; Steiner D. F.; Philipson L. H.. Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships, Anawalt B.; Boyce A.; Chrousos G.; de Herder W. W.; Dhatariya K.; Dungan K.; Grossman A.; Hershman J. M.; Hofland J.; Kalra S.; Kaltsas G.; Koch C.; Kopp P.; Korbonits M.; Kovacs C. S.; Kuohung W.; Laferrère B.; McGee E. A.; McLachlan R.; Morley J. E.; New M.; Purnell J.; Sahay R.; Singer F.; Stratakis C. A.; Trence D. L.; Feingold K. R., Eds.; MDText.com, Inc.: South Dartmouth (MA), 2000. [PubMed] [Google Scholar]
- Yang Y.; Petkova A.; Huang K.; Xu B.; Hua Q.-X.; Ye I.-J.; Chu Y.-C.; Hu S.-Q.; Phillips N. B.; Whittaker J.; Ismail-Beigi F.; Mackin R. B.; Katsoyannis P. G.; Tycko R.; Weiss M. A. An Achilles’ Heel in an Amyloidogenic Protein and Its Repair: Insulin Fibrillation and Therapeutic Design. J. Biol. Chem. 2010, 285, 10806–10821. 10.1074/jbc.M109.067850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong H.; He Z.; Peng A.; Zhang X.; Cheng B.; Sun Y.; Zheng L.; Huang K. Effects of Several Quinones on Insulin Aggregation. Sci. Rep. 2014, 4, 5648 10.1038/srep05648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scigelova M.; Green P. S.; Giannakopulos A. E.; Rodger A.; Crout D. H. G.; Derrick P. J. A Pratical Protocol for the Reduction of Disulfide Bonds in Proteins Prior to Analysis by Mass Spectrometry. Eur. J. Mass Spectrom. 2001, 7, 29–34. 10.1255/ejms.385. [DOI] [Google Scholar]
- Gundry R. L.; White M. Y.; Murray C. I.; Kane L. A.; Fu Q.; Stanley B. A.; Van Eyk J. E. Preparation of Proteins and Peptides for Mass Spectrometry Analysis in a Bottom-Up Proteomics Workflow. Curr. Protoc. Mol. Biol. 2010, 90, 10.25.1–10.25.23. 10.1002/0471142727.mb1025s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.; Chang J. Y. Unfolding and Breakdown of Insulin in the Presence of Endogenous Thiols. FEBS Lett. 2005, 579, 3927–3931. 10.1016/j.febslet.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Wakankar M. S.; Krishnasastry M. V.; Jaokar T. M.; Patel K. A.; Gaikwad S. M. Solution and in Silico Studies on the Recombinant Lectin from Cicer Arietinum Seeds. Int. J. Biol. Macromol. 2013, 56, 149–155. 10.1016/j.ijbiomac.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Yadav P.; Shahane G.; Ramasamy S.; Sengupta D.; Gaikwad S. Structural-Functional Insights and Studies on Saccharide Binding of Sophora Japonica Seed Lectin. Int. J. Biol. Macromol. 2016, 91, 75–84. 10.1016/j.ijbiomac.2016.05.047. [DOI] [PubMed] [Google Scholar]
- Pouchucq L.; Lobos-Ruiz P.; Araya G.; Valpuesta J. M.; Monasterio O. The Chaperonin CCT Promotes the Formation of Fibrillar Aggregates of γ-Tubulin. Biochim. Biophys. Acta, Proteins Proteomics 1866, 2018, 519–526. 10.1016/j.bbapap.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Villegas V.; Zurdo J.; Filimonov V. V.; Avilés F. X.; Dobson C. M.; Serrano L. Protein Engineering as a Strategy to Avoid Formation of Amyloid Fibrils. Protein Sci. 2000, 9, 1700–1708. 10.1110/ps.9.9.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Rahimi F.; Sinha S.; Maiti P.; Bitan G.; Murakami K.. Amyloids and Protein Aggregation-Analytical Methods. In Encyclopedia of Analytical Chemistry; Wiley, 2006. [Google Scholar]
- Namioka S.; Yoshida N.; Konno H.; Makabe K. Residue-Specific Binding Mechanisms of Thioflavin T to a Surface of Flat β-Sheets within a Peptide Self-Assembly Mimic. Biochemistry 2020, 59, 2782–2787. 10.1021/acs.biochem.0c00280. [DOI] [PubMed] [Google Scholar]
- Akbarian M.; Yousefi R.; Moosavi-Movahedi A. A.; Ahmad A.; Uversky V. N. Modulating Insulin Fibrillation Using Engineered B-Chains with Mutated C-Termini. Biophys. J. 2019, 117, 1626–1641. 10.1016/j.bpj.2019.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciszak E.; Beals J. M.; Frank B. H.; Baker J. C.; Carter N. D.; Smith G. D. Role of C-Terminal B-Chain Residues in Insulin Assembly: The Structure of Hexameric LysB28ProB29-Human Insulin. Structure 1995, 3, 615–622. 10.1016/S0969-2126(01)00195-2. [DOI] [PubMed] [Google Scholar]
- Nguyen H. D.; Hall C. K. Molecular Dynamics Simulations of Spontaneous Fibril Formation by Random-Coil Peptides. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 16180–16185. 10.1073/pnas.0407273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M.; Nowotka M.; Papadatos G.; Dedman N.; Gaulton A.; Atkinson F.; Bellis L.; Overington J. P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612–W620. 10.1093/nar/gkv352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V.; Cuendet M. A.; Grosdidier A.; Michielin O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Banavali N.; Foloppe N. Development and Current Status of the CHARMM Force Field for Nucleic Acids. Biopolymers 2000, 56, 257–265. . [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.