

Abstract
Acylated homoserine lactone molecules are used by a number of gram-negative bacteria to regulate cell density-dependent gene expression by a mechanism known as quorum sensing (QS). In Pseudomonas aeruginosa, QS or cell-to-cell signaling controls expression of a number of virulence factors, as well as biofilm differentiation. In this study, we investigated the role played by the las and rhl QS systems during the early stages of static biofilm formation when cells are adhering to a surface and forming microcolonies. These studies revealed a marked difference in biofilm formation between the PAO1 parent and the QS mutants when glucose, but not citrate, was used as the sole carbon source. To further elucidate the contribution of lasI and rhlI to biofilm maturation, we utilized fusions to unstable green fluorescent protein in concert with confocal microscopy to perform real-time temporal and spatial studies of these genes in a flowing environment. During the course of 8-day biofilm development, lasI expression was found to progressively decrease over time. Conversely, rhlI expression remained steady throughout biofilm development but occurred in a lower percentage of cells. Spatial analysis revealed that lasI and rhlI were maximally expressed in cells located at the substratum and that expression decreased with increasing biofilm height. Because QS was shown previously to be involved in biofilm differentiation, these findings have important implications for the design of biofilm prevention and eradication strategies.
Bacteria have an innate propensity to stick to surfaces, and the bacteria growing in adherent, slime-encased communities known as biofilms constitute a major portion of the bacterial biomass present in nature. This phenomenon can be advantageous or deleterious depending on one's perspective. Biofilms established by commensal bacteria in the gastrointestinal tract or biofilms that contribute to the process of bioremediation represent examples of beneficial biofilms. However, in many clinical and industrial settings, biofilms represent a hazardous and costly problem. For these reasons, biofilm control has become an area of intense study. Clearly, regulation of biofilm formation requires a better understanding of the molecular mechanisms underlying the complex and dynamic processes of biofilm development.
Many bacteria are capable of forming biofilms, and Pseudomonas aeruginosa is one of the most commonly studied. Recent work has begun to uncover some of the genetic and molecular mechanisms underlying biofilm production by this organism. In a static system, during the early stages of biofilm development P. aeruginosa cells deficient in flagellar motility exhibit poor surface attachment, while cells lacking type IV pili are unable to form microcolonies (17). As the biofilm matures, microcolonies differentiate into a more complex architecture, often consisting of mushroom and pillar-like structures with intervening water channels. Recently, it was discovered that in a flowing environment, P. aeruginosa quorum-sensing (QS) signal molecules termed autoinducers (AI) play an important role in the differentiation process (6). AI molecules have been detected in biofilms formed on rocks in streams (15) and on catheter tubing from patients (27), proving that this phenomenon takes place in naturally occurring biofilms.
QS systems make use of a transcriptional activator protein that acts in concert with a small AI signaling molecule to stimulate expression of target genes. As the cell population increases, so does the concentration of AI, which provides a means of monitoring cell density. After a threshold level of an AI is reached, the AI binds to the transcriptional activator, enabling it to induce expression of target genes. P. aeruginosa has two well-studied QS systems, las and rhl. The las system is comprised of the transcriptional activator LasR and the AI synthase enzyme LasI, which directs synthesis of the signal molecule N-(3-oxododecanoyl) homoserine lactone (3O-C12-HSL). Similarly, the rhl system is comprised of the transcriptional activator RhlR and the RhlI AI synthase that synthesizes N-butyryl homoserine lactone (C4-HSL). In studies examining QS and P. aeruginosa biofilm formation, strains deficient in production of the las signal molecule, 3O-C12-HSL, formed very thin biofilms that lacked the three-dimensional architecture observed with the parent (6). In addition, while the wild-type biofilm was resistant to sodium dodecyl sulfate (SDS), the biofilm formed by the lasI mutant was easily dispersed upon exposure to SDS. A rhlI mutant biofilm, in contrast, closely resembled the parent biofilm, suggesting that the las QS system, but not the rhl QS system, is important for P. aeruginosa biofilm development.
A number of genes and gene products involved in P. aeruginosa virulence are regulated by either the las QS system or the rhl QS system or both (for a review, see reference 21). In addition, the rhl QS system is under regulatory control of the las system at both the transcriptional level (14, 20) and the posttranslational level (20). Thus, the two regulatory circuits are intimately intertwined. In this investigation, we examined the roles played by the las and rhl QS systems during the initial stages of biofilm formation, when cells are attaching to a surface and forming microcolonies. Furthermore, we sought to elucidate the temporal and spatial expression of two key QS genes, lasI and rhlI, during biofilm development by using gfp reporter fusions. Because the lasI and rhlI genes encode enzymes that ultimately generate the AI signal molecules, understanding the patterns of lasI and rhlI gene expression may reveal important clues about the role played by intercellular communication during biofilm formation.
In the present study, we demonstrated that environmental conditions such as static versus flowing systems and medium composition have a dramatic effect on biofilm production. These findings should be considered during the design and interpretation of P. aeruginosa biofilm studies since factors that influence biofilm formation under one set of conditions may not do so under all circumstances.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacterial strains and plasmids used are listed in Table 1. Plasmid pTdK-GFP was constructed by cloning the 0.775-kb EcoRI-HindIII fragment (containing the GFPmut3 gene) from pFPV25 into EcoRI-HindIII-cut pUCP22. To generate the transcriptional fusion vector pTdK-LVAgfp, pJK1 was digested with SphI, and the ends were made flush with the Klenow fragment, followed by XbaI digestion. The unstable LVAgfp gene from pJBA111 was digested with NotI, treated with the Klenow fragment, and digested with XbaI before being cloned into pJK1. Plasmid prhlI-LVAgfp was constructed from intermediate plasmid pTdK-rhlIGFP. pTdK-rhlIGFP contains the rhlI promoter on a 900-bp EcoRI-XbaI fragment from pLPRI cloned into the same sites of pJKI. To replace the stable green fluorescent protein (GFP) gene with LVAgfp, pTdK-rhlIGFP was digested with SphI, treated with the Klenow fragment, and then digested with XbaI, which removed gfp. In its place we cloned the unstable gfp gene previously isolated from pJBA111 by NotI digestion, treatment with the Klenow fragment, and then XbaI digestion. To construct plasI-LVAgfp, the lasI promoter from p116>97Mn#1 was cloned on a 300-bp EcoRI-BamHI fragment into the same sites of pTdK-LVAgfp. This lasI promoter contains an A-to-G substitution 36 bp upstream of the lasI translational start site and was used because of the short half-life of LVAgfp. The estimated half-life of this GFP is 40 min in Pseudomonas putida and 60 min in Eschericia coli (1), which necessitates the use of strong promoters for sufficient GFP accumulation. The A-to-G substitution increases the promoter strength approximately 10-fold, but the promoter and its expression pattern are otherwise identical to those of the wild type (25).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant genotype or phenotype | Source or reference |
|---|---|---|
| P. aeruginosa strains | ||
| PAO1 | Wild type | 13 |
| PAO-JP1 | ΔlasI::Tet strain PAO1 derivative | 19 |
| PDO-100 | ΔrhlI::Tn501-2 strain PAO1 derivative | 3 |
| PAO-JP2 | ΔlasI::Tet ΔrhlI::Tn501-2 strain PAO1 derivative | 19 |
| Plasmids | ||
| pFPV25 | Promoterless GFPmut3 transcriptional fusion vector, Ampr | 5 |
| pUCP22 | Broad-host-range cloning vector, Ampr | 30 |
| pTdK-GFP | GFPmut3.1 gene under control of the lac promoter, contains an origin of replication for both P. aeruginosa and E. coli, Ampr | This study |
| pJK1 | Promoterless GFPmut3.1 transcriptional fusion vector, contains an origin of replication for both P. aeruginosa and E. coli Ampr | Our laboratory |
| pJBA111 | Source of the LVAgfp gene | 1 |
| pLPRI | rhlI-lacZ transcriptional fusion, Ampr | 29 |
| p116>97Mn#1 | lasI-lasZ transcriptional fusion (with an A-to-G substitution 36 bp upstream of the lasI start of translation, Ampr | Our laboratory |
| pTdK-LVAgfp | Promoterless LVA-gfp transcriptional fusion vector, Ampr | This study |
| plasI-LVAgfp | pTdK-LVAgfp containing an lasI-LVAgfp transcriptional fusion, Ampr | This study |
| pTdK-rhlIGFP | pJK1 containing an rhlI-gfp transcriptional fusion, Ampr | This study |
| prhlI-LVAgfp | pTdK-LVAgfp containing an rhlI- LVAgfp transcriptional fusion, Ampr | This study |
DNA techniques.
Purification, cloning, electrophoresis, and other manipulations of DNA were performed by using standard techniques (11, 24).
Growth media.
The M9 growth medium used for the static biofilm studies consisted of the following: 47.7 mM Na2HPO4 · 7H2O, 21.7 mM KH2PO4, 8.6 mM NaCl, 18.7 mM NH4Cl, and 0.5% (wt/vol) Casamino Acids. Stock solutions of glucose and MgSO4 were filter sterilized and added to final concentrations of 11.1 and 1 mM, respectively, after autoclaving. The FAB medium that was used for both the static and flowthrough biofilm studies consisted of minimal salts [0.1 mM CaCl2, 0.01 mM Fe-EDTA, 0.15 mM (NH4)SO4, 0.33 mM Na2HPO4, 0.2 mM KH2PO4, 0.5 mM NaCl, 0.5% (wt/vol) Casamino Acids, and 1 mM MgCl2, added after autoclaving) and 10 mM sodium citrate as the carbon source. Carbenicillin was added at a concentration of 200 μg/ml for plasmid maintenance.
Static biofilm experiments.
Strains PAO1, PAO-JP1, PDO-100, and PAO-JP2 harboring plasmid pTdK-GFP (gfp constitutively expressed from the lac promoter) were grown in shell vials containing glass coverslips (ViroMed Laboratories, Minneapolis, Minn.). Vials were inoculated with 0.5-ml portions of an overnight culture grown in M9 or FAB medium supplemented with carbenicillin (200 μg/ml) and diluted to a concentration of 107 CFU/ml with the same medium. As indicated below, 1 μM synthetic 3O-C12-HSL or 5 μM synthetic C4-HSL, generated as previously described (18), was added to the culture medium. After 18 h of incubation at 37°C, the glass coverslips were rinsed in phosphate-buffered saline and then examined microscopically (BX-FLA microscope; Olympus America Inc., Lake Success, N.Y.).
Twitching motility assays.
The type IV-mediated twitching motility of PAO1, PAO-JP1, PDO-100, and PAO-JP2 was assayed by using M9 and FAB medium plates containing 1% agar. Overnight cultures grown on M9 and FAB media were stabbed into the bottoms of the plates, and after 48 h of incubation at 37°C, the twitch zones were measured. Assays were performed in triplicate.
Flagellar motility assays.
The flagellum-mediated motility of PAO1, PAO-JP1, PDO-100, and PAO-JP2 was assessed on M9 and FAB medium motility plates containing 0.3% agar. A 1-μl aliquot of an overnight culture grown in M9 or FAB broth was inoculated into the agar, and after 10 h of incubation at 37°C, the diameter of the swim zone was measured. All assays were performed in triplicate.
Western blot analysis of LPS.
Lipopolysaccharide (LPS) was prepared by the whole-cell lysis method of Hitchcock and Brown (12). A 15-μl aliquot of each LPS sample was separated on an SDS–12% polyacrylamide gel and transferred to a nitrocellulose membrane by standard techniques (24). Chemiluminescent detection of the LPS was carried out by using monoclonal antibody (MAb) N1F10 (specific for A-band LPS) and MAb MF15-4 (specific for serotype O5 B-band LPS) and the protocol supplied with an ECL chemiluminescence Western blotting kit (Amersham Life Science Inc., Arlington Heights, Ill.).
Flow chamber experiments.
Polycarbonate flowcells (Protofab, Bozeman, Mont.) were used for biofilm cultivation and microscopic analyses. The flowcells each had a channel (1.6 by 11.1 by 38 mm) with tapered ends and an overall volume of 1.36 ml. Glass coverslips (no. 1; 40 by 60 mm) were used in the flowcells as the biofilm substratum, and silicon tubing was utilized throughout the remainder of the system. A peristaltic pump was employed to maintain a flow rate of 0.21 ml/min, which yielded a residence time within the flowcells of 6.5 min. A polycarbonate bubble trap was used on each line between the pump and the flowcell to eliminate disruption of the biofilm by air bubbles (4). A 2-ml aliquot of an overnight culture (optical density at 660 nm, 1.3 to 1.6) was inoculated directly into each flowcell. Inoculated flowcells were kept static for 1 h before the flow was initiated. Three flowcells were used per experiment, and they were examined on days 4, 6, and 8. At each sampling time, a flowcell was stained with a 2 μM propidium iodide–5 μM Syto85 emulsion (Molecular Probes, Eugene, Oreg.). The emulsion (2 ml per flowcell) was slowly injected immediately upstream of the flowcell, allowed to stain cells for 30 min under static conditions, and then flushed out with fresh medium for 30 min. After flushing, the flowcell was permanently disconnected from the system prior to microscopic analysis. Each experiment was done in duplicate.
Microscopy.
All flowcells were nondestructively analyzed by using a scanning confocal laser microscope (SCLM) equipped with dual photon lasers (Leica Lasertechnik GmbH, Heidelberg, Germany). Images were collected at 488 and 545 nm to record the number of bacteria emitting GFP and the total number of cells stained with propidium iodide-Syto85, respectively. Scans were made through the biofilm that had accumulated on each glass surface at different depths from the substratum to the biofilm surface. Typically, optical sections were obtained every 0.5 μm. Three areas were analyzed per flowcell. In each area, a depth profile was constructed based on the compiled scans.
Image analysis.
Each set of confocal images for an area was analyzed further by image analysis. Percent expression levels were calculated by dividing the pixels above background in the GFP (green) signal by the pixels above background in the propidium iodide-Syto85 (red) signal at the same location. These values were generated for each slice of the biofilm and then compiled to create a vertical distribution of genetic expression. This analysis was done by using CellComp software (University of Rochester, Rochester, N.Y.). This software measured the percentage of similarity between corresponding GFP and propidium iodide-Syto85 optical sections. The images were first converted to binary data according to user-specified thresholds. A pixel was considered active if its intensity exceeded the threshold intensity and passive if it did not. Once the images were converted to binary data, the correspondence was computed for each scan depth by using the following formula:
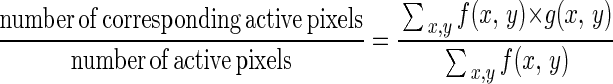 |
where f(x, y) and g(x, y) are the binary functions of the propidium iodide-Syto85 and GFP scans, respectively. The output of CellComp was then fed into a Microsoft Excel Visual Basic macro that graphed correspondence (percent expression) versus height.
Normalization.
Percent expression values were normalized due to the difference in promoter strength between the lasI and rhlI constructs, as well as the difference in signal intensities recorded in the 488- and 545-nm channels. To determine the normalized value for each plasmid construct, 1-ml aliquots from a 24-h culture harboring each construct were washed in phosphate-buffered saline, resuspended in the propidium iodide-Syto 85 emulsion for 30 min, and then fixed with formalin. Wet mounts were analyzed by confocal microscopy. Ten images were collected for each construct. The images were processed as described above. Correspondence values were then considered to be the maximal values attainable for each construct. Based on these numbers, percent expression values from the flowcell studies were adjusted accordingly.
RESULTS
Role of the las and rhl QS systems in static biofilm formation.
To assess the role played by the las and rhl QS systems during the initial stages of biofilm development, PAO1, PAO-JP1 (ΔlasI), PDO100 (ΔrhlI), and PAO-JP2 (ΔlasIrhlI) were grown in shell vials containing glass coverslips for 18 h. Each of the strains harbored plasmids with gfp constitutively expressed from the lac promoter to facilitate microscopic visualization. We examined two minimal media commonly used in biofilm studies, M9 medium containing glucose and FAB medium containing citrate, to evaluate what affect media had on biofilm formation. It should be noted that no differences in the growth rates of PAO1, PAO-JP1, PDO-100, and PAO-JP2 were observed and that the strains grew equally well in M9 and FAB media (data not shown). As Fig. 1 shows, after 18 h of incubation in M9 medium, PAO1 had formed a thick, multilayer biofilm. In contrast to the parent, PAO-JP1 and PDO-100 adhered sparsely to the coverslips and any microcolonies present were only a few cells deep, while the biofilm formed by PAO-JP2 was noticeably thinner. However, when the mutant strains were grown in the presence of an exogenous supply of the deficient AI, the biofilms were much thicker, resembling the parent biofilm (Fig. 1). In contrast, the PAO1 biofilm grown in FAB medium was only a sparse monolayer. Similarly, few cells from the AI-deficient mutants attached to the coverslips in FAB medium in either the presence or the absence of exogenous AI (Fig. 1). These observations suggest that when glucose is used as the carbon source, QS is a regulating factor(s) involved in biofilm initiation; however, in FAB-citrate medium, the QS-regulated component is repressed. Besides carbon source, there are other compositional differences between the two media used, and these differences may have affected biofilm formation as well. O'Toole and Kolter (17) have reported that both type IV pili and flagellar motility are required for the initial stages of P. aeruginosa biofilm formation. In a previous study it was discovered that PDO-100 has markedly decreased type IV pili on its surface and that PAO-JP1, while expressing pili, exhibits abnormal twitching motility when it is grown on LA plates (10). In this study, we examined the twitching motility of strains PAO1, PAO-JP1, PDO-100, and PAO-JP2 in both M9 and FAB media. The results of these analyses showed that in M9 medium strain PAO1 exhibited good twitching motility (2.91 ± 0.24 mm); however, the PAO1 twitch zone was markedly smaller in FAB medium (0.77 ± 0.38 mm). Twitching motility could not be detected in any of the QS mutants grown on either M9 or FAB medium (0 mm). These findings suggest that type IV twitching motility plays a role in static biofilm formation and is consistent with previous findings (17).
FIG. 1.

Static biofilms formed by strains PAO1, PAO-JP1, PDO-100, and PAO-JP2 in M9 medium containing glucose and FAB medium containing citrate. The QS mutants were grown either in the presence or in the absence of exogenous AI. The concentrations of AI added to the media are as follows: 1 μM 3O-C12-HSL for PAO-JP1, 5 μM C4-HSL for PDO-100, and 1 μM 3O-C12-HSL and 5 μM C4-HSL for PAO-JP2.
When we compared the flagellar motility of PAO1 and the flagellar motility of the QS mutants, the mutants exhibited only slightly decreased flagellar motility in M9 medium (Table 2). In FAB medium, the QS mutants exhibited similar or marginally increased motility compared to PAO1; however, the diameters of the swim zones for all four strains in FAB medium were less than one-half those observed for the same strains in M9 medium. Thus, it is possible that decreased flagellar motility contributed to poor attachment by PAO1 in the static biofilm experiments when the organism was grown in FAB medium.
TABLE 2.
Flagellar motility assays in M9 and FAB media
| Strain | Swim zone diam (cm) in:
|
|
|---|---|---|
| M9 medium | FAB medium | |
| PAO1 | 1.78 (0.08)a | 0.55 (0.05) |
| PAO-JP1 | 1.55 (0.05) | 0.55 (0.05) |
| PDO-100 | 1.4 (0.05) | 0.7 (0.1) |
| PAO-JP2 | 1.53 (0.12) | 0.68 (0.104) |
Mean (standard deviation).
LPS expression is not regulated by QS.
LPS is the predominant antigen on the surface of P. aeruginosa cells. The abundance of this antigen, combined with the finding that LPS can play a role in attachment to surface (8, 23), prompted us to investigate whether the LPS expressed by PAO1 and the QS mutants contributed to binding differences in the static biofilms. Most strains of P. aeruginosa express two chemically and immunologically distinct types of LPS, termed A-band LPS and B-band LPS. B-band LPS is composed of long-chain O antigen that determines the serotype specificity of a strain, whereas A-band LPS is a shorter-chain, antigenically conserved molecule made up of α1→2, α1→3, α1→3-linked d-rhamnose residues (2). LPS was isolated from P. aeruginosa PAO1, PAO-JP1, PDO-100, and PAO-JP2 grown in either M9 or FAB medium. Western blots of LPS were then reacted with A-band LPS- and B-band LPS-specific MAbs. As shown in Fig. 2, all of the strains tested produced A-band LPS and B-band LPS in both M9 and FAB media. Our findings reveal that LPS was not influential in decreased biofilm formation by the QS mutants in M9 medium. Furthermore, QS does not appear to regulate LPS expression in P. aeruginosa.
FIG. 2.

Western blots of LPS isolated from PAO1, PAO-JP1, PDO-100, and PAO-JP2 (lanes 1 through 4, respectively) grown in M9-glucose and FAB-citrate media. Western blots were reacted with A-band LPS- and B-band LPS-specific MAbs. Note that in both M9 and FAB media all four strains produced similar amounts of A-band LPS and B-band LPS.
Analysis of lasI expression during PAO1 biofilm formation.
To better understand the role that intercellular communication plays in biofilm development, we fused the lasI promoter to a gfp reporter. The fact that the wild-type and original mutated GFP are very stable, with half-lives greater than 24 h (28), makes this protein less than ideal for real-time expression studies. To circumvent this problem, we utilized a gfp construct that encodes an unstable GFP (LVAgfp) with a half-life of 40 min in P. putida and 60 min in E. coli (1). PAO1 cells harboring the lasI-LVAgfp fusion were grown in flowcells and analyzed for lasI gene expression during biofilm development. On days 4, 6, and 8, the biofilms were examined microscopically, and the percentage of cells expressing lasI-LVAgfp was determined relative to the total number of cells. As shown in Fig. 3A, a large number of cells expressed lasI on day 4. However, as biofilm formation progressed (by days 6 and 8), fewer cells appeared to express lasI. To more precisely address differences in lasI gene expression, we subjected each slice of the biofilm to quantitative analysis by utilizing CellComp software. Using this software, we were able to measure the percentage of cells expressing GFP relative to the total number of cells in any optical section. The results of the temporal and spatial analysis are shown in Fig. 3B. On day 4, approximately 50% of the cells at the substratum (depth, 0 μm) expressed lasI; however, by days 6 and 8, the percentage of cells expressing lasI declined to 16 and 8%, respectively (Fig. 3B). Therefore, our results indicate that lasI expression decreases over the course of biofilm development. The CellComp software also enabled us to ascertain gene expression patterns with respect to the vertical height of the biofilm. On all three days examined, lasI gene expression was greatest at the substratum and decreased with increasing biofilm height (Fig. 3B).
FIG. 3.


(A) Scanning confocal micrographs of biofilms formed by PAO1(lasI-LVAgfp) grown for 8 days in a flowthrough chamber. Biofilms were examined on days 4, 6, and 8 to determine the number of cells expressing lasI-LVAgfp relative to the total number of cells. Magnification, ×360. (B) Percentage of cells expressing lasI plotted as a function of biofilm height. For the quantitative analysis, images obtained by SCLM were analyzed by using CellComp software.
It is interesting that with both strain PAO1(lasI-LVAgfp) and strain PAO1(rhlI-LVAgfp) there was a marked change in biofilm morphology between days 4 and 6. On day 4, the PAO1 biofilms formed in the flowcells consisted predominantly of microcolonies approximately 20 to 30 μm thick. By day 6, however, the biofilms were thicker, and distinct mushrooms and pillars with intervening water channels were visible (Fig. 3 and 4).
FIG. 4.


(A) Scanning confocal micrographs of biofilms formed by PAO1(rhlI-LVAgfp) grown for 8 days in a flowthrough chamber. Biofilms were examined on days 4, 6, and 8 to determine the number of cells expressing rhlI-LVAgfp relative to the total number of cells. Magnification, ×360. (B) Percentage of cells expressing rhlI plotted as a function of biofilm height.
Analysis of rhlI expression during PAO1 biofilm formation.
Similar to the lasI analysis, we constructed a rhlI-LVAgfp fusion to monitor expression of the rhlI gene during PAO1 biofilm development. As shown in Fig. 4A, rhlI gene expression appeared to be fairly constant, and the levels of expression observed on days 4, 6, and 8 differed very little (Fig. 4A). This finding was substantiated by the quantitative analysis shown in Fig. 4B. Thus, it appears that rhlI expression is generally steady throughout biofilm development. It is interesting that on all three days examined, rhlI was expressed in fewer cells than lasI. As observed for lasI gene expression, rhlI gene expression was maximal at the substratum and decreased with increasing biofilm height (Fig. 4B).
Effect of media on biofilm formation in a flowing system.
When we compared PAO1 biofilm formations in M9 and FAB media, the biofilms formed in FAB medium (Fig. 3A and 4A) were virtually indistinguishable from those formed in M9 medium (data not shown). Thus, it appears that in our flowthrough system the difference in medium composition did not markedly affect biofilm development.
DISCUSSION
P. aeruginosa utilizes QS to regulate expression of a number of virulence genes and gene products in a density-dependent fashion, presumably to ensure that during an infection P. aeruginosa can achieve a high cell density before activating host defenses. There is mounting evidence which suggests that this complex regulatory cascade is also involved in P. aeruginosa biofilm development. In previous work it was discovered that in a flowthrough system, the las QS system is important for development of fully differentiated biofilms (6). Our data suggest that QS is also involved during the early stages of biofilm development, which are characterized by attachment and microcolony formation. In static cultures, when glucose is used as the carbon source, QS mutants are significantly impaired in the ability to attach to glass coverslips compared to that of the PAO1 parent (Fig. 1). Intriguingly, the difference in static biofilm formation between PAO1 and the QS mutants was not observed in FAB medium, in which attachment of all four strains was markedly diminished. Type IV pilus expression, flagellar motility, and LPS have all been shown to influence P. aeruginosa biofilm formation (8, 17, 23). Our studies revealed that while the parent strain exhibited significant twitching motility in M9 medium, the QS mutants were twitching deficient. Conversely, in FAB medium, both the mutants and the parent exhibited poor twitching motility. In a recent study, expression of the P. aeruginosa pilA gene, which encodes the pilin subunit, was found to be regulated by the global regulator of carbon metabolism, Crc (16). Furthermore, in the presence of citrate, pilA expression was notably repressed compared to expression in cells grown in glucose (S. L. Kuchma and G. A. O'Toole, personal communication). From these findings it is reasonable to assume that the observed difference between PAO1 biofilm formation in glucose-containing media and PAO1 biofilm formation in citrate-containing media is linked, at least in part, to pilA expression. Therefore, we hypothesize that in FAB medium repression of pilA greatly decreases surface pilus expression, resulting in poor twitching motility of both the mutants and the parent, whereas in glucose-containing media pilA is highly expressed; hence, reduced biofilm formation by the QS mutants can be partially attributed to defective pilus production and/or twitching motility due to the QS mutations. Flagellum-mediated motility may have modestly affected biofilm formation as well because the motility of strain PAO1 was decreased in citrate-containing media compared to that in glucose-containing media. Furthermore, when grown in the presence of glucose, the QS mutants exhibited a slight reduction in flagellar motility compared to that of PAO1. In the case of LPS, no differences were observed between the parent and the QS mutants and no differences were observed with the different carbon sources, suggesting that this surface molecule did not influence biofilm formation. Thus, it appears that type IV pilus expression and, to a lesser degree, flagellar motility contributed to the differences in static biofilm formation observed in this study. Because QS controls a wide range of cellular processes in P. aeruginosa, it is quite possible that additional QS-regulated factors also affected biofilm formation.
Previously, it was discovered that in a static system flagellar motility is important for P. aeruginosa surface attachment, whereas type IV pili facilitate microcolony formation (17). We made the surprising discovery that type IV pili and decreased flagellar motility do not significantly affect biofilm formation in our flowthrough system. When we compared PAO1 biofilm production in flowcells with that in cells grown in M9-glucose and FAB-citrate media, the biofilms formed in FAB medium were virtually identical to those formed in M9 medium. Moreover, in a previous study Davies et al. (6) found that the lasI mutant strain PAO-JP1, which is defective in twitching motility, was similar to the parent in the ability to initiate biofilm formation but was defective in the development of mature biofilms. Taken together, these findings suggest that type IV pili are important during static biofilm formation and may be important in very low-flow or intermittently flowing environments; however, in a constantly flowing system, other factors have a greater influence on biofilm initiation. During the first hour, before the flow is initiated, similar events are expected to occur in both flowing and static systems as cells are attaching to the substratum via their flagella and possibly other surface appendages. We speculate that after the flow is started, the cells are subjected to a shear force that limits surface movement via type IV twitching motility. As a result, microcolonies formed in the flowing system result primarily from cellular division rather than a combination of cell clustering and division. Consequently, the ability to move across the substratum via type IV pili has a much greater affect on the biofilms formed in the static system. From the findings presented here, it is apparent that experimental parameters markedly affect P. aeruginosa biofilm formation. Because a great deal of work has been focused on elucidating the molecular mechanisms of biofilm development, the effects of static conditions versus flowing conditions and medium composition should be taken into consideration in the design and interpretation of biofilm studies.
It is widely accepted that cells growing in a biofilm exhibit marked physiological differences from their planktonic counterparts. Nevertheless, the regulatory mechanisms underlying the switch to the biofilm phenotype and the actual cellular metamorphoses that occur remain unknown. We do know that these changes, for the most part, are not caused by mutations since growth of biofilm cells under planktonic conditions results in reversion to the planktonic phenotype. To further examine the role of QS in biofilm formation, we generated lasI and rhlI fusions to an unstable GFP reporter that allowed us to monitor real-time expression of these genes. Using SCLM, we were able to determine both temporal and spatial gene expression patterns throughout biofilm development. Our findings revealed that of the three days analyzed, lasI gene expression was maximal on day 4 and decreased progressively on days 6 and 8. This result is intriguing in light of the fact that we observed a marked change in biofilm morphology between days 4 and 6 in the metamorphosis from microcolonies to the three-dimensional architecture. On day 4 the biofilm formed in the flowcells consisted predominantly of microcolonies, but by day 6 distinct mushroom- and pillar-like structures with intervening water channels were present. Thus, it is possible that prior to day 6, elevated expression of lasI resulted in activation of an as-yet-unidentified gene(s) required for differentiation into these three-dimensional structures. In contrast, expression of rhlI, which was previously reported to be of little significance in biofilm differentiation (6), fluctuated very little throughout biofilm development, and this gene was expressed in a much lower percentage of cells.
In terms of spatial expression throughout the stratified biofilm, lasI and rhlI gene activity was maximal at the substratum and decreased with increasing biofilm height. This observation is not surprising since both the lasI and rhlI genes are autoregulated; lasI requires 3O-C12-HSL (26), and rhlI depends on the presence of both 3O-C12-HSL and C4-HSL for maximal expression (14, 20). Cells at the substratum are close to one another as well as the adherent surface; thus, at this location there would be less opportunity for the AI to diffuse away from cells. In all probability, increased accumulation of AI at the substratum results in increased expression of both of these QS genes.
Currently, much attention is focused on strategies designed to prevent biofilm formation on both industrial and medical surfaces. One means of doing this involves embedding biofilm-retardant compounds in the surface material or coating the surface material with such compounds. In nature, the seaweed Delisea pulchra produces furanones that have antifouling characteristics (7, 22) and are structurally very similar to acylated homoserine lactone molecules (9). Researchers have found that acylated homoserine lactone-regulated phenotypes, including bacterial swarming in Serratia liquifaciens and bioluminescence by Vibrio fischeri and Vibrio harveyi, are inhibited either by coating surfaces with the furanones or preincubating cells with these compounds (9). The results of our analyses show that expression of the P. aeruginosa AI synthase genes is greatest at the substratum, where AI concentrations are presumably high. It should be noted that in the P. aeruginosa las QS system, activation of genes occurs in a hierarchical fashion, with lasI induced at a lower 3O-C12-HSL concentration than other genes (26). This results in rapid amplification of the 3O-C12-HSL signal before other las-controlled genes are activated. If surface-associated AI analogs or other QS-inhibitory compounds could shut down lasI and rhlI expression, then all of the other genes in the QS cascade would be shut down as well, including those involved in biofilm formation. Our observation that lasI and rhlI expression is maximal at the substratum supports the contention that treatment of surfaces with compounds capable of interfering with QS could be an effective means of preventing P. aeruginosa biofilm formation. In the future, uncovering the molecular mechanisms governing biofilm formation will be a huge undertaking. However, studying expression of genes critical for these processes will give us a better understanding of the genotypic and phenotypic changes associated with the biofilm mode of growth, and this understanding may ultimately lead to novel strategies for controlling problematic and recalcitrant biofilms.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health research grant AI133713 to B.H.I. and by a postdoctoral fellowship from the Canadian Cystic Fibrosis Foundation to T.R.D.
We thank M. Givskov for providing plasmid pJBA111 and J. Lam for the kind gift of MAbs N1F10 and MF15-4.
REFERENCES
- 1.Andersen J B, Sternberg C, Poulsen L K, Bjorn S P, Givskov M, Molin S. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol. 1998;64:2240–2246. doi: 10.1128/aem.64.6.2240-2246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arsenault T L, Hughes D W, MacLean D B, Szarek W A, Kropinski A M B, Lam J S. Structural studies on the polysaccharide portion of “A-band” lipopolysaccharide from a mutant (AK1401) of Pseudomonas aeruginosa strain PAO1. Can J Chem. 1991;69:1273–1280. [Google Scholar]
- 3.Brint J M, Ohman D E. Synthesis of multiple exoproducts in Pseudomonas aeruginosa is under the control of RhlR-RhlI, another set of regulators in strain PAO1 with homology to the autoinducer-responsive LuxR-LuxI family. J Bacteriol. 1995;177:7155–7163. doi: 10.1128/jb.177.24.7155-7163.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christensen B B, Sternberg C, Andersen J B, Palmer R J, Jr, Nielsen A T, Givskov M, Molin S. Molecular tools for study of biofilm physiology. Methods Enzymol. 1999;310:20–42. doi: 10.1016/s0076-6879(99)10004-1. [DOI] [PubMed] [Google Scholar]
- 5.Cormack B P, Valdivia R H, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 6.Davies D G, Parsek M R, Pearson J P, Iglewski B H, Costerton J W, Greenberg E P. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- 7.de Nys R, Steinberg P D, Willemsen P, Dworjanyn S A, Gabelish C L, King R J. Broad spectrum effects of secondary metabolites from the red alga Delisea pulchra in antifouling assays. Biofouling. 1995;8:259–271. [Google Scholar]
- 8.Flemming C A, Palmer R J, Jr, Arrage A A, Van de Mei H C, White D C. Cell surface physiochemistry alters biofilm development of Pseudomonas aeruginosa lipopolysaccharide mutants. Biofouling. 1998;13:213–231. [Google Scholar]
- 9.Givskov M, de Nys R, Manefield M, Gram L, Maximilien R, Eberl L, Molin S, Steinberg P D, Kjelleberg S. Eukaryotic interference with homoserine lactone-mediated prokaryotic signalling. J Bacteriol. 1996;178:6618–6622. doi: 10.1128/jb.178.22.6618-6622.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glessner A, Smith R S, Iglewski B H, Robinson J B. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of twitching motility. J Bacteriol. 1999;181:1623–1629. doi: 10.1128/jb.181.5.1623-1629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendrickson W G, Misra T K. Nucleic acid analysis. In: Gerhardt P, Murray R G E, Krieg N R, editors. Methods for general and molecular biology. Washington, D.C.: American Society for Microbiology; 1994. pp. 436–460. [Google Scholar]
- 12.Hitchcock P J, Brown T M. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol. 1983;154:269–277. doi: 10.1128/jb.154.1.269-277.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holloway B W, Krishnapillai V, Morgan A F. Chromosomal genetics of Pseudomonas. Microbiol Rev. 1979;43:73–102. doi: 10.1128/mr.43.1.73-102.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol Microbiol. 1996;21:1137–1146. doi: 10.1046/j.1365-2958.1996.00063.x. [DOI] [PubMed] [Google Scholar]
- 15.McLean R J, Whiteley M, Stickler D J, Fuqua W C. Evidence of autoinducer activity in naturally occurring biofilms. FEMS Microbiol Lett. 1997;154:259–263. doi: 10.1111/j.1574-6968.1997.tb12653.x. [DOI] [PubMed] [Google Scholar]
- 16.O'Toole G A, Gibbs K A, Hager P W, Phibbs P V, Jr, Kolter R. The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J Bacteriol. 2000;182:425–431. doi: 10.1128/jb.182.2.425-431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Toole G A, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 18.Passador L, Tucker K D, Guertin K, Journet M, Kende A S, Iglewski B H. Functional analysis of the Pseudomonas aeruginosa autoinducer, PAI. J Bacteriol. 1996;178:5995–6000. doi: 10.1128/jb.178.20.5995-6000.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearson J P, Pesci E C, Iglewski B H. Role of Pseudomonas aeruginosa las and rhl quorum-sensing systems in the control of elastase and rhamnolipid biosynthesis genes. J Bacteriol. 1997;179:5756–5767. doi: 10.1128/jb.179.18.5756-5767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pesci E C, Pearson J P, Seed P C, Iglewski B H. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol. 1997;179:3127–3132. doi: 10.1128/jb.179.10.3127-3132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pesci E H, Iglewski B H. Quorum sensing in Pseudomonas aeruginosa. In: Dunny G M, Winans S C, editors. Cell-cell signalling in bacteria. Washington, D.C.: ASM Press; 1999. pp. 147–155. [Google Scholar]
- 22.Reichelt J L, Borowitzka M A. Antimicrobial activity from marine algae: results of a large-scale screening programme. Hydrobiology. 1984;116/117:158–168. [Google Scholar]
- 23.Rocchetta H L, Burrows L L, Lam J S. Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 1999;63:523–553. doi: 10.1128/mmbr.63.3.523-553.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 25.Seed P C. Ph.D. thesis. Rochester, N.Y: University of Rochester; 1996. [Google Scholar]
- 26.Seed P C, Passador L, Iglewski B H. Activation of the Pseudomonas aeruginosa lasI gene by LasR and the Pseudomonas autoinducer PAI: an autoinduction regulatory hierarchy. J Bacteriol. 1995;177:654–659. doi: 10.1128/jb.177.3.654-659.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stickler D J, Morris N S, McLean R J, Fuqua C. Biofilms on indwelling urethral catheters produce quorum-sensing signal molecules in situ and in vitro. Appl Environ Microbiol. 1998;64:3486–3490. doi: 10.1128/aem.64.9.3486-3490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tombolini R, Unge A, Davey M E, de Bruijn F J, Jansson J K. Flow cytometric and microscopic analysis of GFP-tagged Pseudomonas fluorescens bacteria. FEMS Microbiol Ecol. 1997;22:17–28. [Google Scholar]
- 29.Van Delden C, Pesci E C, Pearson J P, Iglewski B H. Starvation selection restores elastase and rhamnolipid production in a Pseudomonas aeruginosa quorum-sensing mutant. Infect Immun. 1998;66:4499–4502. doi: 10.1128/iai.66.9.4499-4502.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West S E, Schweizer H P, Dall C, Sample A K, Runyen-Janecky L J. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene. 1994;148:81–86. doi: 10.1016/0378-1119(94)90237-2. [DOI] [PubMed] [Google Scholar]