

Abstract
Adeno-associated virus (AAV) has shown great potential in gene therapy due to its low immunogenicity, lack of pathogenicity to humans, and ability to provide long-term gene expression in vivo. However, there is currently a need for fast, high-throughput characterization systems that require low volumes for the determination of its sample composition in terms of full and empty capsids since empty capsids are a natural byproduct of AAV synthesis. To address this need, the following study proposes a high-throughput electrophoresis-mediated microfluidics approach that is independent of sample input concentration to estimate the composition of a given sample by combining its protein and ssDNA information relative to a standard. Using this novel approach, we were able to estimate the percentage of full capsids of six AAV8 samples with an average deviation from the actual percentage of 4%. The experiments used for these estimations were conducted with samples of varying percentages of full capsids (21–75%) and varying concentrations (5 × 1011–1 × 1012 VP/mL) with a total volume requirement of 3–10 μL for triplicate analysis of the sample. This method offers a rapid way to evaluate the quality and purity of AAV products. We believe that our method addresses the critical need as recognized by the gene and molecular therapy community.
Introduction
Genetics is intrinsically involved in most common diseases, either as a causative agent or as a facilitator. While not generally associated with each other, the diseases responsible for most of the morbidity and mortality in developed regions, such as coronary heart disease, diabetes, different types of cancer, and depression, have, in fact, a significant genetic component.1 Some of the most common variations in our DNA that result in genetic disorders include single-gene disorders, chromosomal imbalances, and epigenetics.2 Consequently, for the past few decades, great importance has been given to advancements in human genome discoveries and the development of sequencing technologies due to their potential to improve health and prevent diseases.3
To address these genetic variations, in particular single gene disorders, significant improvements have been made to gene therapy since its early conceptualization over 40 years ago. Improvements range from the first publications of nonviral gene therapy in the 1980s to the successful use of viral and nonviral gene therapies in the treatment of cardiovascular diseases, autoimmune disorders, obesity, diabetes, and central nervous system disorders, among others.4 Among the current gene therapy vectors, adeno-associated virus (AAV) has become of particular interest as it has been demonstrated to provide successful, long-term gene transfer in vivo.5 To date, there are two FDA-approved AAV gene therapies: Luxturna for the treatment of a rare inherited retinal dystrophy and Zolgensma for the treatment of spinal muscular atrophy.6,7 In addition, dozens of treatments are currently in clinical trial, and another therapy for the treatment of adenosine deaminase-severe combined immunodeficiency, Strimvelis, has been approved by the European Medicines Agency.7
AAV is a single-stranded DNA (ssDNA) nonenveloped virus that belongs to the parvovirus family and measures approximately 25 nm in diameter.8,9 There are currently nine serotypes of AAV, each with slightly different tropisms which include retina, lung, muscle, liver, and brain cells. The virus is only composed of protein and DNA and has three repeating capsid proteins, VP1, VP2, and VP3, at an expected ratio of 1:1:10, which may vary across serotypes and even within the particles of a given batch.10,11 It is able to infect both dividing and nondividing cells depending on the tropism of the given serotype.12
The simplicity of the AAV genome makes recombinant AAV (rAAV) a great candidate for gene delivery. While AAV generally requires a helper virus, such as an adenovirus or herpes simplex virus, to successfully replicate in human cells, a key change in the development of its design was the use of plasmids for rAAV production. The most common method is the use of the triple transfection of HEK293 cells, where one plasmid includes the gene of interest, another the helper genes, and a third the packaging genes.13,14 The integration of the helper virus functional genes allows for the helper virus-independent manufacturing of recombinant AAV vectors. Additionally, while its restricted packaging limit of 4.7 kb has previously limited its scope, recent studies have been aimed at exploring the potential and integrity of oversized rAAV vectors.15 Another recent innovation in the next generation of AAV vectors is the production of capsid-modified, genome-modified, and both capsid- and genome-modified vectors to increase their efficiencies at reduced doses.16 Despite its great potential, there are still several limitations that prevent the safe and widespread use of this therapy, including the large-scale manufacturing of high-quality rAAV vectors. In addition, a streamlined quality control system for the rapid characterization of viable particles has not been developed. As a result, there is currently a need for rapid and robust assays for rAAV testing for human gene therapy.17−19 The natural byproducts of AAV synthesis are capsids that have not been packaged with DNA, also referred to as empty capsids, and capsids with undesired loading of either fragmented ssDNA, referred to as partially full, or host DNA. It is not completely clear how the presence of empty capsids impacts the therapeutic properties of rAAV, but their removal has shown an increase in transgene expression.20 They have been shown to inhibit the transduction of target cells by exacerbating the immune response to rAAV and by competing with full capsids for cell binding sites, but they have also demonstrated beneficial effects by acting as decoys, which reduce the neutralization of full AAV vectors.21 Regardless of their impact on therapeutic properties, it is important to have effective methods to differentiate full capsids from empty capsids. Despite the purification steps that have been integrated into the rAAV production chain, empty particles can still account for most of the particles in a batch.22 Hence, quality control methods must be designed to monitor the production and purification of AAV suspensions.
Currently, several methods have been utilized in the purification process in attempt to determine the capsid content ratio (full vs empty capsids), as summarized in Table 1. Some of the most prevalent include anion-exchange chromatography (AEX), optical density (OD), transmission electron microscopy (TEM), charge detection mass spectrometry (CDMS), size-exclusion chromatography multiangle light scattering (SEC-MALS), enzyme-linked immunosorbent assay (ELISA) in tandem with quantitative polymerase chain reaction (qPCR), and analytical ultracentrifugation (AUC), in no particular oder.23−25
Table 1. Comparison of Analytical Methods Used to Differentiate between Empty and Full AAV Capsids.
| Analytical Methods | Fundamental Basis | Characteristics |
|---|---|---|
| Anion exchange chromatography | Surface charge density | High accuracy, complex workflow, medium turnaround time (30 min/sample), difficult to establish optimal method applicable to all rAAV serotypes |
| Optical Density | UV absorbance (density analysis) | Fast turnaround time (15 min/sample), requires highly purified AAV to minimize interference with UV absorbance, varying concentration requirements (5 × 1011–1 × 1013 GC/mL) |
| Transmission electron microscopy | Image analysis | Direct characterization method, statistically small sample image size, high coefficient of variation, time-consuming (6 h/sample) |
| Charge detection mass spectrometry | Mass to charge ratio | High accuracy, requires extensive preparation, time-consuming (2 h/sample), low throughput, not easily accessible |
| Size-exclusion chromatography, multiangle light scattering | Size exclusion and static light scattering | High accuracy, medium turnaround time (20–30 min/sample) requires long column equilibration times, relatively high volumes of sample (30–50 μL per run), high concentration requirements (1 × 1013 VP/mL) |
| ELISA + qPCR | Antibody specificity to full and empty capsids, used in tandem with qPCR | Expensive, not scalable, lacks accuracy and precision, compounded error |
| Analytical ultracentrifugation | Separates capsid sedimentation rate of particles (buoyant densities) | High accuracy, large volume of samples (300–400 μL), not scalable, time-consuming (6 h/sample) |
| LabChip electrophoresis (Our method) | Total protein/ssDNA ratio | Scalable, fast turnaround time (6–15 min/sample), high throughput, requires low volumes (3–10 μL) and concentrations (>1 × 1011 GC/mL) |
Ion-exchange-based chromatography techniques, especially AEX, are often used in series and allow for enrichment of full capsids via the use of ion exchange resins, membranes, and monolithic columns; however, this approach can be time-consuming, requiring an analysis time of 30 min per sample and column equilibration between injections.26−28 Although this approach remains time-consuming and each serotype requires condition optimization, it must be noted that recently several groups have been working on the optimization of the technique across different serotypes.29,30 OD, or UV absorbance spectroscopy, utilizes the extinction coefficient (an indicator of light absorbance at a given wavelength) of capsid proteins and DNA, and it estimates the ratio of capsid particle to vector genome based upon the absorbance ratio.31 OD analysis time can be as low as 15 min/sample; however, it has very high purification requirements to prevent interference with the UV absorbance. Next is TEM which, when combined with a deep learning algorithm, allows for the visualization of the overall morphology of AAV samples via capsid staining. Those which are empty exhibit a stained electron-dense core, while those which are full exhibit a bright core.32 CDMS has been used to differentiate between full, partially full, and empty capsids, but to achieve this, a complex and time-consuming workflow needs to be followed, and access to this technology remains limited.33 SEC-MALS is another promising technique for the determination of full and empty capsids, which uses a form of static light scattering to determine the size and weight of the AAV molecules post size-exclusion. While this approach may provide rapid and highly accurate estimations, the process requires extensive SEC column calibration and relatively high volumes of sample (∼50 μL) per individual run. An alternative approach exploits the genome to capsid ratio and requires the simultaneous use of ELISA to determine the capsid protein titer, in tandem with qPCR, to determine viral genome (ssDNA) titer. The empty particle content is determined by subtracting the viral genome titer from the capsid titer. This method risks compounded error from the utilization of two independent systems and assays and has only been optimized for certain serotypes.34 AUC typically requires the samples to have a normalized concentration and requires extensive data acquisition (∼60 scans) and data processing.35 The robustness of AUC makes it the quantitative golden standard in the field for AAV quantification despite its high volume, concentration, and time requirements. Note that these are some of the most established techniques currently available for capsid packaging characterization, but other novel approaches have been gaining traction and have shown great promise as alternative analytical platforms such as mass photometry, capillary isoelectric focusing, and Stunner (UV260/280).36−38
Despite the great potential of each of these techniques, as highlighted above, they are limited by their requirement of significant sample preparation or processing times.23,28,33,39 In fact, sample analysis times for these techniques can range between 15 min (OD) and 6 h (AUC and TEM) per sample, depending on the technique, and require extensive resources, which significantly limits the number of samples that can be processed in a day, making most approaches low throughput.23 In addition, it must be noted that oftentimes the reported analytical times associated with each method excludes the time of sample preparation, whereas the turnaround time that is reported for our proposed method combines the two.
Thus, the goal of this study is to develop a microfluidic technique that mathematically determines the ratio of full to empty particles in an unknown sample by using the genome to protein ratio of the unknown sample normalized to a standard. This approach is largely based on the fact that during AAV production capsids are synthesized prior to being packaged with their genome. Therefore, for a single serotype production, capsids will contain the same amount of protein regardless of their packaging, as represented in Figure S1. In other words, regardless of the presence and nature of genetic material within the capsids, the protein profiles of full and empty capsids should be the same. Please note that for the purpose of this study only full and empty capsids will be considered since together they represent >97% of the total AAV sample, especially in purified samples such as the ones used in this study.23
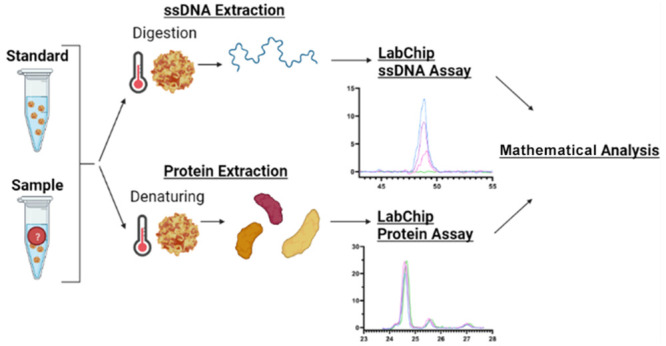
In the context of differentiating between full and empty rAAV capsids for gene therapy, microfluidic techniques can be utilized to characterize its capsids based on the genome to protein ratio of a given sample. Here, the goal is to use microfluidic electrophoresis that can quantify the presence of AAV single-stranded genomic DNA within full rAAV capsids via the application of voltages to digested viral capsids. This study aims to evaluate the ssDNA content in full capsids in tandem with the protein content of full and empty capsids to determine the percentage of full AAV capsids in a given sample of rAAV relative to a standard with a known fraction of full capsids, as highlighted in Figure 1. By utilizing microfluidic technology to create a reliable, high-throughput, and efficient composition assessment method for AAV capsid differentiation, this study will aim to make AAV an even more attractive candidate for gene therapy and allow for streamlined identification of successful versus unsuccessful insertion of genetic material.
Figure 1.

Workflow for AAV sample capsid content analysis. Here, the samples are mixed in a 384-well plate or in PCR tubes, heated following their respective protocols, and transferred onto the LabChip System. The sipper chip in the LabChip platform gets prepared following the protocol with a gel-dye matrix and a marker, together with the provided buffer and ladder tubes. Once the samples and chip are ready, the appropriate assay gets selected on the system, and the results get analyzed using the provided mathematical approach (eq 6).
Materials and Methods
Samples and Sample Preparation
The AAV8 full and empty reference standards were purchased from Vigene Biosciences (Vigene Biosciences, Rockville, MD). The full reference standard had a titer of 7.97 × 1011 genome copies (GC)/mL at a full fraction of 75%, which is the equivalent to 1.07 × 1012 viral particles (VP)/mL. The latter was estimated by accounting for the empty particles, which would not contribute to the genome copies. On the other hand, the empty reference standard was purchased with a titer of 1.44 × 1012 VP/mL at an empty fraction of 96%. Since this value includes both full and empty particles, the titer of only empty particles in this sample was estimated to be 1.38 × 1012 VP/mL. In the case of both the full and the empty reference standards, per the certificate of analysis issued by the provider (Vigene Biosciences), the quality control was conducted via SYBR Green qPCR and ELISA, combined with TEM to determine the percentage of full capsids in the sample. While the vendor did not provide a standard deviation for their estimations, a 10% deviation will be assumed, when needed, for statistical analysis, which may be consistent with the analytical tools used for their capsid ratio estimation. For the experiments comparing full to empty samples, the stock samples were used; however, the ones that used different percentages of full capsids were prepared accounting for the empty particles in the full reference standard and the full particles in the empty reference standard. Furthermore, samples were stored in single-use aliquots at −80 °C to ensure sample stability and degradation did not affect the outcomes of the experiments.
Methods
In this study, the GX Touch II LabChip system (PerkinElmer, Waltham, MA) was used to characterize the AAV samples. For the capsid protein experiments, the standard protocol of the LabChip ProteinExpress assay was used, as seen in Figure 1. Specifically, the high sensitivity protocol was followed, which requires 5 μL of sample, using the optional reducing buffer. Each sample was analyzed three times, and each time 20 nL was removed from the well plate onto the detection chip. For the ssDNA analysis, a custom ssDNA chip assay was used (PerkinElmer). First, the AAV samples were digested 1:1 (5 μL of AAV and 5 μL of the digestion mixture) with a proteinase K mixture (10 μL of proteinase K were diluted with 90 μL of 2 M urea) for 60 min at 55 °C, followed by a proteinase K deactivation for 20 min at 95 °C, as seen in Figure 1. Here, it must be noted that while we are focusing on the detection of ssDNA, AAV could also contain self-complementary DNA, which may also be detectable with the simple insertion of an intercalating dye in the microchip gel formulation. Lastly, the samples were analyzed with a customized ssDNA assay script to amplify the signal and allow for the detection of samples with lower genomic content (PerkinElmer). Each sample was analyzed three times, and each time 20 nL was removed from the well plate onto the detection chip. Note that while 5 μL of sample was used for each assay, if needed, this volume could be reduced significantly without interfering with the assay. Lastly, the statistical analysis for this study was conducted using GraphPad Prism 9, and the figures were made using GraphPad and/or BioRender.com.
Mathematical Formulation to Estimate the Percentage of Full Capsids
The empty, full, and partial AAV capsids have icosahedral symmetry. They are assembled from viral proteins (VP1, VP2, and VP3) and genomic material (ssDNA). To fully characterize the AAV samples, quantitation of both proteins and ssDNA is needed. Before we describe our experimental findings, the following section describes a mathematical formulation to relate protein and DNA concentrations in the samples and an AAV standard.
Here, we describe a new method for the determination of the percentage of full capsids in an AAV sample. The method requires only a few microliters (3–10 μL) of a standard (S) with a known fraction of full capsids. Let us denote the total number of AAV capsids in the sample and standard by N and Ns, respectively. Hence, the number of full and empty capsids can be described using the following equations
| 1 |
| 2 |
Here, f and e refer to full and empty, respectively. Note that the number of partially filled AAVs can be included in this analysis, but since their concentration is below our limit of detection (LOD) and their percentage negligible (<3%) in purified samples such as these, it was excluded from the analysis for simplicity. Since the subunits (VP1–3) of the AAV capsids are the same for both full and empty capsids, it is safe to assume that each capsid is composed of α μg of total proteins. Hence, the ratio of the concentration (c) of proteins in the sample and standard can be expressed as
| 3 |
Similarly, we assume a single ssDNA insert per full AAV capsid to obtain the following ratio for the concentration of ssDNA in the sample and standard:
| 4 |
Since the standard comes with a known percent of full capsids, βs, which can be defined as
| 5 |
we obtain the fraction of full capsids, β, using the following relation:
| 6 |
Hence, we obtain the percentage of the full AAV estimate by measurement of concentrations of total protein and ssDNA using microfluidics electrophoresis with samples and a standard. It is important to note that our method is independent of the total capsid concentrations in samples or standards. This is often the most significant limitation for other techniques summarized in Table 1. The error in the estimated percentage of full AAV as determined by eq 6 only arises from the concentration ratio accuracy errors in the electrophoresis method, not from the resolution accuracy of the method. In other words, rather than obtaining the total protein concentration of a sample from the protein assay (full and empty) and subtracting the concentration obtained from the DNA assay (only full), which would have a compounded error rate from the use of two different assays, the proposed method uses a sample, or standard, of known concentration to normalize both assays. As an example, using the 75% full AAV8 sample, we can normalize the assay for each run to reduce the error introduced by the use of multiple assays and the difference in units between the protein and DNA areas. Another key advantage of the ratio measurements is that as long as all samples within the assay are treated in the same way (diluted by the same factor or heated for the same time) the proposed method is independent of the concentration of the sample since the protein area will account for a difference in concentration.
Results and Discussion
Capsid Protein Profile Characterization
The first step in the validation of the mathematical formulation to estimate the percentage of full capsids in a sample was to independently assess the protein and DNA profiles of AAV8 full (75%) and empty (96%, or 4% full) reference standards. At this stage, it was of particular interest to confirm that the amount of protein in the full and empty samples at a given concentration of capsids was the same. To do this, the AAV standards were denatured using a reducing buffer containing DTT (refer to Figure 1 and Methods). The concentration of the empty reference standard was adjusted to match the concentration of the full reference standard (1.07 × 1012 VP/mL) by diluting it with 1X PBS. The dilution was conducted keeping both the viral concentration and the salt composition, which can affect labeling efficiency and sample conductivity, constant.
On the basis of the electropherogram shown in Figure 2a, and as seen in the summarized corresponding areas under the curve for VP1, VP2, and VP3 (Figure 2b), there was no statistical difference between the full and empty samples VP peaks. Moreover, on the basis of the literature results and normalizing it to their respective molecular weights, VP1 is expected to represent 11.3% of the total capsid area, VP2 9.5%, and VP3 79.2%. In our analysis, the full sample VP1 represents 6.7%, 11.3% for VP2, and 82.1% for VP3. Similarly, the empty sample VP1 represents 6.3%, VP2 11.6%, and VP3 82.1%. The presence of these VP ratios that differ from the expected 1:1:10 ratio highlights the stochastic nature of the VP capsid protein ratios. Therefore, to mitigate the impact of the varying VP ratios across samples and even within a given batch, we decided to integrate the area under the three VP peaks to have a method that is independent of the VP ratios.
Figure 2.

Analysis of capsid proteins from full (purple) and empty (green) AAV8 capsids. (a) Electropherogram representative of VP3, VP2, and VP1 capsid proteins, from lowest to highest molecular weight (left to right), of both full and empty samples. (b) Summarized area under the curve for each main protein peak, showing the statistical difference between the VP peaks yielded by the full and empty capsids.
Moreover, if the sample and standard (usually provided) concentrations are known, these values can be input as the concentration in a modified eq 3 instead of the protein area obtained in the Protein Express assay detailed above (refer to eqs 7 and 8). However, if the concentration is unknown, this approach can be used not only to estimate the percentage of full capsids in the sample but also to estimate the total concentration of the sample, as highlighted in Table 2. Using the area under the curve of the VP protein peaks of the standard and of the sample, as well as the concentration of the standard, we were able to estimate the total sample concentration with an error rate of 1–16% and an average error rate of 6%. Moreover, while additional experiments are needed to determine the LOD of the protein assay, we believe the limit lies between 5 × 1011 and 1 × 1012 VP/mL. As will be discussed later, the current LOD for the ssDNA assay has been estimated to be >1 × 1011 GC/mL, which will generally place the total protein content within the desired range.
Table 2. Estimation of Sample Total Protein Concentration Based on the VP Peak Area under the Curve of the Standard (Known Concentration) and the Area under the Curve of the Samplea.
| Sample No. | Total Concentration (VP/mL) | Protein Area | Predicted Total Concentration (VP/mL) | Prediction Error Rate (%) |
|---|---|---|---|---|
| Set 1 | ||||
| 1–0 | 1.07 × 1012 | 41.71 ± 1.52 | Standard | Standard |
| 1–1 | 1.07 × 1012 | 42.30 ± 3.04 | 1.08 × 1012 | 1 |
| 1–2 | 1.07 × 1012 | 43.67 ± 3.40 | 1.12 × 1012 | 5 |
| Set 2 | ||||
| 2–0 | 1.07 × 1012 | 39.60 ± 3.01 | Standard | Standard |
| 2–1 | 1.20 × 1012 | 41.16 ± 2.37 | 1.11 × 1012 | 7 |
| 2–2 | 1.33 × 1012 | 42.90 ± 2.68 | 1.12 × 1012 | 16 |
| Set 3 | ||||
| 3–0 | 1.07 × 1012 | 31.32 ± 1.68 | Standard | Standard |
| 3–1 | 1.07 × 1012 | 31.86 ± 1.08 | 1.08 × 1012 | 2 |
| 3–2 | 1.07 × 1012 | 33.52 ± 2.77 | 1.14 × 1012 | 7 |
ssDNA Profile Characterization
After confirming that the amount of capsid protein is preserved between full and empty capsids, the next step was to assess the ssDNA content of the capsids (Figure 3). To do this, the sample concentration was normalized to 1.07 × 1012 VP/mL and then digested using a standard Proteinase K digestion protocol (refer to Figure 1 and Methods). As seen in Figure 3, the full reference standard (75% full) exhibits a peak at around 62 s, while the empty standard (96% empty) failed to produce a peak. While it is not surprising that the empty sample did not have a peak, it must be noted that the empty sample should still contain approximately 4.26 × 1010 full capsids, which implies that the LOD is greater than that. Additional experiments were conducted establishing the L.O.D. at approximately 1 × 1011 GC/mL.
Figure 3.

Analysis of genomic material from full (purple) and empty (green) AAV8 capsids. (a) Representative electropherogram of full and empty genomic material where the full sample has a peak and the empty sample does not. (b) Summarized area under the curve for the genetic material peak of each sample; note that for all three runs the empty AAV sample did not produce a peak, showing a significant statistical difference between the ssDNA profiles of the two samples.
Like the total protein estimation shown in Table 2, the DNA information collected with this assay can be input into eq 4 to estimate the number of full particles (or genome copies, GC) in the sample, as seen in Table 3. Using this approach, the prediction error rate ranged from 2 to 20%, with an average error rate of 8%. Moreover, it must be noted that this information can be of particular interest for in vivo studies as only the full capsids will be carrying the genetic material of interest for gene therapy purposes. However, as previously mentioned, the use of this approach to obtain absolute values rather than relative values normalized to the standard will provide a lower accuracy and is meant to be used as a reference.
Table 3. Estimation of Sample Genomic Concentration Based on the ssDNA Area under the Curve of the Standard (Known Concentration) and the Area under the Curve of the Samplea.
| Sample No. | Genomic Concentration (GC/mL) | DNA Area | Predicted Genomic Concentration (GC/mL) | Prediction Error Rate (%) |
|---|---|---|---|---|
| Set 1 | ||||
| 1-0 | 7.97 × 1011 | 11.32 ± 0.33 | Standard | Standard |
| 1-1 | 5.33 × 1011 | 8.02 ± 0.64 | 5.64 × 1011 | 6 |
| 1-2 | 2.66 × 1011 | 3.58 ± 0.52 | 2.52 × 1011 | 5 |
| Set 2 | ||||
| 2-0 | 7.97 × 1011 | 10.76 ± 0.32 | Standard | Standard |
| 2-1 | 5.38 × 1011 | 7.12 ± 0.70 | 5.27 × 1011 | 2 |
| 2-2 | 2.77 × 1011 | 3.64 ± 0.60 | 2.70 × 1011 | 3 |
| Set 3 | ||||
| 3-0 | 7.97 × 1011 | 22.47 ± 2.57 | Standard | Standard |
| 3-1 | 5.33 × 1011 | 13.55 ± 2.00 | 4.80 × 1011 | 10 |
| 3-2 | 2.66 × 1011 | 6.00 ± 1.61 | 2.13 × 1011 | 20 |
Capsid Protein and ssDNA Profiles of Samples with Varying Full Percentages
Next, in order to develop a robust method for estimating the percentage of full capsids in an unknown AAV8 sample, information was collected from samples with varying percentages of full capsids, from 75% to 4% (Figure 4). To do this, both samples were normalized to a total protein concentration of 1.07 × 1012 VP/mL, and the full reference standard was mixed with the empty standard at different ratios to achieve the desired percentages of full. As expected, while the protein concentration remains approximately the same with an average total VP area of 28.6–34.16 (Figure 4a,b), the ssDNA concentration decreases as the number of full capsids decreases, with average areas going from 11.4 to 0.0 (Figure 4c,d).
Figure 4.

Protein and genetic analysis of samples with different percentages of full capsids, including 75% (blue), 50% (purple), 25% (pink), and 4% (green). (a) Electropherogram representative of VP3, VP2, and VP1 capsid proteins from smallest to largest weight (left to right) of both full and empty samples. (b) Summarized integrated area under the curve of the VP protein peaks for the four samples of varying full capsid percentages. (c) Representative electropherogram of the genetic material of the four samples, where the ssDNA peak is observed to decrease as the percentage of full capsids decreases. (d) Summarized area under the curve for the genetic material peak of each sample, for all three runs the empty AAV sample did not produce a peak.
Experimental data was collected on three independent sets of protein and ssDNA assays, analyzed in triplicate. Using the protein and ssDNA information from the full reference standard (75% full) and from the samples, eq 6 was used to estimate β, or the predicted percentage of full capsids. The results for each set can be seen in Table 4.
Table 4. Compiled Protein and ssDNA Data Collected and Analyzed from Three Different Sets of Experiments.
| Sample No. | Percentage Full (%) | Protein Area | DNAArea | Predicted Percentage Full (%) | Prediction Error (%) |
|---|---|---|---|---|---|
| Set 1 | |||||
| 1-0 | 75 | 41.71 ± 1.52 | 11.32 ± 0.33 | Standard | Standard |
| 1-1 | 50 | 42.30 ± 3.04 | 8.02 ± 0.64 | 52 | 2 |
| 1-2 | 25 | 43.67 ± 3.40 | 3.58 ± 0.52 | 23 | 2 |
| Set 2 | |||||
| 2-0 | 75 | 23.76 ± 2.33 | 10.76 ± 0.32 | Standard | Standard |
| 2-1 | 45 | 23.97 ± 2.63 | 7.12 ± 0.70 | 48 | 3 |
| 2-2 | 21 | 24.01 ± 1.67 | 3.64 ± 0.60 | 24 | 3 |
| Set 3 | |||||
| 3-0 | 75 | 22.16 ± 1.12 | 22.47 ± 2.57 | Standard | Standard |
| 3-1 | 50 | 22.94 ± 1.15 | 13.55 ± 2.00 | 44 | 6 |
| 3-2 | 25 | 24.93 ± 2.36 | 6.00 ± 1.61 | 19 | 6 |
Currently, the average prediction deviation of the model from the actual percentage is ±4%, ranging from 2 to 6% with a standard deviation of 2%. Alternatively, if the sample concentration is known within an acceptable margin of error, the error and sample analysis time of the system can be significantly decreased by bypassing the protein assay. Assuming an error rate of 10% for the provided concentrations and full percentages of the samples and consequently of our predictions since they are based on these values and the reported error rate of the detection platform, we were able to use the following modified version of eq 6 to estimate the percentage of full capsids using the ssDNA area and the provided concentrations
| 7 |
| 8 |
where Rconcentration is used to replace Rp since, as suggested by the literature and Table 3, the sample concentration is related to the protein area.
To assess the specificity of this alternative approach, the actual (reported) percentage of full capsids of each sample was compared to the predictions obtained using eq 6, referred to as “Protein”, and eq 8, referred to as “Concentration”, as highlighted in Figure 5. Note that while this method bypasses the need for protein analysis, it is still highly dependent upon the ssDNA analysis, as suggested by eq 8.
Figure 5.

Comparison of prediction accuracy between the protein area prediction method based on eq 6 and the concentration prediction method based on eq 8 in comparison to the actual or reported percentage of full capsids of each sample. In the legend on the right, the sample numbers correspond to those described in Tables 2–4. Moreover, all samples include an error bar of 10% (in each direction) of the total value to account for inaccuracies in the reported standards used to prepare both the standard and samples used in this study.
After analyzing the results obtained using both approaches, a decrease from ±4% (protein area prediction method) to ±3% in average prediction deviation was observed when using the concentration prediction method, ranging from 1 to 9%. In other words, when the total sample concentration is provided, both the error and turnaround time can be decreased. Importantly, the decrease in turnaround time will be limited since the longest incubation is needed for the extraction of ssDNA from the capsid. However, it must be noted that when the known concentrations were used the standard deviation of the predictions increased slightly from 2% to 3%.
Conclusions
Initially, a single electrophoresis-based microfluidic assay capable of differentiating full from empty capsids was considered in which the difference in their respective isoelectric points (pI) was exploited using a charge variant assay. While both full and empty capsids are assembled in the same fashion, it is believed that the presence of the ssDNA genome in full capsids induces a conformational change that affects its external properties.40 However, when the full and the empty reference standards were analyzed simultaneously, despite their difference in pI, the samples coeluted even when numerous external factors were used (Table S1–2 and Figure S1 from the Supporting Information). Therefore, the goal of this study shifted to understanding the relationship between the protein and ssDNA profiles of full and empty standards to develop a mathematical model to estimate the percentage of full capsids in an AAV8 sample of unknown concentration and composition.
First, our primary assays were validated by comparing our findings to the literature. As expected, for a given concentration, the protein profiles of full and empty AAV8 did not show a significant difference between them while the ssDNA assay showed a peak representing the ssDNA genome for the full standard and an absence of peak for the empty standard. A major benefit of running these two assays simultaneously is the ability to obtain both the total sample concentration and the total genomic concentration of the sample, as reported in Tables 2 and 3, in addition to the fraction of full capsids.
Once the individual methods were validated, the relationship between the protein and ssDNA profiles was assessed to develop a mathematical model to characterize the percentage of full capsids in a given sample. While AUC is still the golden standard when it comes to accuracy, its extensive turnaround time and sample usage leaves room for improvement. The proposed method offers a fast, high-throughput alternative that can be used to quickly iterate through batches of sample without the need for highly specialized equipment that requires significant levels of training. Within 2–3 h, including the denaturing and digestion times, depending on the number of samples being analyzed, the proposed method can predict the percentage of full capsids with an average prediction error of ±4% using a total volume of less than 10 μL per sample (including triplicate analysis). For instance, if 10 samples are being analyzed, it would take an average of 15 min/sample, and if run in triplicate it would take 6 min/sample, which is significantly faster than the current methods (15 min −6 h/sample). Due to the high throughput nature of the proposed method, the latter value would decrease as the number of samples increases, which is particularly relevant for the analysis of samples at different stages of manufacturing (i.e., to assess the effect of each step) and for batch-to-batch analysis. Over the course of these experiments, the LOD of the protein assay was estimated to be between 5 × 1011 – 1 × 1012 VP/mL, while the LOD of the ssDNA assay was estimated to be >1 × 1011 GC/mL. However, additional experiments are needed to further narrow down these ranges and determine the limit of quantitation. Moreover, ongoing preliminary experiments with AAV9 (Table S3 and Figure S3) suggest the method may be compatible with additional serotypes. However, additional experiments are needed to fully assess the cross-serotype compatibility.
Acknowledgments
This work was in part supported by PerkinElmer’s research grant to Brown University. Additionally, we acknowledge James White and others from PerkinElmer for their guidance and material supplies. The proprietary subjects disclosed herein are patent-pending property of PerkinElmer Health Sciences, Inc.
Glossary
Abbreviations
- AAV
adeno-associated virus
- AEX
anion-exchange chromatography
- CDMS
charge detection mass spectrometry
- qPCR
quantitative polymerase chain reaction
- AUC
analytical ultracentrifugation
- ssDNA
single-stranded DNA
- rAAV
recombinant AAV
- ddPCR
droplet digital PCR
Supporting Information Available
The supplementary file includes: The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01813.
Materials and methods for protein charge variant experiments, Table S1 (Effect of Different pH Conditions on AAV2 Differentiation), Figure S1 (Electropherograms of AAV2 Full, Empty, and Combined Samples), Table S2 (Effect of Different Compounds on AAV2 Differentiation), Figure S2 (Electropherograms of AAV2 and AAV8 Samples), Table S3 (AAV9 Compiled protein and ssDNA data collected and analyzed using mathematical prediction method), and Figure S3 (Electropherograms of AAV9 protein and ssDNA assays) (PDF)
A.T. is a paid scientific advisor/consultant and lecturer for PerkinElmer.
The authors declare no competing financial interest.
Supplementary Material
References
- King R. A.; Rotter J. I.; Motulsky A. G.. The Genetic Basis of Common Diseases; Oxford Monographs on Medical Genetics; Oxford University Press: Oxford, 2002; Vol. 2. [Google Scholar]
- Jackson M.; Marks L.; May G. H. W.; Wilson J. B. The Genetic Basis of Disease. Essays Biochem 2018, 62 (5), 643–723. 10.1042/EBC20170053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury M. J.; Little J.; Burke W.. Human Genome Epidemiology: A Scientific Foundation for Using Genetic Information to Improve Health and Prevent Disease; Oxford University Press: Oxford, 2004. [Google Scholar]
- Herzog R. W.; Zolotukhin S.. A Guide To Human Gene Therapy; World Scientific: NJ, 2010. 10.1142/7406. [DOI] [Google Scholar]
- Mingozzi F.; High K. A. Therapeutic in Vivo Gene Transfer for Genetic Disease Using AAV: Progress and Challenges. Nat. Rev. Genet 2011, 12 (5), 341–355. 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Keeler A. M.; Flotte T. R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here?. Annual Review of Virology 2019, 6, 601–621. 10.1146/annurev-virology-092818-015530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell J. R.; Al-Zaidy S. A.; Rodino-Klapac L. R.; Goodspeed K.; Gray S. J.; Kay C. N.; Boye S. L.; Boye S. E.; George L. A.; Salabarria S.; Corti M.; Byrne B. J.; Tremblay J. P. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Molecular Therapy 2021, 29 (2), 464–488. 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agbandje-McKenna M.; McKenna R.. Production and Purification of Viruses for Structural Studies. In Structural Virology; C Biomolecular Sciences; The Royal Society of Chemistry, 2011; pp P001–371. 10.1039/9781849732239. [DOI] [Google Scholar]
- Grieger J. C.; Samulski R. J.. Adeno-Associated Virus Vectorology, Manufacturing, and Clinical Applications. In Gene Transfer Vectors for Clinical Application; Friedmann T., Ed.; Academic Press, 2012; Vol. 507, pp 229–254. 10.1016/B978-0-12-386509-0.00012-0. [DOI] [PubMed] [Google Scholar]
- Wörner T. P.; Bennett A.; Habka S.; Snijder J.; Friese O.; Powers T.; Agbandje-McKenna M.; Heck A. J. R. Adeno-Associated Virus Capsid Assembly Is Divergent and Stochastic. Nature Communications 2021, 12 (1), 1–9. 10.1038/s41467-021-21935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama H.; Ishii K.; Maruno T.; Torisu T.; Uchiyama S. Characterization of Adeno-Associated Virus Capsid Proteins with Two Types of VP3-Related Components by Capillary Gel Electrophoresis and Mass Spectrometry. Hum. Gene Ther. 2021, 32 (21–22), 1403–1416. 10.1089/HUM.2021.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth W. W.; Lewin A. S.; Zolotukhin S.; Muzyczka N.. Production and Purification of Recombinant Adeno-Associated Virus. In Vertebrate Phototransduction and the Visual Cycle, Part B; Academic Press, 2000; Vol. 316, pp 743–761. 10.1016/S0076-6879(00)16760-6. [DOI] [PubMed] [Google Scholar]
- Naso M. F.; Tomkowicz B.; Perry W. L.; Strohl W. R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. Biodrugs 2017, 31 (4), 317. 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. N. T.; Sha S.; Hong M. S.; Maloney A. J.; Barone P. W.; Neufeld C.; Wolfrum J.; Springs S. L.; Sinskey A. J.; Braatz R. D. Mechanistic Model for Production of Recombinant Adeno-Associated Virus via Triple Transfection of HEK293 Cells. Molecular Therapy - Methods and Clinical Development 2021, 21, 642–655. 10.1016/j.omtm.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B.; Nakai H.; Xiao W. Characterization of Genome Integrity for Oversized Recombinant AAV Vector. Molecular Therapy 2010, 18 (1), 87–92. 10.1038/mt.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khimani A. H.; Thirion C.; Srivastava A. AAV Vectors Advance the Frontiers of Gene Therapy. Gen. Eng. Biotechnol. News 2022, 42 (1), 38–40. 10.1089/GEN.42.01.13. [DOI] [Google Scholar]
- Wright J. F. Quality Control Testing, Characterization and Critical Quality Attributes of Adeno-Associated Virus Vectors Used for Human Gene Therapy. Biotechnology Journal 2021, 16 (1), 2000022. 10.1002/biot.202000022. [DOI] [PubMed] [Google Scholar]
- Gavin D. K. FDA Statement Regarding the Use of Adeno-Associated Virus Reference Standard Materials. Human Gene Therapy Methods 2015, 26 (1), 3. 10.1089/hgtb.2015.1501. [DOI] [PubMed] [Google Scholar]
- Ayuso E. Manufacturing of Recombinant Adeno-Associated Viral Vectors: New Technologies Are Welcome. Molecular Therapy - Methods & Clinical Development 2016, 3, 15049. 10.1038/mtm.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urabe M.; Xin K. Q.; Obara Y.; Nakakura T.; Mizukami H.; Kume A.; Okuda K.; Ozawa K. Removal of Empty Capsids from Type 1 Adeno-Associated Virus Vector Stocks by Anion-Exchange Chromatography Potentiates Transgene Expression. Molecular Therapy 2006, 13 (4), 823–828. 10.1016/j.ymthe.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Wright J. F. Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment. Biomedicines 2014, 2 (1), 80–97. 10.3390/BIOMEDICINES2010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaud-Budloo M.; François A.; Clément N.; Ayuso E. Pharmacology of Recombinant Adeno-Associated Virus Production. Molecular Therapy - Methods & Clinical Development 2018, 8, 166–180. 10.1016/j.omtm.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpel A. L.; Katsikis G.; Sha S.; Maloney A. J.; Hong M. S.; Nguyen T. N. T.; Wolfrum J.; Springs S. L.; Sinskey A. J.; Manalis S. R.; Barone P. W.; Braatz R. D. Analytical Methods for Process and Product Characterization of Recombinant Adeno-Associated Virus-Based Gene Therapies. Molecular Therapy - Methods & Clinical Development 2021, 20, 740–754. 10.1016/j.omtm.2021.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh N. L.; Berguig G. Y.; Karim O. A.; Cortesio C. L.; de Angelis R.; Khan A. A.; Gold D.; Maga J. A.; Bhat V. S. Comprehensive Characterization and Quantification of Adeno Associated Vectors by Size Exclusion Chromatography and Multi Angle Light Scattering. Sci. Rep. 2021, 11 (1), 3012. 10.1038/s41598-021-82599-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle A. K.; Powers T. W.; Zobel J. F.; Wappelhorst C. N.; Jarrold M. F.; Lyktey N. A.; Sloan C. D. K.; Wolf A. J.; Adams-Hall S.; Baldus P.; Runnels H. A. Comparison of Analytical Techniques to Quantitate the Capsid Content of Adeno-Associated Viral Vectors. Molecular Therapy - Methods & Clinical Development 2021, 23, 254–262. 10.1016/j.omtm.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burova E.; Ioffe E. Chromatographic Purification of Recombinant Adenoviral and Adeno-Associated Viral Vectors: Methods and Implications. Gene Therapy 2005, 12 (1), S5–S17. 10.1038/sj.gt.3302611. [DOI] [PubMed] [Google Scholar]
- Qu G.; Bahr-Davidson J.; Prado J.; Tai A.; Cataniag F.; McDonnell J.; Zhou J.; Hauck B.; Luna J.; Sommer J. M.; Smith P.; Zhou S.; Colosi P.; High K. A.; Pierce G. F.; Wright J. F. Separation of Adeno-Associated Virus Type 2 Empty Particles from Genome Containing Vectors by Anion-Exchange Column Chromatography. J. Virol Methods 2007, 140 (1–2), 183–192. 10.1016/j.jviromet.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Wang C.; Mulagapati S. H. R.; Chen Z.; Du J.; Zhao X.; Xi G.; Chen L.; Linke T.; Gao C.; Schmelzer A. E.; Liu D. Developing an Anion Exchange Chromatography Assay for Determining Empty and Full Capsid Contents in AAV6.2. Molecular Therapy - Methods & Clinical Development 2019, 15, 257–263. 10.1016/j.omtm.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatwani S. L.; Pavlova A.; Pirot Z. Anion-Exchange HPLC Assay for Separation and Quantification of Empty and Full Capsids in Multiple Adeno-Associated Virus Serotypes. Molecular Therapy - Methods and Clinical Development 2021, 21, 548–558. 10.1016/j.omtm.2021.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Mulagapati S. H. R.; Chen Z.; Du J.; Zhao X.; Xi G.; Chen L.; Linke T.; Gao C.; Schmelzer A. E.; Liu D. Developing an Anion Exchange Chromatography Assay for Determining Empty and Full Capsid Contents in AAV6.2. Molecular Therapy - Methods & Clinical Development 2019, 15, 257–263. 10.1016/j.omtm.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer J. M.; Smith P. H.; Parthasarathy S.; Isaacs J.; Vijay S.; Kieran J.; Powell S. K.; McClelland A.; Wright J. F. Quantification of Adeno-Associated Virus Particles and Empty Capsids by Optical Density Measurement. Molecular Therapy 2003, 7 (1), 122–128. 10.1016/S1525-0016(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Dobnik D.; Kogovšek P.; Jakomin T.; Košir N.; Žnidarič M. T.; Leskovec M.; Kaminsky S. M.; Mostrom J.; Lee H.; Ravnikar M.. Accurate Quantification and Characterization of Adeno-Associated Viral Vectors. Frontiers in Microbiology 2019, 10 ( (July), ), 1570. 10.3389/FMICB.2019.01570/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson E. E.; Keifer D. Z.; Asokan A.; Jarrold M. F. Resolving Adeno-Associated Viral Particle Diversity With Charge Detection Mass Spectrometry. Anal. Chem. 2016, 88 (13), 6718–6725. 10.1021/acs.analchem.6b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock M.; Alvira M. R.; Wilson J. M. Analysis of Particle Content of Recombinant Adeno-Associated Virus Serotype 8 Vectors by Ion-Exchange Chromatography. Hum Gene Ther Methods 2012, 23 (1), 56–64. 10.1089/hgtb.2011.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham B.; Nass S.; Kong E.; Mattingly M.; Woodcock D.; Song A.; Wadsworth S.; Cheng S. H.; Scaria A.; O’Riordan C. R. Analytical Ultracentrifugation as an Approach to Characterize Recombinant Adeno-Associated Viral Vectors. Hum Gene Ther Methods 2015, 26 (6), 228–242. 10.1089/hgtb.2015.048. [DOI] [PubMed] [Google Scholar]
- Wu D.; Hwang P.; Li T.; Piszczek G. Rapid Characterization of Adeno-Associated Virus (AAV) Gene Therapy Vectors by Mass Photometry. Gene Therapy 2021 2022, 1–7. 10.1038/s41434-021-00311-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Gao T.; Chen H.; Pekker P.; Menyhart A.; Guttman A. Rapid Determination of Full and Empty Adeno-Associated Virus Capsid Ratio by Capillary Isoelectric Focusing. Current Molecular Medicine 2021, 20 (10), 814–820. 10.2174/1566524020666200915105456. [DOI] [PubMed] [Google Scholar]
- Trivedi P. D.; Yu C.; Chaudhuri P.; Johnson E. J.; Caton T.; Adamson L.; Byrne B. J.; Paulk N. K.; Clément N. Comparison of Highly Pure RAAV9 Vector Stocks Produced in Suspension by PEI Transfection or HSV Infection Reveals Striking Quantitative and Qualitative Differences. Molecular Therapy - Methods & Clinical Development 2022, 24, 154–170. 10.1016/j.omtm.2021.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz E. D.; Rahman K. S.; Bower B. D.; Dismuke D. J.; Falvo M. R.; Griffith J. D.; Harvey S. C.; Asokan A. Biophysical and Ultrastructural Characterization of Adeno-Associated Virus Capsid Uncoating and Genome Release. J. Virol 2013, 87 (6), 2994–3002. 10.1128/JVI.03017-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan B.; Yarbrough J.; Domsic J.; Bennett A.; Bothner B.; Kozyreva O. G.; Samulski R. J.; Muzyczka N.; McKenna R.; Agbandje-McKenna M. Structure and Dynamics of Adeno-Associated Virus Serotype 1 VP1-Unique N-Terminal Domain and Its Role in Capsid Trafficking. J. Virol 2013, 87 (9), 4974–4984. 10.1128/JVI.02524-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.