

Abstract
Nonconventional NMR spin-coupling constants were investigated to determine their potential as conformational constraints in MA’AT modeling of the O-glycosidic linkages of oligosaccharides. Four (1JC1′,H1′, 1JC1′,C2′, 2JC1′,H2′, and 2JC2′,H1′) and eight (1JC4,H4, 1JC3,C4, 1JC4,C5, 2JC3,H4, 2JC4,H3, 2JC5,H4, 2JC4,H5, and 2JC3,C5) spin-couplings in methyl β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside (methyl β-lactoside) were calculated using density functional theory (DFT) to determine their dependencies on O-glycosidic linkage C–O torsion angles, ϕ and ψ, respectively. Long-range 4JH1′,H4 was also examined as a potential conformational constraint of either ϕ or ψ. Secondary effects of exocyclic (hydroxyl) C–O bond rotation within or proximal to these coupling pathways were investigated. Based on the findings of methyl β-lactoside, analogous J-couplings were studied in five additional two-bond O-glycosidic linkages [βGlcNAc-(1→4)-βMan, 2-deoxy-βGlc-(1→4)-βGlc, αMan-(1→3)-βMan, αMan-(1→2)-αMan, and βGlcNAc(1→2)-αMan] to determine whether the coupling behaviors observed in methyl β-lactoside were more broadly observed. Of the 13 nonconventional J-couplings studied, 7 exhibit properties that may be useful in future MA’AT modeling of O-glycosidic linkages, none of which involve coupling pathways that include the linkage C–O bonds. The findings also provide new insights into the general effects of exocyclic C–O bond conformation on the magnitude of experimental spin-couplings in saccharides and other hydroxyl-containing molecules.
1. Introduction
Determinations of oligosaccharide structure in solution require, in part, assessments of the geometries of their constituent O-glycosidic linkages.1−3 Theoretical methods such as aqueous molecular dynamics (MD) simulations4−8 can provide this information if their underlying force fields are parameterized to accurately and quantitatively recapitulate all of the covalent and non-covalent interactions that dictate geometry in solution. Conformational models obtained by MD simulations are only as reliable as the force field that underpins them. However, it has been difficult to obtain rigorous independent experimental validation of MD-derived models because current experimental methods do not provide continuous models of molecular torsion angles that can be superimposed on those obtained by MD. Recent developments using redundant experimental NMR spin-coupling constants and MA’AT analysis9−12 offer a solution to this problem. MA’AT analysis allows the testing of uni- and multi-modal conformational models, but a significantly greater number of experimental observables is required to test the latter. For example, rotational models about the C5–C6 bond in methyl β-d-glucopyranoside (1) typically involve three states, each representing an idealized staggered rotamer of the C4–C5–C6–O6 torsion angle ω (Scheme 1). Testing this model of ω using MA’AT analysis requires at least 7 redundant spin-coupling constants, and, in this case, >12 are available,13 rendering the treatment feasible mathematically.
Scheme 1. Conformational Model of the C4–C5–C6–O6 Torsion Angle in Methyl β-d-Glucopyranoside (1) in Solution, Comprising Three Idealized Staggered Rotamers gg, gt, and tg in Chemical Exchange.

Conventional modeling of O-glycosidic linkages using MA’AT analysis typically employs six trans-O-glycosidic J-couplings to characterize ϕ and ψ, which includes three redundant couplings to model ϕ and three to model ψ (Scheme 2).14 Recent work has shown that these six J-values associated with C–O–C–H, C–O–C–C, and C–O–C coupling pathways across O-glycosidic linkages are sufficient for unimodal modeling of each torsion angle.10−12,15 These treatments yield mean torsion angles and circular standard deviations that are in good agreement with other techniques, the latter reporting on the degree of librational motion of each angle. Determinations of root mean squared deviations (RMSDs) of these models and inspections of parameter space plots yield information on how well unimodal models fit the available experimental J-couplings and whether the model is a unique fit of the experimental data, respectively. In the linkages studied to date, RMSDs are small (<0.3 Hz), and the majority produce unique fits, indicating that ϕ and ψ adopt highly preferred single states, although considerable averaging around the mean torsion angles is observed. MA’AT modeling of ϕ in different types of O-glycosidic linkages suggests that current MD models underestimate the librational motion about this glycosidic torsion angle.15 Despite these advances, however, it would be desirable to increase the number of redundant spin-coupling constants used in MA’AT analyses of linkage conformation (or include other types of NMR constraints) to test unimodal models more rigorously and/or to render feasible modeling beyond a single state.
Scheme 2. Conventional Trans-O-Glycosidic J-coupling Pathways in β-(1→4) Disaccharide 2, Showing Three J-Couplings Sensitive to φ (3JH1′,C4, 2JC1′,C4, and 3JC2′,C4) and Three Sensitive to ψ (3JC1′,H4, 3JC1′,C3, and 3JC1′,C5).

Atom numbering and glycosidic torsion angles φ and ψ are shown. Coupling pathways are highlighted in blue.
The results of the studies described herein address the structural dependences of several nonconventional NMR spin-coupling constants that may serve as potential constraints in MA’AT analyses of O-glycosidic linkages. The primary objective of the work was to determine which of these J-couplings have properties conducive to their use in MA’AT analysis. The structural dependencies were determined by density functional theory (DFT) calculations in four different types of linkages (α-(1→2); β-(1→2); α-(1→3); and β-(1→4); Scheme 3) found in disaccharides 2–7 to determine the extent to which the results are generalizable. We show that several coupling pathways, some of which are peripheral to the linkages themselves and do not include linkage bonds explicitly, yield J-couplings that may prove useful in future MA’AT treatments of O-glycosidic linkages. This work also reveals the importance of understanding the conformational behaviors of exocyclic C–O bonds of saccharides in solution since they impact the values and structural interpretations of both conventional and nonconventional experimental J-couplings.
Scheme 3. Chemical Structures of Methyl β-d-Galactopyranosyl-(1→4)-β-d-glucopyranoside (2), Methyl 2-Acetamido-2-deoxy-β-d-glucopyranosyl-(1→4)-β-d-mannopyranoside (3), Methyl 2-Deoxy-β-d-arabino-hexopyranosyl-(1→4)-β-d-glucopyranoside(4), Methyl α-d-mannopyranosyl-(1→3)-β-d-mannopyranoside (5), Methyl α-d-mannopyranosyl-(1→2)-α-d-mannopyranoside (6), and Methyl 2-Acetamido-2-deoxy-β-d-glucopyranosyl-(1→2)-α-d-mannopyranoside (7), Showing Atom Numbering in the Pyranosyl Rings and Identification of the “a” and “b” Residues of Each Disaccharide.

The O-glycosidic torsion angles phi (φ) and psi (ψ) are shown in each structure.
2. Calculations
2.1. Geometry Optimizations
DFT calculations were conducted on fully substituted model structures 2–7 (Scheme 3) within Gaussian0916 using the B3LYP functional17,18 and the 6-31G* basis set.19 In models 5 and 7, several sites of deoxygenation were introduced at carbons remote from the coupling pathways of interest (Schemes S6 and S8, Supporting Information; see explanation below). In all geometric optimizations, the effects of solvent water were treated using the self-consistent reaction field (SCRF)20 and the integral equation formalism (polarizable continuum) model (IEFPCM).21 For calculations on 2, the phi (ϕ) and psi (ψ) O-glycosidic torsion angles, defined as O5′–C1′–O1′–C4 and C1′–O1′–C4–C3, respectively, were each rotated in 15° increments through 360°, giving a 24 × 24 matrix or 576 optimized structures (Scheme 4). Torsion angles C1′–C2′–O2′–H (θ1), C3–C2–O2–H (θ2), and C4–C3–O3–H (θ3) were fixed at 180°. The remaining seven exocyclic torsion angles in 2 were held constant or were set at an initial value and optimized, as summarized in Scheme 4. Six additional datasets were generated for 2, in which θ1, θ2, or θ3 was fixed at either 60 or 300°, while the other ring C−O torsion angles were fixed at 180°, each set containing 576 structures.
Scheme 4. Torsion Angle Constraints Used in DFT Calculations of Nonconventional Spin-Coupling Constants in 2.

Exocyclic torsion angles θ1, θ2, and θ3 were sampled in three perfectly staggered geometries, yielding 27 datasets containing incremental rotations of φ and ψ through 360°. See text for details.
For structures 3–7, the ϕ and ψ torsion angles were each rotated in 15° increments through 360°, giving 24 × 24 matrices (576 structures). Only one set of exocyclic torsion angles was investigated in 3–7 as summarized in Schemes S4–S8 (Supporting Information). In DFT calculations of 5 and 7, deoxy analogs were used to simplify the structures and/or avoid unfavorable steric interactions when the ϕ and ψ torsion angles were rotated. The deoxy sites were kept to a minimum and judged based on prior work to be sufficiently distant from the coupling pathways of interest to not influence their calculated behaviors.
2.2. Calculations of NMR Spin-Coupling Constants
A group of JHH, JCH, and JCCspin-coupling constants with potentially useful dependencies on ϕ or ψ was calculated in geometry optimized structures of 2–7 by DFT using the B3LYP functional17,18 in Gaussian09.16 The Fermi contact,22−24 diamagnetic and paramagnetic spin–orbit, and spin-dipole terms22 were recovered using a tailored basis set, [5s2p1d|3s1p],25,26 and raw (unscaled) calculated spin-couplings are reported and are similar to experimental values to within ±0.2–0.3 Hz based on prior work.26,27 SCRF20 and the IEFPCM21 were again used to treat the effects of solvent water during J-coupling calculations.
DFT calculations of 1JCH, 1JCC, 3JCH, and 3JCC values gave positive signs as expected, whereas those of 2JCH and 2JCC values gave either positive or negative signs. Experimental sign determinations of the latter geminal J-couplings have been reported in prior work,28−32 and ample comparisons have been made between experimental and calculated signs to validate those obtained by DFT calculations.
2.3. Spin-Coupling Constant Equation Parameterization
Equations relating DFT-calculated JHH, JCH, and JCC values to ϕ or ψ in 2–7 were parameterized to a trigonometric polynomial using R. Equations were parameterized using J-values calculated in a subpopulation of conformers that was selected using a 10 kcal/mol energy cut-off to remove a limited number of highly structurally strained conformers.10,12 A secondary constraint was also applied when necessary to remove DFT-optimized structures containing distorted aldohexopyranosyl rings; Cremer-Pople puckering parameters were calculated from DFT-generated Cartesian coordinates, and a θ value of 35° was used as the cut-off.10,12 The goodness-of-fit of each equation is reported as a root mean squared deviation (RMSD). Equation parameterization was further evaluated using the Akaike information criterion,33 resulting in truncated forms of two equations.
3. Results and Discussion
3.1. Potential Nonconventional Spin-Coupling Constraints for ϕ
Criteria to determine whether a specific J-coupling may have value in MA’AT analyses of ϕ and ψ include: (1) dynamic range; (2) character of the torsion angle versus J-value plot (e.g., does the plot contain multiple maxima and minima?); (3) whether the J-value exhibits a significant secondary dependence on either ϕ or ψ; (4) whether the J-value exhibits other significant structural dependencies in addition to either ϕ or ψ (e.g., dependencies on proximal exocyclic C–O torsion angles); and (5) whether the experimental J-value can be conveniently and accurately measured and its sign determined if required, preferably without the need for stable isotopic labeling. Secondary dependencies or effects (criterion 3) refer to J-coupling behaviors where J-coupling depends primarily on one torsion angle but also shows dependencies on a second or third torsion angle. In the present work, a J-coupling might depend heavily on ϕ but also show some dependence on ψ or vice versa. Secondary dependencies can complicate the structural interpretations of J-couplings if they are not well understood and accounted for. In the following discussion, potential nonconventional J-couplings are identified that satisfy most, if not all, of these criteria.
Five nonconventional J-couplings to evaluate ϕ are shown in Scheme 5, illustrated in 2 although analogous J-couplings exist in disaccharides 3–7 (Scheme 3). These couplings include 1JC1′,H1′, 1JC1′,C2′, 2JC1′,H2′, 2JC2′,H1′, and 4JH1′,H4. 1JC1′,H1′ and 1JC1′,C2′ are expected to depend on ϕ, based on anticipated vicinal lone-pair effects on C–H and C–C bond lengths caused by rotation of the C1′–O1′ bond (ϕ). The C1′–H1′ and C1′–C2′ bond lengths are affected by n→σ* donation when a lone-pair orbital on O1′ is anti to the bond, in general leading to bond elongation34,35 (in the ensuing discussion, oxygen atoms are assumed to bear two sp3-hybridized lone-pair orbitals although other lone-pair arrangements may pertain36−38). Presumably the percent s-character of both bonds is greater (shorter bonds) for geometries in which both O1′ lone-pairs are gauche to them. For 1JC1′,C2′, overlapping vicinal lone-pair effects from C2′–O2′ bond rotation (θ1) also pertain. Thus, at least two structural factors influence 1JC1′,C2′, namely, rotation about ϕ and θ1. Superimposed on these stereoelectronic effects may be bond angle effects caused by the rotation of ϕ and θ1.
Scheme 5. Nonconventional Spin-Coupling Constants That May Depend on φ, Illustrated for the Internal O-Glycosidic Linkage in 2.

Coupling pathways are highlighted in blue. Torsion angles θ1, θ2, and θ3 denote rotations about the C2′–O2′, C3–O3, and C5–C6 bonds, respectively.
Geminal 2JC1′,H2′ and 2JC2′,H1′ values are expected to have strong configurational dependencies superimposed on conformational dependencies on ϕ and θ1. Consider the generalized Ca–Cb–Hb coupling pathway shown in Scheme 6. Rotation of the Ca–Cb bond (γ1) describes the configurational dependence of 2JCa,Hb, manifested in both coupling magnitude and sign. For 2JC1′,H2′ and 2JC2′,H1′ in 2, rotation about the analogous C1′–C2′ bond is constrained by the pyranosyl ring (i.e., the configuration is fixed) such that this constraint determines their baseline magnitudes and signs. C–O Bond rotations γ2 and γ3 are mainly responsible for the conformational dependencies of 2JCa,Hb. In general, for the Ca–Cb–Hb pathway, the effect of rotation of γ3 is greater than that for rotation of γ2; that is, rotation of the C–O bond involving the carbon bearing the coupled hydrogen exerts a greater effect on 2JCa,Hb than rotation of the C–O bond involving the coupled carbon. The difference in sensitivity is ∼2–3 fold.39,40 Thus, while 2JC1′,H2′ might be a potential constraint on ϕ (Scheme 5), rotation of ϕ exerts a smaller effect on the coupling than rotation of θ1, reducing the dynamic range and consequently its usefulness as a ϕ constraint. Conversely, rotation of ϕ exerts a greater effect on 2JC2′,H1′ than rotation of θ1. Thus, all else being equal, 2JC2′,H1′ is likely to be the better probe of ϕ than 2JC1′,H2′. However, even in this case, contributions from θ1 to 2JC2′,H1′ cannot be ignored (i.e., independent information on θ1 would be desirable to determine its contribution to the magnitude of experimental 2JC2′,H1′ values in solution).
Scheme 6. Bond Torsions γ1, γ2, and γ3 That Influence the Magnitude and Sign of 2JCa,Hb (Pathway Highlighted in Blue).

R1 and R2 are assumed to be sp3-hybridized carbons as found in typical saccharide carbon scaffolds.
4JH1′,H4 is likely to exhibit sensitivities to both ϕ and ψ.40−42 If this behavior is confirmed by DFT calculations, 4JH1′,H4 values may serve only as independent tests of the conformational models of ϕ and ψ determined from more robust J-values (see discussion below). Even in this role, the very small dynamic range of 4JH1′,H4 will limit its use in assignments of linkage conformation.
3.2. Potential Nonconventional Spin-Coupling Constraints for ψ
In 2, nine nonconventional J-couplings have the potential to serve as constraints on ψ (Scheme 7): 1JC4,H4, 1JC3,C4, 1JC4,C5, 2JC3,H4, 2JC4,H3, 2JC5,H4, 2JC4,H5, 2JC3,C5, and 4JH1′,H4. Analogous J-values exist in disaccharides 3–7. The structural dependencies of the 1JCH, 1JCC, and 2JCCH values are expected to mimic those sensitive to ϕ (Scheme 5). For example, 1JC3,C4 will depend on the rotameric properties of the C3–C4, C3–O3 (θ2), and C4–O1′ (ψ) bonds.43 Since rotation about the C3–C4 bond is constrained by the pyranosyl ring, only the remaining C–O bonds will largely determine 1JC3,C4 in solution. Likewise, 2JC3,H4 and 2JC5,H4 are likely to be better constraints on ψ than 2JC4,H3 and 2JC4,H5 (see above discussion of Scheme 6). 4JH1′,H4 is likely to serve as an independent test of the conformational models of ψ (and ϕ) determined from more robust J-values.
Scheme 7. Potential Nonconventional Spin-Coupling Constants That May Depend on ψ, Illustrated for the Internal O-Glycosidic Linkage in 2.
Coupling pathways are highlighted in blue. Torsion angles θ1, θ2, and θ3 denote rotations about the C2′–O2′, C3–O3, and C5–C6 bonds, respectively.
The geminal 13C–13C spin-coupling, 2JC3,C5, is expected to show configurational and conformational dependencies analogous to those of 2JC1,C3 in aldopyranosyl rings.40,44 Configuration at the terminal coupled carbons affects its magnitude and sign, especially O3 orientation (axial vs equatorial), while the effect of configuration at the intervening C4 carbon is expected to be small. Conformational effects derive from rotation of the C3–O3 (θ2), C5–C6 (θ3), and C4–O1′ (ψ) bonds, with ψ exerting a greater effect than θ2 and θ3. In 2, the configurational effects are fixed and determine the baseline value of 2JC3,C5 in solution. This value will vary in response to the conformational effects. Consequently, the reliability of 2JC3,C5 as a probe of ψ in solution will depend on the degree to which the contributions from θ2 and θ3 can be determined.
3.3. DFT Calculations of Nonconventional ϕ-Dependent Spin-Coupling Constants in 2
1JC1′,H1′,2JC1′,H2′, 2JC2′,H1′, and 1JC1′,C2′ were calculated as a function of ϕ in 2 (Figure 1). Scatter along the y-axis at discrete values of ϕ shows the secondary effect of ψ. The plots for 1JC1′,H1′ and 2JC1′,H2′ contain significant scatter, indicating that neither J-value will be a reliable independent constraint for ϕ. The dynamic range for 2JC1′,H2′ is also small (∼1 Hz), further reducing its usefulness. The shape of the curve can be understood by noting the calculated 2JC1′,H2′ values in the three idealized staggered rotamers of ϕ (rotamers I–III, Scheme 8) and inspecting the corresponding Newman projections. Less negative values correlate with ϕ rotamers in which a lone-pair orbital on O1′ is anti to the C1′–C2′ bond (rotamers II and III). In contrast to 2JC1′,H2′, 2JC2′,H1′ displays a larger dynamic range (4–5 Hz) when its sign is taken into account, rendering it a better probe of ϕ than 2JC1′,H2′ (Figure 1C). The 2JC2′,H1′ plot also has considerably less y-axis scatter, especially at ϕ values of 0–180° (i.e., 2JC2′,H1′ has less y-axis scatter overall than observed for 2JC1′,H2′). In the three idealized staggered ϕ rotamers shown in Scheme 8, the calculated 2JC2′,H1′ is more positive in III (∼3 Hz) than in I and II (∼0–1 Hz). In III, the C1′–H1′ and C1′–C2′ bonds are both anti to an O1′ lone-pair orbital, whereas in I and II, a lone-pair orbital is anti to only one of these bonds. These lone-pair orbital effects on 2JCCH are consistent with those reported previously.39,40
Figure 1.

Calculated dependencies of 1JC1′,H1′ (A), 2JC1′,H2′ (B), 2JC2′,H1′ (C) and 1JC1′,C2′ (D) on ϕ in 2. Black circles, full dataset; green circles, trimmed dataset. Solid green and black lines represent best fits to the green and black datasets, respectively. See Scheme 8 for the definitions of ϕ rotamers I–III identified in each plot. The scatter observed at discrete ϕ values in these plots reveals the secondary dependencies on ψ.
Scheme 8. Idealized Staggered Rotamers I–III of the C1′–O1′ Bond φ in 2.

The exo-anomeric effect favors rotamer I in the β-linkage of 2. One lone-pair orbital on O1′ is anti to the C1′–C2′ bond in II and III, whereas both orbitals are gauche to this bond in I.
Like 2JC2′,H1′, the plot for 1JC1′,C2′ shows modest y-axis scatter and a relatively large dynamic range (∼6 Hz). Rotamers II and III (Scheme 8) contain an O1′ lone-pair orbital anti to the C1′–C2′ bond, and both give 1JC1′,C2′ values of ∼49 Hz. In contrast, both O1′ lone-pair orbitals are gauche to the C1′–C2′ bond in 1, yielding a 1JC1′,C2′ of ∼53 Hz. These observations are consistent qualitatively with the discussion above, namely, that rotamers II and III are likely to contain longer C1′–C2′ bonds than rotamer I, resulting in less s-character and a smaller 1JC1′,C2′.
3.4. DFT Calculations of Nonconventional ψ-Dependent Spin-Coupling Constants in 2
The dependencies of 1JC4,H4, 1JC3,C4, and 1JC4,C5 on ψ in 2 are shown in Figure 2. 1JC4,H4 behaves similarly to 1JC1′,H1′ (Figure 1A) with regard to y-axis scatter, indicating that, like 1JC1′,H1′, 1JC4,H4 is unlikely to be a useful constraint for ψ. 1JC3,C4 and 1JC4,C5 are more promising, with both showing moderate secondary dependencies on ϕ and adequate dynamic ranges (6–7 Hz). Newman projections of the O1′–C4 bond in 2 for the three idealized staggered rotamers IV–VI of ψ (Scheme 9) provide structural rationales for the behaviors of 1JC3,C4 and 1JC4,C5. 1JC3,C4 in rotamers V and VI (∼43 Hz) is smaller than that in rotamer IV (∼47 Hz), while 1JC4,C5 is smaller in rotamers IV and V (∼41 Hz) than in rotamer VI (∼46 Hz). Rotamers V and VI contain an O1′ lone-pair orbital anti to the C3–C4 bond, but rotamer IV does not, leading to a smaller rC3,C4 in IV relative to V and VI and presumably to the larger 1JC3,C4 in IV. Similarly, rotamers IV and V contain an O1′ lone-pair orbital anti to the C4–C5 bond, whereas rotamer VI does not, resulting in a plot of 1JC4,C5 that mimics that observed for 1JC3,C4 (plots B and C in Figure 2 are essentially phase-shifted along the x-axis).
Figure 2.

Calculated dependencies of 1JC4,H4 (A), 1JC3,C4 (B), and 1JC4,C5 (C) on ψ in 2. See Figure 1 for the definitions of the green and black data points and lines. Scheme 9 contains the definitions of ψ rotamers IV–VI. The scatter observed at discrete ψ values in these plots reveals the secondary dependencies on ϕ.
Scheme 9. Idealized Staggered Rotamers IV–VI of the C4–O1′ Bond ψ in 2.

The dependences of the four 2JCCH values in Scheme 7 on ψ in 2 are shown in Figure 3. The dynamic ranges for 2JC4,H3 (∼1.5 Hz) and 2JC4,H5 (∼1.5 Hz) are small, and neither J-coupling is likely to be a useful constraint for ψ. In contrast, the dynamic ranges of 2JC3,H4 (∼2 Hz) and 2JC5,H4 (∼5 Hz) are larger, especially the latter. The plot of 2JC5,H4 indicates more negative values in rotamers V and VI relative to rotamer IV. In rotamer IV, O1′ lone-pair orbitals are anti to both the C4–H4 and C4–C5 bonds, whereas in rotamers V and VI, a lone-pair orbital is anti to only one of these bonds. This situation mimics that observed for 2JC2′,H1′ where the less negative (more positive) J-coupling is associated with rotamer III (Figure 1C, Scheme 8). Applying the same analysis to 2JC3,H4 (Figure 3A) leads to the prediction that rotamer VI should give a less negative (more positive) J-coupling than rotamers IV and V since it contains O1′ lone-pairs anti to both the C3–C4 and C4–H4 bonds (Scheme 9). The plot in Figure 3A supports this prediction, although rotamers IV and V yield different, albeit more negative values. Orienting an O1′ lone-pair anti to the C3–C4 bond (rotamer V) appears to reduce 2JC3,H4 more significantly than orienting a lone-pair anti to the C4–H4 bond (rotamer IV).
Figure 3.

Calculated dependencies of 2JC3,H4 (A), 2JC4,H3 (B), 2JC5,H4 (C), and 2JC4,H5 (D) on ψ in 2. See Figure 1 for definitions of the green and black data points and lines, and Scheme 9 for the definitions of ψ rotamers IV–VI. The scatter observed at discrete ψ values in these plots reveals the secondary dependencies on ϕ.
Rotamer IV is associated with a more negative 2JC4,H5 than rotamer V even though both contain an O1′ lone-pair orbital anti to the C4–C5 bond, but the most negative value, as expected, is associated with rotamer VI which lacks this interaction (Scheme 9). Likewise, rotamers V and VI are expected to be associated with more positive 2JC4,H3 values than rotamer IV since the former contain an O1′ lone-pair orbital anti to the C3–C4 bond and the latter does not. In addition to vicinal lone-pair effects, longer-range 1,3 lone-pair effects40 from O1′ on the C3–H3 or C5–H5 bond lengths may influence the behaviors of 2JC4,H3 and 2JC4,H5, but these effects cannot be visualized using the projections in Scheme 9.
Collectively, the nonconventional 2JCCH values in 2 behave similarly with respect to the effects of oxygen vicinal lone-pair orbitals on coupling magnitude and sign. The presence of an oxygen lone-pair anti to either the C–C or C–H bond of the coupling pathway, or to both bonds, shifts the coupling to a more positive (less negative) value relative to the coupling observed in conformations lacking these anti arrangements. The lone-pair effects appear additive, although they are probably not equivalent, that is, the effect of a lone-pair anti to the C–C bond may be larger than when anti to the C–H bond. These conclusions are consistent with those drawn in an earlier study of the conformational dependencies of 2JCCH values in saccharides.39,40
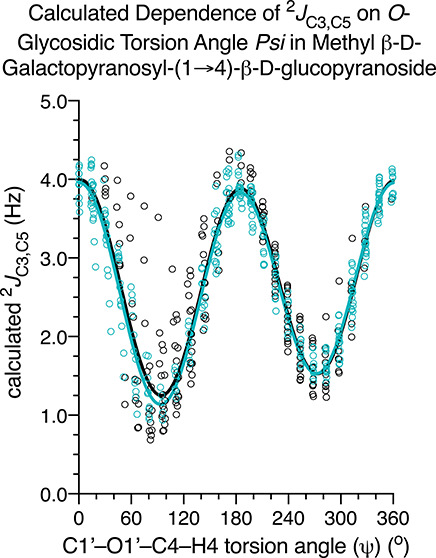
2JC3,C5 shows a bimodal dependence on ψ and is positive in sign with a dynamic range of ∼3 Hz (Figure 4). Rotamers IV and VI correlate with smaller values (∼2 Hz) than rotamer V (∼4 Hz). In rotamer V, the two lone-pair orbitals on O1′ are anti to the C3–C4 and C4–C5 bonds (the two bonds comprising the coupling pathway), whereas in rotamers IV and VI only one of these lone-pair interactions exists (the remaining lone-pair orbital is anti to the C4–H4 bond). Prior studies have shown that the lone-pair arrangements in rotamer V shift 2JCCC to more positive values in saccharides (i.e., this relationship pertains to 2JC1,C3, 2JC2,C4, and 2JC4,C6 in aldohexopyranosyl rings).39,40
Figure 4.

Calculated dependency of 2JC3,C5 on ψ in 2. See Figure 1 for the definitions of the green and black data points and lines, and Scheme 9 for the definitions of ψ rotamers IV–VI. The scatter observed at discrete ψ values reveals the secondary dependence on ϕ.
Based on evaluations in Sections 3.3 and 3.4 above, the following seven nonconventional spin-coupling constants in Schemes 5 and 7 are good candidates for MA’AT modeling of ϕ and ψ in 2: for ϕ, 1JC1′,C2′ and 2JC2′,H1′; for ψ, 1JC3,C4, 1JC4,C5, 2JC3,H4 (marginal), 2JC5,H4, and 2JC3,C5. By extension, related J-couplings in other two-bond O-glycosidic linkages (e.g., those in 3–7, Scheme 3) should prove useful for MA’AT modeling of their linkages, as discussed below.
3.5. Effects of Exocyclic C–O Bond Conformation on ϕ- and ψ-Dependent Nonconventional Spin-Couplings Constants in 2
Having identified seven potentially useful nonconventional ϕ- and ψ-dependent J-couplings, secondary conformational effects that influence them were evaluated. In the treatments discussed above, the effects of ϕ or ψ on these J-couplings were studied. All other exocyclic conformational features were fixed or highly constrained in geometries dictated by the ten torsion angles shown in Scheme 4. Superimposed on these ϕ and ψ dependencies are secondary conformational dependences whose magnitudes need to be understood and quantified in order to apply these nonconventional J-couplings to structural studies. These secondary dependencies arise largely from exocyclic C–O bond torsion angles θ1–θ3 (Scheme 5), and Scheme 10 shows which of these angles affect specific ϕ- and ψ-dependent non-conventional J-couplings. For example, for 1JC1′,C2′ and 2JC2′,H1′, rotation about θ1 affects their magnitudes and/or signs, in addition to rotation about ϕ. In a similar vein, 1JC3,C4 and 2JC3,H4 are affected by θ2 in addition to ψ. In the following discussion, the secondary effects of θ1, θ2, and θ3 are discussed in more detail.
Scheme 10. Secondary Effects of Torsion Angles θ1, θ2, and θ3 on Seven Nonconventional Spin-Coupling Constants Sensitive to φ and ψ in 2.

For each set of J-couplings shown on the right, the torsion angle(s) shown in blue affect their magnitudes and/or signs.
3.5.1. 1JC1′,C2′ and 2JC2′,H1′ (ϕ-Dependent)
Plots of 1JC1′,C2′ as a function of ϕ adopt the same overall shape regardless of the values of θ1, θ2, and θ3 (Figure 5A). However, a y-axis displacement of ∼5 Hz to smaller 1JC1′,C2′ is observed for θ1 rotamers VII and IX relative to the remaining curves, including that for θ1 rotamer VIII (reference state) (Scheme S1, Supporting Information). An inspection of θ1 rotamers shows that an O2′ lone-pair orbital is anti to C1′ in rotamers VII and IX but not in VIII. The former geometry is expected to lengthen the C1′–C2′ bond (vicinal lone-pair interaction), leading to smaller 1JC1′,C2′ values.
Figure 5.

Effects of θ1–θ3 on the dependencies of 1JC1′,C2′ (A) and 2JC2′,H1′ (B) on ϕ in 2. For each plot: black line, reference state; green dots, rotamer VII; green line, rotamer IX; blue dots, rotamer X; blue line, rotamer XII; red dots, rotamer XIII; red line, rotamer XV. For the definitions of rotamers and reference states, see Schemes S1–S3, Supporting Information.
The effects of θ1–θ3 on 2JC2′,H1′ are modest (∼0.5 Hz) except for the curve for rotamer XV (Scheme S3, Supporting Information) near ϕ values of 300° (Figure 5B). 2JC2′,H1′ values are most positive when the two lone-pair orbitals on O2′ are anti to the C1′–C2′ and C2′–C3′ bonds (θ1 rotamer IX). The behavior in rotamer XV at ϕ near 300° probably results from greater data scatter from the secondary ψ dependence and its effect on parameterization since in these ϕ geometries, O2′ and O6 are in close proximity in some ψ rotamers. These findings show that rotation of the C–O bond involving the coupled carbon (in this case, θ1) exerts a smaller effect on 2JCCH than rotation of the C–O bond involving the carbon bearing the coupled hydrogen (in this case, ϕ) (Scheme 6).
3.5.2. 1JC3,C4 and 1JC4,C5 (ψ-Dependent)
The dependence of 1JC3,C4 on ψ (Figure 6A) mimics that of 1JC1′,C2′ on ϕ (Figure 5A) in that both curves are unimodal, although the former J-couplings are smaller by 6−7 Hz (the C3−C4 fragment bears only two oxygen substituents, whereas the C1′–C2′ fragment bears three). 1JCC decreases in magnitude as the number of oxygen substituents on the C–C fragment declines. Secondary effects may pertain when two oxygen substituents are involved in terms of whether both reside on the same carbon or different carbons. The effects of θ1–θ3 on curve shape are small, but the curves associated with θ2 rotamers X and XII are shifted to smaller values by ∼5 Hz relative to θ2 rotamer XI (reference state). An inspection of Scheme S2 (Supporting Information) shows an O3 vicinal lone-pair interaction with the C3–C4 bond in rotamers X and XII but not in rotamer XI. As found for 1JC1′,C2′, lengthening of the C3–C4 bond in the former two rotamers leads to smaller 1JCC values.
Figure 6.

Effects of θ1–θ3 on the dependencies of 1JC3,C4 (A) and 1JC4,C5 (B) on ψ in 2. For each plot: black line, reference state; green dots, rotamer VII; green line, rotamer IX; blue dots, rotamer X; blue line, rotamer XII; red dots, rotamer XIII; red line, rotamer XV. For the definitions of rotamers and reference states, see Schemes S1–S3, Supporting Information.
The curves for 1JC4,C5 are unimodal (Figure 6B) and are affected to a limited extent by θ1–θ3. The effect of θ1 is small but not that of θ2, especially for rotamer X. Exocyclic hydroxymethyl group conformation affects 1JC4,C5, with the curve for rotamer XV (tg) differing from those for rotamers XIII (gg) and XIV (gt). The hydroxymethyl group effects are probably caused by significant data scatter at ψ values of 60–180° where strong steric interactions between O5′ and the C6–O6 fragment occur, especially in the tg rotamer. This scatter may influence curve parameterization and render comparisons unreliable. For ψ values devoid of steric interactions (values of 0–60° and 180–360°), the curves are very similar (y-axis displacements of ≤1 Hz).
3.5.3. 2JC3,H4, 2JC5,H4, and 2JC3,C5 (ψ-Dependent)
The effects of θ1–θ3 on the dependence of 2JC3,H4 on ψ are modest (differences of ∼0.5 Hz) (Figure 7A). The single outlier is θ3 rotamer XV (tg; Scheme S3, Supporting Information), and aberrant parameterization caused by significant data scatter at ψ values of 150–200° (steric clashes) is probably the cause. The effect of θ1 is negligible (remote rotation), but a small θ2 effect (rotation of the C3–O3 bond) is observed. 2JC3,H4 values associated with rotamers X and XII are more positive (less negative) by ∼0.5 Hz than those associated with rotamer XI (reference state). C3–O3 Rotamers containing O3 lone-pair orbitals anti to the C2–C3 and C3–C4 bonds (θ2 rotamer XII) or one O3 lone-pair orbital anti to the C3–C4 bond (θ2 rotamer X) yield slightly more positive (less negative) 2JC3,H4 values than rotamer XI that lacks either of these arrangements. Similar behavior was observed for 2JC2′,H1 (Figure 5B).
Figure 7.

Effects of θ1–θ3 on the dependencies of 2JC3,H4 (A), 2JC5,H4 (B), and 2JC3,C5 (C) on ψ in 2. For each plot: black line, reference state; green dots, rotamer VII; green line, rotamer IX; blue dots, rotamer X; blue line, rotamer XII; red dots, rotamer XIII; red line, rotamer XV. For the definitions of rotamers and reference states, see Schemes S1–S3, Supporting Information.
The effects of θ1–θ3 on the dependencies of 2JC5,H4 and 2JC3,C5 on ψ (Figure 7B,C) are small (<1 Hz); curve shapes are highly conserved. Rotation of θ3 does not affect 2JC5,H4 despite the proximity of the exocyclic hydroxymethyl group to the C5–C4–H4 coupling pathway. Rotation of θ2 also does not much affect 2JC5,H4 despite potential 1,3-lone-pair interactions between O3 lone-pair orbitals and the proximal C4–H4 bond.
Rotations of θ2 and θ3 exert small effects on 2JC3,C5. The available data indicate that θ2 rotamers that orient one of the O3 lone-pair orbitals anti to the C3–C4 bond (rotamers X and XII; Scheme S2, Supporting Information) shift 2JC3,C5 to slightly less positive values relative to rotamer XI that lacks this interaction (Figure 7C). This result contrasts with the effect of ψ on 2JC3,C5 (Figures 4 and 7C) where the most positive 2JC3,C5 value correlates with rotamer V in which the lone-pair orbitals on O1′ are anti to the C3–C4 and C4–C5 bonds. The effects of oxygen lone-pair orbitals on 2JC3,C5 depend on whether the lone-pair resides on an oxygen atom attached to C3 or to C4.
3.6. Summary of 2JCCH Behavior in 2 and Saccharides in General
The results of studies on the effects of exocyclic C–O bond conformation on 2JCCH values in 2 are summarized in Scheme 11. Two types of C–O bond rotations pertain to C–C–H coupling pathways, those associated with the coupled carbon and those associated with the carbon bearing the coupled hydrogen. The latter C–O rotations exert the greater effect on 2JCCH values by a factor of 4–5. Thus, from the standpoint of conformational probes, the latter types of rotations, arising either from exocyclic C–O bonds of hydroxyl groups or C–O bonds involved in O-glycosidic linkages, are more likely to be interrogated successfully by 2JCCH values than the former.
Scheme 11. Summary of Oxygen Lone-Pair Effects on 2JCCH Values in Saccharides, Using 2JC2′,H1′ and 2JC3,H4 in 2 as Examples.

(A) Effect of the C–O bond rotation on the carbon bearing the coupled hydrogen. The φ (for 2JC2′,H1′) or ψ (for 2JC3,H4) rotamers that orient both oxygen lone-pair orbitals anti to both bonds in the C–C–H coupling pathway correlate with the most positive J-values (these interactions are expected to lengthen both bonds). (B) Effect of C–O bond rotation on the coupled carbon. The value of θ1 (for 2JC2′,H1′) and θ2 (for 2JC3,H4) rotamers in which the O–H bond is anti to the C–C bond in the coupling pathway, and one oxygen lone-pair orbital experiences a 1,3 interaction with the coupled hydrogen and correlates with the most negative J-values (both effects are expected to reduce the C–C and C–H bond lengths in the coupling pathway). See Schemes 8, 9, S1, and S2 for rotamers III, VI, VIII and XI, respectively.
In the preceding analysis, six 2JCCH were treated: 2JC1′,H2′, 2JC2′,H1′, 2JC3,H4, 2JC4,H3, 2JC5,H4, and 2JC4,H5. DFT calculations show that all but 2JC2′,H1′ have negative signs, regardless of C–O bond conformation at either carbon in the C–C–H coupling pathway. Application of the empirical projection rule of Pedersen and coworkers28 gives signs that are generally consistent with the DFT calculations. The five DFT-calculated 2JCCH that have negative signs give projection sums of either −0.5 or 0, whereas the single 2JCCH having a positive sign (2JC2′,H1′) gives a projection sum of +0.5. The former sums correlate with 2JCCH values ranging from −3 to −5 Hz, whereas a +0.5 sum correlates with 2JCCH values of ∼0 Hz (Figure S2, Supporting Information). Inspection of the 2JC2′,H1′ curves in Figures 1C and 5B shows that both positive and negative signs are possible depending on ϕ and θ1, giving an average value that is small in magnitude and positive in sign. The projection method only takes into account the configuration of the C–C–H coupling pathway and does not include contributions from C–O bond rotations at both carbons, which are significant.
3.7. Extensions to O-Glycosidic Linkages in Disaccharides 3–7
The preceding discussion of spin-coupling constants in 2 revealed several nonconventional 1J and 2J values that may be useful in MA’AT analyses of the phi and psi torsion angles in O-glycosidic linkages. Do the conclusions drawn from studies of 2 apply to β-(1→4) linkages involving modified residues and to (1→2) and (1→3) O-glycosidic linkages? To address these questions, calculations were conducted on five model disaccharides: methyl 2-acetamido-2-deoxy-β-d-glucopyranosyl-(1→4)-β-d-mannopyranoside (3), methyl 2-deoxy-β-d-arabino-hexopyranosyl-(1→4)-β-d-glucopyranoside (4), methyl α-d-mannopyranosyl-(1→3)-β-d-mannopyranoside (5), methyl α-d-mannopyranosyl-(1→2)-α-d-mannopyranoside (6), and methyl 2-acetamido-2-deoxy-β-d-glucopyranosyl-(1→2)-α-d-mannopyranoside (7) (Scheme 3). The behaviors of seven non-conventional J-couplings in 3–7 were compared to those of analogous J-couplings in 2. The results are shown in Figures 8–10. For 1JC1′,C2′, the overall curve shape is conserved in 2–4 and 7, in which the C1′−O1′ bond is equatorial and the O1′–CX bond (CX is the aglycone carbon) is either axial (7) or equatorial (2–4) (Figure 8A). Absolute values of 1JC1′,C2′ decrease progressively in the order 2 > 3 ≈ 7 > 4. The introduction of an N-acetyl side-chain on C2′ shifts 1JC1′,C2′ to smaller values, and removal of an electronegative substituent at C2′ further reduces 1JC1′,C2′. These curves differ appreciably from those for 5 and 6 that contain α-linkages in which the O1′–CX bond is either axial (6) or equatorial (5). The curve for 5 is an approximate mirror image of that for 2, whereas that for 6 differs from that for 5 in the appearance of a local minimum near ϕ = 240°. The dynamic ranges of the curves for 3–7 are similar to that of the curve for 2, supporting the contention that 1JC1′,C2′ should be a useful probe of ϕ in 3–7. Separate equation parameterizations, however, will be required to treat 1JC1′,C2′ quantitatively in 2–7.
Figure 8.

Calculated dependencies of 1JC1′,C2′ (A) and 2JC2′,H1′ (B) on the H1′–C1′–O1′–CX torsion angle (ϕ) in disaccharides 2–7, where CX represents the aglycone carbon. Bold black curve, 2. Purple curve, 3. Lime green curve, 4. Blue curve, 5. Teal curve, 6. Red curve, 7.
Figure 10.

Calculated dependencies of 2JCX–1,HX (A), 2JCX+1,HX (B), and 2JCX–1,CX+1 (C) on the C1′–O1′–CX–HX torsion angle (ψ) in disaccharides 2–7, where CX represents the aglycone carbon. For example, in 5, 2JCX–1,HX = 2JC2,H3, 2JCX+1,HX = 2JC4,H3, and 2JCX–1,CX+1 = 2JC2,C4. Bold black curve, 2. Purple curve, 3. Lime green curve, 4. Blue curve, 5. Teal curve, 6. Red curve, 7.
The dependencies of 2JC2′,H1′ on ϕ in 2–4 and 7 are very similar, differing only in shifts along the y-axis (Figure 8B). 2JC2′,H1′ becomes increasingly more positive in the order 2 < 3 ≈ 7 < 4. Curves for the α-linked disaccharides 5 and 6 are similar and are approximate mirror images of those for 2–4 and 7. 2JC2′,H1′ values are almost uniformly negative for the two α-linkages, whereas those for the β-linkages are almost uniformly positive, as observed for αMan and βGlc monosaccharides.45 The dynamic ranges of all curves are similar, suggesting that 2JC2′,H1′ may be uniformly applicable in MA’AT analyses of ϕ. However, separate equation parameterizations will be required to apply 2JC2′,H1′ in MA’AT analyses.
One-bond 13C–13C spin-couplings sensitive to ψ are shown in Figure 9. The 1JCX,CX–1 curves (Figure 9A) adopt similar shapes for 5–7, whereas those for 2–4 overlap. The latter structures contain β-(1→4) linkages to aglycone residues bearing equatorial C–O bonds at C4 and C3. The side-chain structure at C2′ exerts little effect on this 1JCC nor does C–O bond orientation at the remote C2 (axial in 3 and equatorial in 2 and 4). 1JCX,CX–1 plots for 6 and 7 are similar and nearly overlap, and both are significantly shifted to larger 1JCC relative to the plot for 5. Inspection of 6 and 7 shows that both contain an axial O1′–C2 bond and an axial C1–O1 bond, whereas corresponding bonds in 5 are equatorial and axial, respectively. The relative disposition of oxygen substituents appended to the coupled carbons over one bond (i.e., the dihedral angle subtended by the vicinal oxygen substituents; ∼180° in 6 and 7 and ∼−60° in 5) is an important determinant of 1JCC values in saccharides.43 The orientation of the C1′–O1′ bond, which is axial in 6 and equatorial in 7, does not appear to influence 1JCX,CX–1 values appreciably. The dynamic range of the curve for 5 is somewhat smaller than that for 2–4 and 6–7, which may render this 1JCC value less useful for MA’AT analyses of ψ in structures like 5.
Figure 9.
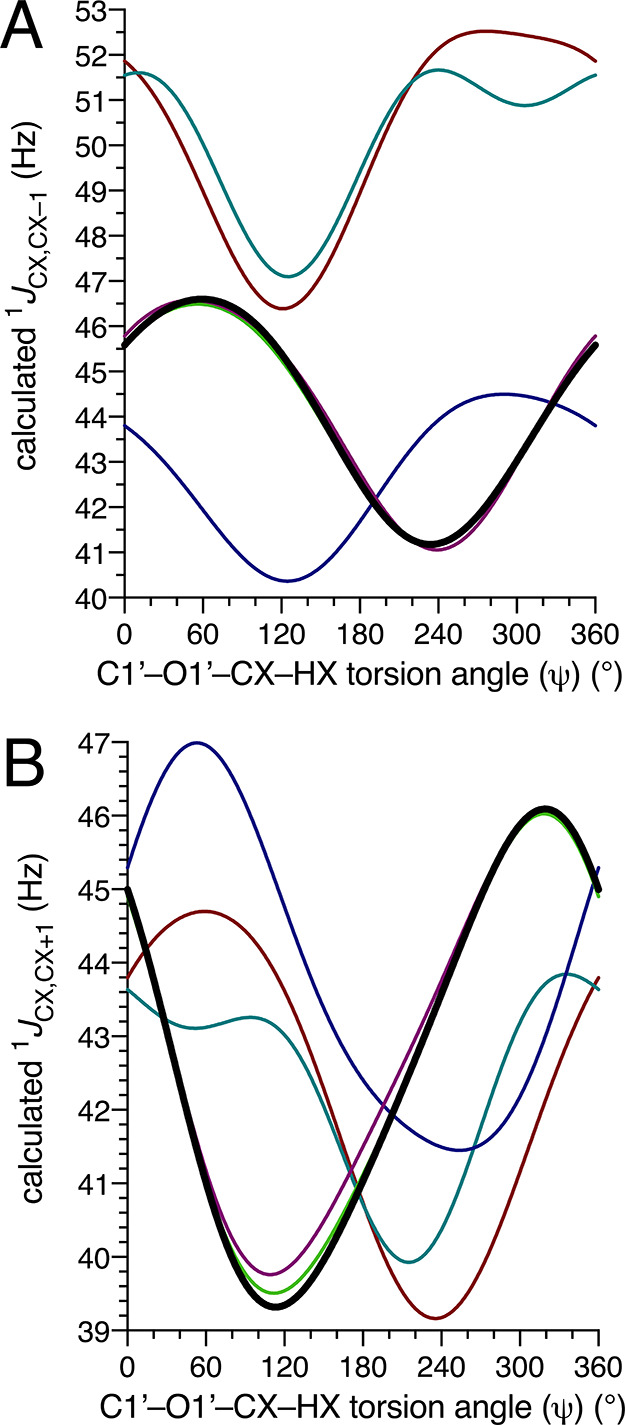
Calculated dependencies of 1JCX,CX–1 (A) and 1JCX,CX+1 (B) on the C1′–O1′–CX–HX torsion angle (ψ) in disaccharides 2–7, where CX represents the aglycone carbon. For example, in 5, 1JCX,CX–1 = 1JC3,C2 and 1JCX,CX+1 = 1JC3,C4. Bold black curve, 2. Purple curve, 3. Lime green curve, 4. Blue curve, 5. Teal curve, 6. Red curve, 7.
The dependencies of 1JCX,CX+1 on ψ in 2–4 are virtually identical, producing overlapping curves (Figure 9B). The latter structures contain β-(1→4) linkages to aglycone residues bearing equatorial bonds at C4 and C5. The side-chain structure at C2′ exerts little effect on this 1JCC nor does C–O bond orientation at the remote C2 (axial in 3 and equatorial in 2 and 4). The curves for 5–7, which contain α anomeric linkages, have roughly similar shapes that are distinct from those for 2–4. For 5, the C–O bonds at C3 and C4 are both equatorial. In contrast, the C–O bonds at C2 and C3 are axial and equatorial, respectively, in 6 and 7. These structural differences in the CX–CX+1 coupling pathway may explain why the curves for 6 and 7 are similar and distinct from that for 5.
Geminal 13C–1H and 13C–13C spin-couplings in 2–7 are shown in Figure 10, all of which are sensitive to ψ. The curve shapes for 2JCX–1,HX in 5–7 are conserved but are shifted along the y-axis, whereas the curves for 2–4 overlap and have shapes distinct from those found for 5–7 (Figure 10A). 2JCX–1,HX is positive or negative in sign in 6–7 depending on ψ, whereas 2JCX–1,HX is uniformly positive in 5. In contrast, 2JCX–1,HX values in 2–4 are strongly negative. These observations are consistent with predictions based on the projection rule,28 where a projection of 0 is associated with the C3–C4–H4 pathways in 2–4, and a projection of 1.5 is associated with the C2–C3–H3 pathway in 5, yielding predicted 2JCCH values of ∼−3 and ∼+5 Hz, respectively (see Figure S2 in Supporting Information), in qualitative agreement with the DFT results. The dynamic ranges of 2JCX–1,HX in 5–7 may render them more useful for MA’AT analysis of ψ than those of 2JCX–1,HX in 2–4. Similar behavior is observed for 2JCX+1,HX (Figure 10B) in that curves of similar shape are observed for 5–7 but are shifted along the y-axis, whereas the curves for 2–4 overlap and have shapes distinct from those for 5–7. 2JCX+1,HX values in 2–7 are negative in sign and all exhibit a dynamic range suitable for MA’AT analysis of ψ.
The geminal 2JCX–1,CX+1 shows a remarkable consistency in its dependency on ψ (Figure 10C), regardless of the location of the pathway in the pyranosyl ring. Two sets of curves are observed. One set includes 2JCX–1,CX+1 values that have uniformly positive signs and is associated with pathways in which the oxygens on the terminal coupled carbons are both equatorial (structures 2–4). The second set includes 2JCX–1,CX+1 values with signs near zero and is associated with pathways in which the oxygens on the terminal coupled carbons are axial and equatorial (structures 5–7). C–O Bond orientation (axial vs equatorial) at the intervening carbon in these C–C–C pathways appears to exert a minimal effect on 2JCCC. The dynamic range of pathways having two equatorial terminal oxygens is larger than that of pathways having an axial-equatorial arrangement, leading to the expectation that 2JCX–1,CX+1 values in the former will be more robust MA’AT constraints on ψ than 2JCX–1,CX+1 values in the latter.
3.8. Behavior of 4JH1′,H4 in Disaccharides 2, 5, and 6
Trans-O-Glycoside four-bond spin-coupling between H1′ and HX (HX is the hydrogen attached to the aglycone carbon) is not expected to be a useful probe in MA’AT analyses of O-glycosidic linkages. Nevertheless, it was investigated to gain a better understanding of its dependence on linkage conformation. Plots of calculated 4JH1’,H4 in 2 as a function of ϕ and ψ are shown in Figure 11. 4JH1′,H4 values are small in magnitude and have a dynamic range of ∼2 Hz. 4JH1′,H4 is equally sensitive to ϕ and ψ, as indicated by the data scatter at discrete values of ϕ in Figure 11A and ψ in Figure 11B. This scatter reaches a minimum at a ϕ/ψ combination of 180/180°, that is, when the five atoms in the H1′–C1′–O1′–C4–H4 coupling pathway are coplanar and adopt a W-shaped arrangement.40,46 Recent MA’AT analyses of 2(10) have yielded mean values of ϕ and ψ of 28° and −8°, respectively, where secondary effects on 4JH1′,H4 are significant, thereby rendering 4JH1′,H4 useless as an independent probe of either torsion angle. However, 4JH1′,H4 could play a confirmatory role if ϕ and ψ in a given linkage were determined from MA’AT analyses of other J-couplings. In this case, complicating secondary effects are reduced, and 4JH1′,H4 can be used as an ancillary test of the conformational assignment. This scenario is illustrated in Figure 12 for 4JH1′,H4 in 2. Using MA’AT-determined mean values of ϕ and ψ, a 4JH1′,H4 of ∼0.9 Hz should be observed, a value with a small uncertainty based on the imposed limited scatter from secondary effects. In practice, however, while coupling trends predicted by DFT are probably reliable, it remains to be established whether 4JH1′,H4 values calculated by DFT are accurate; that is, the curves in Figures 11 and 12 may need to be shifted on the y-axes to bring calculated values in line with experiment.
Figure 11.

Calculated dependencies of 4JH1′,H4 on (A) H1′–C1′–O1′–C4 (ϕ) and (B) C1′–O1′–C4–H4 (ψ) torsion angles in 2. In each plot, the black curve represents the best fit of the data. Point scatter at discrete values of ϕ in (A) and ψ in (B) indicate the extent to which ψ and ϕ, respectively, affect the calculated coupling.
Figure 12.

Calculated dependencies of 4JH1′,H4 on (A) H1′–C1′–O1′–C4 (ϕ) and (B) C1′–O1′–C4–H4 (ψ) torsion angles in 2. In each plot, the black curve represents the best fit of the data. Point scatter at discrete values of ϕ in (A) and ψ in (B) indicate the extent to which ψ and ϕ, respectively, affect the calculated coupling, using restricted ranges of ψ (−30 to 15°) and ϕ (15–45°). The vertical dashed line in each plot identifies the mean value of ϕ (28°) and ψ (−8°) in 2 determined from prior MA’AT analysis.10
The behavior of 4JH1′,H4 in 2 as a function of ϕ and ψ (Figure 11) appears to be independent of the type of O-glycosidic linkage. In 2, the C1′–O1′ and C4–O1′ bonds are both equatorial. In 5, the C1′–O1′ bond is axial and the C3–O1′ bond is equatorial, and in 6, the C1′–O1′ and C2–O1′ bonds are both axial. Plots similar to those shown in Figure 11 for 2 are shown in Figures S3 and S4 for 5 and 6, respectively (see Supporting Information). The latter plots are very similar to those found for 2 with respect to the location of the minimum, dynamic range, the absolute values of the calculated J-couplings, and the nature of the secondary dependencies. Thus, the conclusions drawn regarding the ancillary role of 4JH1′,H4 in evaluating ϕ and ψ in 2 also apply to the corresponding trans-glycosidic 4JHH values in 5 and 6 and probably to other types of O-glycosidic linkages.
3.9. DFT-Parameterized Spin-Coupling Equations for Structures 2–7
Plots of J-couplings in structures 2–7 as a function of either the phi (ϕ) or psi (ψ) O-glycosidic torsion angles (Figures 1–12) were parameterized and are found in the Supporting Information (eqs S1–S105). The curves were fit to the following modified Karplus-like equation (eq 1) using R.
| 1 |
This generalized form of the Karplus-like equation was first described by Pachler.47 He proposed the use of this trigonometric function to account for asymmetry in the Karplus curve caused by substitution of a hydrogen atom in the coupling pathway. This trigonometric polynomial form was adopted because it provides the best parameterization to the DFT data with the smallest number of terms. This form of the equation is also amenable to simple integration, making it compatible with MA’AT analysis9−12 for modeling torsional populations in solution.
4. Conclusions
MA’AT modeling of conformational equilibria and dynamics of saccharides depends in part on reliable equations that relate specific spin-coupling constants to molecular torsion angles.9−12 It has been customary to use 3J values (e.g., 3JHH, 3JCH, and 3JCC) in this modeling for three reasons: (1) these J-couplings typically exhibit strong dependencies on molecular torsion angles (i.e., have large dynamic ranges); (2) they are less prone to secondary structural effects, especially those associated with the rotation of proximal bonds bearing electronegative substituents (e.g., C–O bonds); and (3) the conformational dependencies of other types of spin-coupling constants (e.g., 1J, 2J and 4J) are less well understood. Saccharides like other types of molecules are rich in 1J, 2J, and 3J values involving carbon and hydrogen as coupled nuclei. However, restricting MA’AT analyses to 3J values eliminates nearly 60% of the available J-couplings in an aldohexopyranosyl ring, excluding those involving hydroxyl hydrogens.40 For unimodal modeling, access to 3–4 redundant J-couplings (i.e., those sensitive to the same molecular torsion angle) is sufficient for successful MA’AT modeling.9−12 For multimodal modeling, however, significantly more redundant J-couplings are needed, rendering the use of 1J and/or 2J values important to wider applications of the method. In the absence of using 1J and/or 2J values, additional types of NMR parameters (e.g., residual dipolar couplings or nuclear Overhauser effects) might also enable multimodal modeling.
This study investigated a group of nonconventional J-couplings (mainly 2J values) that are not commonly used in conformational analysis of the O-glycosidic linkages of oligosaccharides. Thirteen J-couplings in six different types of O-glycosidic linkages were investigated. The results show that seven of these J-couplings may prove useful in MA’AT modeling of either ϕ or ψ under certain conditions, regardless of the type of O-glycosidic linkage (e.g., 1→2 vs 1→3 linkages; α vs β linkages). The latter conditions are likely to include prior knowledge of the conformational behaviors of exocyclic C–O bonds proximal to the coupling pathway. This knowledge would reduce the negative impacts of secondary effects on MA’AT modeling of O-glycosidic linkage conformation when nonconventional J-couplings are used as conformational constraints. This knowledge would also improve, although more modestly, the reliability of MA’AT modeling of ϕ and ψ based on conventional 3J values. Experimental studies of the conformational properties of exocyclic C–O bonds in solution have been reported,48−53 and MD simulations provide additional information even though current force fields are not parameterized to provide this information quantitatively. MD simulations of exocyclic hydroxyl C–O bonds not participating in hydrogen bonding show three idealized staggered rotamers to be most stable, although often in different populations. Whether this behavior replicates that found in solution accurately awaits more rigorous experimental study.
While the present study investigated new nonconventional J-couplings of importance to MA’AT modeling of O-glycosidic linkage conformation and dynamics, nonconventional spin-couplings may also prove useful in MA’AT modeling of other conformational properties of saccharides in solution. Unlike O-glycosidic linkages that often adopt single-state conformations about ϕ and ψ, multi-state conformational equilibria may pertain to other conformational behaviors (e.g., exocyclic hydroxyl and hydroxymethyl group conformations). Multi-state modeling of hydroxyl group conformations by MA’AT analysis would be enabled by use of nonconventional J-couplings like those discussed in this report. This modeling not only would advance our current rudimentary understanding of exocyclic C–O bond conformational behavior in solution but would also improve MA’AT modeling in general by reducing or eliminating secondary effects displayed by some redundant J-couplings used in MA’AT analyses.
Acknowledgments
This work was supported by the National Science Foundation (CHE 1707660 and CHE 2002625 to A. S.) and by Omicron Biochemicals, Inc., South Bend, IN. The Notre Dame Radiation Laboratory is supported by the Department of Energy Office of Science, Office of Basic Energy Sciences, under Award Number DE-FC02-04ER15533. This is document number NDRL 5326.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c02793.
Three idealized staggered rotamers VII–IX about the C2′–O2′ bond (θ1) in 2; three idealized staggered rotamers X–XII about the C3–O3 bond (θ2) in 2; three idealized staggered rotamers XIII–XV about the C5–C6 bond (θ3) in 2; plots of calculated 1JC1′,H1′, 2JC1′,H2′, 1JC4,H4, 2JC4,H3, and 2JC4,H5 values as a function of either the H1′–C1′–O1′–C4 (ϕ) or the C1′–O1′–C4–H4 (ψ) torsion angle for different values of θ1, θ2, and θ3 in 2; and a discussion of these plots; torsion angle constraints in DFT calculations of 3–7; plot of projection sums used to predict the magnitudes and signs of 2JCCH values in saccharides; plots of calculated 4JH1′,H3 in 5 as functions of ϕ and ψ; plots of calculated 4JH1′,H2 in 6 as functions of ϕ and ψ; DFT-parameterized spin-coupling equations [S1]–[S105]; and representative Cartesian coordinates for 2–7 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yu Y.; Delbianco M. Conformational Studies of Oligosaccharides. Chem. - Eur. J. 2020, 26, 9814–9825. 10.1002/chem.202001370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Yamaguchi T.; Kato K. New NMR Tools for Characterizing the Dynamic Conformations and Interactions of Oligosaccharides. Chem. Lett. 2013, 42, 1455–1462. 10.1246/cl.130789. [DOI] [Google Scholar]
- Watanabe T.; Yagi H.; Yanaka S.; Yamaguchi T.; Kato K. Comprehensive Characterization of Oligosaccharide Conformational Ensembles with Conformer Classification by Free-energy Landscape via Reproductive Kernel Hilbert Space. Phys. Chem. Chem. Phys. 2021, 23, 9753–9760. 10.1039/d0cp06448c. [DOI] [PubMed] [Google Scholar]
- Woods R. J. Predicting the Structures of Glycans, Glycoproteins, and Their Complexes. Chem. Rev. 2018, 118, 8005–8024. 10.1021/acs.chemrev.8b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson Q.; Lindsay R.; Petridis L.; Shen T. Investigation of Carbohydrate Recognition via Computer Simulation. Molecules 2015, 20, 7700–7718. 10.3390/molecules20057700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plazinska A.; Plazinski W. Comparison of Carbohydrate Force Fields in Molecular Dynamics Simulations of Protein-Carbohydrate Complexes. J. Chem. Theory Comput. 2021, 17, 2575–2585. 10.1021/acs.jctc.1c00071. [DOI] [PubMed] [Google Scholar]
- Park S.-J.; Lee J.; Qi Y.; Kern N. R.; Lee H. S.; Jo S.; Joung I.; Joo K.; Lee J.; Im W. CHARMM-GUI Glycan Modeler for Modeling and Simulation of Carbohydrates and Glycoconjugates. Glycobiology 2019, 29, 320–331. 10.1093/glycob/cwz003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; MacKerell A. D. Jr. Conformational Sampling of Oligosaccharides Using Hamiltonian Replica Exchange with Two-Dimensional Dihedral Biasing Potentials and the Weighted Histogram Analysis Method (WHAM). J. Chem. Theory Comput. 2015, 11, 788–799. 10.1021/ct500993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turney T.; Pan Q.; Sernau L.; Carmichael I.; Zhang W.; Wang X.; Woods R. J.; Serianni A. S. O-Acetyl Side-Chains in Monosaccharides: Redundant NMR Spin-Couplings and Statistical Models for Acetate Ester Conformational Analysis. J. Phys. Chem. B 2017, 121, 66–77. 10.1021/acs.jpcb.6b10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Turney T.; Meredith R.; Pan Q.; Sernau L.; Wang X.; Hu X.; Woods R. J.; Carmichael I.; Serianni A. S. Conformational Populations of β-(1→4) O-Glycosidic Linkages Using Redundant NMR J-Couplings and Circular Statistics. J. Phys. Chem. B 2017, 121, 3042–3058. 10.1021/acs.jpcb.7b02252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Meredith R.; Yoon M.-K.; Wang X.; Woods R. J.; Carmichael I.; Serianni A. S. Synthesis and O-Glycosidic Linkage Conformational Analysis of 13C-Labeled Oligosaccharide Fragments of an Antifreeze Glycolipid. J. Org. Chem. 2019, 84, 1706–1724. 10.1021/acs.joc.8b01411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Meredith R.; Pan Q.; Wang X.; Woods R. J.; Carmichael I.; Serianni A. S. Use of Circular Statistics To Model αMan-(1→2)-αMan and αMan-(1→3)α/βMan O-Glycosidic Linkage Conformation in 13C-Labeled Disaccharides and High-Mannose Oligosaccharides. Biochemistry 2019, 58, 546–560. 10.1021/acs.biochem.8b01050. [DOI] [PubMed] [Google Scholar]
- Thibaudeau C.; Stenutz R.; Hertz B.; Klepach T.; Zhao S.; Wu Q.; Carmichael I.; Serianni A. S. Correlated C–C and C–O Bond Conformations in Saccharide Hydroxymethyl Groups: Parameterization and Application of Redundant 1H-1H, 13C-1H and 13C-13C NMR J-couplings. J. Am. Chem. Soc. 2004, 126, 15668–15685. 10.1021/ja0306718. [DOI] [PubMed] [Google Scholar]
- Bose B.; Zhao S.; Stenutz R.; Cloran F.; Bondo P. B.; Bondo G.; Hertz B.; Carmichael I.; Serianni A. S. Three-Bond C–O–C–C Spin-Coupling Constants in Carbohydrates: Development of a Karplus Relationship. J. Am. Chem. Soc. 1998, 120, 11158–11173. 10.1021/ja981127f. [DOI] [Google Scholar]
- Meredith R. J.; Woods R. J.; Carmichael I.; Serianni A. S. Reconciling MA′AT and Molecular Dynamics Models of Linkage Conformation in Oligosaccharides. Phys. Chem. Chem. Phys. 2020, 22, 14454–14457. 10.1039/d0cp01389g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; et al. Gaussian09, Revision D; Gaussian Inc.: Wallingford, CT, 2009.
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Becke A. D. New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]
- Cancès E.; Mennucci B.; Tomasi J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications To Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. 10.1063/1.474659. [DOI] [Google Scholar]
- Cammi R.; Mennucci B.; Tomasi J. Fast Evaluation of Geometries and Properties of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-Dimethyl-aminobenzonitrile. J. Phys. Chem. A 2000, 104, 5631–5637. 10.1021/jp000156l. [DOI] [Google Scholar]
- Sychrovský V.; Gräfenstein J.; Cremer D. Nuclear Magnetic Resonance Spin–Spin Coupling Constants from Coupled Perturbed Density Functional Theory. J. Chem. Phys. 2000, 113, 3530–3547. 10.1063/1.1286806. [DOI] [Google Scholar]
- Helgaker T.; Watson M.; Handy N. C. Analytical Calculation of Nuclear Magnetic Resonance Indirect Spin–Spin Coupling Constants at the Generalized Gradient Approximation and Hybrid Levels of Density-Functional Theory. J. Chem. Phys. 2000, 113, 9402–9409. 10.1063/1.1321296. [DOI] [Google Scholar]
- Barone V.; Peralta J. E.; Contreras R. H.; Snyder J. P. DFT Calculation of NMR JFF Spin Spin Coupling Constants in Fluorinated Pyridines. J. Phys. Chem. A 2002, 106, 5607–5612. 10.1021/jp020212d. [DOI] [Google Scholar]
- Klepach T.; Zhao H.; Hu X.; Zhang W.; Stenutz R.; Hadad M. J.; Carmichael I.; Serianni A. S.. Informing Saccharide Structural NMR Studies with Density Functional Theory Calculations. In Glycoinformatics: Methods in Molecular Biology; Lütteke T., Frank M., Eds.; Springer: New York, 2015, pp 289–331. [DOI] [PubMed] [Google Scholar]
- Stenutz R.; Carmichael I.; Widmalm G.; Serianni A. S. Hydroxymethyl Group Conformation in Saccharides: Structural Dependencies of 2JHH, 3JHH and 1JCH Spin-Spin Coupling Constants. J. Org. Chem. 2002, 67, 949–958. 10.1021/jo010985i. [DOI] [PubMed] [Google Scholar]
- Bose-Basu B.; Klepach T.; Bondo G.; Bondo P. B.; Zhang W.; Carmichael I.; Serianni A. S. 13C-13C NMR Spin-Spin Coupling Constants in Saccharides: Structural Correlations Involving All Carbons In Aldopyranosyl Rings. J. Org. Chem. 2007, 72, 7511–7522. 10.1021/jo0706776. [DOI] [PubMed] [Google Scholar]
- Pedersen C.; Bock K.; Popoff T.; Theander O.; Nørskov L.; Schroll G. Two- and Three-bond 13C-1H Couplings in Some Carbohydrates. Acta Chem. Scand. 1977, 31b, 354–358. 10.3891/acta.chem.scand.31b-0354. [DOI] [Google Scholar]
- Serianni A. S.; Podlasek C. A. 13C-1H Spin-coupling Constants in Carbohydrates: Magnitude and Sign Determinations via 2D NMR Methods. Carbohydr. Res. 1994, 259, 277–282. 10.1016/0008-6215(94)84062-8. [DOI] [Google Scholar]
- Church T.; Carmichael I.; Serianni A. S. Two-bond 13C-13C Spin-coupling Constants in Carbohydrates: Effect of Structure on Coupling Magnitude and Sign. Carbohydr. Res. 1996, 280, 177–186. 10.1016/0008-6215(95)00311-8. [DOI] [Google Scholar]
- Serianni A. S.; Bondo P. B.; Zajicek J. Verification of the Projection Resultant Method for Two-bond 13C-13C Coupling Sign Determinations in Carbohydrates. J. Magn. Reson. 1996, 112, 69–74. 10.1006/jmrb.1996.0111. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Bondo G.; Zajicek J.; Serianni A. S. Two-bond 13C-13C Spin-coupling Constants in Carbohydrates: New Measurements of Coupling Signs. Carbohydr. Res. 1998, 309, 145–152. 10.1016/s0008-6215(98)00123-2. [DOI] [Google Scholar]
- Akaike H. New Look at the Statistical Model Identification. IEEE Trans. Autom. Control 1974, 19, 716–723. 10.1109/tac.1974.1100705. [DOI] [Google Scholar]
- Freymann R.; Gueron J. Absorption Spectra in the Near Infra Red of Systems Constituted of Chlorhydric Gas and an Organic Oxygen Solvant. C. R. Acad. Sci. 1937, 205, 859–861. [Google Scholar]
- Wolfe S.; Pinto B. M.; Varma V.; Leung R. Y. N. The Perlin Effect: Bond Lengths, Bond Strengths, and the Origins of Stereoelectronic Effects Upon One-Bond C–H Coupling Constants. Can. J. Chem. 1990, 68, 1051–1062. 10.1139/v90-164. [DOI] [Google Scholar]
- Juaristi E.; Cuevas G.. The Anomeric Effect; CRC Press: Boca Raton, 1995, pp 95–111. [Google Scholar]
- Clauss A. D.; Nelsen S. F.; Ayoub M.; Moore J. W.; Landis C. R.; Weinhold F. Rabbit-ears Hybrids, VSEPR Sterics, and Other Orbital Anachronisms. Chem. Educ. Res. Pract. 2014, 15, 417–434. 10.1039/c4rp00057a. [DOI] [Google Scholar]
- Stewart J. J. P. An Examination of the Nature of Localized Molecular Orbitals and Their Value In Understanding Various Phenomena That Occur In Organic Chemistry. J. Mol. Model. 2019, 25, 7–23. 10.1007/s00894-018-3880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepach T. E.; Carmichael I.; Serianni A. S. Geminal 2JCCH Spin-Spin Coupling Constants as Probes of the ϕ Glycosidic Torsion Angle in Oligosaccharides. J. Am. Chem. Soc. 2005, 127, 9781–9793. 10.1021/ja040251y. [DOI] [PubMed] [Google Scholar]
- Hadad M. J.; Zhang W.; Turney T.; Sernau L.; Wang W.; Woods R. J.; Incandela A.; Surjancev I.; Wang A.; Yoon M.-K.; Coscia A.; Euell C.; Meredith R.; Carmichael I.; Serianni A. S.. NMR Spin-Couplings in Saccharides: Relationships Between Structure, Conformation and the Magnitudes of JHH, JCH and JCC Values. In New Developments in NMR 10: NMR in Glycoscience and Glycotechnology; Peters T., Kato K., Eds.; Royal Society of Chemistry, 2017, pp 20–100. [Google Scholar]
- Otter A.; Bundle D. R. Long-range 4J and 5J, Including Interglycosidic Correlations in Gradient-enhanced Homonuclear COSY Experiments of Oligosaccharides. J. Magn. Reson., Ser. B 1995, 109, 194–201. 10.1006/jmrb.1995.0009. [DOI] [PubMed] [Google Scholar]
- Sheng S.; Cherniak R.; van Halbeek H. A 1H NMR Spectroscopic Approach to the Unambiguous Determination of Glycosyl Linkage Positions in Oligosaccharides. Anal. Biochem. 1998, 256, 63–66. 10.1006/abio.1997.2454. [DOI] [PubMed] [Google Scholar]
- Carmichael I.; Chipman D. M.; Podlasek C. A.; Serianni A. S. Torsional Effects on the One-Bond 13C-13C Spin Coupling Constant in Ethylene Glycol: Insights Into the Behavior of 1JCC in Carbohydrates. J. Am. Chem. Soc. 1993, 115, 10863–10870. 10.1021/ja00076a050. [DOI] [Google Scholar]
- Zhang W.; Yoon M.-K.; Meredith R. J.; Zajicek J.; Oliver A. G.; Hadad M.; Frey M. H.; Carmichael I.; Serianni A. S. 13C–13C Spin-Coupling Constants in Crystalline 13C-Labeled Saccharides: Conformational Effects Interrogated by Solid-State 13C NMR Spectroscopy. Phys. Chem. Chem. Phys. 2019, 21, 23576–23588. 10.1039/c9cp03228b. [DOI] [PubMed] [Google Scholar]
- Podlasek C. A.; Wu J.; Stripe W. A.; Bondo P. B.; Serianni A. S. [13C]Enriched Methyl Aldopyranosides: Structural Interpretations of 13C-1H Spin-Coupling Constants and 1H Chemical Shifts. J. Am. Chem. Soc. 1995, 117, 8635–8644. 10.1021/ja00138a020. [DOI] [Google Scholar]
- Barfield M.; Dean A. M.; Fallick C. J.; Spear R. J.; Sternhell S.; Westerman P. W. Conformational Dependence and Mechanisms For Long-range Hydrogen-Hydrogen Coupling Constants Over Four Bonds. J. Am. Chem. Soc. 1975, 97, 1482–1492. 10.1021/ja00839a035. [DOI] [Google Scholar]
- Pachler K. G. R. Extended Hückel Theory MO Calculations of Proton-Proton Coupling Constants–II: The Effect of Substituents on Vicinal Couplings in Monosubstituted Ethanes. Tetrahedron 1971, 27, 187–199. 10.1016/s0040-4020(01)92410-x. [DOI] [Google Scholar]
- Kindahl L.; Sandström C.; Norberg T.; Kenne L. 1H NMR Studies of Hydroxy Protons of Asn- and Ser-Linked Disaccharides in Aqueous Solution. J. Carbohydr. Chem. 2000, 19, 1291–1303. 10.1080/07328300008544152. [DOI] [Google Scholar]
- Poppe L.; van Halbeek H. NMR Spectroscopy of Hydroxyl Protons in Supercooled Carbohydrates. Nat. Struct. Biol. 1994, 1, 215–216. 10.1038/nsb0494-215. [DOI] [PubMed] [Google Scholar]
- Adams B.; Lerner L. Observation of Hydroxyl Protons of Sucrose in Aqueous Solution: No Evidence for Persistent Intramolecular Hydrogen Bonds. J. Am. Chem. Soc. 1992, 114, 4827–4829. 10.1021/ja00038a055. [DOI] [Google Scholar]
- Sandström C.; Baumann H.; Kenne L. NMR Spectroscopy of Hydroxy Protons of 3,4-Disubstituted Methyl α-D-Galactopyranosides in Aqueous Solution. J. Chem. Soc., Perkin Trans. 1988, 2, 809–816. 10.1039/a708550h. [DOI] [Google Scholar]
- Batta G.; Kövér K. E. Heteronuclear Coupling Constants of Hydroxyl Protons in a Water Solution of Oligosaccharides: Trehalose and Sucrose. Carbohydr. Res. 1999, 320, 267–272. 10.1016/s0008-6215(99)00183-4. [DOI] [Google Scholar]
- Zhao H.; Pan Q.; Zhang W.; Carmichael I.; Serianni A. S. DFT and NMR Studies of 2JCOH, 3JHCOH, and 3JCCOH Spin-Couplings in Saccharides: C–O Torsional Bias and H-Bonding in Aqueous Solution. J. Org. Chem. 2007, 72, 7071–7082. 10.1021/jo0619884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.