

Abstract
Progress in cryo-electron microscopy has provided the potential for large-size protein structure determination. However, the success rate for solving multi-domain proteins remains low because of the difficulty in modelling inter-domain orientations. Here we developed domain enhanced modeling using cryo-electron microscopy (DEMO-EM), an automatic method to assemble multi-domain structures from cryo-electron microscopy maps through a progressive structural refinement procedure combining rigid-body domain fitting and flexible assembly simulations with deep-neural-network inter-domain distance profiles. The method was tested on a large-scale benchmark set of proteins containing up to 12 continuous and discontinuous domains with medium- to low-resolution density maps, where DEMO-EM produced models with correct inter-domain orientations (template modeling score (TM-score) >0.5) for 97% of cases and outperformed state-of-the-art methods. DEMO-EM was applied to the severe acute respiratory syndrome coronavirus 2 genome and generated models with average TM-score and root-mean-square deviation of 0.97 and 1.3 Å, respectively, with respect to the deposited structures. These results demonstrate an efficient pipeline that enables automated and reliable large-scale multi-domain protein structure modelling from cryo-electron microscopy maps.
Single-particle cryo-electron microscopy (cryo-EM) has emerged as a powerful means in modelling macromolecular structures at near-atomic resolution (3–5 Å)1. While high-resolution density maps enable the direct construction of atomic structures with limited conformation sampling using software programs traditionally used for X-ray crystallography2,3, the performance of these programs is poor when the resolution of the density map is relatively low (for example, >3 Å)4. For these challenging cases, a common approach is to fit a homologous structure to the density map, followed by atomic-level structural refinement5,6. However, the success of this approach depends strongly on the quality of the starting models, while for many proteins, no previously solved structures for homologous proteins are available.
This difficulty becomes particularly critical for multi-domain proteins consisting of multiple, structurally autonomous subunits. In fact, such multi-domain proteins are common in nature, and statistics has shown that more than two-thirds of prokaryote proteins and four-fifths of eukaryote proteins are composed of two or more domains,7 whereas only one-third of the structures in the Protein Data Bank (PDB)8 contain multiple domains (Supplementary Fig. 1a). Due to this lack of multi-domain templates and the difficulty of ab initio domain orientation modelling, the field of computational structural biology has traditionally focused on the study of individual domains, including the community-wide Critical Assessment of protein Structure Prediction experiments assessing the quality of protein structure predictions mainly on individual domains9. Therefore, although cryo-EM provides great potential for determining large-size proteins1 and there are a considerably greater portion of multi-domain proteins in the Electron Microscopy Data Bank (EMDB)10 than in the PDB (Supplementary Fig. 1b), it is usually difficult to apply homology modelling to create appropriate frameworks for density-map fitting and structural refinements of multi-domain proteins. These factors represent a significant challenge for multi-domain structural modelling based on cryo-EM maps, and currently only less than half of the cryo-EM density maps in the EMDB have atomic structures (Supplementary Fig. 1c). An additional barrier to large-scale cryo-EM structural modelling is that almost all structure fitting and refinement tools are not fully automated, even with given homologous models. For example, many approaches require human interventions in the initial model-to-map fitting, a procedure that often impacts significantly on the quality of the final models11. Hence, the development of advanced cryo-EM methods that could automatically yet reliably assemble multi-domain structures becomes increasingly urgent given the rapid progress of cryo-EM structural biology.
Here, we propose an automated approach, termed domain enhanced modeling using cryo-EM (DEMO-EM), to create accurate full-length structural models for multi-domain proteins from cryo-EM density maps. In addition to its unique dedication to multi-domain proteins, DEMO-EM has several novelties and advantages compared with many existing methods: (1) DEMO-EM integrates the single-domain structural modelling from iterative threading assembly refinement (I-TASSER)12 with deep-neural-network restraints to enhance the modelling accuracy for regions that lack density maps or have low-resolution data, (2) the hierarchical protocol starting with separate domain modelling followed by distance-profile-guided inter-domain structure reassembly simulation enables hybrid multi-domain protein structure prediction without requiring homologous full-length template structures and (3) the procedure is fully automated and can start from the protein sequence alone with no additional information or manual settings required. While a previous method, DEMO13, was proposed to model inter-domain orientations through rigid-body docking guided with analogous templates, a critical differentiating feature of DEMO-EM is its ability to model domain orientations directly from cryo-EM density maps without the need for templates and the efficiency of utilizing map data for atomic-level flexible structural refinement of the entire chain of multi-domain proteins. To systematically examine its strengths and weaknesses, DEMO-EM was tested on a large-scale benchmark dataset consisting of various numbers of continuous and discontinuous domains over synthesized and experimental density maps. The results demonstrate the advantages of DEMO-EM for cryo-EM-guided domain structure assembly and refinement compared with state-of-the-art approaches in the field.
Results
Method overview.
DEMO-EM uses cryo-EM density maps to obtain structural models for multi-domain proteins through a progressive domain assembly and refinement procedure (Fig. 1). The pipeline can start from either experimentally determined domain structures or amino acid sequences. When starting from amino acid sequences, DEMO-EM first constructs multiple sequence alignments from metagenome sequence databases and splits the query into domains using FUpred14 and ThreaDom15, and then generates an initial structural model for each domain using distance-guided iterative threading assembly refinement (D-I-TASSER)16, a new version of I-TASSER12, by incorporating deep-learning-based spatial restraints. Meanwhile, a deep convolutional neural-network predictor DomainDist is extended from our residue-contact prediction method TripletRes17 to predict inter-domain distance maps. To create full-length structural models, DEMO-EM performs a quasi-Newton search for the initial domain and cryo-EM density map fitting, followed by multiple steps of rigid-body domain structure assembly and atomic-level flexible structure refinements. The domain assembly and refinement simulations are primarily guided by the model-density correlations, assisted with a knowledge-based force field and the DomainDist inter-domain distance map predictions, where the final models are selected from the low-energy conformations and further refined by fragment-guided molecule dynamics (FG-MD) simulations18.
Fig. 1 |. Flowchart of DEMO-EM.
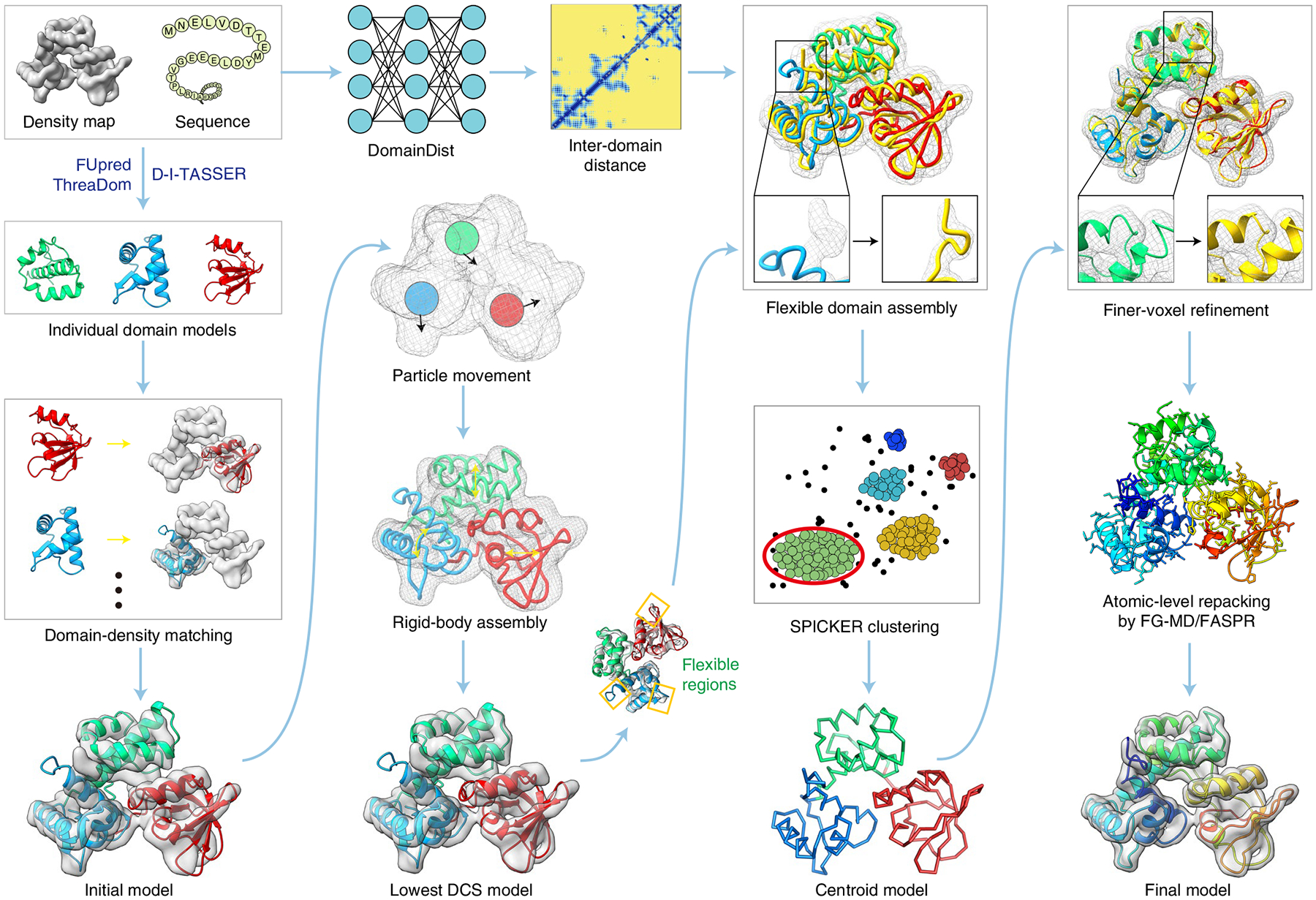
The flowchart is illustrated with a three-domain protein from the iron-dependent regulator of Mycobacterium tuberculosis (pDB ID 1fx7A). Starting from the query sequence, domain boundaries are first predicted by FUpred14 and ThreaDom15, and models of each domain are generated by D-I-TASSEr16. Meanwhile, inter-domain distances are predicted with a deep convolutional neural-network predictor DomainDist. Second, each of the domain models is independently fit to the density map by quasi-Newton searching. Third, the initial full-length models are optimized by a two-step rigid-body rEMC simulation to minimize the DCS) between the density map and full-length model (equation (1)). Fourth, the lowest DCS model selected from the rigid-body assembly simulations is refined by flexible assembly with atom-, segment- and domain-level refinements using rEMC simulation guided by the DCS, inter-domain distance profiles and a knowledge-based force field, with the resulting decoy conformations clustered by SpICKEr53 to obtain a centroid model. Finally, the flexible assembly simulation is performed again for the full-atomic model with constraints from centroid models adding to the energy, and the final model is created from the lowest-energy model after side-chain repacking with FASpr54 and FG-MD18.
Multi-domain structure construction from synthesized maps.
Table 1 and Fig. 2 present a summary of the DEMO-EM models assembled using experimental domain structures and domain models predicted by D-I-TASSER16, respectively, for a benchmark set of 357 non-redundant proteins (Supplementary Section 1), where the density maps are simulated according to the experimental structures by EMAN219 (Supplementary Section 2). When the experimentally determined domain structures are used, DEMO-EM was able to assemble nearly perfect full-length models for almost all the targets, resulting in an average template modeling score (TM-score) of 0.99 and root-mean-square deviation (RMSD) of 0.6 Å (Fig. 2a,b). Importantly, the individual domain structures were well folded in final full-length models with an average TM-score of 0.98 and average RMSD of 0.6 Å, despite the fact that the atomic structure of the full-length models is kept completely flexible in the domain assembly simulations. This suggests that the combination of the inherent DEMO-EM force field and the density-map data is capable of recognizing and maintaining correct folded domain structures. Here, the TM-score is a metric defined to evaluate the topological similarity between protein structures (Supplementary Section 3), taking values (0, 1], where a higher value indicates closer structural similarity20.
Table 1 |.
Results for the 357 test proteins using synthesized density maps
| MDFF | Rosetta | MAINMAST | DEMO-EM | |
|---|---|---|---|---|
| Experimental domain structure assembly | ||||
| TM-score | 0.86 (0.20) | 0.79 (0.23) | - | 0.99 (0.01) |
| RMSD (Å) | 7.1 (9.4) | 8.1 (9.9) | - | 0.6 (0.3) |
| Predicted domain model assembly | ||||
| TM-score | 0.53 (0.22) | 0.45 (0.22) | 0.35 (0.26) | 0.85 (0.17) |
| RMSD | 16.6 (8.1) | 21.2 (10.9) | 18.3 (8.6) | 5.9 (6.4) |
| TM-score (domain)a | 0.63 (0.22) | 0.48 (0.26) | 0.32 (0.25) | 0.83 (0.16) |
| RMSD (domain)b | 5.9 (3.8) | 9.3 (7.1) | 13.7 (6.9) | 3.9 (3.8) |
| Rama favoured (%)c | 75.8 (8.5) | 84.9 (7.1) | 38.5 (14.5) | 91.0 (4.1) |
| Rotamer outliers (%) | 7.1 (4.2) | 1.3 (3.4) | 41.3 (16.2) | 1.2 (1.0) |
| Clash score | 4.4 (4.9) | 36.6 (50.7) | 628.7 (611.7) | 3.3 (4.0) |
| MolProbity score | 2.35 (0.61) | 2.38 (0.58) | 5.17 (0.72) | 1.61 (0.57) |
| EMringer score | 0.32 (0.35) | 1.18 (0.91) | 1.14 (0.76) | 1.48 (0.98) |
| iFSC | 0.31 (0.19) | 0.34 (0.19) | 0.44 (0.16) | 0.67 (0.16) |
Values presented as average (s.d.) with the best result in each category highlighted in bold.
TM-score of individual domain models in full-length models.
rMSD of individual domains models in full-length models.
percentage of ‘ramachandran favoured’ residues.
Fig. 2 |. Results of proteins using synthesized density maps.
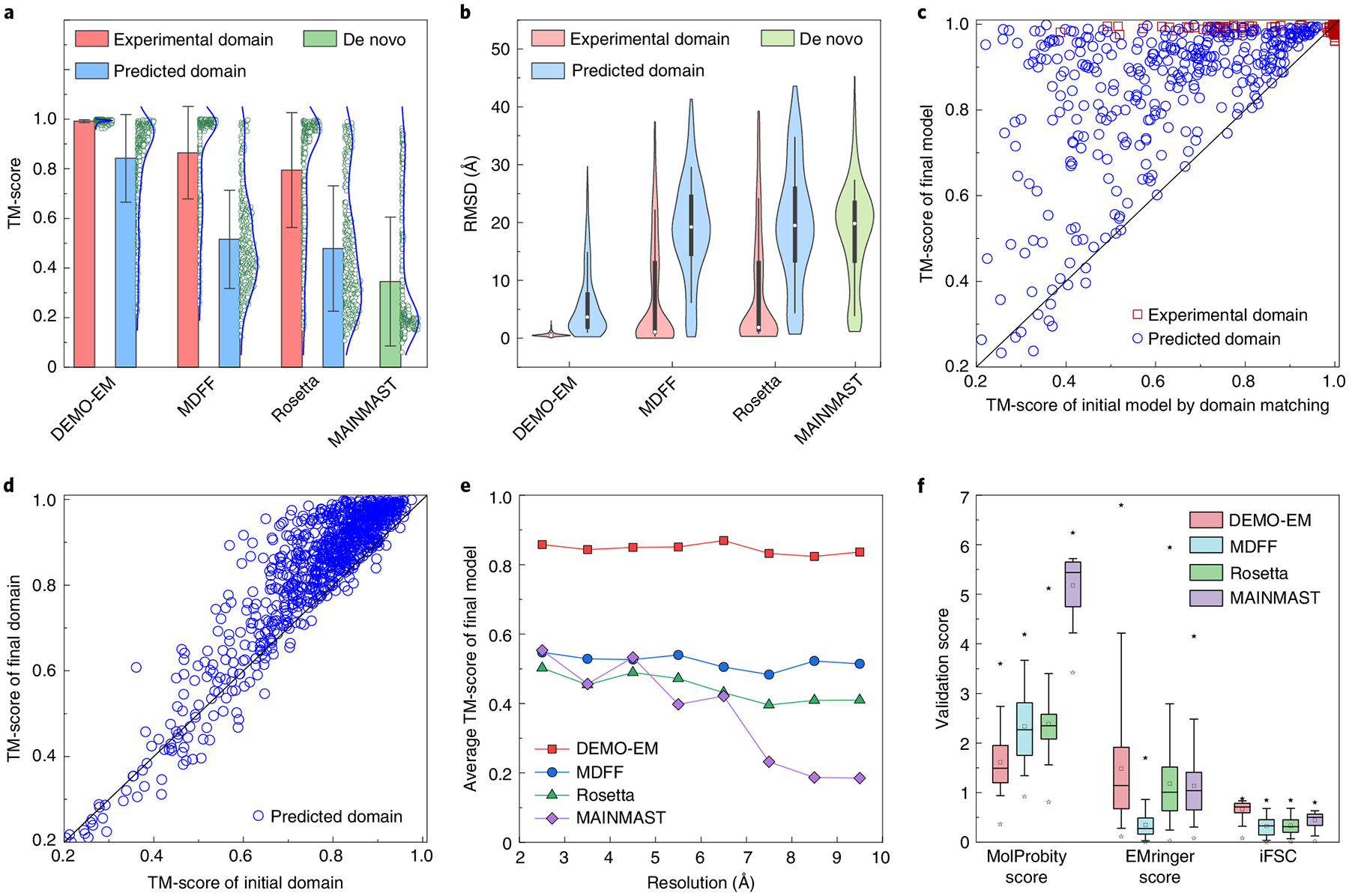
a, Mean ± s.d. (columns with error bars) and distribution (to the right of each column, n = 357 proteins) of the TM-score of the DEMO-EM, MDFF, rosetta and MAINMAST models. b, Boxplot and distribution of the rMSD for the DEMO-EM, MDFF, rosetta and MAINMAST models. The black box indicates the lower and upper quartile, the white dot in the box indicates the median, the whiskers show the 10th and 90th percentiles and the shape of the violin plot indicates the distribution. c, Head-to-head comparison between the TM-score of the initial models obtained by domain matching and that of the final models after rigid-body assembly, flexible assembly and refinement. d, Comparison between the TM-score of the individual domain models from D-I-TASSEr and that of the final full-length models obtained by DEMO-EM. e, The TM-score versus the resolution of the density map for the different methods. f, Boxplot of the validation scores of the models obtained from the different methods. The box represents the lower to upper quartiles, the horizontal line and square in the box represent the median and mean, respectively, the whiskers indicate the 5th and 95th percentiles while the solid and hollow star mark the maximum and minimum value, respectively.
When predicted domain models are used, local structure errors and incorrect domain models can negatively impact full-length model assembly simulations. Nevertheless, DEMO-EM successfully assembled full-length models with a correct global fold (that is, TM-score >0.5)21 for 94.1% of the test cases (Fig. 2a, blue histogram). Figure 2c presents a head-to-head TM-score comparison between the initial model obtained by matching predicted domains with the cryo-EM maps versus the final DEMO-EM model, showing that the TM-score of the final model was improved in nearly all test cases (with an average increase from 0.62 to 0.85, corresponding to P = 1.5 × 10−47 in Student’s t-test). Because a model with a high TM-score must achieve correct modelling of both individual domains and inter-domain orientations, the data in Fig. 2c indicate that DEMO-EM domain assembly simulations could significantly improve the inter-domain orientations. Since the domain structures are kept flexible in DEMO-EM, part of this increase in the TM-score of the full-length model may also result from an improvement in the quality of individual domain structures. To examine this, Fig. 2d compares the TM-score of the initial individual domains with that of the final models provided by DEMO-EM, revealing an improvement for the latter in 810 out of 890 individual domains. On average, the TM-score of individual domains increased from 0.77 to 0.83 (P = 1.3 × 10−22, Student’s t-test), indicating that the domain-level structural improvements brought about by DEMO-EM are statistically significant. The remaining 80 domain models for which the TM-score decreased after flexible assembly are studied in Supplementary Section 4. In addition, the performance of DEMO-EM on large proteins, cases with discontinuous domains and proteins with incorrect domain models is discussed in Supplementary Section 5.
Table 1 presents a comparison of the results obtained from the molecular dynamics flexible fitting (MDFF)5,22 and Rosetta23 models, which are widely used for modelling guided by cryo-EM density maps (Supplementary Section 6). Since both of these methods must start from full-length models, we built initial full-length models by fitting each domain model to density maps using Situs24, one of the best publicly available structure–density map programs. A quick Monte Carlo simulation procedure was also performed to rebuild the broken inter-domain linkers of the initial Situs models (Supplementary Section 6). Note that there are many different advanced MDFF protocols, such as cascade MDFF (cMDFF)5, resolution-exchange MDFF5, MultiMap25 and CryoFold26. In our experiments, we applied the direct MDFF and cMDFF protocols for proteins with cryo-EM density maps with resolution of ≥5 Å and <5 Å, respectively. As shown in Table 1, Fig. 2a,b and Supplementary Fig. 4a,b, DEMO-EM outperformed both MDFF and Rosetta by a margin, with the average TM-score of the full-length models with experimental domains being 15.1% and 25.3% higher than that of MDFF and Rosetta, respectively. The P value in Student’s t-test was 2.9 × 10−34 and 4.5 × 10−44, respectively, suggesting that the difference is statistically significant.
When predicted domains were used, the TM-score improvement when using DEMO-EM increases to 60.4% relative to MDFF and 88.9% to Rosetta, corresponding to Student’s t-test P values of 7.7 × 10−95 and 2.4 × 10−124, respectively. DEMO-EM also made more significant improvements in the domain-level structures. When starting from the predicted domains, DEMO-EM improved the TM-score of individual domains in 91.0% of cases (Fig. 2d), while MDFF and Rosetta did so in only 27.3% and 29.4% of cases, respectively (Supplementary Fig. 4d,e). We also compared DEMO-EM with a de novo method, MAINMAST27, for cryo-EM density map modelling. For full-length models, DEMO-EM achieved an average TM-score that was 142.8% higher than that of MAINMAST (Table 1 and Supplementary Fig. 4c), corresponding to a P value of 3.2 × 10−127 in Student’s t-test. DEMO-EM also obtained better domain models than MAINMAST, with a 159.3% higher average TM-score than that of MAINMAST, which corresponds to a Student’s t-test P value of 5.8 × 10−296.
Figure 2e shows the correlation between the map resolution and the TM-scores of the full-length models created by the different methods using predicted domain modelling or de novo modelling. The performance of DEMO-EM is not significantly affected by a reduction in the resolution of the density maps, retaining a TM-score above 0.8 throughout the resolution range. While the performance of MDFF and Rosetta decreased slightly when using lower-resolution maps, the average TM-score of DEMO-EM remained significantly higher than those of MDFF and Rosetta. In contrast, the performance of MAINMAST dropped sharply with a decrease in resolution. In addition, Table 1 and Supplementary Table 3 summarize the validation scores of the models created by using the different methods. Compared with the models generated by the three control methods, the DEMO-EM models achieve better Molprobity28 and EMringer29 scores (Fig. 2f), thus indicating that they have better model geometry and density fit at the side-chain level. The better global topology and local geometry thus allow DEMO-EM models to achieve an integrated Fourier Shell Correlation (iFSC)11 of 0.67, which is also higher than the values obtained by the control methods.
There are three reasons for the better performance of DEMO-EM over the control methods, including better initial model matching, hierarchical rigid-body domain assembly and deep-learning-guided flexible structure refinement (see Supplementary Section 7 for detailed analyses). Figure 3a summarizes the progress of the model accuracy at each step of DEMO-EM, compared with that of the two control methods. While MDFF and Rosetta start with initial models from Situs (with a TM-score of 0.53) and produce final models that are comparable to or even worse than the initial models, DEMO-EM builds better initial models and its TM-score increases at each of the subsequent structural assembly and refinement steps. Figure 3b,c also presents two examples illustrating the construction process in DEMO-EM, reinforcing its advantages for assembling multi-domain protein complex structures (see Supplementary Section 8 for detailed analyses).
Fig. 3 |. Representative examples showing the process of DEMO-EM.
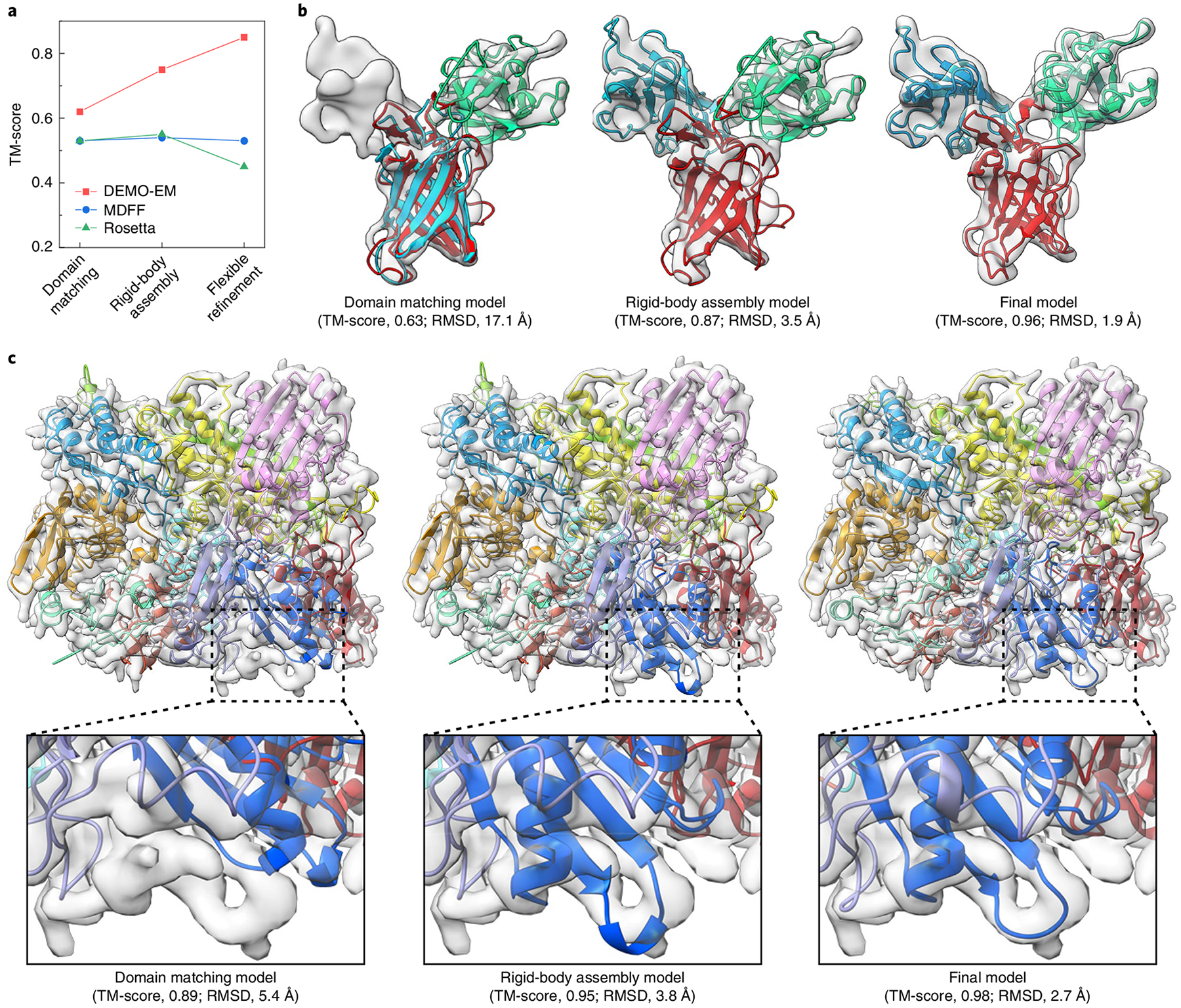
a, Summary of the evolution of the modelling accuracy along the process of the different programs. b,c, Examples of the DEMO-EM process for 1q25A, a three-domain protein, using a simulated density map with a resolution of 9.9 Å (b) and 2e1qC, a protein with ten domains (eight continuous and two discontinuous) using a simulated density map with a resolution of 5.3 Å (c). panels b and c show density maps (grey shadow) and DEMO-EM models (cartoons) with different colours indicating different domains, with enlargements below.
Assembly of structures from experimental density maps.
We further tested DEMO-EM on 51 cases with experimental density maps, with the domain boundary predicted by FUpred14 and ThreaDom15 and the individual domain structures modelled by D-I-TASSER (Supplementary Section 9). As shown in Fig. 4a, DEMO-EM did an acceptable job at domain boundary prediction, and the predicted number of domains is consistent with that determined by DomainParser for 43 of the 51 test proteins. In 82.4% of cases, the domain overlap rate was >80% compared with the DomainParser assignment on the target structure, resulting in an average normalized domain overlap (NDO) score of 0.91. The average TM-score of these domain models by D-I-TASSER was 0.78, with 96.3% of them having a correct fold with a TM-score >0.5 (Supplementary Fig. 9b). After the DEMO-EM assembly, the full-length models had an average TM-score of 0.88 (Fig. 4b) and an average RMSD of 3.2 Å (Table 2), with a correct global fold (TM-score >0.5) achieved in 98.0% of the cases. Meanwhile, the average TM-score of individual domains in the full-length models was increased from 0.78 to 0.84, with 90.2% of the domains being improved (Fig. 4c), again demonstrating the ability of DEMO-EM at the levels of both domain and full-length structure refinements. In addition, we also systematically study the impact of the domain assignment and map segmentation on the accuracy of the final model in Supplementary Section 10.
Fig. 4 |. Results for proteins using experimental density maps.
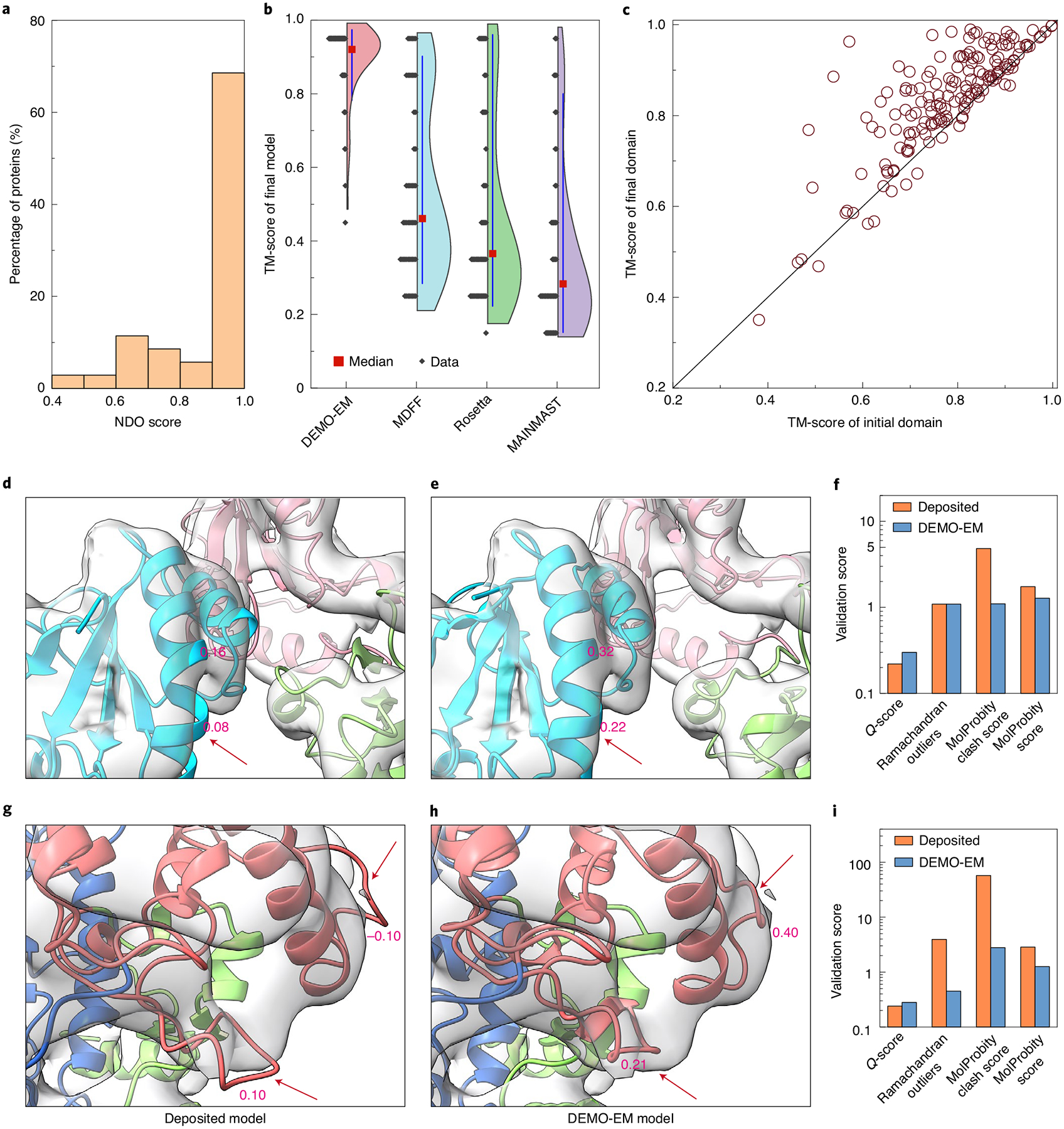
a, The distribution of the NDO score of the domain boundaries predicted by DEMO-EM for 51 proteins. b, The distribution of the TM-scores for full-length models constructed by DEMO-EM, MDFF, rosetta and MAINMAST (n = 51 proteins). The vertical lines represent the 10th to 90th percentile, the red square indicates the median, the shape of the violin plot shows the distribution and the diamond marks the TM-score corresponding to the distribution. c, Head-to-head TM-score comparison of the initial individual models by D-I-TASSEr and of the final full-length models by DEMO-EM. d,e, The model deposited in pDB (pDB ID 6eny) (d) and the model reconstructed by DEMO-EM (e) for the human pLC editing module, where different colours represent different domains, and the value is the average Q-score of the region. f, Model quality of 6eny evaluated by Q-score and Molprobity. g,h, The model deposited in pDB (pDB ID 5fj6) (f) and the model reproduced by DEMO-EM (g) for the p2 polymerase inside in vitro assembled bacteriophage phi6 polymerase complex. i, Model quality of 5fj6 assessed by Q-score and Molprobity.
Table 2 |.
Results for 51 proteins with experimental cryo-EM density maps
| MDFF | Rosetta | MAINMAST | DEMo-EM | |
|---|---|---|---|---|
| TM-score | 0.55 (0.24) | 0.47 (0.28) | 0.36 (0.24) | 0.88 (0.09) |
| RMSD (Å) | 17.7 (11.0) | 24.1 (16.6) | 23.0 (10.3) | 4.2 (3.2) |
| TM-score (domain)a | 0.56 (0.21) | 0.47 (0.26) | 0.42 (0.32) | 0.84 (0.13) |
| RMSD (domain) (Å)b | 7.2 (4.2) | 11.4 (10.0) | 12.0 (8.1) | 3.2 (2.8) |
| Rama favoured (%)c | 72.0 (7.4) | 88.1 (5.2) | 47.4 (28.8) | 86.1 (5.9) |
| Rotamer outliers (%) | 8.6 (3.3) | 0.5 (0.4) | 20.3 (21.4) | 3.9 (2.2) |
| Clash score | 3.9 (4.0) | 9.8 (7.1) | 821.5 (1,137.3) | 2.1 (0.2) |
| MolProbity score | 2.49 (0.41) | 2.02 (0.36) | 4.74 (0.68) | 1.66 (0.55) |
| EMringer score | 0.33 (0.31) | 1.08 (0.78) | 0.79 (0.69) | 1.45 (0.95) |
| iFSC | 0.30 (0.15) | 0.37 (0.20) | 0.35 (0.17) | 0.55 (0.23) |
Values presented as average (s.d.), with the best results in each category in bold.
TM-score of individual domain models in full-length models.
rMSD of individual domains models in full-length models.
percentage of ‘ramachandran favoured’ residues.
The average full-length TM-score (0.88) is slightly higher than that of the benchmark results on the synthesized density maps (0.85), which is probably due to the fact that this dataset contains a lower number of predicted domain models with incorrect folds (3.7%) than the former benchmark dataset (6.4%). Nevertheless, the TM-score of the individual domain models was improved by 7.7% after the flexible assembly, which is comparable to the former benchmark (7.8%). These results are largely consistent with the former benchmark data on synthesized density maps, which demonstrate the robustness of DEMO-EM, whose performance does not depend on the source of the density maps, that is, from synthesis or experiment. Furthermore, the average runtime required by the whole pipeline of DEMO-EM for all 51 test proteins is 8.15 h. Supplementary Fig. 10 shows the runtime of each protein, which increases nearly linearly with sequence length.
For comparison, Table 2 also presents the results obtained from MDFF and Rosetta when starting from the same set of predicted domain models with the initial conformation assembled by Situs and that by MAINMAST modelling. These data again show that DEMO-EM outperformed MDFF, Rosetta and MAINMAST, with the average TM-score of the full-length models being 60.0%, 87.2% and 144.4% higher than that of MDFF, Rosetta and MAINMST, respectively. These results are further confirmed by the head-to-head TM-score comparison shown in Supplementary Fig. 13, where DEMO-EM achieves a higher TM-score for nearly all the targets. When the resolution of the density maps decreases, the TM-scores of DEMO-EM, MDFF and Rosetta are not significantly affected, while the TM-score of MAINMAST drops obviously (Supplementary Fig. 14a). Furthermore, the final models constructed by DEMO-EM achieve higher quality in terms of the model geometry and the density fit, with better MolProbity score, EMringer score and iFSC (Supplementary Fig. 14b) than the control methods. Supplementary Fig. 14a also presents the modelling results of DeepTracer30, a deep-learning-based method for fast de novo protein complex structural modelling from high-resolution density maps. Although DeepTracer outperforms other control methods for the targets with high resolution (<4 Å), its performance is inferior to that of DEMO-EM in all resolution ranges. In particular, the TM-score of the DeepTracer models decreases rapidly when the resolution is worse than 4.5 Å, resulting in an overall average TM-score (0.33) that is 167% lower than that of DEMO-EM.
Figure 4d–i shows two representative examples with density maps taken from EMDB. First, Fig. 4d shows the model of the human PLC editing module deposited in the PDB (PDB ID 6enyD, with the density map from EMD-3906 in EMDB), which was created by fitting a homology model (from PDB ID 3f8uC) with the cryo-EM density map at a resolution of 5.8 Å (ref. 31) using FlexEM32 and Chimera33. Although the deposited model (Fig. 4d) shows close similarity to the DEMO-EM model (Fig. 4e) with a TM-score of 0.96 and RMSD of 1.7 Å, many regions of the deposited model are exposed to the outside of the density map (for example, the helix indicated by the arrow in Fig. 4d), which resulted in an iFSC of 0.64. In the DEMO-EM model, almost all these exposed regions were corrected, where the atom resolvability evaluated by the Q-score34 was consistently improved (see the Q-score comparisons of the two models in Supplementary Fig. 15c,d). Accordingly, the iFSC of the DEMO-EM model was improved to 0.66 (see the entire DEMO-EM model shown in Supplementary Fig. 15a). In addition, the DEMO-EM model has a better local geometry, with the MolProbity score being improved from 1.74 to 1.28 (Fig. 4f), and the side-chain was constructed in the DEMO-EM model while the deposited model contains only the backbone.
Figure 4g shows the deposited model of another example from the P2 polymerase inside in vitro assembled bacteriophage phi6 polymerase complex (PDB ID 5fj6A, with the density map from EMD-3186), which contains two continuous domains mediated by a discontinuous domain. The deposited model was produced by fitting a homology structure (PDB ID 1hhsA) with the density map at a resolution of 7.9 Å (ref. 35) using Chimera33 and Phenix36. Again, the DEMO-EM model is closely consistent with the deposited model, with a TM-score of 0.97 and RMSD of 1.5 Å (Supplementary Fig. 15b). As there are many significant noisy grid points in the experimental density map, some regions of the deposited model (for example, the loops indicated by arrows in Fig. 4g) were incorrectly modelled because they were not wrapped in the density map, which resulted in low Q-scores (−0.10 and 0.10, respectively). The model created by DEMO-EM (Fig. 4h) using the same density map data fixed all these local errors with an improvement in the Q-score to 0.40 and 0.21, respectively, at these two exposed sites. Overall, the average Q-score of the DEMO-EM model increased from 0.25 to 0.29, where 75.5% atoms had an improved Q-score (see Supplementary Figs. 15e and 15f for the Q-score of each atom in the deposited model and the DEMO-EM model, respectively). Furthermore, the DEMO-EM model has a better model geometry compared with the deposited model, with the MolProbity score being improved from 2.85 to 1.26 and the EMringer score from −0.28 to −0.13 (Fig. 4i). Interestingly, despite the improved Q-score and local model quality, the iFSC score of the DEMO-EM model is decreased slightly (from 0.35 to 0.31) compared with the deposited model. As shown in Supplementary Fig. 15e, many regions that were fit to the map had negative Q-scores in the deposited model, suggesting that this higher iFSC score might be a result of overfitting.
Finally, we compared DEMO-EM with the most advanced end-to-end deep-learning structure prediction method, AlphaFold237, on all 51 cases. As shown in Supplementary Table 6, although the individual domains predicted by AlphaFold2 have a higher TM-score (0.89) than that of DEMO-EM (0.84), which is probably because of the lower quality of the domain models built by D-I-TASSER, the quality of the overall full-length models built by DEMO-EM (with a TM-score of 0.88) is better than that achieved by AlphaFold2 (with a TM-score of 0.84), with DEMO-EM obtaining a higher TM-score than AlphaFold2 on 28 out of 51 proteins. We also fed the same full-length models constructed by AlphaFold2 into MDFF, Rosetta and DEMO-EM to examine the performance of the flexible assembly and refinement process. All the methods improved the initial full-length model, showing the usefulness of cryo-EM data even for the best-predicted models. DEMO-EM obtained a clearly higher average TM-score (0.93) than MDFF (0.89) or Rosetta (0.88), again demonstrating the effectiveness of the DEMO-EM refinement simulations (Supplementary Table 6). Furthermore, probably because of the inaccurate local cryo-EM density restraints from the low-resolution density map, the average TM-score of the individual domain models (0.89) was decreased after refinement by MDFF (0.86) and Rosetta (0.85), while only DEMO-EM slightly improved the individual domain quality, resulting in an average TM-score of 0.90. These results again demonstrate the ability of DEMO-EM at the levels of both the domain and full-length structure refinements.
Application to structural modelling of the SARS-CoV-2 genome.
Extended Data Fig. 1 shows the full-length structural models constructed by DEMO-EM for all six severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) proteins38 with cryo-EM data deposited in EMDB. Based on the FUpred and ThreaDom predictions, five proteins contain multiple domains and two of them include discontinuous domains (Supplementary Table 7). Compared with the deposited models, many of which contained missed residues due to the loss of density data, the DEMO-EM models have an average TM-score and RMSD of 0.97 and 1.3 Å, respectively, for the regions where the deposited models have structure. Furthermore, the DEMO-EM models exhibit better model geometry and local quality as measured by different validation scores (Supplementary Table 8 and Supplementary Section 11).
The X-ray structure of the receptor binding domain of the spike protein, which SARS-CoV-2 uses to bind angiotensin-converting enzyme 2 to invade host cells, was released recently (PDB ID 7bz5A)39. Extended Data Fig. 1g shows a comparison of the structural model built by DEMO-EM versus the released X-ray structure, where the DEMO-EM model has a TM-score of 0.97 and RMSD of 0.92 Å, slightly better than those of the deposited model (with a TM-score of 0.96 and RMSD of 0.97 Å). Furthermore, we also constructed the complex structures of these SARS-CoV-2 proteins using a simply extended version of DEMO-EM in which each chain is treated as a virtual ‘domain’ but the connectivity requirement between the virtual ‘domains’ is ignored. As shown in Supplementary Fig. 17, the complex models achieve an average TM-score of 0.97 and an RMSD of 1.1 Å versus the experimental structures, showing the feasibility of extending DEMO-EM for protein–protein complex structural modelling. A systematic test of more advanced cryo-EM-based protein complex structural modelling strategies will be published elsewhere. These results suggest that although DEMO-EM was designed and mainly tested for multi-domain proteins, it can be used to build models of single- and multi-domain proteins and protein–protein complexes.
Discussion
Due to the scarcity of multi-domain template structures in the PDB, automated determination of multi-domain protein structures from cryo-EM density becomes a significant challenge, as most approaches in the community rely on fitting and refinement of homology models. To address this issue, we developed a method, DEMO-EM, dedicated to structure assembly of multi-domain proteins from cryo-EM density maps. Without relying on global homologous templates, the method integrates single-domain modelling and deep residual network learning techniques with progressive rigid-body and flexible Monte Carlo simulations into a hierarchical pipeline that is ready for automated and large-scale multi-domain protein structure prediction.
The good performance of DEMO-EM stems partly from its ability for quick and reliably framework construction, which is enabled by the unique single-domain structural modelling from D-I-TASSER and the coarse-grained density-map space enumeration driven by the quasi-Newton search process. Next, the domain-level rigid-body assembly simulation is capable of correcting domain positions and inter-domain orientations by combining density map restraints with inter-domain potentials, even when domain poses are occasionally incorrectly assigned in the initial frameworks. Finally, the atomic-level flexible structural assembly simulations couple density-map correlations with deep-learning-based inter-domain distance profiles, which helps to fine-tune local structural packing and inter-domain orientations simultaneously and resulted in consistent improvement of both local and global structures. Note that DEMO-EM does not rely on D-I-TASSER, and domain structures constructed by any methods could be assembled by DEMO-EM.
Despite the promising domain assembly results, the applicability and accuracy of DEMO-EM could be further improved in several aspects. First, most of the density maps in our tests are segmented from the full density map by Chimera33. Although manual segmentation is often straightforward, the automatic map segmentation techniques (for example, the methods in Phenix and MAINMAST) could be introduced into DEMO-EM because the segmented map is helpful to improve the accuracy and reduce the computational time. Second, all the individual domain models are directly produced by D-I-TASSER without guidance from the density data. An incorrect initial domain model may lead to a poor final model because it will affect the ability of the algorithm to identify correct poses for initial framework constructions. Therefore, combining the restraints from density data with potentials for individual domain model generation will be helpful to improve the accuracy of the final models. Studies along these lines are in progress.
Methods
DEMO-EM is a hierarchical approach to multi-domain protein structure determination based on cryo-EM maps, consisting of four consecutive steps: (1) determining domain boundaries and modelling individual domains, (2) matching domain models with a density map for the initial framework generation, (3) rigid-body domain structure assembly for domain position and orientation optimization and (4) flexible structure assembly and refinement simulation of full-length structural models (Fig. 1).
Domain parsing and individual domain structure folding.
Starting from the query amino acid sequence, we first run LOMETS40 to create multiple template alignments from the PDB, where ThreaDom15,41 is employed to predict the domain boundary according to the domain conservation score. If the protein is defined as an ‘easy’ target by LOMETS and the alignment coverage is >95%, the domain definition predicted by ThreaDom is applied. Otherwise, the domain boundary is predicted through FUpred14 by maximizing the number of intradomain contacts and minimizing the number of inter-domain contacts on the contact map predicted by a deep-learning-based neural network program, ResPRE42. Next, the structural model of each domain is generated using D-I-TASSER16, which is a version of I-TASSER12 updated by incorporating the interresidue contact and distance maps and hydrogen-boding potentials predicted by deep learning into the iterative threading assembly simulations. According to the sequence of each domain, D-I-TASSER firstly constructs the multiple sequence alignments (MSAs) through DeepMSA43 by iteratively searching the whole-genome and metagenome sequence databases. The top MSAs are then selected based on the contacts predicted by TripletRes44 and inputted into the deep residual neural network-based predictor extended from ResPre42 and TripletRes44 to predict the distance maps, hydrogen-bonding networks and torsion angles. These predicted restraints are integrated into the I-TASSER force field to guide the replica-exchange Monte Carlo (REMC) simulation, and the final model is clustered by SPICKER and refined by FG-MD. For discontinuous domains that contain two or more segments from separate regions of the query sequence, the domain models are obtained by sequentially connecting the sequences of all segments.
Deep neural network-based inter-domain distance prediction.
To help guide the domain orientation assembly, an inter-domain distance map is predicted by a deep residual neural-network algorithm, DomainDist, whose architecture is outlined in Supplementary Fig. 18. DomainDist is an extension of TripletRes17, which was originally developed to predict interresidue contact maps based on a triplet of coevolutionary matrices but is extended here to predict the probability of interresidue distance within 36 bins in the range of 2–20 Å. The DomainDist program was trained on a non-redundant dataset of 26,151 proteins collected from the PDB, where the MSA for each protein was constructed using HHblits45 searching against the Uniclust30 sequence database46. In addition to the two-dimensional (2D) coevolutionary features employed in TripletRes, three one-dimensional (1D) features, including a hidden Markov model, one-hot representation of sequence and field parameters of Potts model, were adopted and tiled to two dimensions and concatenated with the 2D coevolutionary features. The neural network structure was designed following convolutional strategies, using ResNet basic blocks47. The neural network model was trained by the Adam optimization algorithm to marginally minimize the cross-entropy loss. Both intra- and inter-domain distance information was considered during the training, although only inter-domain distance information was considered by DEMO-EM.
Quasi-Newton-based matching of domain and cryo-EM density map.
For each individual domain model from D-I-TASSER, we used limited-memory Broyden– Fletcher–Goldfarb–Shanno (L-BFGS), a quasi-Newton optimization algorithm with six-dimensional (6D) translation–rotation degrees of freedom, to identify the best location and orientation of the domain with the highest correlation with the density map (Supplementary Fig. 19a). Since L-BFGS is a local optimization method whose results depend on the initial solutions, we started the L-BFGS simulation from multiple initial positions (translation vector) and orientations (rotation angle) by enumerating all combinations of Euler angles (ϕ, θ and ψ) with a step size of Srot_ang across the density-map space (Supplementary Fig. 19b). For a given domain pose, a density correlation score (DCS) calculated as
| (1) |
is used to guide the L-BFGS simulations. Here, Nvol is the number of voxels (grid is the number of voxels (grid points) in the density map and ρEM (vi) is the experimental density of the ith voxel vi The density probed from the decoy structure is calculated as
where xj is the position of the jth atom in the decoy, m is its mass and R is the resolution of the density map48. To speed up the matching process, a density map with voxel size of 2 Å interpolated from the original density map is employed. After the L-BFGS simulation, all poses for each domain with DCS <0.5 (or the top ten poses when more than ten poses have DCS of <0.5) are pooled and combined with the top poses of other domains to form the initial models of the full-length models. The combination is made by permutating the initial poses of all the domains and allows for domain overlaps, where the top 30 full-length models with the lowest DCS are selected for the next step of rigid-body domain matching and assembly. Here, Srot_ang is set to 30°, which is well within the large basin of attraction of the search and can balance precision and efficiency according to the recommendation in Situs24.
Rigid-body domain assembly.
Two rounds of rigid-body domain assembly simulations are performed to optimize the domain positions and orientations. In the first round, the domains are treated as particles and a quick REMC simulation is carried out to adjust the positions of the individual domains based on the global model-density correlations. In this step, the energy function contains only DCS (equation (1)), where the movements include rigid-body translation and rotation around each domain’s centre of mass (Supplementary Fig. 20a,b). Note here that the DCS is calculated for the full chain model, which should lead to a more optimal model result compared with the previous step where the optimization was based on the DCS of individual domains. The density map with a voxel size of 3 Å interpolated from the original map is applied to reduce the computational cost. Thirty replicas are sampled in parallel, with the temperature ranging from 0.1 to 15, and a global swap movement between two neighbouring replicas is performed for every 200 Monte Carlo movements. The simulation is terminated when the number of swaps reaches 20 × Ndom, where Ndom is the number of domains. The top 30 models according to the DCS are selected for the next round.
The second round of rigid-body REMC simulation is applied to fine-tune the domain poses with a more detailed energy force field as defined in equations (2)–(5), where a more elaborate density map with a voxel size of 2 Å is interpolated from the original density map for the assembly. Besides the translation and rotation movements used in the first round, three new movements are added (Supplementary Fig. 20c–e), including self-rotation around the N-to-C axis of each domain, translation along the neighbouring domains in the sequence and pose exchange between two domains with similar structures (that is, with TM-score ≥ 0.75) according to TM-align49, which is designed to reduce the case where domains with similar topology are swapped in their initial positions. a similar parameter setting as the first round is employed for the REMC simulation, but the top 40 models according to the DCS are selected for the next step.
Energy function for rigid-body simulation.
Conformations in the rigid-body assembly are assessed by using an energy function with four terms
| (2) |
where the first term is the density correlation score and is defined as in equation (1), but here used for the full-length model.
The second term is the radius-of-gyration restraint, defined as
| (3) |
where Rgdecoy is the radius of gyration of the decoy structure, and Rgmax and Rgmin are the maximum and minimum estimated radius of gyration, respectively. The former is calculated as from the voxels with density ≥0.05 after normalizing the density values to the range of 0–1, where is the centre point of these voxels. Rgmin = 2.849L0.319 (where L is the query sequence length) is the statistical radius of gyration based on the known multi-domain protein models in the PDB, which has a Pearson correlation coefficient of 0.995 with real values (Supplementary Fig. 21).
The third term is the domain boundary connectivity, which is designed to constrain the connectivity of two neighbouring domains along the sequence (m < n) and is calculated as
where bmn is the Cα atom distance between the C-terminal residue of the mth domain and the N-terminal of the nth domain. For the case including discontinuous domains, bmn = (d1 + d2)/2 is the average of two linker distances connecting the continuous domain with the discontinuous segments (Supplementary Fig. 22). b0 = 3.8 Å is the standard distance between neighbouring Cα atoms.
The last term describes steric clashes and penalizes domain pairs occupying the same space, being defined as
| (5) |
where Lm and Ln represent the sequence length of the mth and nth domain, respectively. is the distance between the ith Cα atom of the mth domain and the jth Cα atom of the nth domain in the decoy structure. dcut = 3.75 Å is the distance cutoff to define a clash.
The weighting factors in Erigid are optimized based on a training set of 425 proteins that has sequence identity <30% with the test proteins, by maximizing the correlation between the total energy and RMSD of the decoy models with respect to the native using the differential evolution algorithm50,51. This resulted in values of wdcs = 300, wrg = 1.13, wbc = 0.55 and wsc = 0.91.
Atomic-level flexible domain assembly and refinement.
The process of flexible domain assembly and refinement contains two stages of simulations with progressive voxel resolutions and sampling focuses. In the first stage, six different movements are implemented (Supplementary Fig. 23): (1) LMProt52 perturbation, (2) segment rotation around the axis connecting two terminus, (3) conformational shift of segments along the sequence, (4) rigid-body segment translation, (5) rigid-body tail rotation and (6) rigid-body domain-level translation and rotation. To enhance the efficiency, a nine-residue sliding window is used to determine which region needs more aggressive conformation sampling, where a local score (LCi) for the sliding window of the centre residue (i) is computed as the average correlation coefficient between the nine-residue fragment and the entire density map. The probability for the ith residue to be selected for movement is set as
| (6) |
where LCmax and LCmin (= 0.05) represent the maximum and minimum local score, respectively. As illustrated in Supplementary Fig. 24, the setting in equation (6) helps ensure that the residues that are poorly correlated with the density map can receive more sampling than others. An atomic-level force field (equations (7)–(13)) is designed to guide the REMC simulation at this stage, where a density map with voxel size of 3 Å interpolated from the original density map is applied to reduce the computation cost and the DCS is calculated based on backbone atoms. Similarly, 40 replicas with temperature ranging from 0.01 to 15 are sampled in parallel. The global swap movement between two neighbouring replicas is performed for every movements, where the simulation stops when the number of swaps reaches 200. All accepted decoys in the simulation are clustered by SPICKER53, and the centroid model in the first cluster is selected as a reference model for the second stage.
In the second stage, a finer density map with voxel size of 2 Å is implemented with the DCS computed on all atoms. In addition, all residues have equal probability to be selected for movement and sampling. The REMC simulation is guided by the same force field as defined in equations (7)–(13), but the reference model in equation (10) is replaced by the centroid structure of the first cluster determined by SPICKER in the first stage. The simulation is terminated when the number of swaps reaches 100. The lowest-energy decoy is selected to construct the final model, with the side-chain atoms repacked by FASPR54 followed by FG-MD18 refinement.
DEMO-EM force field for flexible assembly simulation.
The flexible domain assembly simulations are implemented at a semi-atomic level, with each residue represented by N, Cα, C, O, Cβ, H and side-chain centre of mass (SC). Among the seven modelling units, only the three backbone atoms (N, Cα and C) have coordinates determined directly in conformation sampling, while the other four are determined based on their positions relative to the three backbone atoms using the parameters presented in Supplementary Table 9. The simulations are guided by a composite force field consisting of seven energy terms
| (7) |
The first term accounts for the density correlation, having the same form as equation (1) but calculated for the full-length model.
The second term is the inter-domain Cβ distance map as predicted by DomainDist
| (8) |
where is the distance between the ith Cβ(Cα for glycine) atom in the mth domain and jth Cβ atom in the nth domain, is the predicted probability of the distance located in the kth distance bin and ε = 1 × 10−4 is the pseudo count to offset low-probability bins. In the calculation, we only consider atom pairs with probability peak located in [2 Å, 20 Å], excluding those atom pairs with predicted probabilities >0.5 in the last bin [>20 Å], which represents a low prediction confidence in [2 Å, 20 Å].
The third term accounts for torsion angle variations by
| (9) |
where ϕi and ψi represent the backbone torsion angle pair of the ith residue, Ai is the amino acid type of the ith residue, Si is the secondary structure type of the ith residue as predicted by PSSpred55 and P(ϕi, ψi|Ai, Si) is the conditional probability calculated based on the Ramachandran map of 6,023 high-resolution protein structures culled from the PDB using the PISCES server56 based on a resolution cutoff of 1.8 Å, identity cutoff of 25% and R-factor cutoff of 0.25.
The fourth term is the domain structure restraint to prevent topologies of individual domains deviating too far from the initial structures generated by D-I-TASSER
| (10) |
where Lm is the sequence length of the mth domain, xi,m represents the ith Cα atom in the mth domain of the decoy after superposing the domain onto the reference model by D-I-TASSER and is the corresponding atom in the reference model.
The fifth term describes the excluded volume interaction and is defined as
| (11) |
where dij is the distance between the ith and jth atoms from different residues and σij is the sum of the van der Waals radius of the atom pairs taken from QUARK57,58 (Supplementary Table 10).
The sixth term is the hydrogen bonding extended from QUARK57,58. As shown in Supplementary Fig. 25, only backbone H-bonds between residues (i and j) are considered, where four geometric features, that is, the distance between Oi and Hj (D(Oi, Hj)), the internal angle between Ci, Oi and Hj (A(Ci, Oi, Hj)), the internal angle between Oi, Hj and Nj (A(Oi, Hj, Nj)) and the torsion angle between Ci, Oi, Hj and Nj (T(Ci, Oi, Hj, Nj)), are selected to evaluate the bonding. We consider four types of hydrogen bonds (T1: helix, j = i + 4; T2: helix, j = i + 3; T3: parallel β-sheets; and T4: antiparallel β-sheets). The energy term of a single backbone hydrogen bond is thus calculated as
| (12) |
where Tk represents the kth type of hydrogen bond, fl (i, j) denotes the lth feature of the decoy structure and μkl and δkl are the mean and standard deviation of the lth feature in the kth-type hydrogen bond, which were precalculated from the high-resolution PDB structures and are listed in Supplementary Table 11.
The last term is the generic side-chain-atom contact potential and is used to evaluate the contacts between SC in one residue (i) and N, Cα, C, O, Cβ and SC atoms in another residue (j) as
| (13) |
where Ai (or Aj) is the amino acid type of residue i (or j), Mi (or Mj) represents the atom type of the ith (or jth) residue, dij is the distance between the SC of the ith residue and the Mj atom of the jth residue and Uplo(Ai, Aj, Mi, Mj, dij) is the corresponding polarity potential precalculated from 6,500 non-redundant high-resolution PDB structures (https://zhanggroup.org/DEMO-EM/potential.html).
Similarly, the weighting parameters in equation (7) are determined by maximizing the correlation between the total energy and RMSD of the structure decoys of the 425 training proteins. This results in values of wdcs = 320, wta = 0.3, wdr = 1.5, wdt = 0.15, wev = 0.1, whb = 0.05 and wgsc = 0.1.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings and conclusions of this study are available within the paper and its Supplementary Information. The experimental domain models together with the simulated density maps used for training and testing, the models constructed by DEMO-EM from experimental cryo-EM density maps and the models of the SARS-CoV-2 coronavirus genome built by DEMO-EM are available at https://zhanggroup.org/DEMO-EM/. The full experimental cryo-EM density maps can be downloaded from EMDB (http://www.emdataresource.org/) using the code provided in Supplementary Table 5. All data are also available at Zenodo59. Source data for Tables 1–2, Figs. 2–4 and Extended data Fig. 1 are provided with this paper.
Extended Data
Extended Data Fig. 1 |. Overlay of structural models by DEMO-EM on the cryo-EM density maps for the six proteins in SARS-CoV-2 genome.
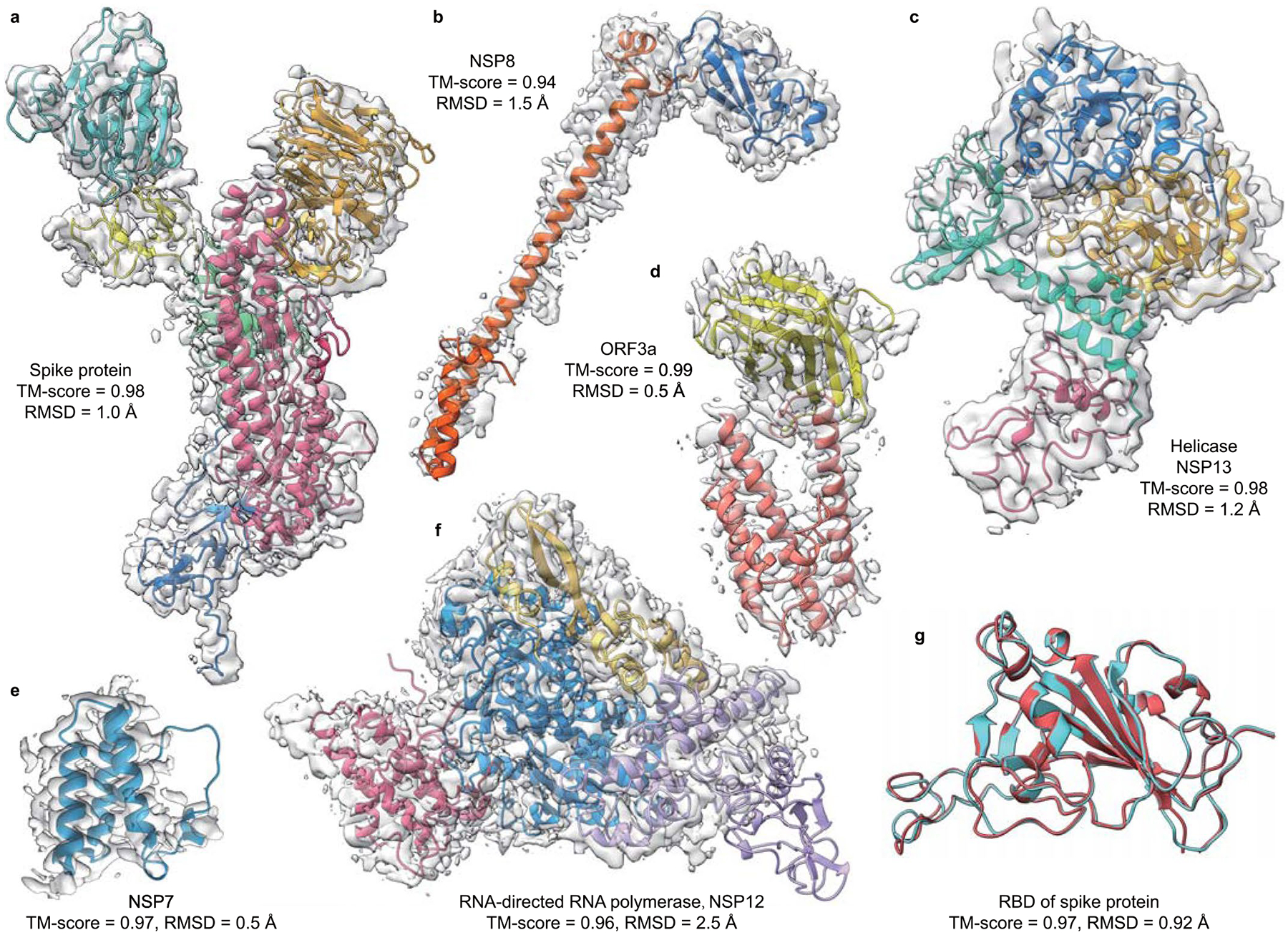
(a) Spike protein (density map from EMD-21375). (b) NSp8 (EMD-11007). (c) Helicase/NSp13 (EMD-22160). (d) OrF3a (EMD-22136). (e) NSp7 (EMD-11007). (f) rNA-directed rNA polymerase/NSp12 (EMD-11007). (g) Comparison of the Spike rBD domain by DEMO-EM (cyan) with the X-ray structure (red, pDB 7bz5A).
Supplementary Material
Acknowledgements
We thank E. Ramírez-Aportela for helpful discussion on the FSC-Q calculation and C. Peng for helping us install the FSC-Q software. This work is supported in part by the National Institute of General Medical Sciences (GM136422 and S10OD026825 to Y.Z.), National Institute of Allergy and Infectious Diseases (AI134678 to Y.Z.), National Science Foundation (IIS1901191 and DBI2030790 to Y.Z.), National Nature Science Foundation of China (62173304 and 61773346 to G.Z.) and Key Project of Zhejiang Provincial Natural Science Foundation of China (LZ20F030002 to G.Z.). This work used the Extreme Science and Engineering Discovery Environment (XSEDE)60, which is supported by the National Science Foundation (ACI1548562).
Footnotes
Code availability
The source code is freely available for academic use at https://zhanggroup.org/DEMO-EM/ and Zenodo59.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s43588-022-00232-1.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43588-022-00232-1.
References
- 1.Kuhlbrandt W The resolution revolution. Science 343, 1443–1444 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Cowtan K The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D 62, 1002–1011 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glaeser RM How good can cryo-EM become? Nat. Methods 13, 28–32 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Singharoy A et al. Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps. eLife 5, e16105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang B, Zhang X, Pearce R, Shen H-B & Zhang Y A new protocol for atomic-level protein structure modeling and refinement using low-to-medium resolution cryo-EM density maps. J. Mol. Biol 432, 5365–5377 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chothia C, Gough J, Vogel C & Teichmann SA Evolution of the protein repertoire. Science 300, 1701–1703 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Bernstein FC et al. The Protein Data Bank: a computer‐based archival file for macromolecular structures. Eur. J. Biochem 80, 319–324 (1977). [DOI] [PubMed] [Google Scholar]
- 9.Kinch LN, Kryshtafovych A, Monastyrskyy B & Grishin NV CASP13 target classification into tertiary structure prediction categories. Proteins 87, 1021–1036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawson CL et al. EMDataBank.org: unified data resource for CryoEM. Nucleic Acids Res. 39, D456–D464 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiMaio F et al. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J et al. The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou XG, Hu J, Zhang CX, Zhang GJ & Zhang Y Assembling multidomain protein structures through analogous global structural alignments. Proc. Natl Acad. Sci. U. S. A 116, 15930–15938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng W et al. FUpred: detecting protein domains through deep-learning based contact map prediction. Bioinformatics 36, 3749–3757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y et al. ThreaDomEx: a unified platform for predicting continuous and discontinuous protein domains by multiple-threading and segment assembly. Nucleic Acids Res. 45, W400–W407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng W et al. Protein structure prediction using deep learning distance and hydrogen‐bonding restraints in CASP14. Proteins 89, 1734–1751 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y et al. Deducing high-accuracy protein contact-maps from a triplet of coevolutionary matrices through deep residual convolutional networks. PLoS Comput. Biol 17, e1008865 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Liang Y & Zhang Y Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 19, 1784–1795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang G et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y & Skolnick J Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Xu J & Zhang Y How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26, 889–895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trabuco LG, Villa E, Mitra K, Frank J & Schulten K Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang RY-R et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chacón P & Wriggers W Multi-resolution contour-based fitting of macromolecular structures. J. Mol. Biol 317, 375–384 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Vant JW et al. Data-guided multi-map variables for ensemble refinement of molecular movies. J. Chem. Phys 153, 214102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shekhar M et al. CryoFold: determining protein structures and data-guided ensembles from cryo-EM density maps. Matter 4, 3195–3216 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terashi G & Kihara D De novo main-chain modeling for EM maps using MAINMAST. Nat. Commun 9, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barad BA et al. EMRinger: side chain–directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfab J, Phan NM & Si D DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes. Proc. Natl Acad. Sci. USA 118, e2017525118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blees A et al. Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Topf M et al. Protein structure fitting and refinement guided by cryo-EM density. Structure 16, 295–307 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pettersen EF et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Pintilie G et al. Measurement of atom resolvability in cryo-EM maps with Q-scores. Nat. Methods 17, 328–334 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilca SL et al. Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun 6, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou P et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng W et al. LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res. 47, W429–W436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue Z, Xu D, Wang Y & Zhang Y ThreaDom: extracting protein domain boundary information from multiple threading alignments. Bioinformatics 29, i247–i256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Hu J, Zhang C, Yu D-J & Zhang Y ResPRE: high-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics 35, 4647–4655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C, Zheng W, Mortuza S, Li Y & Zhang Y DeepMSA: constructing deep multiple sequence alignment to improve contact prediction and fold-recognition for distant-homology proteins. Bioinformatics 36, 2105–2112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Zhang C, Bell EW, Yu DJ & Zhang Y Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13. Proteins 87, 1082–1091 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Remmert M, Biegert A, Hauser A & Söding J HHblits: lightning-fast iterative protein sequence searching by HMM–HMM alignment. Nat. Methods 9, 173–175 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Mirdita M et al. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He K, Zhang X, Ren S & Sun J Deep residual learning for image recognition. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 770–778 (IEEE, 2016). [Google Scholar]
- 48.DiMaio F, Tyka MD, Baker ML, Chiu W & Baker D Refinement of protein structures into low-resolution density maps using rosetta. J. Mol. Biol 392, 181–190 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y & Skolnick J TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storn R & Price K Differential evolution–a simple and efficient heuristic for global optimization over continuous spaces. J. Global Optim 11, 341–359 (1997). [Google Scholar]
- 51.Zhou XG, Peng CX, Liu J, Zhang Y & Zhang GJ Underestimation-assisted global-local cooperative differential evolution and the application to protein structure prediction. IEEE Trans. Evol. Comput 24, 536–550 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.da Silva RA, Degrève L & Caliri A LMProt: an efficient algorithm for Monte Carlo sampling of protein conformational space. Biophys. J 87, 1567–1577 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y & Skolnick J SPICKER: a clustering approach to identify near‐native protein folds. J. Comput. Chem 25, 865–871 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Huang X, Pearce R & Zhang Y FASPR: an open-source tool for fast and accurate protein side-chain packing. Bioinformatics 36, 3758–3765 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan R, Xu D, Yang J, Walker S & Zhang Y A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci. Rep 3, 2619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang G & Dunbrack RL Jr PISCES: a protein sequence culling server. Bioinformatics 19, 1589–1591 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Xu D & Zhang Y Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mortuza S et al. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions. Nat. Commun 12, 5011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou X et al. Source code and data for the paper ‘Progressive assembly of multi-domain protein structures from cryo-EM density maps’. Zenodo https://zenodo.org/record/6363839 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Towns J et al. XSEDE: accelerating scientific discovery. Comput. Sci. Eng 16, 62–74 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings and conclusions of this study are available within the paper and its Supplementary Information. The experimental domain models together with the simulated density maps used for training and testing, the models constructed by DEMO-EM from experimental cryo-EM density maps and the models of the SARS-CoV-2 coronavirus genome built by DEMO-EM are available at https://zhanggroup.org/DEMO-EM/. The full experimental cryo-EM density maps can be downloaded from EMDB (http://www.emdataresource.org/) using the code provided in Supplementary Table 5. All data are also available at Zenodo59. Source data for Tables 1–2, Figs. 2–4 and Extended data Fig. 1 are provided with this paper.