

Abstract
New Schiff bases functionalized with amide and phenolic groups synthesized by the condensation of 2-hydroxybenzaldehyde and 2-hydroxyacetophenone with amino acid amides which in turn were prepared in two steps from N-Boc-amino acids and homoveraltrylamine through intermediate compounds N-Boc-amino acids amides. The compounds were characterized by elemental analysis, FT-IR, UV–Vis, and NMR spectroscopy. The crystal structures of three Schiff bases were determined by single crystal X-ray diffraction. There exists O–HN, N–HO, and C–HO types of hydrogen bonds and C–H secondary bonding interactions in these crystalline solids. The Schiff bases have been screened for anticoagulant and antiplatelet aggregation activities. All the compounds showed procoagulant activity which shortens the clotting time of citrated human plasma in both platelet-rich plasma and platelet-poor plasma except the derivatives of L-methionine which showed anticoagulant activity by prolonging the clotting time. In addition, the compounds derived from benzyl cysteine and phenylalanine showed adenosine diphosphate induced antiplatelet aggregation activity, whereas others did not show any role. Moreover, all these compounds revealed non-hemolytic activity with red blood cells.
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s00706-022-02936-6.
Keywords: Amino acids, Anticoagulant, Antiplatelet, Dopamine, Procoagulant, Schiff bases
Introduction
Amino acids and proteins are the building blocks of life. Amino acids are neurotransmitters and participate in a number of processes mainly in biosynthesis [1]. Peptides are compounds derived from amino acids which have gained enormous popularity and relevance in the recent years particularly, with the emergence of unnatural analogs as components of molecules with therapeutic potential [2–7]. Many biologically active peptides has been discovered and characterized during the last 30 years [8]. Recently, the replacement of natural amino acids in peptides with non-proteinogenic derivatives to obtain drug-like target molecules has become an important goal in synthetic organic chemistry [9, 10]. One of the interesting areas of research in drug design has been the synthesis of peptidomimetic molecules that are expected to have the same therapeutic effects like natural peptide counterparts with an added advantage of metabolic stability [11, 12].
Dopamine (DA) and their derivatives are active compounds for stress, anxiety, and related behaviors. DA dysfunction found to play a key role in social-anxiety disorders [13–19]. 2-(3,4-Dimethoxyphenyl)ethan-1-amine also known as homoveratrylamine is a dimethyl derivative of DA and is one of the starting material for number of nuerotransmitting agents as well as other biologically important compounds [20–22].
Schiff bases are characterized by an imine (> C=N–) linkage typically obtained by facile condensation of carbonyl compounds (mostly aromatic aldehydes and ketones) with primary amines. Schiff bases have been found versatile ligands as they can form stable complexes with most of the transition metals [23]. Schiff bases found applications in catalysis [24] and material science [25]. Furthermore, Schiff bases and their metal complexes are excellent pharmacophore [26–29] such as antibacterial, antifungal, antiviral, antimalarial, antiinflammatory, antioxidant, anticancer, cytotoxic, enzyme inhibitory therapeutics and including anti-COVID-19 [30–37]. Especially, Schiff bases derived from biologically active starting materials namely isatin [30], 2-azetidinone [31], cephalothin [32], and dopamine [13–16] showed interesting biological activities.
Thrombotic disorders increase the risk of stroke and heart attack. Hence, mortality and morbidity rate have been tremendously increased worldwide [38–43]. The underlying cause involved in the pathophysiology of thrombosis is hyperactivation of platelets and coagulation factors [44, 45]. Known procoagulants for the treatment of haemophilia, anticoagulant (Ex. apixaban, dabigatran, edoxaban, enoxaparin, heparin, rivaroxaban, warfarin) and antiplatelet (Ex. clopidogrel, ticagrelor, prasugrel, dipyridamole, eptifibatide, dipyridamole, aspirin, ticlopidine) drugs for thrombotic disorders, the life-threatening side effects such as internal bleeding, miscarriage, nausea and headache limit their usage [42, 43]. Therefore, identification of the anticoagulant and antiplatelet agents with least side effects is of current interest. Schiff bases based on thiosemicarbazones [46–48], hydrazones [49–53], quercetin, catechol, 1,3-indandione, coumarin [54–58] and several others [59–64] have been reported to be effective antiplatelet aggregation agents. Recently reported that anticoagulants derived from snake venom (Ancrod), fungi aspergillusoryzae (Brinase), sweet clover, saliva of leach and extracts of Flax seeds and Jackfruit seeds [38–43, 65, 66], silver nanoparticles [67, 68] and pyrazinyl pyridine [69].
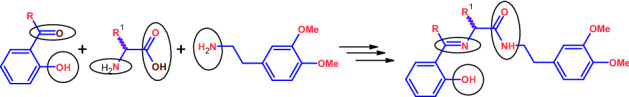
In this regard, we herein reported the synthesis and characterization of new amide and phenol functionalized Schiff’s bases 13–18 derived from amino acids and homoveraltrylamine. These Schiff’s bases have been explored for antithrombotic and antiplatelet aggregation activities.
Results and discussion
Synthesis
The N-Boc-amino acid amides (2S)-tert-butyl [1-[(3,4-dimethoxyphenethyl)amino]-4-(methylthio)-1-oxobutan-2-yl]carbamate (5), (2S)-tert-butyl [3-(benzylthio)-1-[(3,4-dimethoxyphenethyl)amino]-1-oxopropan-2-yl]carbamate (6), (2S)-tert-butyl [1-[(3,4-dimethoxyphenethyl)amino]-1-oxopropan-2-yl]carbamate (7), and (2R)-tert-butyl [1-[(3,4-dimethoxyphenethyl)amino]-1-oxo-3-phenylpropan-2-yl]carbamate (8) were synthesized by the procedure reported in the literature [70–73] by condensation reaction of 2-(3,4-dimethoxyphenyl)ethan-1-amine with the corresponding N-Boc-(S)-amino acids such as (2S)-2-[(tert-butoxycarbonyl)amino]-4-(methylsulfanyl)butanoic acid (1), (2S)-3-(benzylsulfanyl)-2-[(tert-butoxycarbonyl)amino]propanoic acid (2), (2S)-2-[(tert-butoxycarbonyl)amino]propanoic acid (3), and (2R)-2-[(tert-butoxycarbonyl)amino]-3-phenylpropanoic acid (4), respectively, in presence of DCC as a coupling agent in DCM at 0 °C. Deprotection of Boc from 5 to 8 with TFA resulted in the corresponding N-[2-(3,4-dimethoxyphenyl)ethyl] functionalized amino acids amides 9–12 such as (2S)-2-amino-N-[2-(3,4-dimethoxyphenyl)ethyl]-4-(methylsulfanyl)butanamide (9), (2S)-2-amino-N-[2-(3,4-dimethoxyphenyl)ethyl]-3-(benzylsulfanyl)propanamide (10), (2S)-2-amino-N-[2-(3,4-dimethoxyphenyl)ethyl]propanamide (11), and (2R)-2-amino-N-[2-(3,4-dimethoxyphenyl)ethyl]-3-phenylpropanamide (12). The compounds 5, 7, 8, 9, 11, and 12 were confirmed by determining their melting points which were found in the range of their literature values [70–73].
Schiff’s bases 13–16 such as (S)-N-(3,4-dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]-4-(methylthio)butanamide (13), (S)-3-(benzylthio)-N-(3,4-dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]propanamide (14), (S)-N-(3,4-dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]propanamide (15), (R)-N-(3,4-dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]-3-phenylpropanamide (16), (S)-N-(3,4-dimethyoxyphenylethyl)-2-[[1-(2-hydroxyphenyl)ethylidene]amino]-4-(methylthio)butanamide (17), and (S)-3-(benzylthio)-N-(3,4-dimethyoxyphenylethyl)-2-[[1-(2-hydroxyphenyl)ethylidene]amino]propanamide (18) were obtained by the condensation reaction between amino acids amides 9–12 and 2-hydroxybenzaldehyde taken in equimolar ratio in dry methanol at RT [74–79]. The Schiff bases 17 and 18 were similarly obtained by the reaction of 9 and 10 with 2-hydroxyacetophenone. The reaction mixtures were stirred for 3–4 h. The resulted crude yellow solids on recrystallization in a 1:1 of mixture of chloroform and n-hexane form desired pure crystalline Schiff bases. The equations for the reactions involving the synthesis of compounds 5–18 are shown in Scheme 1. The elemental analysis of 5, 10, and 13–18 agreed with their molecular formulae and confirmed the formation all these compounds.

UV–Vis spectroscopy
UV–Vis spectra (Fig. 1) of the newly synthesized compounds 13–18 have been recorded for their 2.5 × 10–5 M solutions in DMSO. The intense high-energy absorption bands observed in the range of λmax = 257–260 and 278–280 nm were assigned to the –* transitions of the aromatic rings. The low energy and less intense bands appeared between λmax = 319–321 nm were attributed to the * transitions within the delocalized system [74–79].
Fig. 1.
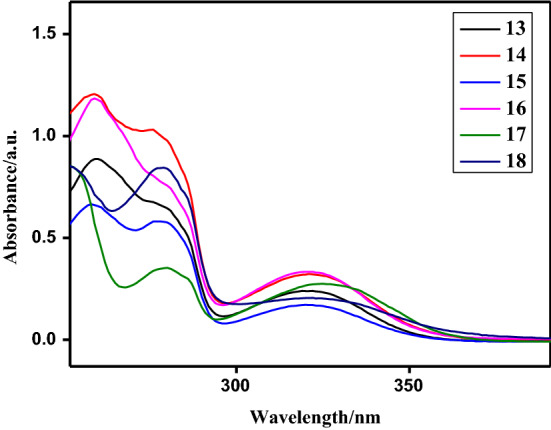
UV–Vis spectra of compounds 13–18
FT-IR spectroscopy
In the FT-IR spectra of compounds 6 and 10, the bands observed in the region of ν, 3322 and 3363 cm−1 were due to N–H stretching and the bands appeared in the range = 2800 to 2900 cm−1 were assigned to aromatic C–H stretching, respectively. In 6, sharp peak appeared at 2100 cm−1 was due to OCONH stretching which indicates Boc (t-Bu) group attached to the amine, which is absent in 10 which indicates its formation. A band appeared at 1684 and 1670 cm−1 in the IR spectra of compounds 6 and 10, respectively, was assigned to CONH stretching. The FT-IR spectra of compounds 13–18 have been compared with the spectra of similar known compounds and the spectra found in agreement with the literature reports [74–79]. The bands observed in the region of 3326–3340 cm−1 attributed to the O–H stretching. The strong bands appeared at 1625–1630 cm−1 were assigned to stretching of the C=N (imine) group present in all these compounds. The stretching bands for amide CONH group observed at around 1650–1655 cm−1. These compounds also displayed bands at 1260 and 1240 cm−1 which were attributed to the C-O stretching. The bands other functionalities appeared at standard wave number regions of their IR spectra. The representative IR spectra of 13–18 are given in Fig. S1 to Fig. S6, respectively (ESI).
NMR spectroscopy
The 1H NMR spectra of compounds 6, 10, and 13–18 were found characteristic [74–79]. In the 1H NMR spectra of both 6 and 10, the aromatic protons appeared in range of δ = 6.69 to 7.26 ppm and the methoxy protons appeared as a singlet at 3.84 and 3.83 ppm, respectively. In the compound 6, the NHCO proton observed as a triplet in the range of 6.34–6.37 ppm and Boc [Me3C] protons appeared as a singlet at 1.45 ppm. While in 10, the NHCO proton shifted downfield and appeared as a broad singlet at 7.38 ppm and the free NH2 protons appeared as a broad peak at 1.80 ppm.
In the 1H NMR spectra of Schiff bases 13–18, the signal for phenolic OH proton observed at about 12.2 ppm and was deshielded due to the presence of intramolecular O–HN hydrogen bonding even in solution. In these compounds, the ArCH2 and CH2N protons appeared as triplets in the range of 2.74–2.77 and 3.46–3.59 ppm, respectively. The methoxy protons observed as independent singlets at 3.81 and 3.78 ppm, respectively. The signal for azomethine (HC=N) proton observed as a singlet in the range of 7.90–8.35 ppm in all the compounds 13–18. The amide proton (CONH) appeared as a triplet at 6.0–6.1 ppm in 13–18 on coupling with neighboring CH2 protons. The signals for aromatic protons appeared in the region 6.6–7.3 ppm. The representative 1H NMR spectra of 6, 10, and 13–18 are given in Fig. S7 to Fig. S25, respectively (ESI).
The 13C{1H} NMR spectra compounds 6, 10, and 13–18 were found characteristic [74–79]. In the 13C NMR spectra of 6 and 10, the peak for CONH carbon appeared at δ = 175 and 177 ppm, respectively, and the peaks for methoxy (OCH3) carbons appeared at ~ 60 ppm. In 6, the signal for Boc carbonyl carbon appeared at 159.9 ppm. In the 13C NMR spectra of 13–18, the signal for ArCH2 carbon appeared at 34.4 ± 0.2 ppm whereas the signal for NCH2 carbon found deshielded and observed at about 40.4 ppm. The peaks for phenolate (ArCO−), azomethine (C=N), and amide (CONH) carbons appeared at around 160, 166, and 170 ppm, respectively, in all these Schiff base compounds. The peaks for both OCH3 carbons appeared independently at slightly different chemical shifts at around 55.3 ± 0.2 ppm in all these compounds. The observations from NMR spectra of 13–18 confirmed their formation. The representative 13C{1H} NMR spectra of compounds 13–18 are given in Fig. S26 to Fig. S32, respectively (ESI).
Mass spectrometry
The amide functionalized Schiff bases 13–18 were characterized by mass spectrometry using electro spray ionization in combined with liquid chromatography (ESI-LC–MS) method. The ESI-LC–MS spectra of 13–18 showed molecular ion (M+) peaks at m/z = 417, 479, 357, 432, 431, and 492.9, respectively, and were agreed with their calculated molecular weights. Hence, mass spectral data confirmed the formation of the respective compounds 13–18. The representative mass spectra of compounds 13–18 are given in Fig. S33 to Fig. S38 (ESI).
Single-crystal X-ray diffraction
The single-crystal X-ray diffraction data for 13, 15, and 17 collected and their molecular structures in solid state were determined. The selected crystal data and structure refinement parameters of 13, 15, and 17 are given in Table 1 (complete data is given in Table S1 in ESI) and their selected bond parameters are given in Table 2. The molecular structures of the compounds 13, 15, and 17 as ORTEP diagrams are shown in Figs. 2, 3, and 4, respectively. The compounds 13, 15, and 17 crystallize in monoclinic system and non-centrosymmetric P21 space group with Z = 2. All the molecules consist of a chiral carbon atom with S-configuration and the two molecules differ in conformations of the two chains connected to these chiral carbons. There exists an intramolecular O–HN hydrogen bond in each compound which is a characteristic for 2-hydroxyaryl Schiff bases [80].
Table 1.
Selected crystal data and structure refinement parameters for 13, 15, and 17
| Compound | 13 | 15 | 17 |
|---|---|---|---|
| Empirical formula | C22H28N2O4S | C20H24N2O4 | C23H30N2O4S |
| Formula weight/g mol−1 | 416.52 | 356.42 | 430.55 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group, Z | P21, 2 | P21, 2 | P21, 2 |
| a/Å | 8.482(3) | 9.692(2) | 4.9379(3) |
| b/Å | 5.1415(15) | 5.1081(8) | 14.2499(9) |
| c/Å | 24.614(7) | 19.469(4) | 15.9154(9) |
| α, β, γ/° | 90, 99.92, 90 | 90, 102.77(2), 90 | 90, 94.354(5), 90 |
| Volume/Å3 | 1057.4(5) | 940.0(3) | 1116.65(12) |
| ρcalc/g cm−3 | 1.308 | 1.2591 | 1.281 |
| μ/mm−1 | 0.184 | 0.088 | 0.176 |
| F (000) | 444.0 | 380.2 | 460 |
| Reflections collected | 7980 | 3686 | 3966 |
| Independent reflections |
3533 [Rint = 0.1003, Rsigma = 0.2199] |
2643 [Rint = 0.0160, Rsigma = 0.0690] |
2920 [Rint = 0.0129] |
| Data/restraints/parameters | 3533/1/267 | 2643/3/238 | 2920/30/299 |
| GOF on F2 | 0.921 | 0.994 | 1.098 |
| Final R indexes [I > = 2σ (I)] |
R1 = 0.0572, wR2 = 0.0999 |
R1 = 0.0419, wR2 = 0.0647 |
R1 = 0.0365, wR2 = 0.0676 |
| Final R indexes [all data] |
R1 = 0.1306, wR2 = 0.1169 |
R1 = 0.0947, wR2 = 0.0767 |
R1 = 0.0521, wR2 = 0.0732 |
| CCDC No | 1,999,894 | 1,999,895 | 2,031,920 |
Table 2.
Selected bond lengths and bond angles of 13, 15, and 17
| 13 | 15 | 17 | |||
|---|---|---|---|---|---|
| Bond lengths/Å | |||||
| S(0AA)-C(23) | 1.803 (5) | – | – | S(1)-C(20) | 1.791(4) |
| S(0AA)-C(24) | 1.810 (5) | – | – | S(1)-C(19) | 1.801(4) |
| C(10)-O(25) | 1.231(6) | O(2)-C(9) | 1.234(4) | O(2)-C(9) | 1.224(3) |
| C(19)-O(21) | 1.354 (5) | O(1)-C(1) | 1.344(4) | O(1)-C(1) | 1.346(4) |
| N(12)-C(13) | 1.294 (6) | N(1)-C(7) | 1.270(4) | N(1)-C(7) | 1.280(4) |
| N(12)-C(11) | 1.460 (6) | N(1)-C(8) | 1.462(4) | N(1)-C(8) | 1.469(4) |
| N(9)-C(10) | 1.339 (6) | N(2)-C(9) | 1.332(4) | N(2)-C(9) | 1.324(4) |
| N(9)-C(8) | 1.447 (5) | N(2)-C(10) | 1.462(4) | N(2)-C(10) | 1.474(8) |
| – | – | – | – | N(2)-C(10') | 1.487(8) |
| Bond angles/° | |||||
| C(14)-C(13)-N(12) | 122.9(4) | C(6)-C(7)-N(1) | 122.1(3) | C(6)-C(7)-N(1) | 116.8(3) |
| H(13)-C(13)-N(12) | 118.5(0) | H(7)-C(7)-N(1) | 118.95(2) | C(21)-C(7)-N(1) | 125.1(4) |
| C(13)-N(12)-C(11) | 114.9 (4) | C(7)-N(1)-C(8) | 118.0(3) | C(7)-N(1)-C(8) | 121.8(3) |
| C(10)-N(9)-C(8) | 123.4 (4) | C(9)-N(2)-C(10) | 123.4(3) | C(9)-N(2)-C(10) | 124.7(4) |
| – | – | – | – | C(9)-N(2)-C(10') | 117.2(4) |
| C(23)-S(0AA)-C(24) | 98.2 (3) | – | – | C(20)-S(1)-C(19) | 100.4(2) |
Fig. 2.
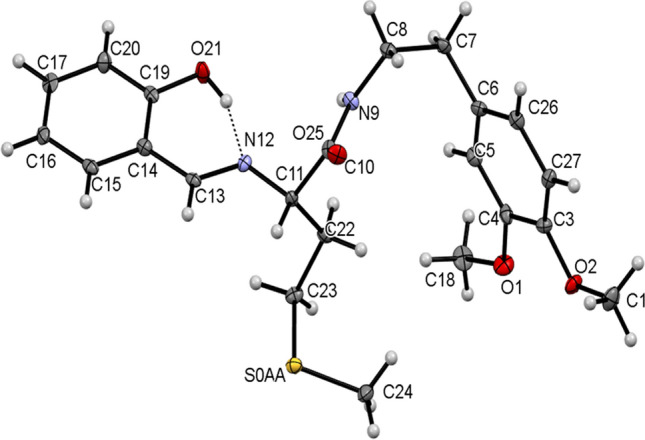
ORTEP view of 13 with thermal ellipsoids drawn at 30% probability. Intramolecular O–HN hydrogen bond is shown as thin dashed lines
Fig. 3.
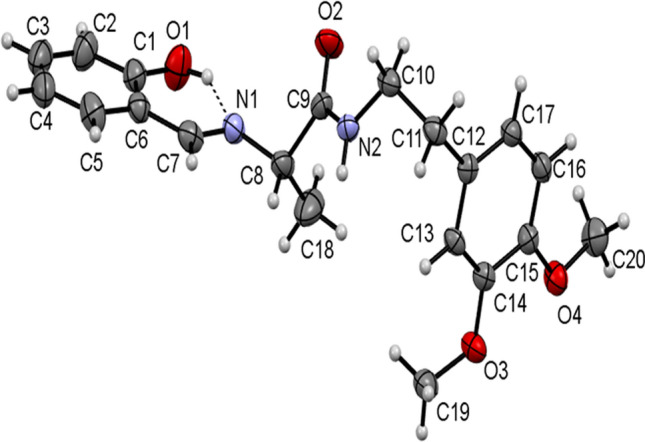
ORTEP view of 15 with thermal ellipsoids drawn at 30% probability. Intramolecular O–HN hydrogen bond is shown as thin dashed lines
Fig. 4.
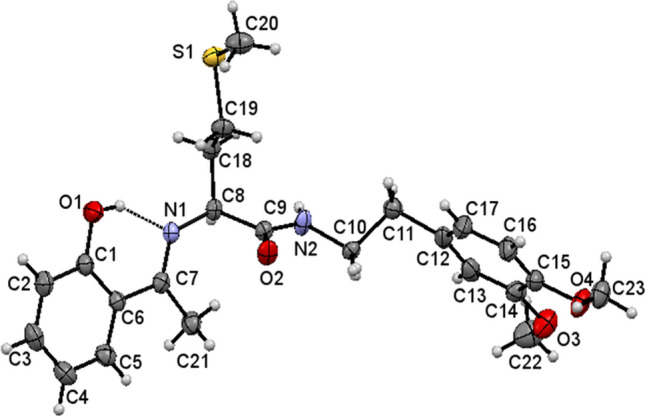
ORTEP view of 17 with thermal ellipsoids drawn at 30% probability. Intramolecular O–HN hydrogen bond is shown as thin dashed lines
The crystal structure of 13 possess N9-H9O25 hydrogen bonded C4 chains linking the molecules along b-axis (Fig. 5, Table 3). The crystal structure of 15 similarly features N2-H2NO2 intermolecular hydrogen bonds linking the molecules into C4 chains running parallel to b-axis. These adjacent parallel chains in 17 are further interlinked via C19-H19Cπ and C20-H20Bπ interactions to form a one-dimensional ribbon parallel to b-axis (Fig. 6, Table 3). Contrary to 15, in 13, the adjacent parallel chains are further interlinked through C24-H24CO21 intermolecular interactions running as C11 chains parallel to a-axis, thereby, forming a two-dimensional (2D) sheet parallel to the ab-plane. Therefore, different architectures are observed in both the crystal structures.
Fig. 5.

A partial view of the crystal packing displaying two-dimensional sheets in 13 through NHO and CHO hydrogen bonding
Table 3.
Geometric parameters for hydrogen bonds and other intermolecular contacts (Å,°) operating in the crystal structures of 13, 15, and 17
| D-HA | D-H | HA | DA | D-HA | |
|---|---|---|---|---|---|
| 13 | O21-H21N12# | 0.82 | 1.94 | 2.6086 | 139 |
| N9-H9O25i | 0.86 | 2.18 | 3.0084 | 162 | |
| C24-H24CO21ii | 0.96 | 2.56 | 3.3292 | 137 | |
| 15 | O1-H1ON1# | 0.84 | 1.86 | 2.568(4) | 141 |
| N2-H2NO2v | 0.86 | 2.32 | 3.165(4) | 171 | |
| C19-H19Cπv,a | 0.96 | 2.75 | 3.610(5) | 149 | |
| C20-H20Bπvi,a | 0.96 | 2.88 | 3.717(4) | 147 | |
| 17 | O1-H1ON1# | 0.85(4) | 1.77(4) | 2.531(4) | 147(4) |
| N2-H2NO2iii | 0.85(2) | 2.10(2) | 2.940(3) | 174(4) | |
| C11-H11AO4iv | 0.97 | 2.59 | 3.371(10) | 137 |
#Intra; i: x, 1 + y, z; ii: 1 + x, − 1 + y, z; iii: − 1 + x,y,z; iv: –x,–1/2 + y,1–z; v: x, –1 + y, z; vi: 1–x, 1/2 + y, 1–z; a:Centroid of C12-C17 aromatic ring
Fig. 6.

A partial view of the crystal packing displaying ribbon-like framework in 15 formed via NHO hydrogen bonds and CHπ interactions
The crystal structure of 17 also features N2-H2O2 intermolecular hydrogen bonds linking the molecules into C4 chains running parallel to a-axis similar to that observed in the other two structures. The adjacent parallel chains are further interlinked through C11-H11AO4 intermolecular interactions running as C7 chains parallel to b-axis, thereby, forming a two-dimensional (2D) sheet parallel to the ab-plane (Fig. 7, Table 3). The crystal structures of 13 and 17 are similar, except for the variation of the length of C-H O chains in the two structures: C11 in 13 and C7 in 17.
Fig. 7.

A partial view of the crystal packing displaying two-dimensional sheets in 17 through NHO and CHO hydrogen bonding
The dihedral angle between the two aromatic rings is 85.0(1)° in 13, 85.74(18)° in 17, and 89.2(2)° in 15. The two methoxy groups and the attached aromatic ring in 13 are in the same plane with the torsions C27-C3-O2-C1 and C5-C4-O1-C18 having values of 1.3(7)° and − 2.0(8)°. The corresponding torsions C13-C14-O3-C22 and C16-C15-O4-C23 in 17 have values of 4.0(6)° and 2.8(6)°, respectively, while, in 15, torsions C13-C14-O3-C19 and C16-C15-O4-C20 have values of 5.8(5)° and − 10.4(5)°, respectively, indicating that the two methoxy groups are in the same plane as that of the attached ring. The –CH2–CH2– groups in all the three molecules have staggered conformation and the molecular conformations of both the molecules are stabilized by intramolecular O–HN hydrogen bonds between azomethine N and phenolic H closing into a S(6) motif. In 17, the ethylene –CH2–CH2– group is disordered over the C–C bond in the ratio 0.5:0.5.
Role of compounds 13–18 in blood coagulation cascade
To check the probable therapeutic potency on thrombotic disorders, the newly synthesized Schiff bases 13–18 were scrutinized for their possible role in blood coagulation cascade and platelet function. The plasma re-calcification time experiment briefly involved that the compounds 13–18 (0 to 40 µg) were initially pre-incubated with 0.1 cm3 of citrated human platelets rich plasma (PRP) in presence of 10 mM Tris–HCl (10 mm3) buffer pH 7.4 for 5 min at 37 °C. Then, 20 mm3 of 0.25 M CaCl2 was added to the pre-incubated mixture and the clotting time was recorded in s. Plots were drawn for clotting time against amount of compounds (µg) and are shown in Fig. 8.
Fig. 8.

Plasma re-calcification time of 13–18 using human plasma as PRP (pink) and PPP (green)
All the compounds exhibited an effect on coagulation cascade in both PRP and PPP which increases with increasing the amount of any compound. Among the compounds screened, the Schiff bases of methionine with 2-hydroxybezaldehyde (13) and 2-hydroxyacetophenone (17) showed an anticoagulation effect by extending the clotting time from control 210 ± 3.5 to 270 ± 1.6, 210 ± 3.5 to 315 ± 1.5 s in PRP and 221 ± 1.5 to 283 ± 2.5, 220 ± 1.3 to 325 ± 1.6 s in PPP, respectively, at a maximum amount of 40 µg of any compound as represented in Fig. 9 when compared with a standard drug heparin (1 µM) as a positive control. Contrary to the above, the Schiff bases derived from benzyl cysteine (14 and 17), alanine (15) and phenyl alanine (16) showed procoagulant effect by reducing the clotting time from the control 189 ± 0.2 to 68 ± 3, 221 ± 1.6 to 122 ± 1.3, 243 ± 1.5 to 155 ± 1.2 and 210 ± 1.2 to 60 ± 3 s in PRP and 200 ± 3.5 to 80 ± 0.9, 230 ± 1.6 to 131 ± 1.3, 246 ± 1.5 to 160, and 220 ± 2.5 to 62 ± 0.3 s in PPP, respectively. Furthermore, the salicyladehyde Schiff bases of benzyl cysteine (14) and phenylalanine (16) showed strong procoagulant activity as revealed by their magnitude of decrease in clotting time from control in both PRP and PPP by about 120 and 160 s than that of alanine (15) which exhibited 90 s respectively. While procoagulant activity of the Schiff base 17 of 2-hydroxy acetophenone with benzyl cysteine showed 100 s which is about 20 s less than the Schiff base 14 of salicylaldehyde. But, in case of methionine, the Schiff base of 2-hydroxyacetophenone showed an anticoagulant activity by increment of 100 s in both PRP and PPP than that of the salicyladehyde Schiff base which showed 60 s.
Fig. 9.
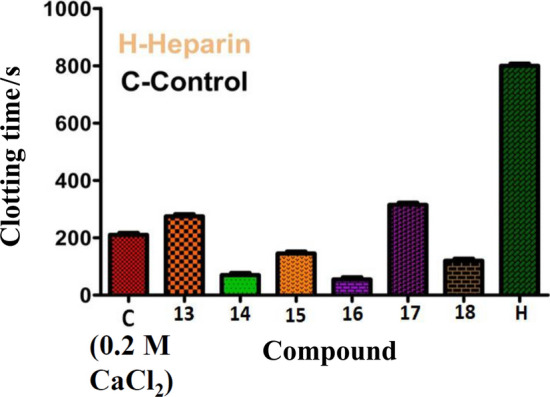
Effect of compounds 13–18 in blood coagulation cascade at a maximum amount of 40 µg
Role of compounds 13–18 in platelet aggregation function
Platelets are anucleated cells which possess numerous membrane-bound receptors for physiological agonists such as collagen, ADP, thrombin, epinephrine, arachidonic acid (AA) and platelet activating factor [49–53]. Binding of an agonist to the receptors alter the platelet morphology leading to their activation and aggregation. Thus, platelet activation/aggregation plays a key role in the pathophysiology of thrombotic disorders [43]. Therefore, the new amide and phenol functionalized Schiff base compounds 13–18 were examined for their interference in the platelet function using PRP and PPP in the presence and absence of an agonist. The experiment was performed on an aggregometer connected with a pen recorder. As platelets aggregate, light transmission increases progressively producing a trace on the recorder which would be the plot of light transmission between PRP and PPP corresponds to the 0 and 100% aggregation respectively against time in s. The pre-incubated compounds 13–18 (10 to 30 µg) with 0.45 cm3 PRP or PPP for 3 min were added ADP (5 µM), an agonist and the aggregation was followed for 6 min. Curiously, among the compounds tested, the Schiff bases derived from benzyl cysteine (14 and 18) and phenylalanine (16) augmented the platelet aggregation induced by a potent agonist ADP as shown in Figs. 10b, 11b, and 12b, respectively. While, the compounds synthesized from methionine (13 and 17) and alanine (15) did not interfere at all in agonist induced platelet function. The percentage of platelet activation by the compounds 14, 16, and 18 was found to be 29, 41, and 45%, respectively. These observations demonstrated that the compounds 14, 16, and 18 derived from benzyl cysteine and phenylalanine containing an aromatic and bulkier group at chiral carbon exhibited strong procoagulant property and augmentation of platelet activation.
Fig. 10.

a Activation of ADP (5 µM) induced platelet aggregation without (Trace 1) and with increasing amount (10–30 µg) of compound 14 (Traces 3, 5, and 7) and b dose (10–30 µg)-dependent aggregation (bar graph) by compound 14
Fig. 11.

a Activation of ADP (5 μM) induced platelet aggregation without (Trace 1) and with increasing amount (10–30 µg) of compound 16 (Traces 3, 5, and 7) and b dose (10–30 µg)-dependent aggregation (bar graph) by compound 16
Fig. 12.

a Activation of ADP (5 μM) induced platelet aggregation without (Trace 1) and with increasing amount (10–30 µg) of compound 18 (Traces 3, 5, and 7) and b dose (10–30 µg)-dependent aggregation (bar graph) by compound 18
Non-hemolytic activity of compounds 13–18
To identify the probable interaction of synthesized compounds on red blood cell (RBC) membrane, hemolytic assay was carried out using RBCs. Strikingly, all the compounds incapable of lysing the RBC membrane, as intact RBCs were identified in compounds treated with RBC sample, while, a positive control water caused haemolysis and PBS buffer as a negative control which as shown in Fig. 13.
Fig. 13.
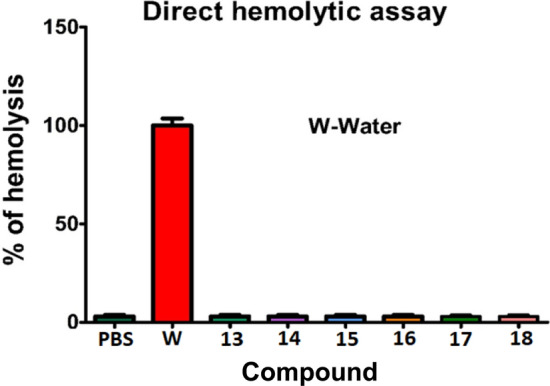
Hemolytic activity of compounds 13–18 measured at 540 nm against water (positive control) and PBS buffer (negative control)
Conclusions
Six new chiral Schiff bases functionalized with peptide and phenol groups 13–18 have been synthesized and characterized by FT-IR, UV–Vis, 1H and 13C{1H} NMR spectroscopy, and LC–MS. The crystal structures of three Schiff bases were determined by single crystal X-ray diffraction. These crystalline solids, the molecules are found to associate by moderate to strong OHN intramolecular, N–HO, and C–H⋯O types of intermolecular hydrogen bonds as well as C–H⋯π intermolecular secondary bonding interactions in 15 forming the supramolecular assemblies. These compounds have been demonstrated to interfere in blood coagulation cascade and platelet aggregation function. Among which benzyl cysteine (14 and 18), alanine (15), and phenylalanine (16)-based Schiff bases exhibited procoagulant activity, whereas methionine (13)-based compound showed a mild anticoagulant activity. Surprisingly, among all the compounds tested, only benzyl cysteine (14 and 18) and phenylalanine (16)-based Schiff bases showed platelet activating potency. These compounds also showed non-hemolytic activity towards RBCs. Thus, these compounds may serve as better prototype in identifying new drugs with least side effects. Thus, it requires further conformational studies in this prospective. Therefore, understanding of their mechanism of action on coagulation cascade and platelet aggregation function may provide more insight into therapeutic usage of said compounds in the management of thrombotic disorders.
Experimental
2-(3,4-Dimethoxyphenyl)ethan-1-amine (97%), N,N’-dicyclohexyl carbodiimide (DCC) (99%), 2-hydroxyacetophenone (99%), N-Boc-L-methionine, and N-Boc-L-benzylcysteine were purchased from Sigma Aldrich, USA. 2-Hydroxybenzaldehyde (99%), N-Boc-L-alanine and N-Boc-L-phenylalanine were obtained from Spectrochem Ind. Pvt. Ltd. Solvents such as methanol (MeOH), petroleum ether (40–60 °C), dichloromethane (DCM), chloroform (CHCl3), n-hexane, diethylether, and other chemicals of commercial analytical reagent grade were purchased from Merck India Ltd., and used as received. Melting points were determined in open capillary tubes closed at one end using a RAGA melting point apparatus. Elemental analysis (C, H, and N) was performed on a LECO–CHNSO–9320 type elemental analyzer. UV–Vis absorption spectra were recorded in ethanol using Agilent Cary-60 spectrophotometer in the spectral window of 200–800 nm. FT-IR spectra were recorded on a PerkinElmer Frontier MIR/FIR spectrometer using ATR. 1H and 13C{1H} NMR spectra were recorded on Agilent VNMRS-400 (6 and 10) and Bruker WM-400 (13–18) 400 MHz NMR spectrometers using tetramethylsilane (TMS) as an internal standard. Single-crystal X-ray diffraction data were collected on Bruker APEX2 (13) and Oxford Diffraction Xcalibur (TM) (15 and 17) single crystal X-ray diffractometers with CCD detector. The structures were solved by SHELXT [81] and refined using SHELXL [82] available within Olex software [83, 84]. The compound 15 is a weak anomalous scatterer. We feel that the presented structural information on this compound is reliable enough to unambiguously solve and refine the structure reliably. In 17, though Friedel Pair Coverage is low, the Flack parameter is enough and presented structural information on this compound is reliable in order to unambiguously solve and refine the structure reliably. Thermo Scientific BioMate-6 UV–Visible spectrophotometer was used for determination of haemolytic assay. Lumi aggregation system of Model-700 was used for the determination of platelet aggregation. Blood samples were collected from healthy voluntary donors.
(S)-tert-Butyl [3-(benzylthio)-1-[(3,4-dimethoxyphenethyl)amino]-1-oxopropan-2-yl]carbamate (6, C25H34N2O5S)
A mixture of 10 cm3 dry CH2Cl2, 1000 mg N-Boc-L-benzylcysteine (2, 3.2 mmol), 582 mg 2-(3,4-dimethoxyphenyl)ethan-1-amine (3.2 mmol), and 0.662 mg DCC (3.2 mmol) was maintained for 3 h at 0 °C in an ice bath with constant stirring. The reaction was monitored by TLC using a mixture of petroleum ether and ethyl acetate as an eluent. The mixture was then filtered to separate the solid N,N’-dicyclohexyl urea formed from DCC and was discarded. The filtrate was evaporated under reduced pressure on a rotary evaporator. The resulting oily mass was treated with 20 cm3 anhydrous ice-cooled acetone to separate additional N,N’-dicyclohexyl urea. The acetone solution was evaporated under reduced pressure and the resulting crude product was purified by crystallization from n-hexane and chloroform. White solid; yield: 920 mg (60%); m.p.: 160–162 °C; FT-IR (ATR): = 3322, 2932, 2856, 2108, 1684, 1654, 1513, 1450, 1239, 1150, 1012, 840, 814, 698 cm−1; 1H NMR (399.82 MHz, CDCl3): δ = 7.26–7.29 (m, 3H, H21,22), 7.20–7.23 (m, 1H, H23), 6.76–6.78 (d, 3 J = 8.4 Hz, 1H, H7), 6.69–6.71 (d, 3 J = 7.2 Hz, 2H, H11,10), 6.34–6.37 (t, 1H, NH), 5.27 (s, 1H, NH-Boc), 4.16–4.17 (b, 1H, CH), 3.84 (s, 3H, OCH3), 3.83 (s, 1H, OCH3), 3.69–3.70 (d, 2 J = 1.9 Hz, 2H, SCH2Ph), 3.44–3.50 (q, 2H, NCH2), 2.80–2.85 (dd, 3 J = 13.99 Hz, 2 J = 5.6 Hz, 1H, CH2S), 2.71–2.75 (t, 2H, CH2Ar), 2.67–2.70 (dd, 3 J = 13.99 Hz, 2 J = 5.6 Hz, 1H, CH2S), 1.41 (s, 9H, t-Bu) ppm; 13C{1H} NMR (99.995 MHz, CDCl3): δ = 175.1 (C2), 159.9 (t-BuOCO), 153.7 (C7), 152.4 (C8), 142.52 (C20), 135.7 (C5), 133.6 (C21), 133.2 (C22), 131.8 (C23), 125.3 (C10), 116.5 (C6), 116.0 (C9), 84.9 (t-BuC), 60.5 (OCH3), 60.3 (OCH3), 58.4 (C1), 41.1 (C3), 39.8 (C4), 38.5 (C19), 32.9 (C18), 30.2 (t-Bu), 29.6 (t-Bu), 29.3 (t-Bu) ppm.
(2S)-2-Amino-N-[2-(3,4-dimethoxyphenyl)ethyl]-3-(benzylsulfanyl)propanamide (10, C20H26N2O3S)
The compound 6 (500 mg, 1.10 mmol) in 5 cm3 dry CH2Cl2 taken in a round bottom flask and this solution was stirred for 10 min at around 0 °C using an ice-bath. A solution of 5 cm3 trifluroacetic acid (TFA) in 5 cm3 CH2Cl2 was added to the above solution. The mixture was allowed to react maintaining the above temperature under continuous stirring further for 4 h. The reaction was monitored by TLC using ethyl acetate and petroleum ether (50:50) as an eluent. At the completion of the reaction, the mixture was added drop-wise a saturated aqueous solution of NaHCO3 until the pH 8 reached. The organic phase separated and was treated with solid anhydrous Na2SO4 to eliminate water residues, filtered, and evaporated under reduced pressure. Yellow oily liquid; yield: 355 mg (90%); FT-IR (ATR): = 3363, 2938, 2850, 2838, 1670, 1519, 1451, 1231, 1143, 1026, 814, 765, 697 cm−1; 1H NMR (399.82 MHz, CDCl3): δ = 7.38 (bs, 1H, NH), 7.26–7.29 (m, 4H, H21,22), 7.207–7.23 (m, 1H, H23), 6.76–6.78 (d, 3 J = 8.4 Hz, 1H, H6), 6.69–6.73 (m, 2H, H9,10), 3.82 (s, 6H, OCH3), 3.67 (s, 2H, SCH2Ph), 3.42–3.48 (q, 2H, NCH2), 3.36–3.39 (dd, 3 J = 14.0 Hz, 2 J = 4.0 Hz, 1H, CH), 2.90–2.95 (dd, 3 J = 13.8 Hz, 2 J = 3.9 Hz, 1H, CH2S), 2.71–2.74 (t, 2H, CH2Ar), 2.58–2.63 (dd, 3 J = 13.6 Hz, 2 J = 8.4 Hz, 1H, CH2S), 1.77–1.80 (bs, 2H, NH2) ppm; 13C{1H} NMR (99.995 MHz, CDCl3): δ = 177.8 (C2), 153.6 (C7), 152.3 (C8), 142.7 (C20), 136.0 (C5), 133.6 (C21), 133.5 (C21’), 133.2 (C22), 131.8 (C23), 125.3 (C6), 116.6 (C10), 116.0 (C9), 60.5 (OCH3), 60.3 (OCH3), 58.5 (C1), 41.9 (C3), 41.0 (C4), 35.5 (C19), 35.5 (C18) ppm.
Synthesis of Schiff bases of amides of amino acids (13–16)
The compound 9/10/11/12 (256/306/206/269 mg, 1.0 mmol) was dissolved in 20 cm3 dry methanol and the resulting solution was stirred for 0.5 h at room temperature (RT). 2-Hydroxybenzaldehyde (100 mg, 1 mmol) in 20 cm3 dry methanol was added to the above solution drop-wise with stirring. The mixture was stirred further for 2–3 h at RT during which time the solution changes to yellow. The progress of the reaction was monitored on TLC using petroleum-ether and ethyl acetate as a mobile phase. After completion, the reaction solution was concentrated on a rotary evaporator to result a yellow precipitate. The yellow precipitate was washed twice with n-hexane (10 cm3 × 2) and then dried under vacuum. The dry solid was recrystallized from a mixture of chloroform and n-hexane (1:1) resulting yellow crystalline solids of 13/14/15/16, respectively.
(S)-N-(3,4-Dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]-4-(methylthio)butanamide (13, C22H28N2O4S)
Yellow crystalline solid; yield: 325 mg (95.3%); m.p.: 87–89 °C;(c = 0.1, CHCl3); FT-IR (ATR): = 3340, 2930, 2850, 1650, 1631, 1550, 1513, 1260, 1235, 1140, 1030, 809, 760, 640 cm−1; UV–Vis: λmax (ε) = 260 (17,972), 279 (13,294), 321 (4910) nm (M−1 cm−1); 1H NMR (399.65 MHz, CDCl3): δ = 12.30 (s, 1H, OH), 8.35 (s, 1H, CH = N), 7.38–7.42 (td, 1H, H16), 7.29–7.31 (dd, 2 J = 1.6 Hz, 3 J = 8.0 Hz, 1H, H17), 6.99–7.02 (d, 3 J = 8.0 Hz, 1H, H14), 6.94–6.97 (td, 1H, H15), 6.61–6.67 (m, 3H, H6, 9,10), 6.05 (bs, 1H, CONH), 4.04–4.07 (dd, 3 J = 9.2 Hz, 2 J = 4.0 Hz,1H, CH), 3.82 (s, 1H, OCH3), 3.78 (s, 1H, OCH3), 3.45–3.59 (m, 2H, NCH2), 2.73–2.77 (t, 3 J = 6.8 Hz, 2H, CH2Ar), 2.51–2.58 (m, 1H, CH2), 2.29–2.42 (m, 2H, CH2S), 2.09–2.14 (m, 1H, CH2), 2.08 (s, 3H, SCH3) ppm; 13C{1H} NMR (99.99 MHz, CDCl3): δ = 169.9 (C2), 166.7 (C11), 160.1 (C13), 148.6 (C7), 147.2 (C8), 132.7 (C15), 131.9 (C17), 131.7 (C5), 120.5 (C12), 118.8 (C10), 118.7 (C16), 116.5 (C14), 112.4 (C6), 111.7 (C9), 70.7 (C1) 55.5 (OCH3), 55.3 (OCH3), 40.3 (C3), 34.5 (C4), 29.5 (C19), 24.5 (C18), 14.5 (CH3) ppm; LC–MS (ESI): m/z calcd. 416.53 (M+), found 417.
(S)-3-(Benzylthio)-N-(3,4-dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]propanamide (14, C27H30N2O4S)
Yellow solid; yield: 370 mg (94.6%); m.p.: 80–83 °C;(c = 0.1, CHCl3); FT-IR (ATR): = 3340, 2925, 2835, 1655, 1630, 1543, 1510, 1255, 1226, 1132, 1030, 804, 755, 640, 530 cm−1; UV–Vis: λmax (ε) = 258 (24,398), 277 (20,756), 321 (6,390) nm (M−1 cm−1); 1H NMR (399.65 MHz, CDCl3): δ = 12.20 (s, 1H, OH), 8.16 (s, 1H, CH = N), 7.40–7.42 (td, 1H, H16), 7.25–7.38 (m, 5H, H21, 22), 7.22–7.24 (m, 1H, H17), 6.70–7.02 (d, 3 J = 8.0 Hz, 1H, H14), 6.94–6.97 (t, 3 J = 7.6 Hz, 1H, H15), 6.63–6.67 (m, 3H, H6, 9,10), 6.11–6.14 (s, 1H, CONH), 4.08 (bs, 1H, CH), 3.81 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.68–3.69 (m, 2H, SCH2Ph), 3.41–3.60 (m, 2H, NCH2), 3.16–3.20 (dd, 2 J = 13.6 Hz, 3 J = 3.6 Hz, 1H, CH2S), 2.79–2.85 (dd, 2 J = 14.0 Hz, 3 J = 8.8 Hz, 1H, CH2S), 2.73–2.76 (t, 3 J = 6.8 Hz, 2H, CH2Ar) ppm; 13C{1H} NMR (CDCl3): δ = 169.1 (C2), 166.9 (C11), 160.1 (C13), 156.6 (C20), 148.6 (C7), 147.2 (C8), 138.3 (C5), 132.7 (C12), 131.8 (C15), 131.7 (C17), 128.8 (C21), 128.3 (C22), 126.8 (C23), 120.5 (C10), 118.8 (C12), 116.5 (C14), 112.8 (C6), 111.8 (C9), 71.3 (C1), 55.4 (OCH3), 55.3 (OCH3), 40.5 (C3), 34.4 (C4), 33.3 (C20), 25.3 (C18) ppm; LC–MS (ESI): m/z calcd. 478.6 (M+), found 479.
(S)-N-(3,4-Dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]propanamide (15, C20H24N2O4)
Yellow crystalline solid; yield: 282 mg (97%); m.p.: 80–82 °C;(c = 0.1, CHCl3); FT-IR (ATR): = 3343, 2981, 2927, 2869, 1650, 1630, 1579, 1542, 1522, 1459, 1419, 1267, 1229, 1138, 1026, 873, 804, 763, 635, 572 cm−1; UV–Vis: λmax (ε) = 257 (13,788), 278 (11,800), 319 (3,876) nm (M−1 cm−1); 1H NMR (399.65 MHz, CDCl3): δ = 12.36 (s, 1H, OH), 8.34 (s, 1H, CH = N), 7.39–7.41 (td, 1H, H16), 7.27–7.30 (d, 3 J = 8.0 Hz, 1H, H17), 6.99–7.01 (d, 3 J = 8.0 Hz, 1H, H14), 6.92–6.96 (t, 3 J = 7.2 Hz, 1H, H15), 6.65–6.68 (m, 3H, H6, 9, 10), 6.06 (s, 1H, CONH), 3.95–4.00 (q, 1H, CH), 3.82 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.44–3.56 (m, 2H, NCH2), 2.74–2.77 (t, 3 J = 7.2 Hz, 2H, CH2Ar), 1.52–1.54 (d, 3 J = 3.4 Hz, 3H, CH3) ppm; 13C{1H} NMR (CDCl3): δ = 170.9 (C2), 165.8 (C11), 160.3 (C13), 148.6 (C7), 147.2 (C8), 132.5 (C5), 131.8 (C15), 131.8 (C17), 120.5 (C12), 118.9 (C16), 118.7 (C14), 116.8 (C10), 112.4 (C6), 111.7 (C9), 66.6 (C1), 55.4 (OCH3), 54.9 (OCH3), 40.4 (C3), 34.6 (C4), 19.7 (CH3) ppm; LC–MS (ESI): m/z calcd. 356.41 (M+), found 357.
(R)-N-(3,4-Dimethyoxyphenylethyl)-2-[(2-hydroxybenzylidene)amino]-3-phenylpropanamide (16, C26H28N2O4)
Yellow solid; yield: 340 mg (96.0%); m.p.: 89–90 °C;(c = 0.1, CHCl3); FT-IR (ATR): = 3326, 2923, 2850, 1655, 1625, 1516, 1451, 1237, 1143, 1019, 805, 760, 705, 636, 540 cm−1; UV–Vis: λmax (ε) = 259 (24,000), 280 (15,420), 319 (6,840) nm (M−1 cm−1); 1H NMR (399.65 MHz, DMSO-d6): δ = 12.21 (s, 1H, OH), 7.89 (s, 1H, CH = N), 7.33–7.35 (td, 1H, H16), 7.24–7.22 (d, 3 J = 7.6 Hz, 1H, H17), 7.26–7.09 (m, 5H, H20-23), 6.96–6.99 (d, 3 J = 8.4 Hz, 1H, H14), 6.85–6.89 (t, 3 J = 7.6 Hz, 1H, H15), 6.63–6.65 (m, 2H, H9,10), 6.57–6.58 (m, 1H, H6), 6.01 (bs, 1H, CONH), 3.99–4.02 (dd, 3 J = 8.8 Hz, 3 J = 4.0 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.464–3.59 (m, 2H, NCH2), 3.35–3.40 (dd, 3 J = 13.6 Hz, 2 J = 3.4 Hz, 1H, H18), 3.06–3.11 (dd, 3 J = 13.2 Hz, 2 J = 8.8 Hz, 1H, H18), 2.63–2.76 (m, 2H, CH2Ar) ppm; LC–MS (ESI): m/z calcd. 432.5 (M+), found 432.
Synthesis of Schiff bases of amides of amino acids (17 and 18)
Compound 9/10 (229/275 mg, 0.73 mmol) was dissolved in 20 cm3 dry methanol and stirred the solution for 0.5 h at room temperature (RT). A solution of 100 mg 2-hydroxyacetophenone (0.73 mmol) in 20 cm3 dry methanol was added to the above solution drop-wise under continuous stirring. The resulting mixture was stirred further for 2–3 h at RT during which time the solution changes to yellow. The progress of the reaction was monitored on TLC using petroleum-ether and ethyl acetate (50:50) as mobile phase. The yellow precipitate left, on evaporation of solvent by a rotary evaporator, was washed twice with n-hexane (10 cm3 × 2) and then dried under vacuum. The dry solid was recrystallized from a mixture of chloroform and n-hexane (1:1) resulting a yellow crystalline solids of 17/18, respectively.
(S)-N-(3,4-Dimethyoxyphenylethyl)-2-[[1-(2-hydroxyphenyl)ethylidene]amino]-4-(methylthio)butanamide (17, C22H28N2O4S)
Yellow crystalline solid; yield: 284 mg (90%); m.p.: 91–93 °C;(c = 0.1, CHCl3); FT-IR (ATR): = 3304, 3063, 2930, 2850, 1644, 1619, 1543, 1516, 1465, 1452, 1374, 1307, 1236, 1158, 1091, 1028, 855, 803, 752, 675, 562, 524, 429 cm−1; UV–Vis: λmax (ε) = 280 (14,250), 326 (5.109) nm (M−1 cm−1); 1H NMR (399.82 MHz, DMSO-d6): δ = 14.99 (s, 1H, OH), 7.55–7.57 (d, 3 J = 7.6 Hz, 1H, H17), 7.35–7.39 (t, 3 J = 7.6 Hz, 1H, H16), 6.97–6.99 (d, 3 J = 8.0 Hz, 1H, H14), 6.87–6.91 (t, 3 J = 7.2 Hz, 1H, H15), 6.54–6.64 (m, 3H, H6, 9, 10), 5.90 (s, 1H, CONH), 4.52–4.55 (q, 1H, CH), 3.81 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.47–3.52 (q, 2H, NCH2), 2.71–2.75 (q, 2H, CH2S), 2.57–2.63 (m, 1H, CHCH2S), 2.41–2.47 (m, 1H, CHCH2S), 2.34–2.40 (m, 2H, CH2Ar), 2.31 (s, 3H, SCH3), 2.09 (s, 3H, CH3) ppm; 13C{1H} NMR (99.99 MHz, CDCl3): δ = 173.1 (C9), 169.9 (C2), 162.7 (C7), 148.6 (C14), 147.2 (C15), 132.4 (C4), 131.7 (C6), 128.9 (C12), 120.5 (C1), 119.3 (C17), 117.7 (C5), 117.1 (C3), 112.5 (C13), 111.8 (C16), 61.6 (C8), 55.5 (OCH3), 55.3 (OCH3), 34.4 (C10), 33.2 (C11), 29.6 (C19), 24.4 (C18), 14.9 (SCH3), 14.5 (CH3) ppm; LC–MS (ESI): m/z calcd. 430.56 (M+), found 431.
(S)-3-(Benzylthio)-N-(3,4-dimethyoxyphenylethyl)-2-[[1-(2-hydroxyphenyl)ethylidene]amino]propanamide (18, C28H32N2O4S)
Yellow solid; yield: 329 mg (91%); m.p.: 95–96 °C;(c = 0.1, CHCl3); FT-IR (ATR): = 3281, 2960, 2837, 1651, 1610, 1552, 1537, 1516, 1504, 1438, 1413, 1330, 1261, 1192, 1136, 1118, 1028, 910, 812, 756, 706 cm−1; UV–Vis: λmax (ε) = 279 (20,823), 322 (7,400) nm (M−1 cm−1); 1H NMR (399.82 MHz, DMSO-d6): δ = 15.49 (s, 1H, OH), 8.042 (s, 1H, H6), 7.62–7.64 (d, 1H, H9), 7.20–7.28 (m, 5H, Ph), 6.72–6.80 (m, 4H, H14, H15, H16, H17), 6.63–6.65 (d, 3 J = 6.4 Hz, 2H, H10), 6.63–6.65 (d, 1H, H12), 5.52–5.54 (s, 1H, CONH), 4.48–4.51 (t, 3 J = 6.4 Hz, 1H, CH), 3.70–3.74 (m, 2H, SCH2Ph), 3.67 (s, 3H, OCH3), 3.66 (s, 3H, OCH3), 3.27–3.30 (m, 2H, NCH2), 2.67–2.74 (dd, 2 J = 13.2 Hz, 3 J = 7.6 Hz, 1H, CH2S), 2.69–2.72 (dd, 2 J = 13.2 Hz, 3 J = 5.6 Hz, 1H, CH2S), 2.61–2.65 (t, 2H, CH2Ar), 2.70 (s, 3H, CH3) ppm; 13C{1H} NMR (99.99 MHz, DMSO-d6): δ = 173.2 (C9), 169.3 (C2), 162.3 (C7), 156.6 (C20), 148.6 (C14), 147.2 (C15), 138.3 (C12), 132.5 (C1), 131.7 (C4), 129.1 (C6), 128.5 (C21, C25), 128.4 (C22, C24), 126.8 (C23), 120.5 (C17), 117.7 (C5), 117.4 (C3), 112.5 (C13), 111.8 (C16), 62.5 (C8), 55.6 (OCH3), 55.3 (OCH3), 35.5 (C10), 34.8 (C11), 33.4 (C19), 24.7 (C18), 15.2 (CH3) ppm; LC–MS (ESI): m/z calcd. 492.63 (M+), found 492.9.
Preparation of platelet-rich plasma (PRP) and platelet-poor plasma (PPP)
The platelet concentration of PRP was adjusted to 3.1 × 108 platelets/cm3 with PPP. The PRP maintained at 37 °C and was used within 2 h for the aggregation process. All the above preparations were carried out using plastic wares or siliconized glass wares.
Plasma re-calcification time
The plasma re-calcification time experiment was carried out according to the reported method [67–69]. Briefly, the compounds 13–18 (2.5 to 40 µM) were pre-incubated with 0.1 cm3 of citrated human PRP in presence of 10 mM Tris–HCl (10 mm3) buffer of pH 7.4 for 5 min at 37 °C. 20 mm3 of 0.25 M CaCl2 was added to the pre-incubated mixture and then the clotting time was recorded in seconds.
Platelets’ aggregation
The platelets’ aggregation experiment was carried out by turbidometric method [67–69] using a Chronolog dual channel aggregometer connected to an Omni scribedual pen recorder to record the light transmission as a function of time. The 500 mm3 of reaction volume containing PRP (0.45 cm3) and newly synthesized compounds 13–18 (10 to 30 µM) was pre-incubated for 3 min in a cylindrical glass cuvette under constant stirring. The aggregation was initiated by the addition of adenosine diphosphate (ADP, 5 µM) an agonist and followed for 6 min. Furthermore, these new Schiff bases were checked for their effect on platelet aggregation function in the absence of any agonist. As platelets aggregate, light transmission increases progressively producing an aggregation trace on the recorder which was a plot of light transmission between PRP and PPP corresponding to the 0 and 100% aggregation against time in s.
Non-haemolytic activity
The effect of new Schiff bases 13–18 on red blood cells was studied as per the previously described procedure [67–69]. Hemolytic activity was determined using washed human erythrocytes. Briefly, packed human erythrocytes and phosphate buffered saline (PBS) (1:9 v/v) were mixed. 1 cm3 of this suspension was incubated independently with the various amounts of Schiff bases (0–100 µg) for 1 h at 37 °C. The reaction was stopped by adding 9 cm3 of ice-cold PBS and then centrifuged at 1000 g for 10 min at 37 °C. The amount of hemoglobin released in the supernatant was measured at 540 nm on UV–Vis spectrophotometer. The activity was expressed as percent of haemolysis against 100% lysis of cells due to addition of water that served as a positive control and PBS served as a negative control.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by CSIR, New Delhi, India, Grant number No. 01(2701)/12/EMR-II). P. Raghavendra Kumar has received this research support.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Vale N, Ferreira A, Matos J, Fresco P, Gouveia MJ. Molecules. 2018;23:2318. doi: 10.3390/molecules23092318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giannis A, Kolter T. Angew Chem Int Ed. 1993;32:1244. doi: 10.1002/anie.199312441. [DOI] [Google Scholar]
- 3.Hanessian S, McNaughton-Smith G, Lombart H-G, Lubell W. Tetrahedron. 1997;53:12789. doi: 10.1016/S0040-4020(97)00476-6. [DOI] [Google Scholar]
- 4.Chakraborty TK, Ghosh S, Jayaprakash S. Curr Med Chem. 2002;9:421. doi: 10.2174/0929867023370941. [DOI] [PubMed] [Google Scholar]
- 5.Rainaldi M, Moretto V, Crisma M, Peggion E, Mammi S, Toniolo C, Cavicchioni C. J Pept Sci. 2002;8:241. doi: 10.1002/psc.392. [DOI] [PubMed] [Google Scholar]
- 6.Mann E, Chana A, Sanchez-Sancho F, Puerta C, Garcia-Merino A, Herradon B. Adv Synth Catal. 2002;344:855. doi: 10.1002/1615-4169(200209)344:8<855::AID-ADSC855>3.0.CO;2-1. [DOI] [Google Scholar]
- 7.Kee S, Jois SDS. Curr Pharm Des. 2003;9:1209. doi: 10.2174/1381612033454900. [DOI] [PubMed] [Google Scholar]
- 8.Braga AL, Lüdtke DS, Paixão MW, Alberto EE, Stefani HA, Juliano L. Eur J Org Chem. 2005 doi: 10.1002/ejoc.200500530. [DOI] [Google Scholar]
- 9.Saitton S, Del TAL, Mohell N, Vollinga RC, Boström D, Kihlberg J, Luthman K. J Med Chem. 2004;47:6595. doi: 10.1021/jm049484q. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y. Curr Opin Chem Biol. 2005;9:118. doi: 10.1016/j.cbpa.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 11.Giannis A, Rubsam F. Adv Drug Res. 1997;29:1. doi: 10.1016/S0065-2490(97)80013-5. [DOI] [Google Scholar]
- 12.Ripka AS, Rich DH. Curr Opin Chem Biol. 1998;2:441. doi: 10.1016/S1367-5931(98)80119-1. [DOI] [PubMed] [Google Scholar]
- 13.Cony EJ, Link JO. J Org Chem. 1991;56:442. doi: 10.1021/jo00001a084. [DOI] [Google Scholar]
- 14.Gu JX, Li ZY, Lin G-Q. Chin J Chem. 1995;13:475. doi: 10.1002/cjoc.19950130516. [DOI] [Google Scholar]
- 15.Argyropoulos SV, Sandford JJ, Nutt D. Pharmacol Ther. 2000;88:213. doi: 10.1016/S0163-7258(00)00083-8. [DOI] [PubMed] [Google Scholar]
- 16.Rozwadowskar MD, Sulima A. Tetrahedron. 2001;57:3499. doi: 10.1016/S0040-4020(01)00224-1. [DOI] [Google Scholar]
- 17.Nutt D, Ballenger J. Anxiety disorders. Oxford: Blackwell; 2003. [Google Scholar]
- 18.Neuvonen K, Fulop F, Neuvonen H, Koch A, Kleinpeter E, Pihlaja K. J Org Chem. 2005;70:10670. doi: 10.1021/jo0507946. [DOI] [PubMed] [Google Scholar]
- 19.Quevedo R, Baquero E, Rodriguez M. Tetrahedron Lett. 2010;51:1774. doi: 10.1016/j.tetlet.2010.01.115. [DOI] [Google Scholar]
- 20.Gluck MR, Zeevalk GD. J Neurochem. 2004;91:788. doi: 10.1111/j.1471-4159.2004.02747.x. [DOI] [PubMed] [Google Scholar]
- 21.Abd-Elzaher MM. Appl Organomet Chem. 2004;18:149. doi: 10.1002/aoc.608. [DOI] [Google Scholar]
- 22.Oliva-Madrid MJ, Garcıa-Lopez J-A, Saura Llamas I, Bautista D, Vicente J. Organometallics. 2012;31:3647. doi: 10.1021/om300141b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giovannantonio MD, Kosmala T, Bonanni B, Serrano G, Zema N, Turchini S, Catone D, Wandelt K, Pasini D, Contini G, Goletti C. J Phys Chem C. 2015;119:19228. doi: 10.1021/acs.jpcc.5b05547. [DOI] [Google Scholar]
- 24.Boiocchi M, Bonizzoni M, Moletti A, Pasini D, Taglietti A. New J Chem. 2007;31:352. doi: 10.1039/b616492g. [DOI] [Google Scholar]
- 25.Colombo S, Coluccini C, Caricato M, Gargiulli C, Gattuso G, Pasini D. Tetrahedron. 2010;66:4206. doi: 10.1016/j.tet.2010.03.102. [DOI] [Google Scholar]
- 26.Kajal A, Bala S, Kamboj S, Sharma N, Saini V. J Cat. 2013;2013:893512. [Google Scholar]
- 27.Abu-Dief AM, Mohamed IMA. Beni-suef Univ J Basic App Sci. 2015;4:119. doi: 10.1016/j.bjbas.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.da Silva CM, da Silva DL, Modolo LV, Alves RB, de Resende MA, Martins CVB, de Fatima A. J Adv Res. 2011;2:1. doi: 10.1016/j.jare.2010.05.004. [DOI] [Google Scholar]
- 29.Khalaf RA, Awad M, Al-Essa L, Mefeh S, Sabbah D, Al-Shalabi E, Shabeeb I. Mol Diversity. 2022;26:1213. doi: 10.1007/s11030-021-10253-z. [DOI] [PubMed] [Google Scholar]
- 30.Vicini P, Geronikaki A, Incerti M, Busonera B, Poni G, Cabras CA, Colla PL. Bioorg Med Chem. 2003;11:4785. doi: 10.1016/S0968-0896(03)00493-0. [DOI] [PubMed] [Google Scholar]
- 31.Kumar KS, Ganguly S, Veerasamy R, Clercq E. Eur J Med Chem. 2010;45:5474. doi: 10.1016/j.ejmech.2010.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pandey A, Dewangan D, Verma S, Mishra A, Dubey RD. Int J Chem Tech Res. 2011;3:178. [Google Scholar]
- 33.Sztanke K, Maziarka A, Osinka A, Sztanke M. Bioorg Med Chem. 2013;21:3648. doi: 10.1016/j.bmc.2013.04.037. [DOI] [PubMed] [Google Scholar]
- 34.Aziz AN, Taha M, Ismail NH, Anouar EH, Yousuf S, Jamil W, Awang K, Ahmat N, Khan KM, Kashif SM. Molecules. 2014;19:8414. doi: 10.3390/molecules19068414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amgad MR. Chem Pap. 2021;75:4669. doi: 10.1007/s11696-021-01640-9. [DOI] [Google Scholar]
- 36.Younis A, Rakha T, El-Gamil M, El-Reash G. J Mol Struct. 2021 doi: 10.1016/j.molstruc.2021.131110. [DOI] [Google Scholar]
- 37.Jan MM, Sheikh AM, Aabid HS. Rev Inorg Chem. 2021;41:199. doi: 10.1515/revic-2020-0020. [DOI] [Google Scholar]
- 38.Denson KWE. Toxicon. 1969;5:5. doi: 10.1016/0041-0101(69)90154-8. [DOI] [PubMed] [Google Scholar]
- 39.Zanetti VC, da Silveria RB, Dreyfuss JL, Haoach J, Mangili C, Veiga SS. Fibrinolysis. 2002;13:135. doi: 10.1097/00001721-200203000-00009. [DOI] [PubMed] [Google Scholar]
- 40.Devaraja S, Nagaraju S, Mahadeshwarasawmy YH, Girish KS, Kemparaju K. Toxicon. 2008;52:130. doi: 10.1016/j.toxicon.2008.04.168. [DOI] [PubMed] [Google Scholar]
- 41.Rajesh R, Raghavendra CD, Nataraju A, Dhanjay DL, Vishwanath BS. Toxicon. 2005;46:84. doi: 10.1016/j.toxicon.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 42.Kumar MNS, Jayanna K, Chethana R, Ashwini S, Chandramma, Girish KS, Kemparaju K, Devaraja S. Pharmacog Mag. 2018;14:175. doi: 10.4103/pm.pm_320_17. [DOI] [Google Scholar]
- 43.Ren Y, Patel K, Crane T. J Extra Corpor Technol. 2010;42:103. [PMC free article] [PubMed] [Google Scholar]
- 44.Ishwar BK, Mishra SK, Jainey PJ, Shastry CS. J Chem Pharm Res. 2011;3:114. [Google Scholar]
- 45.Anacona JR, Noriega N, Camus J. Spectrochim Acta Part A Mol Biomol Spectrosc. 2015;137:16. doi: 10.1016/j.saa.2014.07.091. [DOI] [PubMed] [Google Scholar]
- 46.Tehrani KHME, Sardari S, Mashayekhi V, Zadeh ME, Azerang P, Kobarfard F. Chem Pharm Bull. 2013;61:160. doi: 10.1248/cpb.c12-00651. [DOI] [PubMed] [Google Scholar]
- 47.Al-Saad HN, Mahmood MAR, AlBayati RI. Orient J Chem. 2019;35:829. doi: 10.13005/ojc/350246. [DOI] [Google Scholar]
- 48.Lima LM, Ormelli CB, Fraga CA, Miranda AL, Barreiro EJ. J Brazil Chem Soc. 1999;10:421. doi: 10.1590/S0103-50531999000500013. [DOI] [Google Scholar]
- 49.Bermudez LE, Reynolds R, Kolonoski P, Aralar P, Inderlied CB, Young LS. Antimicrob Agents Chemother. 2003;47:2685. doi: 10.1128/AAC.47.8.2685-2687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shahab FM, Kobarfard F, Shafaghi B, Dadashzadeh S. Xenobiotica. 2010;40:225. doi: 10.3109/00498250903461411. [DOI] [PubMed] [Google Scholar]
- 51.Lima LM, Frattani FS, Dos Santos JL, Castro HC, Fraga CAM, Zingali RB, Barreiro EJ. Eur J Med Chem. 2008;43:348. doi: 10.1016/j.ejmech.2007.03.032. [DOI] [PubMed] [Google Scholar]
- 52.Cunha AC, Figueiredo JM, Tributino JLM, Miranda ALP, Castro HC, Zingali RB, Fraga CAM, de Souza MCBV, Ferreira VF, Barreiro EJ. Bioorg Med Chem. 2003;11:2051. doi: 10.1016/S0968-0896(03)00055-5. [DOI] [PubMed] [Google Scholar]
- 53.Silveira IA, Paulo LG, Miranda AL, Rocha SO, Freitas AC, Barreiro EJ. J Pharm Pharmacol. 1993;45:646. doi: 10.1111/j.2042-7158.1993.tb05670.x. [DOI] [PubMed] [Google Scholar]
- 54.Asif M, Husain A. J App Chem. 2013 doi: 10.1155/2013/247203. [DOI] [Google Scholar]
- 55.Mashayekhi V, Tehrani KHME, Amidi S, Kobarfard F. Chem Pharm Bull. 2013;61:144. doi: 10.1248/cpb.c12-00597. [DOI] [PubMed] [Google Scholar]
- 56.Amidi S, Esfahanizadeh M, Tabib K, Soleimani Z, Kobarfard F. ChemMedChem. 2017;12:962. doi: 10.1002/cmdc.201700123. [DOI] [PubMed] [Google Scholar]
- 57.Dutra LA, Guanaes JFO, Johmann N, Pires MEL, Chin CM, Marcondes S, Santos JLD. Bioorg Med Chem Lett. 2017;27:2450. doi: 10.1016/j.bmcl.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 58.Chen G, Wang C, Zhang Z, Liu X. Med Chem Res. 2019;28:1413. doi: 10.1007/s00044-019-02381-x. [DOI] [Google Scholar]
- 59.Amidi S, Kobarfard F, Moghaddam AB, Tabib K, Soleymani Z. Iran J Pharm Res. 2013;12:91. [PMC free article] [PubMed] [Google Scholar]
- 60.Akhlaghi MF, Amidi S, Esfahanizadeh M, Daeihamed M, Kobarfar F. Iran J Pharm Res. 2014;13:35. [PMC free article] [PubMed] [Google Scholar]
- 61.Tehrani KHME, Zadeh ME, Mashayekhi V, Hashemi M, Kobarfar F, Gharebaghi F, Mohebbi S. Iran J Pharm Res. 2015;14:1077. [PMC free article] [PubMed] [Google Scholar]
- 62.Wanga C, Wanga Y, Denga Q, Liua X. Iran J Pharm Res. 2019;18:1803. doi: 10.22037/ijpr.2019.1100856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalhor N, Mardani M, Abdollahzadeh S, Vakof M, Zadeh ME, Tehrani KHME, Kobarfard F, Mohebbi S. Bull Korean Chem Soc. 2015;36:2632. doi: 10.1002/bkcs.10531. [DOI] [Google Scholar]
- 64.Mirfazli SS, Khoshneviszadeh M, Jeiroudi M, Foroumadi A, Kobarfard F, Shafiee A. Med Chem Res. 2016;25:1. doi: 10.1007/s00044-015-1440-7. [DOI] [Google Scholar]
- 65.Kumar MNS, Kengaiah J, Ramachandraiah C, Shivaiah A, Chandramma, Girish KS, Kemparaju K, Sannaningaiah D. Pharmacog Mag. 2018;14:175. doi: 10.4103/pm.pm_320_17. [DOI] [Google Scholar]
- 66.Sowmyashree G, Bhagyalakshmi M, Girish KS, Kemparaju K, Rangaiah SM, Jane HP. Pharmacogn J. 2015;7:171. doi: 10.5530/pj.2015.3.5. [DOI] [Google Scholar]
- 67.Marulasiddeshwara MB, Dakshayani SS, Kumar MNS, Chethana R, Kumar PR, Devaraja S (2017) Mater Sci Eng C 81182 [DOI] [PubMed]
- 68.Dakshayani SS, Marulasiddeshwara MB, Kumar MNS, Ramesh G, Kumar PR, Devaraja S, Hosamani R. Int J Biol Macromol. 2019;131:787. doi: 10.1016/j.ijbiomac.2019.01.222. [DOI] [PubMed] [Google Scholar]
- 69.Ramesh G, Kumar MNS, Kumar PR, Suchetan PA, Devaraja S, Foro S, Nagaraju G. J Mol Struct. 2020;1200:127040. doi: 10.1016/j.molstruc.2019.127040. [DOI] [Google Scholar]
- 70.Slebioda M, Wodecki Z, Kolodziejczyk AM. Int J Pept Protein Res. 1990;35:539. doi: 10.1111/j.1399-3011.1990.tb00258.x. [DOI] [PubMed] [Google Scholar]
- 71.Giannola LI, De Caro V, Giandalia G, Siragusa MG, Lamartina L. Pharmazie. 2008;63:704. [PubMed] [Google Scholar]
- 72.Escorihuela J, Altava B, Burguete MI, Luis SV. Tetrahedron. 2013;69:551. doi: 10.1016/j.tet.2012.11.025. [DOI] [Google Scholar]
- 73.Anna Z, Andrzej L, Jan K M, Krystyna W, Aleksandra S, Dariusz B, Zbigniew C (2003) Eur J Org Chem 2443
- 74.Kumar PR, Singh AK, Butcher RJ, Sharma P, Toscano RA (2004) Eur J Inorg Chem 1107
- 75.Gupta KC, Sutar AK. Coord Chem Rev. 2008;252:1420. doi: 10.1016/j.ccr.2007.09.005. [DOI] [Google Scholar]
- 76.Vitale AA, Stahl AE, Claro PCS, Addato MAF, Diez RP, Jubert AH. J Mol Struct. 2008;881:167. doi: 10.1016/j.molstruc.2007.09.019. [DOI] [Google Scholar]
- 77.Dong X, Li Y, Li Z, Cui Y, Zhu H. J Inorg Biochem. 2012;108:22. doi: 10.1016/j.jinorgbio.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 78.Satheesh CE, Kumar PR, Sharma P, Lingaraju K, Palakshamurthy BS, Rajanaika H. Inorg Chim Acta. 2016;442:1. doi: 10.1016/j.ica.2015.11.017. [DOI] [Google Scholar]
- 79.Satheesh CE, Kumar PR, Shivakumar N, Lingaraju K, Krishna PM, Rajanaika H, Hosamani A. Inorg Chim Acta. 2019;495:118929. doi: 10.1016/j.ica.2019.05.028. [DOI] [Google Scholar]
- 80.Filarowski A. J Phys Org Chem. 2005;18:686. doi: 10.1002/poc.940. [DOI] [Google Scholar]
- 81.Sheldrick GM. Acta Cryst. 2015;A71:3. [Google Scholar]
- 82.Sheldrick GM. Acta Cryst. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [Google Scholar]
- 83.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. J Appl Cryst. 2009;42:339. doi: 10.1107/S0021889808042726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bourhis LJ, Dolomanov OV, Gildea RJ, Howard JAK, Puschmann H. Acta Cryst. 2015;A71:59. doi: 10.1107/S2053273314022207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.