

Abstract
o-Carboranyl compounds contain specific geometries, ranging from planar to orthogonally distorted biphenyl rings. Herein, 13 o-carboranyl compounds, 1HF–13PP, were synthesized and fully characterized to determine the impact of structural formation of the aromatic group appended with the o-carborane to estimate the efficiency of their radiative decay process. All the compounds exhibited significant intramolecular charge transfer (ICT)-based emission in the crystalline state at 298 K. Remarkably, increasing the distorted dihedral angles between biphenyl rings gradually decreased the emission efficiencies. Furthermore, their radiative decay constants decreased linearly with increasing dihedral angles, which demonstrated the inversely proportional relationship between these two factors. These findings distinctly suggest that the planar or distorted geometry of substituted aryl groups can strongly affect the efficiency of the ICT-based radiative process in o-carboranyl luminophores.
Introduction
Unique photophysical properties1 of organic luminophores bearing ortho-closo-carborane (C2B10H10), a well-known icosahedral boron cluster viewed as a three-dimensional variant of benzene,2,3 have recently provided insights for applying the optoelectronic materials in the field of organic light-emitting diodes,4−6 organic thin-film transistors,7,8 and photovoltaic cells.9,10 In particular, investigations for enhancing their emissive characteristics derived from intramolecular charge transfer (ICT) transition via a strong electron-withdrawing inductive effect on C-substituents and high polarizability of their σ-aromatic framework have consistently proceeded in a decade.11−35 The crucial factors that severely affect the efficiency of the ICT-based radiative mechanism are mainly divided into two categories: (1) structural fluctuations around the o-carborane cage, such as the rotation of the cage26−28 and elongation of the C–C bond into the cage29−33 due to its electron-withdrawing character, and (2) geometrical characteristics of the aromatic group substituted in the cage, such as the orthogonality between the C–C bond and substituted aryl plane34 and the planarity of the aryl rings.35−37 Most factors, excluding the impact of planarity, have been significantly examined by an empirical comparison between the structural isomers of various o-carboranyl compounds and their theoretical analysis based on time-dependent density functional theory (TD-DFT) calculations.38 Our group has been focusing on the impact of molecular geometry, particularly the planarity of the substituted aryl rings, on ICT transition in o-carboranyl luminophores. We recently determined that this impact could be verified experimentally. A comparison of the photophysical properties of o-carboranyl biphenyl and fluorene compounds35 or perfectly planar and distorted para-terphenyl compounds36 revealed that radiative ICT transitions depended on the planarity of the substituted phenyl rings. Several studies have investigated the critical role of such geometrical arrangements in controlling the intrinsic electronic characteristics of o-carboranyl compounds. However, quantitative and in-depth analyses of the relationship between the photophysical properties and structural geometry of the luminescent o-carboranes bearing various aromatic groups are still rare.
Thus, 13 species of biphenyl-based o-carboranes (1HF–13PP, Chart 1), possessing different distortion angles between the biphenyl rings, were strategically designed and prepared to ascertain the relationship between the geometrical arrangement and the ICT-based radiative decay process of the o-carboranyl compounds and further establish the specific molecular geometry, which could maximize their radiative efficiency. The quantitative comparison of their ICT-based radiative decay efficiency in fixed geometry (crystalline solid structure) distinctly provided the structural key factor to enhance the luminescent behaviors. To the best of our knowledge, this is the first study where such a relationship is determined quantitatively. The detailed synthetic procedures, characterization, photophysical property analysis, and theoretical calculation results have been described further.
Chart 1. Molecular structures of biphenyl-based o-carboranyl compounds (1HF–13PP) in the present study.

Results and Discussion
Synthesis and Characterization
Detailed synthetic procedures for biphenyl-based o-carboranyl compounds, 1HF–13PP, possessing structurally different distortion angles between the biphenyl rings, are illustrated in Scheme 1 of the Supporting Information. Each acetylene precursor for o-carboranyl compounds was prepared by a palladium-catalyzed reaction, in other words, by Sonogashira coupling between phenylacetylene and either of the corresponding bromo compounds (1HFA, 2HTA, 4HNA, 5HHA, 7HDA, and 8HPA) or Suzuki–Miyaura coupling using boronic acid derivatives (6HCA and 9HMA–13PPA). The cage-forming reaction39 of decaborane (B10H14) with each acetylene precursor afforded o-carborane compounds 1HF, 2HT, and 4HN–13PP in relatively moderate yields (37–70%, Scheme S1). In particular, subsequent treatments using 4HN with excess oxidizing agents such as 2,3-dichloro-5,6-dicyano-p-benzoquinone converted an ethyl bridge to an ethylene moiety, that is, 3HB in high yield (85%). All synthesized precursors and biphenyl-based o-carboranyl compounds were characterized by multinuclear nuclear magnetic resonance (NMR) spectroscopy (Figures S1–S40 in Supporting Information) and elemental analyses. The broad 1H{11B decoupled} peaks in the upfield region (3.5–2.5 ppm) of the o-carboranyl compounds could be assigned to total 10H atoms, and three peaks between −2 and −11 ppm in the 11B{1H} NMR spectra distinctly confirmed the presence of a closo-o-carborane cage.
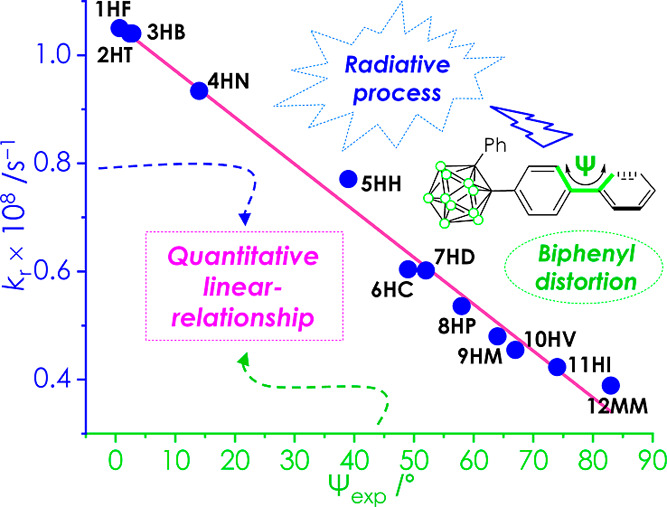
The single-crystal X-ray diffraction (XRD) analysis of 1HF–13PP demonstrates their solid-state molecular structures (Figure 1). Detailed parameters, including selected bond lengths and angles, are illustrated in Tables S1 and S2 of Supporting Information. Remarkably, the results suggested that the dihedral angles (Ψexp = C5–C6–C9–C10, Figure 1 and Table 1) between the biphenyl rings could be altered by the intramolecular steric hindrance owing to the bulkiness of the substituted functional group in the biphenyl moiety. Consequently, distorted geometries were observed for biphenyl rings of o-carboranyl compounds, with planar (1HF: Ψexp = 0.7°) to nearly perpendicular structures (12MM and 13PP: Ψexp = 83°). These values were crucial factors in verifying the relationship between the structural geometry and the efficiency of the radiative process in the o-carboranyl compounds.
Figure 1.

Molecular structures of biphenyl-based o-carboranyl compounds (30% thermal ellipsoids, hydrogen atoms were omitted for clarity) and their dihedral angles (Ψexp = C5–C6–C9–C10, green-dash box) between biphenyl rings.
Table 1. Photophysical and Structural Data for Biphenyl-Based o-Carboranyl Compounds (1HF–13PP).
| λabsa/nm (ε × 10–3 M–1 cm–1) | λex/nm | λemb/nm | Φemb,c | τobsb/ns | krb,d/s–1 × 108 | knrb,e/s–1 × 108 | Ψexpf/deg | Ψcalcg/deg | |
|---|---|---|---|---|---|---|---|---|---|
| 1HF | 282 (19.2), 306 (18.7) | 307 | 477 | 0.341 | 3.25 | 1.05 | 2.03 | 0.67 | 0.12 |
| 2HT | 285 (16.0), 311 (6.3) | 292 | 542 | 0.317 | 3.05 | 1.04 | 2.24 | 2.4 | 0.74 |
| 3HB | 264 (64.2), 290 (19.2) | 296 | 483 | 0.303 | 2.91 | 1.04 | 2.40 | 2.8 | 0.29 |
| 4HN | 286 (58.9), 308 (37.7) | 308 | 461 | 0.281 | 3.01 | 0.934 | 2.39 | 14 | 20 |
| 5HH | 269 (34.5) | 290 | 453 | 0.229 | 2.97 | 0.771 | 2.60 | 39 | 37 |
| 6HC | 266 (17.6) | 291 | 479 | 0.227 | 3.76 | 0.604 | 2.06 | 49 | 48 |
| 7HD | 266 (17.9) | 286 | 469 | 0.251 | 4.17 | 0.602 | 1.80 | 52 | 49 |
| 8HP | 238 (26.3), 270 (13.2) | 300 | 480 | 0.241 | 4.50 | 0.536 | 1.69 | 58 | 51 |
| 9HM | 256 (24.3) | 283 | 493 | 0.182 | 3.79 | 0.480 | 2.16 | 64 | 54 |
| 10HV | 237 (31.9), 266 (19.9) | 296 | 451 | 0.115 | 2.53 | 0.455 | 3.50 | 67 | 57 |
| 11HI | 251 (15.5) | 281 | 452 | 0.074 | 1.75 | 0.423 | 5.29 | 74 | 59 |
| 12MM | 229 (24.9), 272 (6.5) | 275 | 469 | 0.112 | 2.88 | 0.389 | 3.08 | 83 | 89 |
| 13PP | 225 (25.6), 271 (3.3) | 270 | 492 | <0.01 | h | 83 | 90 |
Measured in THF (3.0 × 10–5 M).
Measured in a crystalline state at 298 K.
Absolute PL quantum yield.
kr = Φem/τobs.
knr = kr(1/Φem–1).
Experimental dihedral angle between biphenyl rings from each X-ray crystal structure.
Calculated dihedral angle between the biphenyl rings from each optimized structure in the ground (S0) state.
Not observed due to weak emission.
Analysis of Optical Properties Based on Theoretical Calculations
UV–vis absorption and PL spectroscopy experiments were conducted under various conditions for o-carboranyl compounds 1HF–13PP, which provided information of each photophysical characteristic (Figures 2 and S41, Tables 1, and S3). All the compounds exhibited strong absorption bands centered at λabs = 251–311 nm (Table 1) due to the vibronic structures undergoing spin-allowed π–π* transitions on the biphenyl group. The major bands were slightly red-shifted compared to the absorption center (Figure S42) of 1,1′-biphenyl because introducing o-carborane led to a large stabilization of the lowest unoccupied molecular orbital (LUMO) level via a strong inductive effect.40 Specific absorption features of these o-carboranyl compounds include substantially tailing absorption bands above 300 nm, which are attributed to typical ICT transitions in the o-carborane cages, which differ from 1,1′-biphenyl. The theoretical results, which were calculated by TD-DFT based on the B3LYP/6-31G(d,p) level of theory, could demonstrate the origin of such characteristics. The ground (S0)-state calculation for all the compounds revealed that the lowest-energy electronic transitions were mainly associated with the highest occupied molecular orbital (HOMO) → LUMO transitions (Figure S43). The HOMOs of each compound were predominantly localized on the biphenyl group (>96% in all compounds, insets of Figure S43); however, the LUMOs were substantially distributed over the o-carborane cages (21–38%). These S0-calculation results indicate that the low-energy electronic transitions of the o-carboranyl compounds originate from substantial ICT transitions, corresponding to the o-carborane cage.
Figure 2.
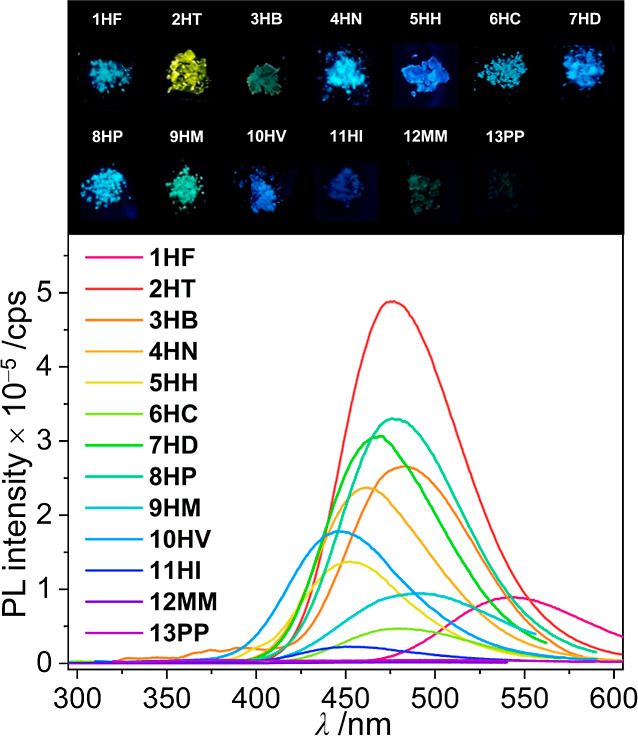
PL spectra for biphenyl-based o-carboranyl compounds (1HF–13PP) in a crystalline state. The inset illustrates the emission color observed under a hand-held ultraviolet lamp (λex = 265 nm).
The emissive properties of 1HF–13PP in the crystalline state (Figure 2 and Table 1) were examined by PL spectroscopy to investigate the radiative process in the specifically fixed molecular geometry. All the compounds exhibited strong (for 1HF–12MM) or moderate (for 13PP) emission in the range from λem = 451 nm; blue region (for 10HV) to 542 nm; yellow region (for 2HT). The emissive bands differed distinctly from those (λem = 308, 316 nm, Figure S42) of 1,1′-biphenyl based on the π–π* transition. Therefore, these could be conducted to reflect the ICT-based emissions involving the o-carborane cage. The results calculated for the S1-optimized structures of the o-carboranyl compounds suggested that the LUMO–HOMO type transitions (λem,calc = 498–529 nm, Figure 3) primarily contributed to the emissions. Interestingly, the LUMOs and HOMOs were significantly localized on the o-carborane cage (72–75%, insets of Figure 3) and biphenyl rings (>90%), respectively. These computational results confirm that the experimentally observed emissions in the crystalline state mainly originate from a radiative decay process based on ICT transitions between o-carborane and biphenyl rings. Similarly, such ICT-based emissions could be observed for o-carboranyl compounds in the rigid state in the region of 450–550 nm (λem = 459–487 nm in tetrahydrofuran (THF) solution at 77 K and 470–506 nm in the film state; bluish-green emission, Figure S41 and Table S3). However, the PL spectra of o-carboranyl compounds in THF solution at 298 K did not exhibit any emissive trace in this low-energy region; furthermore, the compounds either did not emit in various organic solvents (cyclohexane, dichloromethane (DCM), ethyl acetate, and acetone) at 298 K or only demonstrated a significantly weak emissive trace. This quenched ICT-based radiative decay was ascribed to the elongation of the C–C bond of the o-carborane cage in the solution state at an ambient temperature.11,12,16,18,26−28,31,34,37
Figure 3.

Frontier molecular orbitals of biphenyl-based o-carboranyl compounds 1HF–13PP at the first-excited singlet state (S1) with their relative energies from DFT calculations (isovalue = 0.04 a.u.) and molecular orbital distributions on the o-carborane moieties of the LUMO levels (%) and on the biphenyl group of the HOMO level. The transition energy (in nm) was calculated using the TD-B3LYP method with 6-31G(d,p) basis sets.
The estimated C–C bond lengths of the o-carborane cage in all the S1-optimized structures were approximately 2.39 Å (Table 2) and significantly exceeded those of the S0-structures (approximately 1.77 Å) and the experimental values of 1.71–1.73 Å. Such a feature was similarly observed in calculation results for diphenyl-ortho-carborane (1,2-Ph2-1,2-C2B10H10, 1);41 C–C bond length for 1 = 1.76 Å and [1]− (radical anion) = 2.39 Å. The fluctuation of the C–C bond elongation prevented the ICT-based radiative decay process, resulting in no emissive trace in PL spectra at 298 K.
Table 2. Experimental and Theoretical C–C Bond Lengths (Å) in the o-Carborane Cage for Biphenyl-Based o-Carboranyl Compounds (1HF–13PP).
| 1HF | 2HT | 3HB | 4HN | 5HH | ||
|---|---|---|---|---|---|---|
| exp.a | 1.73 | 1.73 | 1.72 | 1.72 | 1.72 | |
| calc.b | S0 | 1.77 | 1.76 | 1.76 | 1.77 | 1.76 |
| S1 | 2.39 | 2.40 | 2.40 | 2.39 | 2.39 | |
| 6HC | 7HD | 8HP | 9HM | 10HV | ||
| exp.a | 1.72 | 1.73 | 1.72 | 1.72 | 1.73 | |
| calc.b | S0 | 1.76 | 1.77 | 1.77 | 1.77 | 1.77 |
| S1 | 2.38 | 2.38 | 2.38 | 2.39 | 2.38 | |
| 11HI | 12MM | 13PP | ||||
| exp.a | 1.72 | 1.72 | 1.71 | |||
| calc.b | S0 | 1.77 | 1.76 | 1.76 | ||
| S1 | 2.39 | 2.39 | 2.40 |
Experimental values from their X-ray crystal structures (C1–C2 bond).
Calculated values from their ground (S0) and the first-excited singlet state (S1) optimized structures.
Intriguingly, the high-energy emissive trace in the 340–380 nm region could be assigned to the spin-allowed π–π*transition-based emission, that is, locally excited (LE)-based emission on the biphenyl moiety. Emissive patterns similar to those of 1,1-biphenyl (Figure S42) were observed in the PL spectra of 5HH and 10HV–13PP in all states (Figure S41 and Table S3). Moreover, the LE-based emission of 13PP was more intense than the ICT-based emission. These features confirm that the distorted geometry of the biphenyl rings plays an important role in alternating the intramolecular electronic transitions and the corresponding radiative decay mechanism.
Quantitative Analysis for the Relationship between Distorted Geometry and the ICT-Based Radiative Process in the Crystalline State
To quantitatively obtain insights into the relationship between the geometric structure of the distorted biphenyl ring and the radiative decay mechanism for ICT-based emissions, the quantum efficiencies (Φem) and decay lifetimes (τobs) of the o-carboranyl compounds were estimated in the crystalline state at 298 K (Table 1 and Figure S44). The emission decay lifetimes of the ICT-based emissions were 1.7–4.5 ns, indicating that the observed emission corresponded to fluorescence. The Φem values for the ICT-based emission in the crystalline state gradually decreased (Φem for 1HF = 34.1% decreased to 7.4% for 11HI, 11.2% for 12MM, and <1% for 13PP), upon increasing the distortional angle between the biphenyl rings (Ψexp for 1HF = 0.67° increased to 83° for 12MM and 13PP). Interestingly, the radiative (kr, Table 1) and nonradiative (knr) decay constants calculated from Φem and τobs revealed the relationship between the efficiency of the radiative process for ICT-based emission and biphenyl geometry. The kr values (1.05 × 108 s–1 for 1HF to 3.89 × 107 s–1 for 12MM, Table 1) for all the compounds exhibited an inversely proportional relationship with the corresponding Ψexp value (Figure 4, blue-dot and orange linear-fitting line), thereby estimating the accuracy of the linear-fitting equation [kr = (−8.64 × 105) × Ψexp + 1.06] as 99% (square R value, R2 = 0.9899). However, the knr values were almost within the range of the 1.7–2.6 × 107 s–1 box pattern, except for those of 10HV–12MM, as they possessed significantly low Φem values (<12%). Additionally, this relationship was graphically illustrated using the calculated values of the dihedral angle (Ψcalc, Table 1) between the biphenyl rings from each S0-optimized structure (Figure S45). The kr of 1HF–11HI was inversely related to the Ψcalc values ([kr = (−1.03 × 106) × Ψcalc + 1.07], R2 = 0.9706). These results strongly indicate that the biphenyl geometry, particularly the distorted structure, diminishes the radiative decay process based on ICT transitions corresponding to o-carborane. Thus, the planarity of the biphenyl group appended to the o-carborane cage is a crucial factor for controlling the efficiency of the ICT-based emission.
Figure 4.
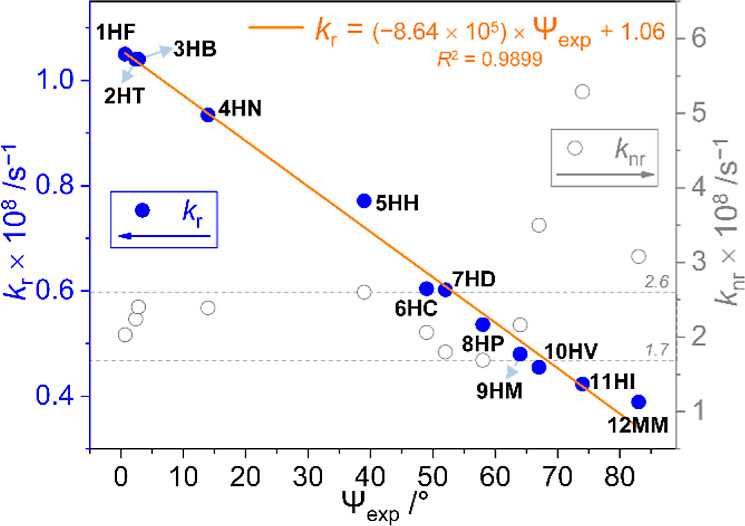
Radiative (kr, filled blue-circle, left) and nonradiative decay constants (knr, hollow gray circle, right) for the biphenyl-based o-carboranyl compounds (1HF–13PP) in the crystalline state as a function of the experimental dihedral angles (Ψexp) from each X-ray crystal structure. The orange line is its linear fitting for the kr values vs Ψexp.
Theoretical Modeling for the Effect of Distorted Geometry on Orbital Localization
The impact of the structural geometry of the biphenyl rings on the ICT-based radiative decay process was further verified by calculating the low-energy transitions assignable to ICT within o-carborane (LUMO → HOMO in THF) for 5HH in the S1 state as Ψ varied from 0 to 90° (Figure 5). The results revealed that the oscillator strengths (fcalc) of the transition dramatically decreased as the biphenyl rings became orthogonally distorted (fcalc = 0.5439 at Ψ = 0° to 0.2181 at 90°, Figure 5a and Table S4). Moreover, the computed emission wavelengths (λcalc) gradually changed to the values attributed to the LE-based high-energy emission upon increasing Ψ (λcalc = 536.95 nm at Ψ = 0° to 430.11 nm at 90°). In addition, the molecular orbital contribution of the o-carborane cage (zone 1, Figure 5a) and terminal phenyl ring (zone 3) to the LUMO and HOMO, respectively, was maximized (71 → 74% and 46 → 51%, respectively, Figure 5b). However, the molecular orbital contribution of the bridged phenyl ring (zone 2) to the HOMO decreased (44 → 36%) as the distortion of the biphenyl ring became perpendicular (Ψ = 0 to 90°). These observations imply that the distortion of biphenyl rings leads to the isolation (inset in Figure 5b when Ψ = 90°) of the orbital contribution for LUMO and HOMO, resulting in the prevention of the ICT transition between o-carborane and the appended phenyl group. All the calculation results strongly suggest that the distorted geometry of the biphenyl rings prohibits the efficient ICT transition corresponding to the o-carborane cage. Consequently, the combined experimental and theoretical findings verify that the molecular geometry, particularly the planarity of aromatic rings linked to the o-carborane cage, is a critical factor in regulating the efficiency of the ICT-based radiative decay mechanism in o-carborane luminophores.
Figure 5.

(a) Computed emission wavelengths (λcalc) and oscillator strengths (fcalc) as a function of the dihedral angle (Ψ) in each first-excited (S1) state. (b) Effects of Ψ in S1-optimized structures on the contributions of the o-carborane cage to the LUMO (zone 1; green-line), bridged phenyl ring to the HOMO (zone 2; purple-line), and terminal phenyl ring to the HOMO (zone 3; dark yellow-line). Insets are frontier orbitals at Ψ = 0 and 90°.
Conclusions
In conclusion, the biphenyl-based o-carborane luminophores of the 13 species of 1HF–13PP, with various distortion geometries on the biphenyl group, were prepared and structurally characterized to establish the relationship between the geometric conformations and photophysical features. Intriguingly, their radiative decay efficiencies for ICT-based emissions in the crystalline state linearly decreased in accordance with the increasing dihedral angles between the biphenyl rings in the crystal structures. Consequently, the results confirm that the planarity of the aromatic groups linked to o-carborane is crucial for controlling the ICT-based radiative decay of o-carborane. Thus, the results of this study make a significant contribution to developing o-carborane-based organic luminophores, exhibiting specific photophysical characteristics.
Experimental Section
General Considerations
All the operations were performed in an inert nitrogen atmosphere using standard Schlenk and glovebox techniques. Anhydrous solvents (acetonitrile (ACN), THF, toluene, trimethylamine (NEt3), dichloroethane (DCE), and DCM), purchased from Sigma-Aldrich, were dried by passing through an activated alumina column and stored over activated molecular sieves (5 Å). Spectrophotometric-grade solvents (cyclohexane, n-hexane, methanol (MeOH), ethyl acetate, toluene, THF, DCM, and acetone) were used as received from Alfa Aesar. Commercial reagents purchased from Sigma-Aldrich (N-bromosuccinimide (NBS), p-toluenesulfonic acid monohydrate (TsOH), 2-isopropoxy-4,4,5,5-tetramethyl[1,3,2]-dioxaborolane, 1,4-dibromobenzene, 2-iodobiphenyl, phenylacetylene, 2-bromo-9H-fluorene, 2-bromotriphenylene, 4-bromobiphenyl, 4-bromophenylboronic acid, 2-bromotoluene, 2-bromostyrene, 2-bromocumene, 2,6-dimethylbenzeneboronic acid, 2,6-diisopropylbenzeneboronic acid, 1.6 M n-butyllithium in hexane (n-BuLi), copper(I) iodide (CuI), potassium carbonate (K2CO3), sodium thiosulfate (Na2S2O3), magnesium sulfate (MgSO4), diethyl sulfide (Et2S), 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), saturated HCl aqueous solution, and poly(methyl methacrylate) (PMMA), 1,1′-biphenyl), decaborane (B10H14) (Alfa Aesar), TCI (9,10-dihydrophenanthrene, tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4)), and bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2) were used without any further purification. 2-bromo-2′-methyl-1,1′-biphenyl (precursor for 6HCA),42 2′-bromo-2,6-dimethyl-1,1′-biphenyl (precursor for 7Br),43 (4-(phenylethynyl)phenyl)boronic acid (precursor for 6HCA, 9HMA, 10HVA, and 11HIA),37 and 1-bromo-4-(phenylethynyl)benzene (precursor for 12MMA and 13PPA)37 were prepared according to previously reported procedures. Deuterated solvents, chloroform-d1 (CDCl3), and dichloromethane-d2 (CD2Cl2) were purchased from Cambridge Isotope Laboratories Inc., and dried over activated molecular sieves (5 Å). All NMR spectra were recorded on a Bruker Avance 400 MHz NMR spectrometer (Bruker; 400.13 MHz for 1H and 1H{11B}, 100.62 MHz for 13C, and 128.38 MHz for 11B{1H}) at an ambient temperature. Chemical shifts are expressed in ppm and referenced against external tetramethylsilane (Me4Si) (1H, 1H{11B}, and 13C) or BF3·Et2O (11B{1H}). Elemental analyses were performed on an EA3000 instrument (EuroVector) at the Central Laboratory of Kangwon National University.
Synthesis of 2-Bromo-9,10-dihydrophenanthrene, 4Br
ACN (25 mL) was added to a mixture of 9,10-dihydrophenanthrene (0.84 mL, 5.0 mmol), NBS (0.98 g, 5.5 mmol), and TsOH, (29 mg, 0.15 mmol) at 50 °C, and the reaction mixture was subsequently refluxed for 12 h. After quenching with excess saturated aqueous Na2S2O3, the mixture was extracted with ethyl acetate (50 mL × 3). The organic layer was dried with anhydrous MgSO4, and the volatiles were removed under vacuum. Purification via recrystallization from MeOH gave 4Br (0.67 g) as a white solid (yield = 52%). Except for elemental analyses, other characterization techniques were not performed due to its poor solubility in any organic solvents. Anal. calcd for C14H11Br: C, 64.89; H, 4.28. Found: C, 64.55; H, 4.01.
Synthesis of 4″-Bromo-2,6-dimethyl-1,1′:2′,1″-terphenyl, 7Br
Boronification: n-BuLi (1.6 M in n-hexane, 1.4 mL, 2.2 mmol) was added to a solution of 2′-bromo-2,6-dimethyl-1,1′-biphenyl (0.49 g, 1.9 mmol) in THF (20 mL) at −78 °C. After stirring for 3 h, 2-isopropoxy-4,4,5,5-tetramethyl[1,3,2]-dioxaborolane (0.42 g, 2.2 mmol) in THF (10 mL) was added dropwise. The reaction mixture was gradually heated to 25 °C and further stirred for 12 h. After quenching with concentrated HCl aqueous solution (20 mL), the mixture was extracted with ethyl acetate (50 mL × 3). The organic layer was dried over anhydrous MgSO4, and the solvents were removed through rotary evaporation. The boronificated crude product was used in situ for the next step without any purification. Suzuki–Miyaura coupling reaction: a cannula was utilized to add toluene (8.0 mL) and distilled water (1.6 mL) to a mixture of the boronificated product (0.49g, 1.6 mmol), 1,4-dibromobenzene (0.75 g, 3.2 mmol), Pd(PPh3)4 (92 mg, 80 μmol), and K2CO3 (0.44 g, 3.2 mmol). The reaction mixture was refluxed at 120 °C for 4 h. After extracting with ethyl acetate (10 mL × 3), the organic layer was dried over anhydrous MgSO4, and the volatiles were removed through rotary evaporation to give a dark brown residue, which was purified by column chromatography on silica gel (eluent: n-hexane only) to afford 7Br (0.17 g) as a white solid. Yield = 32%. 1H NMR (CD2Cl2): δ 7.43 (d, J = 8.0 Hz, 2H), 7.42 (m, 1H), 7.29 (d, J = 8.0 Hz, 2H), 7.17 (m, 1H), 7.08 (t, J = 8.2 Hz, 1H), 6.98 (d, J = 8.2 Hz, 4H), 1.92 (s, 6H, −CH3). 13C NMR (CD2Cl2): δ 140.84, 140.80, 139.87, 139.26, 136.36, 131.09, 130.91, 130.88, 130.30, 128.17, 127.96, 127.57, 127.46, 121.11, 20.88 (−CH3). Anal. calcd for C26H17Br: C, 71.23; H, 5.08. Found: C, 71.55; H, 5.12.
Synthesis of 4-Bromo-1,1′:2′,1″-terphenyl, 8Br
THF (60 mL) and distilled water (15 mL) were injected via a cannula to a mixture of 2-iodobiphenyl (1.7 mL, 10 mmol), 4-bromophenylboronic acid (2.0 g, 10 mmol), Pd(PPh3)4 (1.1 g, 1.0 mmol), and K2CO3 (4.1 g, 30 mmol) at 90 °C and then stirred for 12 h at the same temperature. The reaction mixture was quenched with distilled water (50 mL) and extracted with ethyl acetate (50 mL × 3). After drying over anhydrous MgSO4, the organic volatiles were removed under vacuum to afford a dark brown oil. The residue was purified by column chromatography on silica gel (eluent: n-hexane only) to yield 8Br (0.62 g) as a white solid. Yield = 29%. 1H NMR (CD2Cl2): δ 7.42 (t, J = 4.8 Hz, 3H), 7.40 (m, 1H), 7.35 (d, J = 7.8 Hz, 2H), 7.24 (m, 3H), 7.14 (m, 2H), 7.03 (d, J = 8.0 Hz, 2H). 13C NMR (CD2Cl2): δ 141.65, 141.06, 140.95, 139.66, 131.99, 131.34, 131.08, 130.73, 130.24, 128.39, 128.23, 127.99, 127.02, 120.99. Anal. calcd for C18H13Br: C, 69.92; H, 4.24. Found: C, 69.89; H, 4.10.
General Synthetic Routes for Acetylene Derivatives: 1HFA, 2HTA, 4HTA, 5HHA, 7HDA, and 8HPA
The acetylene derivatives (1HFA, 2HTA, 4HTA, 5HHA, 7HDA, and 8HPA) were synthesized by following the scheme with an adequate amount of starting materials. Toluene (14 mL) and NEt3 (7.0 mL) were added via a cannula to the mixture of each bromo precursor (Scheme S1), CuI, and Pd(PPh3)2Cl2 at 110 °C. After stirring for 15 min, injection of phenylacetylene of three equivalents per bromo precursor made the resulting dark brown slurry. Subsequently, the reaction mixture was refluxed for 24 h. The volatiles were removed with a rotary evaporator, affording a dark gray residue. After washing with MeOH, followed by n-hexane, column chromatography on silica gel (eluent: n-hexane only) was performed to purify acetylene derivatives from the crude products.
Data for 2-(Phenylethynyl)-9H-fluorene, 1HFA
2-Bromo-9H-fluorene (0.74 g, 3.0 mmol), CuI (48 mg, 0.24 mmol), Pd(PPh3)2Cl2 (190 mg, 0.26 mmol), and phenylacetylene (1.0 mL, 9.0 mmol) provided 1HFA (0.63 g) as an ivory solid. Yield = 79%. 1H NMR (CDCl3): δ 7.80 (d, J = 6.4 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.57 (dd, J = 7.1, 3.6 Hz, 4H), 7.40 (t, J = 8.2 Hz, 1H), 7.36 (d, J = 7.8 Hz, 3H), 7.32 (d, J = 8.0 Hz, 1H), 3.92 (s, 2H, −CH2−). 13C NMR (CDCl3): δ 143.72, 143.35, 142.03, 141.25, 131.72, 130.66, 128.50, 128.32, 128.29, 127.31, 127.07, 125.24, 123.61, 121.43, 120.34, 119.94, 90.31 (acetylene–C), 89.53 (acetylene–C), 36.90 (−CH2−). Anal. calcd for C21H14: C, 94.70; H, 5.30. Found: C, 94.70; H, 5.28.
Data for 2-(Phenylethynyl)triphenylene, 2HTA
2-Bromotriphenylene (0.31 g, 1.0 mmol), CuI (19 mg, 0.10 mmol), Pd(PPh3)2Cl2 (70 mg, 0.10 mmol), and phenylacetylene (0.35 mL, 3.0 mmol) provided 2HTA (0.25 g) as a white solid. Yield = 76%. 1H NMR (CDCl3): δ 8.85 (s, 1H), 8.65 (m, 5H), 7.80 (dd, J = 8.5, 1.5 Hz, 1H), 7.68 (m, 4H), 7.65 (dd, J = 8.0, 2.4 Hz, 2H), 7.41 (m, 3H). 13C NMR (CDCl3): δ 131.85, 130.19, 130.11, 130.08, 129.87, 129.69, 129.46, 129.25, 128.59, 128.56, 127.76, 127.74, 127.54, 127.52, 126.99, 123.66, 123.60, 123.57, 123.51, 123.46, 123.38, 122.05, 90.34 (acetylene–C), 89.95 (acetylene–C). Anal. calcd for C26H16: C, 95.09; H, 4.91. Found: C, 94.94; H, 4.90.
Data for 2-(Phenylethynyl)-9,10-dihydrophenanthrene, 4HNA
4Br (0.47 g, 1.8 mmol), CuI (12 mg, 0.14 mmol), Pd(PPh3)2Cl2 (110 mg, 0.16 mmol), and phenylacetylene (0.59 mL, 5.4 mmol) provided 4HNA (0.25 g) as a white solid. Yield = 49%. 1H NMR (CD2Cl2): δ 7.76 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.56 (t, J = 6.4 Hz, 1H), 7.55 (d, J = 4.8 Hz, 1H), 7.48 (dd, J = 8.0, 1.7 Hz, 1H), 7.43 (s, 1H), 7.40 (d, J = 8.5 Hz, 1H), 7.37 (m, 2H), 7.32 (m, 1H), 7.26 (d, J = 4.2 Hz, 2H), 2.89 (s, 4H, −CH2CH2−). 13C NMR (CD2Cl2): δ 138.04, 135.06, 134.18, 131.91, 131.51, 130.54, 128.88, 128.83, 128.69, 128.62, 128.26, 127.43, 124.18, 124.08, 123.71, 122.27, 89.99 (acetylene–C), 89.91 (acetylene–C), 29.23 (−CH2−), 29.20 (−CH2−). Anal. calcd for C20H14: C, 94.45; H, 5.45. Found: C, 94.28; H, 5.33.
Data for 4-(Phenylethynyl)-1,1′-biphenyl, 5HHA
4-Bromobiphenyl (0.70 g, 3.0 mmol), CuI (48 mg, 0.24 mmol), Pd(PPh3)2Cl2 (190 mg, 0.26 mmol), and phenylacetylene (1.0 mL, 9.0 mmol) provided 5HHA (0.67 g) as a white solid. Yield = 87%. 1H NMR (CDCl3): δ 7.62 (d, J = 7.8 Hz, 2H), 7.61 (s, 4H), 7.56 (t, J = 2.4 Hz, 1H), 7.55 (d, J = 1.8 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.38 (t, J = 2.4 Hz, 1H), 7.35 (m, 3H). 13C NMR (CDCl3): δ 141.10, 140.49, 132.17, 131.76, 129.01, 128.52, 128.43, 127.78, 127.17, 123.42, 122.30, 90.19 (acetylene–C), 89.43 (acetylene–C). Anal. calcd for C20H14: C, 94.45; H, 5.55. Found: C, 94.12; H, 5.48.
Data for 2,6-Dimethyl-4″-(phenylethynyl)-1,1′:2′,1″-terphenyl, 7HDA
7Br (0.54 g, 1.6 mmol), CuI (24 mg, 0.12 mmol), Pd(PPh3)2Cl2 (100 mg, 0.14 mmol), and phenylacetylene (0.53 mL, 4.8 mmol) provided 7HDA (0.34 g) as a white solid. Yield = 59%. 1H NMR (CDCl3): δ 7.48 (t, J = 8.2 Hz, 3H), 7.42 (m, 2H), 7.33 (dd, J = 5.3, 2.0 Hz, 5H), 7.19 (d, J = 7.3 Hz, 1H), 7.08 (t, J = 8.2 Hz, 3H), 6.98 (d, J = 7.5 Hz, 2H), 1.94 (s, 6H, −CH3). 13C NMR (CDCl3): δ 141.49, 140.63, 140.17, 139.10, 136.17, 131.68, 131.12, 130.63, 130.09, 128.93, 128.48, 128.35, 127.82, 127.68, 127.40, 127.22, 123.40, 121.44, 89.81 (acetylene–C), 89.49 (acetylene–C), 20.95 (−CH3). Anal. calcd. for C28H22: C, 93.81; H, 6.19. Found: C, 93.72; H, 6.08.
Data for 4-(Phenylethynyl)-1,1′:2′,1″-terphenyl, 8HPA
8Br (0.93 g, 3.0 mmol), CuI (57 mg, 0.30 mmol), Pd(PPh3)2Cl2 (250 mg, 0.36 mmol), and phenylacetylene (1.1 mL, 9.0 mmol) provided 8HPA (0.20 g) as a white solid. Yield = 20%. 1H NMR (CD2Cl2): δ 7.53 (d, J = 4.2 Hz, 1H), 7.51 (d, J = 2.4 Hz, 1H), 7.44 (s, 4H), 7.39 (d, J = 7.8 Hz, 2H), 7.35 (m, 3H), 7.23 (t, J = 7.6 Hz, 3H), 7.14 (t, J = 8.8 Hz, 4H). 13C NMR (CD2Cl2): δ 141.74, 141.37, 140.70, 139.96, 131.71, 131.32, 130.86, 130.57, 130.01, 130.01, 128.49, 128.37, 128.16, 127.93, 127.72, 126.76, 123.41, 121.40, 89.78 (acetylene–C), 89.52 (acetylene–C). Anal. calcd. for C26H18: C, 94.51; H, 5.49. Found: C, 94.88; H, 5.12.
General Synthetic Routes for Acetylene Derivatives, 6HCA, and 9HMA–13PPA
The acetylene derivatives (6HCA and 9HMA–13PPA) were synthesized by following the scheme with an adequate amount of starting materials. THF (10 mL) and distilled water (5.0 mL) were added via a cannula to a mixture of each bromo and boronic acid precursor (Scheme S1), Pd(PPh3)4, and K2CO3 at 90 °C. In addition, the reaction mixture was stirred for 12 h at the same temperature, quenched by distilled water (20 mL), and then extracted with ethyl acetate (50 mL × 3). The organic layer was dried over anhydrous MgSO4, and the organic solvents were removed under vacuum to give a dark brown oil. The crude product was purified by column chromatography on silica gel (eluent: n-hexane only) to afford acetylene derivatives.
Data for 2-Methyl-4″-(phenylethynyl)-1,1′:2′,1″-terphenyl, 6HCA
2-Bromo-2′-methyl-1,1′-biphenyl (1.3 g, 5.4 mmol), (4-(phenylethynyl)phenyl)boronic acid (0.53 g, 2.4 mmol), Pd(PPh3)4 (0.30 mg, 0.43 mmol), and K2CO3 (2.2 g, 16 mmol) provided 6HCA (0.87 g) as a white solid. Yield = 47%. 1H NMR (CDCl3): δ 7.48 (dd, J = 8.9 Hz, 4H), 7.42 (t, J = 8.4 Hz, 1H), 7.33 (m, 6H), 7.17 (m, 1H), 7.15 (t, J = 7.6 Hz, 2H), 7.09 (t, J = 8.0 Hz, 3H), 1.91 (s, 3H, −CH3). 13C NMR (CDCl3): δ 141.64, 141.15, 140.44, 140.41, 135.88, 131.69, 131.16, 130.96, 130.69, 130.03, 129.91, 129.51, 128.47, 128.34, 127.75, 127.55, 127.31, 125.56, 123.43, 121.37, 89.80 (acetylene–C), 89.53 (acetylene–C), 20.18 (−CH3). Anal. calcd for C27H20: C, 94.15; H, 5.85. Found: C, 93.85; H, 6.10.
Data for 2-Methyl-4′-(phenylethynyl)-1,1′-biphenyl, 9HMA
2-Bromotoluene (0.34 g, 2.0 mmol), (4-(phenylethynyl)phenyl)boronic acid (0.53 g, 2.4 mmol), Pd(PPh3)4 (230 mg, 0.20 mmol), and K2CO3 (0.83 g, 6.0 mmol) provided 9HMA (0.30 g) as a white solid. Yield = 57%. 1H NMR (CD2Cl2): δ 7.61 (s, 1H), 7.58 (m, 2H), 7.56 (d, J = 2.4 Hz, 1H), 7.40 (m, 1H), 7.38 (d, J = 4.8 Hz, 2H), 7.35 (s, 1H), 7.33 (s, 1H), 7.28 (m, 2H), 7.24 (m, 2H), 2.29 (s, 3H, −CH3). 13C NMR (CD2Cl2): δ 142.50, 141.56, 135.71, 131.92, 131.65, 130.80, 129.92, 129.73, 128.82, 128.73, 127.91, 126.22, 123.64, 122.01, 89.84 (acetylene–C), 89.58 (acetylene–C), 20.57 (−CH3). Anal. calcd for C21H16: C, 93.99; H, 6.01. Found: C, 94.94; H, 5.96.
Data for 4′-(Phenylethynyl)-2-vinyl-1,1′-biphenyl, 10HVA
2-Bromostyrene (0.18 g, 1.0 mmol), (4-(phenylethynyl)phenyl)boronic acid (0.27 g, 1.2 mmol), Pd(PPh3)4 (120 mg, 0.10 mmol), and K2CO3 (0.41 g, 3.0 mmol) provided 10HVA (0.23 g) as a white solid. Yield = 83%. 1H NMR (CDCl3): δ 7.67 (dd, J = 8.4, 2.4 Hz, 1H), 7.61 (s, 1H), 7.59 (m, 2H), 7.57 (d, J = 4.8 Hz, 1H), 7.40 (dd, J = 8.0, 2.4 Hz, 1H), 7.38 (m, 4H), 7.35 (t, J = 3.8 Hz, 2H), 7.32 (td, J = 8.4, 2.4 Hz, 1H), 6.73 (dd, J = 17.5, 11.0 Hz, 1H, −CHCH2), 5.73 (dd, J = 17.5, 1.3 Hz, 1H, −CHCH2), 5.23 (dd, J = 11.0, 1.3 Hz, 1H, −CHCH2). 13C NMR (CDCl3): δ 140.95, 140.20, 135.88, 135.85, 131.76, 131.41, 130.04, 129.97, 128.50, 128.41, 127.89, 127.88, 126.04, 123.42, 122.12, 115.19, 91.56 (acetylene–C), 91.56 (acetylene–C). Anal. calcd for C22H16: C, 94.25; H, 5.75. Found: C, 94.11; H, 5.69.
Data for 2-Isopropyl-4′-(phenylethynyl)-1,1′-biphenyl, 11HIA
2-Bromocumene (0.31 mL, 2.0 mmol), (4-(phenylethynyl)phenyl)boronic acid (0.53 g, 2.4 mmol), Pd(PPh3)4 (230 mg, 0.20 mmol), and K2CO3 (0.83 g, 6 mmol) provided 11HIA (0.30 g) as a white solid. Yield = 50%. 1H NMR (CD2Cl2): δ 7.58 (t, J = 7.8 Hz, 4H), 7.39 (m, 5H), 7.31 (d, J = 8.0 Hz, 2H), 7.22 (t, J = 7.8 Hz, 1H), 7.18 (t, J = 7.8 Hz, 1H), 3.06 (m, 1H, −CH(CH3)2), 1.17 (d, J = 6.9, 6H, −CH(CH3)2). 13C NMR (CD2Cl2): δ 146.78, 142.68, 140.81, 131.94, 131.57, 130.07, 129.89, 128.84, 128.74, 128.33, 126.02, 125.77, 123.65, 121.98, 89.84 (acetylene–C), 89.56 (acetylene–C), 29.81 (−CH(CH3)2), 24.37 (−CH(CH3)2). Anal. calcd for C23H20: C, 93.20; H, 6.80. Found: C, 93.15; H, 6.75.
Data for 2,6-Dimethyl-4′-(phenylethynyl)-1,1′-biphenyl, 12MMA
1-Bromo-4-(phenylethynyl)benzene (0.77 g, 3.0 mmol), 2,6-dimethylbenzeneboronic acid (0.54 g, 3.6 mmol), Pd(PPh3)4 (350 mg, 0.30 mmol), and K2CO3 (1.2 g, 9.0 mmol) provided 12MMA (0.23 g) as a white solid. Yield = 27%. 1H NMR (CD2Cl2): δ 7.62 (d, J = 8.0 Hz, 2H), 7.57 (dd, J = 8.4, 2.8 Hz, 2H), 7.39 (m, 3H), 7.16 (d, J = 8.4, Hz, 3H), 7.11 (d, J = 6.4 Hz, 2H), 2.03 (s, 6H, −CH3). 13C NMR (CD2Cl2): δ 141.86, 141.58, 136.23, 132.04, 131.94, 129.76, 128.83, 128.72, 127.70, 127.63, 123.68, 121.89, 89.67 (acetylene–C), 89.57 (acetylene–C), 20.90 (−CH3). Anal. calcd for C22H18: C, 93.57; H, 6.43. Found: C, 93.45; H, 6.35.
Data for 2,6-Diisopropyl-4′-(phenylethynyl)-1,1′-biphenyl, 13PPA
1-Bromo-4-(phenylethynyl)benzene (0.54 g, 2.1 mmol), 2,6-diisopropylbenzeneboronic acid (0.51 g, 2.5 mmol), Pd(PPh3)4 (240 mg, 0.21 mmol), and K2CO3 (0.87 g, 6.3 mmol) provided 13PPA (71 mg) as a white solid. Yield = 10%. 1H NMR (CDCl3): 7.59 (d, J = 8.0 Hz, 2H), 7.56 (dd, J = 8.4, 2.4 Hz, 2H), 7.37 (m, 4H), 7.21 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 2.60 (m, 2H, −CH(CH3)2), 1.08 (d, J = 6.9 Hz, 12H, −CH(CH3)2). 13C NMR (CDCl3): δ 146.84, 141.12, 139.00, 131.76, 131.46, 129.80, 128.53, 128.42, 128.18, 123.46, 122.71, 121.63, 89.63 (acetylene–C), 89.41 (acetylene–C), 30.41 (−CH(CH3)2), 24.25 (−CH(CH3)2). Anal. calcd for C26H26: C, 92.26; H, 7.74. Found: C, 92.21; H, 7.69.
General Synthetic Routes for the Biphenyl-Based o-carborane Compounds: 1HF, 2HT, and 4HN–13PP
An excess Et2S (3.0 equivalent of an acetylene derivative) was added to a toluene solution (30 mL) of decaborane (B10H14, 1.5 equivalent per acetylene derivative) and each acetylene derivative (Scheme 1). After stirring at 120 °C, the reaction mixture was further refluxed for 24 h. The solvent was removed with a rotary evaporate. Purification through column chromatography on basic aluminum oxide (eluent: toluene only) and recrystallization was performed to afford biphenyl-based o-carborane compounds as a white solid.
Scheme 1. Synthetic Routes for Biphenyl-Based o-Carboranyl Compounds (1HF–13PP).
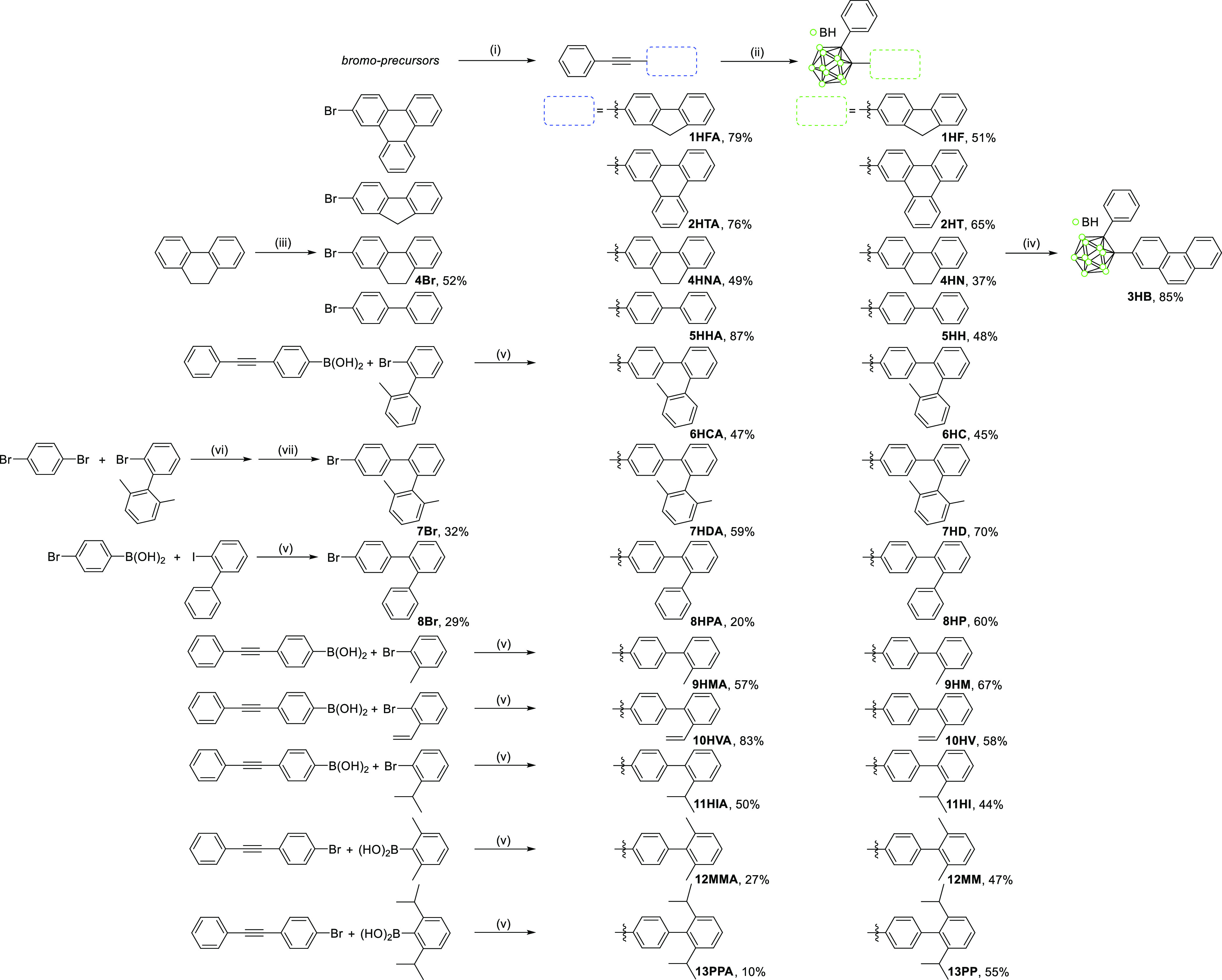
Reaction conditions: (i) phenylacetylene, Pd(PPh3)2Cl2, CuI, NEt3/toluene (1/2, v/v), 110 °C, 24 h; (ii) B10H14, Et2S, toluene, 120 °C, 24 h; (iii) NBS, TsOH, ACN, 50 °C, 12 h; (iv) TsOH, DDQ, DCE, 90 °C, 12 h; (v) Pd(PPh3)4, K2CO3, THF/H2O (2/1, v/v), 90 °C, 12 h; (vi) 2-isopropoxy-4,4,5,5-tetramethyl[1,3,2]-dioxaborolane, THF, 25 °C, 12 h; and (vii) Pd(PPh3)4, K2CO3, toluene/H2O (5/1, v/v), 120 °C, 4 h.
Data for 1HF
Decaborane (0.42 g, 3.5 mmol), Et2S (0.74 mL, 6.9 mmol), and 1HFA (0.61 g, 2.3 mmol) in toluene afforded 1HF as a white solid. Recrystallization from a mixture of DCM/n-hexane provided 0.45 g (1.2 mmol) of crystalline 1HF. Yield = 51%. 1H{11B} NMR (CDCl3): δ 7.69 (d, J = 7.2 Hz, 1H), 7.59 (s, 1H), 7.51 (d, J = 7.7 Hz, 2H), 7.46 (d, J = 6.8 Hz, 3H), 7.33 (m, 2H), 7.19 (t, J = 7.1 Hz, 1H), 7.11 (t, J = 7.3 Hz, 2H), 3.76 (s, 2H, −CH2−), 3.33 (br s, 2H, CB–BH), 2.56 (br s, 5H, CB–BH), 2.36 (br s, 3H, CB–BH). 13C NMR (CDCl3): δ 143.92, 143.68, 143.19, 140.28, 130.86, 130.77, 130.27, 129.78, 129.05, 128.39, 127.83, 127.42, 127.13, 125.28, 120.62, 199.47, 86.16 (CB–C), 85.63 (CB–C), 36.87 (−CH2−). 11B{1H} NMR (CDCl3): δ −3.48 (br s, 2B), −10.10 (br s, 4B), −11.50 (br s, 4B). Anal. calcd for C21H24B10: C, 65.60; H, 6.29. Found: C, 65.56; H, 6.21.
Data for 2HT
Decaborane (0.55 g, 4.5 mmol), Et2S (0.97 mL, 9.0 mmol), and 2HTA (0.98 g, 3.0 mmol) in toluene afforded 2HT as a white solid. Recrystallization from a mixture of DCM/n-hexane provided 0.87 g (2.0 mmol) of crystalline 2HT. Yield = 65%. 1H{11B} NMR (CDCl3): δ 8.70 (s, 1H), 8.58 (t, J = 7.4 Hz, 2H), 8.45 (t, J = 8.1 Hz, 2H), 8.35 (d, J = 8.8 Hz, 1H), 7.65 (dt, J = 17.7, 8.3 Hz, 5H), 7.53 (d, J = 7.2 Hz, 2H), 7.09 (m, 3H), 3.49 (br s, 1H, CB–BH), 2.73 (br s, 2H, CB–BH), 2.63 (br s, 4H, CB–BH), 2.46 (br s, 3H, CB–BH). 13C NMR (CDCl3): δ 130.93, 130.80, 130.76, 160.41, 130.36, 130.15, 129.30, 129.09, 128.64, 128.55, 128.50, 128.27, 127.99, 127.66, 127.59, 126.30, 123.70, 123.54, 123.50, 123.46, 123.18, 85.67 (CB–C), 85.53 (CB–C). 11B{1H} NMR (CDCl3): δ −2.31 (br s, 2B), −8.92 (br s, 4B), −10.23 (br s, 4B). Anal. calcd for C26H26B10: C, 69.93; H, 5.87. Found: C, 69.81; H, 5.87.
Data for 4HN
Decaborane (0.16 g, 1.3 mmol), Et2S (0.28 mL, 2.6 mmol), and 4HNA (0.24 g, 0.86 mmol) in toluene afforded 4HN as a white solid. Recrystallization from n-hexane provided 0.13 g (0.32 mmol) of crystalline 4HN. Yield = 37%. 1H{11B} NMR (CD2Cl2): δ 7.62 (d, J = 7.2 Hz, 1H), 7.50 (d, J = 8.0 Hz, 3H), 7.36 (d, J = 8.4 Hz, 1H), 7.31 (s, 1H), 7.24 (m, 3H), 7.17 (t, J = 8.4 Hz, 3H), 3.33 (br s, 2H, CB–BH), 2.74 (tt, J = 7.8, 3.9 Hz, 4H, −CH2CH2−), 2.53 (br s, 5H, CB–BH), 2.33 (br s, 3H, CB–BH). 13C NMR (CD2Cl2): δ 138.08, 137.80, 136.75, 133.27, 131.14, 131.02, 130.90, 130.65, 129.62, 129.57, 128.74, 128.69, 128.62, 127.41, 124.27, 123.71, 86.04 (CB–C), 29.14 (−CH2CH2−), 28.95 (−CH2CH2−). 11B{1H} NMR (CD2Cl2): δ −2.70 (br s, 2B), −9.14 (br s, 4B), −10.57 (br s, 4B). Anal. calcd for C22H26B10: C, 66.30; H, 6.58. Found: C, 65.98; H, 6.32.
Data for 5HH
Decaborane (0.48 g, 3.9 mmol), Et2S (0.84 mL, 7.8 mmol), and 5HHA (0.67 g, 2.6 mmol) in toluene afforded 5HH as a white solid. Recrystallization from n-hexane provided 0.47 g (1.2 mmol) of crystalline 5HH. Yield = 48%. 1H{11B} NMR (CD2Cl2): δ 7.51 (t, J = 8.0 Hz, 6H), 7.41 (m, 4H), 7.34 (t, J = 7.2 Hz, 1H), 7.27 (t, J = 7.4 Hz, 1H), 7.18 (t, J = 7.6 Hz, 2H), 3.34 (br s, 1H, CB–BH), 2.53 (br s, 6H, CB–BH), 2.33 (br s, 3H, CB–BH). 13C NMR (CD2Cl2): δ 143.18, 139.47, 131.53, 131.12, 130.95, 130.71, 129.92, 129.27, 128.76, 128.50, 127.29, 127.09, 86.00 (CB–C), 85.73 (CB–C). 11B{1H} NMR (CD2Cl2): δ −2.56 (br s, 2B), −9.06 (br s, 4B), −10.47 (br s, 4B). Anal. calcd for C20H24B10: C, 64.49; H, 6.49. Found: C, 64.21; H, 6.28.
Data for 6HC
Decaborane (0.46 g, 3.8 mmol), Et2S (0.81 mL, 7.5 mmol), and 6HCA (0.86 g, 2.5 mmol) in toluene afforded 6HC as a white solid. Recrystallization from a mixture of DCM/n-hexane provided 0.52 g (1.1 mmol) of crystalline 6HC. Yield = 45%. 1H{11B} NMR (CD2Cl2): δ 7.39 (d, J = 8.0 Hz, 4H), 7.33 (t, J = 7.2 Hz, 2H), 7.23 (d, J = 8.3 Hz, 3H), 7.16 (t, J = 7.2 Hz, 3H), 7.06 (t, J = 7.4 Hz, 1H), 7.00 (d, J = 7.4 Hz, 1H), 6.94 (d, J = 7.4 Hz, 1H), 6.88 (d, J = 8.3 Hz, 2H), 3.23 (br s, 2H, CB–BH), 2.47 (br s, 6H, CB–BH), 2.28 (br s, 2H, CB–BH), 1.56 (s, 3H, −CH3). 13C NMR (CD2Cl2): δ 143.97, 140.98, 140.63, 139.59, 135.83, 131.23, 130.98, 130.82, 130.73, 130.62, 130.45, 130.16, 129.93, 129.57, 129.00, 128.65, 128.13, 127.99, 127.46, 125.75, 85.90 (CB–C), 85.83 (CB–C), 19.82 (−CH3). 11B{1H} NMR (CD2Cl2): δ −2.63 (br s, 2B), −9.21 (br s, 4B), −10.61 (br s, 4B). Anal. calcd for C27H30B10: C, 70.10; H, 6.54. Found: C, 70.05; H, 6.25.
Data for 7HD
Decaborane (0.17 g, 1.4 mmol), Et2S (0.29 mL, 2.7 mmol), and 7HDA (0.32 g, 0.90 mmol) in toluene afforded 7HD as a white solid. Recrystallization from DCM provided 0.30 g (0.63 mmol) of crystalline 7HD. Yield = 70%. 1H{11B} NMR (CD2Cl2): δ 7.37 (d, J = 8.0 Hz, 3H), 7.32 (d, J = 8.0 Hz, 3H), 7.21 (t, J = 7.4 Hz, 2H), 7.12 (m, 3H), 7.05 (d, J = 7.8 Hz, 1H), 6.87 (m, 4H), 3.20 (br s, 1H, CB–BH), 2.46 (br s, 6H, CB–BH), 2.27 (br s, 3H, CB–BH), 1.72 (s, 6H, −CH3). 13C NMR (CD2Cl2): δ 143.77, 140.45, 139.38, 139.27, 136.13, 131.01, 130.89, 130.73, 130.61, 130.39, 130.14, 129.04, 129.02, 128.61, 128.37, 127.94, 127.59, 127.34, 85.85 (CB–C), 85.79 (CB–C), 20.71 (−CH3). 11B{1H} NMR (CD2Cl2): δ −2.64 (br s, 2B), −9.22 (br s, 4B), −10.62 (br s, 4B). Anal. calcd for C28H32B10: C, 70.55; H, 6.77. Found: C, 70.35; H, 6.51.
Data for 8HP
Decaborane (0.55 g, 4.5 mmol), Et2S (0.97 mL, 9.0 mmol), and 8HPA (0.99 g, 3.0 mmol) in toluene afforded 8HP as a white solid. Recrystallization from DCM provided 0.81 g (1.8 mmol) of crystalline 8HP. Yield = 60%. 1H{11B} NMR (CD2Cl2): δ 7.45 (d, J = 8.0 Hz, 2H), 7.39 (m, 3H), 7.34 (d, J = 6.8 Hz, 1H), 7.28 (t, J = 7.4 Hz, 3H), 7.21 (t, J = 8.0 Hz, 2H), 7.18 (d, J = 7.8 Hz, 1H), 7.10 (t, J = 7.5 Hz, 2H), 6.92 (d, J = 7.9 Hz, 4H), 3.25 (br s, 1H, CB–BH), 2.49 (br s, 6H, CB–BH), 2.30 (br s, 3H, CB–BH). 13C NMR (CD2Cl2): δ 144.14, 141.24, 140.84, 139.16, 131.10, 131.03, 130.92, 130.67, 130.67, 130.63, 130.10, 130.03, 129.11, 128.70, 128.49, 128.34, 127.95, 126.91, 85.92 (CB–C), 85.83 (CB–C). 11B{1H} NMR (CD2Cl2): δ −2.66 (br s, 2B), −9.15 (br s, 4B), −10.57 (br s, 4B). Anal. calcd for C26H28B10: C, 69.61; H, 6.29. Found: C, 69.55; H, 6.11.
Data for 9HM
Decaborane (0.18 g, 1.4 mmol), Et2S (0.31 mL, 2.9 mmol), and 9HMA (0.26 g, 0.96 mmol) in toluene afforded 9HM as a white solid. Recrystallization from a mixture of acetone/MeOH provided 0.25 g (0.64 mmol) of crystalline 9HM. Yield = 67%. 1H{11B} NMR (CD2Cl2): δ 7.45 (d, J = 8.0 Hz, 4H), 7.24 (t, J = 8.2 Hz, 1H), 7.19 (d, J = 7.4 Hz, 2H), 7.14 (t, J = 7.8 Hz, 3H), 7.07 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 8.0 Hz, 1H), 3.30 (br s, 2H, CB–BH), 2.53 (br s, 5H, CB–BH), 2.31 (br s, 3H, CB–BH), 2.04 (s, 3H, −CH3). 13C NMR (CD2Cl2): δ 144.39, 140.58, 135.57, 131.10, 130.92, 130.86, 130.76, 130.62, 129.63, 129.43, 129.40, 128.64, 128.16, 126.19, 85.91 (CB–C), 85.79 (CB–C), 20.31 (−CH3). 11B{1H} NMR (CD2Cl2): δ −2.57 (br s, 2B), −9.14 (br s, 4B), −10.52 (br s, 4B). Anal. calcd for C21H26B10: C, 65.25; H, 6.78. Found: C, 65.15; H, 6.62.
Data for 10HV
Decaborane (0.15 g, 1.3 mmol), Et2S (0.27 mL, 2.5 mmol), and 10HVA (0.23 g, 0.83 mmol) in toluene afforded 10HV as a white solid. Recrystallization from n-hexane provided 0.19 g (0.48 mmol) of crystalline 10HV. Yield = 58%. 1H{11B} NMR (CD2Cl2): δ 7.59 (d, J = 7.6 Hz, 1H), 7.48 (d, J = 8.0 Hz, 4H), 7.33 (t, J = 8.0 Hz, 4H), 7.28 (d, J = 7.8 Hz, 4H), 7.18 (t, J = 7.7 Hz, 2H), 7.13 (t, J = 6.6 Hz, 3H), 6.35 (dd, J = 11.0, 8.4 Hz, 1H), 5.62 (d, J = 8.8 Hz, 1H), 5.13 (d, J = 8.8 Hz, 1H), 3.33 (br s, 2H, CB–BH), 2.56 (br s, 6H, CB–BH), 2.34 (br s, 2H, CB–BH). 13C NMR (CD2Cl2): δ 143.28, 139.41, 136.02, 135.61, 131.12, 130.90, 130.80, 130.62, 129.99, 129.97, 129.65, 128.67, 128.44, 128.11, 126.24, 115.54, 85.92 (CB–C), 85.68 (CB–C). 11B{1H} NMR (CD2Cl2): δ −2.61 (br s, 2B), −9.19 (br s, 4B), −10.54 (br s, 4B). Anal. calcd for C22H26B10: C, 66.30; H, 6.58. Found: C, 66.15; H, 6.52.
Data for 11HI
Decaborane (0.18 g, 1.5 mmol), Et2S (0.32 mL, 3.0 mmol), and 11HIA (0.30 g, 1.0 mmol) in toluene afforded 11HI as a white solid. Recrystallization from n-hexane provided 0.18 g (0.44 mmol) of crystalline 11HI. Yield = 44%. 1H{11B} NMR (CDCl3): δ 7.43 (dd, J = 7.6, 3.5 Hz, 4H), 7.32 (d, J = 3.6 Hz, 2H), 7.23 (d, J = 7.3 Hz, 1H), 7.14 (t, J = 7.7 Hz, 3H), 7.04 (d, J = 7.8 Hz, 2H), 7.00 (d, J = 7.8 Hz, 1H), 3.29 (br s, 2H, CB–BH), 2.65 (m, 1H, −CH(CH3)2), 2.57 (br s, 5H, CB–BH), 2.37 (br s, 3H, CB–BH), 1.04 (d, J = 6.8 Hz, 6H, −CH(CH3)2). 13C NMR (CDCl3): δ 146.21, 144.16, 139.51, 130.81, 130.78, 130.43, 130.13, 129.48, 129.20, 128.30, 128.28, 125.70, 125.51, 85.32 (CB–C), 85.23 (CB–C), 29.32 (−CH(CH3)2), 24.16 (−CH(CH3)2). 11B{1H} NMR (CDCl3): δ −3.33 (br s, 2B), −10.15 (br s, 4B), −11.40 (br s, 4B). Anal. calcd for C23H30B10: C, 66.63; H, 7.29. Found: C, 66.58; H, 7.21.
Data for 12MM
Decaborane (0.15 g, 1.2 mmol), Et2S (0.26 mL, 2.4 mmol), and 12MMA (0.23 g, 0.81 mmol) in toluene afforded 12MM as a white solid. Recrystallization from DCM provided 0.15 g (0.38 mmol) of crystalline 12MM. Yield = 47%. 1H{11B} NMR (CDCl3): δ 7.44 (dd, J = 8.8, 4.0 Hz, 4H), 7.22 (t, J = 7.3 Hz, 1H), 7.13 (q, J = 7.3 Hz, 3H), 7.05 (d, J = 7.4 Hz, 2H), 6.90 (d, J = 8.0 Hz, 2H), 3.29 (br s, 2H, CB–BH), 2.58 (br s, 5H, CB–BH), 2.37 (br s, 3H, CB–BH), 1.80 (s, 6H, −CH3). 13C NMR (CDCl3): δ 143.32, 140.28, 135.66, 131.01, 130.76, 130.10, 129.12, 129.03, 128.23, 127.58, 127.46, 85.26 (CB–C), 85.18 (CB–C), 20.64 (−CH3). 11B{1H} NMR (CDCl3): δ −3.37 (br s, 2B), −10.36 (br s, 4B), −11.46 (br s, 4B). Anal. calcd for C22H28B10: C, 65.97; H, 7.05. Found: C, 65.91; H, 7.04.
Data for 13PP
Decaborane (51 mg, 0.42 mmol), Et2S (90 μL, 0.84 mmol), and 13PPA (95 mg, 0.28 mmol) in toluene afforded 13PP as a white solid. Recrystallization from a mixture of DCM/n-hexane provided 70 mg (0.15 mmol) of crystalline 13PP. Yield = 55%. 1H{11B} NMR (CD2Cl2): δ 7.45 (t, J = 6.8 Hz, 4H), 7.27 (q, J = 7.6 Hz, 2H), 7.14 (t, J = 8.8 Hz, 4H), 6.92 (d, J = 8.2 Hz, 2H), 3.33 (br s, 2H, CB–BH), 2.56 (br s, 6H, CB–BH), 2.34 (br s, 2H, CB–BH), 2.16 (m, 2H, −CH(CH3)2), 0.95 (d, J = 6.8 Hz, 12H, −CH(CH3)2). 13C NMR (CD2Cl2): δ 146.83, 143.48, 138.30, 131.11, 130.89, 130.81, 130.42, 129.84, 129.15, 128.55, 128.47, 122.85, 85.94 (CB–C), 85.90 (CB–C), 30.45 (−CH(CH3)2), 24.12 (−CH(CH3)2). 11B{1H} NMR (CD2Cl2): δ −2.46 (br s, 2B), −9.29 (br s, 4B), −10.61 (br s, 4B). Anal. calcd for C26H36B10: C, 68.38; H, 7.95. Found: C, 68.31; H, 7.90.
Synthesis of 3HB
DCE (5.0 mL) was added via a cannula to a mixture of 4HN (0.20 g, 0.50 mmol), TsOH (95 mg, 0.50 mmol), and DDQ (0.34 g, 1.5 mmol). The reaction mixture was then refluxed at 90 °C for 12 h. The volatiles were removed under vacuum to afford a dark green residue. Purification through column chromatography on silica gel (eluent: ethyl acetate/n-hexane = 1/20, v/v) gave 3HB (0.17 g) as a white solid. Yield = 85%. 1H{11B} NMR (CDCl3): δ 8.54 (d, J = 7.7 Hz, 1H), 8.41 (d, J = 8.8 Hz, 1H), 7.96 (s, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.72 (d, J = 8.9 Hz, 1H), 7.62 (m, 4H), 7.48 (d, J = 7.8 Hz, 2H), 7.11 (dt, J = 8.4, 4.6 Hz, 3H), 3.43 (br s, 2H, CB–BH), 2.66 (br s, 3H, CB–BH), 2.59 (br s, 3H, CB–BH), 2.42 (br s, 2H, CB–BH). 13C NMR (CDCl3): δ 132.63, 131.47, 131.34, 131.17, 130.77, 130.74, 130.32, 129.44, 128.80, 128.78, 128.43, 128.18, 127.90, 127.67, 127.17, 126.80, 123.05, 122.97, 85.65 (CB–C), 85.30 (CB–C). 11B{1H} NMR (CDCl3): δ −2.26 (br s, 2B), −8.94 (br s, 4B), −10.28 (br s, 4B). Anal. calcd for C22H24B10: C, 66.64; H, 6.10. Found: C, 66.48; H, 6.19.
Ultraviolet–Visible Absorption and Photoluminescence Measurements
Ultraviolet–visible (UV–vis) absorption and photoluminescence (PL) spectra of all biphenyl-based o-carboranyl compounds 1HF–13PP were acquired using V530 (Jasco) and FluoroMax-4P (HORIBA) spectrophotometers, respectively. The UV–vis absorption and PL measurements for the solution were performed at 298 K in oxygen-free and anhydrous THF with a 1 cm quartz cuvette (3.0 × 10–5 M). Furthermore, the PL spectra were measured in THF solution (3.0 × 10–5 M) at 77 K, in THF/distilled water mixtures (3.0 × 10–5 M) at 298 K, in the crystalline state at 298 K, and in the film state at 298 K. Thin films of o-carborane compounds in PMMA were obtained by spin coating THF solution (1 mL) of PMMA (50 mg) containing each o-carborane compound (5 wt % vs PMMA) on a quartz plate of dimensions 10 mm × 10 mm (thickness = 1 mm). The absolute PL quantum yields (PLQYs) in the crystalline state, THF solution (3.0 × 10–5 M), and film state were obtained at 298 K on a Fluoromax-4P spectrophotometer equipped with a 3.2 inch integrating sphere (FM-sphere, HORIBA). Fluorescence decay lifetimes of o-carborane compounds in the crystalline and film states were measured using a time-correlated single-photon counting spectrometer (FLS920-Edinburgh instrument at the Central Laboratory of Kangwon National University) equipped with an EPL-375ps pulsed semiconductor diode laser excitation source and a microchannel plate photomultiplier tube (MCP-PMT, 200–850 nm) detector.
X-ray Crystallography
Single X-ray quality 1HF–13PP crystals were grown from each mixture of organic solvents for X-ray diffractometry. Each single crystal was coated with Paratone oil and mounted on a glass capillary. Crystallographic measurements were performed using a Bruker D8 Quest CCD area detector diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures of o-carboranyl compounds were assessed using direct methods, and all non-hydrogen atoms were subjected to anisotropic refinement with a full-matrix least-squares method on F2 using a SHELXTL/PC software package. X-ray crystallographic data are available in the CIF format (CCDC 2142550–2142562 for 1HF–13PP) and are provided free of charge by the Cambridge Crystallographic Data Centre. Hydrogen atoms were placed at their geometrically calculated positions and refined using a riding model on the corresponding carbon atoms with isotropic thermal parameters. Detailed crystallographic data and selected bond lengths and angles are provided in Tables S1 and S2.
Computational Calculation Studies
The geometries of all biphenyl-based o-carboranyl compounds 1HF–13PP in their ground (S0) and first-excited (S1) states in THF were optimized at the B3LYP/6-31G(d,p)44 level of theory. Vertical excitation energies in the optimized S0 and S1 state geometries were calculated using the TD-DFT method38 at the same level of theory. The solvent effects were evaluated using a self-consistent reaction field method based on the integral equation formalism of the polarizable continuum model with THF as the solvent.45 Most stable geometries were determined by constructing one-dimensional potential energy surfaces as a function of each dihedral angle (Ψ, Figure 5 and Table S4) by rotating a phenyl ring of each biphenyl group between approximately 0 and 90° at intervals of 5° to yield 19 initial conformations for each compound. Conformations that exhibited physically impossible atomic overlaps were excluded from further geometric optimizations. The dihedral angle was fixed, while other geometric variables were fully relaxed for geometry optimization and energy calculation of the resulting initial conformations using the Gaussian 16 software program.46 The contribution (%) of a group in a molecule for each molecular orbital was calculated using the GaussSum 3.0 software program.47
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF-2020R1A2C1006400 and NRF-2021M3H4A1A02055684 for K. M. Lee) funded by the Ministry of Science and ICT. We thank Dr. Dongwook Kim (Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS)) for assistance with XRD experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03344.
1H, 1H{11B}, 13C, and 11B{1H} NMR spectra for the o-carboranyl compounds and their precursors; crystallographic data and parameters; UV–vis absorption and PL spectra for the o-carboranyl compounds and 1,1′-biphenyl; emission decay curves; and computational calculation details (PDF)
Crystallographic data for 13PP (CIF)
Crystallographic data for 12MM (CIF)
Crystallographic data for 11HI (CIF)
Crystallographic data for 10HV (CIF)
Crystallographic data for 9HM (CIF)
Crystallographic data for 3HB(CIF)
Crystallographic data for 8HP(CIF)
Crystallographic data for 7HD(CIF)
Crystallographic data for 6HC(CIF)
Crystallographic data for 4HN(CIF)
Crystallographic data for 2HT(CIF)
Crystallographic data for 1HF (CIF)
Crystallographic data for 5HH (CIF)
Author Contributions
All authors have given approval to the final version of the article.
The authors declare no competing financial interest.
Supplementary Material
References
- Bregadze V. I. Dicarba-closo-dodecaboranes C2B10H12 and Their Derivatives. Chem. Rev. 1992, 92, 209–223. 10.1021/cr00010a002. [DOI] [Google Scholar]
- Poater J.; Solà M.; Viñas C.; Teixidor F. π Aromaticity and Three-Dimensional Aromaticity: Two sides of the Same Coin?. Angew. Chem., Int. Ed. 2014, 53, 12191–12195. 10.1002/anie.201407359. [DOI] [PubMed] [Google Scholar]
- Poater J.; Viñas C.; Bennour I.; Escayola S.; Solà M.; Teixidor F. Too Persistent to Give Up: Aromatic in Boron Clusters Survives Radical Structural Changes. J. Am. Chem. Soc. 2020, 142, 9396–9407. 10.1021/jacs.0c02228. [DOI] [PubMed] [Google Scholar]
- Wee K.-R.; Cho Y.-J.; Jeong S.; Kwon S.; Lee J.-D.; Suh I.-H.; Kang S. O. Carborane-Based Optoelectronically Active Organic Molecules: Wide Band Gap Host Materials for Blue Phosphorescence. J. Am. Chem. Soc. 2012, 134, 17982–17990. 10.1021/ja3066623. [DOI] [PubMed] [Google Scholar]
- Furue R.; Nishimoto T.; Park I. S.; Lee J.; Yasuda T. Aggregation-Induced Delayed Fluorescence Based on Donor/Acceptor-Tethered Janus Carborane Triads: Unique Photophysical Properties of Nondoped OLEDs. Angew. Chem., Int. Ed. 2016, 55, 7171–7175. 10.1002/anie.201603232. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Park S.; Lee Y. H.; Jung J.; Yoo S.; Lee M. H. Homoleptic Tris-Cyclometalated Iridium Complexes with Substituted o-Carboranes: Green Phosphorescent Emitters for Highly Efficient Solution-Processed Organic Light-Emitting Diodes. Inorg. Chem. 2016, 55, 909–917. 10.1021/acs.inorgchem.5b02444. [DOI] [PubMed] [Google Scholar]
- Guo J.; Liu D.; Zhang J.; Zhang J.; Miao Q.; Xie Z. o-Carborane functionalized pentacenes: synthesis, molecular packing and ambipolar organic thin-film transistors. Chem. Commun. 2015, 51, 12004–12007. 10.1039/c5cc03608a. [DOI] [PubMed] [Google Scholar]
- Nar I.; Atsay A.; Altındal A.; Hamuryudan E. o-Carborane, Ferrocene, and Phthalocyanine Triad for High-Mobility Organic Field-Effect Transistors. Inorg. Chem. 2018, 57, 2199–2208. 10.1021/acs.inorgchem.7b03097. [DOI] [PubMed] [Google Scholar]
- Spokoyny A. M.; Li T. C.; Farha O. K.; Machan C. W.; She C.; Stern C. L.; Marks T. J.; Hupp J. T.; Mirkin C. A. Electronic Tuning of Nickel-Based Bis(dicarbollide) Redox Shuttles in Dye-Sensitized Solar Cells. Angew. Chem., Int. Ed. 2010, 49, 5339–5343. 10.1002/anie.201002181. [DOI] [PubMed] [Google Scholar]
- Aniés F.; Qiao Z.; Nugraha M. I.; Basu A.; Anthopoulos T. D.; Gasparini N.; Heeney M. N-type polymer semiconductors incorporating para, meta, and ortho-carborane in the conjugated backbone. Polymer 2022, 240, 124481. 10.1016/j.polymer.2021.124481. [DOI] [Google Scholar]
- Dash B. P.; Satapathy R.; Gaillard E. R.; Maguire J. A.; Hosmane N. S. Synthesis and Properties of CarboraneAppended C3-Symmetrical Extended π Systems. J. Am. Chem. Soc. 2010, 132, 6578–6587. 10.1021/ja101845m. [DOI] [PubMed] [Google Scholar]
- Weber L.; Kahlert J.; Brockhinke R.; Böhling L.; Brockhinke A.; Stammler H. G.; Neumann B.; Harder R. A.; Fox M. A. Luminescence Properties of C-Diazaborolyl-ortho-Carboranes as Donor–Acceptor Systems. Chem.—Eur. J. 2012, 18, 8347–8357. 10.1002/chem.201200390. [DOI] [PubMed] [Google Scholar]
- Ferrer-Ugalde A.; Juárez-Pérez E. J.; Teixidor F.; Viñas C.; Núñez R. Synthesis, Characterization, and Thermal Behavior of Carboranyl-Styrene Decorated Octasilsesquioxanes: Influence of the Carborane Clusters on Photoluminescence. Chem.—Eur. J. 2013, 19, 17021–17030. 10.1002/chem.201302493. [DOI] [PubMed] [Google Scholar]
- Kwon S.; Wee K.-R.; Cho Y.-J.; Kang S. O. Carborane Dyads for Photoinduced Electron Transfer: Photophysical Studies on Carbazole and Phenyl-o-carborane Molecular Assemblies. Chem.—Eur. J. 2014, 20, 5953–5960. 10.1002/chem.201304474. [DOI] [PubMed] [Google Scholar]
- Naito H.; Morisaki Y.; Chujo Y. o-Carborane-Based Anthracene: A Variety of Emission Behaviors. Angew. Chem., Int. Ed. 2015, 54, 5084–5087. 10.1002/anie.201500129. [DOI] [PubMed] [Google Scholar]
- Choi B. H.; Lee J. H.; Hwang H.; Lee K. M.; Park M. H. Novel Dimeric o-Carboranyl Triarylborane: Intriguing Ratiometric Color-Tunable Sensor via Aggregation-Induced Emission by Fluoride Anions. Organometallics 2016, 35, 1771–1777. 10.1021/acs.organomet.6b00172. [DOI] [Google Scholar]
- Núñez R.; Romero I.; Teixidor F.; Viñas C. Icosahedral boron clusters: a perfect tool for the enhancement of polymer features. Chem. Soc. Rev. 2016, 45, 5147–5173. 10.1039/c6cs00159a. [DOI] [PubMed] [Google Scholar]
- Nishino K.; Yamamoto H.; Tanaka K.; Chujo Y. Development of Solid-State Emissive Materials Based on Multifunctional o-Carborane-Pyrene Dyads. Org. Lett. 2016, 18, 4064–4067. 10.1021/acs.orglett.6b01920. [DOI] [PubMed] [Google Scholar]
- Tu D.; Leong P.; Guo S.; Yan H.; Lu C.; Zhao Q. Highly Emissive Organic Single–Molecule White Emitters by Engineering o-Carborane–Based Luminophores. Angew. Chem., Int. Ed. 2017, 56, 11370–11374. 10.1002/anie.201703862. [DOI] [PubMed] [Google Scholar]
- Wu X.; Guo J.; Cao Y.; Zhao J.; Jia W.; Chen Y.; Jia D. Mechanically triggered reversible stepwise tricolor switching and thermochromism of anthracene-o-carborane dyad. Chem. Sci. 2018, 9, 5270–5277. 10.1039/c8sc00833g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K. L.; Smith J. N.; Young E. R.; Carter K. R. Synthetic Emission Tuning of Carborane-Containing Poly(dihexylfluorene)s. Macromolecules 2019, 52, 7951–7960. 10.1021/acs.macromol.9b01325. [DOI] [Google Scholar]
- Ochi J.; Tanaka K.; Chujo Y. Recent Progress in the Development of Solid-state Luminescent o-Carboranes with Stimuli Responsivity. Angew. Chem., Int. Ed. 2020, 59, 9841–9855. 10.1002/anie.201916666. [DOI] [PubMed] [Google Scholar]
- Hong J. H.; Im S.; Seo Y. J.; Kim N. Y.; Ryu C. H.; Kim M.; Lee K. M. Effects of Terminal Biphenyl Ring Geometry on the Photophysical Properties of closo-o-Carboranyl-Anthracene Dyads. J. Mater. Chem. C 2021, 9, 9874–9883. 10.1039/d1tc02051j. [DOI] [Google Scholar]
- Lee S. H.; Mun M. S.; Lee J. H.; Im S.; Lee W.; Hwang H.; Lee K. M. Impact of the electronic environment in carbazole-appended o-carboranyl compounds on the intramolecular-charge-transfer-based radiative decay efficiency. Organometallics 2021, 40, 959–967. 10.1021/acs.organomet.1c00060. [DOI] [Google Scholar]
- Ochi J.; Tanaka K.; Chujo Y. Dimerization-Induced Solid-State Excimer Emission Showing Consecutive Thermochromic Luminescence Based on Acridine-Modified o-Carboranes. Inorg. Chem. 2021, 60, 8990–8997. 10.1021/acs.inorgchem.1c00901. [DOI] [PubMed] [Google Scholar]
- Kim S.; Lee J. H.; So H.; Ryu J.; Lee J.; Hwang H.; Kim Y.; Park M. H.; Lee K. M. Spirobifluorene-Based o-Carboranyl Compounds: Insights into the Rotational Effect of Carborane Cages on Photoluminescence. Chem.—Eur. J. 2020, 26, 548–557. 10.1002/chem.201904491. [DOI] [PubMed] [Google Scholar]
- Mun M. S.; Ryu C. H.; So H.; Kim M.; Lee J. H.; Hwang H.; Lee K. M. Multiple Photoluminescence of Spiro[acridine-fluorene]-based o-Carboranyl Compounds and Potential as a Visual Sensory. J. Mater. Chem. C 2020, 8, 16896–16906. 10.1039/d0tc03801f. [DOI] [Google Scholar]
- You D. K.; So H.; Ryu C. H.; Kim M.; Lee K. M. Strategic Molecular Design of closo-ortho-Carboranyl Luminophore to Manifest Thermally Activated Delayed Fluorescence. Chem. Sci. 2021, 12, 8411–8423. 10.1039/d1sc00791b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixidor F.; Núñez R.; Viñas C.; Sillanpää R.; Kivekäs R. The Distinct Effect of the o-Carboranyl Fragment: Its Influence on the I-I Distance in R3PI2 Complexes. Angew. Chem., Int. Ed. 2000, 39, 4290–4292. . [DOI] [PubMed] [Google Scholar]
- Núñez R.; Farràs P.; Teixidor F.; Viñas C.; Sillanpää R.; Kivekäs R. A Discrete P···I—I···P Assembly: The Large Influence of Weak Interactions on the 31P NMR Spectra of Phosphane-Diiodine Complexes. Angew. Chem., Int. Ed. 2006, 45, 1270–1272. 10.1002/anie.200503007. [DOI] [PubMed] [Google Scholar]
- Wee K.-R.; Cho Y.-J.; Song J. K.; Kang S. O. Multiple Photoluminescence from 1,2-Dinaphthyl-ortho-Carborane. Angew. Chem., Int. Ed. 2013, 52, 9682–9685. 10.1002/anie.201304321. [DOI] [PubMed] [Google Scholar]
- Kahlert J.; Böhling L.; Brockhinke A.; Stammler H.-G.; Neumann B.; Rendina L. M.; Low P. J.; Weber L.; Fox M. A. Syntheses and reductions of C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes. Dalton Trans. 2015, 44, 9766–9781. 10.1039/c5dt00758e. [DOI] [PubMed] [Google Scholar]
- Ochi J.; Tanaka K.; Chujo Y. Experimental proof for emission annihilation through bond elongation at the carbon-carbon bond in o-carborane with fused biphenyl-substituted compounds. Dalton Trans. 2021, 50, 1025–1033. 10.1039/d0dt03618h. [DOI] [PubMed] [Google Scholar]
- Ryu C. H.; Lee S. H.; Yi S.; Hong J. H.; Im S.; Lee K. M. Naphthyl- and quinoline-appended o-carboranyl luminophores: Intramolecular charge transfer-based radiative decay controlled by structural geometry around C-C bond axis. Eur. J. Inorg. Chem. 2021, 46, 4875–4881. 10.1002/ejic.202100768. [DOI] [Google Scholar]
- Shin N.; Yu S.; Lee J. H.; Hwang H.; Lee K. M. Biphenyl- and Fluorene-Based o-Carboranyl Compounds: Alteration of Photophysical Properties by Distortion of Biphenyl Rings. Organometallics 2017, 36, 1522–1529. 10.1021/acs.organomet.7b00093. [DOI] [Google Scholar]
- So H.; Kim J. H.; Lee J. H.; Hwang H.; An D. K.; Lee K. M. Planarity of terphenyl rings possessing o-carborane cages: turning on intramolecular-charge-transfer-based emission. Chem. Commun. 2019, 55, 14518–14521. 10.1039/c9cc07729d. [DOI] [PubMed] [Google Scholar]
- Kim M.; Ryu C. H.; Hong J. H.; Lee J. H.; Hwang H.; Lee K. M. Planarity of N-aryl in Appended 1,2,4-Triazole-based o-Carboranyl Luminophores: Key Factor to Control Intramolecular Charge Transfer. Inorg. Chem. Front. 2020, 7, 4180–4189. 10.1039/d0qi00915f. [DOI] [Google Scholar]
- Runge E.; Gross E. K. U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. 10.1103/physrevlett.52.997. [DOI] [Google Scholar]
- Jiang W.; Knobler C. B.; Hawthorne M. F. Synthesis and Structural Characterization of Bis- and Tris(closo-1,2-C2B10H11-1-yl)-Substituted Biphenyl and Benzene. Inorg. Chem. 1996, 35, 3056–3058. 10.1021/ic951575d. [DOI] [Google Scholar]
- Huh J. O.; Kim H.; Lee K. M.; Lee Y. S.; Do Y.; Lee M. H. o-Carborane-assisted Lewis acidity enhancement of triarylboranes. Chem. Commun. 2010, 46, 1138–1140. 10.1039/b918263b. [DOI] [PubMed] [Google Scholar]
- Fox M. A.; Nervi C.; Crivello A.; Low P. J. Carborane radical anions: spectroscopic and electronic properties of a carborane radical anion with a 2n + 3 skeletal electron count. Chem. Commun. 2007, 23, 2372–2374. 10.1039/b700110j. [DOI] [PubMed] [Google Scholar]
- Zhang Q.-W.; An K.; Liu L.-C.; Guo S.; Jiang C.; Guo H.; He W. Rhodium-Catalyzed Intramolecular C-H Silylation by Silacyclobutanes. Angew. Chem., Int. Ed. 2016, 55, 6319–6323. 10.1002/anie.201602376. [DOI] [PubMed] [Google Scholar]
- Staniland S.; Yuan B.; Giménez-Agulló N.; Marcelli T.; Willies S. C.; Grainger D. M.; Turner N. J.; Clayden J. Enzymatic Desymmetrising Redox Reactions for the Asymmetric Synthesis of Biaryl Atropisomers. Chem.—Eur. J. 2014, 20, 13084–13088. 10.1002/chem.201404509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkley J. S.; Pople J. A.; Hehre W. J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980, 102, 939–947. 10.1021/ja00523a008. [DOI] [Google Scholar]
- Miertuš S.; Scrocco E.; Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. 10.1016/0301-0104(81)85090-2. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; RanasingheZakrzewski D. V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision B.01; Gaussian. Inc.: Wallingford, CT, 2016.computer-program [Google Scholar]
- O’Boyle N. M.; Tenderholt A. L.; Langner K. M. Cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.