

Abstract
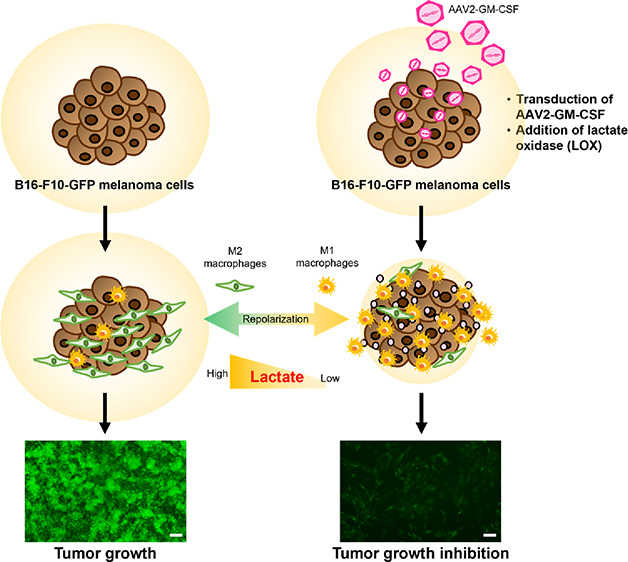
Oncolytic virotherapy was approved as a localized treatment for advanced melanoma by the US Food and Drug Administration (FDA) in 2015. Granulocyte macrophage colony stimulating factor (GM-CSF) encoded by clinical virus-infected tumor cells, acting as a pro-inflammatory cytokine or growth factor, increases tumor antigen presentation, leading to the activation of macrophages and T cells. Notably, tumor-secreted lactate can promote the suppressive functions of M2-polarized tumor-associated macrophages and subsequently promote tumor growth. Furthermore, the consumption of tumor-secreted lactate has been implicated in the beneficial polarization of macrophages. Here, we report that GM-CSF-encoded recombinant adeno-associated virus (AAV2-GM-CSF) infection in B16-F10 mouse melanoma cells combined with lactate oxidase (LOX) leads to the recruitment of M1 macrophages for the inhibition of cancer cell growth. This study suggests that GM-CSF combined with LOX has potential as cancer virotherapy.
Introduction
Oncolytic virotherapy represents a novel drug class in which native or modified viruses mediate tumor regression through the selective replication within and lysis of tumor cells as well as the induction of systemic antitumor immunity capable of eradicating tumors at distant, uninjected sites.1−3 Talimogene laherparepvec (T-VEC) is a type I herpes simplex virus that was genetically modified to preferentially replicate in tumor cells and express granulocyte macrophage colony stimulating factor (GM-CSF) to increase tumor antigen presentation for tumor growth inhibition.3 More interestingly, it is presently the only oncolytic virus approved by the US Food and Drug Administration (FDA) with an indication for advanced melanoma.3 Among various viruses, adeno-associated virus (AAV) has a unique transgene function in clinical trials with low immunogenicity and non-pathogenic properties, showing its advantages as a genetic vector approved by US FDA.4
Generally, GM-CSF functionally stimulates the proliferation of bone marrow progenitor cells and their differentiation into granulocytes and macrophages.5,6 GM-CSF acts as a pro-inflammatory cytokine and key growth factor produced by several immune cells, such as macrophages and activated T cells.7 In particular, GM-CSF drives the polarization of macrophages into an M1 phenotype, as indicated by the development of a pro-inflammatory phenotype in vitro8 and the production of chemokines for leukocyte recruitment9 as well as cytokines for pro-inflammatory actions upon stimulation.10 These molecular signals also contribute to the roles of GM-CSF in the differentiation and activation of T helper cells, further promoting pro-inflammatory events and the clearance of infectious agents.11
Notably, lactate is the primary carbon source for the tricarboxylic acid (TCA) cycle in cancer cells due to the metabolic reprogramming of cancer cells.12 Furthermore, clinical surgical resections from patients show nonhomeostatic glucose metabolism after the infusion of a labeled 13C-glucose infusion, leading to considerably elevated levels of lactate.13 Specifically, lactate can promote the suppressive functions of the M2-like polarization of tumor-associated macrophages and subsequently promote tumor growth.12,14 Overall, the depletion of lactate through lactate oxidase (LOX) activity has been implicated in the potentially beneficial repolarization of macrophages.14,15 We hypothesized that if the GM-CSF produced by cancer cells infected with a recombinant adeno-associated virus (AAV2) combined with LOX can recruit M1 macrophages, then the inhibition of cancer cell proliferation can be improved (Figure 1A). This approach was verified with GM-CSF produced by B16-F10 mouse melanoma cells combined with or without LOX at a neutral or acidic pH; the effects on the macrophage distribution and proliferation of cancer cells achieved with this therapy were compared with those achieved with a GM-CSF standard.
Figure 1.

Macrophage polarization induced by AAV2-GM-CSF. (A) Schematic of GM-CSF production by cancer cells infected with AAV2-GM-CSF combined with lactate oxidase (LOX) for the promotion of tumor-suppressing M1 macrophage recruitment. Specifically, LOX oxidized cancer cell-secreted lactate, leading to the conversion of tumor-promoting M2 macrophages into M1 macrophages. (B) Plasmid map of pAAV-GM-CSF (size: 6165 base pairs). (C) Top: cell viability of B16-F10 cells after infection with AAV2-GM-CSF. Cell viability is given as the percentage of viable cells remaining after treatment for 1, 2, 3, 7, or 13 days compared with the percentage of viable unexposed cells and was determined with a CellTiter 96 AQueous One Solution Cell Proliferation Assay (n.s., not significant; two-tailed unpaired Student’s t test). The bars represent the mean ± standard deviation (n = 6). Bottom: quantitative determination of GM-CSF expression by B16-F10 cells after infection with AAV2-GM-CSF. The bars represent the mean ± standard deviation (n = 4).
Results
Cancer Cells Infected by AAV2
We performed immunovirotherapy utilizing GM-CSF derived from target cancer cells infected with AAV2-GM-CSF (Figure 1B). In vitro characterization and viral transduction assays were performed in complete culture medium (10% fetal bovine serum, 100 U mL–1 penicillin, and 100 μg mL–1 streptomycin). No cytotoxicity was observed for any duration of AAV2-GM-CSF incubation with B16-F10 cells (Figure 1C, top). When cells were transduced with AAV2-GM-CSF for 7 days, cell viability was maintained at 0.98 ± 0.05. Even a longer incubation of 13 days resulted in a cell viability of 0.94 ± 0.07, indicating that AAV2-GM-CSF exhibited low toxicity to the target cells and did not inhibit the proliferation of AAV2-GM-CSF-infected B16-F10 cells.
To evaluate the level of GM-CSF produced by B16-F10 melanoma cells following various transduction periods, we used a Mouse GM-CSF Quantikine ELISA Kit to detect the GM-CSF protein secreted by B16-F10 cells. Viral transduction led to an increase in the level of GM-CSF expressed by cells (Figure 1C, bottom). The results showed that the GM-CSF level increased from 1.12 ± 0.21 to 6.14 ± 0.59 ng mL–1 as the viral transduction time increased from 1 day to 3 days, respectively. Additionally, when the transduction period was increased to 13 days, the GM-CSF level was decreased to 4.75 ± 0.20 ng mL–1. Overall, AAV2-infected B16-F10 cells could sustainably produce GM-CSF over 10 days.
GM-CSF Produced by Cancer Cells by AAV2 Transduction
To validate the functionality of GM-CSF produced by B16-F10 melanoma cells infected with AAV2-GM-CSF, the ability of GM-CSF (25 ng mL–1) to induce M1-macrophage polarization was assessed. We evaluated the populations of M1 (MHC II-expressing macrophages)16 and M2 (CD206-expressing macrophages)14−16 macrophages induced by the GM-CSF standard or GM-CSF encoded by AAV2. As shown in Figure 2A, the populations of M1 macrophages clearly increased in the presence of the GM-CSF standard (∼2.50-fold compared to the negative control) or GM-CSF (∼4.38-fold compared to the negative control). In contrast, the negative control failed to induce polarization. Similar proliferation of M2 macrophages induced by GM-CSF standard or GM-CSF was noted. Specific biomarker staining indicated increased M0, M1, or M2 macrophage levels, which were also verified by confocal microscopy (Figure 2B) and consistent with flow cytometric analysis results.
Figure 2.

Effects of GM-CSF produced by cancer cells on macrophages. (A) The expression of surface markers on M1 or M2 macrophages based on fold changes relative to that on untreated macrophages (§p < 0.0005, ‡p < 0.00005; two-tailed unpaired Student’s t test). Surface biomarkers such as MHC II and CD206 were used to identify M1 and M2 macrophages, respectively. The bars represent the mean ± standard deviation (n = 4). (B) Representative confocal images of macrophages treated under various conditions for 6 days. Scale bar = 100 μm. (C) Quantitative determination of cytokines (TNF-α, IL-6, IL-10, or IL-12) and NO2– concentrations using Cytokine Expression ELISA and Nitrite Assay Kits (*p < 0.05, #p < 0.005; §p < 0.0005, ‡p < 0.00005; n.s., not significant; two-tailed unpaired Student’s t test). The bars represent the mean ± standard deviation (n = 4).
To confirm that M1 macrophages expressed cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-12,10 we measured these cytokines by using a Mouse TNF-α ELISA Kit, Mouse IL-6 ELISA Kit, or Mouse IL-12 ELISA Kit, respectively (Figure 2C). When compared to those treated with the negative control, M0 macrophages treated with GM-CSF at 6 days tended to express higher levels of inflammatory factors (TNF-α, IL-6 and IL-12). Compared to the GM-CSF standard, GM-CSF had a subtle effect on IL-10 in M2 macrophages (not significant by t test). Furthermore, the production of nitric oxide (NO) in M1 macrophages induced by the bacterial endotoxin lipopolysaccharide (LPS) (100 ng mL–1) was characterized,14,15 and the total concentration of nitrite (NO2–) was measured by using a Nitrite Assay Kit (Griess assay).14,15 Surprisingly, NO2– expression was higher in the GM-CSF standard (16.24 ± 0.5 μM) and GM-CSF (23.78 ± 0.6 μM) groups due to the induction of M1 polarization.14,15 Despite these differences, the results for selected surface markers or cytokines were similar when the GM-CSF standard or GM-CSF produced by cancer cells was used.
Effect of GM-CSF Combined with Lactate Oxidase on the Macrophage Distribution and Cancer Cell Growth
Typically, the tumor microenvironment consists of specific produced factors and metabolic products. Thus, the pH changes associated with lactate secretion by cancer cells affect not only immune cells but also tumor growth.12 Overall, tumor-secreted lactate can promote the suppressive functions of the M2-like polarization of tumor-associated macrophages and subsequently promote tumor growth.12,14,15 Previously, we developed matrix-incorporated LOX for the depletion of lactate, which has been implicated in the potentially beneficial repolarization of macrophages.12,14,15
To verify the influence of lactate on macrophage distribution, we used l-lactic acid (1.0 M) to adjust the culture medium from pH 7.4 to 6.7. The setup contained 8.1 μmol of l-lactic acid per milliliter of pH 6.7 culture medium. The pH value was changed from 6.7 to 6.5 after lactate oxidization by LOX. Furthermore, M0 macrophage incubated with pH 7.4, 6.7, or 6.5 medium for 0 or 6 days were evaluated. In comparison with Day 0, the macrophages’ growths at Day 6 were significantly different at various pH values (Figure 3).
Figure 3.
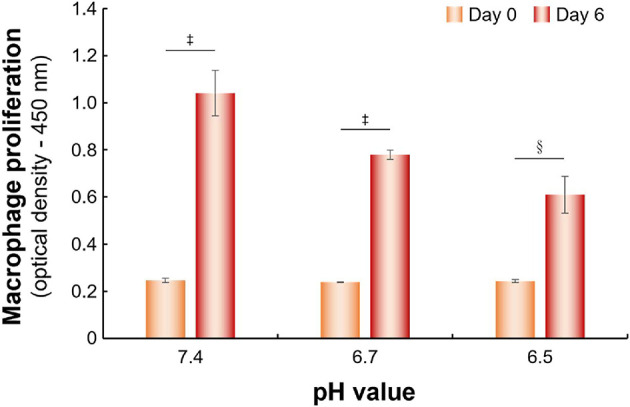
The proliferation of macrophages cultured in different pH condition media. Cell proliferation is given as the optical density (OD) value of macrophages cultured in different pH condition for 0 or 6 days determined by CCK-8 kit (§p < 0.0005, ‡p < 0.00005; two-tailed unpaired Student’s t test). The bars represent the mean ± standard deviation (n = 6).
M0 macrophages in culture medium at pH 7.4 or 6.7 were treated with GM-CSF with or without LOX for 6 days. At pH 6.7, GM-CSF combined with LOX promoted the polarization of M1 macrophages (∼1.27-fold) compared to GM-CSF alone (Figure 4A). As expected, the distribution of M1 macrophages was maintained at acidic pH compared to the neutral conditions when lactate was oxidized. Consistent with these findings, fluorescence micrographs displayed similar staining of F4/80-expressing M0 macrophages, MHC II-expressing M1 macrophages, and CD206-expressing M2 macrophages (Figure 4B). After 6 days of incubation, the signal for MHC II-expressing M1 macrophages was notably detectable in the GM-CSF with LOX group compared with the GM-CSF alone group at pH 6.7.
Figure 4.

Effect of GM-CSF combined with LOX on macrophages or on cancer cell growth. (A) The fold changes in M1 and M2 marker expression relative to untreated macrophages (#p < 0.005, §p < 0.0005; two-tailed unpaired Student’s t test). The bars represent the mean ± standard deviation (n = 4). (B) Representative confocal images of macrophages treated as in (A). Scale bar = 100 μm. (C) Schematic representation of the coculture model established with AAV2-GM-CSF-infected B16-F10-GFP cancer cells (receiver well) and macrophages (membrane insert) for the measurement of cancer cell growth. (D) The cell viability of B16-F10-GFP cells cocultured with macrophages under various conditions using Transwell plates (*p < 0.05, §p < 0.0005, ‡p < 0.00005; two-tailed unpaired Student’s t test). The bars represent the mean ± standard deviation (n = 4). (E) Representative images of B16-F10-GFP cells cocultured with macrophages under various conditions in Transwell plates. Scale bar = 100 μm.
In an assay, M0 macrophages were separated from B16-F10-GFP cells infected with AAV2-GM-CSF via a 0.4 μm porous polyester (PET) membrane (Figure 4C). The growth-inhibitory effect on B16-F10-GFP cells cocultured with M0 macrophages under various conditions was measured. As expected, B16-F10-GFP cells cocultured with macrophages in the GM-CSF combined with LOX group were significantly different, as indicated by GFP fluorescence signals, from cells cocultured without any treatments (Figure 4D,E). Thus, we postulated that M0 macrophages were promptly polarized into M1 macrophages by treating with GM-CSF and lactate consumption by LOX.
Discussion
The US FDA previously approved an engineered virus as an immunodrug for immunotherapy in advanced melanoma.17 Among various cancer therapies, oncolytic virotherapy represents a class of promising cancer therapeutics, with viruses from several families currently being evaluated in clinical trials.18 Furthermore, one of the most significant technical solutions needed in clinical virotherapy is enhanced systemic viral transduction.19,20 Currently, the accurate and specific delivery of genetic material at an appropriate dose is a major challenge.
T-VEC was developed for intratumoral injection in the clinic; this virotherapy produces GM-CSF and enhances local and systemic antitumor immune responses.21 Furthermore, GM-CSF induces M1 macrophages and the subsequent production of pro-inflammatory cytokines, consistent with our results (Figure 2).5−11 In general, macrophages are divided into antitumoral M1 macrophages and protumoral M2 macrophages. Therefore, the modulation of macrophages is an effective approach to suppress cancer cell growth.
On the other hand, tumor-secreted lactate acts as a significant regulator that modulates the immune system.12 Furthermore, the consumption of tumor-secreted lactate has been implicated in the beneficial polarization of macrophages.14,15 In our studies, GM-CSF combined with LOX exhibited a higher level of M1 macrophages (Figure 3) than GM-CSF alone, leading to an expressively improved proportion of M1 macrophages. Consistent with our previous studies,14,15 in low-lactate medium, M2 macrophages can be repolarized into M1 macrophages. However, the regulation of the cancer microenvironment needs to be studied with RAW 264.7 cells and primary macrophages derived in vitro from circulating monocytes, and the obtained results will be evaluated in further in vivo experiments.
Conclusions
Macrophages are involved in the cancer-initiating inflammatory responses. Our work based on virotherapy using AAV2 as a transgene vector demonstrates that GM-CSF produced by cancer cells leads to the recruitment of M1 macrophages. This study also suggests that GM-CSF combined with LOX could have produced a synergistic effect on regulating the distribution of macrophages as an improved virotherapy to potentiate cancer treatment.
Methods
Materials and Cell Culture
Phosphate-buffered saline (PBS, pH 7.4), branched polyethylenimine (bPEI 25 K, MW = 25,000), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), LOX from Aerococcus viridans, LPS, l-lactic acid, and a Nitrite Assay Kit (Griess Reagent) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). A CellTiter 96 Aqueous One Solution Cell Proliferation Assay was purchased from Promega (Madison, WI, USA). A mouse Csf2-tagged ORF clone was purchased from OriGene (Rockville, MD, USA). The plasmids pHelper, pAAV-RC2, and pAAV-MCS and a QuickTiter AAV Quantitation Kit were purchased from Cell Biolabs (San Diego, CA, USA). An AAVpro Purification Kit Maxi (All Serotypes) was purchased from TAKARA Bio Inc. (Shiga, Japan). A Mouse GM-CSF Quantikine ELISA Kit and anti-mouse MMR/CD206 antibody were purchased from R&D Systems (Minneapolis, MN, USA). A recombinant mouse GM-CSF protein, an anti-F4/80 antibody [CI: A3–1], and an anti-MHC class II antibody were purchased from Abcam (Cambridge, MA, USA). A chicken anti-rat IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 647), donkey anti-rabbit IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 555), donkey anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody (Alexa Fluor 488), and donkey anti-goat IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 488) were purchased from Invitrogen (Carlsbad, CA, USA). A Mouse IL-6 ELISA Kit, a Mouse IL-10 ELISA Kit, a Mouse IL-12 ELISA Kit, and a Mouse TNF-α ELISA Kit were purchased from Elabscience (Houston, Texas, USA). A Cell Counting Kit-8 (CCK-8) was purchased from Targetmol (Boston, MA, USA).
The 293T (ATCC CRL-3216), B16-F10 mouse melanoma (BCRC 60031) and RAW 264.7 mouse macrophage (BCRC 60001) cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U mL–1 penicillin, and 100 μg mL–1 streptomycin. Cells were cultured at 37 °C in a 5% CO2 atmosphere.
Virus Production, Purification, and Titration
AAV2-GM-CSF production was performed with an AAV helper-free packaging system (Cell Biolabs, San Diego, CA, USA). AAV2-GM-CSF was produced by bPEI 25 K-mediated cotransfection of plasmid DNAs (50 μg pHelper, 25 μg pAAV-RC2, and 25 μg pAAV-GM-CSF) into 293 T cells in a 15-cm dish. The three plasmids were mixed with 100 μg PEI in serum-free DMEM, vortexed for 30 s and incubated at room temperature for 15–20 mins. The transfection time was 30 mins, and the transfected cells were incubated for 72 h. The purification and titration of AAV2-GM-CSF were performed according to the protocols of an AAVpro Purification Kit Maxi (TAKARA Bio Inc. Shiga, Japan) and a QuickTiter AAV quantitation kit (Cell Biolabs, San Diego, CA, USA). The amount of AAV2-GM-CSF ranged from 1011 to 1012 genome copies (GC) per milliliter for each round (4 × 15-cm dishes) of virus production. Purified viruses were stored at −80 °C before use. To evaluate the AAV2 transduction efficiency in B16-F10 cells, we used an AAV2-GFP (green fluorescent protein) assay detected with flow cytometry (60–70%).
Transduction for GM-CSF Production
B16-F10 cells were seeded in 48-well plates at 3.5 × 104 cells per well and incubated for 24 h before transduction. AAV2-GM-CSF (single dose: 9 × 109 GC per well) was administered every 24 h for a total of three doses. The cytotoxicity of AAV2-GM-CSF was analyzed on days 1, 2, 3, 7, and 13. A CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) was used to measure cell viability. The optical density of untreated cells at 490 nm was set at 1, and the viability of transduced cells is expressed as a ratio to that of untreated cells.
Culture medium was collected every 24 h after transduction. The amount of GM-CSF produced by B16-F10 cells was measured by a Mouse GM-CSF Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA). To avoid any influence of cellular metabolites from B16-F10 cells in the culture medium, the culture medium was concentrated with a 5 kDa desalting column and solvent-exchanged with PBS. The concentrated cancer cell-secreted GM-CSF solution was stored at −20 °C before use.
Macrophage Proliferation in Different pH Value Conditions
RAW264.7 cells were seeded in 24-well plates at 1.4 × 105 cells per well and incubated overnight. The culture medium was changed to pH 7.4, 6.7, or 6.5 adjusted by 1.0 M l-lactic acid. After cultured for 0 or 6 days, the proliferation was assessed by a CCK-8 (Targetmol, Boston, MA, USA).
In Vitro Macrophage Polarization
RAW264.7 cells were seeded in 24-well plates at 1.4 × 105 cells well–1 and incubated overnight. The concentrated cancer cell-secreted GM-CSF solution was diluted with pH 7.4 DMEM (10% FBS, 100 U mL–1 penicillin, and 100 μg mL–1 streptomycin) to a concentration of 25 ng mL–1. A recombinant mouse GM-CSF protein (Abcam, Cambridge, UK) was also diluted to the same concentration and used as the standard control. RAW264.7 cells were treated with GM-CSF for 6 days, and the culture medium was changed every day. The negative control group was RAW264.7 cells without GM-CSF treatment.
To evaluate the polarizing function of GM-CSF in a tumor microenvironment containing lactate, macrophages were incubated with GM-CSF in pH 7.4 or 6.7 culture medium that was adjusted with 1.0 M l-lactic acid for 6 days. LOX (0.025 U well–1) was also added to catalyze lactate conversion into pyruvate, mimicking the removal of lactate in the tumor microenvironment. As expected, the converted pyruvate modulated a lowering of pH (∼6.5) in the culture medium.
Analysis of Macrophage Phenotypes
Treated cells were fixed with 4% paraformaldehyde (PFA), and immunostaining was performed using an anti-F4/80 antibody (Abcam, Cambridge, UK), an anti-MHC class II antibody (Abcam, Cambridge, UK), and an anti-MMR/CD206 antibody (R&D Systems, Minneapolis, MN, USA) for the analysis of macrophage phenotypes. Signal amplification was performed with chicken anti-rat IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 647; Invitrogen, Carlsbad, CA, USA) for F4/80, donkey anti-rabbit IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 555; Invitrogen, Carlsbad, CA, USA) and donkey anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody (Alexa Fluor 488; Invitrogen, Carlsbad, CA, USA) for MHC class II, and donkey anti-goat IgG (H + L) cross-adsorbed secondary antibody (Alexa Fluor 488; Invitrogen, Carlsbad, CA, USA) for CD206. Cells were treated with DAPI to label the nuclei. The immunostained cells were observed under an LSM 700 confocal microscope (Carl Zeiss, Oberkochen, Germany) and quantitatively assessed on an Attune NxT flow cytometer (Thermo Fisher Scientific, Waltham, MA, USA). Untreated cells were used as a negative control.
After polarization with GM-CSF for 6 days, the TNF-α, IL-6, IL-10, and IL-12 protein expression levels of macrophages were quantified with ELISA kits (Elabscience, Houston, Texas, USA). The expression of nitric oxide synthase (iNOS) in polarized macrophages was evaluated after incubation with 100 ng mL–1 LPS for 24 h. The NO2– production level was assessed with a Nitrite Assay Kit (Sigma-Aldrich Co., St. Louis, MO, USA) according to the manufacturer’s protocol.
Inhibitory Effects of Cancer Cells
To verify the effect of treated macrophages on the growth of cancer cells, we generated B16-F10 cells expressing green fluorescent protein (B16-F10-GFP) for the evaluation of cancer cell growth. B16-F10 cells were infected by GFP-lentivirus transduction. Briefly, B16-F10 cells were seeded in 12-well plates at 1.4 × 105 cells per well and incubated for 24 h. Transduction with GFP-lentivirus (National RNAi Core Facility at Academia Sinica, Taipei City, Taiwan) was performed at a dose of 5.5 × 106 RIU per well. After viral infection, the treated cells were cultured in DMEM supplemented with 10% FBS, 100 U mL–1 penicillin, 100 μg mL–1 streptomycin, and 2 μg mL–1 puromycin for 7 days. The final ratio of GFP expression was 98.4%, as measured by an Attune NxT flow cytometer (Thermo Fisher Scientific, Waltham, MA, USA).
A Transwell assay was performed using a Transwell plate. The inserts contained 0.4 μm pore filters, which allowed molecules to be exchanged. M0 macrophages were seeded in the upper chamber at 1.5 × 105 cells per well, and B16-F10-GFP cells were seeded in the lower chamber at 3 × 104 cells per well. After incubating with GM-CSF and LOX in pH 7.4 or 6.7 culture medium for 6 days, the cell viability of B16-F10-GFP cells was observed under an Eclipse Ti-S fluorescence microscope (Nikon) and assessed with a CCK-8 (Targetmol, Boston, MA, USA).
Acknowledgments
The authors thank the Taiwan Ministry of Science and Technology Grant and the NSYSU-KMU Joint Research Project.
This research was funded by Taiwan Ministry of Science and Technology (grant numbers MOST 108–2628-E-110-003-MY3, MOST 108–2628-B-110-001, MOST 109–2628-B-110-003, and MOST 110–2628-B-110-003) and the NSYSU-KMU Joint Research Project (#NSYSUKMU 111-P26).
The authors declare no competing financial interest.
References
- Melcher A.; Harrington K.; Vile R. Oncolytic virotherapy as immunotherapy. Science 2021, 374, 1325–1326. 10.1126/science.abk3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler T.; Wick W. Oncolytic virotherapy: Potentially a game-changing tumor treatment. Cancer Cell 2021, 39, 753–755. 10.1016/j.ccell.2021.05.014. [DOI] [PubMed] [Google Scholar]
- Cassidy T.; Craig M. Determinants of combination GM-CSF immunotherapy and oncolytic virotherapy success identified through in silico treatment personalization. PLoS Comput. Biol. 2019, 15, e1007495 10.1371/journal.pcbi.1007495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Samulski R. J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- van de Laar L.; Coffer P. J.; Woltman A. M. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. 10.1182/blood-2011-11-370130. [DOI] [PubMed] [Google Scholar]
- Lotfi N.; Thome R.; Rezaei N.; Zhang G.-X.; Rezaei A.; Rostami A.; Esmaeil N. Roles of GM-CSF in the Pathogenesis of Autoimmune Diseases: An Update. Front Immunol. 2019, 10, 1265. 10.3389/fimmu.2019.01265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev E. D.; Shriver L. P.; Maresz K.; Pedras-Vasconcelos J.; Verthelyi D.; Dittel B. N. GM-CSF Production by Autoreactive T Cells Is Required for the Activation of Microglial Cells and the Onset of Experimental Autoimmune Encephalomyelitis. The Journal of Immunology 2007, 178, 39–48. 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- Neupane K. R.; McCorkle J. R.; Kopper T. J.; Lakes J. E.; Aryal S. P.; Abdullah M.; Snell A. A.; Gensel J. C.; Kolesar J.; Richards C. I. Macrophage-Engineered Vesicles for Therapeutic Delivery and Bidirectional Reprogramming of Immune Cell Polarization. ACS Omega 2021, 6, 3847–3857. 10.1021/acsomega.0c05632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushach I.; Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukocyte Biol. 2016, 100, 481–489. 10.1189/jlb.3RU0316-144R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya P.; Thiruppathi M.; Elshabrawy H. A.; Alharshawi K.; Kumar P.; Prabhakar B. S. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine+ 2015, 75, 261–271. 10.1016/j.cyto.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J. A. GM-CSF-Dependent Inflammatory Pathways. Front. Immunol. 2019, 10, 2055. 10.3389/fimmu.2019.02055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z.-X.; Kempson I. M.; Hsieh C.-C.; Tseng S. J.; Yang P.-C. Potential therapeutics using tumor-secreted lactate in nonsmall cell lung cancer. Drug Discovery Today 2021, 26, 2508–2514. 10.1016/j.drudis.2021.07.014. [DOI] [PubMed] [Google Scholar]
- Fan T. W. M.; Lane A. N.; Higashi R. M.; Farag M. A.; Gao H.; Bousamra M.; Miller D. M. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Molecular Cancer 2009, 8, 41. 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z.-X.; Fa Y.-C.; Kempson I. M.; Tseng S. J. Repolarization of M2 to M1 Macrophages Triggered by Lactate Oxidase Released from Methylcellulose Hydrogel. Bioconjugate Chem. 2019, 30, 2697–2702. 10.1021/acs.bioconjchem.9b00618. [DOI] [PubMed] [Google Scholar]
- Liao Z.-X.; Ou D.-L.; Hsieh M.-J.; Hsieh C.-C. Synergistic Effect of Repolarization of M2 to M1 Macrophages Induced by Iron Oxide Nanoparticles Combined with Lactate Oxidase. Int. J. Mol. Sci. 2021, 22, 13346. 10.3390/ijms222413346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou D.-L.; Tseng S.-J.; Kempson I. M.; Hsu C.-L.; Yang P.-C.; Liao Z.-X. Enhanced Targeting and Immune Activation of Tumor Microenvironment by Nanomodified Anti-PD1 in Liver Cancer. Adv. Therap. 2021, 4, 2100048. 10.1002/adtp.202100048. [DOI] [Google Scholar]
- Naldini L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- Miest T. S.; Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng S. J.; Huang K.-Y.; Kempson I. M.; Kao S.-H.; Liu M.-C.; Yang S.-C.; Liao Z.-X.; Yang P.-C. Remote Control of Light-Triggered Virotherapy. ACS Nano 2016, 10, 10339–10346. 10.1021/acsnano.6b06051. [DOI] [PubMed] [Google Scholar]
- Tseng S. J.; Kempson I. M.; Huang K.-Y.; Li H.-J.; Fa Y.-C.; Ho Y.-C.; Liao Z.-X.; Yang P.-C. Targeting Tumor Microenvironment by Bioreduction-Activated Nanoparticles for Light-Triggered Virotherapy. ACS Nano 2018, 12, 9894–9902. 10.1021/acsnano.8b02813. [DOI] [PubMed] [Google Scholar]
- Ferrucci P. F.; Pala L.; Conforti F.; Cocorocchio E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. 10.3390/cancers13061383. [DOI] [PMC free article] [PubMed] [Google Scholar]