

Abstract
Atomistic Molecular Dynamics (MD) simulations provide researchers the ability to model biomolecular structures such as proteins and their interactions with drug-like small molecules with greater spatiotemporal resolution than is otherwise possible using experimental methods. MD simulations are notoriously expensive computational endeavors that have traditionally required massive investment in specialized hardware to access biologically relevant spatiotemporal scales. Our goal is to summarize the fundamental algorithms that are employed in the literature to then highlight the challenges that have affected accelerator implementations in practice. We consider three broad categories of accelerators: Graphics Processing Units (GPUs), Field-Programmable Gate Arrays (FPGAs), and Application Specific Integrated Circuits (ASICs). These categories are comparatively studied to facilitate discussion of their relative trade-offs and to gain context for the current state of the art. We conclude by providing insights into the potential of emerging hardware platforms and algorithms for MD.
1. Introduction
Now more than ever, the SARS-CoV-2 pandemic demonstrates the need to rapidly design therapeutic treatments to protect against diseases that pose a grave threat to human health.1 It is well-known that drug design remains an expensive and inefficient process consuming nearly a decade of time on average with total costs regularly cited in the billions of USD to develop a single successful candidate.2,3 Compounding this issue is the fact that the “drug-like” chemical space is itself not well understood as a whole, with estimates of the upper bound on this space varying between 1018–10100 depending on the assumptions made.4 While modern purchasable compound libraries only cover a small fraction of the prospective drug-like chemical space, it is now possible to commercially order molecules from a library of 10s of billions of virtual compounds.5,6 Thus, the scale of molecular interrogation needed in drug discovery necessitates alternatives to purely experimental approaches.
Classical Atomistic Molecular Dynamics (MD) simulations provide researchers the ability to apply a “computational microscope” to study the dynamic properties of biomolecules at spatiotemporal scales that current experimental methods are not able to access.7 The dynamics of interest include protein folding, protein-drug binding, conformational change, and transmembrane transport of substrates.7 MD as a field of study has existed for well over 70 years, with some of the first works reported in the late 1950s and early 1960s, though it was not until 1977 that the first MD simulation of a protein (BPTI) was considered.8−10
MD simulations are also notoriously expensive computational endeavors. By repeatedly integrating Newton’s equations of motion over very small timesteps, trillions of iterations are required before biologically relevant time scales can be reached. In the face of these challenges, hardware accelerators have played a crucial role in allowing MD simulations to unveil biological phenomena that would otherwise be practically infeasible using traditional hardware architectures.11 Over the years, accelerators for MD have grown from Application Specific Integrated Circuits (ASICs) to now include Graphics Processing Units (GPUs) and Field Programmable Gate Arrays (FPGAs). As a product of the end of Moore’s law, richer programming development environments, and algorithmic advances, biologically relevant simulation time scales are becoming more readily available to researchers without access to specialized architectures or High-Performance Computing (HPC) resources.
The goal of this perspective is to review MD hardware acceleration work to facilitate a comparative discussion on current approaches and gain insight into opportunities to improve on the current state of the art. Industry trends suggest that future hardware environments will be heterogeneous motivating the need for a review that explores the trade-offs of different hardware acceleration approaches. The structure of this article is as follows:
We will provide an overview of the MD algorithm and its components in Section 2 to provide a sufficient level of background information for the latter sections. We then discuss the algorithms employed for the primary bottleneck in MD simulations, nonbonded force computations, in Section 3.
Following the coverage of the algorithms, we discuss the implementations of MD engines for various hardware acceleration platforms. We begin this by discussing GPU-based MD acceleration in Section 4. GPUs have been increasingly employed for MD simulation given their successes in applications requiring intensive mathematical operations such as matrix multiplication (for example, Deep Learning). We briefly discuss the history of GPU-based MD engines and then discuss current limitations as well as open areas for research.
FPGA-based MD acceleration work is discussed in Section 5. The discussion is split into three subsections; beginning with the early developments in the area, we then describe the subsequent push toward production-focused MD engines followed by recent developments in FPGA-based MD engines, concluding with a comparative analysis of these three distinct periods.
The final class of ASIC-based architectures is presented in Section 6 where we discuss the early work in the area followed by the two prominent ASIC architectures; Anton by the D.E. Shaw Research group and the MD-GRAPE project by the Riken Institute.
We then conclude with a comparative discussion of the trade-offs for each class of architectures and expand upon future directions for research in MD acceleration.
2. Overview of Molecular Dynamics Algorithm
2.1. Algorithm Description
Molecular Dynamics (MD)7,12−14 considers an “n-body” system of particles (that is, atoms) where each particle possibly exerts a nonzero force on all of the other particles in the system. A description of the relevant biological events is given in Figure 1.
Figure 1.

At each iteration, bonded interactions are first computed. The number of bonded interactions tends to be relatively small, thus this step can be done in time proportional to O(n) with n being the number of atoms. In contrast to the subsequent nonbonded step, the number of interactions can grow to be proportional to O(n2) in the worst case when considering all pairwise combinations. Afterward, the acceleration vectors for each atom are updated followed by their positions, completing one iteration of the simulation loop.
Time steps that are chosen for simulations are typically on the order of femtoseconds (10–15s) for reasons concerning numerical stability and simulation quality. The magnitude of the timesteps are consequently very small in comparison to the biologically relevant time scales.17,18 The number of sequential operations needed to achieve these time scales can grow to be at least 109–1012.7
2.2. Force Computation
The first ingredient
of an atomistic MD simulation of n atoms are their
positions in Euclidean space, given as 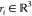 for the ith atom. The full set of n atom positions is given
by the vector r. Each of the n atoms
additionally carries a charge given by qi. Potential energy of the system, given as U, can then be computed using ri and qi for each of the n atoms. The specific form of the
potential energy function can vary (for example, refs (19, 20, and 21)), the model
we consider gives a basic idea of the components.20
for the ith atom. The full set of n atom positions is given
by the vector r. Each of the n atoms
additionally carries a charge given by qi. Potential energy of the system, given as U, can then be computed using ri and qi for each of the n atoms. The specific form of the
potential energy function can vary (for example, refs (19, 20, and 21)), the model
we consider gives a basic idea of the components.20
The forces on each atom are then defined as the negative gradient of the potential energy function:
| 1 |
where F is a vector-valued function, r is a position vector corresponding to a specific atom in the simulation.
The potential energy function that we consider within the context of molecular dynamics simulations is generally defined as
| 2 |
which decomposes the potential energy function into the sum of bonded and nonbonded terms.
2.2.1. Bonded Interactions
The bonded interactions can be further decomposed into the spring potential between atom pairs separated by a single covalent bond (1,2-pairs), the angular bond potential, and the torsion angular potential between atoms connected by 3 covalent bonds (1,4-pairs).
| 3 |
The term Uspring describes the bonded potential energy of 1,2-pairs as a function of the displacement of the bond length from its equilibrium position and is defined as
| 4 |
where 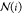 denotes the set of j indices
with covalent bonds to atom i, kb,ij is the spring constant
describing the strength of the bond, r0,ij is the equilibrium bond length, where both kb,ij and r0,ij are specific to the atom
types of the 1,2-pair, and rij = ||ri – rj||.
denotes the set of j indices
with covalent bonds to atom i, kb,ij is the spring constant
describing the strength of the bond, r0,ij is the equilibrium bond length, where both kb,ij and r0,ij are specific to the atom
types of the 1,2-pair, and rij = ||ri – rj||.
Subsequently, Uangular describes the movement of bond angles from their equilibrium positions is defined as
| 5 |
where θij is the angle between vectors rij = rj – ri and rkj = rj – rk, θij,0 is the equilibrium angle, and kθ is the angle constant.
Finally, the 4-body torsion angle potential Udihedral models the presence of steric barriers between the planes formed by the first three and last three atoms of a consecutively bonded (i, j, k, l)-quadruple of atoms (that is, 1,4 pairs):
| 6 |
where ψ is the angle between the (i, j, k)-plane and the (j, k, l)-plane, ϕ is the equilibrium angle between the two planes, and kdihedral is a constant. The exact form of the 4-body torsion potential varies between force field definitions.
2.2.2. Non-Covalent Interactions
While covalent interactions occur over relatively short distances (approximately 1–2 Å), noncovalent interactions may occur over much larger distances that could in theory involve any pair of atoms in the full simulation space.
We can decompose the nonbonded potential energy function Unon-bonded as
| 7 |
which is a sum of the two primary types of interactions, van der Waals and Coulomb electrostatics.
van der Waals interactions (Figure 2) are modeled using a Lennard-Jones potential energy function and describe the competing attractive and repulsive forces between two atoms.These forces fall off quickly with distance so they are typically for atom pairs within a cutoff distance rc. This potential is defined as
| 8 |
where A and C are constants that depend on the types of atoms i, j involved in the interaction, rij is the distance between the atoms, rc is the cutoff radius, and the notation j < rc denotes the set of neighbors within the cutoff radius. This term is commonly truncated at approximately rc = 10–14 Å.
Figure 2.
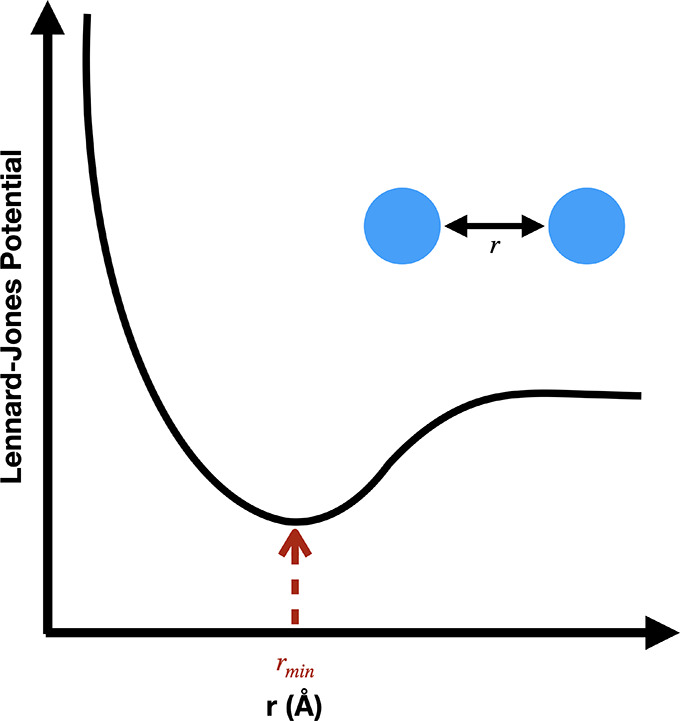
Lennard-Jones potential as a function of interatomic distance for a diatomic system.22
The influence of the Coulomb electrostatic potential, in contrast to the Lennard-Jones potentials, falls off slowly with distance and is defined as
| 9 |
where qi, qj are the point
charges for atoms i and j, rij is the distance between
the atoms, and 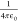 denotes the Coulomb constant. Truncation
of this term is difficult to achieve as atoms can have non-negligible
interactions at arbitrarily long distances (see Section
3).
denotes the Coulomb constant. Truncation
of this term is difficult to achieve as atoms can have non-negligible
interactions at arbitrarily long distances (see Section
3).
2.3. Integration Algorithms
Provided that forces have been computed, an integration algorithm is needed in order to drive forward the dynamics of the system.14 Two criteria used in the determination of the method are the approximate conservation of energy in the system and “time-reversibility”,14 which we will expand upon in Section 2.5. The Verlet algorithm23 achieves both of these criteria and is given by
| 10 |
where O(δt4) is the order of the local error of the calculation due to truncation of the Taylor series expansion about r(t). In order to compute quantities such as kinetic energy K, the velocities v(t) can be computed as
| 11 |
Alternative implementations of the Verlet method exist such as the “leap-frog” scheme that addresses issues around the handling of velocities that might introduce unnecessary error to the calculation. This “leap-frog” method directly computes v(t) at half time-step intervals and is given by
| 12 |
| 13 |
Lastly, we mention the velocity Verlet method as it is algebraically equivalent to the Verlet and leapfrog schemes, it is also implemented in most modern MD packages. The velocity Verlet integration scheme is given by
| 14 |
| 15 |
The integration methods covered here are not exhaustive, modern MD packages support additional algorithms for more sophisticated simulations.24,25
2.4. Role of Sampling in MD
A point that is often understated, especially when discussing MD with those who may not be domain experts, is that the exact motions observed of during an MD simulation are not meant to be taken literally.14,26 Another common question that frequently arises is how much simulation is required for an MD run. In either case it should be clarified that running a single simulation for an incredible amount of time does not guarantee that the biological event we are attempting to simulate even occurs, despite our knowledge about the time scales on which the event tends to occur.27 MD simulations are sensitive to their initial conditions and two nearby initial starting points will produce trajectories that diverge exponentially in time.28
When running simulations, we are concerned with the measurement of an observable O which depends on the microscopic system configuration, or microstate, s. For clarity, si = (ri, pi) where ri is the position of the ith atom and pi is the momentum. The individual frames that are generated sequentially over the course of a simulation can be seen as a distribution of microstates26,28 in phase space. For simplicity, we use s(t) to refer to the microstate of the system at time t. It is possible to compute the value of O using the time average where all possible microstates will have been sampled:
| 16 |
Clearly it is impossible to evaluate eq 16 as in reality we must choose a limit of time steps, tobs, as a computation budget we are willing to exert for an MD run. We now have a discrete form for eq 16:
| 17 |
however when computing O, it is important to understand whether enough exploration in terms of system configuration has been done rather than being purely concerned with the simulation time scale itself.
2.5. Relating Microscopic Behavior to Macroscopic Quantities by Statistical Mechanics
Statistical Mechanics
provides a link between the behavior of a system at the microscopic
level to its macroscopic properties we generally refer to as O, such as energy or entropy.14,26,28 We assume a probability density ρ(s, t) over the phase space that is governed by our
choice of ensemble.14,26,28 The simplest ensemble is known as the Microcanonical or NVE ensemble which refers to constant moles (N), volume (V), and energy (E).26,28 We note here that the simulations covered in this work correspond
to the NVE ensemble unless stated otherwise. The Hamiltonian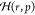 governs the evolution of the system over
time in phase space and represents the sum of the kinetic and potential
energy of the system:
governs the evolution of the system over
time in phase space and represents the sum of the kinetic and potential
energy of the system:
| 18 |
where N is the number of
atoms and mi is the mass
of the ith atom.14,26 It can then be shown that Newton’s second law of motion can
be derived from the Hamiltonian formulation by taking derivatives
of  ,
,
| 19 |
| 20 |
then inserting back into eq 20 to yield Newton’s second law of motion:26
| 21 |
| 22 |
In subsection 2.3, we introduced
the typical integration schemes used in MD, however
we did not make the connection as to why the Verlet methods are preferred
in our case of MD. Briefly stated, a numerical integrator that conserves 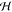 is
known as a symplectic integrator.14,26 This property
allows for the system dynamics to satisfy the conservation
of energy requirement of the NVE ensemble.26 Furthermore, as Hamilton’s eqs 19 and 20 are symmetric
in time, an integrator should be time-reversible.14,26
is
known as a symplectic integrator.14,26 This property
allows for the system dynamics to satisfy the conservation
of energy requirement of the NVE ensemble.26 Furthermore, as Hamilton’s eqs 19 and 20 are symmetric
in time, an integrator should be time-reversible.14,26
The density ρ(s, t) clearly depends on t, however as the system approaches equilibrium in the limit of time (that is, t → ∞), then ∂ρ/∂t = 0 and we arrive at ρeq.14 Given the equilibrium density ρeq and the set of points in phase spaces for which ρeq is nonzero, the system is said to be ergodic if there exists at least one trajectory that, given sufficient time, visits all of the microstates in phase space.14,26,28 This is another way of saying that sampling a single system over infinite time is equivalent to sampling s over many systems frozen at a single point in time, provided the ergodic hypothesis holds.14,26,28 With ρeq, we can then reformulate eq 17 by ignoring time and instead taking a weighted average of O according to the probability density given by ρeq(s):
| 23 |
Thus, the predictive power of the MD method lies within the efficient and effective sampling of phase space, given the choice of ensemble.
3. Algorithms for Non-Bonded Force Computations
Computation of nonbonded interactions comprise the major bottleneck
in MD simulations and according to Amdahl’s law should be the
focus of acceleration efforts.29 Numerous
algorithms have been proposed30,31 that are more efficient
than the naive 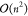 direct computation with greater accuracy
than a cutoff-based method.31 We discuss
methods featured in the literature in the following subsections.
direct computation with greater accuracy
than a cutoff-based method.31 We discuss
methods featured in the literature in the following subsections.
3.1. Particle Mesh Ewald
The Particle Mesh Ewald method, also known as PME, is a prominent algorithm for nonbonded electrostatic force calculations in biological simulations. There are a variety of PME formulations used in the literature.32−34 The common idea among these methods is the splitting of the slowly converging sum of electrostatic contributions in eq 9 into a short-range contribution, smooth long-range contribution and a “self” contribution. At a high level, the PME methods consists of five steps:31
-
1.
Charge assignment: The charges in the simulation real space are “smeared” onto a uniform grid of points using a window function.
-
2.
Grid transformation: The charge grid is then transformed from real-space to a reciprocal space by way of the Fast Fourier Transform (FFT).
-
3.
Multiplication by optimized influence function: The components of the FFT-transformed charge grid are then multiplied by an optimal Green’s function. A Green’s function is a mathematical tool used to solve difficult instances of ODE’s and PDE’s.
-
4.
Transformation from reciprocal space back to real space: An inverse FFT is performed to transform the perturbed charge grid from reciprocal space back to real space.
-
5.
Force assignment: The forces from the charge grid are interpolated onto each atom in the simulation space using the same window function from step 1.
For a more in-depth description, we refer the reader to30 and to the relevant papers.32−34
3.2. Tree-Based Approaches
The Barnes-Hut
algorithm35 and Fast Multipole Method (FMM)36 provide an efficient way of computing nonbonded
electrostatic interactions by using an octree decomposition of the
simulation space. The intuition behind these methods is that beyond
a “well-separated” distance, the electrostatic contributions
of these “far-field” particles to a reference particle
become more similar to increasing distance. Interactions under this
distance are interpreted as “near-field” and are much
more sensitive to distance from the reference particle. The algorithms
differ in the stopping criteria for the decomposition as well as the
form of the potential function that aggregates the interactions of
each “cell” in the octree. Whereas the Barnes-Hut method
uses a simple sum aggregation of the cell, FMM uses “Multipole”
expansion of the spherical harmonics of the cell, which gives a series
of progressively finer angular features, that is “moments”:
The order p of the multipole expansion are functions
of an acceptable level of error ϵ where p is
typically chosen as 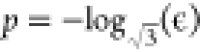 . The core of the Fast Multipole
Method
lies within a set of three translation operations that propagate the
computed interaction information through the simulation tree data
structure, an upward pass of information, and a downward pass stage
as well as three translation operations; translation of a multipole
expansion, conversion of a multipole expansion into a local expansion,
and translation of a local expansion. For a more in-depth description
of the tree-based methods, we refer the reader to the relevant papers.31,35,36
. The core of the Fast Multipole
Method
lies within a set of three translation operations that propagate the
computed interaction information through the simulation tree data
structure, an upward pass of information, and a downward pass stage
as well as three translation operations; translation of a multipole
expansion, conversion of a multipole expansion into a local expansion,
and translation of a local expansion. For a more in-depth description
of the tree-based methods, we refer the reader to the relevant papers.31,35,36
3.3. Multigrid and Multilevel Summation
Alternative hierarchical methods also exist, including the Multigrid37,38 and later Multilevel Summation methods,39 which exhibit linear asymptotic complexity in the number of atoms n. In contrast to the FMM and other tree-based methods, the Multigrid method hierarchically decomposes the simulation into a series of progressively finer potential grids rather than hierarchically decomposing the interaction pairs themselves.
3.4. Quality Measurements
Two metrics to assess the quality of a simulation are consistently mentioned in the literature; the Relative RMS Force Error and the Total Energy Fluctuation.10,34,40,41
The Relative RMS Force Error is used
to validate a new design of an MD simulation which may potentially
differ from a reference MD simulation in some way such as the precision
used to compute forces, the cutoffs used for Lennard-Jones interactions,
and so forth. The forces obtained from the high quality simulation
are given for the ith atom as 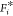 , and the forces from the query simulation
are given respectively as Fi. Thus, the Relative RMS Force Error is defined as
, and the forces from the query simulation
are given respectively as Fi. Thus, the Relative RMS Force Error is defined as
| 24 |
While Relative RMS Force Error can communicate how well a new simulation may reflect the behavior of a reliable reference point, it is difficult to determine whether the results of the simulation are physically plausible from this metric alone.
The Total Energy Fluctuation provides a measurement of physical plausibility by measuring the sum of the relative change in total energy, given by ΔE, of the physical system at each time step. The total energy E of the system is the sum of the kinetic and potential energy of the system:
| 25 |
where Ui is the potential energy for the ith atom, and Ki is the kinetic energy for that atom. The kinetic energy for an atom in the simulation is computed as
| 26 |
where mi is the mass of the ith atom in
the simulation and 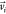 is the atom’s
velocity vector. The
Total Energy Fluctuation is then defined as
is the atom’s
velocity vector. The
Total Energy Fluctuation is then defined as
| 27 |
where E0 is the initial total energy of the system and Ei is the total energy at time step i. In the ideal case, this value should approach 0, indicating that the total energy in the simulation has been conserved and the results are likely to be physically meaningful. In practice, an acceptable upper bound has been proposed as ΔE ≤ 0.003.34 However, the choice of this particular threshold for ΔE appears to be the product of a choice made for a specific simulation and does not necessarily apply to arbitrary simulation systems and or force fields.42−48
3.5. Comparison of Algorithms for Non-Bonded Interactions
Support for the Particle Mesh Ewald method is nearly ubiquitous among modern production MD software packages given that it is well studied, “easier” to implement efficiently compared to some tree-based approaches such as Fast Multipole Method (FMM),36 exhibits good asymptotic runtime in the number of atoms n, and is generally accepted to be “accurate” according to the metrics presented (eqs 24 and 27). Improvements have been made to the original PME method, such as the “Smooth” Particle Mesh (SPME) algorithm,33 which improves the accuracy of the interpolation from the simulation space to the charge grid that is input into the later Fast Fourier Transform calculation by way of B-spline interpolation. Additional variations of PME-based approaches include the k-GSE method, which uses a series of Gaussian kernels to perform the charge spreading step (Table 1).34
Table 1. Various Non-bonded Force Interaction Algorithms Featured in the Literature Covered in This Review.
| abbrev. | name |
|---|---|
| PME | Particle Mesh Ewald |
| SPME | Smooth Particle Mesh Ewald |
| k-GSE | k Gaussian Split Ewald |
| FMM | Fast Multipole Method |
| MGrid | Multigrid |
| BH | Barnes-Hut |
4. GPU-Based Acceleration
4.1. GPU Architecture Overview
Graphics processing units (GPUs) have garnered much attention as accelerators for applications in machine learning and are noted generally for possessing the ability to exploit data parallelism inherent in certain classes of algorithms. Whereas a Central Processing Unit (that is, CPU) is designed to be capable of quickly switching between multiple serial tasks that may differ enormously in their control flow or required resources, GPUs are designed to trade-off the CPU’s cache and control resources per core for a greater number of lightweight compute cores.49
In Figure 3, the components of the NVIDIA Tesla V100 GPU architecture are shown, indicating the GPU Processing Clusters (GPCs), each of which is composed of multiple Texture Processing Clusters (TPCs), composed of multiple Streaming Multiprocessors (SMs). The SMs, depicted in Figure 4, of the GPU are the fundamental computation unit and within these elements are special cores for floating-point and integer arithmetic as well as specialized tensor cores that accelerate matrix and tensor multiplications often encountered in deep neural networks. In order to exploit data parallelism, the V100 GPU utilizes a Single Instruction, Multiple Thread (SIMT) execution model, where a thread can be thought of as an arbitrary execution of the kernel.51 The SIMT execution model then is best thought of as an extension of Single Instruction, Multiple Data (SIMD) to multiple threads.51 For the sake of clarity, when referring to SIMD, we are referring to the array-processor defined in Flynn’s Taxonomy.52 In this model groups of threads are called thread blocks, which are decomposed into execution “warps”. Warp sizes are typically chosen as 32 across recent NVIDIA GPU architectures. Warps are then scheduled for execution across the SMs of the GPU, which can execute multiple warps in parallel among the SM compute cores. The hierarchy of SM groups in the GPU allow for the sharing of memory resources at various levels with L0/L1/L2 cache memories as well as a high bandwidth DRAM. Modern NVIDIA GPUs also feature NVLink interconnect technology, which allows for GPU-to-GPU data transfers at up to 160 Gigabytes/second, providing 5 times as much bidirectional bandwidth as PCIe Gen 3 x16.53
Figure 3.

Description of an NVIDIA GPU architecture.50
Figure 4.
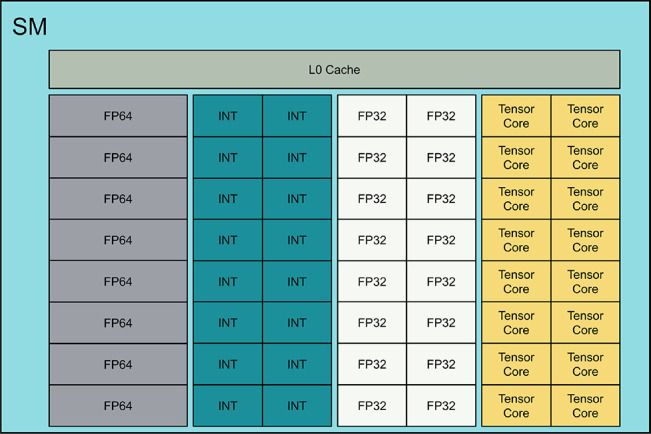
Description of an NVIDIA GPU Streaming Multiprocessor (SM) unit..50
The advantage that GPUs provide relative to CPU implementations of MD simulations is precisely the SIMT model of parallelism. The number of threads that may operate in parallel are well into the thousands for modern GPUs, whereas the number of CPU threads on a consumer device remain limited to single digits. Even considering more advanced CPU architectures such as the IBM Power Series or special extensions to CPU instruction sets such as the Intel Advanced Vector Extensions54 which provide CPUs with enhancements for SIMD processing, GPUs remain far superior in exploiting data parallelism.49
In comparison to FPGAs, which can leverage the same data parallelism that makes GPUs favorable for MD, GPUs lack support for custom data-types that have been used in ASIC and FPGA architectures to maximize resource utilization. For example, ASIC and FPGA architectures allow for the designer to employ custom floating units that can be tuned to the most effective level of precision, while GPUs are limited to computing with 16-bit, 32-bit, and 64-bit floating point units that are included in the architecture. This flexibility in the representation of the data can allow for ASIC and FPGA designs to mix various levels of precision throughout the computation, for example to compute individual forces with 32-bit fixed point and then accumulate forces with 64-bit fixed point using dedicated hardware units (see corresponding discussion in Sections 5 and 6).
4.2. GPU MD Applications
Classical MD simulations were some of the earliest beneficiaries of GPU acceleration.55,56 This adoption was due in part to the availability of the CUDA GPU programming framework, drastically reducing the complexity of mapping a nongraphics application to the GPU.55,57 Additionally, GPUs were posed as solutions to the development of costly specialized processors due to their relatively low cost as a consequence of their popularity as accelerators for gaming.55 Subsequent years show an explosion of interest in the use of GPUs to accelerate Classical MD simulations, with increasing complexity of algorithms studied.58 A recent comprehensive review of the work in this field has been published59 and so we refer the reader there for more detail. In this overview, we focus primarily on the works published since.59
4.3. GPU Software Implementations
There exist numerous packages for MD itself, so we will not exhaustively study all those available in detail. We instead choose to highlight some of the popular packages and discuss their implementations comparatively.
NAMD (NAnoscale Molecular Dynamics)60 was the earliest adopter of GPUs among MD packages, leveraging the programmability provided by the Nvidia CUDA GPU programming library.55 NAMD is built using the C++ programming language and CHARMM++61 parallel computation library. The philosophy of NAMD is to make running simulations a simple process for the user, not to serve as a platform to be modified by the user extensively.62 NAMD is supported across Windows, OSX, and Linux operating systems. In the initial description of the GPU features for NAMD, the PME algorithm55 is presented as the kernel for nonbonded interactions. However, the description clarifies that the reciprocal space calculation for PME is actually implemented to run on the CPU, while a combination of short-range electrostatics and van der Waals interactions are the target for GPU acceleration. In addition to the reciprocal space calculation, all other steps (for example, bonded interactions, integration) of the MD simulation are carried out on the CPU. Subsequent releases of NAMD increasingly moved computation from the CPU to the GPU, notably all forces are now computed on the GPU including all computation related to the PME implementation (SPME).62,63 The latest version 3.0 of NAMD is being actively developed and represents a significant shift in the design philosophy.62,64 The NAMD developers note that the traditional decomposition of the MD algorithm across the CPU and GPU, where the GPU is tasked with force evaluations and the CPU with all other tasks related to integration, leaves higher-end GPUs idling for a significant portion of time.62 Currently, the developers of NAMD are developing a “GPU-resident” version where the MD computation is moved almost entirely from the CPU to the GPU to reduce costs associated with moving data between the CPU and GPU for each iteration of the MD simulation.62 A recent blog post by a collaborative team at Nvidia also details the most recent work in migrating the NAMD code from CPU to GPU.64 Preliminary results show a speedup of up to approximately 1.9 times faster than NAMD v. 2.13 when using the same GPU, the Nvidia V100.64 Lastly, it should be noted that current available version of the 3.0 alpha version of NAMD supports the GPU-resident approach for a single GPU at a time, with the focus being on being able to run more parallel instances of the same MD system, allowing for greater amount of sampling.62 The NAMD software is distributed free of charge with its source code.
GROMACS (GROningen MAchine for Chemical Simulation) is another popular open source MD package.65 The design philosophy of GROMACS is to run in as many different computing environments as possible, ranging from a laptop equipped with only a CPU to massive clusters of heterogeneous servers that may feature multiple GPUs per node. Version 4.5 of GROMACS was the first to feature GPU acceleration, with the entire MD calculation implemented on the GPU, similar to the current goal of the NAMD project we previously described.66 However, in contrast to the approach described by the NAMD authors, the latest iteration of GROMACS is targeting increasingly heterogeneous platforms, making use of both the CPU and the GPU implementation of the MD algorithm in what the authors refer to as a “bottom-up heterogeneous approach”.67 In order to accomplish this approach for acceleration of MD, GROMACS uses a scheduling protocol to allocate work between the CPU threads and GPU(s) efficiently, taking into account the particular topology of the compute environment as well as NUMA (Non-Uniform Memory Access) considerations when placing threads. The GROMACS software is able to assign force calculations to both the CPU and GPU to optimize communication and computation overlap, rather than explicitly placing all force calculations on a single device. Interestingly, this extends to the FFT calculations required for the PME algorithm; specifically, while the truncated real-space nonbonded interactions are carried out on the GPU, the CPU is used to compute the forces for the reciprocal space. Additionally, the GROMACS developers note their use of CUDA streams to allow for nonlocal nonbonded interactions to be sent to high priority queues across the various GPUs, allowing for preemption of the local kernel to return force calculations early. Additionally, GROMACS possesses the ability to offload entire MD iterations to the GPU. In order to minimize data transfer overheads, GROMACS uses the (relative to PCIe bandwidth with host CPU) higher bandwidth GPU-GPU interconnect (NVLink) to leverage data transfer capabilities between GPUs. The developers of GROMACS state further improvement to the load balancing capabilities for CPU and GPU task scheduling as well as continued work to overlap communication and computation as priorities. Recent work has extended GROMACS to use the FMM method, which the authors anticipate will become the algorithm of choice for “the largest parallel runs”.67 An implementation of FMM for GROMACS has since been published.58
Additionally, Amber is one of the most popular MD packages for Drug Design.68 The name Amber often refers to the set of force fields developed by the Amber project.19,69,70 Amber additionally provides extensive support for various types of simulations as well as analysis tools such as PTRAJ and CPPTRAJ.71 For the sake of our discussion, we are concerned with the GPU MD simulation program, pmemd.72 Version 11 of the Amber software was the first to provide support for GPU acceleration of the PME calculations. The most recent published description of the Amber software, corresponding to version 12, states that the entire MD calculation was moved onto the GPU.72 The authors note that their intention with version 12 of the software was to “port the exact equations as they are described in AMBER’s CPU code” to the GPU. A major point of emphasis was the introduction of a numerical precision model for the MD engine termed “SPFP” which combines single-precision floating point and 64-bit fixed point arithmetic.73 The distinction should be made that NVIDIA GPUs have not themselves supported fixed-point arithmetic at the hardware level so the implementation approximates this through software. As opposed to NAMD and GROMACS, the Amber implementation as presented in72 does not use table lookup to compute the forces between the atoms, choosing instead to directly evaluate the equations. The pmemd program supports running simulations using multiple GPUs by use of MPI. In the case of a multi-GPU run, all simulation data structures are replicated on each device to minimize data transfer overhead. The FFT calculation required for reciprocal space force evaluations in PME are implemented using the NVIDIA-developed cuFFT library.74 Additional features have been added to later iterations of Amber, as well as ongoing performance enhancements that are detailed in.75 Amber provides a number of free simulation and analysis tools under AmberTools, which is regularly updated. An extensive manual detailing the functionality available through the package is also regularly updated with each release. More advanced programs such as pmemd are available by way of a licensing mechanism.
4.4. Characterizing Performance of GPUs for MD
There is great interest currently in characterizing the performance of GPUs to better understand their scaling behavior. The Amber MD software package regularly publishes benchmarking data on their Web site.76
In Figure 5, we summarize some of the recent analysis of price to performance ratios for several popular GPUs spanning from “consumer-grade” (NVIDIA GTX 1080TI) to “datacenter-grade” (NVIDIA V100) with respect to the Amber 2016 and Amber 2018 MD packages, a widely popular MD simulation package. The Amber software at each time point is examined across multiple commodity GPUs, and with each device a price-to-performance metric is reported, where the best values would be those approaching 0. In both cases, despite active development of the GPU implementation of the pmemd code,75 the price-to-performance exhibits a marginally decreasing relationship to theoretically more capable devices possessing progressively larger numbers of processing elements and larger global memories. Another observation is that the price-to-performance ratios for the top-end GPU, the NVIDIA V100, increases for all simulation types monitored in this study. It is not explicitly mentioned here why this is the case, analysis for other algorithms is also not included, and it is important to note that other MD software packages along with their specific algorithmic implementations may exhibit different behavior. As the more recent NVIDIA A100 begins to supplant the V100, based on the observations made here, extra performance will come at a premium that does not necessarily scale as favorably as it does for lower-end cards.
Figure 5.

GPU price to performance comparison for Amber MD software for versions 2016 and 2018. The data are collected from the Amber Web site.76
Recent benchmarking of the Gromacs67 software to characterize price-to-performance has also been performed.77 The main conclusions from this work agree with what has been observed for the Amber results; consumer grade GPUs provide the greatest price-to-performance as opposed to professional datacenter-grade GPUs. The conclusions of the work further imply that the growth of GPU capabilities or other unspecified architectures will lead to improved throughput of simulations in future works.
Recently, NAMD was used in a Gordon Bell prize winning work that investigated the use of large scale atomistic MD to illuminate mechanisms of SARS-CoV-2 spike dynamics.78 Two molecular simulations are studied in this case, an 8.5 million atom spike-ACE2 complex and a 305 million atom virion (that is, the complete virus structure). In the case of the smaller spike-ACE2 system, scaling to more GPUs quickly saturates performance, while for the larger virion system, the performance is nearly linear up to 512 compute nodes and then begins to saturate. The PME algorithm is used in both MD simulations, demonstrating the negative impact communication overheads have on simulation performance, resulting in marginally decreasing returns with each additional device added.
As stated previously in Section 4.2, the developers of NAMD are working to improve their implementation by offloading more routines from the CPU to the GPU, such as the numerical integration step. The analysis reported by the NAMD developers,79 shows the simulation throughput (measured in nanoseconds of simulation per day) of the NAMD 2.14 versus NAMD 3.0alpha9 versions running on an NVIDIA DGX-2 as a function of number of GPU devices. The projected performance assumes the ideal optimized version of the software with minimal dependency on the CPU, demonstrating an exponential growth in performance in this case. However, it is unclear at this time what the outcome of this optimization will ultimately achieve. The communication costs imposed with increased number of devices into the thousands will likely continue to limit performance of a GPU-centric implementation for acceleration of a single system, though these observations provide insight into the NAMD designers focus on the GPU-resident design which can be used to accelerate enhanced sampling algorithms.62,80
4.5. Discussion
GPUs have experienced enormous interest as general purpose accelerators given their low unit cost and increased accessibility through the NVIDIA CUDA, OpenCL, and AMD RocM libraries. Great interest remains in understanding the limits of the technology, evidenced across performance analyses for production MD software packages, such as Amber, Gromacs, and NAMD.62,67,81,82 Future work in improving the scaling of GPU implementations will pay particular attention to algorithms that address the efficiency of all-to-all communication patterns inherent to PME-based approaches.58,67,83 Integration with often GPU-centric deep learning tools will also play a significant role in addressing the efficiency of MD simulation.78,84,85
The primary advantage GPUs have exhibited to date compared to competing architectural solutions lie within the robust development resources available to researchers to map the nonbonded force calculations to the SIMT-paradigm GPUs exploit, coupled with their relatively low prices. It is also the case that more complex accurate algorithms, such as PME which depend on an underlying Fast Fourier Transform, have benefited from GPU acceleration. With tools such as the NVIDIA CUDA framework,86 GPUs are more favorable to alternatives such as FPGAs for the relative ease of development. With that said, GPUs lack hardware flexibility that can allow the programmer to define custom data-types that may make better use of the chip resources or allow specific calculations to be done more efficiently over the course of the 109–1012 iterations required.7 GPUs are also defined as general purpose processors and so while instructions can be carried out in parallel on separate data, there remains latency in execution due to control flow overhead such as process synchronization and instruction fetching. Imbalances in computational capacity of the GPU versus the bandwidth for which data can be supplied result in challenges to keep the processors effectively occupied. FPGAs in contrast leverage a spatial architecture in which the data flows through a user defined pipeline that alleviates the aforementioned sources of control flow latency and are also directly equipped with high bandwidth I/O interfaces49,87 (see the corresponding discussion in Section 5).
5. Reconfigurable Architectures for MD
5.1. Overview of FPGA Architecture
At a high level, an FPGA can be described as an array of programmable logic blocks and programmable interconnect between those blocks that can be configured after fabrication to implement arbitrary program logic.88 An example of an Intel FPGA (formerly Altera) architecture is shown in Figure 6.The Adaptive Logic Modules (ALMs) are the fundamental compute units of an FPGA. ALMs consist of a Lookup table (LUT) which is a memory that maps address signals as inputs and the outputs are stored in the corresponding memory entries.88 LUTs can be programmed to compute any n-input Boolean function. Full Adders perform addition and subtraction of binary inputs. Flip Flops (FF) are the basic memory element for the FPGA and serve as registers allowing the ALM to maintain state. The FPGA architecture then interconnects many of the ALMs through programmable interconnections forming an array of processing elements.88 Coupled with this mesh of ALMs are specialized or “hardened” elements such as on-chip Block RAM (BRAM) and Digital Signal Processing (DSP) blocks.88 BRAMs are configurable random access memory modules that can support different memory layouts and interfaces.88 DSP blocks help compute variable precision fixed-point and floating-point operations.49 Additionally, modern FPGAs also include microprocessors that serve as a controller, allowing a user to run an operating system such as Linux in order to leverage facilities such as the device’s communication drivers or running high-level programming languages such as Python.88 Lastly, FPGAs use I/O blocks that allow for direct low-latency interaction with various network, memory, and custom interfaces and protocols.49,88
Figure 6.

Description of an FPGA architecture.
FPGAs, unlike GPUs and CPUs, are a spatial architecture, in that the hardware directly and continuously executes a spatial hardware circuit representation of the software. This eliminates control flow overhead that is encountered in general purpose architectures which require instruction-fetch as well as process scheduling and synchronization which can be detrimental to performance.87 The programmable hardware approach instead allows the data to flow through the pipelines that the designer specifies in software or through Computer Aided Design (CAD) tools.49 In contrast to ASIC architectures, which are not able to be updated once the design has been fabricated, FPGAs are able to be reconfigured with updates to the algorithms as advances are made.
Despite the ability to be reconfigured with arbitrary logic, updating an algorithm design for an FPGA is a significantly slower process than it is for CPUs and GPUs, with compilation time scales varying between minutes to a few days.89 The learning curve for designing FPGA implementations has historically been much steeper than for CPUs and GPUs due to a dearth in higher level development tools.89 However, as industry trends suggest heterogeneous architectures as a solution for the issues faced by general purpose processors in the trade-offs required for power versus efficiency,49,90 a number of tools have been developed to ease the development of programs that exploit the FPGA in anticipation of a heterogeneous processing future.91−97
While early FPGA-based MD implementations were constrained to use vendor-specific toolchains that required VHDL or C-like dialects (SRC MAP C) to program, modern tools include Intel Quartus Prime, Xilinx Vivado, and OpenCL toolchains, which allow for a higher-level C/C++ interface to implement designs. In Table 2, we document the FPGA hardware utilized in MD simulations reported in the literature. The number of ALMs, embedded memories, dedicated multiplier blocks, and transceiver I/O rates have all increased by several orders of magnitude from the initial period of work. In Table 3, we provide a summary of the development tools used in the implementations reported in the literature. Recently, more attention has been paid to FPGAs for MD due to advances in the hardware itself amortizing the relatively higher cost as compared to GPU accelerators and thus addressing a major issue with their viability.98,99
Table 2. Characteristics of FPGAs Featured in Molecular Dynamics Simulations.
| ref | accelerator | ALMs | I/O band | embed. mem. | mult. blocks |
|---|---|---|---|---|---|
| (98, 99) | Intel Stratix 10 | 933 120 | 28.3 gb/s | 253 Mbits | 11 520 |
| (100)a | Xilinx V5 LX 330T | 51 840 | 3.75 gb/s | 11 664 kbits | 192 |
| (29, 101, 102) | Xilinx XC2VP100 | 99 216 | 3.125 gb/s | 7992 kbits | 444 |
| (103, 104) | Xilinx XC2VP70–5 | 74 448 | 3.125 gb/s | 5904 kbits | 328 |
| (29, 102, 105−108) | Xilinx XC2 V6000 | 33 792 | 840 mb/s | 2592 kbits | 144 |
| (109) | Xilinx Virtex-E 2000E | 43 200 | 622 mb/s | 655.36 kbits | 0 |
Denotes FPGA was used for simulation of performance.
Table 3. Description of the Development Environments Featured in the FPGA-Based MD Simulation Literature.
| ref | year | dev. board | dev. framework |
|---|---|---|---|
| (109) | 2004 | TM-3110 | VHDL |
| (103, 111) | 2005 | WildstarII-Pro112 | VHDL |
| (105) | 2006 | SRC 6 MAPstation | Carte |
| (102) | 2006 | SRC 6 MAPstation (E/C) | Carte |
| (106, 107) | 2006 | SRC 6 MAPstation (E) | Carte |
| (104) | 2006 | WildstarII-Pro112 | VHDL |
| (101) | 2007 | SRC 6 MAPstation (E) | Carte |
| (29) | 2008 | SRC 6 MAPstation (E/C) | Carte |
| (113) | 2011 | Gidel PROCStar III | Proc Dev. Kit |
| (98, 99) | 2019 | Intel Stratix 10 | Intel Quartus Prime Pro |
5.2. Early FPGA Work
One of the earliest examples of applying FPGA accelerators to the problem of molecular dynamics is ref (109). The motivation of the work emphasized the power of reconfigurable processors to address the bottlenecks posed by the high costs of ASICs and the expensive memory hierarchies in general purpose processors. This work focuses solely on the short-range Lennard-Jones interactions while ignoring the Couloumb electrostatics to simplify the implementation.
The system consists of the PairGen, which generates the neighbor lists for each atom in the simulation; the Force computer, which computes the forces for the atoms using table look ups to retrieve precomputed parameters; and the acceleration update, which then is forwarded to the Verlet integration update which writes the new atom positions to the onboard memory. Fixed-point representations are used exclusively, each tuned specifically to the dynamic range of each quantity using a two particle system at various distances to precompute their possible values in order to minimize resource consumption. The design was implemented in VHDL. The simulation used for the analysis consisted of 8192 atoms. The FPGA system as configured gives a per-time step performance of 37 s, while the general processor benchmark (Intel Pentium 4 2.4 GHz, single core) is able to achieve 10.8 s. The authors claimed that with improvements to the memory systems as well as migrating to more up to date FPGA hardware with a higher clock rate would improve the speedup from 5.1× to 21×.
The work of ref (103) improves upon the shortcomings of ref (109) with an FPGA architecture that includes both Coulomb and Lennard-Jones forces. The design was implemented on a single Xilinx Virtex-II-Pro XC2VP70 FPGA and used fixed-point arithmetic throughout the implementation, and the force computations were carried out using lookup tables with interpolating polynomials, similar to ref (109). Furthermore, the work improves upon ref (109) by reporting a 57× speedup with respect to a single-core Intel Xeon 2.4 GHz CPU, with a benchmark similar to the previous work. Considering that most of the implementation was similar, the starkest difference was that the FPGA used in ref (103) had nearly twice as many ALMs as those used in ref (109) (74 448 vs 43 200), higher transceiver I/O bandwidth (3.125 gb/s vs 622 mb/s), more embedded memory (5904 kbits vs 655.36 kbits), and dedicated multiplier blocks (328 vs 0). Therefore, by using more capable hardware with a similar force calculation allowed the accelerator to vastly outperform the initial FPGA designs.
5.3. Toward Production FPGA-Based MD
While the initial studies of refs (103, 108, and 109) focused on proof-of-concept implementations, ref (102) investigates how one could go about porting a “production” MD system to an FPGA architecture, citing issues with ref (109) and103 being their focus on a “textbook” data set and the use of simplistic nonbonded force algorithms. Another point of criticism with the previous work was the use of “textbook” MD codes as general processor benchmarks, therefore the NAMD MD software62 is compared with the FPGA results and serves as the starting point for the FPGA implementation. The authors document their design process from extracting the relevant kernels from the NAMD code, implementation on the FPGA using MAP C, optimization of the initial designs using multiple target platforms of the SRC MAPStation, comparing the two featuring the Xilinx XC2 V6000 and Xilinx Virtex-II Pro XC2VP100 FPGAs. Their FPGA implementation is said to closely follow the original implementation (in Charm + +) including the use of SPME for nonbonded force calculations, improving upon the brute-force approaches of refs (103 and 109) Despite the increased design complexity, the work reports a 3× speedup versus the CPU baseline (dual-core Intel Xeon 2.8 GHz) using a data set of 92 224 atoms.
The work of ref (104) also proposes to address some of the issues with the earlier work
by expanding the number of particle types, demonstration of integration
with existing software (that is, ProtoMol114) similarly to,102 and general architectural
improvements such as the introduction of “semi-floating point”
representations for force calculations and optimizations to reduce
the size of the lookup tables. The improved design yields a stable
simulation of the bovine pancreatic trypsin inhibitor (BPTI) when
considering total energy fluctuation (approximately 0.014 J) relative
to the CPU benchmark. However, performance metrics are reported using
the “textbook” data set of 8192 atoms and use the direct  nonbonded force calculation for the most
competitive case, reporting a 15.7× speedup over the CPU benchmark
(dual-core Intel Xeon 2.8 GHz).
nonbonded force calculation for the most
competitive case, reporting a 15.7× speedup over the CPU benchmark
(dual-core Intel Xeon 2.8 GHz).
A later paper100 improves upon ref (104) by implementing a more sophisticated algorithm (Multigrid37) to compute the short-range Lennard-Jones and Coulomb forces. The updated design is then compared against ProtoMol114 and NAMD CPU benchmarks. The FPGA design presented in ref (100) is validated using a 10 000 time step simulation where total energy fluctuation is measured. Compared to the results collected from a ProtoMol CPU implementation (dual-core Intel Xeon 2.8 GHz), a similar energy fluctuation is reported for both the FPGA design100 and ProtoMol.
The work of ref (105) claimed to be the first FPGA implementation of a realistic nonbonded force calculation algorithm in an FPGA-centric hardware acceleration platform, in contrast to the previous work of ref (109) (Lennard-Jones only),103 (simple data set). For the sake of brevity, the culmination of these works is presented in ref (29), which also provides the first study of parallelization of FPGA-based MD in a cluster of “reconfigurable-hardware-accelerated nodes”. The results of the scaling analysis were somewhat disappointing. On the one hand, as the number of nodes increases (using 1 Xilinx Virtex-II XC2 V6000 FPGAs per node), the latency per MD time step decreases. On the other hand, the decrease in latency compared to the CPU benchmark (dual-core Intel Xeon 2.8 GHz) is significantly worse than the CPU benchmark as the speedup from using 1 node to using 8 nodes drops from 2.08× to 1.51 ×. It is worth noting that the authors chose to implement their own software for the MD simulations for the CPU-based and FPGA-based implementations, therefore it is difficult to directly compare to other works while also considering this was the first scaling analysis done for FPGA-based MD.
The work of refs (101 and 115) demonstrates a design that makes use of the two Xilinx XC2VP100 FPGAs available on the SRC Series E MAPstation. Effectively, one of the FPGAs is used as a controller for the nonbonded force calculations, handling memory accesses from the host machine and running the outer loop of the PME calculations which are pipelined between both FPGAs. The second on-board FPGA is exclusively dedicated to the inner loop, the force calculations for a single atom and its “neighbors”. A major focus of this study was to present a highly optimized FPGA-based MD accelerator with what was at the time of publication, outdated resources thus suggesting the improvements in performance shown here (approximately 3×) to upward of 12× to 15×.
5.4. Recent Developments in FGPA-Based MD
The first end-to-end FPGA implementations of MD are presented in refs (98 and 99). The authors first develop an implementation that includes only the direct-evaluation component of the PME algorithm in ref (99) and then go on to include the full PME calculation and its FFT kernel dependencies in ref (98).
One of the primary contributions of both works98,99 is a detailed study of the trade-offs of various implementation designs. Of particular interest are the memory mapping schemes of the simulation “cells”, the distribution of work for the force calculations, and various implementations of the nonbonded force pipelines. For the Dihydrofolate Reductase (DFHR) test case of 23 558 atoms, the best design uses separate memory modules for each simulation “cell” along with a work distribution in which each pipeline works on its own “homecell”. The FPGA implementation achieves an approximately 10% speedup over the best GPU (NVIDIA Titan RTX) performance. Additionally, the FPGA implementation using first-order interpolation is shown to have a similar energy conservation as the Amber CPU benchmark. However, when the authors extend their analysis to include molecular systems that vary in their size and density, there is no design that consistently outperforms the others across all cases.
5.5. Discussion
A quantitative as well as qualitative summary of the work in FPGA-based acceleration for which a simulation was demonstrated is provided in Tables 4, 5, and 6.
Table 4. Quantitative Characteristics of Simulations Approached with FPGA-Based Acceleration.
| ref | year | #atoms | force prec. | box-size |
|---|---|---|---|---|
| (109) | 2004 | 8192 | 50-bit Fixed | not reported |
| (103) | 2005 | 8192 | 48-bit Fixed | not reported |
| (111) | 2005 | 8192 | 35-bit Fixed | not reported |
| (105) | 2006 | 32 932 | 17-bit Fixed | 73.8 × 71.8 × 76.8 Å3 |
| (102) | 2006 | 92 224 | 32-bit Float | 108 × 108 × 72 Å3 |
| (106, 107) | 2006 | 32 932 | 32-bit Float | 73.8 × 71.8 × 76.8 Å3 |
| (104) | 2006 | 8192 | 35-bit Float | 64 × 50 × 50 Å3 |
| (101) | 2007 | 23 558 | 32-bit Float | 62.23 × 62.23 × 62.23 Å3 |
| (29) | 2008 | 32 932 | 32-bit Float | 73.8 × 71.8 × 76.8 Å3 |
| (99) | 2019 | 20 000 | 32-bit Float | 59.5 × 51 × 51 Å3 |
| (99) | 2019 | 20 000 | 32-bit Float | 59.5 × 51 × 51 Å3 |
| (98) | 2019 | 23 558 | 32-bit Float | 62.23 × 62.23 × 62.23 Å3 |
| (98) | 2019 | 23 558 | 32-bit Float | 62.23 × 62.23 × 62.23 Å3 |
Table 5. Quantitative Characteristics of FPGA-Based Simulation Performance.
| ref | year | time/day | speedup | benchmark |
|---|---|---|---|---|
| (109) | 2004 | 2.34 ps | 0.29× | Intel Pentium 4 2.4 GHz |
| (103) | 2005 | 517.4 ps | 57× | Intel Xeon 2.4 GHz |
| (111) | 2005 | 3.8 ps | 51× | Intel Xeon 2.4 GHz |
| (105) | 2006 | 0.188 ns | 2.72× | Intel Xeon 2.8 GHz |
| (102) | 2006 | 28 ps | 3× | Intel Xeon 2.8 GHz |
| (106, 107) | 2006 | 0.22 ns | 1.9× | Intel Xeon 2.8 GHz |
| (104) | 2006 | 0.72 ns | 15.7× | Intel Xeon 2.8 GHz |
| (101) | 2007 | 0.2 ns | 3.19× | Intel Xeon 2.8 GHz |
| (29) | 2008 | 246.86 ps | 2.08× | Intel Xeon 2.8 GHz (dual-core) |
| (99) | 2019 | 1.4 μs | 96.5× | Intel Xeon |
| (99) | 2019 | 1.4 μs | 3.29× | NVIDIA GTX 1080Ti |
| (98) | 2019 | 630.25 ns | 25.3× | Intel Xeon |
| (98) | 2019 | 630.25 ns | 1.1× | NVIDIA GTX 1080Ti |
Table 6. Qualitative Characteristics of Simulations Approached with FPGA-Based Acceleration.
| ref | year | force | LJ | coul. | alg. | arch. |
|---|---|---|---|---|---|---|
| (109) | 2004 | LUT | yes | no | direct | full MD |
| (103, 111) | 2005 | LUT | yes | yes | direct | nonbond only |
| (105) | 2006 | LUT | yes | yes | SPME | nonbond only |
| (102) | 2006 | LUT | yes | yes | SPME | nonbond only |
| (106, 107) | 2006 | direct | yes | yes | SPME | nonbond only |
| (104) | 2006 | LUT | yes | yes | direct | nonbond only |
| (101) | 2007 | direct | yes | yes | PME | nonbond only |
| (29) | 2008 | direct | yes | yes | SPME | nonbond only |
| (113) | 2011 | direct | yes | yes | PME | nonbond only |
| (99) | 2019 | LUT | yes | no | direct | full MD |
| (98) | 2019 | LUT | yes | yes | PME | full MD |
In terms of throughput, measured as nanoseconds per day, time scales achieved by FPGA-based designs have improved by over 6 orders of magnitude.98,99,109 It is important to note however that much of the work reported for FPGA-based MD were not evaluated using production-level software on either the CPU or for the FPGA. Exceptions to this are the work of ref (102) which closely followed the NAMD62 source code and101 which closely followed the Amber81 source code. In terms of the general processor benchmarks, the reported relative speedup versus CPUs has consistently outperformed, though not by a large margin. Considering the throughput performance versus GPUs first reported in refs (98 and 99), while there is evidence of an improvement, it remains modest.
FPGA-based implementations have lacked evaluation in the production setting, considering the majority of the work has been relegated to standard benchmark data sets. Therefore, there has not been an exceptionally large simulation considered using FPGA hardware in the number of atoms or the size of the simulation box.
As work in the field progressed with hardware advances, designs have eschewed Fixed-point arithmetic for Single precision Floating Point in order to target an appropriate level of accuracy for production MD. While it is difficult to estimate the minimum precision required for all simulations, the work of refs (98, 99, and 113) provide the most comprehensive analysis of this. The most current work98,99 demonstrates that in their design, the use of Single Floating-point versus 32-bit Fixed point is more efficient in terms of ALMs and BRAM utilization. The main bottleneck presented by FPGA hardware has instead been resource utilization for force calculation pipelines. As the hardware has progressed, the number of pipelines working in parallel has increased several orders of magnitude.99,102
The vast majority of the FPGA-based designs have served as accelerators connected to a host machine to compute pairwise interactions given a cutoff threshold. Considering that the outer loop of the simulation was generally run using a CPU based code for the more complicated nonbonded interaction algorithms such as PME, this paradigm presented a clear bottleneck for memory accesses. The work of ref (29) addressed this in their design by utilizing the on-chip BRAM to store intermediate force calculations before making updates to the on board memory. Even with designs to optimize memory requirements,29,101,102 it was not feasible to remove the host bottleneck until recent work demonstrated fully end-to-end simulations completely on FPGAs.98,99 Additionally, the work of refs (98 and 99) provided additional investigation into the use of direct force calculations versus the use of look-up tables, deciding to use lookup tables to conserve DSP utilization with minimal loss in accuracy.
Lastly, as the applications have progressed, as have the available development tools. The early work in FPGA-based MD has either made use of VHDL103,104,109,111 or vendor-specific toolchains featuring C or FORTRAN interfaces, namely Carte and Gidel Pro Dev. Kit.101,102,105−107,113 The complexity of development on FPGAs versus other mainstream computing architectures such as CPUs or GPUs has been the primary hurdle to their adaption in a broad class of applications.89,116 More recently, additional tools such as Xilinx Vivado, OpenCL, and Intel Quartus Prime have provided developers with a richer set of tools with C/C++ interfaces to FPGA-based designs. Additional work in scaling FPGA implementations to multiple devices is ongoing. Industry trends suggest that in the near future heterogeneous architectures will become the norm, along with software stacks to ease the deployment of software in these environments.49
6. ASIC-Based Acceleration
Application Specific Integrated Circuits, or ASICs, are specialized hardware that can be tuned to solve specific problems very efficiently. In the context of MD simulations, most of the early work, as well as the most important advances, have been as a result of ASIC designs.117−120
6.1. Early ASIC Development
One of the earliest examples of an MD specific ASIC appeared in 1982121,122 for a condensed matter physics application. At the time of this work, MD simulations were typically constrained to be on the order of a few hundred atoms with the largest only approaching the order of a few thousands of atoms on modest time scales. The Delft Molecular Dynamics Processor (DMDP)121,122 was designed solely to accelerate the nonbonded force calculations with the host computer tasked with all other steps of the simulation. By keeping all particle data in the DMDP’s local memory and using fixed-point arithmetic (24 bits for position data, 32 bits for velocity data), the DMDP was able to achieve performance comparable to a Cray-1 supercomputer123 at reportedly less than 1% of the cost.
6.2. GRAPE (GRAvity PipE) and MDGRAPE
The Riken Institute-developed GRAPE/MD-GRAPE architectures for various instances of the n-body problem have led to numerous Gordon-Bell prizes over the years.124 The first GRAPE VLSI chip was introduced in 1990125,126 reporting performance comparable to the CRAY-XMP/1 supercomputer at a cost reportedly 10 000× less. GRAPE was proposed as an alternative to more complicated algorithms exhibiting favorable asymptotic complexity, by instead choosing the brute-force calculation of the direct interactions between particles with the use of a cutoff. The proposed design125 was similar to that of the DMDP with a host compute node managing the overall simulation with the GRAPE chip exclusively computing the long-range interactions.
While the early demonstrations of GRAPE were primarily focused on celestial systems simulations,126 the authors emphasized the flexibility of the machine to potentially handle arbitrary instances of the n-body problem ranging from the simulation of galaxies to that of biological macro-molecules such as proteins by “simply” replacing the potential energy function. Notably, the first GRAPE was apparently lacking this feature as it was not until several years later that the MD-GRAPE ASIC was introduced127−129 as an extended version of GRAPE that could support an arbitrary potential. The hardware architecture of MD-GRAPE was similar to that of previous ASICs121,122 in that the ASIC would be tasked with performing only the direct force calculations.
Work on improving the MD-GRAPE system continued for some time afterward with MDGRAPE-2130 increasing the number of pipelines in the chip (from 1 to 4), clock frequency, use of double floating point precision instead of 80-bit fixed point, and support for Direct Memory Accesses (DMA), while achieving a sustained performance of 15 billion floating point operations per second (that is, gigaFLOPs or GFLOPS) which was equivalent to the peak performance of other top high-performance computers at the time. Additionally, the overall workflow supported more advanced Ewald-based nonbonded forces. However, MDGRAPE-2 was restricted to “real-space” accelerations. Functionality for computing Fast Fourier transforms was implemented instead as a separate accelerator, “WINE-2”.131,132
The subsequent MDGRAPE-3 chip118 was reported as having a particularly high development cost at approximately USD 20 million. The authors emphasize that at less than $10 USD per GFLOP, the system was cost-effective unlike previous works122 or other notable high-performance general purpose computers such as the IBM BlueGene/L132 (approximately $140 USD per GFLOP). A number of molecular dynamics packages including Amber (version 6)68 and CHARMM (Chemistry at HARvard Macromolecular Mechanics)133 were already ported for MDGRAPE-2 and were also compatible with the MDGRAPE-3 chip.
While the MDGRAPE-3 system was demonstrated as the first quadrillion floating point operations per second (that is, petaFLOPS or PFLOPS) machine, significant bottlenecks remained in its design an accelerator attached to I/O buses of a host. MDGRAPE-4134 integrates the functions of both a host and the accelerator as a system on a chip (SoC), removing host communication bottlenecks. The MDGRAPE-4 chip134 consists of dedicated hardware for force pipelines, general processor cores along with a control general processor, network interfaces units, as well as an FPGA interface and memory units. The general processing cores (GP) are grouped onto each board in 8 blocks, and each general processor has 8 cores. The control general processor (CGP) is situated in the middle of the board and controls the execution flow of the force calculations by communicating with each of the general processors, the force pipelines (PP), and the network interfaces (NIF). Whereas the force evaluation pipeline spanned 80% of the die areas in MDGRAPE-3, it is reduced to only 20% by comparison in the MDGRAPE-4.
Force calculations are computed using mixed precision and formats. Input coordinates are expressed in a 32-bit fixed-point format as the dynamic range is expected to be relatively “small”. Force computations are expressed with a single-precision IEEE-754 floating point. Force summation is done using 32-bit fixed point. MDGRAPE-4 additionally differs from MDGRAPE-3 by calculating Couloumb and van der Waals forces and potentials simultaneously, whereas MDGRAPE-3 can only compute one at a time. Further, MDGRAPE-4 exploits Newton’s third law to reduce the computational cost by half. MDGRAPE-4 has 64 pipelines in eight blocks on each board, can handle 51.2 × 109 interactions per second, with peak system performance equivalent to 2.5 trillion floating point operations per second (that is, teraFLOPS or TFLOPS). MDGRAPE-4 uses the Gaussian Split Ewald method for force calculation34 as it is quoted as possessing a good property for acceleration by specialized pipeline due to the Gaussian spread function having a spherical symmetry. The MDGRAPE-4 is programmed using C and assembly language. The authors in ref (134) ported the GROMACS66 “mdrun” engine to their architecture.
They also leave open in their discussion of what the optimal configuration for global memory should be and point to the fact that strong scaling of MD simulations is becoming more difficult and requires increasing amounts of specialization to improve performance, it may be worthwhile to investigate ways to improve cost/performance. They also point out that systems with a single chip or on a single module will be solutions that could address the cost/compute power concerns.
MDGRAPE-4a was subsequently developed to improve data and message flow efficiency and was applied to simulating potential therapeutics for the SARS-CoV-2 virus.135,136 The developers achieved performance of roughly 1 μs per day for the systems including about 100 thousand atoms using timesteps of 2.5 fs. Despite the improvement however, the authors still point out that the most recent Anton system is estimated to be 1–2 orders of magnitude ahead of the MDGRAPE-4a machine.136 The full MDGRAPE-4a system cost approximately $6.5 million USD to develop, which is significantly less expensive than that of MDGRAPE-3118 which was reported cost approximately $20 million USD but remains well beyond the reach of many researchers in terms of available development resources to replicate a design.
6.3. Anton
Unlike the first iterations of GRAPE/MDGRAPE, the principal task the Anton117 machine was designed for was to accelerate “very-long” time scale trajectories of biological entities such as proteins. In contrast to the MDGRAPE-3 architecture which split the MD tasks between a host microprocessor and a number of accelerator chips, the Anton ASIC utilized a system-on-a-chip SoC design with dedicated hardware to perform all tasks on chip or in a network of chips.
The Anton ASIC consists of 4 subsystems. The high-throughput interaction subsystem (HTIS)137 computes electrostatic and van der Waals interactions. The flexible subsystem138 controls the ASIC while handling tasks such as bonded force calculations, FFT, and integration. The communication subsystem handles communication on chip and between chips with 5.3GB/s communication bandwidth between chips. The memory subsystem provides access to the attached DRAM while also providing special memory write operations that support accumulations of force, energy, potential, and charge spreading. The HTIS is programmable by the use of SRAM lookup tables to allow for changes in force fields in ongoing research. It is not clear however to what extent this module can be modified to handle the recent work in force field development, an active area of research in biochemistry.
At the heart of the HTIS are the pairwise point interaction modules (PPIMs) and their pairwise point interaction pipelines (PPIPs). Anton distributes particle interactions using an efficient method that minimizes communication costs associated with importing and exporting atomic information among the nodes while also achieving scaling benefits with increasing number of nodes.34,139 The particles assigned to each node are referred to in two separate sets; the set of tower particles and the set of plate particles. The determination of a tower versus plate atom is based upon a set of criteria that is specified in detail elsewhere.34 The PPIM is distributed to a set of tower particles T and a set of plate particles P, for which the Cartesian product of the two sets is computed to form the set of possible pairwise interactions. Using a set of multiple match units in parallel, the PPIM filters the particle pairs for criteria assessing their suitability for an interaction evaluation. After filtering, the PPIPs compute the interactions between the particle pairs. The PPIPs feature optimized numerical precision for each functional unit along the datapath, maximizing the use of die area. The HTIS then accumulates the results and writes the results to the memory subsystem.
In terms of hardware capabilities, the subsequent Anton 2 ASIC140 improves upon the original in nearly every conceivable way. The Anton 2 chip is fabricated using 40 nm process technology versus 90 nm for the original. The HTIS supports a maximum of 32 768 atoms, an increase from 6144. The data bandwidth for the torus neighbors has also been increased from 221 Gb/s to 1075 Gb/s. A priority of this updated design was to increase the overlap of communication with computation in order to make more effective utilization of the hardware capabilities. In addition to hardware improvements, all embedded software on Anton 2 is written using C++ using a ported version of the GCC compiler. This is an improvement upon the mix of C and assembly required for the original Anton.
The latest iteration of the Anton series was recently introduced as Anton 3.139 Anton 3 includes improvements to the physical capabilities of the hardware from Anton 2, featuring 1500× increase in transistor count, 107× increase in SERDES Bandwidth (GB/s), 1290× increase in the number of PPIP modules, as well as 264 newly developed specialized bond interaction calculators. Anton 3 also features several improvements in the PPIP modules which feature a special bonded interaction calculator that makes for a higher throughput calculation while requiring one-third the die area of a geometry core which was previously used to compute bonded forces. For nonbonded forces, Anton 3 introduces two specialized PPIP modules, the Big PPIP and the Small PPIP. The Small PPIP module computes interactions with r > 5°A with specialized simpler logic at lower precision (14-bit datapaths) allowing for three Small PPIPs to fit in the area as one Big PPIP. Conversely, the Big PPIP handle interactions with r ≤ 5°A at increased precision (23-bit datapaths). Other significant improvements to the filtering and communication protocols for mapping interactions to PPIPs achieve improved load balancing due to the novel Manhattan Method for distributing the computation of nonbonded interactions, which is introduced in the work describing Anton 3.139
6.4. Discussion
Characteristics of the various ASIC architectures developed for MD are summarized in Table 7. The ASIC designs discussed follow a similar trajectory to FPGA-based development. In the earlier period of the work, the MDGRAPE118,128,130 and MD-Engine120 designs implemented brute-force nonbonded calculations, even if they were part of a larger more complex simulation workflow as is the case in ref (120). The Anton design implemented the full-md simulation on chip coupled with a more sophisticated PME nonbonded force calculation. Subsequent ASICs followed this design closely, moving to SoC-based designs with PME based nonbonded force calculations.
Table 7. Characteristics of ASIC Designs.
| year | name | alg. | arch. | force calc. | force accum. |
|---|---|---|---|---|---|
| 1996 | MD-GRAPE128 | direct | nonbond only | 32-bit float | 80-bit fixed |
| 1999 | MD-Engine120 | direct | nonbond only | 40-bit float | 64-bit float |
| 2003 | MDGRAPE-2130 | direct | nonbond only | 32-bit float | 64-bit float |
| 2003 | MDGRAPE-3118 | direct | nonbond only | 32-bit float | 80-bit fixed |
| 2009 | Anton141 | GSE34 | full MD | 32–36 bit fixed | 86-bit fixed |
| 2014 | MDGRAPE-4134 | GSE34 | full MD | 32-bit float | 32-bit fixed |
| 2014 | Anton 2140 | u-series142 | full MD | 32–36 bit fixed | 86-bit fixed |
| 2021 | Anton 3139 | u-series142 | full MD | 14–23 bit fixed | 14–23 bit fixed |
ASIC implementations of MD remain orders of magnitude “faster” than alternative architectures (for example, Tables 8 and 9). ASICs are also orders of magnitude more expensive to build, with the costs for full clusters given in the millions of USD.118,134
Table 8. Performance of Various Accelerator Configurations to Run a Single Simulation of Dihydrofolate Reductase (DHFR)a.
| accelerator | engine | time scale (ns/day) |
|---|---|---|
| Anton 3 (64-node) (ASIC) | Custom139 | 212 200 |
| Anton 2 (512-node) (ASIC) | Custom140 | 85 800 |
| Intel Stratix 10 (FPGA) | Custom98 | 630 |
| 2x Nvidia Titan-RTX (GPU) | Amber154 | 629.03 |
| NVIDIA V100 SXM (GPU) | Amber154 | 522.20 |
| NVIDIA V100 PCIE (GPU) | Amber154 | 277.14 |
| NVIDIA TITAN X (GPU) | OpenMM155 | 393 |
| NVIDIA TITAN V (GPU) | OpenMM155 | 419 |
| NVIDIA RTX 3090 (GPU) | ACEMD156 | 1308 |
DHFR is a 159-residue protein (suspended in water) target for cancer therapeutics that has been used as a standard benchmark for MD simulation throughput. All simulations reported here employ NVE microcanonical constraints. NVE refers to the set of constraints on MD simulations in which moles (N), volume (V), and energy (E) are conserved in the simulation. All simulations reported here with the exception of Anton 2140 use the PME32 algorithm for non-bonded interactions. Anton 2 uses the μ-series142 algorithm for non-bonded interactions.
Table 9. Selected MD Simulations among the Largest of Those Reported in the Literature.
The justifications for development of ASICs have been cited as being a choice between developing better algorithms versus developing specialized hardware.117,118,120,130 The large upfront costs were amortized by the expected improvement in potential simulation throughput versus general processors which was demonstrated for refs (40 and 141).
As a consequence of the high cost and technical expertise required, development of MD specific ASICs has also been mostly relegated to two institutions, the Riken Institute funded by the Japanese government and the privately funded D.E. Shaw Research group based in the United States. Thus, access to ASIC-based MD engines remains an issue. In response, the Anton machines117,140 have been provided to researchers as part of an NIH grant at the Pittsburgh Supercomputing Center. Recently, MD simulation trajectory data of the SARS-CoV-2 main protease generated by the MDGRAPE-4 series of ASICs has been made publicly available.135,143 Additionally, the MDGRAPE-4 series and Anton 2 ASICs have improved their programmability by implementing a C/C++ interface.
The Anton and MDGRAPE series of ASICs have helped make significant strides in the simulations of fundamental biological processes including protein-folding and protein–ligand binding.135,141,144 In the face of this, much attention is being shifted toward the integration of machine learning into molecular simulation workflows,78 and fundamental research is ongoing in the development of more accurate force fields using deep learning and quantum mechanics.19,85 Future development of ASICs will undoubtedly depend on the outcomes of these efforts.
7. Future Directions
7.1. Heterogeneous Architectures
With increasing complexity of workflows, it is becoming difficult to design a processor which can optimally handle a diverse range of tasks most effectively.78,90,93,145−150 Instead, there is acceptance in the industry that competing architectures can be viewed as complementary devices and potentially can be coupled together in a heterogeneous architecture. The OneAPI project151 is a primary example of the direction the hardware industry will take in the future for design of servers that will come to be used across industry, government, and academia. CPUs excel at instruction level parallelism and are able to handle diverse sets of tasks, GPUs are able to exploit data parallelism with orders of magnitude more cores than a traditional CPU, and FPGAs can approach ASIC performance as they exploit instruction level and data parallelism while being able to be reconfigured thus coming at a lower cost in terms of investment in technical development or cost to acquire. A heterogeneous workflow can ideally map various simulation steps to a specific architecture that are best suited to exploit the optimal performance. Challenges remain in designing tools that can allow developers to design their codes to leverage multiple hardware platforms, which do not all support the same instruction sets.152 The AMD Heterogeneous-Computing Interface for Portability (HIP)153 is a solution for enabling heterogeneous device development, allowing CUDA-based code to run on NVIDIA and AMD devices. In the meantime, it is of great utility to better develop quantitative studies of how the various architectures covered in this work compare overall as well as specific investigation of the various components of the MD workflow can be mapped optimally among the classes of accelerators (Table 10).
Table 10. Qualitative Comparison of Accelerator Classes Covered in the Discussiona.

The time scales quoted here correspond to results collected for a common benchmark study that has been considered among the various architectures discussed in this work, Dihydrofolate Reductase (DHFR), and are not meant to be presented as definitive assessments.
7.2. Synthesis of Machine Learning and Hardware Acceleration for MD
The integration of machine learning and molecular dynamics has picked up much enthusiasm in recent years.160 With the rise of machine learning integration into molecular simulation workflows, especially those of deep-learning, present areas of interest for work include demonstrating how these technologies can be used in tandem with hardware accelerators to better exploit molecular dynamics simulations.161−163 The JAX, M.D. project164 aims to develop a differentiable molecular dynamics workflow by providing a framework that allows for seamless integration of machine learning models with physical simulation code. JAX, MD itself is built upon the JAX python library which integrates Autograd and the Tensorflow accelerated linear algebra library, XLA,165 to allow for high performance codes to be deployed on GPUs and Tensor Processing Units (that is, TPUs) which are AI ASICs designed by Google.166 The TorchMD python library85 was recently introduced and presents another attempt to bridge machine learning and molecular dynamics simulations, providing an interface built upon the PyTorch167 deep learning library. By adhering to a PyTorch backend, the TorchMD tools can also leverage hardware accelerators such as GPUs and TPUs, as well as others that may come to be supported by PyTorch in the future. The initial debut of the TorchMD library compared to a production MD simulation code highlights the need for further optimization of the code itself, but also where the utility of more capable acceleration in terms of software design as well as the target hardware architectures themselves are likely to play a crucial role in enabling the practical use of differentiable MD simulations in the future.
Numerous hardware accelerators for AI are coming to market to meet the insatiable compute demand (measured in floating point operations per second or FLOPs) for machine learning workflows, estimated as doubling every 3.5 months.168 The Cerebras Systems Inc. CS-1/CS-2 wafer scale processor169 and SambaNova Reconfigurable Dataflow Architecture170 are recently proposed alternatives to traditional multicore architectures. Cerebras CS-2 is the largest chip ever built, fabricated from the largest square from a single silicon wafer. The CS-2 is over 56× larger than the largest GPU, features 850 000 programmable compute cores, 40 gigabytes of on-chip SRAM, 20 petabytes/sec of memory bandwidth, and 220 petabits/sec of interconnect bandwidth. The system is additionally supported by custom compilation tools that optimize the reconfigurable dataflow processing units depending on the users application. Cerebras also provides support for Tensorflow and PyTorch APIs, drastically lowering the barrier to entry. The SambaNova DataFlow Accelerator is motivated by a similar use case in machine learning acceleration with its own reconfigurable dataflow architecture, custom compilation tools, and custom “SambaFlow” software stack supporting Tensorflow and PyTorch APIs. The Graphcore Intelligence Processing Unit (IPU) is yet another AI accelerator poised to challenge the status quo of the GPU-centric development environments used in AI and MD simulations today.171 As these and likely additional accelerators come to market, offering advantageous levels of computation relative to energy utilization, the current status quo of a GPU-centric development environment is likely to pivot to utilize these architectures with tight integration with differentiable machine learning libraries.
7.3. Further Improvement of Accuracy and Algorithms
The discussion covered in this work considers the context of NVE-ensemble simulations which have been implemented across all of the various architectures employed. It is also the case that simple integration algorithms such as velocity Verlet are mostly common among the works discussed. With that said, research into improving the accuracy as well as efficiency of MD simulations remains an active area so it is important to highlight some of the work as future systems in heterogeneous environments will need to be designed with such features in mind.
An obvious limitation of the classical MD algorithm as described in this review is the order of the integration time step, which is often chosen as 2 fs in order to capture the most granular behavior, bond vibrations as shown in Figure 1. One technique featured in modern MD engines is Hydrogen Mass Repartitioning (HMR) which allows for the integration time step to be increased by a factor of 2 with minimal loss in accuracy or stability of the simulation trajectory by repartitioning the mass of heavy atoms into the bonded hydrogen atoms.172 Other techniques for improving the efficiency of MD simulations include Multiple time scale MD,45 which partitions the interacting sets of atom neighbors into primary and secondary interactions, and then update force calculations at staggered time intervals. Additionally, Reaction-Field Electrostatics173 is another method to improve the efficiency of long-range electrostatics and is implemented in popular MD packages such as OpenMM.174
Other limitations of the MD algorithms covered in this work are the empirical force fields that are employed in the simulations. Ab initio molecular dynamics (AIMD) treats the electronic structure of the atoms in the simulation explicitly and can provide more accurate description of the dynamics of the system (for example, bond formation and breaking).175 While AIMD has historically been prohibitively expensive to consider for the systems covered in this work, progress has been made in the acceleration of these calculations using deep learning to approximate the interatomic potential energy surface from AIMD simulations.176−178 A recent work demonstrates the application of a heterogeneous computation system (Summit Supercomputer at Oak Ridge National Laboratory) to scale deep-learning based Potential Energy Surface (PES) models in tandem with the LAMMPS MD engine179 to perform studies of very large systems, on the order of 100 million atoms, at the nanosecond time scale with ab initio accuracy.180
Much of the MD acceleration covered in this work has been concerned with accelerating the time scales of the microscopic kinetics of the biomolecules ranging from very short time scales to what are hopefully biologically meaningful time scales. However, even a trajectory extracted from a relatively long-time scale MD simulation is susceptible to becoming “stuck” in one of the huge numbers of energy minima in the high-dimensional potential energy surface due to the presence of high potential energy barriers between these minima.181,182 Lacking an ability to effective sample low-energy states of a biomolecule negatively affects the accuracy of the macroscopic thermodynamic properties such as free energy, a critical measurement especially for applications in drug discovery. Modern MD packages include support for algorithms that allow for improved sampling such as Replica-Exchange Molecular Dynamics (REMD),181 Metadynamics (MTD),182 and Adaptively Biased MD (ABMD).183
Enhanced sampling methods are provided in packages such as PLUMED,184,185 which wraps existing MD simulation engines such as those discussed in Section 4.2. GPU packages such as Amber also provide support for enhanced sampling algorithms.75,81 Anton also has been shown to implement enhanced sampling algorithms,186−188 and the latest iteration of Anton suggested continued development for enhanced sampling methods.139 Some of the challenges in implementing these types of algorithms in specialized hardware have been identified previously.186 Specifically, the memory resource requirements for replica exchange (RE)181 forced the Anton 2 machine to use multiple Anton nodes to run a single replica of a typical solvated protein.186 Considering that REMD may require hundreds of replicas,80 the Anton architecture was seen as being better suited for the simpler Simulated Tempering (ST) algorithm,189 where the entire machine could better accelerate a single simulation.186 Additionally, REMD imposes communication requirements when exchanging systems between nodes. The frequency of exchanges is a parameter that may be tuned, but as the exchanges are attempted more frequently, the communication burden grows. The current generation of Anton (3) is presumed to have greater amounts of SRAM available, given the current description of the machine features an increase of nearly 2 orders of magnitude capacity (atoms) per node.139 Enhanced sampling may also require other functions in addition to the force field to be evaluated. ST requires evaluation of the potential energy function,186 which at the hardware level required updating force calculation parameters to instead compute energies. Lastly, while an FPGA-focused implementation of an enhanced sampling algorithm has not been presented in the literature, ongoing work that studies networks of FPGAs for MD acceleration could potentially be extended to enhanced sampling algorithms.190 Challenges faced by the Anton machine are likely applicable to the case of FPGAs, such as dynamically swapping between evaluation of forces and potential energy. Additional work in applying machine learning to accelerate sampling of atomic systems is ongoing with promising results.191,192
8. Conclusions
We have reviewed three broad categories of accelerators for molecular dynamics simulations that include GPUs, FPGAs, and ASICs. Our discussion details the benefits of rich development environments that have enabled the popularity of GPUs for MD acceleration while also identifying outstanding issues of the hardware itself to scale popular long-range electrostatics algorithms effectively. We then investigated the history of MD acceleration in the context of FPGAs, which exhibit favorable properties such as flexibility in data types in contrast to GPUs, elimination of control and synchronization overheads, while exhibiting instruction-level and data-parallelism. Despite these properties, however, a major hurdle in the successful mainstream adoption of FPGAs as accelerators for MD has been a lack of accessible development tools for researchers. That stated, FPGA hardware as well as development tools have progressed significantly since the first MD applications were developed in the previous decade, and industry trends suggest a future in which servers will become more heterogeneous. Today there are already examples of servers featuring FPGAs and microprocessors on the same chip as well as the top supercomputers in the world regularly featuring GPU coprocessors to accelerate a diverse array of workloads that often combine MD with machine learning and deep learning. While ASIC architectures have historically been cost-effective to reduce the “time to solution” for achieving biologically relevant MD time scales, they are also expensive to develop and lack the flexibility to be reprogrammed to a significant degree. The GPU has been enormously beneficial to the scientific community for its ability to democratize longer-time scale MD, as well as fuel research into more effective MD sampling methods and thermodynamic property calculations. With more specialized architectures for AI appearing, such as the Cerebras Wafer-scale accelerators, SambaNova Reconfigurable DataFlow architecture, and Graphcore Intelligence Processing Unit (IPU), it is not difficult to imagine a future where MD applications which are already beginning to feature tight integration with machine learning and deep learning will be designed to leverage a diverse array of processing capabilities rather than being implemented entirely on one paradigm.
Acknowledgments
All work performed at Lawrence Livermore National Laboratory is performed under the auspices of the U.S. Department of Energy under Contract DE-AC52-07NA27344. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.
This work was supported in part by the Laboratory Directed Research and Development program at Lawrence Livermore National Laboratory (20-ERD-062), LLNL-JRNL-834158. This work was also supported in part by CRISP, one of six centers in JUMP, an SRC program sponsored by DARPA, and in part by NSF grants 2003279, 1911095, 1826967, 2100237, 2112167, and 2052809.
The authors declare no competing financial interest.
References
- WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/ (accessed 10-11-2021).
- Smietana K.; Siatkowski M.; Møller M. Trends in clinical success rates. Nat. Rev. Drug Discovery 2016, 15, 379–380. 10.1038/nrd.2016.85. [DOI] [PubMed] [Google Scholar]
- DiMasi J. A.; Grabowski H. G.; Hansen R. W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Schneider P.; Walters W. P.; Plowright A. T.; Sieroka N.; Listgarten J.; Goodnow R. A. Jr; Fisher J.; Jansen J. M.; Duca J. S.; Rush T. S.; Zentgraf M.; Hill J. E.; Krutoholow E.; Kohler M.; Blaney J.; Funatsu K.; Luebkemann C.; Schneider G. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discovery 2020, 19, 353–364. 10.1038/s41573-019-0050-3. [DOI] [PubMed] [Google Scholar]
- Lucas X.; Grüning B. A.; Bleher S.; Günther S. The purchasable chemical space: a detailed picture. J. Chem. Inf. Model. 2015, 55, 915–924. 10.1021/acs.jcim.5b00116. [DOI] [PubMed] [Google Scholar]
- REAL Compounds—Enamine. https://enamine.net/compound-collections/real-compounds (accessed 10-5-2021).
- Dror R. O.; Dirks R. M.; Grossman J. P.; Xu H.; Shaw D. E. Biomolecular simulation: a computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- Alder B. J.; Wainwright T. E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1957, 27, 1208–1209. 10.1063/1.1743957. [DOI] [Google Scholar]
- Gibson J. B.; Goland A. N.; Milgram M.; Vineyard G. H. Dynamics of Radiation Damage. Phys. Rev. 1960, 120, 1229–1253. 10.1103/PhysRev.120.1229. [DOI] [Google Scholar]
- McCammon J. A.; Gelin B. R.; Karplus M. Dynamics of folded proteins. Nature 1977, 267, 585–590. 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- Shaw D. E.; Maragakis P.; Lindorff-Larsen K.; Piana S.; Dror R. O.; Eastwood M. P.; Bank J. A.; Jumper J. M.; Salmon J. K.; Shan Y.; Wriggers W. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- Adcock S. A.; McCammon J. A. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. (Washington, DC, U. S.) 2006, 106, 1589–1615. 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M.; McCammon J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- Allen M. P. M.; Tildesley D. J.; Allen T. D.. Computer Simulation of Liquids; Clarendon Press: Oxford, 1989. [Google Scholar]
- Shan Y.; Kim E. T.; Eastwood M. P.; Dror R. O.; Seeliger M. A.; Shaw D. E. How does a drug molecule find its target binding site?. J. Am. Chem. Soc. 2011, 133, 9181–9183. 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Pan A. C.; Arlow D. H.; Borhani D. W.; Maragakis P.; Shan Y.; Xu H.; Shaw D. E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13118–13123. 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwier M. C.; Chong L. T. Reaching biological timescales with all-atom molecular dynamics simulations. Curr. Opin. Pharmacol. 2010, 10, 745–752. 10.1016/j.coph.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Tian C.; Kasavajhala K.; Belfon K. A. A.; Raguette L.; Huang H.; Migues A. N.; Bickel J.; Wang Y.; Pincay J.; Wu Q.; Simmerling C. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- Potential Energy Functions. https://www.ks.uiuc.edu/Research/namd/2.9/ug/node22.html (accessed 10-11-2021).
- Bonded interactions—GROMACS 2019 documentation. https://manual.gromacs.org/documentation/2019/reference-manual/functions/bonded-interactions.html (accessed 5-2-2021).
- Yu N.; Polycarpou A. A. Adhesive contact based on the Lennard-Jones potential: a correction to the value of the equilibrium distance as used in the potential. J. Colloid Interface Sci. 2004, 278, 428–435. 10.1016/j.jcis.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Verlet L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. 10.1103/PhysRev.159.98. [DOI] [Google Scholar]
- Molecular Dynamics—GROMACS 2021 documentation. https://manual.gromacs.org/documentation/2021/reference-manual/algorithms/molecular-dynamics.html (accessed 5-25-2021).
- 17Theory Behind OpenMM: Introduction—OpenMM Users Guide 7.5 documentation. http://docs.openmm.org/latest/userguide/theory.html (accessed 5-25-2021).
- Tuckerman M.Statistical Mechanics: Theory and Molecular Simulation; Oxford University Press: Oxford, 2010. [Google Scholar]
- Zuckerman D. M. Equilibrium sampling in biomolecular simulations. Annu. Rev. Biophys. 2011, 40, 41–62. 10.1146/annurev-biophys-042910-155255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.Lectures on Statistical Physics and Protein Folding; World Scientific Publishing: Singapore, 2005. [Google Scholar]
- Scrofano R.; Gokhale M. B.; Trouw F.; Prasanna V. K. Accelerating Molecular Dynamics Simulations with Reconfigurable Computers. IEEE Trans. Parallel Distrib. Syst. 2008, 19, 764–778. 10.1109/TPDS.2007.70777. [DOI] [Google Scholar]
- Arnold A.; Fahrenberger F.; Holm C.; Lenz O.; Bolten M.; Dachsel H.; Halver R.; Kabadshow I.; Gähler F.; Heber F.; Iseringhausen J.; Hofmann M.; Pippig M.; Potts D.; Sutmann G. Comparison of scalable fast methods for long-range interactions. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2013, 88, 063308. 10.1103/PhysRevE.88.063308. [DOI] [PubMed] [Google Scholar]
- Sagui C.; Darden T. A. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155–179. 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Shan Y.; Klepeis J. L.; Eastwood M. P.; Dror R. O.; Shaw D. E. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J. Chem. Phys. 2005, 122, 54101. 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- Barnes J.; Hut P. A hierarchical O(N log N) force-calculation algorithm. Nature 1986, 324, 446–449. 10.1038/324446a0. [DOI] [Google Scholar]
- Greengard L.; Rokhlin V. A fast algorithm for particle simulations. J. Comput. Phys. 1987, 73, 325–348. 10.1016/0021-9991(87)90140-9. [DOI] [Google Scholar]
- Skeel R. D.; Tezcan I.; Hardy D. J. Multiple grid methods for classical molecular dynamics. J. Comput. Chem. 2002, 23, 673–684. 10.1002/jcc.10072. [DOI] [PubMed] [Google Scholar]
- Hardy D. J.; Wu Z.; Phillips J. C.; Stone J. E.; Skeel R. D.; Schulten K. Multilevel summation method for electrostatic force evaluation. J. Chem. Theory Comput. 2015, 11, 766–779. 10.1021/ct5009075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy D. J.; Wolff M. A.; Xia J.; Schulten K.; Skeel R. D. Multilevel summation with B-spline interpolation for pairwise interactions in molecular dynamics simulations. J. Chem. Phys. 2016, 144, 114112. 10.1063/1.4943868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amisaki T.; Fujiwara T.; Kusumi A.; Miyagawa H.; Kitamura K. Error evaluation in the design of a special-purpose processor that calculates nonbonded forces in molecular dynamics simulations. J. Comput. Chem. 1995, 16, 1120–1130. 10.1002/jcc.540160906. [DOI] [Google Scholar]
- Engle R. D.; Skeel R. D.; Drees M. Monitoring energy drift with shadow Hamiltonians. J. Comput. Phys. 2005, 206, 432–452. 10.1016/j.jcp.2004.12.009. [DOI] [Google Scholar]
- Zhou R.; Berne B. J. A new molecular dynamics method combining the reference system propagator algorithm with a fast multipole method for simulating proteins and other complex systems. J. Chem. Phys. 1995, 103, 9444–9459. 10.1063/1.470006. [DOI] [Google Scholar]
- Humphreys D. D.; Friesner R. A.; Berne B. J. Simulated Annealing of a Protein in a Continuum Solvent by Multiple-Time-Step Molecular Dynamics. J. Phys. Chem. 1995, 99, 10674–10685. 10.1021/j100026a035. [DOI] [Google Scholar]
- Watanabe M.; Karplus M. Dynamics of molecules with internal degrees of freedom by multiple time-step methods. J. Chem. Phys. 1993, 99, 8063–8074. 10.1063/1.465633. [DOI] [Google Scholar]
- Tuckerman M.; Berne B. J.; Martyna G. J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. 10.1063/1.463137. [DOI] [Google Scholar]
- Tuckerman M. E.; Berne B. J.; Rossi A. Molecular dynamics algorithm for multiple time scales: Systems with disparate masses. J. Chem. Phys. 1991, 94, 1465–1469. 10.1063/1.460004. [DOI] [Google Scholar]
- Tuckerman M. E.; Martyna G. J.; Berne B. J. Molecular dynamics algorithm for condensed systems with multiple time scales. J. Chem. Phys. 1990, 93, 1287–1291. 10.1063/1.459140. [DOI] [Google Scholar]
- Figueirido F.; Levy R. M.; Zhou R.; Berne B. J. Large scale simulation of macromolecules in solution: Combining the periodic fast multipole method with multiple time step integrators. J. Chem. Phys. 1997, 106, 9835–9849. 10.1063/1.474115. [DOI] [Google Scholar]
- Compare Benefits of CPUs, GPUs, and FPGAs for Different oneAPI. https://software.intel.com/content/www/us/en/develop/articles/comparing-cpus-gpus-and-fpgas-for-oneapi.html (accessed 10-4-2021).
- Nvidia Tesla V100 GPU Architecture. https://images.nvidia.com/content/volta-architecture/pdf/volta-architecture-whitepaper.pdf (accessed 9-9-2021).
- Lindholm E.; Nickolls J.; Oberman S.; Montrym J. NVIDIA Tesla: A Unified Graphics and Computing Architecture. IEEE Micro 2008, 28, 39–55. 10.1109/MM.2008.31. [DOI] [Google Scholar]
- Flynn M. J. Some Computer Organizations and Their Effectiveness. IEEE Trans. Comput. 1972, C-21, 948–960. 10.1109/TC.1972.5009071. [DOI] [Google Scholar]
- Nvidia Tesla P100 GPU Architecture. https://images.nvidia.com/content/pdf/tesla/whitepaper/pascal-architecture-whitepaper.pdf (accessed 9-9-2021).
- Intel Advanced Vector Extensions 512 (Intel AVX-512) Overview. https://www.intel.com/content/www/us/en/architecture-and-technology/avx-512-overview.html (accessed 11-28-2021).
- Stone J. E.; Phillips J. C.; Freddolino P. L.; Hardy D. J.; Trabuco L. G.; Schulten K. Accelerating molecular modeling applications with graphics processors. J. Comput. Chem. 2007, 28, 2618–2640. 10.1002/jcc.20829. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Stone J. E.; Schulten K.. Adapting a message-driven parallel application to GPU-accelerated clusters. SC ‘08: International Conference for High Performance Computing, Networking, Storage and Analysis; 2008; pp 4244–2835. [Google Scholar]
- Stone J. E.; Hardy D. J.; Ufimtsev I. S.; Schulten K. GPU-accelerated molecular modeling coming of age. J. Mol. Graphics Modell. 2010, 29, 116–125. 10.1016/j.jmgm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohnke B.; Kutzner C.; Grubmüller H. A GPU-Accelerated Fast Multipole Method for GROMACS: Performance and Accuracy. J. Chem. Theory Comput. 2020, 16, 6938–6949. 10.1021/acs.jctc.0c00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jász Á.; Rák Á.; Ladjánszki I.; Cserey G. Classical molecular dynamics on graphics processing unit architectures. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2020, 10, 27. 10.1002/wcms.1444. [DOI] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acun B.; Gupta A.; Jain N.; Langer A.; Menon H.; Mikida E.; Ni X.; Robson M.; Sun Y.; Totoni E.; Wesolowski L.; Kale L.. Parallel programming with migratable objects: Charm++ in practice. SC ‘14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; 2014; pp 647–658. [Google Scholar]
- Phillips J. C.; Hardy D. J.; Maia J. D. C.; Stone J. E.; Ribeiro J. V.; Bernardi R. C.; Buch R.; Fiorin G.; Hénin J.; Jiang W.; McGreevy R.; Melo M. C. R.; Radak B. K.; Skeel R. D.; Singharoy A.; Wang Y.; Roux B.; Aksimentiev A.; Luthey-Schulten Z.; Kalé L. V.; Schulten K.; Chipot C.; Tajkhorshid E. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 44130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J. E.; Hynninen A.-P.; Phillips J. C.; Schulten K. Early Experiences Porting the NAMD and VMD Molecular Simulation and Analysis Software to GPU-Accelerated OpenPOWER Platforms. International Workshop on OpenPower for High Performance Computing 2016, 9945, 188–206. 10.1007/978-3-319-46079-6_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delivering up to 9X the Throughput with NAMD v3 and NVIDIA A100 GPU. https://developer.nvidia.com/blog/delivering-up-to-9x-throughput-with-namd-v3-and-a100-gpu/ (accessed 2-14-2022).
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Páll S.; Zhmurov A.; Bauer P.; Abraham M.; Lundborg M.; Gray A.; Hess B.; Lindahl E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. 10.1063/5.0018516. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E. 3rd; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Götz A. W.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Le Grand S.; Götz A. W.; Walker R. C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- cuFFT. https://docs.nvidia.com/cuda/cufft/index.html (accessed 2-17-2022).
- Lee T.-S.; Cerutti D. S.; Mermelstein D.; Lin C.; LeGrand S.; Giese T. J.; Roitberg A.; Case D. A.; Walker R. C.; York D. M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model. 2018, 58, 2043–2050. 10.1021/acs.jcim.8b00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- pmemd.cuda GPU Implementation. https://ambermd.org/GPUPerformance.php (accessed 11-28-2021).
- Kutzner C.; Pall S.; Fechner M.; Esztermann A.; Groot B. L.; Grubmuller H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. 10.1002/jcc.26011. [DOI] [PubMed] [Google Scholar]
- Casalino L.; Dommer A. C.; Gaieb Z.; Barros E. P.; Sztain T.; Ahn S.-H.; Trifan A.; Brace A.; Bogetti A. T.; Clyde A.; Ma H.; Lee H.; Turilli M.; Khalid S.; Chong L. T.; Simmerling C.; Hardy D. J.; Maia J. D. C.; Phillips J. C.; Kurth T.; Stern A. C.; Huang L.; McCalpin J. D.; Tatineni M.; Gibbs T.; Stone J. E.; Jha S.; Ramanathan A.; Amaro R. E. AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics. Int. J. High Perform. Comput. Appl. 2021, 35, 432–451. 10.1177/10943420211006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAMD Performance. http://www.ks.uiuc.edu/Research/namd/benchmarks/ (accessed 5-4-2021).
- Yang Y. I.; Shao Q.; Zhang J.; Yang L.; Gao Y. Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 151, 070902. 10.1063/1.5109531. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Aktulga H. M.; Belfon K.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Giambasu G.; Gilson M. K.; Gohlke H.; Goetz A. W.; Harris R.; Izadi S.; Izmailov S. A.; Jin C.; Kasavajhala K.; Kaymak M. C.; King E.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Machado M.; Man V.; Manathunga M.; Merz K. M.; Miao Y.; Mikhailovskii O.; Monard G.; Nguyen H.; O’Hearn K. A.; Pantano; Qi R.; Rahnamoun A.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Skrynnikov N. R.; Smith J.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xue Y.; York D. M.; Zhao S.; Kollman P. A.. Amber 2021. https://ambermd.org/doc12/Amber21.pdf (accessed 5-4-2021).
- Welton B.; Miller B.. Exposing Hidden Performance Opportunities in High Performance GPU Applications. 18th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing; CCGRID, 2018; pp 301–310. [Google Scholar]
- Kohnke B.; Kutzner C.; Beckmann A.; Lube G.; Kabadshow I.; Dachsel H.; Grubmüller H. A CUDA fast multipole method with highly efficient M2L far field evaluation. Int. J. High Perform. Comput. Appl. 2021, 35, 97–117. 10.1177/1094342020964857. [DOI] [Google Scholar]
- Noé F.; Tkatchenko A.; Müller K.-R.; Clementi C. Machine Learning for Molecular Simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. 10.1146/annurev-physchem-042018-052331. [DOI] [PubMed] [Google Scholar]
- Doerr S.; Majewski M.; Pérez A.; Krämer A.; Clementi C.; Noe F.; Giorgino T.; De Fabritiis G. TorchMD: A Deep Learning Framework for Molecular Simulations. J. Chem. Theory Comput. 2021, 17, 2355–2363. 10.1021/acs.jctc.0c01343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUDA Toolkit. https://developer.nvidia.com/cuda-toolkit (accessed 11-29-2021).
- Sirowy S.; Forin A.. Where’s the beef? Why FPGAs are so fast. https://www.microsoft.com/en-us/research/wp-content/uploads/2016/02/tr-2008-130.pdf (accessed 9-30-2021), 2008.
- Kastner R.; Matai J.; Neuendorffer S.. Parallel Programming for FPGAs. 2018, arXiv preprint arXiv:1805.03648. https://arxiv.org/abs/1805.03648 (accessed 9-30-2021).
- Bacon D. F.; Rabbah R.; Shukla S. FPGA programming for the masses. Commun. ACM 2013, 56, 56–63. 10.1145/2436256.2436271. [DOI] [Google Scholar]
- Vetter J. S.; Brightwell R.; Gokhale M.; Mccormick P.; Ross R.; Shalf J.; Antypas K.; Donofrio D.; Humble T.; Schuman C.; Van Essen B.; Yoo S.; Aiken A.; Bernholdt D.; Byna S.; Cameron K.; Cappello F.; Chapman B.; Chien A.; Hall M.; Hartman-Baker R.; Lan Z.; Lang M.; Leidel J.; Li S.; Lucas R.; Mellor-Crummey J.; Peltz P. Jr; Peterka T.; Strout M.; Wilke J.. Extreme Heterogeneity 2018—Productive Computational Science in the Era of Extreme Heterogeneity: Report for DOE ASCR Workshop on Extreme Heterogeneity; 2018. [Google Scholar]
- Auerbach J.; Bacon D. F.; Burcea I.; Cheng P.; Fink S. J.; Rabbah R.; Shukla S.. A compiler and runtime for heterogeneous computing. Proceedings of the 49th Annual Design Automation Conference; New York, NY, 2012; pp 271–276. [Google Scholar]
- Auerbach J.; Bacon D. F.; Cheng P.; Rabbah R.. Lime: a Java-compatible and synthesizable language for heterogeneous architectures. Proceedings of the ACM International Conference on Object Oriented Programming Systems Languages and Applications; New York, NY, 2010; pp 89–108. [Google Scholar]
- Chamberlain R. D. Architecturally truly diverse systems: A review. Future Gener. Comput. Syst. 2020, 110, 33–44. 10.1016/j.future.2020.03.061. [DOI] [Google Scholar]
- Czajkowski T. S.; Aydonat U.; Denisenko D.; Freeman J.; Kinsner M.; Neto D.; Wong J.; Yiannacouras P.; Singh D. P.. From Opencl to High-Performance Hardware on FPGAS. 22nd International Conference on Field Programmable Logic and Applications (FPL); 2012; pp 531–534. [Google Scholar]
- An Independent Evaluation of: The AutoESL AutoPilot High-Level Synthesis Tool; Berkeley Design Technology, Inc.: Berkely, CA, 2010. [Google Scholar]
- Greaves D.; Singh S.. Designing application specific circuits with concurrent C# programs. Eighth ACM/IEEE International Conference on Formal Methods and Models for Codesign; MEMOCODE, 2010; pp 21–30. [Google Scholar]
- Bachrach J.; Vo H.; Richards B.; Lee Y.; Waterman A.; Avižienis R.; Wawrzynek J.; Asanovic K.. Chisel: Constructing Hardware in a Scala Embedded Language. https://people.eecs.berkeley.edu/~jrb/papers/chisel-dac-2012-corrected.pdf (accessed 10-19-2021).
- Yang C.; Geng T.; Wang T.; Patel R.; Xiong Q.; Sanaullah A.; Wu C.; Sheng J.; Lin C.; Sachdeva V.; Sherman W.; Herbordt M.. Fully integrated FPGA molecular dynamics simulations. SC ‘19: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. 2019; pp 1–31. [Google Scholar]
- Yang C.; Geng T.; Wang T.; Lin C.; Sheng J.; Sachdeva V.; Sherman W.; Herbordt M.. Molecular Dynamics Range-Limited Force Evaluation Optimized for FPGAs. IEEE 30th International Conference on Application-specific Systems, Architectures and Processors (ASAP). 2019; pp 263–271. [Google Scholar]
- Gu Y.; Vancourt T.; Herbordt M. C. Explicit Design of FPGA-Based Coprocessors for Short-Range Force Computations in Molecular Dynamics Simulations. Parallel Comput 2008, 34, 261–277. 10.1016/j.parco.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam S. R.; Agarwal P. K.; Smith M. C.; Vetter J. S.; Caliga D. Using FPGA Devices to Accelerate Biomolecular Simulations. Computer 2007, 40, 66–73. 10.1109/MC.2007.108. [DOI] [Google Scholar]
- Kindratenko V.; Pointer D.. A case study in porting a production scientific supercomputing application to a reconfigurable computer. 14th Annual IEEE Symposium on Field-Programmable Custom Computing Machines; FCCM, 2006; pp 13–22. [Google Scholar]
- Gu Y.; Van Court T.; DiSabello D.; Herbordt M. C.. Preliminary report: FPGA acceleration of molecular dynamics computations. 13th Annual IEEE Symposium on Field-Programmable Custom Computing Machines; FCCM; ,2005; pp 269–270. [Google Scholar]
- Gu Y.; VanCourt T.; Herbordt M. C.. Improved Interpolation and System Integration for FPGA-Based Molecular Dynamics Simulations. 2006 International Conference on Field Programmable Logic and Applications. 2006; pp 1–8. [Google Scholar]
- Scrofano R.; Prasanna V. K.. Preliminary Investigation of Advanced Electrostatics in Molecular Dynamics on Reconfigurable Computers. SC ’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing. 2006; pp 45–45. [Google Scholar]
- Scrofano R.; Gokhale M.; Trouw F.; Prasanna V. K.. Hardware/Software Approach to Molecular Dynamics on Reconfigurable Computers. 2006 14th Annual IEEE Symposium on Field-Programmable Custom Computing Machines. 2006; pp 23–34. [Google Scholar]
- Gokhale M. B.; Rickett C. D.; Hsu C. H. Promises and Pitfalls of Reconfigurable Supercomputing. Proceedings of the International Conference on Engineering of Reconfigurable Systems & Algorithms (ERSA) 2006, 11–20. [Google Scholar]
- Scrofano R.; Prasanna V. K. Computing Lennard-Jones Potentials and Forces with Reconfigurable Hardware. Proceedings of the International Conference on Engineering of Reconfigurable Systems & Algorithms (ERSA) 2004, 284–292. [Google Scholar]
- Azizi N.; Kuon I.; Egier A.; Darabiha A.; Chow P.. Reconfigurable molecular dynamics simulator. 12th Annual IEEE Symposium on Field-Programmable Custom Computing Machines; FCCM, 2004; pp 197–206. [Google Scholar]
- TM-3 documentation. https://www.eecg.utoronto.ca/~tm3/ (accessed 10-10-2021).
- Gu Y.; VanCourt T.; Herbordt M. C.. Accelerating molecular dynamics simulations with configurable circuits. International Conference on Field Programmable Logic and Applications. 2005; pp 475–480. [Google Scholar]
- Annapolis Micro Systems, Inc., WILDSTAR II Pro PCI. http://pdf.cloud.opensystemsmedia.com/xtca-systems.com/19243.pdf (accessed 5-13-2021).
- Chiu M.; Khan M. A.; Herbordt M. C.. Efficient Calculation of Pairwise Nonbonded Forces. 2011 IEEE 19th Annual International Symposium on Field-Programmable Custom Computing Machines; FCCM, 2011; pp 73–76. [Google Scholar]
- Matthey T.; Cickovski T.; Hampton S.; Ko A.; Ma Q.; Nyerges M.; Raeder T.; Slabach T.; Izaguirre J. A. ProtoMol, an object-oriented framework for prototyping novel algorithms for molecular dynamics. ACM Trans. Math. Softw. 2004, 30, 237–265. 10.1145/1024074.1024075. [DOI] [Google Scholar]
- Agarwal P. K.; Alam S. R. Biomolecular simulations on petascale: promises and challenges. J. Phys.: Conf. Ser. 2006, 46, 327–333. 10.1088/1742-6596/46/1/046. [DOI] [Google Scholar]
- Villarreal J.; Najjar W. A.. Compiled hardware acceleration of Molecular Dynamics code. International Conference on Field Programmable Logic and Applications. 2008; pp 667–670. [Google Scholar]
- Shaw D. E.; Chao J. C.; Eastwood M. P.; Gagliardo J.; Grossman J. P.; Ho C. R.; Ierardi D. J.; Kolossváry I.; Klepeis J. L.; Layman T.; McLeavey C.; Deneroff M. M.; Moraes M. A.; Mueller R.; Priest E. C.; Shan Y.; Spengler J.; Theobald M.; Towles B.; Wang S. C.; Dror R. O.; Kuskin J. S.; Larson R. H.; Salmon J. K.; Young C.; Batson B.; Bowers K. J.. Anton, a special-purpose machine for molecular dynamics simulation. Proceedings of the 34th Annual International Symposium on Computer Architecture—ISCA ’07, 2007. [Google Scholar]
- Taiji M.; Narumi T.; Ohno Y.; Futatsugi N.; Suenaga A.; Takada N.; Konagaya A.. Protein Explorer: A Petaflops Special-Purpose Computer System for Molecular Dynamics Simulations. SC ‘03: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; New York, NY, 2003; p 15. [Google Scholar]
- Fine R.; Dimmler G.; Levinthal C. FASTRUN: a special purpose, hardwired computer for molecular simulation. Proteins: Struct., Funct., Bioinf. 1991, 11, 242–253. 10.1002/prot.340110403. [DOI] [PubMed] [Google Scholar]
- Toyoda S.; Miyagawa H.; Kitamura K.; Amisaki T.; Hashimoto E.; Ikeda H.; Kusumi A.; Miyakawa N. Development of MD Engine: High-speed accelerator with parallel processor design for molecular dynamics simulations. J. Comput. Chem. 1999, 20, 185–199. . [DOI] [Google Scholar]
- Bakker A. F.; Bruin C.; van Dieren F.; Hilhorst H. J. Molecular dynamics of 16000 Lennard-Jones particles. Phys. Lett. A 1982, 93, 67–69. 10.1016/0375-9601(82)90217-1. [DOI] [Google Scholar]
- Bakker A. F.; Bruin C. In Special Purpose Computers; Alder B. J., Ed.; Academic Press: Cambridge, U.K., 1988; pp 183–232. [Google Scholar]
- Fincham D.; Ralston B.J. Molecular Dynamics Simulation Using the CRAY-i Vector Processing Computer. Comput. Phys. Commun. 1981, 23, 127–134. 10.1016/0010-4655(81)90027-8. [DOI] [Google Scholar]
- Bell G.; Bailey D. H.; Dongarra J.; Karp A. H.; Walsh K. A look back on 30 years of the Gordon Bell Prize. Int. J. High Perform. Comput. Appl. 2017, 31, 469–484. 10.1177/1094342017738610. [DOI] [Google Scholar]
- Sugimoto D.; Chikada Y.; Makino J.; Ito T.; Ebisuzaki T.; Umemura M. A special-purpose computer for gravitational many-body problems. Nature 1990, 345, 33–35. 10.1038/345033a0. [DOI] [Google Scholar]
- Ito T.; Makino J.; Ebisuzaki T.; Sugimoto D. A special-purpose N-body machine GRAPE-1. Comput. Phys. Commun. 1990, 60, 187–194. 10.1016/0010-4655(90)90003-J. [DOI] [Google Scholar]
- Fukushige T.; Taiji M.; Makino J.; Ebisuzaki T.; Sugimoto D. A Highly Parallelized Special-Purpose Computer For Many-Body Simulations With An Arbitrary Central Force: MD-GRAPE. Astrophys. J. 1996, 468, 51. 10.1086/177668. [DOI] [Google Scholar]
- Komeiji Y.; Yokoyama H.; Uebayasi M.; Taiji M.; Fukushige T.; Sugimoto D.; Takata R.; Shimizu A.; Itsukashi K. A high performance system for molecular dynamics simulation of biomolecules using a special-purpose computer. Pac. Symp. Biocomput. 1996, 472–487. [PubMed] [Google Scholar]
- Komeiji Y.; Uebayasi M.; Takata R.; Shimizu A.; Itsukashi K.; Taiji M. Fast and accurate molecular dynamics simulation of a protein using a special-purpose computer. J. Comput. Chem. 1997, 18, 1546–1563. . [DOI] [Google Scholar]
- Susukita R.; Ebisuzaki T.; Elmegreen B. G.; Furusawa H.; Kato K.; Kawai A.; Kobayashi Y.; Koishi T.; McNiven G. D.; Narumi T.; Yasuoka K. Hardware accelerator for molecular dynamics: MDGRAPE-2. Comput. Phys. Commun. 2003, 155, 115–131. 10.1016/S0010-4655(03)00349-7. [DOI] [Google Scholar]
- Narumi T.; Susukita R.; Furusawa H.; Ebisuzaki T.. 46 TFLOPS special-purpose computer for molecular dynamics simulations: WINE-2. Proceedings of the 5th International Conference on Signal Processing; 2000; pp 575–582. [Google Scholar]
- Bhatele A.; Kumar S.; Mei C.; Phillips J. C.; Zheng G.; Kale L. V. Overcoming scaling challenges in biomolecular simulations across multiple platforms. IEEE International Symposium on Parallel and Distributed Processing 2008, 1–12. 10.1109/IPDPS.2008.4536317. [DOI] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. 10.1002/jcc.540040211. [DOI] [Google Scholar]
- Ohmura I.; Morimoto G.; Ohno Y.; Hasegawa A.; Taiji M. MDGRAPE-4: a special-purpose computer system for molecular dynamics simulations. Philos. Trans. R. Soc., A 2014, 372, 20130387. 10.1098/rsta.2013.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T. S.; Okimoto N.; Koyama Y. M.; Hirano Y.; Morimoto G.; Ohno Y.; Taiji M. Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation. Sci. Rep. 2020, 10, 16986. 10.1038/s41598-020-74099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MDGRAPE-4A. https://www.r-ccs.riken.jp/exhibit (accessed 5-9-2021).
- Larson R. H.; Salmon J. K.; Dror R. O.; Deneroff M. M.; Young C.; Grossman J. P.; Shan Y.; Klepeis J. L.; Shaw D. E.. High-throughput pairwise point interactions in Anton, a specialized machine for molecular dynamics simulation. IEEE 14th International Symposium on High Performance Computer Architecture; 2008; pp 331–342. [Google Scholar]
- Kuskin J. S.; Young C.; Grossman J. P.; Batson B.; Deneroff M. M.; Dror R. O.; Shaw D. E.. Incorporating flexibility in Anton, a specialized machine for molecular dynamics simulation. IEEE 14th International Symposium on High Performance Computer Architecture; 2008; pp 343–354. [Google Scholar]
- Shaw D. E.; Adams P. J.; Azaria A.; Bank J. A.; Batson B.; Bell A.; Bergdorf M.; Bhatt J.; Butts J. A.; Correia T.; Dirks R. M.; Dror R. O.; Eastwood M. P.; Edwards B.; Even A.; Feldmann P.; Fenn M.; Fenton C. H.; Forte A.; Gagliardo J.; Gill G.; Gorlatova M.; Greskamp B.; Grossman J. P.; Gullingsrud J.; Harper A.; Hasenplaugh W.; Heily M.; Heshmat B. C.; Hunt J.; Ierardi D. J.; Iserovich L.; Jackson B. L.; Johnson N. P.; Kirk M. M.; Klepeis J. L.; Kuskin J. S.; Mackenzie K. M.; Mader R. J.; McGowen R.; McLaughlin A.; Moraes M. A.; Nasr M. H.; Nociolo L. J.; O’Donnell L.; Parker A.; Peticolas J. L.; Pocina G.; Predescu C.; Quan T.; Salmon J. K.; Schwink C.; Shim K. S.; Siddique N.; Spengler J.; Szalay T.; Tabladillo R.; Tartler R.; Taube A. G.; Theobald M.; Towles B.; Vick W.; Wang S. C.; Wazlowski M.; Weingarten M. J.; Williams J. M.; Yuh K. A.. Anton 3: Twenty Microseconds of Molecular Dynamics Simulation before Lunch. SC ‘21: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; New York, NY, 2021; pp 1–11. [Google Scholar]
- Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. SC ’14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; 2014; pp 41–53. [Google Scholar]
- Shaw D. E.; Dror R. O.; Salmon J. K.; Grossman J. P.; Mackenzie K. M.; Bank J. A.; Young C.; Deneroff M. M.; Batson B.; Bowers K. J.; Chow E.; Eastwood M. P.; Ierardi D. J.; Klepeis J. L.; Kuskin J. S.; Larson R. H.; Lindorff-Larsen K.; Maragakis P.; Moraes M. A.; Piana S.; Shan Y.; Towles B. Millisecond-scale molecular dynamics simulations on Anton. SC ‘09: Proceedings of the Conference on High Performance Computing Networking. Storage and Analysis 2009, 1–11. 10.1145/1654059.1654126. [DOI] [Google Scholar]
- Predescu C.; Lerer A. K.; Lippert R. A.; Towles B.; Grossman J. P.; Dirks R. M.; Shaw D. E. The u-series: A separable decomposition for electrostatics computation with improved accuracy. J. Chem. Phys. 2020, 152, 084113. 10.1063/1.5129393. [DOI] [PubMed] [Google Scholar]
- S K. T.; Noriaki O.; M K. Y.; Yoshinori H.; Gentaro M.; Yousuke O.; Makoto T.. Molecular dynamics trajectories for SARS-CoV-2 Mpro with 7 HIV inhibitors. https://zenodo.org/record/3975394#.YXe99dnMJro (accessed 5-9-2021).
- Okimoto N.; Futatsugi N.; Fuji H.; Suenaga A.; Morimoto G.; Yanai R.; Ohno Y.; Narumi T.; Taiji M. High-performance drug discovery: computational screening by combining docking and molecular dynamics simulations. PLoS Comput. Biol. 2009, 5, e1000528 10.1371/journal.pcbi.1000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhar A. A.; Prasanna V. K.; Shaaban M. E.; Wang C.-L. Heterogeneous computing: challenges and opportunities. Computer 1993, 26, 18–27. 10.1109/2.214439. [DOI] [Google Scholar]
- Brodtkorb A. R.; Dyken C.; Hagen T. R.; Hjelmervik J. M.; Storaasli O. O. State-of-the-art in Heterogeneous Computing. Sci. Program. 2010, 18, 1–33. 10.1155/2010/540159. [DOI] [Google Scholar]
- Mitra T. Heterogeneous Multi-core Architectures. IPSJ. Trans. Syst. LSI Des. Methodol. 2015, 8, 51–62. 10.2197/ipsjtsldm.8.51. [DOI] [Google Scholar]
- Mittal S.; Vetter J. S. A Survey of CPU-GPU Heterogeneous Computing Techniques. ACM Comput. Surv. 2015, 47, 1–35. 10.1145/2788396. [DOI] [Google Scholar]
- Hagleitner C.; Diamantopoulos D.; Ringlein B.; Evangelinos C.; Johns C.; Chang R. N.; D’Amora B.; Kahle J. A.; Sexton J.; Johnston M.; Pyzer-Knapp E.; Ward C.. Heterogeneous Computing Systems for Complex Scientific Discovery Workflows. Proceedings of the Design, Automation Test in Europe Conference (DATE); 2021; pp 13–18. [Google Scholar]
- Di Natale F.; Bhatia H.; Carpenter T. S.; Neale C.; Kokkila-Schumacher S.; Oppelstrup T.; Stanton L.; Zhang X.; Sundram S.; Scogland T. R. W.; Dharuman G.; Surh M. P.; Yang Y.; Misale C.; Schneidenbach L.; Costa C.; Kim C.; D’Amora B.; Gnanakaran S.; Nissley D. V.; Streitz F.; Lightstone F. C.; Bremer P.-T.; Glosli J. N.; Ingólfsson H. I.. A massively parallel infrastructure for adaptive multiscale simulations: modeling RAS initiation pathway for cancer. SC ‘19: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; 2019; pp 1–16. [Google Scholar]
- oneAPI Programming Model. https://www.oneapi.io/ (accessed 10-18-2021), 2021.
- Wang S.; Prakash A.; Mitra T.. Software Support for Heterogeneous Computing. IEEE Computer Society Annual Symposium on VLSI; ISVLSI, 2018; pp 756–762. [Google Scholar]
- AMD ROCm Release Notes v4.1—ROCm Documentation 1.0.0 documentation. https://rocmdocs.amd.com/en/latest/Current_Release_Notes/Current-Release-Notes.html (accessed 3-24-2021).
- pmemd.cuda GPU Implementation. https://ambermd.org/GPUPerformance.php (accessed 1-10-2021).
- OpenMM. https://openmm.org/benchmarks (accessed 1-10-2021).
- Harvey M. J.; Giupponi G.; Fabritiis G. D. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; Kochanek S. E.; Casalino L.; Ieong P. U.; Dommer A. C.; Amaro R. E. Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism. ACS Cent Sci. 2020, 6, 189–196. 10.1021/acscentsci.9b01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.; Nishima W.; Daniels M.; Bascom G.; Kobayashi C.; Adedoyin A.; Wall M.; Lappala A.; Phillips D.; Fischer W.; Tung C.-S.; Schlick T.; Sugita Y.; Sanbonmatsu K. Y. Scaling molecular dynamics beyond 100,000 processor cores for large-scale biophysical simulations. J. Comput. Chem. 2019, 40, 1919–1930. 10.1002/jcc.25840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino P. L.; Arkhipov A. S.; Larson S. B.; McPherson A.; Schulten K. Molecular dynamics simulations of the complete satellite tobacco mosaic virus. Structure 2006, 14, 437–449. 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Noé F. In Machine Learning Meets Quantum Physics; Schütt K. T., Chmiela S., von Lilienfeld O. A., Tkatchenko A., Tsuda K., Müller K.-R., Eds.; Springer: Cambridge, 2020; pp 331–372. [Google Scholar]
- Jones D.; Kim H.; Zhang X.; Zemla A.; Stevenson G.; Bennett W. F. D.; Kirshner D.; Wong S. E.; Lightstone F. C.; Allen J. E. Improved Protein-Ligand Binding Affinity Prediction with Structure-Based Deep Fusion Inference. J. Chem. Inf. Model. 2021, 61, 1583–1592. 10.1021/acs.jcim.0c01306. [DOI] [PubMed] [Google Scholar]
- Lau E. Y.; Negrete O. A.; Bennett W. F. D.; Bennion B. J.; Borucki M.; Bourguet F.; Epstein A.; Franco M.; Harmon B.; He S.; Jones D.; Kim H.; Kirshner D.; Lao V.; Lo J.; McLoughlin K.; Mosesso R.; Murugesh D. K.; Saada E. A.; Segelke B.; Stefan M. A.; Stevenson G. A.; Torres M. W.; Weilhammer D. R.; Wong S.; Yang Y.; Zemla A.; Zhang X.; Zhu F.; Allen J. E.; Lightstone F. C. Discovery of Small-Molecule Inhibitors of SARS-CoV-2 Proteins Using a Computational and Experimental Pipeline. Front. Mol. Biosci. 2021, 8, 678701. 10.3389/fmolb.2021.678701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson G. A.; Jones D.; Kim H.; Bennett W. F. D.; Bennion B. J.; Borucki M.; Bourguet F.; Epstein A.; Franco M.; Harmon B.; He S.; Katz M. P.; Kirshner D.; Lao V.; Lau E. Y.; Lo J.; McLoughlin K.; Mosesso R.; Murugesh D. K.; Negrete O. A.; Saada E. A.; Segelke B.; Stefan M.; Torres M. W.; Weilhammer D.; Wong S.; Yang Y.; Zemla A.; Zhang X.; Zhu F.; Lightstone F. C.; Allen J. E.. High-throughput virtual screening of small molecule inhibitors for SARS-CoV-2 protein targets with deep fusion models. SC ‘21: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; New York, NY, 2021; pp 1–13.
- Schoenholz S. S.; Cubuk E. D.. JAX MD: End-to-End Differentiable, Hardware Accelerated, Molecular Dynamics in Pure Python; 2019. [Google Scholar]
- Bradbury J.; Frostig R.; Hawkins P.; Johnson M. J.; Leary C.; Maclaurin D.; Necula G.; Paszke A.; VanderPlas J.; Wanderman-Milne S.; Zhang Q.. JAX: composable transformations of Python+NumPy programs. http://github.com/google/jax (accessed 11-30-2021).
- Jouppi N. P.; Yoon D. H.; Kurian G.; Li S.; Patil N.; Laudon J.; Young C.; Patterson D. A domain-specific supercomputer for training deep neural networks. Commun. ACM 2020, 63, 67–78. 10.1145/3360307. [DOI] [Google Scholar]
- Paszke A.; Gross S.; Massa F.; Lerer A.; Bradbury J.; Chanan G.; Killeen T.; Lin Z.; Gimelshein N.; Antiga L.; Desmaison A.; Kopf A.; Yang E.; DeVito Z.; Raison M.; Tejani A.; Chilamkurthy S.; Steiner B.; Fang L.; Bai J.; Chintala S. In Advances in Neural Information Processing Systems 32; Wallach H., Larochelle H., Beygelzimer A., d’ Alché-Buc F., Fox E., Garnett R., Eds.; Curran Associates, Inc.: Red Hook, NY, 2019; pp 8024–8035. [Google Scholar]
- Amodei D.; Hernandez D.; Sastry G.; Clark J.; Brockman G.; Sutskever I.. AI and Compute. https://openai.com/blog/ai-and-compute/ (accessed 5-6-2021).
- Cerebras Systems: Achieving Industry Best AI Performance Through A Systems Approach. https://cerebras.net/wp-content/uploads/2021/04/Cerebras-CS-2-Whitepaper.pdf (accessed 5-6-2021).
- Accelerated Computing with a Reconfigurable Dataflow Architecture. https://sambanova.ai/wp-content/uploads/2021/06/SambaNova_RDA_Whitepaper_English.pdf (accessed 5-6-2021).
- Jia Z.; Tillman B.; Maggioni M.; Scarpazza D. P.. Dissecting the Graphcore IPU Architecture via Microbenchmarking. arXiv preprint arXiv:1912.03413. https://arxiv.org/abs/1912.03413 (accessed 9-30-2021).
- Hopkins C. W.; Le Grand S.; Walker R. C.; Roitberg A. E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. 10.1021/ct5010406. [DOI] [PubMed] [Google Scholar]
- Tironi I. G.; Sperb R.; Smith P. E.; van Gunsteren W. F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995, 102, 5451–5459. 10.1063/1.469273. [DOI] [Google Scholar]
- Eastman P.; Pande V. S. OpenMM: A Hardware Independent Framework for Molecular Simulations. Comput. Sci. Eng. 2010, 12, 34–39. 10.1109/MCSE.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Car R.; Parrinello M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471–2474. 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Han J.; Wang H.; Saidi W. A.; Car R.; Weinan E.. End-to-end Symmetry Preserving Inter-atomic Potential Energy Model for Finite and Extended Systems. arXiv preprint arXiv:1805.09003. https://arxiv.org/abs/1805.09003 (accessed 9-30-2021).
- Zhang L.; Han J.; Wang H.; Car R.; E W. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys. Rev. Lett. 2018, 120, 143001. 10.1103/PhysRevLett.120.143001. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wang H.; Chen W.; Zeng J.; Zhang L.; Wang H.; Weinan E. DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models. Comput. Phys. Commun. 2020, 253, 107206. 10.1016/j.cpc.2020.107206. [DOI] [Google Scholar]
- Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- Lu D.; Wang H.; Chen M.; Lin L.; Car R.; E W.; Jia W.; Zhang L. 86 PFLOPS Deep Potential Molecular Dynamics simulation of 100 million atoms with ab initio accuracy. Comput. Phys. Commun. 2021, 259, 107624. 10.1016/j.cpc.2020.107624. [DOI] [Google Scholar]
- Sugita Y.; Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. 10.1016/S0009-2614(99)01123-9. [DOI] [Google Scholar]
- Laio A.; Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babin V.; Roland C.; Sagui C. Adaptively biased molecular dynamics for free energy calculations. J. Chem. Phys. 2008, 128, 134101. 10.1063/1.2844595. [DOI] [PubMed] [Google Scholar]
- PLUMED consortium, Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. 10.1038/s41592-019-0506-8 [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Scarpazza D. P.; Ierardi D. J.; Lerer A. K.; Mackenzie K. M.; Pan A. C.; Bank J. A.; Chow E.; Dror R. O.; Grossman J. P.; Killebrew D.; Moraes M. A.; Predescu C.; Salmon J. K.; Shaw D. E. Extending the Generality of Molecular Dynamics Simulations on a Special-Purpose Machine. IEEE 27th International Symposium on Parallel and Distributed Processing 2013, 933–945. 10.1109/IPDPS.2013.93. [DOI] [Google Scholar]
- Pan A. C.; Weinreich T. M.; Piana S.; Shaw D. E. Demonstrating an Order-of-Magnitude Sampling Enhancement in Molecular Dynamics Simulations of Complex Protein Systems. J. Chem. Theory Comput. 2016, 12, 1360–1367. 10.1021/acs.jctc.5b00913. [DOI] [PubMed] [Google Scholar]
- Pan A. C.; Jacobson D.; Yatsenko K.; Sritharan D.; Weinreich T. M.; Shaw D. E. Atomic-level characterization of protein-protein association. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 4244–4249. 10.1073/pnas.1815431116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinari E.; Parisi G. Simulated tempering: A new Monte Carlo scheme. EPL 1992, 19, 451–458. 10.1209/0295-5075/19/6/002. [DOI] [Google Scholar]
- Wu C.; Geng T.; Yang C.; Sachdeva V.; Sherman W.; Herbordt M.. A Communication-Efficient Multi-Chip Design for Range-Limited Molecular Dynamics. IEEE High Performance Extreme Computing Conference; HPEC, 2020; pp 1–8. [Google Scholar]
- Zhang J.; Yang Y. I.; Noé F. Targeted Adversarial Learning Optimized Sampling. J. Phys. Chem. Lett. 2019, 10, 5791–5797. 10.1021/acs.jpclett.9b02173. [DOI] [PubMed] [Google Scholar]
- Noé F.; Olsson S.; Köhler J.; Wu H.. Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning. Science 2019, 365. 10.1126/science.aaw1147 [DOI] [PubMed] [Google Scholar]