

SUMMARY
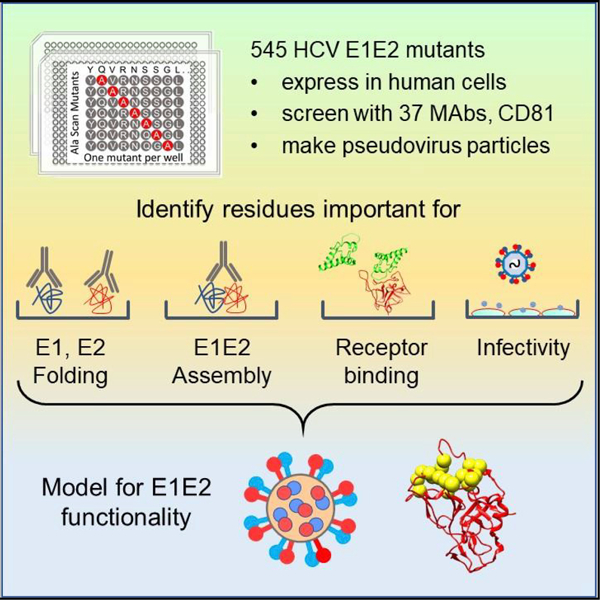
The E1 and E2 envelope proteins of hepatitis C virus (HCV) form a heterodimer that drives virus-host membrane fusion. Here, we analyze the role of each amino acid in E1E2 function, expressing 545 individual alanine mutants of E1E2 in human cells, incorporating them into infectious viral pseudoparticles, and testing them against 37 different monoclonal antibodies (MAbs) to ascertain full-length translation, folding, heterodimer assembly, CD81 binding, viral pseudoparticle incorporation, and infectivity. We propose a model describing the role of each critical residue in E1E2 functionality and use it to examine how MAbs neutralize infection by exploiting functionally critical sites of vulnerability on E1E2. Our results suggest that E1E2 is a surprisingly fragile protein complex where even a single alanine mutation at 92% of positions disrupts its function. The amino-acid-level targets identified are highly conserved and functionally critical and can be exploited for improved therapies and vaccines.
Graphical abstract
In brief
Pfaff-Kilgore et al. describe the role of individual amino acids in hepatis C virus E1E2 protein function by generating a comprehensive E1E2 mutation library and testing 545 clones for E1E2 folding, assembly, and infectivity. Their model describes the role of residues in E1E2 functionality and examines how antibodies neutralize infection.
INTRODUCTION
Hepatitis C virus (HCV) causes acute or chronic infection and liver disease, which can progress to liver cirrhosis, cancer, and death. Despite effective treatments for HCV infection, no vaccine has been approved to prevent HCV. Difficulties for HCV vaccines include extreme sequence variability in the virus envelope (Env) proteins, a high mutation frequency in the virus genome, and limited animal models for HCV infection (Drummer, 2014a). HCV is extremely variable in sequence, with seven genotypes (1–7) and sequence variability within each genotype.
The Env-protein complex of HCV comprises two highly glycosylated transmembrane proteins E1 and E2, cleaved from the genome-encoded polyprotein, which form a heterodimer (E1E2) and mediate cell entry and virus-host membrane fusion. The limited E1E2 structural and functional information impedes the engineering and use of E1E2 for vaccines. E1 is a 192-residue protein (Y192 to A383 in the polyprotein), currently represented by a single crystal structure covering the N-terminal 76 residues and a 29-residue fragment obtained by nuclear magnetic resonance studies (El Omari et al., 2014; Spadaccini et al., 2010). Neither E1 structure includes important regions such as the putative internal fusion loop (IFL), a critical structure believed to mediate fusion between the host cell and virion membranes.
E2 consists of 363 residues (E384 to A746) with several regions that display broad sequence variability, primarily the hypervariable region HVR1 (residues 384 to 411), the variable region VR2 (460–486), and a proposed VR3 (570–580), also referred to as the intergenotypic variable region (igVR). Many regions within E1 and E2 are highly conserved, but their functional roles are not completely understood. Partial E2 structures obtained as co-crystals with antibody Fab fragments (Flyak et al., 2018; Khan et al., 2014; Kong et al., 2013) cover most of its main globular region (up to 68% of E2, residues 410 to 647) (Flyak et al., 2018).
We have a limited understanding of the interaction between E1 and E2 or of their arrangement on the virion surface. There is evidence that E1E2 heterodimers exist as E1E2 trimers on the virus surface (Falson et al., 2015). We and others have proposed specific residues and regions as important for E1E2 interaction (Douam et al., 2014; Gopal et al., 2017), and several models have been proposed for the E1E2 heterodimer derived from antibody epitope data and computational analyses of E1E2 secondary and tertiary structures (Cao et al., 2019; Castelli et al., 2017b; Freedman et al., 2017; Patel et al., 2001).
The E1E2 proteins drive fusion of the HCV membrane with the host-cell endosomal membrane in a pH-dependent manner (Hsu et al., 2003), although how E1 and E2 achieve this fusion event is poorly understood. Since HCV is a member of the Flaviviridae family, a class II fusion-protein structure might be expected but is not present in the available structures of the E2 core domains and the E1 N-terminal region (El Omari et al., 2014; Kong et al., 2013). Evidence points to E1 being responsible for membrane fusion, as E1 has a heptad repeat (HR) motif (residues 330–347) that is characteristic of fusion proteins and a conserved region (residues 272 to 286) that resembles an IFL (Drummer et al., 2007; Lavillette et al., 2007; Li et al., 2009; Tong et al., 2017).
HCV entry into hepatocytes is a multi-step process initiated by low-affinity interactions of E1 and E2 with cell-surface attachment factors, including low-density lipoprotein receptor (LDLR) and glycosaminoglycans (GAGs) (Gerold et al., 2020). E2 then engages with scavenger receptor BI (SR-BI) and then with CD81 (Kumar et al., 2021), initiating signaling that promotes HCV-CD81 movement to tight-junction regions of cell-cell contact, where interaction with claudin 1 (CLDN1) and occludin facilitate viral internalization (Benedicto et al., 2009; Ploss et al., 2009).
The E1E2 glycoproteins are targets for the immune system. Studies of numerous monoclonal antibodies (MAbs) have characterized distinct E2 antigenic sites, designated as domains I–III, A–D, or antigenic regions AR1–5 (Drummer, 2014b; Fuerst et al., 2017; Giang et al., 2012; Pierce et al., 2016; Wang et al., 2011). These MAbs include numerous virus neutralizing Abs (NAbs) (Colbert et al., 2019; Giang et al., 2012; Sautto et al., 2013; Wang et al., 2011), including NAbs targeting AR4 and AR5 that bind only when E1 and E2 are complexed together (Giang et al., 2012). However, NAb mechanisms of action are not always readily apparent due to the limited understanding of E1E2 structure. Domain B/AR3 MAbs are presumed to act by preventing interaction with CD81, whose binding site on E2 overlaps with these antigenic sites (Kong et al., 2013).
Here, we investigated how amino acids across the entire E1E2 contribute to structure and function by individually mutating 545 of the 555 E1E2 residues to alanine and expressing each mutant protein in mammalian cells. We used 37 distinct MAbs to assess full-length translation, E1 and E2 folding, and E1E2 heterodimer assembly. We also assessed the ability of each Env mutant to bind CD81, incorporate into pseudoparticles, and mediate infection of target cells. Based on these data, and in light of the current understanding of E1E2 structure and the HCV-infection pathway, we propose a model describing the mechanistic role played by residues across the entire E1E2 protein. Our model suggests a mechanism by which MAbs targeting functionally critical sites within E1E2 neutralize HCV infection. Our analysis also reveals that HCV E1E2 is a surprisingly fragile protein complex in which even a single alanine substitution at most residues can severely compromise E1E2 function. This comprehensive mutagenic analysis of HCV E1E2 provides detailed insights into the infective mechanism used by HCV and identifies conserved amino-acid target sites for therapies, vaccines, and engineering strategies to combat this virus.
RESULTS
Identification of amino acids critical to HCV E1E2 folding and assembly
To understand how HCV E1E2 folds and functions on an amino-acid level, we generated an alanine scan library of mutants covering E1E2. We used a parental expression construct for E1E2 (HCV genotype 1a, strain H77) with a V5 tag at the E2 C terminus and introduced an alanine substitution at each of 545 E1E2 residues. Existing alanine residues were changed to serine. All clones (545/555 residues, 98% coverage) were individually transfected into HEK-293T cells and tested by flow cytometry for binding to 37 different MAbs and to a soluble fragment of CD81 (CD81-LEL), identifying the epitopes for these MAbs and CD81, most of which have been reported (Bailey et al., 2017; Castelli et al., 2017b; Colbert et al., 2019; Gopal et al., 2017; Keck et al., 2004; Kong et al., 2015). The known properties of the 37 MAbs used are summarized in Table S1.
Each MAb was also characterized previously for its ability to bind E1, E2, or E1E2 heterodimers. While MAbs were individually characterized in previous reports, the complete dataset was used here in aggregate to understand the steps required for E1 and E2 folding, assembly of the E1E2 heterodimer, and interaction with CD81. Here, we defined an amino acid as being “critical” for a specific structure-function step if its mutation to alanine caused failure of that step (mean reactivity with MAbs ≤30% of WT E1E2) but not previous steps (summarized in Figure 1).
Figure 1. Experimental and data analysis processes.
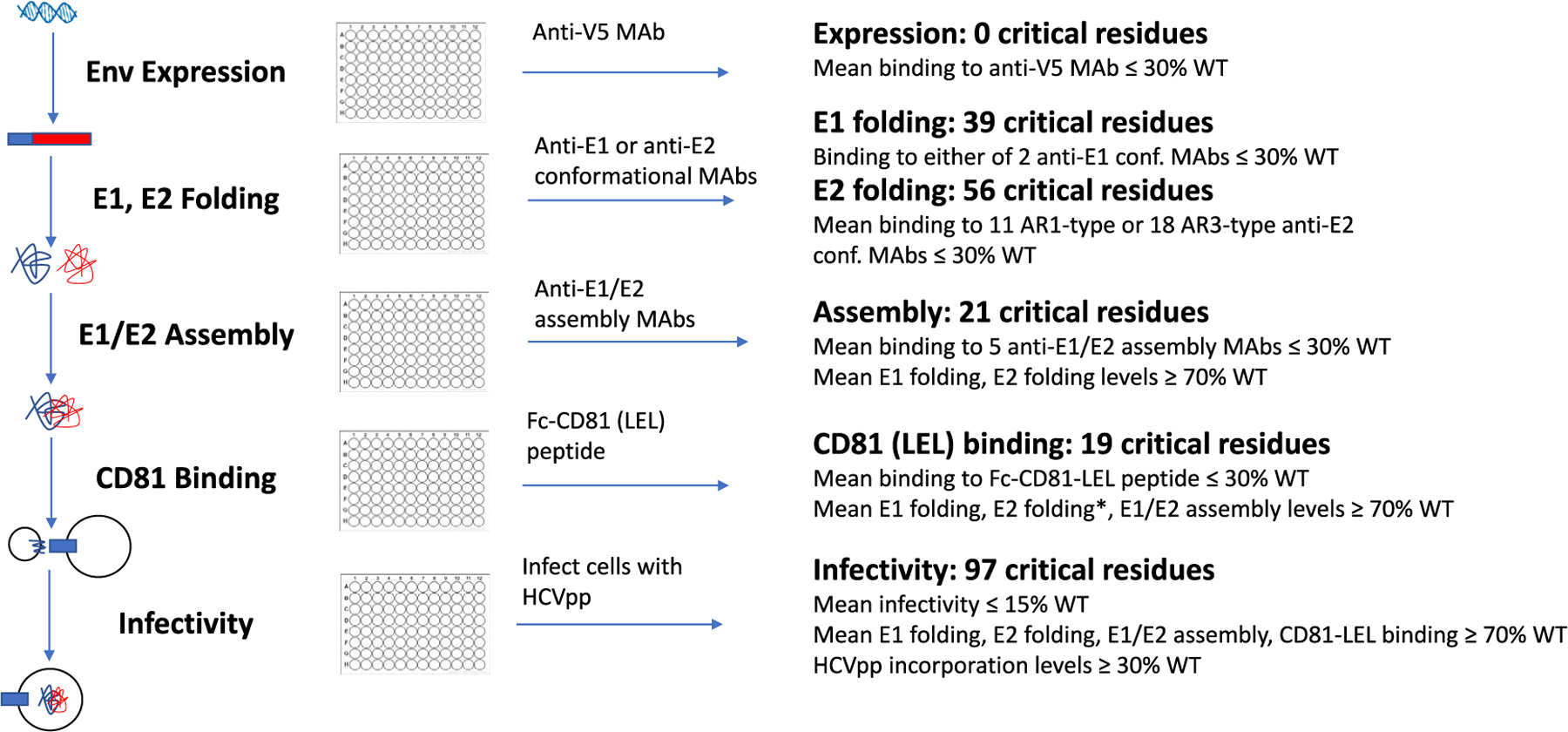
Sequential analysis of E1E2 folding and function enabled amino acids involved in each step to be identified. This screening strategy also facilitates the exclusion of mutants that are locally misfolded or that have an expression defect (Davidson and Doranz, 2014; Paes et al., 2009). Residues identified as contributing directly to MAb epitopes were excluded from the corresponding structure-function analysis using said MAbs, as they impart no structural or functional information about E1E2.
We confirmed that the mutant Env proteins were expressed comparably to WT levels by detecting the V5-tagged E2 protein with an anti-V5 MAb using flow cytometry. Each clone demonstrated a V5 level greater than 40% of the expression with WT E1E2, with an average expression of 86% of wild type (WT) (Figure S1A). Thus, nearly all mutants were translated at levels sufficient for structure-function analysis. The complete dataset for all 545 mutations binding to the 37 MAbs is provided in Table S3.
To visualize critical residues on E1E2, we created a composite structure by combining an E1 N-terminal ectodomain crystal structure (4UOI [El Omari et al., 2014]), an E1 partial NMR structure (2KNU [Spadaccini et al., 2010]), and the most complete E2 core structure (6MEI [Flyak et al., 2018]). We used a published E1E2 heterocomplex model (Castelli et al., 2017a) to complete regions of the protein where no structure was available. This composite structure is used for visualization of critical residues but was not used for analysis of data and so does not impact our conclusions; future improved structures of E1E2 will help better visualize the results of our analysis. All critical residues identified are summarized in Table S2.
Amino acids critical for E1 folding
We analyzed the complete dataset to understand how E1E2 folds and functions, visualizing the results on an E1E2 composite structure (Figure 2A). To identify residues critical for E1 folding, we analyzed the E1E2 mutation results for clones that did not bind to conformation-dependent anti-E1 MAbs. Anti-E1 conformational MAbs are rare; we used HEPC112 (Colbert et al., 2019) and HCVE1-C1, reported here, isolated by Integral Molecular. Both MAbs recognize conformational epitopes with distinct critical residues for binding and so bind only the folded E1 protein (Figure S2). The threshold for designating a critical residue for E1 folding was mean reactivity ≤30% to either of the conformational anti-E1 MAbs.
Figure 2. Identification of residues critical for E1E2 folding and assembly.
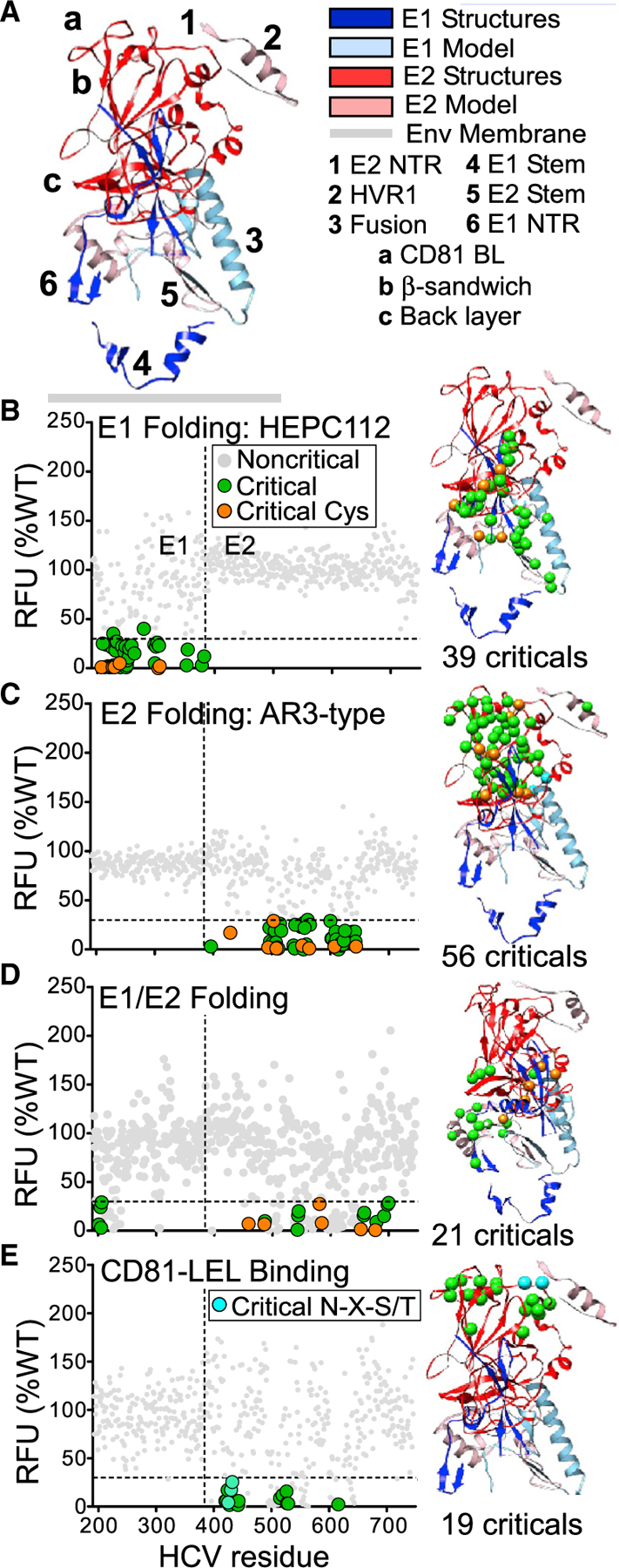
The 545 mutants in the HCV E1E2 mutation library were tested for reactivity to MAbs to identify residues important for folding and assembly. The average MAb reactivity data (in relative fluorescence units [RFUs], normalized to reactivity with WT E1E2, mean of at least two measurements) were plotted for each clone. In all graphs, the horizontal dashed line indicates the threshold for identifying critical residues (≤30% of WT, representing ≥2 SD below WT). Critical residues for each folding step are shown as colored circles and noncritical residues as gray circles. Critical residues are plotted on an E1E2 composite structure (described below) using the same colors as in the graph.
(A) Composite structure of the HCV E1E2 heterodimer. A composite structure of the E1E2 heterodimer was constructed. Dark ribbons represent crystal structures (dark blue for E1, red for E2; E1, PDB: 4UOI (Y192-P294) and 2KNU (T314-Q342); red - E2, PDB: 6MEI; R410-G647). Light-colored ribbons represent the predictive model (light blue for E1, pink for E2). Orange spheres represent cysteines, cyan represents glycosylation sequences, and green represents all other critical residues. Structural regions are (1) E1 N-terminal region (NTR) E384-Q409, (2) hypervariable region 1 (HVR1) E384-I411, (3) E1 fusion peptide-like region C272-L286, (4) E1 stem region (G315-A349), (5) E2 stem region (R648-W716), and (6) E2 NTR Y192-D206. (a) CD81 binding loop (BL) T519-D535, (b) β-sandwich R492-A566, and (c) back layer C597-N645.
(B) Critical residues for E1 folding. E1 folding was tested using anti-E1 conformational MAbs HEPC-112 (shown here) and HCVE1-C1 (data in Table S3). Critical residues were defined as clones for which either MAb bound with reactivity ≤30% of WT level. Colored circles just above the threshold line correspond to clones showing >30% reactivity to HEPC112 but ≤30% reactivity to HCVE1-C1.
(C) Critical residues for E2 folding. E2 folding was tested using 32 anti-E2 conformational anti-AR3 MAbs (shown here) or anti-AR1 MAbs (data in Table S3). Residues were considered critical if the corresponding clone showed mean reactivity ≤30% to the 10 AR1-or 23 AR3-type MAbs. Colored circles above the threshold correspond to clones with >30% reactivity to AR3-type MAbs but ≤30% reactivity to AR1-type MAbs.
(D) Critical residues for E1E2 heterodimer assembly. E1E2 assembly was tested using 5 E1E2 heterodimer-specific MAbs. Gray circles below the 30% threshold correspond to clones showing mean reactivity <70% WT levels of reactivity in previous functional steps.
(E) Critical residues for binding of the CD81-LEL peptide. The E1E2 mutation library was tested for binding to CD81-LEL. Gray circles below the 30% threshold correspond to clones showing mean reactivity <70% WT levels of reactivity in one or more previous functional steps.
We identified 39 E1 residues that were critical for E1 folding (Figure 2B): four lie in the E1 transmembrane (TM) domain, including D382 immediately adjacent to the E1E2 cleavage site (383–384), and one (D279) in the proposed E1 IFL (C272-L286). Other E1 critical residues include six cysteine residues (C207, C226, C229, C238, C304, and C306), four of which contribute to disulfide bonds in a crystal structure of the E1 N terminus (C207-C226, and C229–C238) (El Omari et al., 2014). The only E1 cysteines not critical for E1 folding were the two cysteines (C272 and C281) in the proposed IFL. No residues within E2 were identified as critical for E1 folding.
Amino acids critical for E2 folding
To identify residues critical for E2 folding, we analyzed the E1E2 mutation results for clones that were incapable of binding to anti-E2 conformational MAbs. We defined a critical residue for E2 folding as one whose mutation resulted in an inability to bind either 11 AR1- or 18 AR3-type MAbs (excluding the MAbs’ epitope residues). The threshold for a critical residue for E2 folding was a mean reactivity ≤30% to either MAb type. AR2 was excluded for this purpose because we screened only a single MAb, whose binding data suggest that AR2 MAbs are not affected by mutations other than those we identified for AR1 and AR3 MAbs. MAb conformational dependence was confirmed by epitope mapping (Gopal et al., 2017) and by reducing or non-reducing SDS-PAGE/western-blot analysis (Figure S2).
We identified 56 E2 residues as critical for E2 folding (Figure 2C). These segregated into four distinct regions on the E2 structure: C494-V516 and F537-G565 located in the β-sandwich region, C597-C644 in the back layer, T425-C429 in the front layer, and T518-D535 in the CD81 binding loop. Nine cysteines were identified as critical, including the constituents of 4 disulfide bonds (C429-C503, C494-C564, C508-C552, and C607-C644).No E1 residues were identified as critical for E2 folding.
Amino acids critical for E1E2 heterodimerization
Without an available structure of the E1E2 heterodimer, we have had a poor understanding of which amino acids mediate the E1E2 heterodimer assembly and function. To identify residues critical for E1E2 assembly, we analyzed the E1E2 mutation results for clones that were incapable of binding 5 MAbs that bind only when E1 and E2 form a complete heterodimer (AR4A, AR5A, S1, AB, and HEPC111) (Colbert et al., 2019; Giang et al., 2012). The threshold for designating a critical residue for E1E2 heterodimerization was a mean reactivity ≤30% to the 5 E1-E2 MAbs but ≥70% of mean WT folding levels of E1 and E2.
We identified 21 residues as critical for E1E2 assembly (Figure 2D). Four E1 residues were critical (Y201, T204, N205, and D206), located in the E1 N-terminal region and predicted to lie near the E1E2 membrane. Eight critical residues were located in the E2 stem region (C652, R657, D658, C677, F679, L692, D698, and Q700, including the C652-C677 disulfide bond) and form the binding sites for neutralizing E1E2 MAbs (Colbert et al., 2019; Keck et al., 2019; Velazquez-Moctezuma et al., 2017). These E1 and E2 residues are consistent with a recently posted description of a cryoelectron microscopy (cryo-EM) structure of a membrane-extracted full-length E1E2 heterodimer, with an E1-E2 interface that included interactions between E1 residues Y192-N205 and E2 residues E655-I690 (Torrents de la Peña et al., 2021). Four critical residues lie on the E2 core structure in different regions of the β-sandwich: R543, P544, P545, W549, and W487. Residues R543, P544, P545, and W549 lie in a loop and contribute to the epitopes of several AR1-type MAbs (Flyak et al., 2018; Gopal et al., 2017). Other critical residues include cysteine pairs that form disulfide bonds in the back layer of E2 (C459-C486 and C581-C585). Strikingly, all assembly critical residues were either fully conserved (19 residues) or highly conserved (D658 and F679) across reference sequences of all HCV genotypes (Figure S3). This highlights the importance of these residues for E1E2 assembly and suggests druggable points of vulnerability in the E1E2 structure.
Amino acids critical for host-cell receptor CD81 binding
We next identified residues critical for binding to the CD81 coreceptor by reanalyzing a published dataset for our E1E2 mutation library binding to the CD81 long extracellular loop (LEL) region (fused to the Fc region of immunoglobulin G [IgG]) (Gopal et al., 2017). Clones were defined as critical for CD81 binding if they showed ≤30% reactivity with CD81 but ≥70% of mean WT folding levels of E1, E2, and the E1E2 heterodimer. We excluded binding data for AR3-targeting MAbs from this analysis as these MAbs bind to the same region as CD81.
We identified 19 residues as critical for CD81 binding (Figure 2E), most of them in one of two distinct E2 regions: the front layer, residues 420–453, and the CD81 binding loop, residues 519–535 (designated “a” in Figure 2A). These locations are consistent with previous localization of the CD81-binding site from mutagenesis, electron microscopy, and X-ray crystallography studies (Gopal et al., 2017; Kong et al., 2013; Kumar et al., 2021; Owsianka et al., 2006).
Identification of amino acids critical for infectivity
To determine the effect of each mutation on infectivity, we generated HCV pseudoviral particles (HCVpps) from each E1E2 library mutant. We verified that each mutant E1E2 protein incorporated into pseudoparticles (Figure S1B). Nearly all mutants were incorporated into HCVpps at sufficient levels for structure-function analysis; only 10 clones demonstrated HCVpp incorporation levels lower than 30% of the WT E1E2 level, with average incorporation across the library at 88% of the WT level.
The mutation library HCVpps were tested for infectivity (Figure 3A), and we defined critical residues for infectivity as those whose mutation gave infectivity ≤15% of WT infectivity (>2 SD below WT) but had high levels (≥70% of WT) of mean reactivity for E1 and E2 folding, E1E2 assembly, CD81-LEL binding, and sufficient (≥30% of WT) incorporation into HCVpps. Such clones have no apparent structural defect yet are not capable of mediating infection. Other residues are also likely involved in infectivity but were not identified because they did not fold correctly when mutated. We identified 97 critical residues for infectivity, 32 in E1 and 65 in E2 (Figure 3B), with 35% (34/97) completely conserved across reference sequences from all HCV genotypes (Figure S3). This supports their importance for HCV infection and again suggests druggable points of vulnerability in the E1E2 structure.
Figure 3. Identification of E1E2 amino acids critical for infectivity.
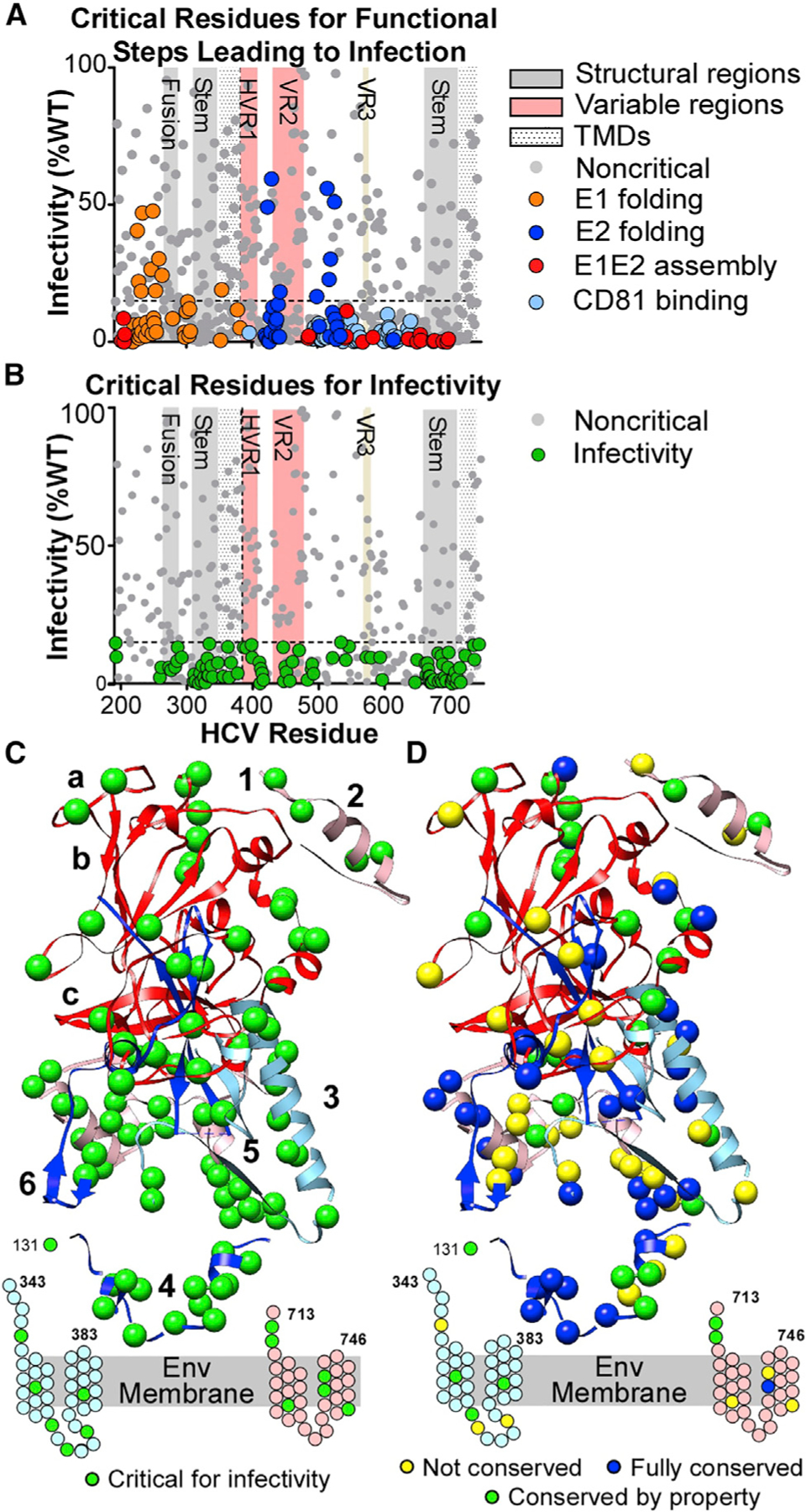
(A) Infectivity values (mean of 5 experiments) for E1E2 clones with mutations critical for E1 or E2 folding, E1E2 heterodimer assembly, and CD81 binding.
(B) Infectivity values (mean of 5 experiments) for clones not critical for functional steps (i.e., excluding clones colored in A). Green circles are critical residues for infectivity (clones with mean infectivity ≤15% and with ≥70% for E1E2 folding, E1E2 heterodimer assembly, and CD81-LEL binding). Gray circles below the threshold line represent clones with mean reactivity <70% of WT in previous functional steps. Important structural regions are marked, including HVR1, VR2, and VR3.
(C) Critical residues for infectivity are shown as green spheres on the E1E2 composite structure. Residues in the stem and TM regions of E1 and E2 that lack structures are shown as cartoons. Annotations are as in Figure 1A.
(D) Structural representation of E1E2 critical residues for infectivity, characterized by their conservation across the 7 HCV genotypes. Residues fully conserved across all 7 genotypes are shown as dark blue spheres. Residues with conserved properties are shown as green spheres. Non-conserved residues are shown as yellow spheres.
The critical residues for infectivity localized primarily in the E1 and E2 TM and stem regions and near the E1E2 membrane (Figures 3C and 3D). The TM regions, containing 9 critical residues, are important for HCV heterodimerization and membrane fusion, as for other viral Env proteins (Ciczora et al., 2007; Falson et al., 2015). The stem regions contained 45% (44/97) of the critical residues, 14 in E1 and 30 in E2. In many viral Env proteins, the stem regions act in concert with the Env fusion loop to drive virus-host membrane fusion (Hsieh et al., 2011; Lin et al., 2011). We identified 5 infectivity-critical residues (S273, Y276, G282, F285, and L286) in E1 residues 272–286 (region “3”), a region proposed to contain the IFL that drives membrane fusion (Drummer et al., 2007; Lavillette et al., 2007). Infectivity-critical residues Y192 and Q193 on E1 are at the site of cleavage of E1 from the polyprotein. Five critical residues for infectivity were found in HVR1 (residues 384–410), which is thought to be important for HCV interaction with SR-BI and CD81 to facilitate HCV attachment (Bartosch et al., 2003).
A model for the role of individual amino acids in HCV E1E2 folding and function
Our comprehensive analysis of HCV E1E2 allows us to propose a functional model that takes into account the current understanding of the HCV entry pathway (Gerold et al., 2020) and proposes a mechanism by which individual amino acids contribute to E1E2 folding and function. Specifically, our data suggest that E1 folding involves a set of 39 critical residues, E2 folding involves a set of 56 residues, and E1E2 assembly involves a set of 21 residues (Figure 4A). Folding-critical residues include 6 cysteines for E1 folding, 9 cysteines for E2 folding, and 6 E1 cysteines for E1E2 heterodimerization. Once folded, infectivity is initiated by the interaction of 3 E2 N-terminal residues (G390, G398, and L399) with receptors SR-BI, LDLR, or VLDLR (Gerold et al., 2020) to promote virus uptake (Figure 4B, stage 1). Since HVR1 partially covers the E2 CD81-binding region, it is proposed that interaction with these receptors induces a conformational change in E2, exposing the CD81-binding site (Gerold et al., 2020) (Bankwitz et al., 2010). Next, 19 E2 residues interact with CD81 on the target cell surface (stage 2). In the late endosome, protonation of E2-H691, E2-H693, and E1-H261 drives remodeling of the E1 structure (the role of histidines is discussed below). This structural rearrangement releases the presumed E1 IFL (residues 272–286) to form an extended fusion structure, likely a trimer of E1E2 (stage 3). Five critical residues in the fusion loop drive its insertion into the endosomal membrane. The extended E1 protein then begins to fold in on itself to form a “hairpin” structure between residues E318 and M347 (stage 4). Next, 14 residues in the E1 stem region pack along the sides of the fusion loop trimer to form a trimer hairpin that pulls the virus and host membranes together (stage 5). The fusion of the two membranes is stabilized by 9 E1E2 residues interacting in the TMs and 6 critical IFL residues that lie behind the hydrophobic IFL tip, as well as 8 HR critical-residue interactions in the internal trimer core. This functional model reconciles our data here with the structural information currently solved for E1 and E2 as well as other functional studies over the past two decades that have studied isolated mutations of E1 and E2.
Figure 4. Model for the roles of individual amino acids in E1E2 function.
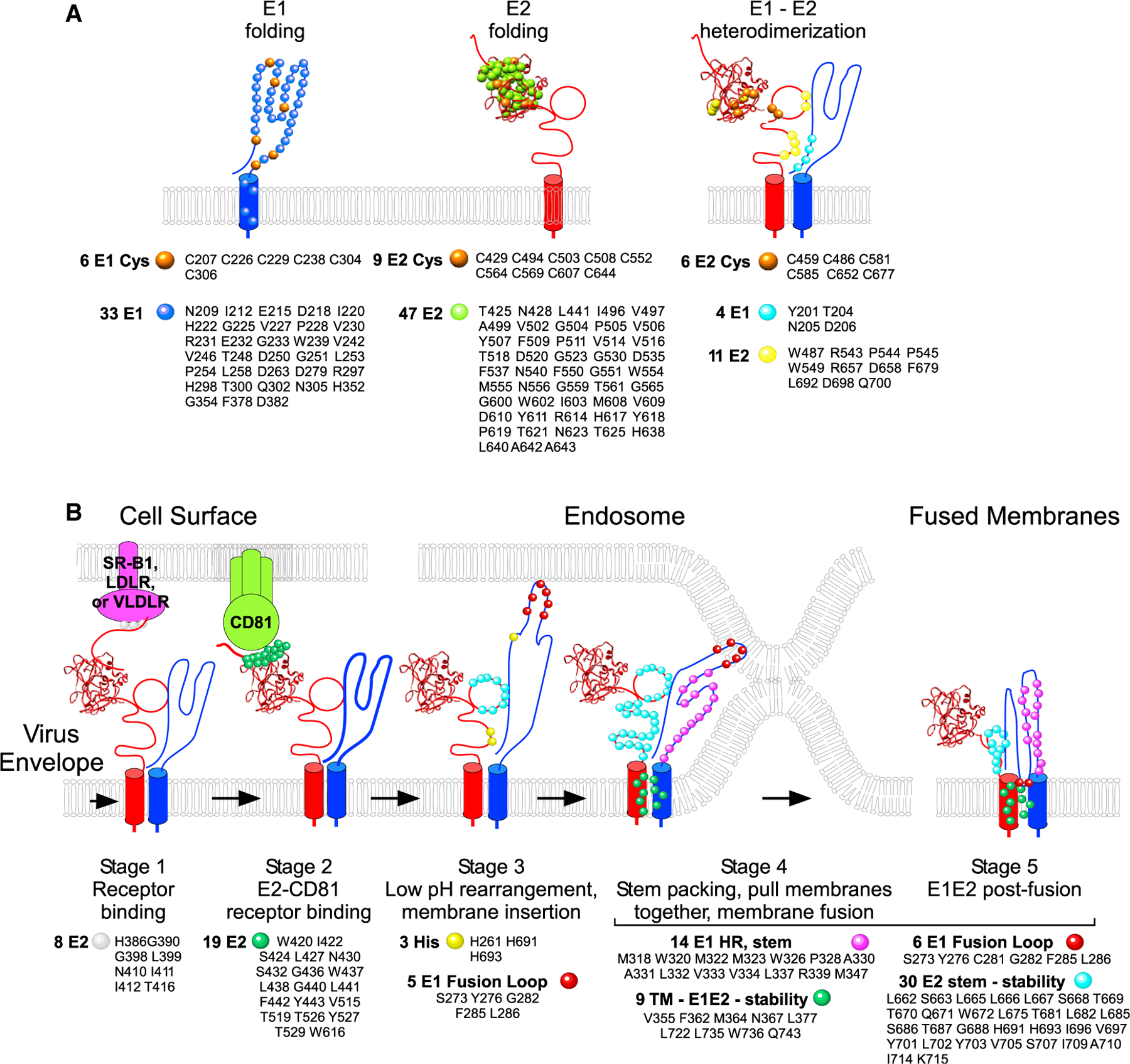
Proposed functional model describing the mechanism by which individual amino acids contribute to E1E2 function.
(A) Critical residues for E1 and E2 folding and heterodimerization are shown on simplified representations of the protein structure.
(B) Critical residues for HCV infectivity are assigned to their likely stage in the HCV entry pathway, based on available information of the pathway.
Correlation of critical residues with NAb epitopes and mechanisms of action
NAbs inhibit viral infectivity, but for many, their mechanisms of neutralization are unknown due to the limited understanding of E1E2 structure and function. To ascertain if NAbs act by binding to functionally important regions of the E1E2 protein, we compared the locations and conservation of epitope residues for the NAbs (including 18 broadly NAbs [bNAbs]) and non-NAbs used in our analyses, along with other anti-HCV MAbs (Table S1). The locations and properties of the MAb epitopes are summarized in Figure 5.
Figure 5. Epitope locations for bNAbs, NAbs, and non-neutralizing MAbs (non-NAbs).
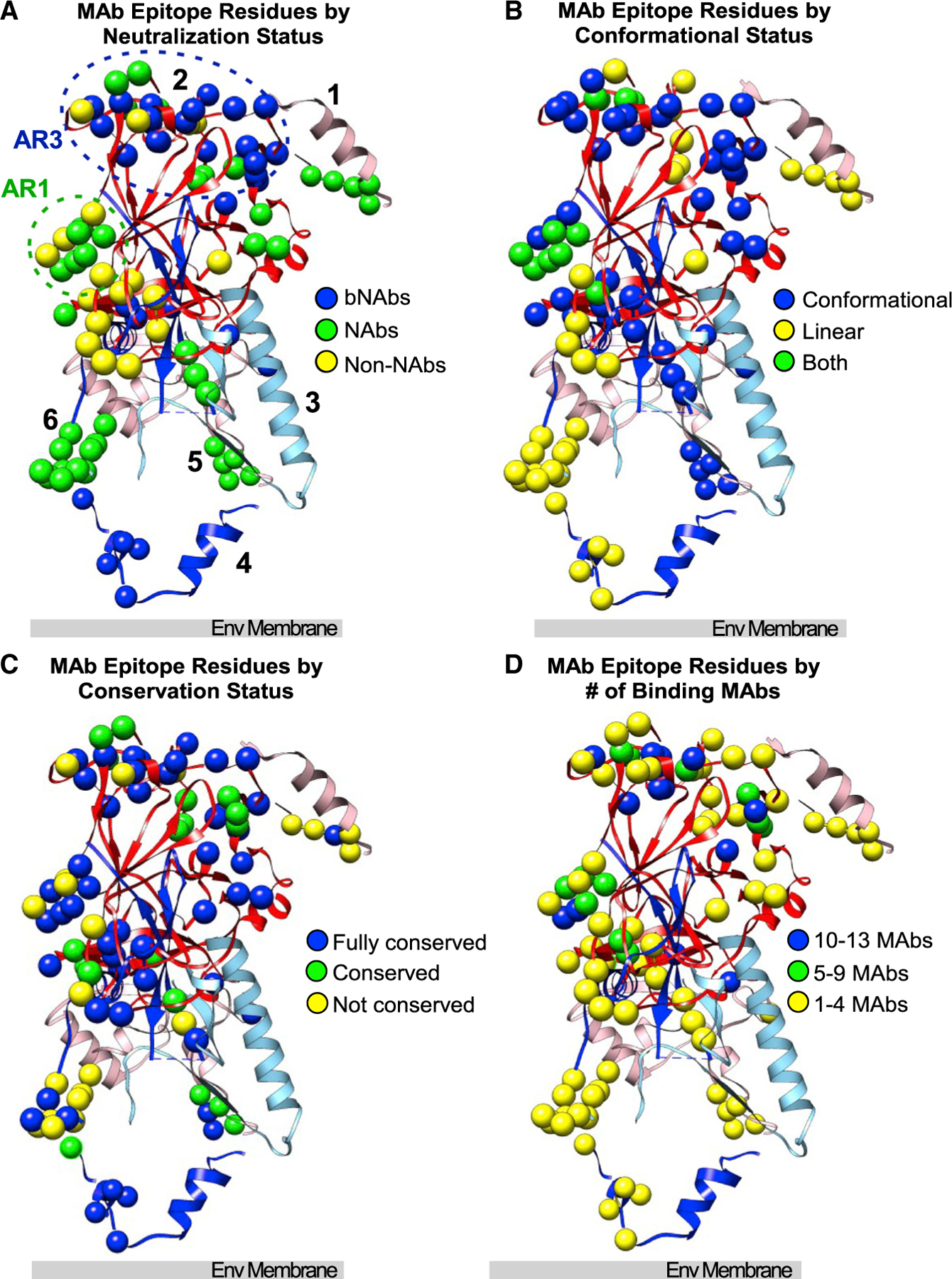
(A–D) MAb epitope residues are plotted on the composite E1E2 structure, categorized by (A) MAb neutralization status, (B) residues that contribute to MAb epitopes that are conformational, linear, or both, (C) conservation of the epitope residue, or (D) number of MAbs binding to that residue. Numbers and orientation correspond to Figure 1A.
Overlap between functionally critical residues and NAb epitopes helps explain NAb mechanism of action (Figure 6). AR3-type NAb epitopes overlap with CD81-binding-site residues, as well as infectivity-critical residue T534, consistent with a NAb mechanism of action that blocks E2 interaction with CD81. Two infectivity-critical residues (N410 and I411) in E2 HVR-1- and −2-adjacent residues in the conserved N-terminal region (cNTR) (I414, T416) are bNAb epitopes (for H77.16 and HEPC98 [Sabo et al., 2011]), which likely function by preventing E2 interactions with other receptors. Four E2 residues critical for E1E2 assembly (R543, P544, P545, and W549) overlap with epitopes of anti-E2 NAbs binding in the AR1/β-sandwich region (HEPC108, HEPC158, HEPC151–2, and H77.28 [Colbert et al., 2019; Sabo et al., 2011]), suggesting their mechanism as inhibiting the interaction of or remodeling between E1 and E2. The NAb epitopes defined to date do not overlap with any residues critical for E1 or E2 folding, consistent with most folding-critical residues not being accessible on the protein surface.
Figure 6. Co-localization of NAb epitopes with residues critical for E1E2 folding or function.
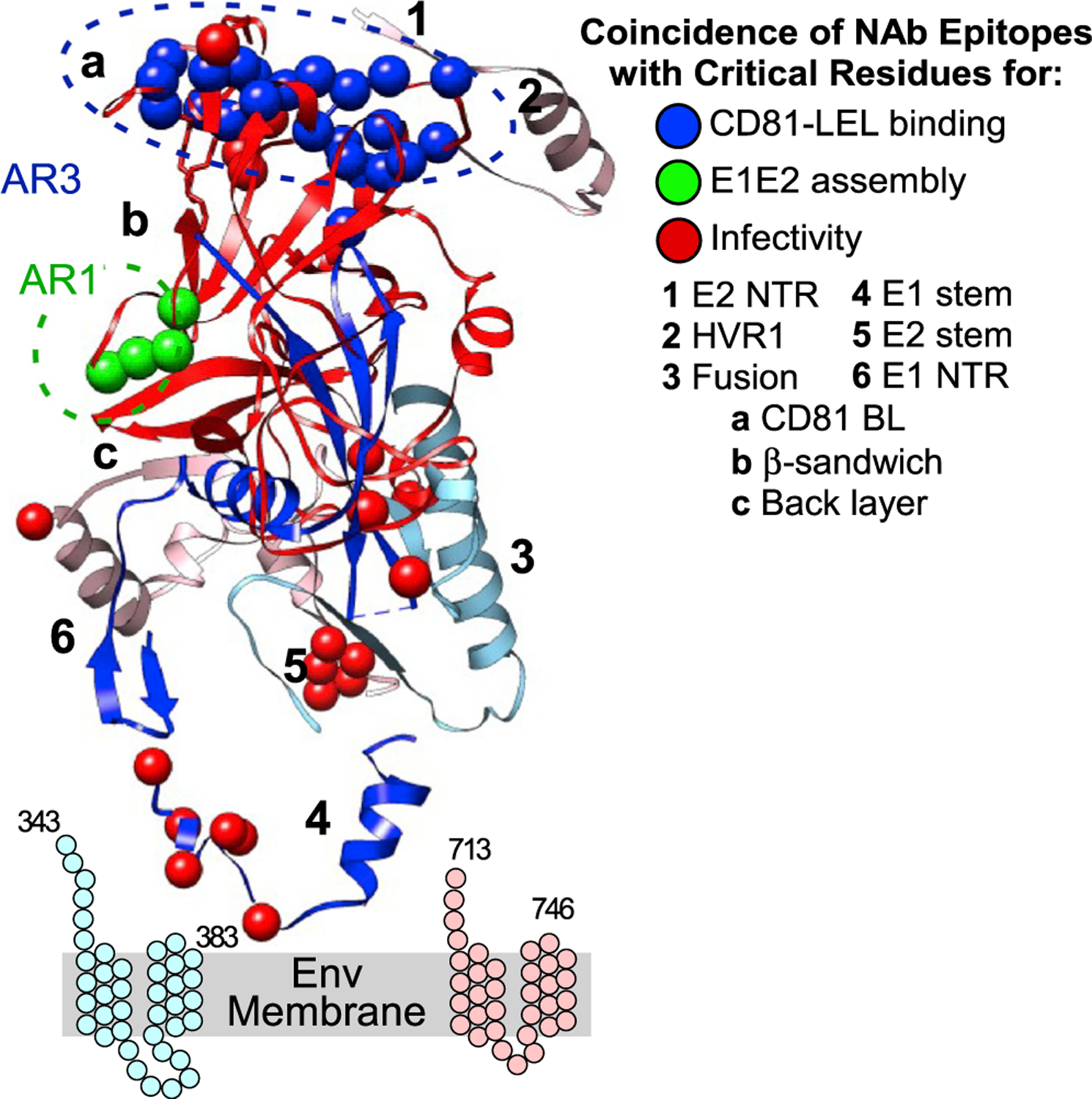
NAb epitope residues that are also critical for functional steps are shown. The stem and TM regions of E1 and E2 are shown as cartoons.
The NAb panel also included conformational anti-E1E2 NAbs (AR4A, AR5A, S1, and HEPC111) that bind only when E2 is complexed with E1 (Giang et al., 2012) and which appear to bind only to E2 (at AR4 and AR5 in the E2 C-terminal region, which includes the stem). AR4A and HEPC111 bind the E2 stem, while AR5A binds an epitope distinct from AR5A in the back layer and the stem (Colbert et al., 2019; Keck et al., 2019; Giang et al., 2012; Velazquez-Moctezuma et al., 2017). The epitopes of these NAbs overlap with infectivity-critical residues in the E2 stem region (L665, L666, and W672). This finding is consistent with these NAbs impeding the function of the stem region during the late stages of membrane fusion.
Several anti-E1 NAb epitope residues overlap with E1 infectivity-critical residues, including E1 stem-region epitope residues I313-P328 (region “4”). This suggests that the anti-E1 NAbs IGH505 and IGH526 block infectivity by perturbing a crucial function of the E1 stem, likely during hairpin formation, which is consistent with their known mechanism of acting at a post-binding stage (Haberstroh et al., 2008; Meunier et al., 2008; Wahid and Dubuisson, 2013).
Of the identified 97 critical infectivity residues, 78 do not colocalize directly on NAb epitopes. Most of these 78 residues are hydrophobic (44/80, 56%) and poorly exposed on the available E1 or E2 structures or lie in the E1 or E2 stem and TM regions. These residues are points of unexploited vulnerability and potential targets for drugs, as their predicted interior locations suggest that they are hidden from humoral immunity.
DISCUSSION
To identify specific amino acids that define the mechanism of HCV E1E2 folding and function, we individually mutated each E1E2 residue and tested for their binding to 37 different MAbs or CD81, as well as their ability to infect cells. This allowed us to determine the contribution of each amino acid to distinct steps of E1E2 folding and infectivity involving Env expression, E1 and E2 folding, E1E2 heterodimer assembly, virion incorporation, CD81 receptor binding, and viral fusion. Many of the identified functional residues of E1E2 are highly conserved across genotypes, making them ideal targets for therapeutic or vaccine targeting. Indeed, co-location of NAbs with these functional residues explains many of their mechanisms of action.
Structure-function features of E1E2
Several lines of evidence point to E1 being responsible for membrane fusion, including a conserved IFL-like domain on E1 (residues 272 to 286) (Drummer et al., 2007; Lavillette et al., 2007; Li et al., 2009; Tong et al., 2017). Consistent with this, the E1 infectivity-critical residues we identified fell primarily in three locations: the putative fusion peptide (272–286), the E1 stem region (315–349), or the E1 TM (350–383), all regions that would be considered crucial for the functioning of a class II fusion protein (Chi et al., 2016; Douam et al., 2018; Ma et al., 2002). We identified 5 residues critical for infectivity (S273, Y276, G282, F285, and L286), all highly or completely conserved among HCV genotypes, and some previously identified as important for infectivity, consistent with this region functioning as a fusion loop (Drummer et al., 2007; Lavillette et al., 2007). A number of residues critical for infectivity within E1 span residues 267 to 312 that include the IFL (264–286) and that can suppress the activity of cationic amphiphilic drugs (CADs) (Banda et al., 2019; Perin et al., 2016). CADs are thought to act by perturbing membrane lipids (Gunesch et al., 2020), suggesting that mutations which suppress the effect of CADs do so by altering protein interactions with the membrane, consistent with the involvement of E1 residues in membrane fusion.
The majority of identified infectivity-critical residues (65/97) lie in E2, with 52% (34/65) in the E2 stem or TM regions, highlighting their importance in E2 functionality. The role of the E2 stem and the TM in fusion is consistent with previous findings that the E2 back layer/stem region is important for virus-host fusion. It has been proposed that HCV E1 represents a novel class of virus membrane fusion protein machinery (Li and Modis, 2014), which would be further distinguished from known flavivirus fusion mechanisms by the contribution of HCV E2 to this process.
A number of cellular proteins naturally interact with E1E2 as receptors or attachment factors. While we identified residues that interact with CD81, it is likely that other cellular proteins interact with additional residues in E1E2. We identified 3 HVR1 residues (G390, G398, and L399) as critical for infectivity, potentially acting through interactions with receptors other than CD81, as seen for HVR1 with SR-BI (Bankwitz et al., 2014). The HVR1 residues at positions 390 and 398 have been suggested to play a role in E2 binding to receptors or attachment factors on the cell surface due to their location in a conformationally flexible, surface-exposed subregion that tends to be basic and hydrophilic (Penin et al., 2001). HVR1 residue 399 tends to be hydrophobic, and so its role may be to maintain HVR1 conformation and anchoring to the E2 core.
Previous analyses by us and others identified residues potentially involved in E1E2 heterodimerization (Cao et al., 2019; Ciczora et al., 2005; Cocquerel et al., 2000; Douam et al., 2014; Drummer and Poumbourios, 2004; Falson et al., 2015; Gopal et al., 2017; Jusoh et al., 2010; McCaffrey et al., 2011; Moustafa et al., 2018). Our analysis here refines these previous results and adds additional MAbs, infectivity testing, and includes conformational anti-E1 MAbs that differentiate residues critical for E1 folding. As would be expected, misfolded E1 or E2 proteins demonstrated deficits in assembly; mutations at 68% (38/56) of E2 folding-critical residues were deficient in E1E2 assembly. In contrast, only 28% (11/39) of mutations at E1 folding-critical residues were deficient in assembly, suggesting that although largely requiring a properly folded E2 protein, heterodimerization may tolerate some local misfolds of E1. Related to this, 23% (9/39) of residues that eliminated E1 folding demonstrated infectivity values above the 15% cutoff, suggesting that complete protein folding of at least some parts of E1 may not be essential for viral infectivity. This conclusion is limited, however, by the current existence of only two conformation-dependent E1 MAbs, one first described here that provides more insight into E1 than previously possible. Of the 17 E2 assembly critical residues, 8 lie in the E2 C-terminal region (C652, R657, D658, C677, F679, L692, D698, and Q700), including stem (672–715), supporting previous evidence that a C652-C677 disulfide-bond-stabilized structure is important for E1E2 assembly (McCaffrey et al., 2012).
Despite the predicted involvement of TM regions in E1E2 assembly (Ciczora et al., 2005, 2007), no TM residues were identified as critical for E1E2 assembly. Of previously identified residues, only D278A showed a major decrease in MAb reactivity (average 39% binding by E1E2 MAbs), consistent with a role in E1E2 assembly. Another previous model predicted seven E1E2 residue pairs interacting to heterodimerize E1E2 (Cao et al., 2019). In our study none of these mutations were critical for E1E2 interaction, as determined by binding with E1E2 dimerdependent MAbs. Only one residue (L654A) showed a major reduction, with an average binding by E1E2 MAbs of 40% of binding to WT. Another study implicated three E1 residues involved in interactions with E2 (Douam et al., 2014). Here, none of these residues were identified as critical for interaction with E2, but I308 was critical for infectivity.
Role of specific residue types in functional steps
Our data provide insights into the role of different residue types that mediate the structure and function of HCV E1E2. Nearly all cysteines were critical for E1 or E2 folding or E1E2 heterodimer formation. The only exception was E1 C281, which is predicted to form a disulfide bond in the proposed fusion loop and whose mutation greatly decreased infectivity. It is likely that disulfide partner C272 would also be important for infectivity, but C272 was one of the few residues not included in our mutation library.
Mutations at glycosylation sites generally had a deleterious effect, with infectivity below 15% of WT for mutations at 11/15 predicted glycosylated Asn residues. However, there were no consensus sequences for which both positions in the glycosylation motif (N-X-S/T) were critical residues for infectivity using our metrics. Rather, mutation of individual glycosylation motif residues (but not necessarily both) affected folding steps prior to infectivity. For example, N209 and N305 were critical for E1 folding, while T425, N540, N556, N623, and T625 were critical for E2 folding. In other cases, the residues were not critical for folding steps but cumulatively resulted in reductions in folding that were below the 70% threshold needed to validate a residue as critical to infectivity. For example, both N645/T647 mutations eliminated infectivity but also decreased E2 folding and CD81 binding sufficiently so that this site was not identified as critical for E1E2 heterodimerization. Mutation of the N430/S432 glycosylation consensus sequence reduced CD81 binding to HCV, similar to a previous finding (Alhammad et al., 2015). Except for this N430 glycosylation, our findings do not suggest a direct functional role for glycosylation in E1E2 infectivity.
In this study we used HEK-293T cells, which may result in different glycosylation patterns compared with that obtained in hepatoma-derived cells. For example, characterization of E1E2 glycosylation in Huh7.5-derived virions suggests that E1E2 is predominantly modified by high-mannose N-linked glycans (Guo et al., 2018), whereas a soluble form of E2 generated in HEK-293T cells contained a more heterogeneous mixture of both complex and high-mannose glycan types (Urbanowicz et al., 2019a, 2019b). However, mutation of glycosylation sites did not impact E1E2 function (with the exception of N430), so we do not expect the use of HEK-293T cells to impact the main conclusions of our study.
For many viral Env proteins, a positive charge resulting from low-pH protonation of histidines induces conformational changes that initiate membrane fusion with the host cell (Mair et al., 2014; Sánchez-San Martín et al., 2009). Since HCV fusion depends on low pH in the endosome, histidine residues are likely important in this process (Boo et al., 2012; Hsu et al., 2003; Tscherne et al., 2006). Individual mutation of 10 of the 15 histidine residues in HCV E1E2 resulted in complete loss of infectivity (≤15% of WT infectivity). Three of these histidines (H222, H298, and H352) lie in E1 and were critical for E1 folding, and two E2 histidines (H617 and H638) were critical for E2 folding, consistent with an assembly defect when mutated (Boo et al., 2012), and four histidines (H261, H386, H691, and H693) were critical for infectivity. One absolutely conserved E2 histidine (H488) was not classified as critical for infectivity because its mean reactivity to E1E2 assembly sensitive MAbs (67% of WT) was just below our cutoff threshold (≥70% of WT). H261, conserved in all but genotype 5a, is located on E1 close to the predicted fusion loop. H691 and H693 are in the E2 stem and are absolutely conserved and important for infectivity (Boo et al., 2012). These results are consistent with histidine residues playing a significant role in pH-induced E1E2 conformational changes that contribute to both the folding and function of E1E2.
HCV E1E2 is a remarkably fragile Env protein
HCV E1E2 exhibits considerable sequence heterogeneity among genotypes and isolates, exemplified by E2 containing three VRs. HCV accumulates mutations during replication, and the virus exists as a population of closely related quasispecies (Martell et al., 1992). This high mutation frequency results from the HCV RNA-dependent RNA polymerase lacking proofreading ability, exhibiting high error rates of approximately 10−3 per site for G:U/U:G mismatches (Powdrill et al., 2011). For a virus that mutates so readily, it would be reasonable to expect that its Env protein might be relatively accommodating to mutations. However, our results suggest that HCV E1E2 is a remarkably fragile Env protein where even a single alanine substitution at 92% of its residues can have severe consequences on virus functionality. Of the 545 individual E1E2 mutations tested, 311 (57%) resulted in completely non-functional Env protein (<15% WT level infectivity), while 190 mutations (35%) supported moderate infectivity (15%–70%). Only 44 of 545 mutations (8%) supported full infectivity (≥70% of WT level infectivity; Figure 7). The corresponding residues (19 in E1 and 25 in E2) include 12 residues in HVRs and 2 residues at cleavage sites. Despite its high fragility, HCV exhibits remarkable natural diversity, suggesting that the virus’s high replication rate can compensate for this fragility to maintain infectivity in the face of detrimental variability. Our results are also consistent with others’ studies where 40% of random mutations within vesicular stomatitis virus (VSV) result in defective viruses (Sanjuán et al., 2004).
Figure 7. HCV E1E2 is remarkably fragile.
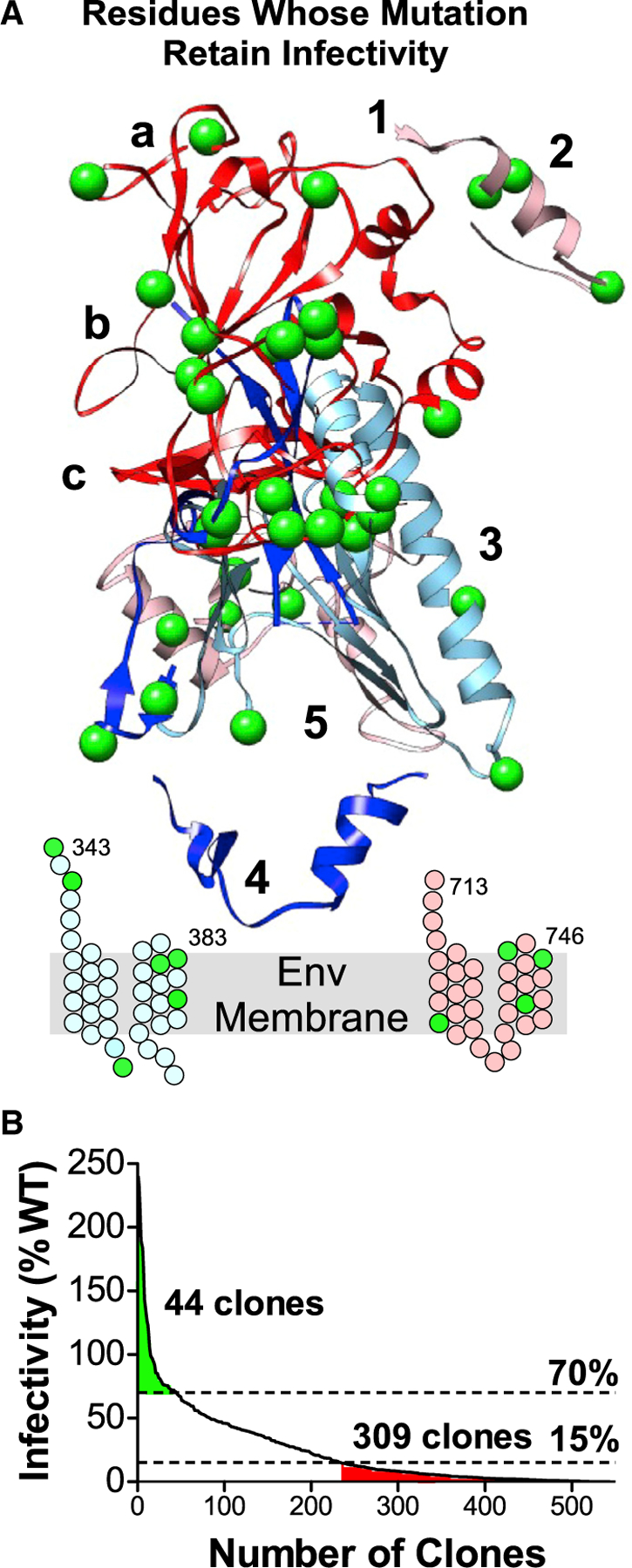
(A) HCV E1E2 residues whose mutation to alanine had little effect on infectivity (>70% of WT levels) are shown as green spheres. Residues in the stem and TM regions of E1 and E2 are shown as cartoons.
(B) E1E2 mutation library clones arranged in order of decreasing infectivity. Mutations in 309/545 clones (57%) eliminated infectivity (≤15% of WT level infectivity). Only 44 clones (8%) retained infectivity after mutation (>70% of WT levels).
Our conclusions, naturally, can only be applied to the alanine mutations that we studied here; alanine was chosen as the substitute amino acid for our studies due to its minimal side chain, which informs the function of the side chain at each residue position. Serine, threonine, and alanine (which was mutated to serine) comprised nearly 60% (26/44) of E1E2 residues that could be mutated without functional consequence. These residues are located in hydrophobic stretches of the sequence, so their mutation to alanine would not affect protein hydrophobicity. Nevertheless, the choice of alanine does not alone explain the remarkable fragility of HCV Env. In similar studies with other viral Env proteins (dengue virus [DENV], HIV-1, chikungunya virus [CHIKV], Zika virus [ZIKV], Ebola virus [EBOV], severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]) (Christian et al., 2013; Sullivan et al., 2017; J.M.P.-K., E.D., and B.D., unpublished data), HCV E1E2 remains the most fragile of all viral Env proteins that we have studied to date. Similar studies of non-viral cellular proteins (G protein-coupled receptors [GPCRs], ion channels, and transporters) also have been highly accommodating of alanine changes (Greene et al., 2011; Thomas et al., 2017; Tucker et al., 2018; Wescott et al., 2016), suggesting that HCV Env is remarkably fragile.
Conclusion
Our model of HCV E1E2 structure and function proposes how specific residues contribute to E1E2 folding and infectivity, complementing our current understanding of E1E2. This model also helps explain the mechanisms by which NAbs inhibit the virus. The functional identification of residues that directly contribute to E1E2 folding may be useful for engineering more stable variants of E1E2 for vaccines or structural studies. Targeting functionally important and conserved sites on E1E2, either in its native structure or using E1E2 variants engineered to preferentially expose these sites, may result in improved vaccine candidates capable of broad protection. Our results also suggest that E1E2 is a remarkably fragile Env protein that does not easily maintain function after single amino-acid changes, and we determined which amino-acid side chains can be deleted with or without functional consequence.
Limitations of the study
A limitation of our study has been the absence of a complete structure for E1, E2, and the E1E2 complex. This led us to analyze our extensive antibody-binding data and to propose residues important for the folding and interaction of the two proteins. A preprint describing a full E1E2 cryo-EM structure (Torrents de la Peña et al., 2021) indicates that a structure will soon be available, allowing insights from comparison with our data.
Our expressed E1E2 protein, in epitope mapping and infectivity studies, included a V5 tag at the E2 C terminus, with the potential for structural perturbations. However, since this tag lies on the opposite side of the membrane from the MAb-targeted E1E2 ectodomains, it is unlikely to affect MAb binding to most regions of E1 and E2, supported by our analysis of numerous MAbs demonstrating strong binding to cellularly expressed E1 and E2. We have also observed robust binding by MAbs whose binding is specific to E1E2 in complex, and the results enabled us to propose residues important for E1-E2 assembly.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Benjamin Doranz (bdoranz@integralmolecular.com).
Materials availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and code availability
The data used for the analyses described here, antibody and CD81-LEL binding and HCVpp infectivity, for each mutation in the HCV E1E2 library, are included in Table S3 and have been deposited in the Mendeley Data repository. DOI is listed in the Key resources table.
This paper also analyzes existing, publicly available protein structural data. The accession numbers for these datasets are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| AP33 | Owsianka et al., 2005 | N/A |
| AR2A | Law et al., 2008 | N/A |
| AR3A | Law et al., 2008 | N/A |
| AR3B | Law et al., 2008 | N/A |
| AR3C | Law et al., 2008 | N/A |
| AR3D | Law et al., 2008 | N/A |
| AR4A | Giang et al., 2012 | N/A |
| AR5A | Giang et al., 2012 | N/A |
| e20 | Mancini et al., 2009 | N/A |
| e137 | Perotti et al., 2008 | N/A |
| H77.39 | Sabo et al., 2011 | N/A |
| HCV1 | Broering et al., 2009 | N/A |
| HEPC3 | Bailey et al., 2017 | N/A |
| HEPC13 | Bailey et al., 2017 | N/A |
| HEPC43 | Bailey et al., 2017 | N/A |
| HEPC74 | Bailey et al., 2017 | N/A |
| IGH505 | Meunier et al., 2008 | N/A |
| IGH526 | Meunier et al., 2008 | N/A |
| e301 | Burioni et al., 2002 | N/A |
| e509 | Sautto et al., 2012 | N/A |
| H77.16 | Sabo et al., 2011 | N/A |
| H77.28 | Sabo et al., 2011 | N/A |
| HEPC98 | Bailey et al., 2017 | N/A |
| HEPC108 | Colbert et al., 2019 | N/A |
| HEPC111 | Colbert et al., 2019 | N/A |
| HEPC112 | Colbert et al., 2019 | N/A |
| HEPC122 | Colbert et al., 2019 | N/A |
| HEPC146 | Colbert et al., 2019 | N/A |
| HEPC 151–1 | Colbert et al., 2019 | N/A |
| HEPC 151–2 | Colbert et al., 2019 | N/A |
| HEPC153 | Colbert et al., 2019 | N/A |
| HEPC154 | Colbert et al., 2019 | N/A |
| HEPC158 | Colbert et al., 2019 | N/A |
| AR1A | Law et al., 2008 | N/A |
| AR1B | Law et al., 2008 | N/A |
| e8 | Castelli et al., 2014 | N/A |
| e10 | Bugli et al., 2001 | N/A |
| H60 | Castelli et al., 2014 | N/A |
| H77.47 | Sabo et al., 2011 | N/A |
| HEPC50 | Bailey et al., 2017 | N/A |
| HEPC46 | Bailey et al., 2017 | N/A |
| S1 | Giang et al., 2012 | N/A |
| AB | Giang et al., 2012 | N/A |
| HCVE1-C1 | this manuscript | N/A |
| CD81-LEL fused to immunoglobulin Fc fragment | Gopal et al., 2017 | N/A |
| HRP conjugated secondary Ab | Southern Biotech | Cat# 040–05 |
| Anti CD81 antibody JS-81 | BD Biosciences | Cat# 555676 |
| Alexa Fluor 488-conjugated goat anti-human secondary antibody | Jackson ImmunoResearch Laboratories | Cat# 109–545-003 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E.coli NEB 5-alpha | New England Biolabs | Cat# C987H |
|
| ||
| Critical commercial assays | ||
|
| ||
| Intellicyt high throughput flow cytometer | Sartorius | iQue3 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific | Cat# 34580 |
| Renilla-Glo Luciferase assay | Promega | Cat# E2820 |
| Pierce Femto kit | Pierce | Cat# 34095 |
| Imaging System | Bio-Rad | ChemiDoc |
|
| ||
| Deposited data | ||
|
| ||
| HCV partial E2 structure 6MEI | Flyaketal., 2018 | PDB 6MEI |
| HCV partial E1 structure 4UOI | El Omari et al., 2014 | PDB 4UOI |
| HCV partial E1 structure 2KNU | Spadaccini et al., 2010 | PDB 2KNU |
| HCV1 Model structure | Castelli et al., 2017a | N/A |
| E1E2 mutation library clones, with values for | Mendeley Data | 2 |
| Ab binding, infectivity values, Table S3 | ||
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK-293T | ATCC | Cat# CRL-3216 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCMV-dR8.74, lentiviral packaging plasmid | Naldini et al., 1996 | N/A |
| pNL-luc, encoding Renilla luciferase | Connor et al., 1995 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Structure Visualization Software | https://www.cgl.ucsf.edu/chimera | UCSF Chimera |
| GraphPad Prism Graphing software | GraphPad | v 9.0.0 |
|
| ||
| Other | ||
|
| ||
| Saponin | VWR | Cat# TCS0019–025G |
| DMEM | Fisher Scientific | Cat# MT10017CM |
| Sodium pyruvate | VWR | Cat# 45000–710 |
| L-Alanyl-L-Glutamine | VWR | Cat# 45001–086 |
| HEPES | VWR | Cat# 45000–690 |
| Penicillin-Streptomycin | Fisher Scientific | Cat# 15–140-163 |
| Paraformaldehyde | VWR | Cat# 100496–496 |
| Donor Goat Serum | Atlanta Biologicals | Cat# S13150 |
| DPBS(++) | Cytiva | Cat# SH30264.02 |
| Cellstripper | Corning | Cat# 25–056-CI |
| Goat Serum | R&D Systems | Cat# S13150 |
| PVDF membrane | VWR | Cat# 28165–512 |
| Precision plus Protein Dual Color Standards | BioRad | Cat# 61–0374 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK-293T cells were obtained from ATCC. Cells were maintained at 37°C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, penicillin-streptom ycin (100 U/ml), 10 mM HEPES pH7.4, 1 mM sodium pyruvate, and 2 mM L-alanyl-L-glutamine
METHOD DETAILS
Construction of HCV E1E2 mutation library
Comprehensive high-throughput alanine scanning mutagenesis was carried out on an expression plasmid for HCV E1E2 (genotype 1a, strain H77; reference sequence NC_004102), encoding a C-terminal V5 epitope tag, as previously described (Castelli et al., 2017b). All E1E2 residues were mutated individually to create a library of individual point mutants, each changing the parental sequence residue to alanine, with alanine residues changed to serine. Individual mutations were confirmed by DNA sequencing (Macrogen), and clones were arrayed into 384-well plates, one mutant per well. The resulting HCV E1E2 alaninescan library covered 98% of target residues (545/555) on E1E2. Ten residues were not mutated because they were not obtained over multiple mutagenesis attempts. The parental sequence used to construct this library (including the V5 tag) has been used in infectivity assays and epitope mapping of over 100 MAbs, including those with highly conformational epitopes that recognize only structurally intact E1E2, suggesting that this parental sequence accurately represents native E1E2 folding, conformation, and function.
Expression of the HCV E1E2 library for mapping
The HCV E1E2 mutation library, arrayed in 384-well microplates, was transfected into HEK-293T cells and allowed to express for 22 hours. Prior to MAb staining, cells were fixed in 4% paraformaldehyde (VWR) and permeabilized with 0.1% w/v saponin (VWR) in PBS plus calcium and magnesium. Cells were stained with MAbs (0.33 to 2.0 µg/mL) diluted in 10% normal goat serum (NGS, R&D Systems), 0.1% w/v saponin, pH 9.0. MAb screening concentrations were determined using an independent immunofluorescence titration curve against wild-type HCV E1E2 to ensure that signals were within the linear range of detection. The cells were incubated with anti-HCV antibody for 1 hour at 20°C, or overnight at 4°C, followed by a 30-minute incubation with Alexa Fluor 488-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Westgrove, PA) in 10% NGS. Cells were washed twice with PBS without calcium or magnesium and resuspended in Cellstripper (Corning) plus 0.1% BSA (Sigma-Aldrich, St. Louis, MO). Cellular fluorescence was detected using an Intellicyt high-throughput flow cytometer (Sartorius). Background fluorescence was determined by fluorescence measurement of vector-transfected control cells. MAb reactivities against each mutant HCV E1E2 clone were calculated relative to wild-type E1E2 reactivity by subtracting the signal from vector-transfected controls and normalizing to the signal from wild-type HCV E1E2 transfected controls.
HCV pseudovirus production
HCV mutant reporter pseudoviruses (HCVpp) were produced in 384-well format using the HCV library. Lentiviral reporter pseudotypes with HCV E1E2 were produced essentially as described previously (Fong et al., 2014) by cotransfecting each individual library mutant plasmid with plasmids encoding HIV-1 core (gag-pol, based on (Naldini et al., 1996)) and Renilla luciferase (pNL-luc, based on pNL4–3-R−E− (Connor et al., 1995)). Cells were incubated at 37°C in 5% CO2 to allow for transfection and pseudotype production. Supernatants were harvested 48 h after transfection, diluted 1:1 with DMEM containing 2X Dextran, and stored at −80°C.
HCV pseudovirus budding
The ability of each library E1E2 mutation to support HCVpp formation was determined by ELISA, using HCVpp produced in each well of the E1E2 library plates. ELISA plates were precoated with a mouse anti-human antibody against CD81 (JS81) to capture HCVpp particles that bud off from the plasma membrane (HEK-293 cells contain high levels of CD81 (Kalemera et al., 2021). The supernatants produced from each library mutant were thawed and 25 µL was added to the plates and incubated overnight for HCVpp capture on the antibody-coated surface. The plates were blocked with 3% BSA (Sigma-Aldrich, St. Louis, MO) for 15 min at room temperature. Detection MAb HCV1 (which recognizes a linear epitope on E2) was added to the plates at 2 µg/mL and allowed to incubate for 1 h at room temperature. The plates were washed three times with PBS−/−, and then horseradish peroxidase (HRP)-conjugated rabbit anti-human secondary antibody diluted at 1:5,000 was added for 30 min at room temperature. The plates were washed three times with PBS−/−, and reactivity was detected on a Perkin-Elmer Envision using SuperSignal West Pico chemiluminescent substrate (ThermoScientific, Waltham, MA).
HCV pseudovirus infectivity experiments
HCVpp produced from each mutant of the E1E2 library were used to infect HEK-293T cells, which were transfected with a plasmid expressing HCV receptor Claudin 1 and expressed for 22 h pre-infection. Pseudovirus was then added to cells and a spinoculation was performed at 2,000 rpm for 60 min at 20°C, followed by incubation at 37°C for 48 h. The supernatant was removed, cells were lysed with 25 µL of lysis buffer per well, and 20 µL of cell lysate was transferred to a white 384-well plate before adding 30 µL of Renilla luciferase substrate (Promega, Madison, WI). Luminescence was immediately detected on an Envision (PerkinElmer). Signal was measured and normalized to WT. Infectivity values represent the mean of 5 experiments.
Isolation of a conformational Anti-E1 MAb
To isolate E1-focused MAbs, an expression construct was made for the HCV (H77 strain) E1 ectodomain fused to the CD4 transmembrane region. Virus-like particles (VLPs) displaying HCV E1 proteins (‘Lipoparticles’) were produced by cotransfection of HEK-293T cells with the HCV E1 construct and the retroviral (MLV) Gag protein, as previously described (Hoffman et al., 2000; Willis et al., 2008). ‘Null’ VLPs were produced the same way but without transfection of E1. After immunizing chickens with HCV E1 construct DNA and boosting with E1-VLPs, an scFv phage display library was constructed from B cells from chickens that showed serum reactivity with E1 by flow cytometry. The phage library was panned against E1 VLPs (for positive selection) or null VLPs (for de-selection), as previously described (Tucker et al., 2018). Candidate scFv were converted to human IgG1-Fc format. Briefiy, the scFv region was PCR amplified and cloned using infusion cloning (Clontech) in-frame with a leader sequence and the Fc fragment of human IgG1 to create a scFv-Fc gene. scFv-Fc constructs were transfected into HEK-293T cells by calcium phosphate precipitation. Secreted scFv-Fc were purified from the culture media at 48 to 72 h post-transfection by protein A chromatography, followed by concentration and buffer exchange against PBS. Quantification of the purified scFv-Fc was performed using a bicinchoninic acid (BCA) assay (Thermo Scientific, Waltham, MA). Further analyses of the obtained clones identified MAb HCVE1-C1, which was characterized as a non-neutralizing MAb recognizing a conformational epitope on E1 (Figure S2).
Structural visualization
In the absence of any solved structure for the E1E2 heterodimer (and with only partial structures available for E1 and E2), a “composite” structure was constructed from published crystal structures and a predictive model of the HCV E1E2 heterodimer (Castelli et al., 2017b). To conceptualize how critical residues might be oriented in the E1E2 polyprotein structure, we used the MatchMaker tool in Chimera to align published crystal structures (PDB ID 6MEI) with a published predictive model of the polyprotein structure (Castelli et al., 2017b). In regions of overlap, the crystal structure was preferentially chosen. The final composite structure, used here to visualize critical residues identified independent of structural information, incorporates the crystal structures available for E1 (PDB ID 4UOI and 2KNU) and the most complete structure for E2 core (PDB ID 6MEI) (Flyak et al., 2018).
Sequence conservation of HCV E1E2 across HCV genotypes
Our E1E2 sequence (HCV1a isolate H77) was compared with E1E2 reference sequences for each genotype (International Committee on Taxonomy of Viruses) using ClustalW. Subtype/genotype, isolate/locus, Accession # (Reference): 1b, HPCJCG, D90208 (Kato et al., 1990); 2a, HPCPOLP, D00944 (Han and Houghton, 1992; Hotta et al., 1994; Okamoto et al., 1991); 3a, HPCEGS, D17763 (Sakamoto et al., 1994); 4a, ED43, Y11604 (Chamberlain et al., 1997a); 5a, EUH1480, Y13184 (Chamberlain et al., 1997b); 6a, EUHK2, Y12083 (Adams et al., 1997); 7a, QC69, EF108306 (Murphy et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in the appropriate figure legends. MAb reactivities for each alanine scan mutant are expressed as a percentage of wild-type reactivity, and represent the average of at least two replicate values for each measurement (Figure 2). The binding values for MAbs HEPC112 and HCV-E1C1 binding to specific mutations are also displayed graphically (Figure S2) and are plotted with error bars depicting half the range (highest minus lowest binding value) of at least two measurements. Similarly, the neutralization experiments in Figure S2 were performed with two replicates and values are shown with error bars showing the range of these values. Values from infectivity analyses (Figure 3) represent the average of 5 experiments.
Supplementary Material
Highlights.
Test 545 hepatis C virus (HCV) E1E2 envelope mutants for infectivity, antibody binding
Identify residues important for HCV E1 and E2 folding, E1E2 interaction, infectivity
HCV E1E2 is a fragile protein complex where most mutations compromise function
Functional residues of E1E2 are highly conserved across genotypes
ACKNOWLEDGMENTS
We thank Ginny Feltzin for help in writing this manuscript and Jazmean Williams for performing western blots. This work was supported by NIAID contract HHSN272201400058C to B.J.D. and R01AI127469 to J.R.B. and J.E.C., Jr. M.L. is partly supported by NIH grants AI123365 and AI123861.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110859.
DECLARATION OF INTERESTS
J.M.P.-K., E.D., K.K.-E., M.H., E.R., R.C., and B.J.D. are current or former employees of Integral Molecular. B.J.D. is a shareholder of Integral Molecular. J.E.C., Jr. has served as a consultant for Eli Lilly, GlaxoSmithKline, and Luna Biologics, is a member of the Scientific Advisory Boards of CompuVax and Meissa Vaccines, and is founder of IDBiologics. The J.E.C., Jr. laboratory at Vanderbilt University Medical Center has received sponsored research agreements from Takeda Vaccines, IDBiologics, and AstraZeneca.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Akbar and Jusoh (2013); Ball et al. (2014); Drummer et al. (2006); Flint et al. (1999); Keck et al. (2016); Kong et al. (2012); Mankowski et al. (2018).
REFERENCES
- Adams NJ, Chamberlain RW, Taylor LA, Davidson F, Lin CK, Elliott RM, and Simmonds P (1997). Complete coding sequence of hepatitis C virus genotype 6a. Biochem. Biophys. Res. Commun 234, 393–396. 10.1006/bbrc.1997.6627. [DOI] [PubMed] [Google Scholar]
- Akbar R, and Jusoh SA (2013). Stability, orientation and position preference of the stem region (residues 689–703) in Hepatitis C Virus (HCV) envelope glycoprotein E2: a molecular dynamics study. F1000Research 2, 64. 10.12688/f1000research.2-64.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhammad Y, Gu J, Boo I, Harrison D, McCaffrey K, Vietheer PT, Edwards S, Quinn C, Coulibaly F, Poumbourios P, and Drummer HE (2015). Monoclonal antibodies directed toward the hepatitis C virus glycoprotein E2 detect antigenic differences modulated by the N-terminal hypervariable region 1 (HVR1), HVR2, and intergenotypic variable region. J. Virol 89, 12245–12261. 10.1128/jvi.02070-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JR, Flyak AI, Cohen VJ, Li H, Wasilewski LN, Snider AE, Wang S, Learn GH, Kose N, Loerinc L, et al. (2017). Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI Insight 2, e92872. 10.1172/jci.insight.92872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball JK, Tarr AW, and McKeating JA (2014). The past, present and future of neutralizing antibodies for hepatitis C virus. Antivir. Res 105, 100–111. 10.1016/j.antiviral.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda DH, Perin PM, Brown RJP, Todt D, Solodenko W, Hoffmeyer P, Kumar Sahu K, Houghton M, Meuleman P, Muller R, et al. (2019). A central hydrophobic E1 region controls the pH range of hepatitis C virus membrane fusion and susceptibility to fusion inhibitors. J. Hepatol 70, 1082–1092. 10.1016/j.jhep.2019.01.033. [DOI] [PubMed] [Google Scholar]
- Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M, Herrmann E, Zeisel MB, Baumert TF, Keck ZY, Foung SKH, et al. (2010). Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol 84, 5751–5763. 10.1128/jvi.02200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankwitz D, Vieyres G, Hueging K, Bitzegeio J, Doepke M, Chhatwal P, Haid S, Catanese MT, Zeisel MB, Nicosia A, et al. (2014). Role of hypervariable region 1 for the interplay of hepatitis C virus with entry factors and lipoproteins. J. Virol 88, 12644–12655. 10.1128/jvi.01145-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, and Cosset FL (2003). Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem 278, 41624–41630. 10.1074/jbc.m305289200. [DOI] [PubMed] [Google Scholar]
- Benedicto I, Molina-Jimenez F, Bartosch B, Cosset FL, Lavillette D, Prieto J, Moreno-Otero R, Valenzuela-Fernandez A, Aldabe R, Lopez-Cabrera M, and Majano PL (2009). The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol 83, 8012–8020. 10.1128/jvi.00038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boo I, teWierik K, Douam F, Lavillette D, Poumbourios P, and Drummer HE (2012). Distinct roles in folding, CD81 receptor binding and viral entry for conserved histidine residues of hepatitis C virus glycoprotein E1 and E2. Biochem. J 443, 85–94. 10.1042/bj20110868. [DOI] [PubMed] [Google Scholar]
- Broering TJ, Garrity KA, Boatright NK, Sloan SE, Sandor F, Thomas WD Jr., Szabo G, Finberg RW, Ambrosino DM, and Babcock GJ (2009). Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J. Virol 83, 12473–12482. 10.1128/jvi.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugli F, Mancini N, Kang CY, Di Campli C, Grieco A, Manzin A, Gabrielli A, Gasbarrini A, Fadda G, Varaldo PE, et al. (2001). Mapping B-cell epitopes of hepatitis C virus E2 glycoprotein using human monoclonal antibodies from phage display libraries. J. Virol 75, 9986–9990. 10.1128/jvi.75.20.9986-9990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burioni R, Matsuura Y, Mancini N, Tani H, Miyamura T, Varaldo PE, and Clementi M (2002). Diverging effects of human recombinant anti-hepatitis C virus (HCV) antibody fragments derived from a single patient on the infectivity of a vesicular stomatitis virus/HCV pseudotype. J. Virol 76, 11775–11779. 10.1128/jvi.76.22.11775-11779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Yu B, Kong D, Cong Q, Yu T, Chen Z, Hu Z, Chang H, Zhong J, Baker D, and He Y (2019). Functional expression and characterization of the envelope glycoprotein E1E2 heterodimer of hepatitis C virus. PLoS Pathog 15, e1007759. 10.1371/journal.ppat.1007759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli M, Clementi N, Pfaff J, Sautto GA, Diotti RA, Burioni R, Doranz BJ, Dal Peraro M, Clementi M, and Mancini N (2017a). A biologically-validated HCV E1E2 heterodimer structural model. Sci. Rep 7, 214. 10.1038/s41598-017-00320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli M, Clementi N, Pfaff J, Sautto GA, Diotti RA, Burioni R, Doranz BJ, Dal Peraro M, Clementi M, and Mancini N (2017b). A biologically validated HCV E1E2 heterodimer structural model. Sci. Rep 7, 214. 10.1038/s41598-017-00320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli M, Clementi N, Sautto GA, Pfaff J, Kahle KM, Barnes T, Doranz BJ, Dal Peraro M, Clementi M, Burioni R, and Mancini N (2014). HCV E2 core structures and mAbs: something is still missing. Drug Discov. Today 19, 1964–1970. 10.1016/j.drudis.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain RW, Adams N, Saeed AA, Simmonds P, and Elliott RM (1997a). Complete nucleotide sequence of a type 4 hepatitis C virus variant, the predominant genotype in the Middle East. J. Gen. Virol 78, 1341–1347. 10.1099/0022-1317-78-6-1341. [DOI] [PubMed] [Google Scholar]
- Chamberlain RW, Adams NJ, Taylor LA, Simmonds P, and Elliott RM (1997b). The complete coding sequence of hepatitis C virus genotype 5a, the predominant genotype in South Africa. Biochem. Biophys. Res. Commun 236, 44–49. 10.1006/bbrc.1997.6902. [DOI] [PubMed] [Google Scholar]
- Chi X, Niu Y, Cheng M, Liu X, Feng Y, Zheng F, Fan J, Li X, Jin Q, Zhong J, et al. (2016). Identification of a potent and broad-spectrum hepatitis C virus fusion inhibitory peptide from the E2 stem domain. Sci. Rep 6, 25224. 10.1038/srep25224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian EA, Kahle KM, Mattia K, Puffer BA, Pfaff JM, Miller A, Paes C, Davidson E, and Doranz BJ (2013). Atomic-level functional model of dengue virus Envelope protein infectivity. Proc. Natl. Acad. Sci. U S A 110, 18662–18667. 10.1073/pnas.1310962110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciczora Y, Callens N, Montpellier C, Bartosch B, Cosset FL, De Beeck AO, and Dubuisson J (2005). Contribution of the charged residues of hepatitis C virus glycoprotein E2 transmembrane domain to the functions of the E1E2 heterodimer. J. Gen. Virol 86, 2793–2798. 10.1099/vir.0.81140-0. [DOI] [PubMed] [Google Scholar]
- Ciczora Y, Callens N, Penin F, Pecheur EI, and Dubuisson J (2007). Transmembrane domains of hepatitis C virus envelope glycoproteins: residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J. Virol 81, 2372–2381. 10.1128/jvi.02198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L, Wychowski C, Minner F, Penin F, and Dubuisson J (2000). Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J. Virol 74, 3623–3633. 10.1128/jvi.74.8.3623-3633.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert MD, Flyak AI, Ogega CO, Kinchen VJ, Massaccesi G, Hernandez M, Davidson E, Doranz BJ, Cox AL, Crowe JE Jr., and Bailey JR (2019). Broadly neutralizing antibodies targeting new sites of vulnerability in hepatitis C virus E1E2. J. Virol 93, e02070–e02118. 10.1128/jvi.02070-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, and Landau NR (1995). Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206, 935–944. 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Davidson E, and Doranz BJ (2014). A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 143, 13–20. 10.1111/imm.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douam F, Dao Thi VL, Maurin G, Fresquet J, Mompelat D, Zeisel MB, Baumert TF, Cosset FL, and Lavillette D (2014). Critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 59, 776–788. 10.1002/hep.26733. [DOI] [PubMed] [Google Scholar]
- Douam F, Fusil F, Enguehard M, Dib L, Nadalin F, Schwaller L, Hrebikova G, Mancip J, Mailly L, Montserret R, et al. (2018). A protein coevolution method uncovers critical features of the Hepatitis C Virus fusion mechanism. PLoS Pathog 14, e1006908. 10.1371/journal.ppat.1006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummer HE (2014a). Antibodies to the high-density lipoprotein receptor SR-B1 potently inhibit hepatitis C virus replication in vivo: new avenues for preventing reinfection of the liver following transplantation. Hepatology 60, 1463–1465. 10.1002/hep.27276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummer HE (2014b). Challenges to the development of vaccines to hepatitis C virus that elicit neutralizing antibodies. Front. Microbiol 5, 329. 10.3389/fmicb.2014.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummer HE, Boo I, Maerz AL, and Poumbourios P (2006). A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J. Virol 80, 7844–7853. 10.1128/jvi.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummer HE, Boo I, and Poumbourios P (2007). Mutagenesis of a conserved fusion peptide-like motif and membrane-proximal heptad-repeat region of hepatitis C virus glycoprotein E1. J. Gen. Virol 88, 1144–1148. 10.1099/vir.0.82567-0. [DOI] [PubMed] [Google Scholar]
- Drummer HE, and Poumbourios P (2004). Hepatitis C virus glycoprotein E2 contains a membrane-proximal heptad repeat sequence that is essential for E1E2 glycoprotein heterodimerization and viral entry. J. Biol. Chem 279, 30066–30072. 10.1074/jbc.m405098200. [DOI] [PubMed] [Google Scholar]
- El Omari K, Iourin O, Kadlec J, Sutton G, Harlos K, Grimes JM, and Stuart DI (2014). Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat. Commun 5, 4874. 10.2210/pdb4uoi/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falson P, Bartosch B, Alsaleh K, Tews BA, Loquet A, Ciczora Y, Riva L, Montigny C, Montpellier C, Duverlie G, et al. (2015). Hepatitis C virus envelope glycoprotein E1 forms trimers at the surface of the virion. J. Virol 89, 10333–10346. 10.1128/jvi.00991-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint M, Thomas JM, Maidens CM, Shotton C, Levy S, Barclay WS, and McKeating JA (1999). Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J. Virol 73, 6782–6790. 10.1128/jvi.73.8.6782-6790.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Ruiz S, Colbert MD, Luong T, Crowe JE Jr., Bailey JR, and Bjorkman PJ (2018). HCV broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe 24, 703–716.e3. 10.1016/j.chom.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong RH, Banik SSR, Mattia K, Barnes T, Tucker D, Liss N, Lu K, Selvarajah S, Srinivasan S, Mabila M, et al. (2014). Exposure of epitope residues on the outer face of the chikungunya virus envelope trimer determines antibody neutralizing efficacy. J. Virol 88, 14364–14379. 10.1128/jvi.01943-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman H, Logan MR, Hockman D, Koehler Leman J, Law JLM, and Houghton M (2017). Computational prediction of the heterodimeric and higher-order structure of gpE1/gpE2 envelope glycoproteins encoded by hepatitis C virus. J. Virol 91, e02309–e02316. 10.1128/jvi.02309-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst TR, Pierce BG, Keck ZY, and Foung SKH (2017). Designing a B Cell-Based vaccine against a highly variable hepatitis C virus. Front. Microbiol 8, 2692. 10.3389/fmicb.2017.02692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerold G, Moeller R, and Pietschmann T (2020). Hepatitis C virus entry: protein interactions and fusion determinants governing Productive hepatocyte invasion. Cold Spring Harb. Perspect. Med 10, a036830. 10.1101/cshperspect.a036830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, and Law M (2012). Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. U S A 109, 6205–6210. 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal R, Jackson K, Tzarum N, Kong L, Ettenger A, Guest J, Pfaff JM, Barnes T, Honda A, Giang E, et al. (2017). Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog 13, e1006735. 10.1371/journal.ppat.1006735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene TA, Alarcon S, Thomas A, Berdougo E, Doranz BJ, Breslin PAS, and Rucker JB (2011). Probenecid inhibits the human bitter taste receptor TAS2R16 and suppresses bitter perception of salicin. PLoS One 6, e20123. 10.1371/journal.pone.0020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunesch AP, Zapatero-Belinchon FJ, Pinkert L, Steinmann E, Manns MP, Schneider G, Pietschmann T, Bronstrup M, and von Hahn T (2020). Filovirus antiviral activity of cationic amphiphilic drugs is associated with lipophilicity and ability to induce Phospholipidosis. Antimicrob. Agents Chemother 64, e00143–e00220. 10.1128/aac.00143-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Yu H, Zhong Y, He Y, Qin X, Qin Y, Zhou Y, Zhang P, Zhang Y, Li Z, and Jia Z (2018). Lectin microarray and mass spectrometric analysis of hepatitis C proteins reveals N-linked glycosylation. Medicine (Baltimore) 97, e0208. 10.1097/md.0000000000010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberstroh A, Schnober EK, Zeisel MB, Carolla P, Barth H, Blum HE, Cosset FL, Koutsoudakis G, Bartenschlager R, Union A, et al. (2008). Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 135, 1719–1728.e1. 10.1053/j.gastro.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Han JH, and Houghton M (1992). Group specific sequences and conserved secondary structures at the 3’ end of HCV genome and its implication for viral replication. Nucleic Acids Res 20, 3520. 10.1093/nar/20.13.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman TL, Canziani G, Jia L, Rucker J, and Doms RW (2000). A biosensor assay for studying ligand-membrane receptor interactions: binding of antibodies and HIV-1 Env to chemokine receptors. Proc. Natl. Acad. Sci. U S A 97, 11215–11220. 10.1073/pnas.190274097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta H, Doi H, Hayashi T, Purwanta M, Soemarto W, Mizokami M, Ohba K, and Homma M (1994). Analysis of the core and E1 envelope region sequences of a novel variant of hepatitis C virus obtained in Indonesia. Arch. Virol 136, 53–62. 10.1007/bf01538816. [DOI] [PubMed] [Google Scholar]
- Hsieh SC, Zou G, Tsai WY, Qing M, Chang GJ, Shi PY, and Wang WK (2011). The C-terminal helical domain of dengue virus precursor membrane protein is involved in virus assembly and entry. Virology 410, 170–180. 10.1016/j.virol.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, and McKeating JA (2003). Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U S A 100, 7271–7276. 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusoh SA, Welsch C, Siu SWI, Bockmann RA, and Helms V (2010). Contribution of charged and polar residues for the formation of the E1-E2 heterodimer from Hepatitis C Virus. J. Mol. Model 16, 1625–1637. 10.1007/s00894-010-0672-1. [DOI] [PubMed] [Google Scholar]
- Kalemera MD, Capella-Pujol J, Chumbe A, Underwood A, Bull RA, Schinkel J, Sliepen K, and Grove J (2021). Optimized cell systems for the investigation of hepatitis C virus E1E2 glycoproteins. J. Gen. Virol 102, jgv001512. 10.1099/jgv.0.001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, and Shimotohno K (1990). A structural protein encoded by the 5’ region of the hepatitis C virus genome efficiently detects viral infection. Jpn. J. Cancer Res 81, 1092–1094. 10.1111/j.1349-7006.1990.tb02518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck ZY, Girard-Blanc C, Wang W, Lau P, Zuiani A, Rey FA, Krey T, Diamond MS, and Foung SKH (2016). Antibody response to hypervariable region 1 interferes with broadly neutralizing antibodies to hepatitis C virus. J. Virol 90, 3112–3122. 10.1128/jvi.02458-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck ZY, Pierce BG, Lau P, Lu J, Wang Y, Underwood A, Bull RA, Prentoe J, Velazquez-Moctezuma R, Walker MR, et al. (2019). Broadly neutralizing antibodies from an individual that naturally cleared multiple hepatitis C virus infections uncover molecular determinants for E2 targeting and vaccine design. PLoS Pathog 15, e1007772. 10.1371/journal.ppat.1007772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck ZY, Sung VMH, Perkins S, Rowe J, Paul S, Liang TJ, Lai MMC, and Foung SKH (2004). Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J. Virol 78, 7257–7263. 10.1128/jvi.78.13.7257-7263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AG, Whidby J, Marcotrigiano J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, et al. (2014). Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509, 381–384. 10.2210/pdb4nx3/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, et al. (2013). Hepatitis C virus E2 envelope glycoprotein core structure. Science 342, 1090–1094. 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Giang E, Robbins JB, Stanfield RL, Burton DR, Wilson IA, and Law M (2012). Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. U S A 109, 9499–9504. 10.1073/pnas.1202924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Kadam RU, Giang E, Ruwona TB, Nieusma T, Culhane JC, Stanfield RL, Dawson PE, Wilson IA, and Law M (2015). Structure of hepatitis C virus envelope glycoprotein E1 antigenic site 314–324 in complex with antibody IGH526. J Mol. Biol. 427, 2617–2628. 10.1016/j.jmb.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Hossain RA, Yost SA, Bu W, Wang Y, Dearborn AD, Grakoui A, Cohen JI, and Marcotrigiano J (2021). Structural insights into hepatitis C virus receptor binding and entry. Nature 598, 521–525. 10.1038/s41586-021-03913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillette D, Pecheur EI, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, and Cosset FL (2007). Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol 81, 8752–8765. 10.1128/jvi.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, et al. (2008). Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med 14, 25–27. 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- Li HF, Huang CH, Ai LS, Chuang CK, and Chen SS (2009). Mutagenesis of the fusion peptide-like domain of hepatitis C virus E1 glycoprotein: involvement in cell fusion and virus entry. J. Biomed. Sci 16, 89. 10.1186/1423-0127-16-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, and Modis Y (2014). A novel membrane fusion protein family in Flaviviridae? Trends Microbiol 22, 176–182. 10.1016/j.tim.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SR, Zou G, Hsieh SC, Qing M, Tsai WY, Shi PY, and Wang WK (2011). The helical domains of the stem region of dengue virus envelope protein are involved in both virus assembly and entry. J. Virol 85, 5159–5171. 10.1128/JVI.02099-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HC, Ke CH, Hsieh TY, and Lo SY (2002). The first hydrophobic domain of the hepatitis C virus E1 protein is important for interaction with the capsid protein. J. Gen. Virol 83, 3085–3092. 10.1099/0022-1317-83-12-3085. [DOI] [PubMed] [Google Scholar]
- Mair CM, Meyer T, Schneider K, Huang Q, Veit M, and Herrmann A (2014). A histidine residue of the influenza virus hemagglutinin controls the pH dependence of the conformational change mediating membrane fusion. J. Virol 88, 13189–13200. 10.1128/jvi.01704-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini N, Diotti RA, Perotti M, Sautto G, Clementi N, Nitti G, Patel AH, Ball JK, Clementi M, and Burioni R (2009). Hepatitis C virus (HCV) infection may elicit neutralizing antibodies targeting epitopes conserved in all viral genotypes. PLoS One 4, e8254. 10.1371/journal.pone.0008254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankowski MC, Kinchen VJ, Wasilewski LN, Flyak AI, Ray SC, Crowe JE Jr., and Bailey JR (2018). Synergistic anti-HCV broadly neutralizing human monoclonal antibodies with independent mechanisms. Proc. Natl. Acad. Sci. U S A 115, E82–E91. 10.1073/pnas.1718441115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell M, Esteban JI, Quer J, Genesca J, Weiner A, Esteban R, Guardia J, and Gomez J (1992). Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol 66, 3225–3229. 10.1128/jvi.66.5.3225-3229.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey K, Boo I, Tewierek K, Edmunds ML, Poumbourios P, and Drummer HE (2012). Role of conserved cysteine residues in hepatitis C virus glycoprotein e2 folding and function. J. Virol 86, 3961–3974. 10.1128/jvi.05396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey K, Gouklani H, Boo I, Poumbourios P, and Drummer HE (2011). The variable regions of hepatitis C virus glycoprotein E2 have an essential structural role in glycoprotein assembly and virion infectivity. J. Gen. Virol 92, 112–121. 10.1099/vir.0.026385-0. [DOI] [PubMed] [Google Scholar]
- Meunier JC, Russell RS, Goossens V, Priem S, Walter H, Depla E, Union A, Faulk KN, Bukh J, Emerson SU, and Purcell RH (2008). Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J. Virol 82, 966–973. 10.1128/jvi.01872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa RI, Haddad JG, Linna L, Hanoulle X, Descamps V, Mesalam AA, Baumert TF, Duverlie G, Meuleman P, Dubuisson J, and Lavie M (2018). Functional study of the C-terminal part of the hepatitis C virus E1 ectodomain. J. Virol 92, e00939–e01018. 10.1128/jvi.00939-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DG, Sablon E, Chamberland J, Fournier E, Dandavino R, and Tremblay CL (2015). Hepatitis C virus genotype 7, a new genotype originating from central Africa. J. Clin. Microbiol 53, 967–972. 10.1128/jcm.02831-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, and Trono D (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267. 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Okada S, Sugiyama Y, Kurai K, Iizuka H, Machida A, Miyakawa Y, and Mayumi M (1991). Nucleotide sequence of the genomic RNA of hepatitis C virus isolated from a human carrier: comparison with reported isolates for conserved and divergent regions. J. Gen. Virol 72 (Pt 11), 2697–2704. 10.1099/0022-1317-72-11-2697. [DOI] [PubMed] [Google Scholar]
- Owsianka A, Tarr AW, Juttla VS, Lavillette D, Bartosch B, Cosset FL, Ball JK, and Patel AH (2005). Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Virol 79, 11095–11104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owsianka AM, Timms JM, Tarr AW, Brown RJP, Hickling TP, Szwejk A, Bienkowska-Szewczyk K, Thomson BJ, Patel AH, and Ball JK (2006). Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol 80, 8695–8704. 10.1128/jvi.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paes C, Ingalls J, Kampani K, Sulli C, Kakkar E, Murray M, Kotelnikov V, Greene TA, Rucker JB, and Doranz BJ (2009). Atomic-level mapping of antibody epitopes on a GPCR. J. Am. Chem. Soc 131, 6952–6954. 10.1021/ja900186n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J, Patel AH, and McLauchlan J (2001). The transmembrane domain of the hepatitis C virus E2 glycoprotein is required for correct folding of the E1 glycoprotein and native complex formation. Virology 279, 58–68. 10.1006/viro.2000.0693. [DOI] [PubMed] [Google Scholar]
- Penin F, Combet C, Germanidis G, Frainais PO, Deleage G, and Pawlotsky JM (2001). Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol 75, 5703–5710. 10.1128/jvi.75.12.5703-5710.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin PM, Haid S, Brown RJ, Doerrbecker J, Schulze K, Zeilinger C, von Schaewen M, Heller B, Vercauteren K, Luxenburger E, et al. (2016). Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63, 49–62. 10.1002/hep.28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perotti M, Mancini N, Diotti RA, Tarr AW, Ball JK, Owsianka A, Adair R, Patel AH, Clementi M, and Burioni R (2008). Identification of a broadly cross-reacting and neutralizing human monoclonal antibody directed against the hepatitis C virus E2 protein. J. Virol 82, 1047–1052. 10.1128/jvi.01986-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BG, Keck ZY, Lau P, Fauvelle C, Gowthaman R, Baumert TF, Fuerst TR, Mariuzza RA, and Foung SKH (2016). Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: implications for vaccine design. Proc. Natl. Acad. Sci. U S A 113, E6946–E6954. 10.1073/pnas.1614942113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, and Rice CM (2009). Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886. 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powdrill MH, Tchesnokov EP, Kozak RA, Russell RS, Martin R, Svarovskaia ES, Mo H, Kouyos RD, and Gotte M (2011). Contribution of a mutational bias in hepatitis C virus replication to the genetic barrier in the development of drug resistance. Proc. Natl. Acad. Sci. U S A 108, 20509–20513. 10.1073/pnas.1105797108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo MC, Luca VC, Prentoe J, Hopcraft SE, Blight KJ, Yi M, Lemon SM, Ball JK, Bukh J, Evans MJ, et al. (2011). Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J. Virol 85, 7005–7019. 10.1128/jvi.00586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto M, Akahane Y, Tsuda F, Tanaka T, Woodfield DG, and Okamoto H (1994). Entire nucleotide sequence and characterization of a hepatitis C virus of genotype V/3a. J. Gen. Virol 75, 1761–1768. 10.1099/0022-1317-75-7-1761. [DOI] [PubMed] [Google Scholar]
- Sánchez-San Martín C, Liu CY, and Kielian M (2009). Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol 17, 514–521. 10.1016/j.tim.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R, Moya A, and Elena SF (2004). The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. U S A 101, 8396–8401. 10.1073/pnas.0400146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautto G, Mancini N, Diotti RA, Solforosi L, Clementi M, and Burioni R (2012). Anti-hepatitis C virus E2 (HCV/E2) glycoprotein monoclonal antibodies and neutralization interference. Antivir. Res 96, 82–89. 10.1016/j.antiviral.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Sautto G, Tarr AW, Mancini N, and Clementi M (2013). Structural and antigenic definition of hepatitis C virus E2 glycoprotein epitopes targeted by monoclonal antibodies. Clin. Dev. Immunol 2013, 1–12. 10.1155/2013/450963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadaccini R, D’Errico G, D’Alessio V, Notomista E, Bianchi A, Merola M, and Picone D (2010). Structural characterization of the transmembrane proximal region of the hepatitis C virus E1 glycoprotein. Biochim. Biophys. Acta 1798, 344–353. 10.1016/j.bbamem.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Sullivan JT, Sulli C, Nilo A, Yasmeen A, Ozorowski G, Sanders RW, Ward AB, Klasse PJ, Moore JP, and Doranz BJ (2017). High-throughput protein engineering improves the antigenicity and stability of soluble HIV-1 envelope glycoprotein SOSIP trimers. J. Virol 91, e00862–e00917. 10.1128/jvi.00862-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Sulli C, Davidson E, Berdougo E, Phillips M, Puffer BA, Paes C, Doranz BJ, and Rucker JB (2017). The bitter taste receptor TAS2R16 achieves high specificity and accommodates diverse glycoside ligands by using a two-faced binding pocket. Sci. Rep 7, 7753. 10.1038/s41598-017-07256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Chi X, Yang W, and Zhong J (2017). Functional analysis of hepatitis C virus (HCV) envelope protein E1 using a trans-complementation system reveals a dual role of a putative fusion peptide of E1 in both HCV entry and morphogenesis. J. Virol 91, e02468–e02516. 10.1128/jvi.02468-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrents de la Peña A, Sliepen K, Eshun-Wilson L, Newby M, Allen JD, Koekkoek S, Zon I, Chumbe A, Crispin M, Schinkel J, et al. (2021). Structure of the Hepatitis C Virus E1E2 Glycoprotein Complex. Preprint at BioRxiv 10.1101/2021.12.16.472992. [DOI] [PMC free article] [PubMed]
- Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, and Rice CM (2006). Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol 80, 1734–1741. 10.1128/jvi.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker DF, Sullivan JT, Mattia KA, Fisher CR, Barnes T, Mabila MN, Wilf R, Sulli C, Pitts M, Payne RJ, et al. (2018). Isolation of state-dependent monoclonal antibodies against the 12-transmembrane domain glucose transporter 4 using virus-like particles. Proc. Natl. Acad. Sci. U S A 115, E4990–E4999. 10.1073/pnas.1716788115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanowicz A, Zagozdzon R, and Ciszek M (2019a). Modulation of the immune system in chronic hepatitis C and during antiviral interferon-free therapy. Arch. Immunol. Ther. Exp. (Warsz) 67, 79–88. 10.1007/s00005-018-0532-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanowicz RA, Wang R, Schiel JE, Keck ZY, Kerzic MC, Lau P, Rangarajan S, Garagusi KJ, Tan L, Guest JD, et al. (2019b). Antigenicity and immunogenicity of differentially glycosylated hepatitis C virus E2 envelope proteins expressed in mammalian and insect cells. J. Virol 93, 01403–e01418. 10.1128/jvi.01403-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez-Moctezuma R, Law M, Bukh J, and Prentoe J (2017). Applying antibody-sensitive hypervariable region 1-deleted hepatitis C virus to the study of escape pathways of neutralizing human monoclonal antibody AR5A. PLoS Pathog 13, e1006214. 10.1371/journal.ppat.1006214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahid A, and Dubuisson J (2013). Virus-neutralizing antibodies to hepatitis C virus. J. Viral Hepat 20, 369–376. 10.1111/jvh.12094. [DOI] [PubMed] [Google Scholar]
- Wang Y, Keck ZY, and Foung SKH (2011). Neutralizing antibody response to hepatitis C virus. Viruses 3, 2127–2145. 10.3390/v3112127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wescott MP, Kufareva I, Paes C, Goodman JR, Thaker Y, Puffer BA, Berdougo E, Rucker JB, Handel TM, and Doranz BJ (2016). Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc. Natl. Acad. Sci. U S A 113, 9928–9933. 10.1073/pnas.1601278113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis S, Davidoff C, Schilling J, Wanless A, Doranz BJ, and Rucker J (2008). Virus-like particles as quantitative probes of membrane protein interactions. Biochemistry 47, 6988–6990. 10.1021/bi800540b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used for the analyses described here, antibody and CD81-LEL binding and HCVpp infectivity, for each mutation in the HCV E1E2 library, are included in Table S3 and have been deposited in the Mendeley Data repository. DOI is listed in the Key resources table.
This paper also analyzes existing, publicly available protein structural data. The accession numbers for these datasets are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| AP33 | Owsianka et al., 2005 | N/A |
| AR2A | Law et al., 2008 | N/A |
| AR3A | Law et al., 2008 | N/A |
| AR3B | Law et al., 2008 | N/A |
| AR3C | Law et al., 2008 | N/A |
| AR3D | Law et al., 2008 | N/A |
| AR4A | Giang et al., 2012 | N/A |
| AR5A | Giang et al., 2012 | N/A |
| e20 | Mancini et al., 2009 | N/A |
| e137 | Perotti et al., 2008 | N/A |
| H77.39 | Sabo et al., 2011 | N/A |
| HCV1 | Broering et al., 2009 | N/A |
| HEPC3 | Bailey et al., 2017 | N/A |
| HEPC13 | Bailey et al., 2017 | N/A |
| HEPC43 | Bailey et al., 2017 | N/A |
| HEPC74 | Bailey et al., 2017 | N/A |
| IGH505 | Meunier et al., 2008 | N/A |
| IGH526 | Meunier et al., 2008 | N/A |
| e301 | Burioni et al., 2002 | N/A |
| e509 | Sautto et al., 2012 | N/A |
| H77.16 | Sabo et al., 2011 | N/A |
| H77.28 | Sabo et al., 2011 | N/A |
| HEPC98 | Bailey et al., 2017 | N/A |
| HEPC108 | Colbert et al., 2019 | N/A |
| HEPC111 | Colbert et al., 2019 | N/A |
| HEPC112 | Colbert et al., 2019 | N/A |
| HEPC122 | Colbert et al., 2019 | N/A |
| HEPC146 | Colbert et al., 2019 | N/A |
| HEPC 151–1 | Colbert et al., 2019 | N/A |
| HEPC 151–2 | Colbert et al., 2019 | N/A |
| HEPC153 | Colbert et al., 2019 | N/A |
| HEPC154 | Colbert et al., 2019 | N/A |
| HEPC158 | Colbert et al., 2019 | N/A |
| AR1A | Law et al., 2008 | N/A |
| AR1B | Law et al., 2008 | N/A |
| e8 | Castelli et al., 2014 | N/A |
| e10 | Bugli et al., 2001 | N/A |
| H60 | Castelli et al., 2014 | N/A |
| H77.47 | Sabo et al., 2011 | N/A |
| HEPC50 | Bailey et al., 2017 | N/A |
| HEPC46 | Bailey et al., 2017 | N/A |
| S1 | Giang et al., 2012 | N/A |
| AB | Giang et al., 2012 | N/A |
| HCVE1-C1 | this manuscript | N/A |
| CD81-LEL fused to immunoglobulin Fc fragment | Gopal et al., 2017 | N/A |
| HRP conjugated secondary Ab | Southern Biotech | Cat# 040–05 |
| Anti CD81 antibody JS-81 | BD Biosciences | Cat# 555676 |
| Alexa Fluor 488-conjugated goat anti-human secondary antibody | Jackson ImmunoResearch Laboratories | Cat# 109–545-003 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E.coli NEB 5-alpha | New England Biolabs | Cat# C987H |
|
| ||
| Critical commercial assays | ||
|
| ||
| Intellicyt high throughput flow cytometer | Sartorius | iQue3 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific | Cat# 34580 |
| Renilla-Glo Luciferase assay | Promega | Cat# E2820 |
| Pierce Femto kit | Pierce | Cat# 34095 |
| Imaging System | Bio-Rad | ChemiDoc |
|
| ||
| Deposited data | ||
|
| ||
| HCV partial E2 structure 6MEI | Flyaketal., 2018 | PDB 6MEI |
| HCV partial E1 structure 4UOI | El Omari et al., 2014 | PDB 4UOI |
| HCV partial E1 structure 2KNU | Spadaccini et al., 2010 | PDB 2KNU |
| HCV1 Model structure | Castelli et al., 2017a | N/A |
| E1E2 mutation library clones, with values for | Mendeley Data | 2 |
| Ab binding, infectivity values, Table S3 | ||
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK-293T | ATCC | Cat# CRL-3216 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCMV-dR8.74, lentiviral packaging plasmid | Naldini et al., 1996 | N/A |
| pNL-luc, encoding Renilla luciferase | Connor et al., 1995 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Structure Visualization Software | https://www.cgl.ucsf.edu/chimera | UCSF Chimera |
| GraphPad Prism Graphing software | GraphPad | v 9.0.0 |
|
| ||
| Other | ||
|
| ||
| Saponin | VWR | Cat# TCS0019–025G |
| DMEM | Fisher Scientific | Cat# MT10017CM |
| Sodium pyruvate | VWR | Cat# 45000–710 |
| L-Alanyl-L-Glutamine | VWR | Cat# 45001–086 |
| HEPES | VWR | Cat# 45000–690 |
| Penicillin-Streptomycin | Fisher Scientific | Cat# 15–140-163 |
| Paraformaldehyde | VWR | Cat# 100496–496 |
| Donor Goat Serum | Atlanta Biologicals | Cat# S13150 |
| DPBS(++) | Cytiva | Cat# SH30264.02 |
| Cellstripper | Corning | Cat# 25–056-CI |
| Goat Serum | R&D Systems | Cat# S13150 |
| PVDF membrane | VWR | Cat# 28165–512 |
| Precision plus Protein Dual Color Standards | BioRad | Cat# 61–0374 |